 Tore Ericsson†
Tore Ericsson† Dickson O. Ojwang
Dickson O. Ojwang William R. Brant
William R. Brant- Department of Chemistry—Ångström Laboratory, Ångström Advanced Battery Centre, Uppsala University, Uppsala, Sweden
Prussian white (PW), Na2Fe [Fe(CN)6], is a highly attractive cathode material for sustainable sodium-ion batteries due to its high theoretical capacity of ∼170 mAhg−1 and low-cost synthesis. However, there exists significant variability in the reported electrochemical performance. This variability originates from compositional flexibility possible for all Prussian blue analogs (PBAs) and is exasperated by the difficulty of accurately quantifying the specific composition of PW. This work presents a means of accurately quantifying the vacancy content, valence distribution, and, consequently, the overall composition of PW via Mössbauer spectroscopy. PW cathode material with three different sodium contents was investigated at 295 and 90 K. The observation of only two iron environments for the fully sodiated compound indicated the absence of [Fe(CN)6]4- vacancies. Due to intervalence charge transfer between iron centers at 295 K, accurate determination of valences was not possible. However, by observing the trend of spectral intensities and center shift for the nitrogen-bound and carbon-bound iron, respectively, at 90 K, valence mixing between the iron sites could be quantified. By accounting for valence mixing, the sum of iron valences agreed with the sodium content determined from elemental analysis. Without an agreement between the total valence sum and the determined composition, there exists uncertainty around the accuracy of the elemental analysis and vacancy content determination. Thus, this study offers one more stepping stone toward a more rigorous characterization of composition in PW, which will enable further optimization of properties for battery applications. More broadly, the approach is valuable for characterizing iron-based PBAs in applications where precise composition, valence determination, and control are desired.
Introduction
Sodium-ion batteries (NIBs) are rapidly attracting interest as a viable alternative to lithium-ion batteries primarily based on cost and sustainability. To leverage the competitive advantage that NIBs bring, the various battery components must be designed with sustainability in mind, from elemental components to production. Subsequently, iron- and manganese-based Prussian blue analogs (PBAs) are highly attractive options for cathodes in NIBs. PBAs can be described by the general formula AxM [M′(CN)6]1−y□ynH2O, where A is an alkali metal cation, M and M′ are frequent transition metals, and □ denotes a [M′(CN)6]n− vacancy. The compositional flexibility of PBAs is a boon for designing the material class for a broad range of applications. However, it introduces several challenges when being utilized as a battery material, specifically that the composition is highly sensitive to the synthesis parameters, which can have a negative impact on properties (You et al., 2014; You et al., 2015; Rudola et al., 2017; Chen et al., 2018; Li et al., 2019). For example, Prussian white (PW) with ideal composition Na2Fe [Fe(CN)6] has a theoretical capacity of 170 mAh g−1 and an average voltage output of ∼3.2 V, which is comparable with the specific energy density achievable for LiFePO4 (Wang et al., 2015; Logan et al., 2020). However, achieving this capacity is extremely difficult due to the presence of [Fe(CN)6]n− vacancies (Hurlbutt et al., 2018). Furthermore, the presence of severe phase transitions which damage the structure during operation in a battery is strongly dependent on the A+ cation and [M′(CN)6]n− vacancy content (Rudola et al., 2017; Tapia-Ruiz et al., 2021; Boström and Brant 2022). Despite the importance of PBAs, their accurate and consistent compositions are rarely reported. This was highlighted by a recent meta-study on the reported structures of PBAs as a function of composition (Boström and Brant 2022). When clear trends emerged, quantitative conclusions became difficult to be drawn due to uncertainties on the reported compositions. One means through which the composition determination of PBAs can be improved is to adapt characterization tools to study them. One such method that displays promise for supporting analysis of composition is Mössbauer spectroscopy.
Mössbauer spectroscopy has been particularly advantageous for investigating the presence of vacancies in iron hexacyanoferrates. As already mentioned, quantification of the [M′(CN)6]n− vacancy content is critical for PBAs applied as electrodes in battery applications. If different transition metals occupy the M and M’ sites, this information can be extracted from cation ratios via methods such as inductively coupled plasma optical emission spectrometry (ICP-OES) (Xi and Lu 2021). However, in the case of AxFe[Fe(CN)6], the exact composition becomes more ambiguous. Fortunately, Mössbauer spectroscopy has been able to provide clear evidence for only two octahedral Fe environments in NaxFe [Fe(CN)6]. In other words, one Fe coordinated exclusively by six carbon atoms and the other by six nitrogen atoms (Brant et al., 2019; Ojwang et al., 2020; Ojwang et al., 2021). Two Fe environments are only possible if the vacancy content is lower than the detection limit of the measurement. With sufficient vacancies, other iron environments with a mixed coordination or lower coordination number would be observed (Reguera et al., 1999; Yang et al., 2015; You et al., 2015; Yang et al., 2020). Although identification of unique Fe environments by Mössbauer spectroscopy has become possible, occasionally there has been some ambiguity with regards to the oxidation states on each Fe center. This has led to the disagreement with X-ray diffraction results regarding the exact A-site content required to charge balance the transition metals (Ojwang et al., 2021). Part of this ambiguity may lie in the rapid intervalence charge transfer process between iron centers connected via a cyanide bridge creating a mixed valence configuration at room temperature (Robin and Day 1968). Naturally, if one assumes that no such valence mixing is occurring, extraction of absolute valence contributions will be inaccurate. The issue is further exacerbated by peak overlap and broadening in samples exhibiting valence mixing. However, intervalence charge transfer in PBAs has been shown to be temperature-dependent (Li et al., 2008) with better-resolved spectra observable at lower temperatures (Martínez-García et al., 2006). Therefore, there is a potential for Mössbauer spectroscopy to provide accurate data on both composition and valence distribution in iron-based PBAs.
In the present work, accurate determination of oxidation states in NaxFe [Fe(CN)6] by Mössbauer spectroscopy was explored at lower temperatures. By observing trends in spectral intensities and the center shift between 295 and 90 K and as a function of composition, this work presents a guide for extracting accurate information on the composition of iron hexacyanoferrates.
Experiment
Mössbauer measurements were carried out at 295 and 90 K on a spectrometer with a constant acceleration type of vibrator and a57CoRh source. PW samples prepared previously (Ojwang et al., 2020; Ojwang et al., 2021) were investigated. PW powder was ground together with BN and enclosed into sealed aluminum pockets. The so-formed absorbers had a concentration of

TABLE 1. Sample compositions, structures, and original references.
Results and Discussion
For details on sample composition, structure, and morphology, the reader is directed to previous work performed on the same samples (Table 1). Representative Mössbauer spectra are shown in Figure 1, and fitted results are presented in Table 2. The line intensities reveal no Fe vacancies on any of the two Fe sites within experimental errors. The Fe occupancies, I, at different sites are proportional to the spectral areas, A, in the Fe Mössbauer spectra. However, the spectral areas are also dependent on the Lamb–Mössbauer factor f. In fact, A is proportional to the product I•f. The f-factor is dependent on temperature and the Debye temperatures for the different sites. In the present case, the obtained spectral areas A (FeC) and A (FeN) are equal to 50 (1) % both at 295 and 90 K. This is a strong indication that the f-factors for FeC and FeN are within the experimental error and are the same especially at 90 K. Thus, it is safe to claim that the obtained spectral areas are good measures of the Fe occupancies I, as presented in Table 2.
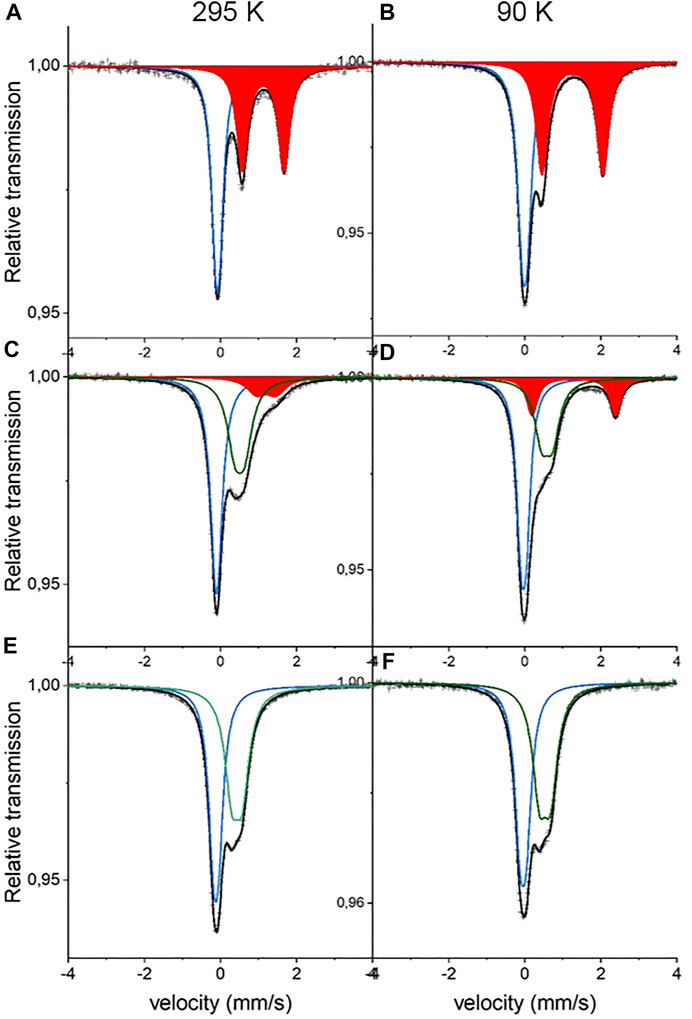
FIGURE 1. Mössbauer spectra of NaxFe [Fe(CN)6].nH2O at 295 K (left panel) and 90 K (right panel). (A,B) are x = 1.8 (C,D) are x = 1.0, and (E,F) are x = 0.5. Blue sub-spectra represent low-spin FeC2+, red sub-spectra represent high-spin FeN2+, and green sub-spectra represent high-spin FeN3+.
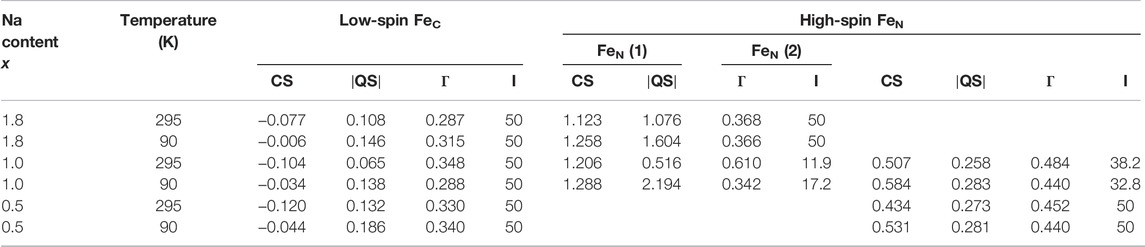
TABLE 2. Results from the fitting of NaxFe [Fe(CN)6].nH2O spectra at 295 K and at 90 K. The center shift CS, the magnitude of the electric quadrupole splitting
The 295-K spectrum in Figure 1 for high Na content,
Considering the intensities and line widths for the sample with x = 1.0, it is clear that the differences between the 295 and 90 K are large. At 295 K, the line widths are broader, and the resolution between the two FeN doublets is worse than that for the 90 K spectrum. Electron valence hopping at 295 K would give rise to the broadening of the resonance lines due to a mixed valency. This would make it difficult to assign the two FeN doublets to a specific Fe valence (Robin and Day 1968). As heating can induce electron valence hopping, the effect can be minimized through cooling (Martínez-García et al., 2006). At 90 K, the electron hopping time is increased and is longer than the Mössbauer observation time of around 100 ns. This gives rise to the better defined Fe spectra and thus provides more reliable ascriptions to Fe valences. Subsequently, the hyperfine parameters for the two FeN doublets are clearly representatives of 2+ and 3+, respectively. In determining the proportions of different valences on the high-spin FeN, the low-temperature intensities are, therefore, more trustworthy. It is interesting to note that there still exists a relatively large amount of FeN2+ at x = 1. The prevalent prediction is that all Fe at the high-spin site would be oxidized to Fe3+ at x = 1 (Wang et al., 2015). Consequently, this prediction has to be modified. From the line intensities in the spectrum for x = 1, a mixture of Fe0.66(2)3+ and Fe0.34(2)2+ at the high-spin FeN site can be calculated.
Turning on to the result for the FeC site, for x = 0, it has been shown that the center shift at 295 K for FeC3+ is −0.17 (1) mm/s (Ojwang et al., 2020), while for

TABLE 3. Result for the Fe valence distribution as measured at 90 K in the studied samples from the analysis given in the text.
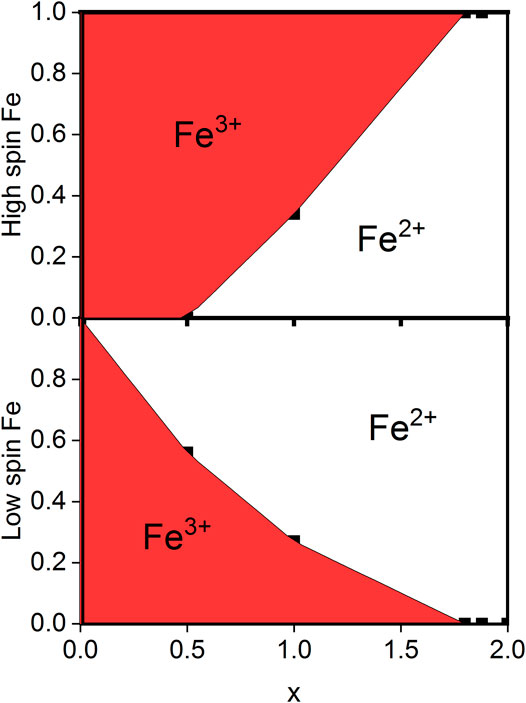
FIGURE 2. Valence distribution found for NaxFe [Fe(CN)6].1.8H2O for varying x-values. At x = 0, all Fe on both sites are Fe3+, while at x > 1.8, all Fe on both sites are Fe2+. Data are measured at 90 K.
The same analysis can be made for the spectrum at x = 0.5, arriving at the valence distribution given in Table 3 and shown in Figure 2. It is worth mentioning that these results lie within two standard deviations of the expected charge balance based on the measured sodium content. In other words, the valence balance from the formulas given in Table 3 are in deficit of -0.20 (13) for x = 1.8, in deficit of −0.34 (33) for x = 1.0, and in excess of +0.05 (34) electronic units for x = 0.5. Furthermore, while the valence distribution shown in Table 3 and Figure 2 was extracted from data at 90 K, it is a representative of the room-temperature valence distribution.
The remarkably large change in
Conclusion
It has been possible from the Mössbauer spectra to reveal the Fe valence distribution in the three studied samples by using the spectral intensities for FeN and the center shift for FeC. The reliability of this result increases strongly if the electronic relaxation time in the samples is slowed by performing the Mössbauer study at low temperature. Consequently, contrary to expectations from electrochemical cycling, a valence distribution across the two iron centers was observed. This distribution between the iron sites is consistent with each other, and their sum is consistent with the sodium content determined via elemental analysis. This study provides another avenue for confirming the composition and studying valence changes in electroactive PBAs. This approach will be essential for adequately characterizing and interpreting the performance of iron-based PBAs for application in sodium-ion batteries.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material; further inquiries can be directed to the corresponding author.
Author Contributions
TE and LH performed all measurements, data fitting and analysis, and wrote the results, discussions, and main conclusion. DOO prepared the samples, was involved in discussions regarding interpretation of the results, and reviewed and edited the final manuscript. WRB supervised the work, wrote part of the original draft, and reviewed and edited the final manuscript.
Funding
This research was funded by the Energimyndigheten project: 45517-1.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The strategic research area StandUp for Energy is also acknowledged.
References
Boström, H. L. B., and Brant, W. R. (2022). Octahedral Tilting in Prussian Blue Analogues. J. Mat. Chem. C. doi:10.1039/D2TC00848C
Brant, W. R., Mogensen, R., Colbin, S., Ojwang, D. O., Schmid, S., Häggström, L., et al. (2019). Selective Control of Composition in Prussian White for Enhanced Material Properties. Chem. Mat. 31 (18), 7203–7211. doi:10.1021/acs.chemmater.9b01494
Chen, Y., Woo, H. J., Kufian, M. Z., Teo, L. P., Wang, F., Zhao, C., et al. (2018). Synthesis of Low Vacancies PB with High Electrochemical Performance Using a Facile Method. Mater. Technol. 35 (11-12), 759–766. doi:10.1080/10667857.2018.1493835
Hurlbutt, K., Wheeler, S., Capone, I., and Pasta, M. (2018). Prussian Blue Analogs as Battery Materials. Joule 2 (10), 1950–1960. doi:10.1016/j.joule.2018.07.017
Li, D., Clérac, R., Roubeau, O., Harté, E., Mathonière, C., Le Bris, R., et al. (2008). Magnetic and Optical Bistability Driven by Thermally and Photoinduced Intramolecular Electron Transfer in a Molecular Cobalt−Iron Prussian Blue Analogue. J. Am. Chem. Soc. 130 (1), 252–258. doi:10.1021/ja0757632
Li, L., Nie, P., Chen, Y., and Wang, J. (2019). Novel Acetic Acid Induced Na-Rich Prussian Blue Nanocubes with Iron Defects as Cathodes for Sodium Ion Batteries. J. Mat. Chem. A 7 (19), 12134–12144. doi:10.1039/C9TA01965K
Logan, E. R., Hebecker, H., Eldesoky, A., Luscombe, A., Johnson, M. B., and Dahn, J. R. (2020). Performance and Degradation of LiFePO4/Graphite Cells: The Impact of Water Contamination and an Evaluation of Common Electrolyte Additives. J. Electrochem. Soc. 167 (13), 130543. doi:10.1149/1945-7111/abbbbe
Martínez-García, R., Knobel, M., Goya, G., Gimenez, M. C., Romero, F. M., and Reguera, E. (2006). Heat-induced Charge Transfer in Cobalt Iron Cyanide. J. Phys. Chem. Solids 67 (11), 2289–2299. doi:10.1016/j.jpcs.2006.05.045
Ojwang, D. O., Häggström, L., Ericsson, T., Ångström, J., and Brant, W. R. (2020). Influence of Sodium Content on the Thermal Behavior of Low Vacancy Prussian White Cathode Material. Dalton Trans. 49 (11), 3570–3579. doi:10.1039/D0DT00014K
Ojwang, D. O., Svensson, M., Njel, C., Mogensen, R., Menon, A. S., Ericsson, T., et al. (2021). Moisture-Driven Degradation Pathways in Prussian White Cathode Material for Sodium-Ion Batteries. ACS Appl. Mat. Interfaces 13 (8), 10054–10063. doi:10.1021/acsami.0c22032
Reguera, E., Fernández-Bertrán, J., and Balmaseda, J. (1999). The Existence of Ferrous Ferricyanide. Transit. Met. Chem. 24 (6), 648–654. doi:10.1023/A:1006942415737
Robin, M. B., and Day, P. (1968). Mixed Valence Chemistry-A Survey and Classification. Adv. Inorg. Chem. Radiochem. 10, 247–422. doi:10.1016/S0065-2792(08)60179-X
Rudola, A., Du, K., and Balaya, P. (2017). Monoclinic Sodium Iron Hexacyanoferrate Cathode and Non-Flammable Glyme-Based Electrolyte for Inexpensive Sodium-Ion Batteries. J. Electrochem. Soc. 164 (6), A1098–A1109. doi:10.1149/2.0701706jes
Tapia-Ruiz, N., Armstrong, A. R., Alptekin, H., Amores, M. A., Au, H., Barker, J., et al. (2021). 2021 Roadmap for Sodium-Ion Batteries. J. Phys. Energy 3 (3), 031503. doi:10.1088/2515-7655/ac01ef
Wang, L., Song, J., Qiao, R., Wray, L. A., Hossain, M. A., Chuang, Y.-D., et al. (2015). Rhombohedral Prussian White as Cathode for Rechargeable Sodium-Ion Batteries. J. Am. Chem. Soc. 137 (7), 2548–2554. doi:10.1021/ja510347s
Xi, Y., and Lu, Y. (2021). Interpretation on a Nonclassical Crystallization Route of Prussian White Nanocrystal Preparation. Cryst. Growth & Des. 21 (2), 1086–1092. doi:10.1021/acs.cgd.0c01409
Yang, D., Xu, J., Liao, X.-Z., Wang, H., He, Y.-S., and Ma, Z.-F. (2015). Prussian Blue without Coordinated Water as a Superior Cathode for Sodium-Ion Batteries. Chem. Commun. 51 (38), 8181–8184. doi:10.1039/C5CC01180A
Yang, L., Liu, Q., Wan, M., Peng, J., Luo, Y., Zhang, H., et al. (2020). Surface Passivation of NaxFe[Fe(CN)6] Cathode to Improve its Electrochemical Kinetics and Stability in Sodium-Ion Batteries. J. Power Sources 448, 227421–227428. doi:10.1016/j.jpowsour.2019.227421
You, Y., Wu, X.-L., Yin, Y.-X., and Guo, Y.-G. (2014). High-quality Prussian Blue Crystals as Superior Cathode Materials for Room-Temperature Sodium-Ion Batteries. Energy Environ. Sci. 7 (5), 1643–1647. doi:10.1039/C3EE44004D
Keywords: Prussian white, Mössbauer spectroscopy, sodium-ion battery, intervalence charge transfer, low-defect, cathode
Citation: Ericsson T, Häggström L, Ojwang DO and Brant WR (2022) Investigation of Valence Mixing in Sodium-Ion Battery Cathode Material Prussian White by Mössbauer Spectroscopy. Front. Energy Res. 10:909549. doi: 10.3389/fenrg.2022.909549
Received: 31 March 2022; Accepted: 17 May 2022;
Published: 04 July 2022.
Edited by:
Ivana Hasa, University of Warwick, United KingdomReviewed by:
Wenli Zhang, Guangdong University of Technology, ChinaFriedrich Wagner, Technical University of Munich, Germany
Copyright © 2022 Ericsson, Häggström, Ojwang and Brant. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William R. Brant, d2lsbGlhbS5icmFudEBrZW1pLnV1LnNl
†These authors have contributed equally to this work and share first authorship