 Mariame Coulibaly
Mariame Coulibaly Nashaat M. Mazrui1,3
Nashaat M. Mazrui1,3 Sofi Jonsson
Sofi Jonsson Robert P. Mason
Robert P. Mason
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Environ. Chem. , 14 June 2021
Sec. Inorganic Pollutants
Volume 2 - 2021 | https://doi.org/10.3389/fenvc.2021.660058
This article is part of the Research Topic Environmental Chemistry of Mercury: Sources, Pathways, Transformations and Impact View all 12 articles
Monomethylmercury (CH3Hg) is a neurotoxic pollutant that biomagnifies in aquatic food webs. In sediments, the production of CH3Hg depends on the bacterial activity of mercury (Hg) methylating bacteria and the amount of bioavailable inorganic divalent mercury (HgII). Biotic and abiotic reduction of HgII to elemental mercury (Hg0) may limit the pool of HgII available for methylation in sediments, and thus the amount of CH3Hg produced. Knowledge about the transformation of HgII is therefore primordial to the understanding of the production of toxic and bioaccumulative CH3Hg. Here, we examined the reduction of HgII by sulfidic minerals (FeS(s) and CdS(s)) in the presence of dissolved iron and dissolved organic matter (DOM) using low, environmentally relevant concentrations of Hg and ratio of HgII:FeS(s). Our results show that the reduction of HgII by Mackinawite (FeS(s)) was lower (<15% of the HgII was reduced after 24 h) than when HgII was reacted with DOM or dissolved iron. We did not observe any formation of Hg0 when HgII was reacted with CdS(s) (experiments done under both acidic and basic conditions for up to four days). While reactions in solution were favorable under the experimental conditions, Hg was rapidly removed from solution by co-precipitation. Thermodynamic calculations suggest that in the presence of FeS(s), reduction of the precipitated HgII is surface catalyzed and likely involves S−II as the electron donor. The lack of reaction with CdS may be due to its stronger M-S bond relative to FeS, and the lower concentrations of sulfide in solution. We conclude that the reaction of Hg with FeS(s) proceeds via a different mechanism from that of Hg with DOM or dissolved iron, and that it is not a major environmental pathway for the formation of Hg0 in anoxic environments.
Mercury (Hg) is considered as a global and high-priority pollutant (Clarkson and Magos, 2006; Mergler et al., 2007). While it is released as elemental or divalent Hg (Hg0 and HgII) from natural and anthropogenic sources (Driscoll et al., 2013), the main concern lies with the accumulation of Hg as monomethylmercury (CH3Hg) in aquatic food webs (Eagles-Smith et al., 2018; Sunderland et al., 2018). Production of CH3Hg in aquatic systems from HgII is facilitated by microorganisms carrying the Hg-methylation genes (HgcA and HgcB-genes) primarily in anoxic environments, such as in sediments, soils or on resuspended particles (Parks et al., 2013; Podar et al., 2015). The production of CH3Hg is controlled by the composition of the bacterial community, bacterial activity and the availability of HgII for bacterial uptake (Benoit et al., 2003; Fitzgerald et al., 2007; Compeau and Bartha, 1985; Gilmour et al., 1992). In environments where Hg methylation rates are typically high, the amount of HgII available to Hg methylating bacteria is controlled by competition between adsorption of Hg to the solid phase, the chemical speciation in the dissolved phase as well as removal processes, such as reduction of HgII to volatile elemental Hg (Hg0).
Under anoxic conditions, Hg can be reduced to Hg0via biotic and abiotic processes (Spangler et al., 1973; Steffan et al., 1988). Abiotic processes include photoreduction (Garcia et al., 2005; O'Driscoll et al., 2006; Whalin et al., 2007), which is likely limited in anoxic environments, and chemical reduction of HgII in the presence of organic matter (Baohua et al., 2011; Zheng et al., 2012; Chakraborty et al., 2015; Jiang et al., 2015) or mineral-associated ferrous iron (Charlet et al., 2002; Jeong et al., 2010; Remy et al., 2015; Richard et al., 2016; O’Loughlin et al., 2003). For the latter pathway, several iron-containing minerals have been suggested to reduce Hg, including hydrous ferric oxide (Richard et al., 2016), siderite (Ha et al., 2017) and clay (Peretyazhko et al., 2006a). Recently, reduction of Hg on iron sulfide mineral surfaces was also suggested (Bone et al., 2014), although the mechanism was not completely determined. In anoxic environments, the competition between inorganic sulfide phases and organic matter likely control the bioavailability of Hg as both complex Hg strongly and likely influence important reactions such as HgII reduction (Skyllberg and Drott, 2010).
The affinity of HgII for mineral surfaces, especially sulfide containing minerals, has been well documented (Jeong et al., 2008; Jeong et al., 2010; Skyllberg and Drott, 2010). Studies examining the sorption to mackinawite showed that Hg can replace iron in the mineral, forming black meta-cinnabar (β-HgS(s)) and red cinnabar (α-HgS(s))-like structures, and this was the primary reaction. Both the sorption and co-precipitation of Hg with FeS(s) has been shown to influence its methylation by bacteria (Rivera et al., 2019). Whether HgII can also be reduced on interaction with iron sulfide minerals remains less clear but has been speculated to occur in anoxic contaminated sediments (Han et al., 2020).
Most researchers who also investigated the reaction between HgII and FeS(s), did not detect Hg0 (Liu et al., 2008; Jeong et al., 2010; Skyllberg and Drott, 2010). However, cinnabar and Hg0 were formed when HgII interacted with pyrite and troilite (Bower et al., 2008). Only one work so far has reported the reduction of mercury by FeS(s) (Bone et al., 2014). This work suggested that Hg0 was generated from the reduction of HgII-S-II species in the presence of FeS(s), but that adsorption of Hg to the solid was not necessary for the reaction, suggesting a reaction involving Hg complexes in solution. Thermodynamically, whether the reaction occurs in solution or at the mineral surface is likely controlled by solution chemistry and the Hg concentration. The relative importance also likely depends on the fractionation of Hg between the dissolved and solid phases, which depends on its concentration, pH and sulfide concentration (Supporting Information (SI), Supplementary Tables S1, S2). Combining the precipitation reaction with that of a major dissolved Hg species in solution under sulfidic conditions results in the overall reaction shown below for Hg co-precipitation:
where the solid is either from solution saturation or from co-precipitation:
One important difference in the studies to date, as noted by Bone et al. (2014), is the difference in the HgII:FeS(s) ratio. In many studies this is higher than the molar ratio found in the environment, which ranges from 3 × 10−3 to ∼10−7 for regionally contaminated and uncontaminated locations. The studies of Bone et al. used a range from 0.4 to 20 × 10−3, which is at the high end of the environmental range, but lower than the ratios of Jeong et al. (2010), for example, (>10−2). We therefore proposed to do our follow-up studies at more environmentally-relevant concentrations to further investigate how this ratio may influence the experimental results.
In contrast to the differences in reaction mechanisms in the presence of FeS(s), reactions of HgII with reduced sulfur have been documented in several studies showing the reduction of Hg by sulfite (Van Loon et al., 2001; Feinberg et al., 2015). According to other previous work, FeII also plays an important role in the reduction of HgII to Hg0 by reduced iron species including magnetite, green rust, haematite and siderite (Ona-Nguema et al., 2002; Peretyazhko et al., 2006b; Wiatrowski et al., 2009; Ha et al., 2017). Given the reactions noted above, and the literature, whether Hg reduction would occur in solution or on the solid surface will depend on the environmental conditions. As noted, most prior studies have been done at high concentrations given the analytical tools used to evaluate the interactions, and this study was therefore designed to examine Hg interactions at low Hg concentrations, and to examine if there was the potential for Hg reduction in such environments. Further, the study was aimed at probing the potential reaction pathways for formation of Hg0 in such systems. The potential reactions include reactions of dissolved or solid-phase Hg with reduced species (Fe(II), S(-II) or other reduced S species). As always, in such systems the interactions are complex as there is the potential for abiotic transformations of Fe and S (e.g., Fe3+ being reduced by HS−).
Besides interactions with inorganic solids, Hg speciation in natural systems is strongly influenced by dissolved organic matter (DOM) (Ravichandran, 2004; Slowey, 2010; Gerbig et al., 2011b; Muresan et al., 2011; Jeremiason et al., 2015). Studies have shown the importance of DOM, not just as a group of Hg-binding ligands, but also due to its impact on HgII–S-II(aq) reactions and on the stability of HgS(s) (Ravichandran et al., 1998; Waples et al., 2005; Deonarine and Hsu-Kim, 2009; Skyllberg and Drott, 2010; Gerbig et al., 2011a). Indeed, it has been reported that HgS(s) nanoparticle dissolution is mediated by DOM (Slowey, 2010). In addition, research indicating the potential for DOM to reduce HgII was shown by a positive correlation between dissolved organic carbon (DOC) concentration and Hg0 production (Rocha et al., 2003; Park et al., 2008). These results are however contradicted by other studies which found a negative correlation between DOC concentration and Hg0 production (Amyot et al., 1997; Garcia et al., 2005; O'Driscoll et al., 2006; Mauclair et al., 2008), which was explained by the influence of complexation on Hg reduction. Some studies have demonstrated that under anoxic dark conditions, DOM can rapidly convert HgII to Hg0 at very low DOM concentrations (up to ∼70% at 0.2 mg/L) (Baohua et al., 2011; Zheng et al., 2012). However, according to others, there is no Hg reduction by DOM in dark environments (Matthiessen, 1998). Photo-reduction is considered the main abiotic process responsible for the conversion of HgII to Hg0 in natural systems, and studies show that this reduction process is enhanced by the presence of DOM (Allard and Arsenie, 1991; Costa and Liss, 2000). However, DOM could also reduce Hg reduction by altering light penetration. It is unlikely that photochemical processes are important in most anoxic environments.
To further understand the potential for Hg reduction in the presence of mineral surfaces, and to examine the potential reduction pathways, we investigated the production of Hg0 from HgII in the presence of two sulfidic minerals, FeS(s) and CdS(s), under anoxic and dark conditions. We hypothesized that under the experimental conditions, Hg would be co-precipitated onto the solid surface and that the Hg reduction reaction will involve a surface interaction. To explore the role of surfaces and S−II or FeII as electron donors for the HgII reduction, Hg0 production rates at different pH values and HgII: FeS(s) ratios were examined, and contrasted to reactions of HgII with dissolved FeII. Additionally, reactions with CdS(s) were examined as this could help interpret the reaction mechanisms. While FeS(s) and pyrite (FeS2) are ubiquitous minerals in environmental settings, the presence of CdS(s) is also likely given its low solubility (Stumm and Morgan, 1996). These results were compared and discussed along with the thermodynamic aspects of the potential reduction pathways. In addition, the effect of DOM on the efficiency of any metal sulfide reactive barriers was examined by looking at the reduction of HgII by sulfidic minerals (FeS(s) and CdS(s)) in presence of dissolved organic matter (DOM).
All solutions used in the experiments were prepared under an inert atmosphere using a glovebox (N2 atmosphere) and using MQ-water (Ω < 18.2) degassed by purging boiling water with N2 for 20 min and as it cooled to room temperature. Sulfide minerals (FeS(s) and CdS(s)) were synthesized and characterized as described elsewhere (Jonsson et al., 2016). Briefly, disordered FeS(s) was synthesized by adding 100 ml of 0.6 M Na2S to 100 ml of 0.6 M Mohr’s salt ((NH4)2Fe(II)(SO4)2∙6H2O); and CdS(s) by adding 25 ml of 0.6 M Na2S to 25 ml of 0.6 M Cd(NO3)2·4H2O.
The minerals were characterized using X-ray Diffraction Crystallography (XRD) and Brunauer–Emmett–Teller (BET) measurements (Jonsson et al., 2016). XRD studies were conducted by Rigaku UltimaIV diffractometer with Cu Kα radiation (λ = 1.5418 Å) operating at a beam voltage of 40 kV and beam current of 45 mA. The patterns were acquired at a scan rate of 2°min−1, from 0 to 80 degrees in the 2θ range. BET surface-area measurements were performed using nitrogen sorption experiments conducted on a Quantochrome Nova 2000e instrument. All the samples were degassed for 5 h before analysis. Specific surface area was calculated using the adsorption isotherm within 0.05 < P/P0 < 0.3 range, where P/P0 is the relative pressure.
The Hg(aq) working standard was prepared from a 1,000 ppm Hg(aq) stock solution (Merck, Allemagne, 1,000 mgL−1 Hg in 1.00 M HNO3) and then adjusted using 2–8 M KOH(aq) to obtain the desired pH. Mercury working solutions were prepared daily for each experiment. The ferrous iron solution was prepared by dissolving Mohr’s salt in MQ-water. The DOM isolates used were extracted from surface waters collected at the shelf break of the North Atlantic Ocean and on the western side of Long Island Sound (United States) (Mazrui et al., 2018). The extraction procedure involved passing 0.2 µm filtered seawater through a modified benzene styrene polymer cartridge (Bond Elut) at a rate of <4 ml/min (Dittmar et al., 2008). The cartridge was then rinsed with dilute HCl, dried and the adsorbed DOM eluted with methanol and acetone. DOM dissolved in organic solvent was dried at 40°C using a Nitrogen evaporator (N-EVAP 111). Stock solutions of DOM were prepared by dissolving approximately 0.1 g of the DOM in 100 ml of degassed purified water. The solutions were then filtered through a 0.02 µm PTFE syringe filter, adjusted to pH 7–8, using dilute HCl/KOH and stored in the dark in airtight containers at 4°C until use.
The reduction of HgII in the presence of FeS(s), CdS(s), FeII(aq) or DOM was tested by adding HgII(aq) to slurries of FeS(s) or CdS(s) or solutions of FeII(aq) or DOM in acid cleaned glass vials (total volume of 10 ml). The samples were then incubated in the glove box under anoxic and dark conditions (foil-wrapped sealed serum bottles) to prevent photochemical reactions. Each experimental set was done in triplicate (n = 3) at room temperature. At the end of each experiment, vials were removed from the glove box and produced Hg0(g) collected onto Goldtrap™(Supelco) traps. For the collection, two tubes were inserted through the septum of the vial. One tube was used to purge the headspace of the vial with Argon (Ar) at a rate of 200 ml/min for 20 min, while the other collected the purged gasses onto a gold trap. Collected Hg0(g) was then analyzed using a Cold Vapor Atomic Fluorescence Spectrophotometer (CVAFS) (Tekran, model 2,500) after thermal desorption of the Hg0 from the gold-traps. A calibration curve was prepared by analyzing 10–200 µL of air saturated with Hg0(g) from a vial containing Hg0(g) at a known temperature.
Based on the BET determined surface area (Jonsson et al., 2016), concentrations of FeS(s) and CdS(s) in the experiments were adjusted to have a concentration, reported as surface area to volume of solution ratio, of 1, 5, and 30 m2L−1. This is equivalent to 0.02, 0.09, and 0.54 g L−1 for FeS(s) and 0.01, 0.07, and 0.41 g L−1 for CdS(s), respectively. Samples containing FeS(s) or CdS(s) and 50 pM HgII were equilibrated for 24 h under dark conditions at pH 5–8 for the initial experiments. The production of purgeable Hg0 was measured after 24 h in the mixtures and control solutions consisting of degassed MQ water and 50 pM HgII. In a similar manner, reduction of HgII by DOM or FeII was tested by incubating 10 ml of experimental solutions containing 5.0 mg C/L of DOM (24 h and pH 7–8) or 1 mM FeII (0–4.5 h and pH 5 and 7.5) and 50 pM of HgII. Experiments were also performed over time at a pH of 7–8 in the presence of FeS(s) at different HgII:FeS(s) ratios to examine the impact on the reaction rate.
After the incubation period, experimental solutions containing FeS(s) slurries were filtered through a 0.02 µm PTFE syringe filter and prepared for FeII analysis inside the glove box. Samples for FeII analysis were removed from the glove box and immediately analyzed for the concentration of aqueous FeII using the ferrozine method (Vollier et al., 2000). Briefly, ferrozine (monosodium salt hydrate of 3-(2-pyridyl)-5, 6-diphenyl-1, 2, 4-triazine-p,p′-disulfonic acid) was reacted with dissolved iron to form a stable magenta complex which absorbs in the visible region at 562 nm. A PharmaSpec UV-1700 UV−Vis spectrometer (Shimadzu) was then used to detect the complex before and after a reduction step with hydroxylamine.
The potential reactions that could occur were examined using calculations of the respective equilibrium constants and the free energy (ΔG) of the reaction under the experimental conditions. The concentration of dissolved Fe (Fe(II)T) and sulfide (S(-II)T), and the individual species (principally Fe2+ and FeS0, with the potential for FeOH+, FeCl+, and FeSO40 being present at higher pH and anion concentrations) was calculated using the solubility model for FeS(s) of Rickard (2006) which considers that the Fe(II) concentration is determined by a solubility reaction and an equilibrium reaction:
where FeS0 represents a series of (FeS)x cluster compounds that form in the presence of FeS(s). The Hg speciation and interaction with FeS(s) was modeled using constants from Skyllberg and Drott (2010), Stumm and Morgan (1996). Equilibrium constants for the redox reactions were from Stumm and Morgan (1996). The results of the thermodynamic calculations are detailed in Supplementary Tables S1, S2; Tables 1–3. Supplementary Table S1 details the solubility of FeS(s) across the pH range used in the experiments, Supplementary Table S2 contains a listing of the examined reactions while Table 1 details the concentrations used in the calculations at pH 7. The calculated free energies of the various reactions are contained in Tables 2, 3. The concentrations of Hg0 were those measured in the experiments and it was assumed that the total concentration of oxidized forms (sulfate and Fe(III)) were low, (respectively, 0.1 µM and 1 nM) given that these were primarily produced by reduction of HgII, or were present as trace constituents in the experimental solution. Their dissolved speciation was taken into account in the calculations.
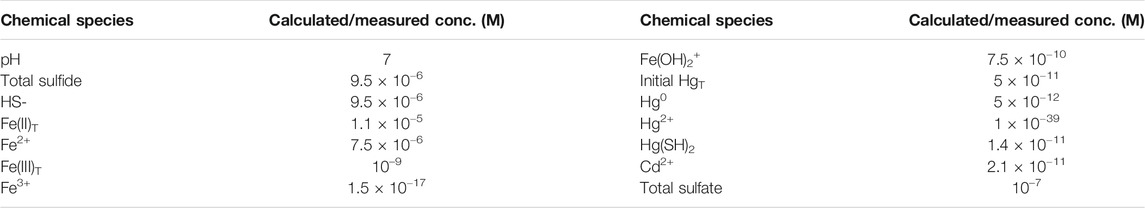
TABLE 1. Concentrations used to determine the free energy of reactions (Tables 2, 3) at pH 7. Values for individual forms of Hg, Fe and S are calculated using the equations in Supplementary Table S2. The HgT and Hg0 concentrations are based on the added and measured Hg concentrations. The total sulfide and Fe(II) concentrations are based on the solubility data of (Rickard, 2006) for FeS(s) (Supplementary Table S1). A total Fe(III) concentration of 1 nM and a sulfate concentration of 0.1 µM is assumed. For Cd, the concentration is derived from the solubility product reaction.
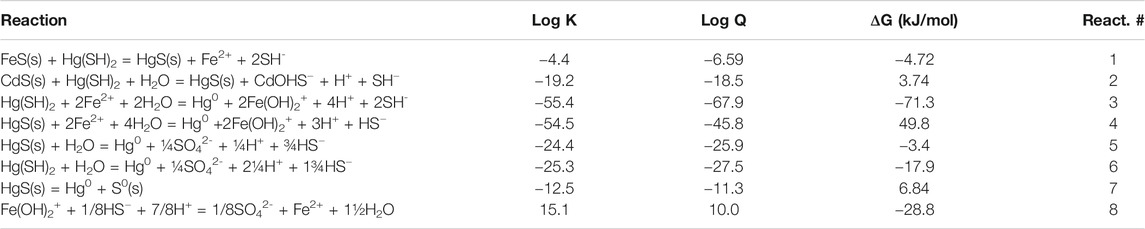
TABLE 2. Calculated free energies of the various potential reactions discussed in the text based on the concentrations in Table 1, and writing the reactions in terms of the major dissolved forms of the metals and sulfide at pH 7. All solids are assumed to have an activity of 1. The redox calculations are done assuming the presence of 5 pM Hg0.

TABLE 3. Calculated free energies for the reactions involving Hg co-precipitation and Hg(II) reduction at the different pH values of the experiments.
The rates of reaction were calculated using the Hg0 data and with an assumption of a pseudo first order reaction as the concentration of Hg0 (<50 pM) is at least five orders of magnitude higher than the concentrations of FeII, S−II in solution or in the solid phase. Additionally, under the experimental conditions, <1% of the solid is dissolved at equilibrium. Therefore, the assumption of a pseudo first order is valid.
To test the reduction of HgII in the presence of FeS(s), we initially quantified the amount of purgeable Hg0 from pH controlled slurries containing 0.09 g/L FeS(s) (corresponding to a surface area concentration of 5 m2L−1) and 50 pM HgII that were incubated under anaerobic and dark conditions. In control samples where no FeS(s) was added (pH ranging from 5 to 8) less than 2% of the initially added HgII was lost as Hg0 after 24 h of incubation (Figure 1). In FeS(s) mineral suspension, however, ∼12–∼15% of the total HgII was reduced, and the amount of Hg0 produced increased with pH. Overall, the amount of Hg0 produced doubled at pH 8 compared to that at pH 5, and this difference was statistically significant, although the differences in the production rate at the higher pH values were not. Overall, the effect of pH was greater at lower pH. However, the production of Hg0 (<15% of the initial HgII) remained very low compared to the levels observed during the interaction of HgII-ferric oxide or HgII DOM experiments (Zheng et al., 2012; Ha et al., 2017), or in the presence of Fe(II) alone in our studies, as discussed further below.
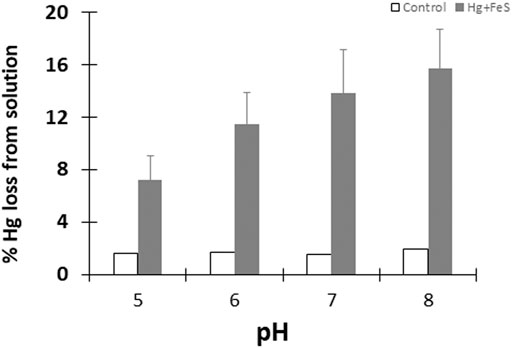
FIGURE 1. Percentage of Hg0 produced after 24 h in the presence (grey bars) and absence (white bars) of FeSm at different pH. Reactions done under dark and anoxic conditions with 50 pM HgII and FeS(s) at a surface area to volume of solution ratio of 5 m2/L. Error bars show mean ± standard deviation (n = 3).
To further explore reaction kinetics, net reduction of HgII was tested as a function of reaction time (1 h–3 days) and mineral surface area concentration (1–30 m2L−1) at a pH of 7–8. At all tested mineral surface area concentrations, the reduction rate of HgII was rapid within the first hour (>1 pmolL−1h−1), and most Hg0 was produced within the first hour of the experiment (Figure 2). The initial average rates of production are compiled in Table 4 assuming the reaction was first order, and while the initial rates appeared to increase with surface area, these differences were not statistically significant as rates were respectively, 0.78 ± 0.50, 0.92 ± 0.07 and 1.09 ± 0.16 h−1. The concentration of Hg0 formed then gradually increased at a slower rate (<0.2 pmolL−1h−1) to reach a maximum after 48 h. At this equilibration point, the production of Hg0 was slow relative to that in the first hour, and accumulated Hg0 concentrations reached a plateau concentration. After the first hour, the rates of reduction were an order of magnitude lower (Table 4) and the rates appeared more related to the relative FeS(s) surface area, increasing with the amount of FeS(s) present. At FeS(s) surface concentration of 1, 5 and 30 m2L−1 of FeS(s) suspensions, respectively, ∼12, ∼15 and ∼17% of the HgII was reduced over the course of 24 h.
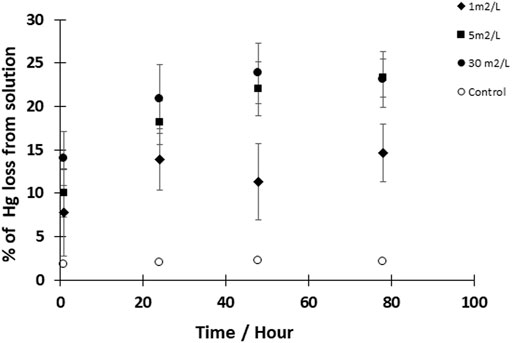
FIGURE 2. Kinetics of Hgll reduction by FeS(s). Experimental solutions contained 50 pM HgII and FeS (s) at a concentration of 1, 5, and 30 m2/L (surface area to volume of solution ratio). Reactions were performed at pH 7–8 under dark and anoxic conditions. Error bars represent mean ± standard deviation (n = 3).
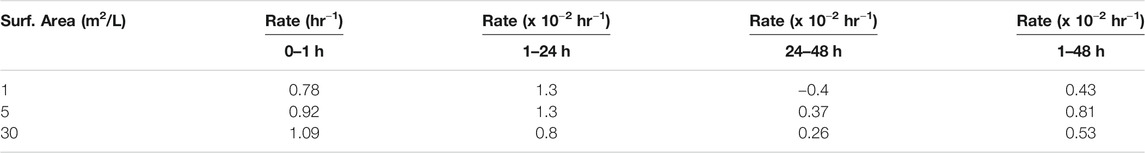
TABLE 4. Calculated rates of reaction in the presence of different amounts of FeS(s) and over time at a pH of 7–8.
While the formation of Hg0 increased with surface area, the relationship was not linear. Several studies on the reduction of HgII in the presence of iron oxide minerals have shown that the minimum equilibration time necessary for the production of Hg0 was 24 h (Wiatrowski et al., 2009; Ha et al., 2017), and our results also suggest that the system is approaching steady state over a similar time period, even though our studies were done at much lower ratios of HgII:FeS(s). The initial high rate of reduction followed by slower formation of Hg0 suggests that competing reactions are occurring. Initially there would be high concentrations of dissolved Hg in solution but given the experimental conditions, the dissolved Hg would rapidly decrease due to co-precipitation of HgS(s) on the FeS(s) surface, or through surface complexation, as discussed below.
The reduction of HgII by cadmium sulfide (CdS(s)) was investigated at a CdS(s) concentration corresponding to a surface area concentration of 5 m2L−1 and HgII concentration of 50 pM. During the entire duration of the experiment (up to 4 days), measurements indicated that less than 2% of the total Hg was reduced with CdS(s) (Figure 3). The fraction of HgII reduced to Hg0 was thus similar to the reduction observed in controls, suggesting that the presence of CdS(s) did not significantly enhance Hg reduction.
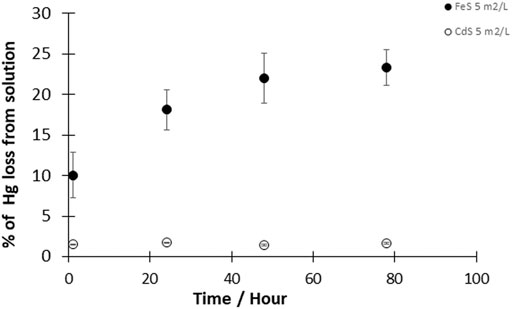
FIGURE 3. Kinetics of the reaction of Hgll with CdS(s) and FeS(s). Experimental solutions contained 50pM HgII and 5 m2/L CdS(s) or 5 m2/L FeS(s) (given as surface area to volume of solution ratio). Reactions were performed at pH 7–8 under dark and anoxic conditions. Error bars represent the mean ± standard deviation (n = 3).
To examine the impact of DOM on the reduction of Hg, aqueous solutions of HgII were reacted with two different DOM extracts, obtained from waters collected at the shelf break of the North Atlantic Ocean (DOM1) and from western Long Island Sound (DOM2) (Mazrui et al., 2018). The two DOM were characterized by determining their optical properties (Supplementary Table S3). The Specific Ultraviolet Absorption (SUVA254), calculated as absorption at 254 nm divided by the DOC concentration, is a measure of the aromaticity of the DOM. The absorption ratio (ratio of absorbance at 250–365 nm), on the other hand, is a measure of the molecular weight of the DOM. Since high molecular weight DOM absorbs more strongly at longer wavelengths than low molecular weight DOM, a lower absorption ratio indicates that the DOM has a higher relative molecular weight. Here, we found that DOM2 had a lower absorption ratio and a higher SUVA254 than DOM1. We also found that DOM1 had a proteinaceous fluorescence signal (intense emission at a lower wavelength) similar to tyrosine and tryptophan emissions while DOM2 had a humic-like fluorescence signal (Supplementary Figure S1), which likely reflects differences in the amount of allochthonous vs. autochthonous DOM sources. Thus, DOM2 had more humic characteristics, i.e., of more allochthonous origin, hydrophobic and aromatic, with a lower nitrogen content and a higher phenolic and sulfur content than DOM1.
At pH 7–8, 17% of the added HgII in the DOM1 sample was reduced to Hg0 after 24 h, whereas ∼12% was reduced by DOM2 (Figure 4), indicating a slower reaction rate of the latter. The maximum reduction was obtained after 48 h, with ∼25% of the HgII reduced by DOM1. This result is consistent with some previous studies of HgII reduction by DOM in the absence of light (Zheng et al., 2012; Chakraborty et al., 2015).
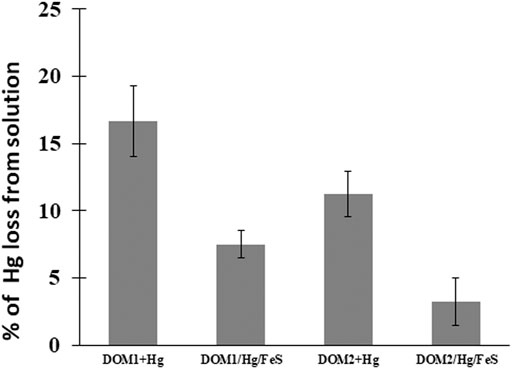
FIGURE 4. Percent HgII converted to Hg0 after 24 h of reacting 50 pM HgII, 5 mg C/L DOM and 5 m2L−1FeS(s) at pH 7–8 under dark and anoxic conditions. Error bars represent mean ± standard deviation (n = 3).
The reducing capacity of FeS(s) in the presence of the two different DOM was further investigated to examine the impact of both as thiols ligands can affect both the dissolved concentration and the speciation of HgII (Figure 4). The total concentrations of Hg0 produced decreased in the presence of FeS(s) for both DOM1 (55% decrease) and DOM2 (71% decrease). In contrast to reduction of HgII at pH 7–8 within 24 h with DOM only, added FeS(s) decreased Hg0 production. The produced Hg0 was lower however than in the presence of FeS(s) alone (Figure 1), indicating that the presence of DOM hindered the reduction when a solid surface was present, but enhancing the reduction in its absence. This may suggest that at pH 7–8 the reaction likely involves Hg associated with the FeS(s) and not dissolved Hg, but the influence of DOM may also be due to its binding to the FeS(s) surface, thereby reducing the extent of the reaction.
Finally, to examine the role of dissolved vs. solid phase reactions, Hg0 production was evaluated in the presence of dissolved Fe(II) at two different pH values. The rate of production was higher in these homogeneous solutions (e.g., 1.32 h−1 for the first hr at pH 7) than in the presence of FeS(s) (0.78–1.09 h−1; Table 1) although the dissolved Fe(II) used in these experiments was higher than that found in equilibrium with the solid.
In most studies looking at the interactions of HgII and FeS(s), the products obtained were the stable species β-HgS(s), and Hg0 was not detected suggesting that the primary interaction was an exchange reaction with the release of Fe2+ from FeS(s) with concomitant β-HgS formation. This is essentially a cation exchange reaction driven by the thermodynamic favorability of precipitating HgS(s) (Jeong et al., 2008; Jeong et al., 2010; Skyllberg and Drott, 2010):
The reaction is favorable under the experimental conditions except at the lower pH (ΔG = −4.72 kJ/mol at pH 7 and 7.8 kJ/mol at pH 5; Table 3). Therefore, reduction of dissolved Hg could only occur initially, before HgII is co-precipitated. Bone et al. (2014), however, suggested that Hg0 was generated from the reduction of HgII by FeS(s). They formulated a reduction hypothesis starting from HgII adsorption to the mineral. Nevertheless, they could not conclusively verify the role of the reductant—S−II or FeII in HgII reduction, and overall, the role of S−II or FeII as electron donors in HgII reduction appears to vary according to the ratio of HgII:FeS(s). Similar to the Bone et al. hypothesis, others (Hua and Deng, 2008; Hyun et al., 2012) suggested that U(VI) reduction by mackinawite or amorphous FeS(s) occurred following U(VI) adsorption onto the mineral surface and simultaneous release of FeII. They proposed that once sorbed to the mackinawite surface, either the surface U(VI) is reduced by S−II at the FeII depleted mackinawite surface or the dissolved U(VI) is reduced by dissolved HS− released by congruent dissolution of mackinawite. Kirsch et al. (2008) come to a similar conclusion for their studies of antimony. However, in contrast to U(VI), Hg is known to have a high affinity for reduced S even in substantially oxic environments (Wolfenden et al., 2005). In our experiments, co-precipitation reduces the dissolved Hg in solution and so while the reactions in the dissolved phase with FeII or S−II may be favorable (Reactions 3 and 6 in Table 2), they are unlikely to be the only reactions occurring over time. Indeed, the reactions were slower in the presence of the FeS(s) suggesting that interaction of Hg with the solid is occurring, as predicted by the thermodynamic calculations at pH 7 and 8 (Tables 2, 3).
The reaction of HgII with the surface is pH dependent, as pH affects both the dissolved speciation of HgII and sulfide, and the surface charge on the mineral. The point of zero charge (PZC) for mackinawite is around 7.5 (Wolthers et al., 2005) and so under the experimental conditions the surface is either positively charged or near neutral, and thus would not hinder the interaction of the dissolved Hg complexes with the surface. At the lower pH values, the uncharged Hg-sulfide complex dominates in solution but becomes less important as the pH increases. Overall, the noted pH effect on the reduction reaction is likely not related to the impact of pH on the interaction of Hg with the mineral surface. However, the precipitation of HgS(s) becomes less favorable at low pH. The reactions on the surface and in solution involving HgII reduction become more favorable at higher pH, primarily due to the decreasing concentrations of Fe(II) and HS− with increasing pH (Table 3). Furthermore, the experimental pH effect is relatively small with the increase in Hg0 production increasing by less than a factor of 2 for a change in [H+] of 103.
Most studies on the interactions between HgII and minerals show that the production of Hg0 increases with the pH of the solution (Wiatrowski et al., 2009; Bone et al., 2014; Ha et al., 2017). The results reported by Andersson (1979) on the interaction between HgII and Fe2O3.nH2O found that the amount of reduced HgII increased with pH, from pH values of 6.2–8.5. The same result has been observed by Patterson et al. (1997) with the interaction between chromium and FeS(s). Thus, our results agree with these studies with the best rate of reduction at pH 7–8. The influence of pH on the production of Hg0 in the range 7–8 could be explained by the formation of the dissolved species FeOH+ in these studies, which increases in relative concentration with pH, as shown by Amirbahman et al. (2013), although this reaction is unlikely to be occurring in our experiments (Table 3). Our results showed an increase in reduction with increased mineral surface area, suggesting that co-precipitated HgS(s) is likely involved in the reaction. Other studies on heterogeneous reduction have demonstrated that the formation of surface complexes is responsible for the enhanced reaction rate (Liu et al., 2008; Pecher et al., 2002; Schwarzenbach and Stone, 2003) and this adsorption depends on the pH (Kim et al., 2004; Miretzky et al., 2005). We conclude that this explains why the increase in pH promotes the reduction of HgII.
At the low concentrations of Hg used in our experiments compared to the other studies mentioned above, the amount of Fe2+ released into solution from the co-precipitation of Hg onto the mineral surface is small compared to the Fe2+ in solution in equilibrium with the solid phase. The following reactions determine the dissolved Fe(II) and total S(-II) concentrations in equilibrium with the solid (Stumm and Morgan, 1996; Wolthers et al., 2005; Rickard, 2006; Supplementary Tables S1, S2):
While FeOH+ is the dominant complex formed in solution in the absence of sulfide, the principal form is the free ion. In the presence of sulfide, FeS0 is also found where this represents a series of cluster compounds with 1:1 stoichiometry (Rickard, 2006), and is present at a fixed concentration in equilibrium with the solid. The total dissolved FeII concentration depends on both the pH and the sulfide concentration (Supplementary Table S1). At pH 5.5, FeOH+ is insignificant but increases to about 10% of the total FeII at pH 8, according to the thermodynamic calculations. The calculations, based on Rickard (2006), predict a dissolved FeII concentration of 182 μM at pH 5 and 3.7 μM at pH 8. Our measurements (Table 5) found slightly higher concentrations at higher pH and less of a pH effect (e.g., 137 μM at pH 7–8 and 166 μM at pH 5–6 for 5 m2/L FeS(s)), and also that the dissolved FeII increased with the amount of FeS(s) added, suggesting that the assumption of a pure solid (activity = 1) is likely not completely valid for our studies, likely due to the amorphous nature of the solid used. While we did not measure sulfide concentrations, the predicted dissolved concentration (total S−II(aq) ∼ FeII(aq)) is not high enough to precipitate the majority of the dissolved Hg as HgS(s) (Table 1), and much of the HgII is in solution initially as Hg(SH)2, and its deprotonated forms (HgS2H− and HgS22−) (Skyllberg and Drott, 2010).
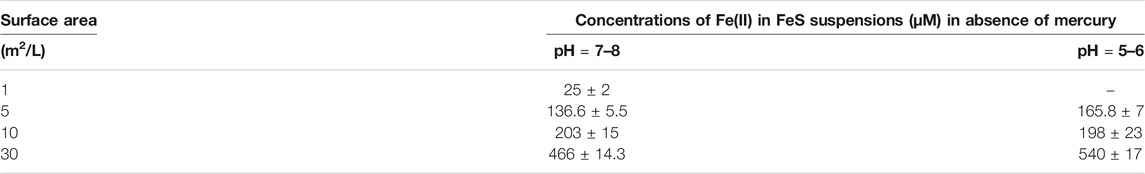
TABLE 5. Measured concentrations of Fell in iron sulfide suspensions.
The source of the electrons for the reduction of HgII is either from a redox reaction on the surface involving the mineral constituents, or a reaction with dissolved reduced ions, either FeII or S−II. If sulfide was being oxidized during the reduction of Hg, then one would predict this should have occurred in the presence of the CdS(s), but no reduction was observed. The equilibrium dissolved S−II and CdII concentrations in the presence of the solid (K = −14.36 for CdS(s) + H+ = Cd2+ + HS−) are lower, however, than in the presence of FeS(s), and thus the reduction in the presence of CdS(s) would be less favorable even if S−II was the reductant. Thermodynamic calculations suggest the concentration of sulfide is at low nM levels in the presence of CdS(s) and that the co-precipitation reaction of Hg with the CdS(s) is not thermodynamically favorable (Table 2). Thus, the lack of reaction in this case does not necessarily negate the role of sulfide oxidation in Hg reduction.
The other potential reductant is FeII for the reactions in the presence of the FeS(s), as it is with the reactions in the presence of dissolved FeII, and no surfaces (Figure 5). Thermodynamically the reaction is favorable in solution (ΔG = −71.3 kJ/mol at pH 7) under the experimental conditions (Tables 2, 3), even given the low concentration of HgII relative to FeII and HS−, and with the assumption that FeIII is low (Table 1):
Thus, while Hg remains in solution, the reaction will proceed and this likely accounts for the initial formation of Hg0 in the initial time period, and could account for some of the trend seen with pH. However, as the concentration of dissolved HgII decreases as Hg is precipitated onto the FeS(s), this reaction will no longer occur. The time series measurements were made at pH 7–8 where co-precipitation is a favorable reaction, thereby decreasing the dissolved concentration of Hg over time. The differences in the rate of reaction in homogeneous solution (Figure 5) and in the presence of FeS(s) (Figure 2) indicates that the majority of the HgII is being co-precipitated or surface absorbed to the solid.
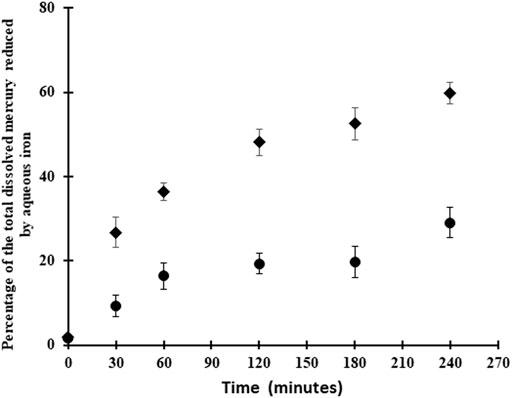
FIGURE 5. Kinetics of HgII (50 pM) reduction by FeII (1 mM) at pH 5 (circle) and 7.5 (square).
Another mechanism is therefore needed to account for the Hg0 formation at later times. The reaction of co-precipitated HgS(s) with Fe(II) is not favorable (Table 3) and therefore the reaction that occurs with the precipitated Hg does not involve Fe(II):
Overall, we conclude that FeII or FeOH+ is not the reductant in our experiments after the Hg has co-precipitated onto the solid. Thus, there is a difference in the mechanisms for the reduction of HgII in the presence of FeS(s) and with dissolved FeII (Jeong et al., 2010; Richard et al., 2016). Note, however, that at pH 5 the precipitation reaction is not thermodynamically favorable and therefore the reactions in solution dominate, with FeII being the primary reductant (Table 3).
Alternatively, the reductant could be S−II, and the reason for the low reaction in the presence of CdS(s) is probably because of the low sulfide concentration in equilibrium with the solid. The potential reaction is thermodynamically favorable, even under the low concentrations of the experimental conditions, for both the reactions in the water and that with the solid, except at the lower pH levels (Table 3):
and
The equations represent either oxidation of dissolved sulfide or of the S−II associated with the HgS(s). Overall, again, these calculations suggest that the reaction later in the experimental time period does not involve dissolved reduced species and that the reduction involves reactions within the solid, with the electrons being provided from the oxidation of S−II by an electron transfer reaction at the surface, followed by the release of Hg0 into solution. Overall, these results suggest that initially in the experiments, the dissolved Hg is being reduced by either Fe(II) or HS−, but that later in the experiment the reaction only involves reduced S. If FeII is being oxidized, the FeIII produced would likely remain adsorbed on the solid, but it is also likely that the Fe(III) would be reduced back to Fe(II) by the sulfide in solution as this reaction is favorable under the experimental conditions (ΔG = −28.8 kJ/mol at pH 7; Tables 2, 3). Thus, once the Hg is co-precipitated onto the FeS(s) surface, whether the reaction involves initially S(−II) or Fe(II) is somewhat academic as the final products will be the same because of the reduction of any Fe(III) produced.
If the reaction involves sulfide oxidation, the fate would depend on the degree of oxidation of S−II. It is likely that some intermediate product, such as elemental sulfur (S0), could result, rather than complete oxidation to sulfate. Indeed, an electron exchange reaction between HgII and S−II could potentially occur with the formation of Hg0 and elemental S. Given the uncertainty in the equilibrium constants (Stumm and Morgan, 1996; Skyllberg and Drott, 2010), and assuming pure solids are formed, the reaction is near equilibrium at pM Hg0 concentrations (i.e., [Hg0] ∼ K; Table 2, Reaction #7):
As mentioned earlier, the lack of a reaction with CdS(s) is likely because of the lack of precipitation of HgS(s) on the surface at the low sulfide concentrations found in equilibrium with the solid phase, and the low sulfide concentration in solution. This is because of the stronger M-S bond in CdS(S) compared to FeS(S).
Furthermore, as noted above, the increase in the amount of mineral surface of mackinawite (Figure 2) slightly influences the quantities of Hg0 produced. With 30 m2L−1 of FeS(s), the reduction reached a maximum of 11 pM of Hg0 after 24 h of reaction, while this maximum was 6.9 pM for 1 m2L−1 of FeS(s). These results indicate that a surface catalytic role of precipitated HgS(s) on mackinawite is involved in the production of Hg0. Wiatrowski et al. (2009) demonstrated that the kinetics of HgII reduction by magnetite systematically varies as a function of magnetite concentration. Amirbahman et al. (2013)’s study on the kinetics of HgII reduction by FeII suggested that the mineral phases are important factors affecting the rate of the mercury reductive pathways, and O’Loughlin et al. (2020) showed that there were differences in the reaction rates in the presence of FeII-containing clays. However, Jeong et al. (2010) have shown that the adsorption of HgII onto the surfaces of mackinawite only occurs below a certain molar ratio of HgII and FeS(s), which implies that the ratio of HgII:FeS(s) could also influence the production of Hg0. This ratio in our study was 5–7 orders of magnitude lower than that of Jeong et al. (2010).
In summary, our study indicated that mercury reduction by FeS(s) is kinetically slow and the production of Hg0 is small compared to other potential reduction pathways in environmental ecosystems, such as HgII reduction in the presence of dissolved FeII or DOM, and also appears to occur via a different mechanism. The experiments were carried out with excess FeS(s) concentrations so the reaction can therefore be described according to pseudo first order kinetics. The overall reaction rate constants obtained are k = 67 × 10−3 h−1; 85 × 10−3 h−1; and 92 × 10−3 h−1, respectively, for 1, 5, and 30 m2/L of FeS(s). These values are similar in terms of the link between reaction rate constant and mineral concentration noted in some studies (Wiatrowski et al., 2009; Amirbahman et al., 2013; Ha et al., 2017). However, our experimental data show that the average net rate of HgII reduction by FeS(s), assuming first order kinetics over the experimental time period, is lower than HgII reduction by humic substances (1.6–2.1 × 10−2 h−1; Chakraborty et al., 2015), minerals such as clay (1.74 × 10−1 h−1) (Peretyazhko et al., 2006a), hematite from phlogopite (6.60 × 10−1 h−1) or magnetite (Wiatrowski et al., 2009). These results confirm that the production rate of Hg0 is a function of the nature of the mineral (i.e., oxide or sulfide), and likely the form of Hg adsorbed or precipitated on the surface of the mineral.
Overall, under our experimental conditions, the homogeneous Hg reduction in presence of aqueous FeII without mineral surfaces was more favorable than the experiments in presence of FeS(s) mineral (Figure 5), which is consistent with the thermodynamic calculations (Table 3). The initial reaction rate was 20–70% higher for the aqueous FeII experiments. However, the concentration of FeII in the homogeneous experiments was much higher than in the mineral studies and this could potentially account for the higher conversion rate to Hg0, although the rate should be similar given that the initial HgII concentration was the same, and the FeII concentrations in both cases is substantially higher and not rate limiting Rather, the mechanisms are likely different for the two situations. Although several authors have shown the role of surface-catalysis by iron minerals on the rate of mercury reduction, our data (Figure 5) shows fast reduction of mercury in presence of aqueous FeII. In contrast, Ha et al. (2017) indicated that mercury reduction by aqueous ferrous iron in the absence of a solid phase was kinetically slow. Pasakarnis et al. (2013), Amirbahman et al. (2013) suggested that HgI sorbed onto the mineral surface during the transformation of HgII to Hg0 and acts as a surface-catalyst in this reaction. Peretyazhko et al. (2006b) demonstrated that adsorption of FeII to the haematite surface created very reactive sites for the reduction of HgII, while in the absence of haematite particles, no production of Hg0 occurred. The difference between this study and previous studies mentioned above might be due to the low concentration of FeII in the FeS(s) suspensions or more likely because the reaction proceeds via a different mechanism once the Hg is co-precipitated.
It is well known that dissolved organic matter (DOM) has a strong interaction with mercury and other trace metals affecting their speciation, mobility and toxicity (Buffle, 1988). Under abiotic dark conditions in aquatic systems, DOM participates in the conversion of HgII to Hg0 but also contributes to the strong complexation of HgII (Ravichandran et al., 1998; Deonarine and Hsu-Kim, 2009; Zheng and Hintelmann 2010; Zheng et al., 2012; Han et al., 2007). This complexation is attributed to reduced sulfur ligands (Waples et al., 2005; Merritt and Amirbahman, 2007). Indeed, DOM is a mixture of molecular organic compounds with a large number of hydrophilic functional groups: carboxylic (COOH), phenolic and/or alcoholic (OH), carbonyl (C=O) and amine groups (NH2). Reduced sulfur groups also exist in different oxidation states (R-SH, R-S=S-R and R-SO3H). Chakraborty et al. (2015) showed that the ratio of the–COOH/−OH groups and the sulfur content in the humic substances reveal a strong competition between complexation and reduction of HgII. They suggested that several parameters such as pH, total sulfur content, the −COOH/−OH ratio and salinity influenced the reduction of HgII in presence of DOM. In our studies, the less humic DOM1 reduced Hg at a higher rate than that with DOM2, and this is consistent with the data of Chakraborty et al. (2015) who showed that the rate of reduction was higher for humic material with less total S, or a higher ratio of carboxylic to thiol groups. As discussed above and shown in Supplementary Figure S1, DOM2 has more humic character while DOM1 is more protein-like in terms of its fluorescence.
We observed that the HgII reduction by DOM was diminished in presence of FeS(s), whatever the characteristics of the experiment (Figure 4). Calculations of the speciation of dissolved Hg in the presence of FeS(s) and DOM at the concentrations used in the experiment, using the RSH:DOM ratios determined by Seelen (2018) for comparable coastal waters and the Hg(SR)2 binding constant from Skyllberg and Drott (2010), suggest that a small fraction of the Hg—5–10% depending on the DOM was organically complexed during the experiments. This is consistent with the results that showed the extent of reduction in the presence of FeS(s) and DOM was lower than that of FeS(s) alone. Overall, we conclude that the presence of DOM increases the barrier to Hg reduction by sulfide surfaces, and likely also has a similar effect for other reductive surfaces.
Mishra et al. (2011) observed that the HgII reduction by magnetite and green rust was severely diminished in the presence of bacterial biomass, suggesting inhibition by surface sulfhydryl groups. These experiments suggest that the conditions of the experiment likely determine whether Hg is primarily bound to the reduced S in DOM or the inorganic reduced sulfide in FeS(s), or is removed by co-precipitation. Furthermore, in most of the studies on the interaction between HgII and FeS(s), the products obtained were the stable solids metacinnabar, cinnabar, and Hg associated with iron sulfides (Jeong et al., 2008; Liu et al., 2008; Skyllberg and Drott, 2010) suggesting FeII present in FeS(s) suspension acts as an electron donor in the production of Hg0. However, in the presence of DOM and FeS(s), this mechanism could be changed as DOM likely keeps the Hg in solution and prevents its interaction with the solid phase, although our calculations show that the extent of complexation was small. However, depending on the pH, the DOM can also interact with the mineral surface and therefore hinder the co-precipitation of HgII and any surface reactions. Dissolved organic matter is known to play a dual role in HgS(s) formation and stabilization (Slowey, 2010; Gerbig et al., 2011a), creating a competition between its complexation of Hg and Hg adsorption to the iron sulfide (Skyllberg and Drott, 2010) and, influencing, through its complexation of dissolved Hg, the dissolution of cinnabar (Ravichandran et al., 1998; Waples et al., 2005). We conclude that our data showing that HgII reduction in presence of both DOM and FeS(s) was less than found in the presence of either DOM or FeS(s) only, is because of the competition between FeS(s) and DOM for complexation and the extent of HgS formation (Skyllberg and Drott, 2010). The HgII would be less available for reduction by DOM, FeII or FeS(s) under these conditions. Zhu et al. (2013) have shown that the strength of FeII as a reducing agent is affected by DOM during the reduction of 2-nitrophenol (2-NP) in TiO2 suspensions. Overall, the HgII reduction in presence of DOM or mineral phases involves complicated reaction pathways but the presence of DOM increases the barrier to reduction.
Our study demonstrates that HgII can be reduced to Hg0 in the presence of FeS(s) but the extent of reduction is slow compared to that found with hydrous ferric oxide, with dissolved Fe(II) and in the presence of DOM. The data presented herein show clearly that in the presence of sulfide surfaces, HgII is less available for reduction. However, our results also showed that there was no Hg0 production in presence of CdS(s) in contrast to FeS(s), suggesting that the presence of a sulfide surface is not sufficient for this reaction to occur (Figure 4). The concentration of sulfide in solution also plays a role in controlling the extent of the reaction. Neither FeS(s) nor CdS(s) enhanced HgII reduction compared to DOM or FeII. Based on thermodynamic calculations (Tables 2, 3; Supplementary Table S2), we suggest that S−II was the likely electron donor for reduction of precipitated HgII in the presence of FeS(s), and its higher concentration in the FeS(s) solutions compared to the CdS(s) solutions accounts for the differences in the Hg0 formation. At low pH in the presence of FeS(S), precipitation of HgII is unlikely to occur and in this instance, reactions in solution are likely controlling the rate of reduction. We therefore suggest that while the Hg does not need to be adsorbed to the surface for the reaction to proceed, this is the likely fate of Hg in the presence of FeS solids under environmental conditions.
Extrapolating these findings to environmental conditions, we suggest that chemical reduction of HgII is complex in anoxic environments, such as sediment, with many potential reaction pathways. This reaction is influenced by ferrous iron, minerals, sulfide, DOM and interactions between the different compounds and solid phases. However, we conclude that the presence of FeS(s) in environmental sediments is not the major driver of the formation of Hg0 in such systems as the reactions are slow once the Hg interacts with the mineral surface. Other reduction pathways are much more favorable with dissolved reductants (reduced Fe and S species). Furthermore, this study shows the influence of DOM on the reaction between Hg and FeS(s) and that its presence needs to be considered because DOM affects mercury transformation and mercury reactivity toward minerals, as shown by Skyllberg and Drott (2010). The type of DOM also influences the rate of reaction, as it does complexation.
Overall, processes that convert HgII to Hg0 under anoxic conditions are important mitigators of the production and bioaccumulation of CH3Hg as reduction potentially removes ionic Hg from the system where it could otherwise be methylated. More research at lower Hg concentrations is needed to further understand the primary reactions that are occurring and the potential role of DOM and pH in controlling the rates of Hg reduction.
All the data related to this study are included in the article/Supplementary Material. The validated concentration datasets for this study will be provided on valid request. The data will be submitted to the University of Connecticut's data archiving facility to make it available upon request. The data will also be made available through Mason's research website: mason.mercury.uconn.edu.
All authors contributed to the design of the study. MC lead and conducted most of the laboratory work. NMM and SJ contributed with material (extracted DOM, lab-synthesized FeS(s) and CdS(s)) and with labwork. MC and RPM wrote the paper with inputs from SJ and NMM.
The research was funded through the Fulbright Visiting Scholar Program to MC which supported her stay in United States (Grantee ID 68150507). SJ acknowledges funding from the Swedish Research Council (International Postdoc grant 637–2014–54). NM and RM acknowledge partial funding from the National Science Foundation Environmental Chemical Science program (Award # 1607913 to RM and J. Zhao).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Staff and students from the labs of RM (University of Connecticut) are acknowledged for valuable discussions during the study. MC thanks the Fulbright Program for providing the scholarship; the Department of Marine Sciences and Chemistry, University of Connecticut for the reception within the department and for the help received; all staff and students of the RM lab for their help.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenvc.2021.660058/full#supplementary-material
Allard, B., and Arsenie, I. (1991). Abiotic Reduction of Mercury by Humic Substances in Aquatic System - an Important Process for the Mercury Cycle. Water Air Soil Pollut. 56 (1), 457–464. doi:10.1007/bf00342291
Amirbahman, A., Kent, D. B., Curtis, G. P., and Marvin-Dipasquale, M. C. (2013). Kinetics of Homogeneous and Surface-Catalyzed Mercury(II) Reduction by Iron(II). Environ. Sci. Technol. 47 (13), 7204–7213. doi:10.1021/es401459p
Amyot, M., Gill, G. A., and Morel, F. M. M. (1997). Production and Loss of Dissolved Gaseous Mercury in Coastal Seawater. Environ. Sci. Technol. 31, 3606–3611. doi:10.1021/es9703685
Andersson, A. (1979). “Mercury in Soils,” in The Biogeochemistry of Mercury in the Environment. Editor O. Nriagu (Amsterdam, The Netherlands: Elsevier, North-Holland Biomedical Press), 79–112.
Baohua, G., Yongrong, B., Carrie, L. M., Wenming, D., Xin, J., and Liang, L. (2011). Mercury Reduction and Complexation by Natural Organic Matter in Anoxic Environments. PNAS 108, 1479–1483. doi:10.1109/iccsn.2011.6014680
Benoit, J. M., Gilmour, C. C., Heyes, A., Mason, R. P., and Miller, C. L. (2003). “Geochemical and Biological Controls over Methylmercury Production and Degradation in Aquatic Ecosystems,” in Biogeochemistry of Environmentally Important Trace Elements. Acs Symposium Series, 262–297.
Bone, S. E., Bargar, J. R., and Sposito, G. (2014). Mackinawite (FeS) Reduces Mercury(II) under Sulfidic Conditions. Environ. Sci. Technol. 48 (18), 10681–10689. doi:10.1021/es501514r
Bower, J., Savage, K. S., Weinman, B., Barnett, M. O., Hamilton, W. P., and Harper, W. F. (2008). Immobilization of Mercury by Pyrite (FeS2). Environ. Pollut. 156 (2), 504–514. doi:10.1016/j.envpol.2008.01.011
Buffle, J. (1988). Complexation Reactions in Aquatic Systems: An Analytical Approach. Chichester: Ellis Horwood Ltd.
Chakraborty, P., Vudamala, K., Coulibaly, M., Ramteke, D., Chennuri, K., and Lean, D. (2015). Reduction of Mercury (II) by Humic Substances-Influence of pH, Salinity of Aquatic System. Environ. Sci. Pollut. Res. 22 (14), 10529–10538. doi:10.1007/s11356-015-4258-4
Charlet, L., Bosbach, D., and Peretyashko, T. (2002). Natural Attenuation of TCE, as, Hg Linked to the Heterogeneous Oxidation of Fe(II): An AFM Study. Chem. Geology 190, 303–319. doi:10.1016/s0009-2541(02)00122-5
Clarkson, T. W., and Magos, L. (2006). The Toxicology of Mercury and its Chemical Compounds. Crit. Rev. Toxicol. 36 (8), 609–662. doi:10.1080/10408440600845619
Compeau, G. C., and Bartha, R. (1985). Sulfate-Reducing Bacteria: Principal Methylators of Mercury in Anoxic Estuarine Sediment. Appl. Environ. Microbiol. 50 (2), 498–502.
Costa, M., and Liss, P. (2000). Photoreduction and Evolution of Mercury from Seawater. Sci. Total Environ. 261 (1-3), 125–135. doi:10.1016/s0048-9697(00)00631-8
Deonarine, A., and Hsu-Kim, H. (2009). Precipitation of Mercuric Sulfide Nanoparticles in NOM-Containing Water: Implications for the Natural Environment. Environ. Sci. Technol. 43 (7), 2368–2373. doi:10.1021/es803130h
Dittmar, T., Koch, B., Hertkorn, N., and Kattner, G. (2008). A Simple and Efficient Method for the Solid-phase Extraction of Dissolved Organic Matter (SPE-DOM) from Seawater. Limnol. Oceanogr. Methods 6, 230–235. doi:10.4319/lom.2008.6.230
Driscoll, C. T., Mason, R. P., Chan, H. M., Jacob, D. J., and Pirrone, N. (2013). Mercury as a Global Pollutant: Sources, Pathways, and Effects. Environ. Sci. Technol. 47 (10), 4967–4983. doi:10.1021/es305071v
Eagles-Smith, C. A., Silbergeld, E. K., Basu, N., Bustamante, P., Diaz-Barriga, F., Hopkins, W. A., et al. (2018). Modulators of Mercury Risk to Wildlife and Humans in the Context of Rapid Global Change. Ambio 47 (2), 170–197. doi:10.1007/s13280-017-1011-x
Feinberg, A. I., Kurien, U., and Ariya, P. A. (2015). The Kinetics of Aqueous Mercury(II) Reduction by Sulfite over an Array of Environmental Conditions. Water Air Soil Pollut. 226 (4). doi:10.1007/s11270-015-2371-0
Fitzgerald, W. F., Lamborg, C. H., and Hammerschmidt, C. R. (2007). Marine Biogeochemical Cycling of Mercury. Chem. Rev. 107 (2), 641–662. doi:10.1021/cr050353m
Garcia, E., Amyot, M., and Ariya, P. A. (2005). Relationship between DOC Photochemistry and Mercury Redox Transformations in Temperate Lakes and Wetlands. Geochi. Cosmochim. Acta 69 (8), 1917–1924. doi:10.1016/j.gca.2004.10.026
Gerbig, C. A., Kim, C. S., Stegemeier, J. P., Ryan, J. N., and Aiken, G. R. (2011a). Formation of Nanocolloidal Metacinnabar in Mercury-DOM-Sulfide Systems. Environ. Sci. Technol. 45 (21), 9180–9187. doi:10.1021/es201837h
Gerbig, C. A., Ryan, J. N., and Aiken, G. R. (2011b). “The Effects of Dissolved Organic Matter on Mercury Biogeochemistry,” in Environmental Chemistry and Toxicology of Mercury Editors Y. Cai, G. Liu, and N. O’Driscoll (New York, NY: Wiley), 259–292. doi:10.1002/9781118146644.ch8
Gilmour, C. C., Henry, A. E., and Mitchell, R. (1992). Sulfate stimulation of mercury methylation in freshwater sediments. Environ. Sci. Technol. 26 (11), 2281–2287. doi:10.1021/es00035a029
Ha, J., Zhao, X., Yu, R., Barkay, T., and Yee, N. (2017). Hg(II) Reduction by Siderite (FeCO3). Appl. Geochem. 78, 211–218. doi:10.1016/j.apgeochem.2016.12.017
Han, S., Lehman, R. D., Choe, K. Y., and Gill, G. A. (2007). Chemical and physical speciation of mercury in Offatts Bayou: A seasonally anoxic bayou in Galveston Bay. Limnol. Oceanogr. 52 (4), 1380–1392. doi:10.4319/lo.2007.52.4.1380
Han, Y.-S., Kim, S.-H., Chon, C.-M., Kwon, S., Kim, J. G., Choi, H. W., et al. (2020). Effect of FeS on Mercury Behavior in Mercury-Contaminated Stream Sediment: A Case Study of Pohang Gumu Creek in South Korea. J. Hazard. Mater. 393, 122373. doi:10.1016/j.jhazmat.2020.122373
Hua, B., and Deng, B. (2008). Reductive Immobilization of Uranium(VI) by Amorphous Iron Sulfide. Environ. Sci. Technol. 42 (23), 8703–8708. doi:10.1021/es801225z
Hyun, S. P., Davis, J. A., Sun, K., and Hayes, K. F. (2012). Uranium(VI) Reduction by Iron(II) Monosulfide Mackinawite. Environ. Sci. Technol. 46 (6), 3369–3376. doi:10.1021/es203786p
Jeong, H. Y., Lee, J. H., and Hayes, K. F. (2008). Characterization of Synthetic Nanocrystalline Mackinawite: Crystal Structure, Particle Size, and Specific Surface Area. Geochimica et Cosmochimica Acta 72 (2), 493–505. doi:10.1016/j.gca.2007.11.008
Jeong, H. Y., Sun, K., and Hayes, K. F. (2010). Microscopic and Spectroscopic Characterization of Hg(II) Immobilization by Mackinawite (FeS). Environ. Sci. Technol. 44 (19), 7476–7483. doi:10.1021/es100808y
Jeremiason, J. D., Portner, J. C., Aiken, G. R., Hiranaka, A. J., Dvorak, M. T., Tran, K. T., et al. (2015). Photoreduction of Hg(II) and Photodemethylation of Methylmercury: The Key Role of Thiol Sites on Dissolved Organic Matter. Environ. Sci. Process. Impacts 17 (11), 1892–1903. doi:10.1039/c5em00305a
Jiang, T., Skyllberg, U., Wei, S., Wang, D., Lu, S., Jiang, Z., et al. (2015). Modeling of the Structure-specific Kinetics of Abiotic, Dark Reduction of Hg(II) Complexed by O/N and S Functional Groups in Humic Acids while Accounting for Time-dependent Structural Rearrangement. Geochim. Cosmochim. Acta 154, 151–167. doi:10.1016/j.gca.2015.01.011
Jonsson, S., Mazrui, N. M., and Mason, R. P. (2016). Dimethylmercury Formation Mediated by Inorganic and Organic Reduced Sulfur Surfaces. Sci. Rep. 6. doi:10.1038/srep27958
Kim, C. S., Rytuba, J. J., and Brown, G. E. (2004). EXAFS Study of Mercury(II) Sorption to Fe- and Al-(hydr)oxides. J. Colloid Interf. Sci. 271 (1), 1–15. doi:10.1016/s0021-9797(03)00330-8
Kirsch, R., Scheinost, A. C., Rossberg, A., Banerjee, D., and Charlet, L. (2008). Reduction of Antimony by Nano-Particulate Magnetite and Mackinawite. Mineral. Mag. 72, 185–189. doi:10.1180/minmag.2008.072.1.185
Liu, J., Valsaraj, K. T., Devai, I., and DeLaune, R. D. (2008). Immobilization of Aqueous Hg(II) by Mackinawite (FeS). J. Hazard. Mater. 157 (2-3), 432–440. doi:10.1016/j.jhazmat.2008.01.006
Matthiessen, A. (1998). Reduction of Divalent Mercury by Humic Substances - Kinetic and Quantitative Aspects. Sci. Total Environ. 213, 177–183. doi:10.1016/s0048-9697(98)00090-4
Mauclair, C., Layshock, J., and Carpi, A. (2008). Quantifying the Effect of Humic Matter on the Emission of Mercury from Artificial Soil Surfaces. Appl. Geochem. 23 (3), 594–601. doi:10.1016/j.apgeochem.2007.12.017
Mazrui, N. M., Seelen, E., King'ondu, C. K., Thota, S., Awino, J., Rouge, J., et al. (2018). The Precipitation, Growth and Stability of Mercury Sulfide Nanoparticles Formed in the Presence of marine Dissolved Organic Matter. Environ. Sci. Process. Impacts 20 (4), 642–656. doi:10.1039/c7em00593h
Mergler, D., Anderson, H. A., Chan, L. H. M., Mahaffey, K. R., Murray, M., Sakamoto, M., et al. (2007). Methylmercury Exposure and Health Effects in Humans: A Worldwide Concern. AMBIO: A J. Hum. Environ. 36 (1), 3–11. doi:10.1579/0044-7447(2007)36[3:meahei]2.0.co;2
Merritt, K. A., and Amirbahman, A. (2007). Mercury Mobilization in Estuarine Sediment Porewaters: A Diffusive Gel Time-Series Study. Environ. Sci. Technol. 41 (3), 717–722. doi:10.1021/es061659t
Miretzky, P., Bisinoti, M. C., and Jardim, W. F. (2005). Sorption of Mercury (II) in Amazon Soils from Column Studies. Chemosphere 60 (11), 1583–1589. doi:10.1016/j.chemosphere.2005.02.050
Mishra, B., O’Loughlin, E. J., Boyanov, M. I., and Kemner, K. M. (2011). Binding of HgIIto High-Affinity Sites on Bacteria Inhibits Reduction to Hg0by Mixed FeII/IIIPhases. Environ. Sci. Technol. 45 (22), 9597–9603. doi:10.1021/es201820c
Muresan, B., Pernet-Coudrier, B., Cossa, D., and Varrault, G. (2011). Measurement and Modeling of Mercury Complexation by Dissolved Organic Matter Isolates from Freshwater and Effluents of a Major Wastewater Treatment Plant. Appl. Geochem. 26 (12), 2057–2063. doi:10.1016/j.apgeochem.2011.07.003
O'Driscoll, N. J., Siciliano, S. D., Lean, D. R. S., and Amyot, M. (2006). Gross Photoreduction Kinetics of Mercury in Temperate Freshwater Lakes and Rivers: Application to a General Model of DGM Dynamics. Environ. Sci. Technol. 40 (3), 837–843. doi:10.1021/es051062y
O’Loughlin, E. J., Kelly, S. D., Kemner, K. M., Csencsits, R., and Cook, E. R. (2003). Reduction of AgI, AuIII, CuII, and HgII by FeII/FeIII hydroxysulfate green rust. Chemosphere 53 (5), 437–446. doi:10.1016/S0045-6535(03)00545-9
O’Loughlin, E. J., Boyanov, M. I., Kemner, K. M., and Thalhammer, K. O. (2020). Reduction of Hg(II) by Fe(II)-bearing Smectite clay Minerals. Minerals 10 (12), 1079. doi:10.3390/min10121079
Ona-Nguema, G., Abdelmoula, M., Jorand, F., Benali, O., Block, J.-C., and Génin, J.-M. R. (2002). Iron(II,III) Hydroxycarbonate green Rust Formation and Stabilization from Lepidocrocite Bioreduction. Environ. Sci. Technol. 36 (1), 16–20. doi:10.1021/es0020456
Park, J.-S., Oh, S., Shin, M.-Y., Kim, M.-K., Yi, S.-M., and Zoh, K.-D. (2008). Seasonal Variation in Dissolved Gaseous Mercury and Total Mercury Concentrations in Juam Reservoir, Korea. Environ. Pollut. 154 (1), 12–20. doi:10.1016/j.envpol.2007.12.002
Parks, J. M., Johs, A., Podar, M., Bridou, R., Hurt, R. A., Smith, S. D., et al. (2013). The Genetic Basis for Bacterial Mercury Methylation. Science 339 (6125), 1332–1335. doi:10.1126/science.1230667
Pasakarnis, T. S., Boyanov, M. I., Kemner, K. M., Mishra, B., O’Loughlin, E. J., Parkin, G., et al. (2013). Influence of Chloride and Fe(II) Content on the Reduction of Hg(II) by Magnetite. Environ. Sci. Technol. 47 (13), 6987–6994. doi:10.1021/es304761u
Patterson, R. R., Fendorf, S., and Fendorf, M. (1997). Reduction of Hexavalent Chromium by Amorphous Iron Sulfide. Environ. Sci. Technol. 31 (7), 2039–2044. doi:10.1021/es960836v
Pecher, K., Haderlein, S. B., and Schwarzenbach, R. P. (2002). Reduction of Polyhalogenated Methanes by Surface-Bound Fe(II) in Aqueous Suspensions of Iron Oxides. Environ. Sci. Technol. 36, 1734–1741. doi:10.1021/es011191o
Peretyazhko, T., Charlet, L., and Grimaldi, M. (2006a). Production of Gaseous Mercury in Tropical Hydromorphic Soils in the Presence of Ferrous Iron: A Laboratory Study. Eur. J. Soil Sci. 57 (2), 190–199. doi:10.1111/j.1365-2389.2005.00729.x
Peretyazhko, T., Charlet, L., Muresan, B., Kazimirov, V., and Cossa, D. (2006b). Formation of Dissolved Gaseous Mercury in a Tropical lake (Petit-Saut Reservoir, French Guiana). Sci. Total Environ. 364 (1-3), 260–271. doi:10.1016/j.scitotenv.2005.06.016
Podar, M., Gilmour, C. C., Brandt, C. C., Soren, A., Brown, S. D., Crable, B. R., et al. (2015). Global Prevalence and Distribution of Genes and Microorganisms Involved in Mercury Methylation. Sci. Adv. 1, e1500675. doi:10.1126/sciadv.1500675
Ravichandran, M., Aiken, G. R., Reddy, M. M., and Ryan, J. N. (1998). Enhanced Dissolution of Cinnabar (Mercuric Sulfide) by Dissolved Organic Matter Isolated from the Florida Everglades. Environ. Sci. Technol. 32, 3305–3311. doi:10.1021/es9804058
Ravichandran, M. (2004). Interactions between Mercury and Dissolved Organic Matter-Aa Review. Chemosphere 55 (3), 319–331. doi:10.1016/j.chemosphere.2003.11.011
Remy, P.-P., Etique, M., Hazotte, A. A., Sergent, A.-S., Estrade, N., Cloquet, C., et al. (2015). Pseudo-first-order Reaction of Chemically and Biologically Formed green Rusts with HgII and C15H15N3O2: Effects of pH and Stabilizing Agents (Phosphate, Silicate, Polyacrylic Acid, and Bacterial Cells). Water Res. 70, 266–278. doi:10.1016/j.watres.2014.12.007
Richard, J.-H., Bischoff, C., Ahrens, C. G. M., and Biester, H. (2016). Mercury (II) Reduction and Co-precipitation of Metallic Mercury on Hydrous Ferric Oxide in Contaminated Groundwater. Sci. Total Environ. 539, 36–44. doi:10.1016/j.scitotenv.2015.08.116
Rickard, D. (2006). The Solubility of FeS. Geochim. Cosmochim. Acta 70, 5779–5789. doi:10.1016/j.gca.2006.02.029
Rivera, N. A., Bippus, P. M., and Hsu-Kim, H. (2019). Relative Reactivity and Bioavailability of Mercury Sorbed to or Coprecipitated with Aged Iron Sulfides. Environ. Sci. Technol. 53 (13), 7391–7399. doi:10.1021/acs.est.9b00768
Rocha, J., Sargentini, E., Zara, L. F., Rosa, A. H., Santos, A. D., and Burba, P. (2003). Reduction of Mercury(II) by Tropical River Humic Substances (Rio Negro)-Part II. Influence of Structural Features (Molecular Size, Aromaticity, Phenolic Groups, Organically Bound Sulfur). Talanta 61 (5), 699–707. doi:10.1016/s0039-9140(03)00351-5
Schwarzenbach, R. P., and Stone, A. T. (2003). Mineral Surface Catalysis of Reactions between FeII and Oxime Carbamate Pesticides. Geochimica et Cosmochimica Acta 67, 2775–2791. doi:10.1016/s0016-7037(03)00281-3
Seelen, E. A. (2018). A Multi-Estuary Appraoch to Better Understand the Sources and Fate of Methylmercury within Estuarine Water Columns. PhD thesis (Storrs, CT:University of Connecticut), 168.
Skyllberg, U., and Drott, A. (2010). Competition between Disordered Iron Sulfide and Natural Organic Matter Associated Thiols for Mercury(II)-An EXAFS Study. Environ. Sci. Technol. 44 (4), 1254–1259. doi:10.1021/es902091w
Slowey, A. J. (2010). Rate of Formation and Dissolution of Mercury Sulfide Nanoparticles: The Dual Role of Natural Organic Matter. Geochimica et Cosmochimica Acta 74 (16), 4693–4708. doi:10.1016/j.gca.2010.05.012
Spangler, W. J., Spigarelli, J. L., Rose, J. M., Flippin, R. S., and Miller, H. H. (1973). Degradation of Methylmercury by Bacteria Isolated from Environmental Samples. Appl. Microbiol. 25 (4), 488–493. doi:10.1128/aem.25.4.488-493.1973
Steffan, R. J., Korthals, E. T., and Winfrey, M. R. (1988). Effects of Acidification on Mercury Methylation, Demethylation, and Volatilization in Sediments from an Acid-Susceptible lake. Appl. Environ. Microbiol. 54 (8), 2003–2009. doi:10.1128/aem.54.8.2003-2009.1988
Sunderland, E. M., Li, M., and Bullard, K. (2018). Decadal Changes in the Edible Supply of Seafood and Methylmercury Exposure in the United States. Environ. Health Perspect. 126 (1), 017006. doi:10.1289/ehp2644
Van Loon, L. L., Mader, E. A., and Scott, S. L. (2001). Sulfite Stabilization and Reduction of the Aqueous Mercuric Ion: Kinetic Determination of Sequential Formation Constants. J. Phys. Chem. A. 105 (13), 3190–3195. doi:10.1021/jp003803h
Vollier, E., Inglett, P. W., Hunter, K., Roychoudhury, A. N., and Cappellen, P. V. (2000). Book Reviews. Housing Stud. 15, 785–790. doi:10.1080/02673030050134619
Waples, J. S., Nagy, K. L., Aiken, G. R., and Ryan, J. N. (2005). Dissolution of Cinnabar (HgS) in the Presence of Natural Organic Matter. Geochim. Cosmochim. Acta 69 (6), 1575–1588. doi:10.1016/j.gca.2004.09.029
Whalin, L., Kim, E.-H., and Mason, R. (2007). Factors Influencing the Oxidation, Reduction, Methylation and Demethylation of Mercury Species in Coastal Waters. Mar. Chem. 107 (3), 278–294. doi:10.1016/j.marchem.2007.04.002
Wiatrowski, H. A., Das, S., Kukkadapu, R., Ilton, E. S., Barkay, T., and Yee, N. (2009). Reduction of Hg(II) to Hg(0) by Magnetite. Environ. Sci. Technol. 43 (14), 5307–5313. doi:10.1021/es9003608
Wolfenden, S., Charnock, J. M., Hilton, J., Livens, F. R., and Vaughan, D. J. (2005). Sulfide Species as a Sink for Mercury in lake Sediments. Environ. Sci. Technol. 39 (17), 6644–6648. doi:10.1021/es048874z
Wolthers, M., Charlet, L., van Der Linde, P. R., Rickard, D., and van Der Weijden, C. H. (2005). Surface Chemistry of Disordered Mackinawite (FeS). Geochimica et Cosmochimica Acta 69 (14), 3469–3481. doi:10.1016/j.gca.2005.01.027
Zheng, W., and Hintelmann, H. (2010). Isotope Fractionation of Mercury during its Photochemical Reduction by Low-Molecular-Weight Organic Compounds. J. Phys. Chem. A. 114 (12), 4246–4253. doi:10.1021/jp9111348
Zheng, W., Liang, L., and Gu, B. (2012). Mercury Reduction and Oxidation by Reduced Natural Organic Matter in Anoxic Environments. Environ. Sci. Technol. 46 (1), 292–299. doi:10.1021/es203402p
Keywords: mercury, reduction, iron sulfide, cadmium sulfide, dissolved organic matter
Citation: Coulibaly M, Mazrui NM, Jonsson S and Mason RP (2021) Abiotic Reduction of Mercury(II) in the Presence of Sulfidic Mineral Suspensions. Front. Environ. Chem. 2:660058. doi: 10.3389/fenvc.2021.660058
Received: 28 January 2021; Accepted: 27 May 2021;
Published: 14 June 2021.
Edited by:
Martin Jiskra, University of Basel, SwitzerlandReviewed by:
Amrika Deonarine, Texas Tech University, United StatesCopyright © 2021 Coulibaly, Mazrui, Jonsson and Mason. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Robert P. Mason, cm9iZXJ0Lm1hc29uQHVjb25uLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.