 Asima Sultana
Asima Sultana Samadhan Lomate
Samadhan Lomate
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Energy Res. , 03 March 2023
Sec. Advanced Clean Fuel Technologies
Volume 11 - 2023 | https://doi.org/10.3389/fenrg.2023.1133514
This study presents an evaluation of catalytic performance of unsupported simple oxide catalysts with varying acid-base properties for the vapor phase hydrogenation of levulinic acid (LA) to γ-valerolactone (GVL) using formic acid (FA) in an aqueous medium. Among the different oxides tested MgO catalyst is found to be highly active. Between the MgO prepared by different methods and commercially obtained, the MgO-UBE showed 100% LA conversion and 100% selectivity to GVL at 270°C. The MgO-UBE catalyst favorably allowed the production of γ-valerolactone in the presence of 50 wt% water relative to the amount of a mixture of levulinic acid and formic acid. Based on the catalytic activity and characterization results, it was concluded that the presence of Mg (OH)2 and a higher number of Lewis acid-base pair sites, Mg2+LCO2−LC are needed to achieve high LA conversion and selectivity to GVL.
The conversion of lignocellulose to sugars, followed by their consecutive dehydration and hydrogenation to levulinic acid (LA) is well-established process. LA is a practically viable starting material for the synthesis of various value-added products, among which γ-valerolactone (GVL) is an important chemical obtained through selective hydrogenation. The GVL derived from abundant and inexpensive lignocellulosic biomass has attracted attention, due to its sustainability and environmentally benign nature for the production of fuel additives, green solvents and intermediates for chemicals and biofuels (Gilkey and Xu et al., 2016; Xu et al., 2020; Lange et al., 2010; Ye et al., 2020; Liu et al., 2022).
Extensive research is carried out to hydrogenate LA to GVL using both the homogeneous and heterogeneous catalysts (Liguori et al., 2015; Yan et al., 2015). Heterogeneous catalysts are especially important owing to the ease of GVL separation from the catalyst and reactant and product mixtures. Among the various heterogeneous catalysts studied, noble metals (e.g., Ru, Pt, Pd, Rh, Ir, and Au) exhibited good performance and in particular Ru catalyst was noticeably the more active (Upare et al., 2011; Wright and Palkovits, 2012). However, the use of precious metals is expected to be limited, if any, due to higher cost. Therefore, the search and development of non-noble metal-based catalysts are highly sought after.
In recent years significant efforts have been devoted to developing low-cost catalysts based on mono (Upare et al., 2011; Putrakumar et al., 2015; Fu et al., 2016; Varkolu et al., 2016; Xu et al., 2016; Mallesham et al., 2018; He et al., 2019; Sakakibara et al., 2019) and bimetallic transition metal oxides (Yan and Chen, 2013; Obregon et al., 2014; Shimizu et al., 2014; Yan and Chen, 2014; Upare et al., 2015; Al-Naji et al., 2016; Gupta and Kantam, 2018; Jones et al., 2018; Yoshida et al., 2018; Dutta et al., 2019; Liu et al., 2019). Most of these studies involve the use of metals such as Cu and Ni instead of precious metals for LA hydrogenation (Upare et al., 2011; Putrakumar et al., 2015; Fu et al., 2016; Varkolu et al., 2016; Xu et al., 2016; Mallesham et al., 2018; He et al., 2019; Sakakibara et al., 2019). Bimetallic catalysts containing Cu and Ni were found to show enhanced activity due to reduced carbon deposits on the catalyst surface (Shimizu et al., 2014; Upare et al., 2015; Al-Naji et al., 2016; Yoshida et al., 2018). The disadvantage of Cu-Ni containing catalyst is high metal loadings which are vulnerable to deactivation due to metal leaching and sintering under corrosive reaction conditions. LA and its esters could be converted to GVL with above 90% selectivity at 200°C using 50 wt% Cu/Al2O3 and Cu/ZrO2 and in water-methanol mixture under liquid phase conditions (Hengne and Rode, 2012). Nanocomposite Cu/SiO2 catalysts have shown outstanding catalytic performance in the hydrocyclisation of levulinic acid using molecular H2 to γ-valerolactone showing 100% LA conversion and 94% GVL selectivity at the reaction temperature of 265°C however the reaction was carried out in 1,4 dioxane solvent (Upare et al., 2011). Chen et al., 2015, reported high catalytic activity of copper supported on γ-Al2O3 catalyst for the hydrogenation of LA in vapor-phase (10% aqueous solution) to GVL. However, it was observed that the conversion decreases from 100% to 41% in 60 h of reaction time. They further investigated Cu supported on ZrO2, Al2O3, SiO2, and TiO2 for the vapor phase conversion of LA to GVL using molecular hydrogen at atmospheric pressure. The 5 wt% copper on ZrO2 catalyst exhibited the highest activity with 81% LA conversion and 83% GVL selectivity and the results were correlated to the dispersion of copper and acidity (Balla et al., 2016).
Previously we have shown promising catalysts (Lomate et al., 2018) based on 6 wt% copper supported on Al2O3, SiO2, TiO2, ZSM-5, and SiO2–Al2O3 catalysts in converting LA (28%) to GVL at 250°C using formic acid as hydrogen source in aqueous media. Among several catalysts investigated, Cu-SiO2 showed the best activity of 56% LA conversion and 87% GVL selectivity. It was identified that the SiO2 support properties that lead to partially oxidized copper species along with the large number of acid sites are responsible for high LA conversion and GVL selectivity (Lomate et al., 2017). Similar observations were made on Ni supported catalysts which are active for the conversion of LA to GVL where it was shown that the support plays a significant role (Fu et al., 2016; Varkolu et al., 2016; Mallesham et al., 2018). The performance of the metal supported catalysts is determined by a set of factors, including, the surface area of the support, particle size and oxidation state of the supported metal, and its interactions with the solid matrix. The above combination of factors poses challenges in designing the catalysts with specific properties to achieve the high activity, selectivity, and stability. The use of simple oxides such as Al2O3 and ZrO2 for GVL synthesis from levulinic acid esters in liquid phase using alcohols as hydrogen donor (Chia and dumesic et al., 2011; Szollosi and Bartok, 1999) showed the potential to overcome some of the drawbacks of supported metal catalysts. It is expected that the simple oxides do not suffer the disadvantage of metal supported oxide catalysts such as metal leaching and sintering and shows the potential for further applied research and development.
Magnesium oxide (MgO) was reported to be one of the most active catalysts in charge transfer reduction (CHTR) of carbonyl compounds and gas phase catalytic transfer hydrogenation (CTH) of ketones (Fang, and Riisager, 2021; Liu et al., 2022). A combination of basic sites and weakly acidic OH groups on MgO are reported to play an important role (Szollosi and Bartok, 1999). In this study, we report an excellent conversion of LA to GVL using the FA as hydrogen source and under mild conditions using an inexpensive simple acidic and basic oxide catalysts including Hydrotalcite, La2O3, MgO, CeO2, ZnO, BaO, SiO2, Al2O3, and ZrO2. Furthermore, the hydrolysis of lignocellulosic biomass leads to the formation of LA and an equimolar mixture of FA and water as by-products (Mehdi et al., 2008) and the use of as-synthesized aqueous mixture as reactants allows economically efficient production of GVL as no subsequent separation is required for further upgrading. FA as a reductant source can also avoid the external hydrogen supply and would be a promising practical approach.
Levulinic acid (99%) is from Alfa Aesar and formic acid (>99.9%) was from WAKO. The catalysts used in this study were procured from different sources. The SiO2 was obtained from Japan chemical society (SiO2-JRC), Al2O3 (GB-45-Mizusawa Chemical Industries), ZrO2 (RC-100-Daiichi Kigenso Kagoku Kogyo CO., Ltd.), Hydrotalcite (Mg/Al = 3-WAKO), La2O3 (WAKO), ZnO (WAKO), and BaO (WAKO). Commercially available magnesium oxides from the different suppliers MgO-WAKO and MgO-UBE were used in this study. For comparison, the MgO catalyst was also prepared through the calcination of Mg (NO3)2.6H2O at 500°C for 3 h and is termed as MgO-CAL and by co-precipitation method (MgO-CPT) in which 51.28 g of Mg (NO3)2.6H2O and 33.17 g of K2CO3 was dissolved in 200 mL water separately. These two solutions were added simultaneously to a beaker containing 200 mL of deionized water under constant stirring at room temperature. The final volume of the solution was adjusted to 3,000 mL by adding water and aged for 24 h followed by filtering and washing with deionized water to remove the potassium from the precipitate. The precipitate was dried in an oven at 110°C for 18 h and calcined at 500°C for 2 h in air.
The catalytic activity for the hydrogenation of LA into GVL was conducted in a vertical downflow fixed bed reactor with an inner diameter of 12 mm and a length of 364 mm at atmospheric pressure. The required amount of catalyst (0.5 g, unless specified) was placed in between two quartz wool beds in the reactor and the reaction temperature was measured by a thermocouple inserted at the center of the catalyst bed. Before the reaction, the reactor was placed in an electric furnace consisting of three heating zones with independent temperature controllers and the catalyst was pre-treated in N2 flow (50 mL min−1) for 1 h at 500°C. For the catalyst activity measurements, a mixture of 50 wt% aqueous solution of LA and FA with a required molar ratio was fed to the reactor through HPLC pump along with nitrogen gas at a flow rate of 50 mL min−1. The liquid products were collected in an ice-cooled trap at the reactor outlet at 1 h intervals. The collected products were analyzed by gas chromatography equipped with a DB WAX (60 M) capillary column and an FID detector.
The BET surface area of the catalysts were measured on a Micromeritics ASAP 2020 instrument by N2 adsorption-desorption at liquid N2 temperature using 0.3–0.5 g of the sample. Prior to the measurement the samples were heated to 100°C with a heating rate of 5°C min−1 and then degassed at this temperature for 1 h followed by which the temperature was increased to 350°C and maintained for 3 h. To calculate the pore volume and average pore size the BJH method was used.
The crystallinity of the powdered catalysts were characterized by powder X-ray diffraction using a Bruker D8 Advance X-ray diffractometer using nickel-filtered CuKα radiation (λ = 0.15406 nm) in the range of 10–70° and a scanning speed of 2° min−1, and a voltage and current of 40 kV and 40 mA, respectively.
The weight loss profiles of the catalysts were studied using thermogravimetric analysis (TGA) and differential thermal analysis (DTA), which were collected with a Shimadzu-50 thermo-analyzer apparatus in Air.
The TPD method was used to measure catalyst basic sites and its nature using a Belcat system. In each measurement, 100 mg of sample was loaded into a quartz reactor and heated in a He flow to 550°C for 1 h with a ramp rate of 10°C min−1 to remove the adsorbed water and other adsorbed species. Thereafter, the catalyst temperature was decreased to 100°C in He flow (50 min−1). The catalyst was exposed to 50 mL min−1 flow of undiluted CO2 for 1 h. After this adsorption step, the physically adsorbed CO2 was removed by purging the reactor with helium flow at 100°C. The temperature programmed desorption of CO2 was carried out by increasing the temperature to 700°C at a rate of 10°C min−1 and the desorbed CO2 was measured using TCD.
FTIR spectra were collected by a JASCO FTIR-610 instrument. About 10 mg of the catalyst was pressed into a disk and set in the FTIR cell. The catalyst was activated under He flow (70 mL/min) at 250°C for 30 min and then cooled down to room temperature before recording the spectra.
To study the nature of acid sites on the catalyst surface, pyridine adsorption was studied using a self-supporting sample disk of about 15 mg cm2 in a high vacuum system using the Fourier transform infrared (FTIR) technique. Before pyridine adsorption, the samples were heated to 500°C at 10°C min−1 under vacuum and then cooled down to room temperature. After the pretreatment, the catalyst was exposed to saturated pyridine vapor for 10 min and infrared spectra were recorded after outgassing at different temperatures (150, 250, 350°C, and 450°C) using a Nicolet 6700 FT-IR spectrometer at a resolution of 4 cm−1.
The UV-vis spectra of the catalyst were recorded using a UV-2600 spectrometer in diffuse reflectance mode between the 200 and 800 nm range at a step of 0.5 nm with a bandwidth of 2 nm. BaSO4 was used as a reference sample to measure the baseline spectrum.
The surface chemical composition and the oxidation state of the species present in the reduced catalysts were characterized by X-ray photoelectron spectroscopy (XPS). XPS analysis was performed using an ESCALAB 250 spectrometer equipped with an Al-Kα X-ray source (1,486.6 eV). The XPS data were corrected with respect to the carbon C 1 s peak at 284.5 eV.
Catalyst surface morphology was examined using Scanning Electron Microscopy (SEM) Hitachi (Model SV 9000). Photoluminescence (PL) studies were made using JASCO (FP-8600) fluorescence spectrometer at room temperature using xenon as the excitation source and for spectral analysis, the Fluor Essence™ software was used.
Table 1 shows the vapor phase hydrogenation activity of LA to GVL over several simple oxide catalysts along with their textural properties, BET surface area, total pore volume, average pore size. Among different oxides tested the MgO catalyst showed the highest catalytic performance of 70% LA conversion and 99% GVL selectivity. The MgO-Al2O3 mixed oxide derived from hydrotalcite showed the similar high performance, 67% LA conversion and 98% GVL selectivity. The other basic oxides La2O3 and BaO showed a small LA conversion and poor GVL selectivity. The relatively more acidic Al2O3 and ZrO2 were moderately active and showed 22% and 32% LA conversion respectively but poor GVL selectivity. On the other hand, SiO2 and SiO2-Al2O3 showed high selectivity of 89% and 59% to byproduct angelica lactone and low selectivity of 3% and 41% to the desired product GVL.

TABLE 1. Catalytic transfer hydrogenation of LA to GVL using different metal oxides.
The oxide catalysts used have widely varying surface area and pore volume and no strong relationship between catalytic activity and textural properties were found. On the other hand, the basic oxides are more active than acidic oxides in the GVL production. Both the higher conversion and better selectivity of various oxide catalysts can be in general explained based on basic and acidic nature of active sites. Two reaction pathways are reported in the literature to produce GVL (Al-Shaal et al., 2012). In the first path, the step 1 is the formation of pseudolevulinic acid by an intramolecular addition of the carboxyl on the carbonyl group. In step 2, pseudolevulinic acid is then dehydrated to α-angelica lactone which is subsequently hydrogenated to GVL. In the second pathway the hydrogenation of LA to 4-hydroxypentanoic acid followed by cyclodehydration leads to the formation of GVL. The acidic sites on the surface of metal oxides are not active for the decomposition of FA into H2 and CO2 which is crucial for the generation of H2 required for hydrogenation of angelica lactone to GVL (Ai, 1977). Whereas, the basic sites, because of their electron-donating ability adsorbs and activates the formic acid and undergoes oxidative dehydrogenation producing CO2 to H2. The formic acid is initially activated by a proton transfer from formic acid to a basic site, a lattice oxygen of the catalyst, resulting in the formation of a formate ion, which stabilizes on a metal cation adjacent to the oxygen. The low selectivity to GVL suggests a lack of hydrogenation activity of angelica lactone over La2O3 and BaO oxides.
It has been reported that the acid catalyzed LA hydrogenation involves dehydration of LA molecules to produce angelica lactone on Lewis acid sites, followed by hydrogenation of angelica lactone to GVL (Kumar et al., 2015). Several authors have studied the interaction and decomposition of FA on MgO and it was established that MgO catalyzes the decomposition of formic acid to CO2 and H2 and H-transfer on carbonyl substrates thus contributing to the high conversion and selectivity of levulinic acid to GVL (Ai, 1977; Mars et al., 1963; Yamamoto et al., 1997).
Even though no strong correlation was found between the textural properties of diverse metal oxides and their performance we expected a correlation to exist for a given class of simple oxide catalysts. Since the MgO catalyst showed promising performance for the conversion of LA to GVL, these catalysts were further explored for their performance optimization and active site characterization. MgO obtained from four different sources, which varied in their physio-chemical properties, were chosen for identifying the relationship between catalyst function, activity, and selectivity (Table 2). All MgO catalysts showed very high selectivity to GVL but significantly varying levels of LA conversion (Figure 1). Among the catalysts, MgO-UBE showed the highest LA conversion of 81%. The conversion of LA to GVL using formic acid as hydrogen source involves two steps 1) Decomposition of formic acid to CO2 + H2 and 2) Hydrogenation of LA with H2 to produce GVL. The activation and decomposition of formic acid is an important step and literature evidence suggests that on bulk MgO formic acid is adsorbed as formate which can easily decompose to CO2 and H2 at temperature >250°C (Noto et al., 1967). In addition, the hydrogenation of LA to GVL involves both the dehydration of LA to angelica lactone and its subsequent hydrogenation to GVL. The presence of Lewis acid-base pair site, Mg2 + LCO2-LC in MgO, as revealed from UV-VIS spectroscopy and Photoluminescence spectroscopy, could assist adsorption of LA and FA on basic sites and LA dehydration on Lewis’s acid sites synergistically and assist in the hydrogenation of LA with FA to selectively produce GVL. Furthermore, the activity of the catalyst was found to be similar using either hydrogen or formic acid as hydrogen source (Figure 1).
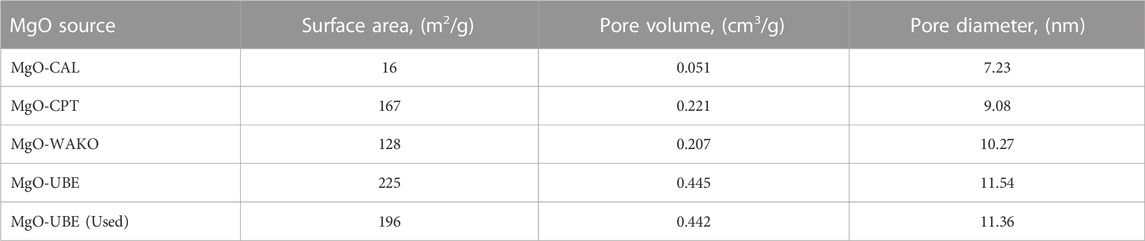
TABLE 2. Physicochemical properties of magnesium oxides.
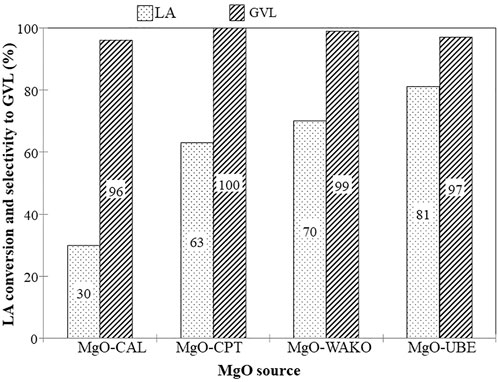
FIGURE 1. LA hydrogenation activity of different Mgo catalysts reaction conditions temperature-250°C: FA weight ratio-1; 2 liquid flow-0.03 mL/min,N2flow-50 cc/min, catalyst weight-0.5 g
During the hydrolysis of biomass, the LA and FA are typically obtained in equimolar ratio (Valentini et al., 2019) and therefore it is important to maximize the yield of the desired product while operating with such reactant mixtures. Moreover, the LA hydrogenation of using FA as a hydrogen source was reported to be strongly dependent on the concentration of FA in the reaction mixture (Varkolu et al., 2016). Since MgO-UBE catalyst exhibited promising catalytic performance towards hydrogenation of LA, further optimization of performance and selectivity were studied by changing reaction conditions such as the LA: FA ratio, reaction temperature and space velocity. Catalyst long-term stability was also evaluated. The effect of FA concentration on LA conversion and GVL selectivity over MgO-UBE catalyst was studied at 250 °C using different molar ratios of LA to FA between 1:0.5 to 1:2 (Figure 2). When the FA concentration is stoichiometrically low, LA: FA ratio of 1: 0.5, the LA conversion of 44% and GVL selectivity of 100% was obtained and the level of LA conversion was limited by the FA reactant availability as expected. Between LA: FA ratio of 1: 1 to 1: 2, the LA conversion and GVL selectivity reached to >98%. The MgO-UBE catalyst can convert the LA completely to GVL and remains stable up 55 h under LA to FA ratio 1:1, a more realistic condition and such performance was not seen over MgO in the past. Although Hussain et al., 2018, reported that MgO shows high LA conversion in the initial hours of the reaction with GVL selectivity of 98%, but then the activity decreased to 42% in 3 h.
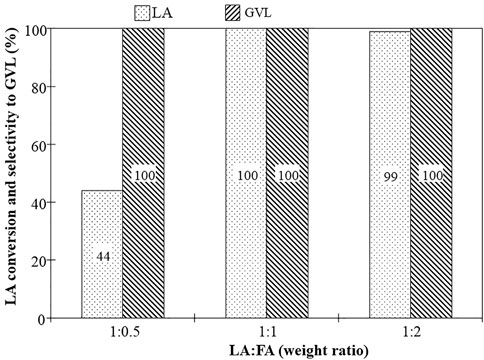
FIGURE 2. Influence of LA: FA ratio on LA conversion and GVL selectivity, reaction conditions: Catalyst-Mgo-UBE, temperature-250°C, liquid flow-0.015 mL/min, N2 flow-25 cc/min, catalyst weight-0.5 g
The impact of reaction temperature on the activity and selectivity is presented in Figure 3. At the reaction temperature of 210°C, the LA conversion (27%) and GVL selectivity (93%) were lower compared to 230°C which showed above 94% conversion of LA and selectivity to GVL reached 100%. Further increase in temperature up to 300 °C did not negatively impact the conversion and selectivity under the specific SV conditions. The effect of space velocity was studied by changing the N2 flow while keeping the flow rate of the other constituents the same (Table 3). The LA conversion significantly decreased from 84% to 75% upon increasing the SV from 2.25 to 6.75 h−1 and had no impact on selectivity to GVL.
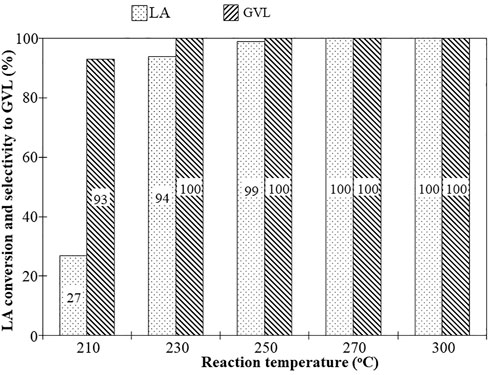
FIGURE 3. Influence of reaction tempreature on LA conversion and GVL selectivity. Reaction conditions: Catalyst-MgO-UBE, LA:FA weight ratio-1:2, liquid flow-0.015 mL/min, N2 flow-25 cc/min, catalyst weight-0.5 g
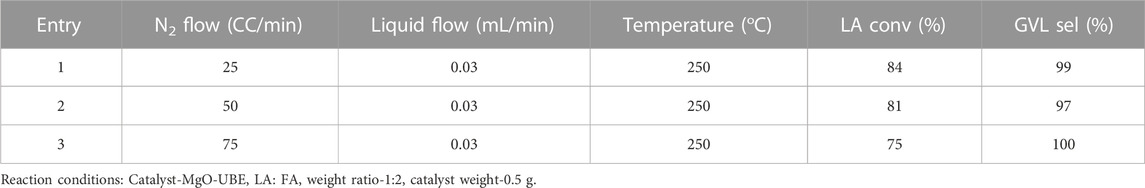
TABLE 3. Influence of space velocity on conversion of LA to GVL.
The effect of LA concentration was evaluated, by adjusting the H2O concentration and while keeping the LA: FA ratio constant. Upon increase in LA concentration from 10% to 100% the LA conversion increased from 56% to 99% (Table 4).
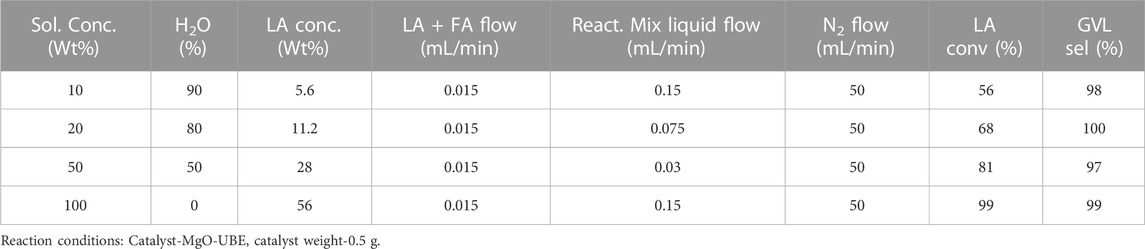
TABLE 4. Effect of variation of LA and H2O concentration on conversion selectivity.
The time on stream profiles of LA conversion and GVL selectivity of the MgO-UBE catalyst is presented in Figure 4. For the 50 h time on stream conversion studied at 250°C, the MgO-UBE catalyst maintained high activity and selectivity to GVL. Further investigations are needed to understand the better stability of MgO-UBE catalyst compared to other catalyst reported in the literature (Hussain et al., 2018). Hussain et al. tested the MgO catalyst for hydrogenation of LA to GVL in the absence of water, whereas in present investigation catalytic activity was tested in the presence of water. The BET surface (Table 4) and SEM images (pictures not included) of the used catalyst suggest no significant MgO corrosion. Furthermore, Ashokraju et al. (2018) reported that the presence of water played a crucial role in obtaining a higher yield of γ-valerolactone and catalyst stability over Cu/Fe2O3 catalyst.
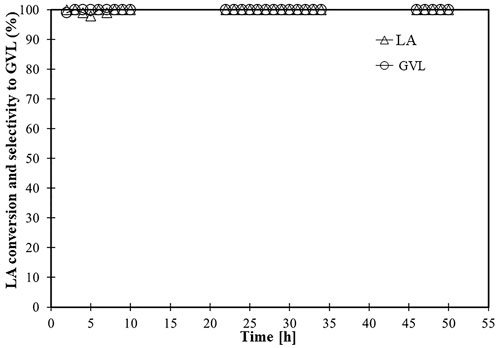
FIGURE 4. Time on stream activity of MgO-UBE catalysts for the conversion of LA to GVL conversion reaction conditions: Catalyst-MgO-UBE, temperature-250°C, LA, FA weight ratio-1:2, liquid flow-0.015 mL/min, N2 flow-25 cc/min, catalyst weight-0.5 g
Supplementary Figure S1 Depicts the crystallinity and phases of the MgO as examined by XRD. The diffraction patterns appearing at 2θ 18.6, 32.9, 38.1, 58.7, and 37.0, 43.0, 50.4 are characteristic of MgO and Mg (OH)2 phases respectively (Selvam et al., 2011). The MgO-UBE, MgO-WAKO, MgO-CPT and MgO-CAL catalysts showed phases corresponding to both Mg (OH)2 and MgO. The relative intensities indicate that the MgO-UBE and MgO-WAKO have a larger contribution of Mg (OH)2 phase compared to MgO-CAL and MgO-CPT catalysts. The BET surface area (Table 1) compliments the XRD observations, the catalysts with more crystalline MgO phase MgO-CAL and MgO-WAKO have lower surface area. Similarly, the catalysts containing more amorphous MgO phase, MgO-CPT and MgO-UBE have large surface area.
Thermogravimetric analysis (TGA-DTA) of MgO catalysts, the weight loss and heat change as a function of temperature are shown in Figure 5. The weight loss below 200°C is attributable to the desorption of H2O from the catalysts. The weight loss beyond 200°C is attributed to the H2O desorbed during the transformation of Mg (OH)2 to MgO (Montero et al., 2017). The MgO-UBE and MgO-WAKO showed prominent weight loss around 370°C corresponding to the transformation of Mg (OH)2 to MgO indicating a larger amount of its presence compared to MgO-CPT and MgO-CAL. The MgO-CAL catalyst as expected showed minimal amount of weight loss around 370°C indicating mostly the presence of MgO. The TGA results further compliment the outcome of BET and XRD analysis.
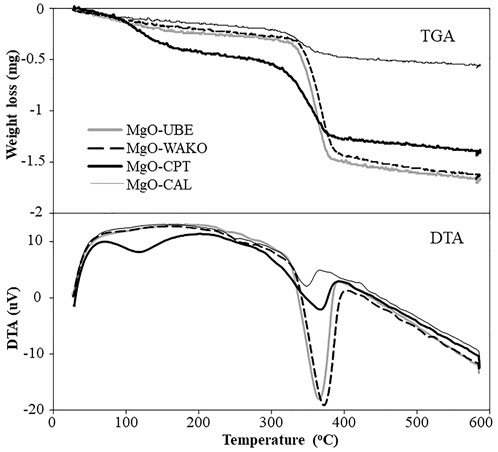
FIGURE 5. TGA-DTA curves of different MgO catalysts.
The XPS spectra corresponding to Mg2P and O1 S binding energies of different MgO catalysts are shown in Figure 6. The lowest BE peak at 49.2 eV can be assigned to the magnesium ions binding with hydroxyl groups (Mg-OH) and 50.2 eV BE corresponding to the magnesium ions binding with oxygen (Mg-O) (Li et al., 2014; Khamkongkaeo et al., 2017). Based on these known attributes and in accordance with other techniques, the presence of MgO and Mg (OH)2 phases are confirmed. In O1 s spectra a clear peak is observed around 531 eV along with a shoulder around 534 eV is observed in all MgO catalysts. In general, three characteristics BEs corresponding to 529.6, 531.2, and 533.1 eV are reported for MgO. The peak at 529.6 eV is assigned to the lattice oxygen bound to magnesium ions (Mg-O). The peaks at 531.2 and 533.1 eV are assigned to the hydroxyl groups of Mg (OH)2 and adsorbed water, respectively. The presence of a clear peak around 531 eV with a shoulder around 534 eV suggests the presence of Mg(OH)2 which increased in the following order MgO-UBE > MgO-WAKO > MgO-CPT > MgO-CAL. The O1 s results are consistent with the Mg2p spectra and indicate that the MgO catalysts have a mixture of MgO and Mg(OH)2. However, MgO-UBE and MgO-WAKO have noticeably higher amount of Mg(OH)2 component compared to MgO-CAL and Mg-CPT.
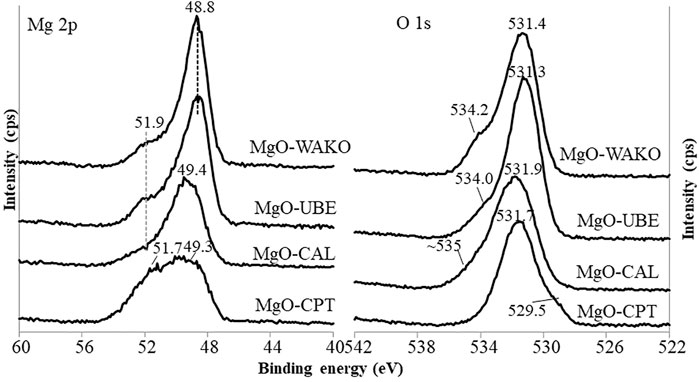
FIGURE 6. XPS profiles of different MgO catalysts.
The catalysts were further characterized by in situ FTIR after treating the samples at 250°C in He (Supplementary Figure S2). The sharp band centered at around 3,700 cm−1 is attributed to the presence of hydroxyl groups at the low-coordination sites or defects in the Mg (OH)2 structure (Kumar and Kumar, 2008; Kumar et al., 2015). The spectra also showed a band between 400 to 650 cm−1, which is attributed to the Mg-O bond. The MgO-UBE sample, as expected based on previous characterization results, has more OH groups and the MgO-CAL has more MgO oxides. The intensity of 400–650 cm−1 peak is higher in MgO-CAL and MgO-CPT indicating moreMgO species in these two samples compared to MgO-UBE and MgO-WAKO. The strength of basic and acidic sites was further characterized by CO2-TPD (Figure 7). The CO2 desorption profiles varying in intensity and position with respect to temperature were observed indicating different basic strength of the active sites on MgO. Presence of different types of OH groups with the decrease in base strength is in the following order: hydroxyl group < oxygen in Mg2+ and O2−pairs < low coordination oxygen anions are reported in the literature (Hu et al., 2007; Chen et al., 2015).
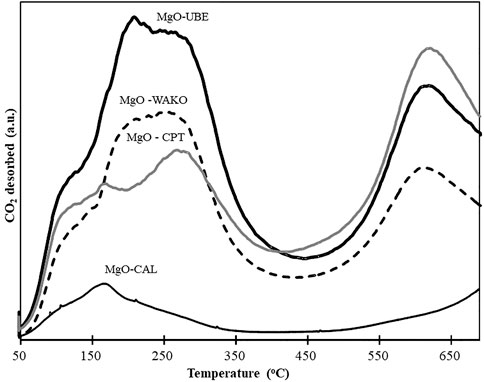
FIGURE 7. CO2-TPD profiles of different MgO catalysts.
As seen in Figure 7 typically, there were three dominant peaks that appeared at below 200, 200–300 and 500°C–700°C temperature windows, which are attributed to weak, medium and strong basic sites, respectively. The results reveal that MgO-UBE possesses a higher number of medium and strong basic sites compared to other MgO indicating the presence of a higher number of Mg2+-O2−pairs or more number low coordinated sites in MgO-UBE. The total amount of basic sites was found to be in the following order MgO-UBE > MgO-WAKO > MgO-CPT > MgO-CAL.
The coordination states of Mg2+ were characterized by UV-VIS spectroscopy (Figure 8). UV-VIS spectra of all samples were recorded in the of 200–800 nm wavelength range. The UV spectra of all the MgO showed bands around 200–250 and 260–300 nm, which are assigned to the excitation of O2− surface anions on the edge (4-fold coordinated) and on the corner (3-fold coordinated) of MgO (Schwach et al., 2015). The intensity of both the bands is higher in MgO-UBE and MgO-WAKO compared to MgO-CAL and MgO-CPT. However, the relative intensity of 260–300 nm band is higher MgO-CAL and MgO-CPT. This observation indicates the presence of a higher number of low coordination sites or defects in MgO-UBE and MgO-WAKO samples.
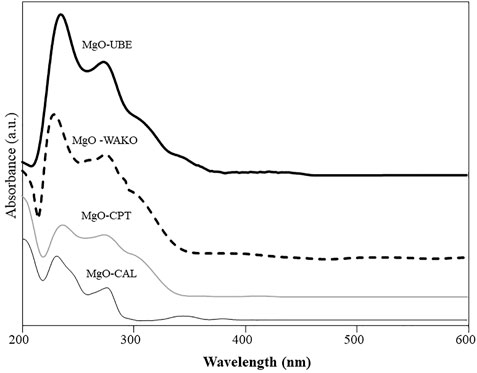
FIGURE 8. Diffuse reflectance UV-Vis spectra of different MgO catalysts.
Differences in the coordination number of surface ions due to defects or oxygen vacancies change the acceptor-donor (acid-base) behavior of MgO and play a significant role in its catalytic behavior. Photoluminescence (PL) spectroscopy is used to probe the defects and low-coordinated edge or corner sites. With this technique, luminescent species are characterized by a couple of excitations (λexc) and emission (λem) wavelengths. PL of different MgO catalysts is taken at two different excitations, 250 and 292 nm and the emission spectra are shown in Supplementary Figure S3. Excitation at these wavelengths generates several emission bands at 338, 352, 450 nm and 340, 365, 490 nm respectively, which are reported due to surface defects, namely, oxygen vacancies (Chizallet et al., 2008). For the MgO spectra two group’s assignments are proposed in the literature such as (λexc—240 nm; λem—380 nm) and (λexc—280 nm; λem—470 nm) couples, which were assigned to O2-4°C and O2-3°C ions, respectively. The band around 350 nm in both the 250 and 292 nm excited spectra is apparent only in MgO-UBE and MgO-WAKO indicating that O2-4°C is present only in these catalysts. Whereas the bands corresponding to O2-3°C ions appearing between 450 and 490 nm is present in all the samples. The other weaker emissions may be due to energy transfer from one anion to another present on low coordination sites on the surface. The PL results of different MgO catalyst indicates that the defects at the surface of MgO depend on its electronic properties, i.e., on the coordination number and the local topology.
FTIR spectra of different MgO catalysts after pyridine adsorption are shown in Figure 9. The spectra showed bands at 1,440, 1,480, 1,575, and 1,595 cm-1 corresponding to Lewis acid sites and all the MgO catalysts analyzed exhibited a measurable concentration of Lewis acid sites (LAS). The intensity of all the corresponding bands significantly decreased with an increase in the evacuation temperature indicating that it possessed relatively weak Lewis acid sites, as expected. Among the MgO catalysts, the intensity of the bands evacuated at different temperatures is much higher in MgO-UBE compared to other MgO catalysts. It is reported that in a Mg2+LCO2−LC pair, the relative acidity of a Mg2+LC cation and Lewis basicity of O2−LC anion depends on the coordination number and Lewis acid-base pair site, Mg2+LCO2−LC increase as their coordination number decreases (Chizallet et al., 2006).
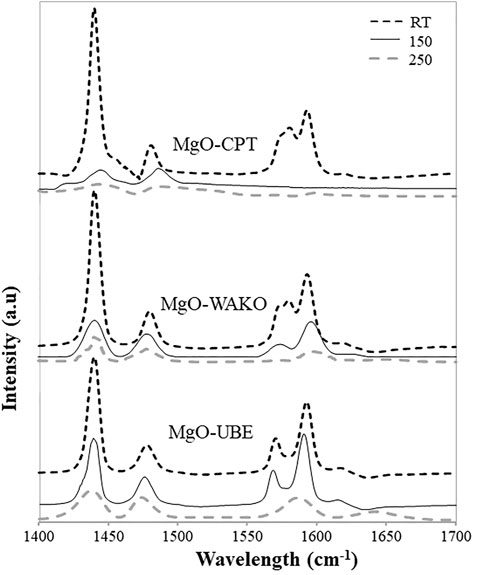
FIGURE 9. Pyridine adsorbed drift spectra of different MgO catalysts.
The selectivity to GVL can take place through α-angelica lactone or 4-hydroxypentanoic acid intermediate. In vapor phase, the reaction is expected to take place through angelica lactone intermediate (Al-Shaal et al., 2012). For the catalyst to be active and selective, it is expected to possess both the hydration and hydrogenation functions. In this work, we attempted to show that oxides without transition or precious group metals can possess both these functions which can inherently have advantages over metal supported catalysts. Additional benefits of the use of simple oxides, without transition or precious group metals, showed stable performance over time on stream as shown in Figure 4. The absence of redox elements, transition or precious group metals leads to avoidance of potential metal leaching that will lead to a decrease in conversion overtime on stream (Putrakumar et al., 2015). Among the several oxide catalysts studied, we identified MgO to be active and selective for the above reactions. From the different MgO catalysts evaluated based on the surface area and XRD, it is expected to possess different textural and surface properties. The catalysts textural properties, BET surface area, total pore volume, average pore size diameter did not show a strong correlation to activity and selectivity, even between the catalysts with similar functions.
It was expected that the catalyst possesses various acid base properties with minimal if any, redox properties. The MgO-UBE and MgO-Wako contained MgO/Mg(OH)2 phases, whereas other MgO with low surface area is more crystalline and mainly contains MgO phase as identified from XRD. Weight loss curves in TGA showed MgO-UBE and MgO-Wako corresponding to a large amount of hydroxyl groups complimenting the XRD observations. From XRD, TGA and XPS even though it was not possible to distinguish the bulk and surface hydroxyl groups, based on weight loss and surface area it is reasonable to assume that MgO-UBE and MgO-WAKO contain relatively more surface hydroxyl groups. The UV and PL analysis showed MgO-UBE and MgO-Wako contains low coordination edge or corner sites, which are attributed to the pair of Lewis acid-base sites (Mg2+LCO2−LC) (Chizallet et al., 2008; Schwach et al., 2015).
Further investigation of the nature of surface acid-base properties by FTIR after pyridine adsorption and CO2-TPD indicated the presence of a relatively large number of Lewis acid sites and many strong basic sites in MgO-UBE catalyst compared to other MgO catalysts. The presence of a large number of basic sites in MgO-UBE facilitates the adsorption of acidic molecules LA and FA. On the other hand, the presence of Lewis acid sites promotes the dehydration of LA (Kumar et al., 2016). In the LA hydrogenation using formic acid as hydrogen source, the activation and decomposition of FA is a key step for the reaction path. In general, formic acid exhibits two modes of decomposition predominantly to give CO and H2O (dehydration, decarbonylation) or to CO2 and H2 (decarboxylation) (Ai, 1977). The basic sites of MgO promote the dehydrogenation of FA by O-H bond cleavage leading to the formation of H2 and CO2 via formate intermediate (Mars et al., 1963; Ai, 1977; Yamamoto et al., 1997).
Furthermore, the Mg2+LCO2−LC sites were also shown to be active for the charge transfer hydrogenation reaction of mesityl oxide with 2-propanol (Cosimo et al., 2014). During the charge transfer hydrogenation of 2-butanone on MgO presence of basic (O2–) site and adjacent surface hydroxyl site pairs appear prerequisite for the reaction (Szollosi and Bartok, 1999; Glinski et al., 2008; Szollosi and Bartok. 1999).
The active sites responsible for the different reaction steps in LA hydrogenation with formic acid are present in higher numbers in MgO-UBE and MgO-WAKO leading to high activity and selectivity.
Correlation of catalytic activity with different characterization results revealed that the presence of Mg (OH)2 and a higher number of Lewis acid-base pair sites, Mg2+LCO2−LC are needed to achieve high LA conversion and GVL selectivity. Xu et al. (2016) reported that on acid-base catalysts, rather than a single acid or base site, the Lewis acid-base pair sites, mediates the hydrogenation of carbonyl groups with alcohols as the hydrogen donor.
The current catalytic results for hydrogenation of LA obtained on MgO-UBE catalyst is compared with some of the reported literature results and summarized in Table 5. It is noteworthy that, bare magnesium oxide catalytic system, without the addition of metals, offers a highly selective transformation of LA into GVL using vapor phase reactor at atmospheric pressure. Kopetzki and Antonietti. (2010) demonstrated that transfer hydrogenation from formic acid to levulinic acid under hydrothermal conditions can be catalyzed by bases. MgO based catalytic system is highly efficient for decomposition of formic acid into H2 and CO2 (Mars et al., 1963; Ai, 1977; Yamamoto et al., 1997) and for hydrogenation of LA to GVL. From all the observations it can be said that MgO-UBE catalyst is a highly active and stable catalyst for the conversion of LA to GVL using formic acid as a hydrogen source. Thus, this catalyst could be the most suitable candidate for practical applications.
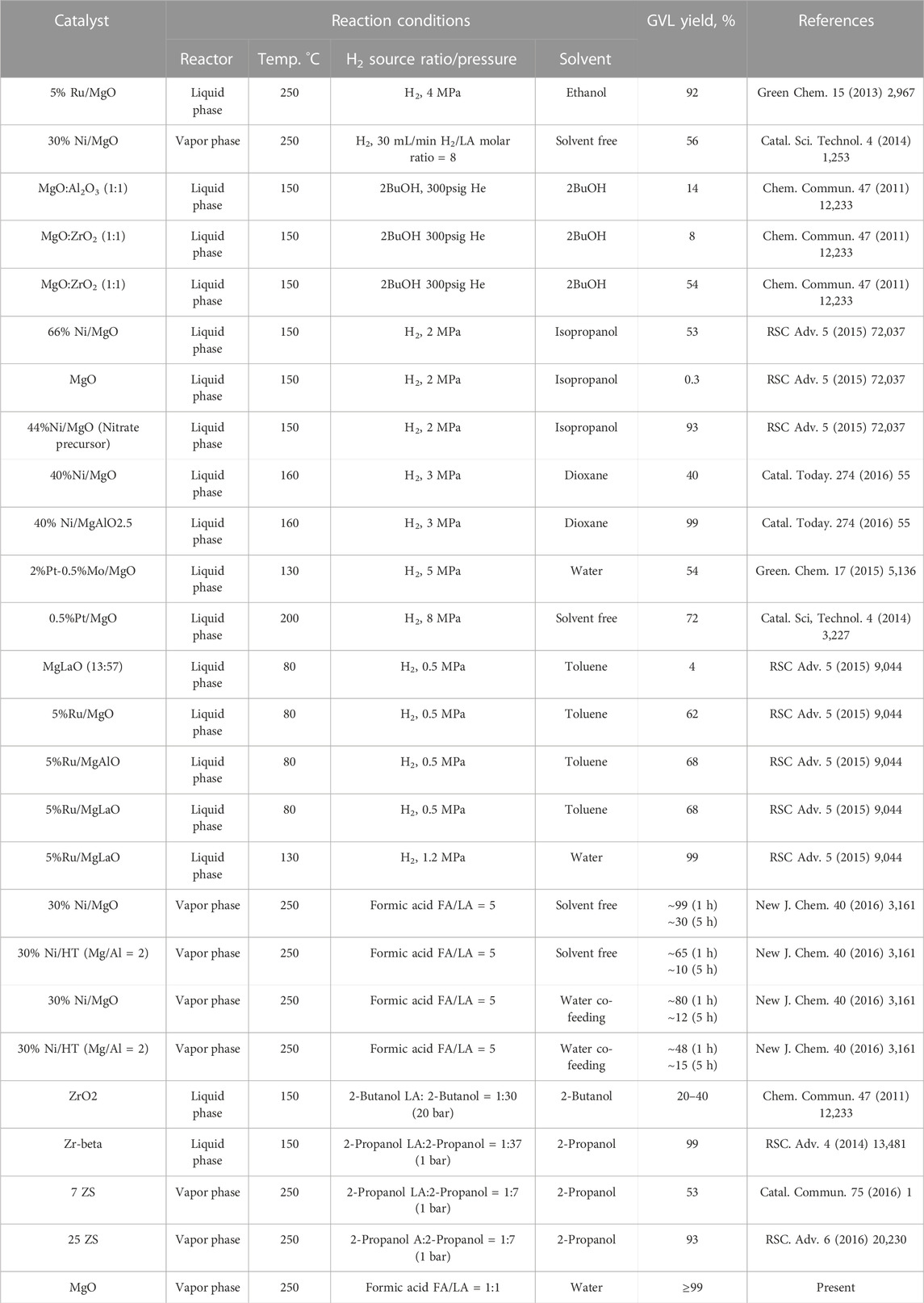
TABLE 5. A comparison of present and literature reported results on conversion of LA to GVL.
The catalytic conversion of levulinic acid (LA) to γ-valerolactone (GVL) can be achieved without using a direct source of molecular hydrogen. In the present study, an industrially viable process for the selective synthesis of γ-valerolactone using bio-based aqueous solution of LA and FA as hydrogen source over a simple solid heterogeneous magnesium oxide (MgO) catalyst is provided. It is found that MgO alone is an active catalyst for LA conversion to GVL using formic acid as hydrogen source and in presence of H2O. This catalyst advantageously allows the production of γ-valerolactone in the presence of 50% (W/W) water relative to the amount of a mixture of levulinic acid and formic acid. Importantly, the decomposition of formic acid and hydrogenation of levulinic acid can be carried out over the same MgO catalyst. This process provides 80%–100% conversion of levulinic acid to γ-valerolactone, with selectivity between 80% and 100%. The coexistence of basic sites for LA and FA adsorption and Lewis acid sites for dehydration and a combination of Lewis acid-base pair site, Mg2+LCO2−LC in MgO particularly in MgO-UBE possibly works synergistically in the hydrogenation of LA using FA to selectively produce GVL. The present invention could lead to a simple process with low cost and high yield.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenrg.2023.1133514/full#supplementary-material
Ai, M. (1977). Activities for the decomposition of formic acid and the acid-base properties of metal oxide catalysts. J. Catal. 50, 291–300. doi:10.1016/0021-9517(77)90038-0
Al-Naji, M., Yepez, A., Balub, A. M., Romero, A. A., Chen, Z., Wilde, N., et al. (2016). Insights into the selective hydrogenation of levulinic acid to γ - valerolactone using supported mono- and bimetallic catalysts. J. Mol. Catal. A Chem. 17, 145–152. doi:10.1016/j.molcata.2016.03.015
Al-Shaal, M. G., Wright, W. R. H., and Palkovits, R. (2012). Exploring the ruthenium catalysed synthesis of γ-valerolactone in alcohols and utilisation of mild solvent-free reaction conditions. Green Chem. 14, 1260–1263. doi:10.1039/c2gc16631c
Ashokraju, M., Mohan, V., Murali, K., Venkat, R. M., David, R. B., and Rao, K. S. (2018). Formic acid assisted hydrogenation of levulinic acid to γ-valerolactone over ordered mesoporous Cu/Fe2O3 catalyst prepared by hard template method. J. Chem. Sci. 130 (16), 1–8. doi:10.1007/s12039-018-1418-3
Balla, P., Perupogu, V., Vanama, P. K., and Komandur, V. R. C. (2016). Hydrogenation of biomass-derived levulinic acid to γ-valerolactone over copper catalysts supported on ZrO2. J. Chem. Technol. Biotechnol. 91, 769–776. doi:10.1002/jctb.4643
Chen, J., Tian, S., Lu, J., and Xiong, Y. (2015). Catalytic performance of MgO with different exposed crystal facets towards the ozonation of 4-chlorophenol. Appl. Catal. A Gen. 506, 118–125. doi:10.1016/j.apcata.2015.09.001
Chia, M., and Dumesic, J. A. (2011). Liquid-phase catalytic transfer hydrogenation and cyclization of levulinic acid and its esters to γ -valerolactone over metal oxide catalysts. Chem. Commun. 47, 12233–12235. doi:10.1039/c1cc14748j
Chizallet, C., Bailly, M. L., Costentin, G., Lauron-Pernot, H., Krafft, J. M., Bazin, P., et al. (2006). Thermodynamic brønsted basicity of clean MgO surfaces determined by their deprotonation ability: Role of Mg2+–O2- pairs. Catal. Today 116, 196–205. doi:10.1016/j.cattod.2006.01.030
Chizallet, C., Costentin, G., Pernot, H. L., Krafft, J. M., Che, M., Delbecq, F., et al. (2008). Assignment of photoluminescence spectra of MgO powders: TD-DFT cluster calculations combined to experiments. Part I: Structure effects on dehydroxylated surfaces. J. Phys. Chem. C 112, 16629–16637. doi:10.1021/jp8045017
Cosimo, J. I. D., Diez, V. K., Ferretti, C., and Apesteguia, C. R. (2014). Basic catalysis on MgO: Generation, characterization, and catalytic properties of active sites. Catalysis 26, 1–28. doi:10.1039/9781782620037-00001
Dutta, S., Yu, I. K. M., Tsang, D. C. W., Ng, Y. H., Ok, Y. S., Sherwood, J., et al. (2019). Green synthesis of gamma-valerolactone (GVL) through hydrogenation of biomass-derived levulinic acid using non-noble metal catalysts: A critical review. Chem. Eng. J. 372, 992–1006. doi:10.1016/j.cej.2019.04.199
Fang, W., and Riisager, A. (2021). Recent advances in heterogeneous catalytic transfer hydrogena -tion/hydrogenolysis for valorization of biomass-derived furanic compounds. Green Chem. 23, 670–688. doi:10.1039/d0gc03931d
Fu, J., Sheng, D., and Lu, X. (2016). Hydrogenation of levulinic acid over nickel catalysts supported on aluminum oxide to prepare γ -Valerolactone. Catalysts 6, 6–10. doi:10.3390/catal6010006
Gilkey, M. J., and Xu, B. (2016). Heterogeneous catalytic transfer hydrogenation as an effective pathway in biomass upgrading. ACS Catal. 6, 1420–1436. doi:10.1021/acscatal.5b02171
Gupta, S. S. R., and Kantam, M. L. (2018). Selective hydrogenation of levulinic acid into γ-valerolactone over Cu/Ni hydrotalcite-derived catalyst. Catal. Today 309, 189–194. doi:10.1016/j.cattod.2017.08.007
He, D., He, Q., Jiang, P., Zhou, G., Hu, R., and Fu, W., (2019). Novel Cu/Al2O3-ZrO2 composite for selective hydrogenation of levulinic acid to γ-valerolactone. Catal. Commun., 125, 82–86. doi:10.1016/j.catcom.2019.03.029
Hengne, A. M., and Rode, V. C. (2012). Cu–ZrO2 nanocomposite catalyst for selective hydrogenation of levulinic acid and its ester to γ-valerolactone. Green Chem. 14, 1064–1072. doi:10.1039/c2gc16558a
Hu, J., Zhu, K., Chen, L., Kubel, C., Richards, R., Ku1bel, C., et al. (2007). MgO(111) Nanosheets with unusual surface activity. J. Phys. Chem. C 111, 12038–12044. doi:10.1021/jp073383x
Hussain, S. K., Velisoju, V. K., Rajan, N. P., and Kumar, B. P., Synthesis of γ-valerolactone from levulinic acid and formic acid over Mg-Al hydrotalcite like compound. (2018). synthesis of g-valerolactone from levulinic acid and formic acid over Mg-Al hydrotalcite like compound. Chem. Sel., 3, 6186, doi:10.1002/slct.201800536
Jones, D. R., Iqbal, S., Thomas, L., Ishikawa, S., Reece, C., Miedziak, P. J., et al. (2018). xNi–yCu–ZrO2 catalysts for the hydrogenation of levulinic acid to gamma valorlactone. Catal. Struct. React 4, 12–23. doi:10.1080/2055074x.2018.1433598
Khamkongkaeo, A., Mothaneeyachart, N., Sriwattana, P., Boonchuduang, T., Phetrattanarangsi, T., Thongchai, C., et al. (2017). Ferromagnetism and diamagnetism behaviors of MgO synthesized via thermal decomposition method. J. Alloys. Compd. 705, 668–674. doi:10.1016/j.jallcom.2017.02.170
Kopetzki, D., and Antonietti, M. (2010). Transfer hydrogenation of levulinic acid under hydrothermal conditions catalyzed by sulfate as a temperature-switchable base. Green Chem. 12, 656. doi:10.1039/B924648G
Kumar, A., and Kumar, J. (2008). On the synthesis and optical absorption studies of nano-size magnesium oxide powder. J. Phys. Chem. Solids 69, 2764–2772. doi:10.1016/j.jpcs.2008.06.143
Kumar, V. V., Naresh, G., Sudhakar, M., Tardio, J., Bhargava, S. K., and Venugopal, A. (2015). Role of Brønsted and Lewis acid sites on Ni/TiO2 catalyst for vapour phase hydrogenation of levulinic acid: Kinetic and mechanistic study. Appl. Catal. A 505, 217–223. doi:10.1016/j.apcata.2015.07.031
Lange, J. P., Price, R., Ayoub, P. M., Louis, J., Petrus, L., Clarke, L., et al. (2010). A platform of cellulosic transportation fuels. Angew. Chem. Int. 49, 4479–4483. doi:10.1002/anie.201000655
Li, L. X., Xu, D., Li, X. Q., Liu, W. C., and Jia, Y. (2014). Excellent fluoride removal properties of porous hollow MgO microspheres. Chem. 38, 5445–5452. doi:10.1039/c4nj01361a
Liguori, F., Marrodan, C. M., and Barbaro, P. (2015). Environmentally friendly synthesis of γ-Valerolactone by direct catalytic conversion of renewable sources. ACS Catal. 5, 1882–1894. doi:10.1021/cs501922e
Liu, M., Li, S., Fan, G., Yang, L., and Li, F. (2019). Hierarchical flower-like bimetallic NiCu catalysts for catalytic transfer hydrogenation of ethyl levulinate into γ-Valerolactone. Ind. Eng. Chem. Res. 58, 10317–10327. doi:10.1021/acs.iecr.9b01774
Liu, Z., Zhang, Z., Wen, Z., and Xue, B. (2022). High efficiency catalytic transfer hydrogenation of furfural to furfuryl alcohol over metallic oxide catalyst. Letts 152, 3537–3547. doi:10.1007/s10562-022-03924-5
Lomate, S., Sultana, A., and Fujitani, T. (2017). Effect of SiO2 support properties on the performance of Cu-SiO2 catalysts for the hydrogenation of levulinic acid to gamma valerolactone using formic acid as a hydrogen source. Catal. Sci. Technol. 7, 3073–3083. doi:10.1039/c7cy00902j
Lomate, S., Sultana, A., and Fujitani, T. (2018). Vapor phase catalytic transfer hydrogenation (CTH) of levulinic acid to γ-Valerolactone over copper supported catalysts using formic acid as hydrogen source. Catal. Lett. 148, 348–358. doi:10.1007/s10562-017-2241-z
Mallesham, B., Sudarsanam, P., Reddy, B. V. S., Rao, B. G., and Reddy, B. M. (2018). Nanostructured nickel/silica catalysts for continuous flow conversion of levulinic acid to γ-Valerolactone. ACS Omega 13, 16839–16849. doi:10.1021/acsomega.8b02008
Mars, P., Scholten, J. J. F., and Zwietering, P. (1963). The catalytic decomposition of formic acid. Adv. Catal 14, 35–113. doi:10.1016/S0360-0564(08)60338-7
Mehdi, H., Fabos, V., Tuba, R., Bodor, A., Mika, L. T., and Horvath, I. T. (2008). Integration of homogeneous and heterogeneous catalytic processes for a multi-step conversion of biomass: From sucrose to levulinic acid, γ-valerolactone, 1,4-pentanediol, 2-Methyl-tetrahydrofuran, and alkanes. Catal 48, 49–54. doi:10.1007/s11244-008-9047-6
Montero, S. G., Alshammari, H., Dalebout, R., Nowicka, E., Morgan, D. J., Shaw, G., et al. (2017). Deactivation studies of bimetallic AuPd nanoparticles supported on MgO during selective aerobic oxidation of alcohols. Appl. Catal. A Gen. 546, 58–66. doi:10.1016/j.apcata.2017.07.045
Noto, Y., Fukuda, K., Onishi, T., and Tamaru, K. (1967). Mechanism of formic acid decomposition over dehydrogenation catalysts. Trans. Faraday Soc. 63, 3081–3087. doi:10.1016/j.cattod.2021.04.011
Obregon, I., Corro, E., Izquierdo, U., Requies, J., and Arias, P. L. (2014). Levulinic acid hydrogenolysis on Al2O3-based Ni-Cu bimetallic catalysts. Chin. J. Catal.35 35, 656–662. doi:10.1016/s1872-2067(14)60051-6
Putrakumar, B., Nagaraju, N., Kumar, V. P., and Chary, K. V. R., (2015). Hydrogenation of levulinic acid to γ -valerolactone over copper catalysts supported on γ -Al2O3. Catal. Today. 250, 209–217. doi:10.1016/j.cattod.2014.07.014
Sakakibara, K., Endo, K., and Osawa, T., (2019). Facile synthesis of γ-valerolactone by transfer hydrogenation of methyllevulinate and levulinic acid over Ni/ZrO2. Catal. Commun., 125, 52 doi:10.1016/j.catcom.2019.03.021
Schwach, P., Hamilton, N., Eichelbaum, M., Thum, L., Lunkenbein, T., Schlogl, R., et al. (2015). Structure sensitivity of the oxidative activation of methane over MgO model catalysts: II. Nature of active sites and reaction mechanism. J. Catal. 329, 574–587. doi:10.1016/j.jcat.2015.05.008
Selvam, N. C. S., Kumar, R. T., Kennedy, L. J., and Vijaya, J. J. (2011). Comparative study of microwave and conventional methods for the preparation and optical properties of novel MgO-micro and nano-structures. J. Alloys. Compd. 509, 9809–9815. doi:10.1016/j.jallcom.2011.08.032
Shimizu, K., Kanno, S., and Kon, K. (2014). Hydrogenation of levulinic acid to γ-valerolactone by Ni and MoOx co-loaded carbon catalysts. Green Chem. 16, 3899–3903. doi:10.1039/c4gc00735b
Szollosi, G., and Bartok, M. (1999). Role of basic and acidic centers of MgO and modified MgO in catalytic transfer hydrogenation of ketones studied by infrared spectroscopy. J. Mol. Struct. 482-2, 13–17. doi:10.1016/s0022-2860(98)00833-3
Upare, P. P., Jeong, M. G., Hwang, Y. K., Kim, D. H., Kim, Y. D., Hwang, D. W., et al. (2015). Nickel-promoted copper–silica nanocomposite catalysts for hydrogenation of levulinic acid to lactones using formic acid as a hydrogen feeder. Appl. Catal. A Gen. 491, 127–135. doi:10.1016/j.apcata.2014.12.007
Upare, P. P., Lee, J. M., Hwang, D. W., Halligudi, S. B., Hwang, Y. K., and Chang, J. S. (2011). Selective hydrogenation of levulinic acid to γ-valerolactone over carbon-supported noble metal catalysts. J. Ind. Eng. Chem. 17, 287–292. doi:10.1016/j.jiec.2011.02.025
Upare, P. P., Lee, J. M., Hwang, Y. K., Hwang, D. W., Lee, J. H., Halligudi, S. B., et al. (2011). Direct hydrocyclization of biomass-derived levulinic acid to 2- methyltetrahydro furan over nanocomposite copper/silica catalysts. ChemSusChem 4, 1749. doi:10.1002/cssc.201100380
Valentini, F., Kozell, V., Petrucci, C., Marrocchi, A., Gu, Y., Gelman, D., et al. (2019). Formic acid, a biomass-derived source of energy and hydrogen for biomass upgrading. Energy Environ. Sci.12 12, 2646–2664. doi:10.1039/c9ee01747j
Varkolu, M., Velpula, V., Burri, D. R., and Kamaraju, S. R. R., (2016) Gas phase hydrogenation of levulinic acid to γ -valerolactone over supported Ni catalysts with formic acid as hydrogen source. New. J. Chem. 40, 3261–3267. doi:10.1039/c5nj02655e
Wright, W. R. H., and Palkovits, R. (2012). Development of heterogeneous catalysts for the conversion of levulinic acid to γ-valerolactone. ChemSusChem. 5, 1657–1667. doi:10.1002/cssc.201200111
Xu, Q., Li, X., Pan, T., Yu, C., Deng, J., Guo, Q., et al. (2016). Supported copper catalysts for highly efficient hydrogenation of biomass-derived levulinic acid and γ-valerolactone. Green Chem. 8, 1287–1294. doi:10.1039/c5gc01454a
Xu, R., Liu, K., Du, H., Liu, H., Cao, X., Zhao, X., et al. (2020). Falling leaves return to their roots: A review on the preparation of γ-valerolactone from lignocellulose and its application in the conversion of lignocellulose. Chem. Eur. 13, 6461. doi:10.1002/cssc.202002008
Yamamoto, H., Watanabe, N., Wada, A., Domen, K., and Hirose, C. (1997). Adsorption and decomposition of formic acid on MgO(001) surface as investigated by temperature programmed desorption and sum-frequency generation spectroscopy: Recurrence induced defect sites. J. Chem. Phys. 106 (11), 4734–4744. doi:10.1063/1.473470
Yan, K., and Chen, A. (2013). Efficient hydrogenation of biomass-derived furfural and levulinic acid on the facilely synthesized noble-metal-free Cu-Cr catalyst. Energy 58, 357–363. doi:10.1016/j.energy.2013.05.035
Yan, K., and Chen, A. (2014). Selective hydrogenation of furfural and levulinic acid to biofuels on the ecofriendly Cu–Fe catalyst. Fuel 115, 101–108. doi:10.1016/j.fuel.2013.06.042
Yan, K., Jarvis, C., Gu, J., and Yan, Y. (2015). Production and catalytic transformation of levulinic acid: A platform for speciality chemicals and fuels. Renew. Sustain Energy Rev. 51, 986–997. doi:10.1016/j.rser.2015.07.021
Ye, L., Han, Y., Fenga, J., and Lua, X. (2020). A review about GVL production from lignocellulose: Focusing on the full components utilization. Industrial Crops Prod. 144, 112031–112048. doi:10.1016/j.indcrop.2019.112031
Keywords: hydrogenation, oxide catalyst, levulinic acid, gamma-valerolactone, formic acid
Citation: Sultana A, Lomate S and Fujitani T (2023) Highly active MgO catalysts for hydrogenation of levulinic acid to γ-valerolactone using formic acid as the hydrogen source. Front. Energy Res. 11:1133514. doi: 10.3389/fenrg.2023.1133514
Received: 29 December 2022; Accepted: 22 February 2023;
Published: 03 March 2023.
Edited by:
Xin Tu, University of Liverpool, United KingdomCopyright © 2023 Sultana, Lomate and Fujitani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Asima Sultana, YXNpbWEuc3VsdGFuYUBhaXN0LmdvLmpw
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.