 Wei Liu1*
Wei Liu1* Fei Yan
Fei Yan
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Energy Res. , 22 September 2023
Sec. Process and Energy Systems Engineering
Volume 11 - 2023 | https://doi.org/10.3389/fenrg.2023.1276626
This article is part of the Research Topic Low-grade Thermal Energy Conversion and Utilization View all 6 articles
The thermal decomposition mechanism of n-hexane is investigated by using density functional theory and ReaxFF force field. The initial decomposition reactions, the effect of temperature on thermal decomposition and first-order kinetics are analyzed. The results show that the C-C bonds in n-hexane molecule are more easily decomposed than that of C-H bonds, and the breakage of C3-C4 bond is the main initial decomposition reaction. The main decomposition products of n-hexane are H2, CH4, C2H2, C2H4, C2H6, and C3H6. The decomposition rate of n-hexane is accelerated by temperature. The apparent activation energy and pre-exponential factor of n-hexane thermal decomposition are 209.8 kJ mol−1 and 1.1 × 1013 s−1, respectively.
With the rapid development of economy, energy shortage and environmental pollution have become the key problems facing mankind (Chen et al., 2019; Wang et al., 2022; Tang et al., 2023). Therefore, the development of environment-friendly energy conversion technology is necessary to improve the energy efficiency of low-grade energy (Zhang et al., 2017; Wang et al., 2018a; Zhang C. et al., 2018). Among the energy conversion technologies, the organic Rankine cycle (ORC), which uses organic fluid as the working fluid, can effectively convert waste heat (Tumen Ozdil et al., 2015; Wang et al., 2020), solar energy (Twomey et al., 2013), biomass energy (Uris et al., 2015), ocean thermal energy (Chen and Huo, 2023; Huo et al., 2023) and geothermal energy (Liu et al., 2015; Wang et al., 2018b) into electricity. Ozahi et al. (Ozahi et al., 2017) investigated the thermodynamic performance of ORC system for usage in municipal solid waste power plant by using n-hexane, n-pentane, HFC-245fa, isobutene and HCFC-141b as working fluids, the results indicated that n-hexane has the highest energetic and energetic efficiencies at all working fluids. Aljundi et al. (Aljundi, 2011) investigated the impact of alternative dry fluids on the performance of ORC and compared them with other working fluids, they found that n-hexane was the best working fluid in this ORC system and HFC-227ea was the worst.
For low-temperature ORCs, lower heat source temperatures usually result in high heat recovery costs and low heat efficiency. Previous results have shown that supercritical ORCs has higher output work and thermal efficiency than subcritical ORCs. Karellas et al. (Karellas and Schuster, 2008) used HFC-134a and HFC-245fa as working fluid to evaluate the performance of ORC, and the results showed that the thermal efficiency of subcritical ORC was much lower than that of supercritical ORC. Oyewunmi et al. (Oyewunmi et al., 2016) used the same working fluid to compare the heat recovery performance of supercritical ORC and subcritical ORC through theoretical calculation, and the results indicated that supercritical ORC had higher thermal efficiency and output work.
However, the supercritical ORC has a high heat source temperature, and the working fluid may decompose in the process of circulation (Huo et al., 2017; Huo et al., 2022a; Huo et al., 2022b). The decomposition products of the working fluid may consist of gaseous, liquid, and solid states, which will affect the performance and even the safety of ORC (Huo et al., 2017; Huo et al., 2019a). The gaseous products will increase the condensing pressure of the ORC system, thereby reducing the performance of the system. The liquid products will mix with the working fluid, and then affect the thermophysical properties of the working fluid. Solid products can clog pipes and components, creating a system safety hazard. For example, Rajabloo et al. (Raja et al., 2017) investigated the effect of the pyrolysis of octamethyltrisiloxane and isopentane on the ORC systems, the results indicated that pyrolysis products have a significant effect on the cycling efficiency, the performance of ORC system will be reduced by the pyrolysis product CH4. The impact of n-pentane decomposition on the supercritical ORC system performance was studied by Dai et al. (Andersen and Bruno, 2005), they found that the condensation pressure was increased by the generation of non-condensable gaseous produces. Therefore, thermal stability of working fluid is one of the key factors to be considered in the screening of working fluids (Pu et al., 2020; Xin et al., 2020; Li et al., 2022; Si et al., 2022).
At present, the thermal stability and decomposition mechanism of alkanes have been studied extensively. Andersen et al. (Andersen and Bruno, 2005) investigated the thermal stability of hydrocarbon working fluids by using a miniature reactor, they found that the temperature can be considered as an indicator of the thermal stability of hydrocarbon working fluids. The thermal stability of cyclopentane was investigated by Ginosar et al. (Ginosar et al., 2011) using a circulation system, the results showed that the 300°C was its safe using temperature, and the presence of air has a negative effect on the thermal stability of cyclopentane. The thermal stability of n-hexane, cyclopentane, isopentane, n-pentane, isobutene and n-butane was studied by Dai et al. (Dai et al., 2013; Dai et al., 2016a) by using reactor, they found that the decomposition temperatures of these working fluids were 260–280°C, 260–280°C, 290–310°C, 280–300°C, 330–350°C, and 300–320°C, respectively. Xin et al. (Xin et al., 2020) investigated the decomposition mechanism of toluene, benzene, n-hexane, isohexane, cyclohexane, n-pentane, isopentane, cyclopentane, n-butane, isobutene and cyclobutane by using ReaxFF molecular dynamic (RMD) and density functional theory (DFT) method, they found that the initial decomposition reactions of hydrocarbon working fluids were the cracking of C-C and C-H bonds, and the C-H bonds of most hydrocarbon working fluids were more difficult to decompose than the C-H bonds, however, the work was mainly concerned with the thermal decomposition mechanisms of cyclo-butane, cyclopentane and cyclohexane, with only occasional mention of n-hexane. As a potential working fluid in ORC system, the thermal decomposition mechanism of n-hexane should be further investigated.
In this paper, the thermal decomposition mechanism of n-hexane is calculated by DFT calculation and RMD simulation, which can be divided into the following three parts. Firstly, detailed DFT calculation and RMD simulation details are provided. Secondly, the initial decomposition reactions of n-hexane and the effect of temperature on the decomposition of n-hexane are studied, and the first-order kinetic analysis is carried out. Finally, a conclusion on the thermal decomposition mechanism of n-hexane is given.
In this paper, M06-2X/6-31 + G (d, p) method (Zhao and Truhlar, 2007; Zhao and Truhlar, 2008) is used to optimize the geometric structures of the reactants and products involved in the initial decomposition reactions. The zero-point energy (ZPE) correction is carried out for all the geometric structures. All the optimized structures are analyzed by frequency calculation, and neither the reactants nor the products have imaginary frequencies. All the reaction paths are calculated by G09W software (Gaussian, 2009).
In this paper, the ReaxFF force field (Chenoweth et al.) applicable to the description of C/H atoms is used to study the thermal decomposition mechanism of n-hexane. A periodic box containing 100 n-hexane molecules is established at 573.15 K and 4.0 MPa. First, the energy of the system is minimized, and then the NVT equilibrium simulation of 50 ps is performed. After equilibrium simulation, the 1,000 ps NVT simulations at the reaction temperature of 2000–3000 K are performed to investigate the effect of temperature on the thermal decomposition of n-hexane, the temperature interval is set as 200 K.
For reactive molecular dynamics simulation, the simulation time scale (ps) is much lower than the experimental time scale (s) due to the limitation of computing power. Arrhenius formula is applicable to the study of thermal decomposition characteristic of hydrocarbon working fluids (Dai et al., 2016b). According to Arrhenius equation, temperature does not affect the apparent activation energy of working fluid decomposition but only the decomposition rate, thus the reaction rate is often accelerated by increasing the simulated temperature (Castro-Marcano and van Duin, 2013; Zhang X. et al., 2018; Xin et al., 2020). Previous studies found that the ReaxFF reactive molecular dynamic simulation results with higher simulation temperatures are consistent with the experimental results. For example, the apparent activation energy of n-pentane decomposition obtained by the ReaxFF reactive molecular dynamic simulations was 228.33 kJ mol−1 (Xin et al., 2020), this value was well consistent with the experimental result of 227.57 kJ mol−1 (Dai et al., 2016b). The apparent activation energies of n-decane decomposition obtained by the ReaxFF reactive molecular dynamic simulations were 57.80–65.43 kcal mol−1 (Wang et al., 2012), the values also well agree with the experimental value of 64.29 ± 2.39 kcal mol−1 (Stewart et al., 1998). Therefore, from the perspective of the whole pyrolysis process of hydrocarbon working fluids, the simulation results are in good agreement with the experimental results. Previous study (Xin et al., 2020) has also shown that increasing temperature only increases the thermal decomposition rates of working fluids and the formation rates of products. Therefore, 2000 K–3000 K are set to the simulation temperatures in this study to accelerate the decomposition rate of n-hexane.
In order to understand the thermal decomposition mechanism of n-hexane, it is necessary to investigate the initial decomposition reactions of n-hexane. The initial decomposition reactions of n-hexane involves a total of 6 reaction paths, Paths 1–3 are the dehydrogenation reactions, and Paths 4–6 are mainly caused by the breakage of C-C bonds. The bond dissociation energies of initial decomposition reactions are calculated by DFT method.
As shown in Figure 1, Path1 refers to the separation of H atom on C1 atom from the n-hexane molecule to generate C6H13-1 and H radicals., and the bond dissociation energy of this reaction is 420.1 kJ mol−1. Paths 2 and 3 occur mainly through the cleavage of C2-H and C3-H with bond dissociation energies of 408.0 and 403.9 kJ mol−1, respectively. The bond dissociation energies of C-C bonds are lower than that of C-H bonds, and the bond dissociation energies of Paths 4, 5, and 6 are 372.7, 367.0, and 362.3 kJ mol−1, respectively. It can be seen that the bond dissociation energy of C3-C4 bond is the lowest, which indicates that Path6 is the kinetic optimal path, which is most likely to occur in the initial decomposition reactions, and then generate C3H7 radicals.
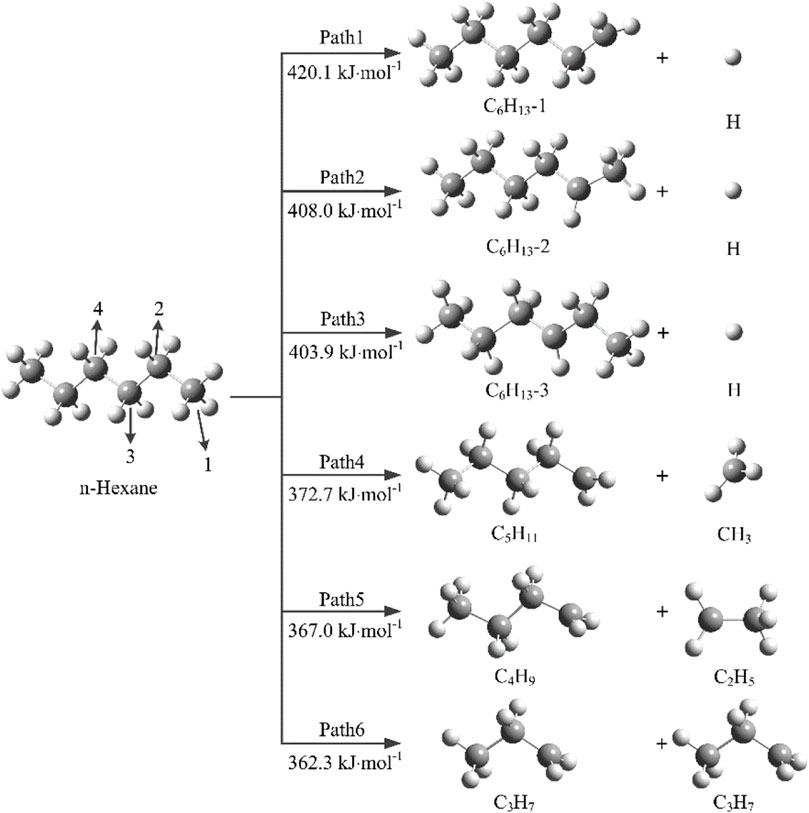
FIGURE 1. Possible initial decomposition reactions and bond dissociation energies of n-hexane.
After the initial decomposition reactions of n-hexane, the corresponding radicals are generated immediately, and these radicals will continue to undergo decomposition reactions or collide with other radicals and n-hexane molecules to generate corresponding products. The thermal decomposition products of n-hexane at 2600 K are shown in Figure 2.
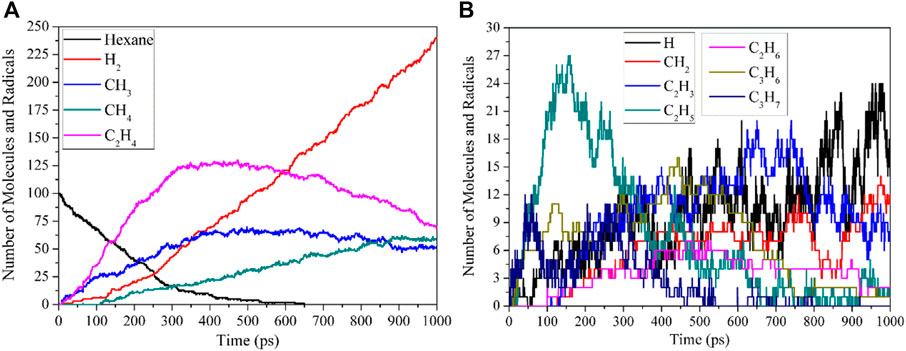
FIGURE 2. Reactant and main products during thermal decomposition of n-hexane at 2600 K. (A, B) both show the number evolution of Molecules and Radicals.
It can be seen from Figure 2 that the thermal decomposition products of n-hexane are H2, CH4, C2H4, and C3H6. The initial generation time of C2H4 and C3H6 is earlier than that of other products, which is also consistent with the research results in Section 2.1. Because the bond dissociation energy of C3-C4 bond is the lowest, C3H7 radical is most easily generated, and C3H7 radicals undergo dehydrogenation and demethylation to produce C3H6 and C2H4 molecules, so these two products are first generated. CH4 molecule is formed mainly by the combination of two CH3 groups. The C2H6 formation path is mainly divided into three categories: the first type is produced by the collision of C2H5 radicals with other radicals or H atoms on the molecules; the second type is produced by the combination reactions between C2H5 radicals and H radicals; the third type is produced by the combination of CH3 radicals and CH3 radicals. In the thermal decomposition process of n-hexane, the reactions of large radicals is mainly in the direction of decomposition, so the paths of forming C3H8, C4H10 and C5H12 molecules are the secondary reaction paths, so these molecular number are small. H2 molecule is mainly produced by the collision of H radicals with other radicals or molecules, and the combination of H radicals. Since the radicals and molecular products produced by the thermal decomposition of n-hexane will continue to decompose to produce alkenes and alkynes, most of the separated H elements are stable in the form of H2 molecules. CH4 molecule starts later than C2H4 and C3H6 because the bond dissociation energy of C1-C2 bond is slightly higher than that of C2-C3 bond and C3-C4 bond. CH4 molecule is mainly produced by the collision of CH3 radicals produced by the decomposition of n-hexane and other fragments with other radicals or H atom on the molecule. In addition, the alkanes and alkenes produced during thermal decomposition will continue to decompose to form alkynes, so the number of C2H2 molecules will continue to increase over time. After analysis, it can be seen that the main thermal decomposition products of hexane are H2, CH4, C2H4, and C3H6, so the influence of temperature on the generation of these products is mainly discussed in the next section.
The reaction temperature has an important influence on the thermal decomposition of the working fluid. The evolution of n-hexane and the number of main products molecules over time at different temperatures is shown in Figure 3.
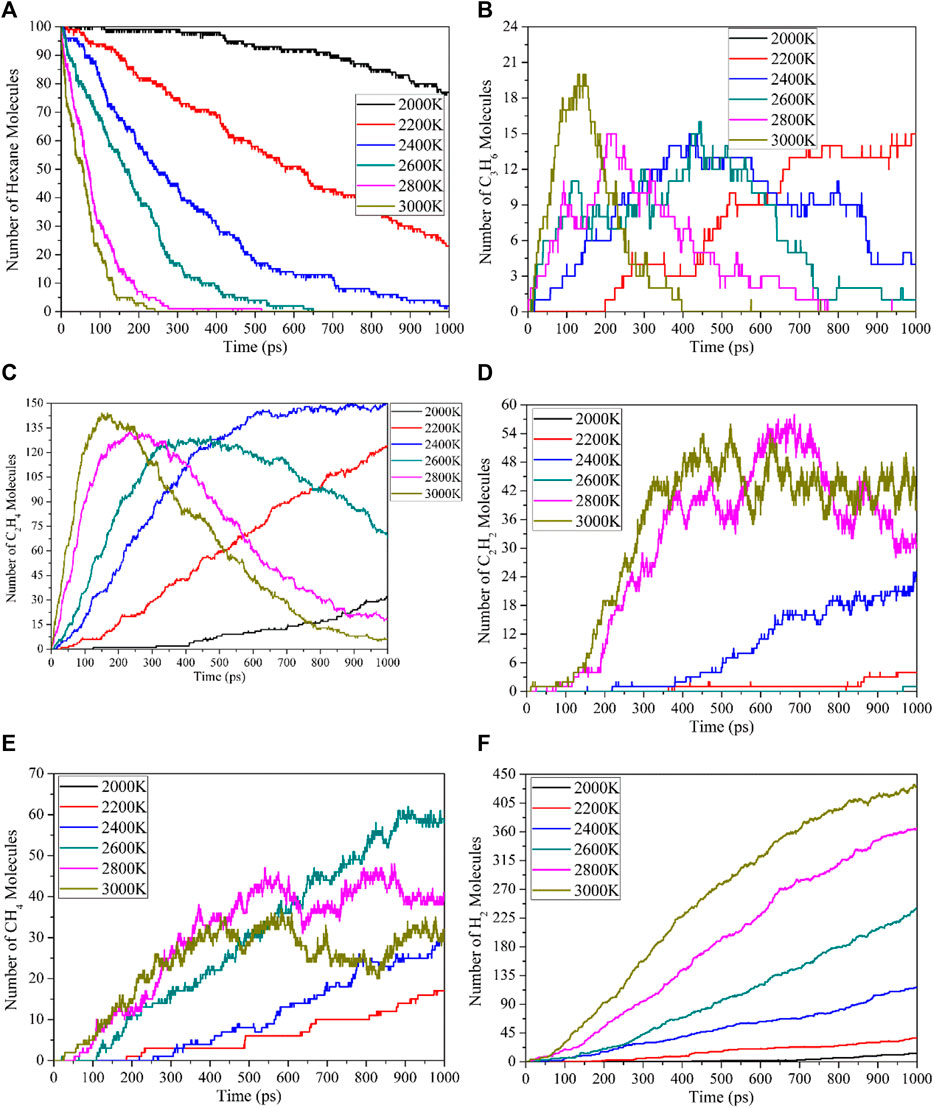
FIGURE 3. Time evolution of number of n-hexane and main products molecules at different temperatures: (A) n-hexane, (B) C3H6, (C) C2H4, (D) C2H2, (E) CH4, (F) H2.
As shown in Figure 3A, the decomposition rate of n-hexane increases with the increase of temperature, and the higher reaction temperature reduces the initial decomposition time of n-hexane, accelerates the decomposition rate and promotes the decomposition of n-hexane. The thermal decomposition of n-hexane mainly generates C3H7 radical, which will undergo dehydrogenation to produce C3H6 molecule, The number of C3H6 molecules generated over time is shown in Figure 3B. In addition, little C3H8 molecules are produced in the reaction system, because the reactions to produce propane is the secondary reactions. C3H8 molecules will continue to decompose to form C2H5 + CH3 and C3H7 + H, and then form the C3H6 and C2H4 molecules. C2H4 and C2H2 molecules are the main C2 molecular products of the thermal decomposition of n-hexane, and their molecular numbers change over time as shown in Figures 3C, D. The C2H4 molecule undergo a dehydrogenation reaction to form C2H2 molecule. In addition, the C3H6 molecule will also undergo demethylation to form C2H2 molecule, so the number of molecules of C2H2 will gradually increase with the increase of time. CH4 is the main small molecular product of the thermal decomposition of n-hexane and the only single carbon molecular product. From Figure 3E, it can be seen that the number of CH4 molecules first increases with time at all temperatures, reaching a peak value at 3000 K and 2800 K, and then slowly decreases. This is due to the decomposition of CH4 molecules into CF3, CF2 and CF radicals, these radicals will combine to form C2H4 and C2H2 molecules. H2 is the most important product of the thermal decomposition of n-hexane, as shown in Figure 3F. Since the thermal decomposition reactions of n-hexane is mainly in the direction of the formation of C=C and C≡C bonds, most of the other H elements except alkenes and alkynes exist in the form of H2 molecules, so H2 is the product with the most molecules. The higher the reaction temperature, the faster the H2 molecule is formed, which also proves the promotion of temperature on the thermal decomposition of n-hexane.
In previous studies on thermal decomposition of working fluid, the decomposition rate of working fluid is often used to study the first-order kinetics of working fluid decomposition (Dai et al., 2016b; Huo et al., 2017). Therefore, this work uses first-order kinetics to characterize the thermal decomposition of n-hexane:
In this work, the number of molecules is used instead of the concentration of n-hexane. The rate constant k at different temperatures is obtained from the linear fitting of Nt and time t:
Where k is the rate constant, t is the time, Nt is the number of n-hexane molecules at time t, and N0 is the number of n-hexane molecules at the beginning.
Then the rate constant k at different temperatures is substituted into the Arrhenius equation:
Where A refers to the pre-exponential factor, R is the gas constant, Ea is the apparent activation energy, and T is the thermodynamic temperature.
Formula (3) shows that lnk is linearly dependent on 1/T because Ea and A are assumed to be constants in the Arrhenius equation, so Ea and A can be obtained by linear fitting. The fitted Arrhenius equation for the thermal decomposition of n-hexane is shown in Figure 4. The calculated Ea is 209.8 kJ mol−1, and the pre-exponential factor is 1.1 × 1013 s−1. The apparent activation energy Ea of n-hexane thermal decomposition obtained from RMD simulations is consistent with the experimental value of Li et al. (Li and Zhao, 2015) (208.15 ± 1.0 kJ mol−1). Therefore, the results obtained from the RMD simulations on the thermal decomposition of working fluid are reliable.
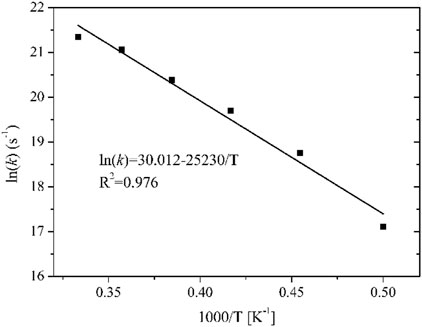
FIGURE 4. First-order kinetic fitting diagram of the rate constant and temperature of n-hexane decomposition at 2000–3000 K.
In this work, the thermal decomposition mechanism of n-hexane is studied by using DFT calculations and RMD simulations. The initial decomposition reactions of n-hexane can be divided into two categories: one is caused by the breakage of the C-H bond, and the other is caused by the cracking of the C-C bond. The C3-C4 bond has the lowest dissociation energy and is the most prone to homolytic reaction. The main decomposition products of n-hexane are H2, CH4, C2H2, C2H4, C2H6 and C3H6. The increase of temperature can accelerate the decomposition rate of n-hexane and promote the formation of decomposition products. The first order kinetic analysis of the thermal decomposition of n-hexane is carried out, and the apparent activation energy is calculated to be 209.8 kJ mol−1.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
WL: Conceptualization, Writing–original draft, Writing–review and editing. NW: Conceptualization, Writing–original draft. JC: Visualization, Writing–original draft. AS: Data curation, Writing–original draft. FY: Methodology, Supervision, Writing–original draft, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by Chongqing Research Program of Basic Research and Frontier Technology (No. cstc2020jcyj-msxmX0840).
The authors would like to acknowledge the colleagues from the State Power Investment Corporation Research Institute for their perspectives and suggestions related to data collection and statistical analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aljundi, I. H. (2011). Effect of dry hydrocarbons and critical point temperature on the efficiencies of organic Rankine cycle. Renew. Energy 36 (4), 1196–1202. doi:10.1016/j.renene.2010.09.022
Andersen, W. C., and Bruno, T. J. (2005). Rapid screening of fluids for chemical stability in organic Rankine cycle applications. Industrial Eng. Chem. Res. 44, 5560–5566. doi:10.1021/ie050351s
Castro-Marcano, F., and van Duin, A. C. T. (2013). Comparison of thermal and catalytic cracking of 1-heptene from ReaxFF reactive molecular dynamics simulations. Combust. Flame 160, 766–775. doi:10.1016/j.combustflame.2012.12.007
Chen, W., and Huo, E. (2023). Opportunities and challenges of ocean thermal energy conversion technology. Front. Energy Res. 11, 1207062. doi:10.3389/fenrg.2023.1207062
Chen, X., Liu, C., Li, Q., Wang, X., and Xu, X. (2019). Dynamic analysis and control strategies of Organic Rankine Cycle system for waste heat recovery using zeotropic mixture as working fluid. Energy Convers. Manag. 192, 321–334. doi:10.1016/j.enconman.2019.04.049
Chenoweth, K., van Duin, A. C. T., and Goddard, W. A. (2008). ReaxFF reactive force field for molecular dynamics simulations of hydrocarbon oxidation. J. Phys. Chem. A 112 (5), 1040–1053. doi:10.1021/jp709896w
Dai, X., Shi, L., An, Q., and Qian, W. (2016b). Chemical kinetics method for evaluating the thermal stability of Organic Rankine Cycle working fluids. Appl. Therm. Eng. 100, 708–713. doi:10.1016/j.applthermaleng.2016.02.091
Dai, X., Shi, L., An, Q., and Qian, W. (2016a). Screening of hydrocarbons as supercritical ORCs working fluids by thermal stability. Energy Convers. Manag. 126, 632–637. doi:10.1016/j.enconman.2016.08.024
Dai, X. Y., An, Q. S., and Shi, L. (2013). Experiment research for the thermal stability of isobutene and isopentane. J. Eng. Thermophys. 34 (8), 1416–1419.
Ginosar, D. M., Petkovic, L. M., and Guillen, D. P. (2011). Thermal stability of cyclopentane as an organic rankine cycle working fluid. Energy & Fuels 25 (9), 4138–4144. doi:10.1021/ef200639r
Huo, E., Chen, W., Deng, Z., Gao, W., and Chen, Y. (2023). Thermodynamic analysis and optimization of a combined cooling and power system using ocean thermal energy and solar energy. Energy 278, 127956. doi:10.1016/j.energy.2023.127956
Huo, E., Hu, Z., Wang, S., Xin, L., and Bai, M. (2022b). Thermal decomposition and interaction mechanism of HFC-227ea/n-hexane as a zeotropic working fluid for organic Rankine cycle. Energy 246, 123435. doi:10.1016/j.energy.2022.123435
Huo, E., Liu, C., Xin, L., Li, X., Xu, X., Li, Q., et al. (2019a). Thermal stability and decomposition mechanism of HFO-1336mzz (Z) as an environmental friendly working fluid: experimental and theoretical study. Int. J. Energy Res. 43 (9), 4630–4643. doi:10.1002/er.4599
Huo, E., Liu, C., Xu, X., and Dang, C. (2017). A ReaxFF-based molecular dynamics study of the pyrolysis mechanism of HFO-1336mzz(Z). Int. J. Refrig. 83, 118–130. doi:10.1016/j.ijrefrig.2017.07.009
Huo, E., Liu, C., Xu, X., Li, Q., and Dang, C. (2018b). A ReaxFF-based molecular dynamics study of the oxidation decomposition mechanism of HFO-1336mzz(Z). Int. J. Refrig. 93, 249–258. doi:10.1016/j.ijrefrig.2018.06.019
Huo, E., Liu, C., Xu, X., Li, Q., and Dang, C. (2018a). Dissociation mechanisms of HFO-1336mzz(Z) on Cu(1 1 1), Cu(1 1 0) and Cu(1 0 0) surfaces: A density functional theory study. Appl. Surf. Sci. 443, 389–400. doi:10.1016/j.apsusc.2018.03.001
Huo, E., Liu, C., Xu, X., Li, Q., Dang, C., Wang, S., et al. (2019c). The oxidation decomposition mechanisms of HFO-1336mzz(Z) as an environmentally friendly refrigerant in O2/H2O environment. Energy 185, 1154–1162. doi:10.1016/j.energy.2019.07.140
Huo, E., Liu, C., Xu, X., Liu, L., and Wang, S. (2019b). Dissociation mechanism of HFC-245fa on Cu(1 1 1) surfaces with and without oxygen-covered: A density functional theory study. Appl. Surf. Sci. 480, 487–496. doi:10.1016/j.apsusc.2019.03.016
Huo, E., Xin, L., Wang, S., and Liu, C. (2021). The impact of H2O on the combustion of n-pentane: A reactive molecular dynamic simulation study. J. Mol. Liq. 345, 117036. doi:10.1016/j.molliq.2021.117036
Huo, E., Xin, L., and Wang, S. (2022a). Thermal stability and pyrolysis mechanism of working fluids for organic rankine cycle: A review. Int. J. Energy Res. 46 (14), 19341–19356. doi:10.1002/er.8518
Huo, E., Xin, L., Zhang, S., Liu, C., Wang, S., and Zhang, L. (2022d). The combustion mechanism of leaking propane (R290) in O2 and O2/H2O environments: reaxFF molecular dynamics and density functional theory study. Process Saf. Environ. Prot. 161, 603–610. doi:10.1016/j.psep.2022.03.080
Huo, E., Zhang, S., Xin, L., Wang, S., Cai, S., Zhang, L., et al. (2022c). Pyrolysis mechanism study of n-heptane as an endothermic hydrocarbon fuel: A reactive molecular dynamic simulation and density functional theory calculation study. Comput. Theor. Chem. 1211, 113696. doi:10.1016/j.comptc.2022.113696
Karellas, S., and Schuster, A. (2008). Supercritical fluid parameters in organic rankine cycle applications. Int. J. Thermodyn. 11 (3), 101–108.
Li, D., and Zhao, Y. (2015). Understanding the chain mechanism of radical reactions in n-hexane pyrolysis. Res. Chem. Intermed. 41, 3507–3529. doi:10.1007/s11164-013-1468-6
Li, Q., Ren, J., Liu, Y., and Zhou, Y. (2022). Prediction of critical properties and boiling point of fluorine/chlorine-containing refrigerants. Int. J. Refrig. 143, 28–36. doi:10.1016/j.ijrefrig.2022.06.024
Liu, Q., Shen, A., and Duan, Y. (2015). Parametric optimization and performance analyses of geothermal organic Rankine cycles using R600a/R601a mixtures as working fluids. Appl. Energy 148, 410–420. doi:10.1016/j.apenergy.2015.03.093
Oyewunmi, O. A., Ferré-Serres, S., Lecompte, S., van den Broek, M., De Paepe, M., and Markides, C. N. (2016). “An assessment of subcritical and trans-critical organic Rankine cycles for waste-heat recovery,” in The International Conference on Applied Energy – ICAE, December 3 to 7, 2023, Beijing, China.
Ozahi, E., Tozlu, A., and Abusoglu, A. (2017). Thermodynamic performance assessment of different fluids in a typical organic Rankine cycle for usage in municipal solid waste power plant. ACTA Phys. POL. A 132, 807–812. doi:10.12693/APhysPolA.132.807
Pu, Y., Liu, C., Li, Q., Xu, X., and Huo, E. (2020). Pyrolysis mechanism of HFO-1234yf with R32 by ReaxFF MD and DFT method. Int. J. Refrig. 109, 82–91. doi:10.1016/j.ijrefrig.2019.09.020
Rajabloo, T., Bonalumi, D., and Iora, P. (2017). Effect of a partial thermal decomposition of the working fluid on the performances of ORC power plants. Energy 133, 1013–1026. doi:10.1016/j.energy.2017.05.129
Si, M., Kuai, L., Huo, E., Li, L., and Bai, M. (2022). Thermal decomposition and interaction mechanism of HFC-134a/HFC-32 mixture in Organic Rankine Cycle. J. Environ. Chem. Eng. 10 (6), 108947. doi:10.1016/j.jece.2022.108947
Song, Z., Bai, M., Yang, Z., Lei, H., Qian, M., Zhao, Y., et al. (2022). Gasification of alpha-O-4 linkage lignin dimer in supercritical water into hydrogen and carbon monoxide: reactive molecular dynamic simulation study. Fuel 329, 125387. doi:10.1016/j.fuel.2022.125387
Stewart, J., Brezinsky, K., and Glassman, I. (1998). Supercritical pyrolysis of decalin, tetralin, and n-decane at 700—800K. Product distribution and reaction mechanism. Combust. Sci. Technol. 136, 373–390. doi:10.1080/00102209808924178
Tang, J., Li, Q., Wang, S., and Yu, H. (2023). Thermo-economic optimization and comparative analysis of different organic flash cycles for the supercritical CO2 recompression Brayton cycle waste heat recovery. Energy 278, 128002. doi:10.1016/j.energy.2023.128002
Tumen Ozdil, N. F., Segmen, M. R., and Tantekin, A. (2015). Thermodynamic analysis of an Organic Rankine Cycle (ORC) based on industrial data. Appl. Therm. Eng. 91, 43–52. doi:10.1016/j.applthermaleng.2015.07.079
Twomey, B., Jacobs, P. A., and Gurgenci, H. (2013). Dynamic performance estimation of small-scale solar cogeneration with an organic Rankine cycle using a scroll expander. Appl. Therm. Eng. 51 (1-2), 1307–1316. doi:10.1016/j.applthermaleng.2012.06.054
Uris, M., Linares, J. I., and Arenas, E. (2015). Size optimization of a biomass-fired cogeneration plant CHP/CCHP (Combined heat and power/Combined heat, cooling and power) based on Organic Rankine Cycle for a district network in Spain. Energy 88, 935–945. doi:10.1016/j.energy.2015.07.054
Wang, Q., Hua, X., Cheng, X., Li, J., and Li, X. (2012). Effects of fuel additives on the thermal cracking of n-decane from reactive molecular dynamics. J. Phys. Chem. A 116, 3794–3801. doi:10.1021/jp300059a
Wang, S., Huo, E., Guan, Z., and Cai, S. (2023). Pyrolysis mechanism of HFO-1234yf/iso-butane mixture: reaxFF reactive molecular dynamic simulation study. Comput. Theor. Chem. 1223, 114098. doi:10.1016/j.comptc.2023.114098
Wang, S., Liu, C., Li, J., Sun, Z., Chen, X., and Wang, X. (2020). Exergoeconomic analysis of a novel trigeneration system containing supercritical CO2 Brayton cycle, organic Rankine cycle and absorption refrigeration cycle for gas turbine waste heat recovery. Energy Convers. Manag. 221, 113064. doi:10.1016/j.enconman.2020.113064
Wang, S., Liu, C., Zhang, C., Xu, X., and Li, Q. (2018b). Thermo-economic evaluations of dual pressure organic Rankine cycle (DPORC) driven by geothermal heat source. J. Renew. Sustain. Energy 10 (6), 063901. doi:10.1063/1.5034062
Wang, S., Liu, C., Zhang, C., and Xu, X. (2018a). Thermodynamic evaluation of leak phenomenon in liquid receiver of ORC systems. Appl. Therm. Eng. 141, 1110–1119. doi:10.1016/j.applthermaleng.2018.06.051
Wang, S., Liu, C., Zhang, S., Li, Q., and Huo, E. (2022). Multi-objective optimization and fluid selection of organic Rankine cycle (ORC) system based on economic-environmental-sustainable analysis. Energy Convers. Manag. 254, 115238. doi:10.1016/j.enconman.2022.115238
Xin, L., Liu, C., Tan, L., Xu, X., Li, Q., Huo, E., et al. (2021). Thermal stability and pyrolysis products of HFO-1234yf as an environment-friendly working fluid for Organic Rankine Cycle. Energy 228, 120564. doi:10.1016/j.energy.2021.120564
Xin, L. Y., Liu, C., Liu, Y., Huo, E., Li, Q., Wang, X., et al. (2020). Thermal decomposition mechanism of some hydrocarbons by ReaxFF-based molecular dynamics and density functional theory study. Fuel 275, 117885. doi:10.1016/j.fuel.2020.117885
Yu, W., Li, Q., Liu, C., Liu, L., and Xu, X. (2023). Decomposition mechanism of hydrofluorocarbon (HFC-245fa) in supercritical water: A ReaxFF-MD and DFT study. Int. J. Hydrogen Energy 48 (3), 864–878. doi:10.1016/j.ijhydene.2022.10.013
Zhang, C., Liu, C., Wang, S., Xu, X., and Li, Q. (2017). Thermo-economic comparison of subcritical organic Rankine cycle based on different heat exchanger configurations. Energy 123, 728–741. doi:10.1016/j.energy.2017.01.132
Zhang, C., Liu, C., Xu, X., Li, Q., Wang, S., and Chen, X. (2018a). Effects of superheat and internal heat exchanger on thermo-economic performance of organic Rankine cycle based on fluid type and heat sources. Energy 159, 482–495. doi:10.1016/j.energy.2018.06.177
Zhang, X., Li, Y., Tian, S., Xiao, S., Chen, D., Tang, J., et al. (2018b). Decomposition mechanism of the C5-PFK/CO2 gas mixture as an alternative gas for SF6. Chem. Eng. J. 336, 38–46. doi:10.1016/j.cej.2017.11.051
Zhao, Y., and Truhlar, D. G. (2008). Exploring the limit of accuracy of the global hybrid meta density functional for main-group thermochemistry, kinetics, and noncovalent interactions. Chem. Theory Comput. 4 (11), 1849–1868. doi:10.1021/ct800246v
Zhao, Y., and Truhlar, D. G. (2007). The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Accounts 120 (1-3), 215–241. doi:10.1007/s00214-007-0310-x
Keywords: n-hexane, thermal decomposition, organic Rankine cycle, ReaxFF reactive molecular dynamic, density functional theory
Citation: Liu W, Wang N, Chen J, Shen A and Yan F (2023) Thermal decomposition of n-hexane in organic Rankine cycle: a study combined ReaxFF reactive molecular dynamic and density functional theory. Front. Energy Res. 11:1276626. doi: 10.3389/fenrg.2023.1276626
Received: 12 August 2023; Accepted: 13 September 2023;
Published: 22 September 2023.
Edited by:
Xiangdong Liu, Yangzhou University, ChinaReviewed by:
Erguang Huo, Suzhou University of Science and Technology, ChinaCopyright © 2023 Liu, Wang, Chen, Shen and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wei Liu, bGl1d2VpQHNwaWMuY29tLmNu; Fei Yan, eWFuZmVpY3F1ZXJAYWx1LmNxdS5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.