 S. Shaqiri
S. Shaqiri D. Kaczmarek1
D. Kaczmarek1 F. vom Lehn
F. vom Lehn T. Kasper
T. Kasper- 1Institute for Combustion and Gas Dynamics—Mass Spectrometry in Reactive Flows, University of Duisburg-Essen, Duisburg, Germany
- 2Institute for Combustion Technology, RWTH Aachen University, Aachen, Germany
Iso-octane is frequently used as a surrogate fuel or as a component in primary reference fuel blends when low-temperature combustion strategies in engines are investigated. To develop control strategies for these engines, the reaction kinetics of iso-octane must be known starting from the low temperatures and intermediate pressures before ignition to the high temperatures and pressures of combustion. This work adds new experimental data sets to the validation data for reaction mechanism development by investigating the oxidation of iso-octane in stoichiometric mixtures in a flow reactor at pressures of p = 1, 10, and 20 bar and 473K ≤ T ≤ 973 K. The experimental data are compared to simulations with recent reaction mechanisms [Atef et al., Combustion and Flame 178, (2017), Bagheri et al., Combustion and Flame 212, (2020), Cai et al., Proceedings of the Combustion Institute 37, (2018), Fang et al., Combustion and Flame 214, (2020)]. The comparison between experimental and simulated mole fractions as function of temperature show reasonable agreement for all investigated pressures. In particular, the experimentally observed onset of low-temperature reactivity above a certain pressure, the shift of the negative temperature coefficient (NTC) regime with increasing pressure to higher temperatures, and the acceleration of the high-temperature chemistry are captured well in the simulations. Deviations between experimental and simulated results are discussed in detail for the reactivity of iso-octane and some key intermediates such as 2,2,4,4-tetramethyl-tetrahydrofuran, iso-butene and acetone at low temperatures.
1 Introduction
Despite the growing share of renewables, advanced combustion strategies present a good bridging technology in the transformation of the transportation sector for those areas in which rapid electrification appears difficult, e.g., in heavy duty, ship, or air traffic. These strategies typically run the combustion process at low temperatures and with fuel-lean mixtures. Under these conditions, complete fuel conversion can be achieved and the formation of soot and nitrogen oxides is avoided (Musculus et al., 2013). Low-temperature combustion (LTC) is used in engines, e.g., in the form of Gasoline-Controlled Auto-Ignition (GCAI) (Nuss et al., 2019) or Premixed Charge Compression Ignition (PCCI) (Korkmaz et al., 2018). In both combustion processes a homogeneous fuel-air mixture is generated, which then burns after auto-ignition. A particular problem in the control of LTC processes are the large fluctuations of temperature, mixture composition, and pressure that can occur in each cycle on a very short time scale due to the superposition of reaction kinetic and fluid mechanical effects. These fluctuations make it difficult to operate the engine stably in a large parameter field and present a particular challenge for control of transient operation (Nuss et al., 2019). Ignition and pollutant formation processes are determined by the kinetics of the substances and reactions involved in the process. Hence, a detailed understanding of the chemical kinetics is essential for control strategies and improvement of the process.
Gasoline and diesel fuel are complex mixtures consisting of numerous species with different molecular structures, e.g., n-alkanes, iso-alkanes, cycloalkanes, and aromatics. Surrogate fuels have similar characteristics as the real fuels but are composed of only a limited number of representative components so that the reaction kinetics are considerably simplified compared to real fuels. Iso-octane is used frequently as a neat surrogate fuel or as a component in a primary reference fuel (PRF) blend (Tanaka, 2003; Lü et al., 2005; Jia and Xie, 2006; Yao et al., 2009; Masurier et al., 2015) to model knocking propensities of engine processes and to determine the octane number of gasoline. Consequently, reliable reaction mechanisms for iso-octane are needed to advance the investigation of LTC in engines. Previous experimental studies of iso-octane oxidation have focused primarily on stoichiometric, high-temperature and moderate-pressure conditions. They include the investigation of auto-ignition in shock tubes (Vermeer et al., 1972; Fieweger et al., 1997; Davidson et al., 2002; Davidson et al., 2005; Shen et al., 2008; Hartmann et al., 2011) and rapid compression machines (Griffiths et al., 1993; Cox et al., 1996; Minetti et al., 1996; He et al., 2005; He et al., 2007; Mittal and Sung, 2007; Kukkadapu et al., 2012; Kukkadapu et al., 2013; Masurier et al., 2015), identification and determination of concentration profiles in jet-stirred (Lignola et al., 1989; D’Anna et al., 1992; Dagaut et al., 1994; Ciajolo et al., 1993; Dagaut et al., 1993) and flow reactors (Dryer and Brezinsky, 1986; Callahan et al., 1996; Chen et al., 2000; Lu et al., 2019), as well as studies in engines (Maynard et al., 1967; Leppard, 1992; Li et al., 1994). Experiments in shock tubes and rapid compression machines typically address conditions relevant for LTC combustion in engines but they often lack information on the concentration histories of species formed at various temperatures and pressures. Speciation measurements are particularly valuable for the development of reaction mechanisms. Corresponding speciation studies at various conditions are summarized in more detail below.
One of the first efforts to identify intermediate species in motored engines has been undertaken by Maynard et al. (1967). Twenty-one intermediate species were identified including 2,2,4,4-tetramethyl-tetrahydrofuran (TMTHF) and iso-butene. Leppard (1992) and Li et al. (1994) performed experiments in a motored engine to identify additional intermediate species and characterize the auto-ignition chemistry. In both studies inlet pressures and temperatures as well as engine speeds were varied.
Jet-stirred and flow reactor studies of the oxidation of iso-octane are mostly focused on the determination of concentration time histories in small pressure and temperature ranges providing valuable data for reaction mechanism development. Dryer and Brezinsky (1986) investigated the oxidation of iso-octane in a flow reactor at 1080 K, atmospheric pressure, and a stoichiometric equivalence ratio (ɸ). Contrary to the studies in engines, dimethyl-pentenes were not detectable. D’Anna et al. (1992) studied the stoichiometric oxidation of iso-octane in a jet stirred reactor at 7 and 9 bar with fixed residence time in a temperature range of 600–760 K. In this work, the formation of dimethyl-pentenes was detected, which are expected to be formed by ß-scission of iso-octyl radicals. Dagaut et al. (1993) and Dagaut et al. (1994)) investigated the oxidation of n-heptane and iso-octane at 10 atm, 550–1150 K, and equivalence ratios of 0.3, 0.5, 1, and 1.5. Various species including all dimethyl-pentenes, C5-olefins, and oxygenated compounds could be identified. Callahan et al. (1996) presented experimental data in a pressurized flow reactor at 12.5 atm in a temperature range of 550–900 K under stoichiometric conditions. Only major products were evaluated. Chen et al. (2000) investigated the lean oxidation of iso-octane at ɸ = 0.005 in a flow reactor at 3, 6, and 9 atm in a small temperature range of 915–950 K. 23 intermediate species have been evaluated as a function of time. Lu et al. (2019) investigated the oxidation of n-heptane, iso-octane, ethanol, and their mixtures in a flow reactor at an equivalence ratio of 0.058, 550–900 K, and 10 bar. The concentration of CO was measured as a function of initial reactor temperature as an indicator of global reactivity and was compared to various PRF mixtures. Wang et al. (2019) evaluated the important pyrolysis pathways of iso-octane from 848 to 1123 K at 30 Torr and from 723 to 998 K at 760 Torr.
Several reaction mechanisms (Halstead et al., 1977; Cox and Cole, 1985; Hu and Keck, 1987; Callahan et al., 1996; Ranzi et al., 1997; Davis and Law, 1998; Ogink and Golovitchev, 2001; Curran, 2002; Glaude et al., 2002; Zheng et al., 2002; Tanaka et al., 2003; Buda et al., 2005) have been developed for the description of iso-octane combustion based on the experimental results. These mechanism developments until 2006 including the temperature, pressure, and equivalence range of the validation data have been summarized by Jia and Xie (2006). In 2011, an additional detailed chemical kinetic mechanism for the simulation of gasoline surrogate components was published by Mehl et al. (2011). The mechanism is based on the previously developed mechanism of Curran (2002). It includes 1550 species and 6000 reactions and was validated by comparison to experimental data obtained in shock tubes and rapid compression machines. An updated iso-octane mechanism was published in 2017 by Atef et al. (2017). This mechanism is mainly based on the mechanism of Mehl et al. (2011), with the addition of a diisobutylene sub-mechanism published by Metcalfe et al. (2007) and also found in AramcoMech 2.0 (Li et al., 2017). More importantly, the thermochemistry data were updated in this mechanism based on new group values of Burke et al. (2015), while the low-temperature kinetics were updated following suggestions of Bugler et al. (2015), including the addition of new alternative isomerization pathways for peroxy hydroperoxide radicals. Furthermore, Cai et al. (2019) presented a skeletal kinetic mechanism for iso-octane using rate rules, which were optimized (Cai et al., 2016) based on the updated rate rules of Bugler et al. (2015), and also using the updated group values (Burke et al., 2015) for thermochemistry estimation.
Even though flow reactor experiments do not reach the same high temperatures and pressures as the LTC combustion processes in engines, they provide relevant kinetic information in a temperature and pressure range that occurs in these engines just before the main ignition. To control the ignition behavior and heat release rates during that phase model-based control strategies as presented by Nuss et al. (2019) for gasoline type controlled auto-igniton (GCAI) and by Korkmaz et al. (2018) for partially premixed compressed ignition (PCCI) rely on that data.
The work presented here extends the data base for model validation in this temperature and pressure regime by investigating the oxidation of iso-octane in a flow reactor 473–973 K, an ɸ = 1 and at 1–20 bar, which is at significantly higher pressure than the flow reactor studies in literature. It focuses on the influence of pressure on the occurrence of the NTC region.
2 Experimental Setup
The oxidation of iso-octane is investigated in a flow reactor with gas-chromatographic analysis of the composition of the mixture at the reactor outlet. The original reactor design was described by Sen et al. (2015), and modifications to the design are summarized in recent papers by Kaczmarek et al. (2019a, 2019b). The reactor was operated in a temperature range of 473K ≤ T ≤ 973 K. For the experiments in this work, the reactor has been modified to withstand higher pressures of up to 20 bar. A brief description of the reactor and the experimental conditions are provided below.
The reactor consists of an inner quartz tube with a length of 65 cm and an internal diameter of 6 mm that is enclosed in an outer tube of Inconel. Compared to the total flow rate a negligible amount of gas permeates into a small gap between the two tubes so that the pressure gradient between atmosphere and reactor pressure is applied to the Inconel and not the quartz tube. To prevent composition changes by contact of gas with the Inconel tube, it is coated by SiO2. The reactor is uniformly heated by external heating tapes and has an isothermal zone of 40 cm. At the inlet and outlet steep temperature gradients occur. Before the measurements at elevated pressure, the temperature profiles for each externally set temperature are measured inside of the reactor and used in the simulation of the experiment. They are provided in the Supplementary Material.
Gas samples are taken from the reactor after expansion of the reacting mixture to 1 bar through a needle valve, which is also used to regulate the pressure in the reactor. Sampling and sample transfer to the gas chromatograph (GC) is performed using a syringe driven by an autosampler. All relevant lines are heated to prevent condensation. The GC uses a MSieve 5A column to separate permanent gases before detection by a thermal conductivity detector, and a RTX 5 fused silica column to separate CO2, H2O, as well as hydrocarbons before detection by a mass spectrometer, flame ionization detector, and thermal conductivity detector. Neat He is used as carrier gas.
Gas flows are metered by Coriolis flow controllers. A controlled flow of liquid iso-octane is provided by a Coriolis flow controller and enters a home-built vaporizer, where it is mixed with one half of the total nitrogen flow to prevent condensation when the iso-octane/nitrogen flows are combined with the other half of the nitrogen and the oxygen flow at the inlet of the reactor. Liquid iso-octane is fed by a pressurized vessel. The total gas flow in all experiments was 1000 sccm. The inlet mole fractions were: ×(C8H18) = 0.00148, ×(O2) = 0.01852, ×(N2) = 0.98. In the temperature range 473 ≤ T ≤ 973 K this flow rate results in variable residence times of 0.31–0.18 s at 1 bar and 6.19–3.57 s at 20 bar. Measurements were performed with stoichiometric mixtures at 1, 10, and 20 bar.
Gases were calibrated using certified calibration mixtures, and home-made binary calibration mixtures for as many compounds as possible. For species not directly calibrated from standards, the effective carbon number method was used to calculate the calibration coefficient (Scanlon and Willis, 1985). In general, uncertainties below 10% could be achieved for calculated mole fractions.
3 Modeling
Simulations were performed using the plug-flow module of (CHEMKIN-PRO 19.2, Reaction Design San Diego, 2017). The measured temperature profiles between inlet and outlet were used for all simulations and are provided in the Supplementary Material. For simulations the reaction mechanism of Atef et al. (2017) was employed because it reproduces the NTC behavior of iso-octane combustion well. In addition, simulations were performed with the mechanisms of Bagheri et al. (2020), Cai et al. (2019), and Fang et al. (2020) to compare reaction pathways for species that could not be predicted well with the reaction mechanism of Atef et al. (2017).
Aramco 2.0 (Zhou et al., 2016) was used for the C0–C4 base chemistry in the kinetic mechanism of Atef et al. (2017), whereas the chemistry of C5–C7 species was taken from LLNL’s gasoline surrogate mechanism (Mehl et al., 2011), with the addition of the sub-mechanism for the diisobutylene isomers by Metcalfe et al. (2007) that are produced during the oxidation of iso-octane. More importantly, the thermochemistry data were updated in the mechanism of Atef et al. (2017) based on new group values of Burke et al. (2015), while the low-temperature kinetics were updated following suggestions of Bugler et al. (2015), including the addition of new alternative isomerization pathways for peroxy hydroperoxide radicals. The detailed chemical kinetic model of Fang et al. (2020) contains species up to C12 and is mainly based on the model of Zhang et al. (2019) for alkanes and the C0–C4 base chemistry of Aramco 2.0. The high temperature chemistry has largely been taken from the work of Mehl et al. (2011). The skeletal kinetic model for PRF mixtures proposed by Cai et al. (2019) was developed using rate rules, which were optimized (Cai et al., 2016) based on the updated rate rules of Bugler et al. (2015), and also using the updated group values (Burke et al., 2015) for thermochemistry estimation.
Nitrogen was used as dilution gas. It serves two purposes: First, it is used as standard for the quantification of the measurements. Second, the high dilution reduces the impact of temperature changes in the reaction zone caused by heat release from exothermic reactions. Simulations using a shear-flow model that also takes the heat release by reactions into account have been performed for selected reaction conditions of this work. The mole fractions found in the simulations at the outlet of the reactor do not differ significantly from results of simulations performed using a plug-flow model of the reactor. Consequently, the less time-consuming plug-flow model was used for all further simulations in this work.
4 Results and Disscusion
4.1 General Overview
Beside the main species of the reaction (iso-octane, O2, H2O, CO, CO2), several oxidation products of the fuel were identified and quantified as a function of temperature and pressure. The products can be classified into cyclic ethers, olefins, and carbonyls. 2,2,4,4-tetramethyl-tetrahydrofuran (TMTHF) could be clearly identified among the cyclic ethers. Olefinic hydrocarbons include iso-butene and 2,4-dimethyl-2-pentene, 4,4-dimethyl-2-pentene, 2,4-dimethyl-1-pentene, 4,4-dimethyl-1-pentene as well as 2,4,4-trimethyl-1-pentene and 2,4,4-trimethyl-2-pentene. Acetone and small amounts of acetaldehyde are among the identifiable carbonyls. In general, the lower the pressure, the lower are the observed concentrations of intermediate species due to the decrease in the consumption of iso-octane. For some intermediates, the mole fractions may even fall below the detection limit of the GC at lower pressures. Mole fraction profiles are reported for all measured and identified species in the Supplementary Material together with the simulation results. To show major aspects of the influence of pressure on the oxidation of iso-octane, results at 1, 10, and 20 bar are shown in the following.
4.2 Overall Pressure-Dependence of the NTC Behavior of Iso-Octane
The temperature dependence of the iso-octane consumption changes markedly with pressure, as can be seen from Figure 1. Considering the experimental results, iso-octane starts to react at approximately 898 K at 1 bar. At a pressure of 10 and 20 bar the iso-octane mole fractions start to decrease near 573 and 548 K, respectively, indicating initial fuel consumption reactions.
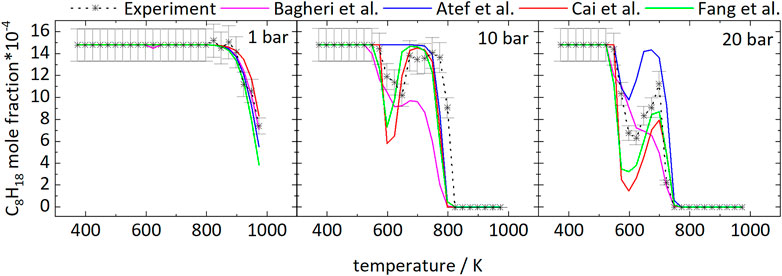
FIGURE 1. Mole fractions of iso-octane vs. reactor-temperature at 1, 10, and 20 bar. Dashed lines with symbols and solid lines correspond to experimental and modeling results, respectively.
The mole fractions decrease until temperatures of 623 and 648 K are reached at 20 and 10 bar, respectively. The subsequent increase in mole fractions indicates a reduction of reactivity of the mixtures with a minimal fuel conversion at 698 K (20 bar) and 748 K (10 bar). At higher temperatures, the mole fractions decrease again. As a result, the curves at higher pressure display a temperature range of 50–100 K with a negative temperature coefficient (NTC). This NTC region shifts to higher temperatures with increasing pressure, as can be seen from Figure 1. All mechanisms used in this work, beside the mechanism of Atef et al. (2017), predict initial fuel consumption in a similar temperature range. The mechanism of Atef et al. (2017) predicts too low reactivity at 10 bar, as indicated by the absence of the NTC region and the start of initial fuel consumption at significantly higher temperatures compared to experimental results and the other mechanisms used in this work. The temperature range of the NTC region is predicted well by the mechanisms of Atef et al. (2017) (at 20 bar), Cai et al. (2019), and Fang et al. (2020). An exception is the mechanism of Bagheri et al. (2020), which predicts the NTC region at slightly higher temperatures. Also, the decrease of reactivity after initial fuel consumption is predicted to be not as pronounced by this mechanism as by the mechanisms of Atef et al. (2017), and Fang et al. (2020).
Figure 2 shows the mole fractions of CO compared to the model predictions. The temperature-dependent changes of the mole fraction of CO are roughly inverse to the mole fraction changes of the fuel. As one of the main stable products in these measurements it is mainly produced at temperatures of 798 and 723 K at 10 and 20 bar, respectively, and the formation is reduced in the NTC region. From Figure 2 it can be seen that even though the NTC region shifts to higher temperatures as pressure increases, the reactivity of the mixture at low temperatures increases at higher pressures, effectively broadening the region of low-temperature reactivity (called LTC region in the following).
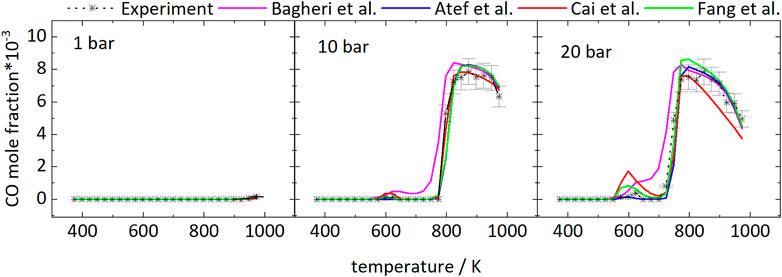
FIGURE 2. Mole fractions of CO vs. reactor-temperature at 1, 10, and 20 bar. Dashed lines with symbols and solid lines correspond to experimental and modeling results, respectively.
In the following discussions all temperatures below the upper end of the NTC region (in the LTC region) will be referred to as “low temperatures” and the rest will be called “intermediate temperatures”.
Figure 3 shows the mole fraction profiles as function of temperature of diisobutylene isomers (DIB) as one important intermediate. As expected, it is mainly formed in the LTC region and at intermediate temperatures above 748 K, where the second peak at intermediate temperatures at 20 bar is significantly smaller. The deviations to the predicted mole fractions are acceptable, besides the prediction of 2,4,4-trimethyl-1-pentene with the model of Cai et al. (2019) at 20 bar. The best prediction is by the Cai et al. (2019) mechanism at 10 bar. The formation pathways are discussed in Section 4.4 using the reaction pathway analyses with the mechanisms of Atef et al. (2017) and Cai et al. (2019).
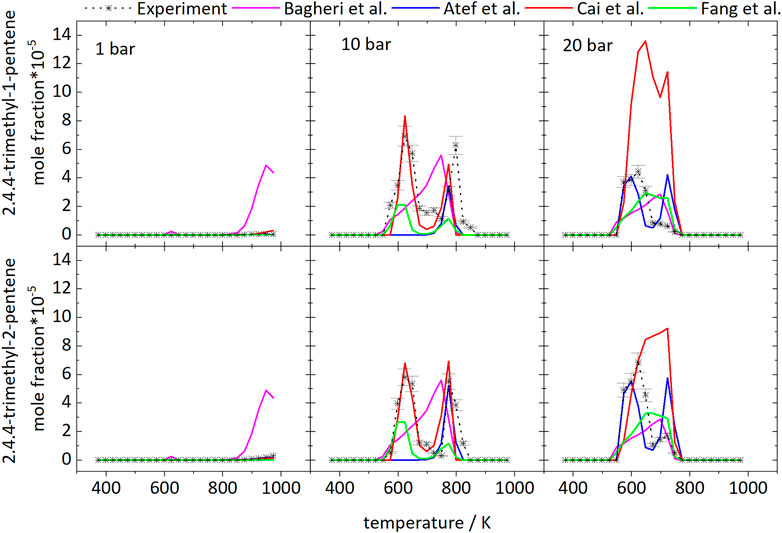
FIGURE 3. Mole fractions of diisobutylene isomers vs. reactor-temperature at 1, 10, and 20 bar. Dashed lines with symbols and solid lines correspond to experimental and modeling results, respectively.
As the residence time increases in the experiments when the pressure increases, the more pronounced LTC region could also be caused by this change. To verify that the observed NTC effect can be attributed to a kinetic effect of pressure, simulations with constant residence times for all pressures were performed using the mechanism of Atef et al. (2017). Intermediate residence times corresponding to a total gas flow of 1000 sccm at 10 bar were chosen for this study. Figure 4 displays the temperature-dependent fuel mole fraction curves generated in these simulations.
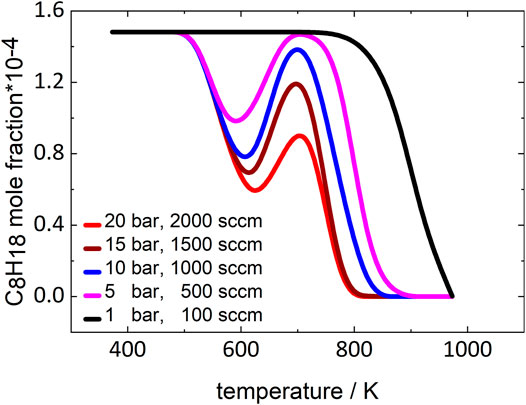
FIGURE 4. Simulated mole fractions of iso-octane with constant residence time at 1, 5, 10, 15, and 20 bar. Simulations performed using the model of Atef et al. (2017).
At 1 bar, no NTC behavior can be observed, while at all higher pressures a distinct NTC region appears in the simulated curves that becomes more pronounced as pressure increases. Consequently, the reactions leading to the NTC region must be more dependent on pressure than on residence time. The reactions that are important for this pronounced pressure dependence are discussed in the following section.
4.3 Overview of the LTC Reaction Pathways of Iso-Octane
Figure 5 shows an overview of expected low-temperature reaction pathways of hydrocarbon fuels from reference (Atef et al., 2017). The fuel is consumed by sequential hydrogen abstractions and oxygen addition to the resulting radical. The peroxides isomerize and form hydroperoxides, which can undergo the same reaction sequence again leading to ketohydroperoxides and their decomposition products. At each stage the intermediates can also decompose and form mainly olefins and cyclic ethers as intermediates. For a large fuel like iso-octane the simple sketch in Figure 5 becomes substantially more complicated.
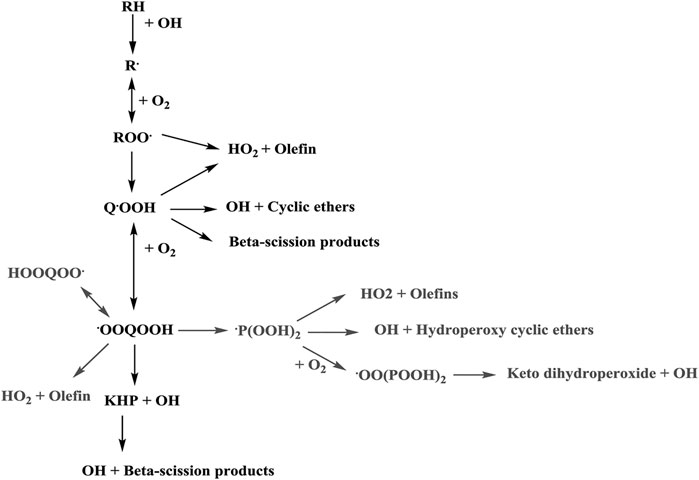
FIGURE 5. General fuel consumption pathway (Atef et al., 2017).
Following Westbrook et al. (1991) the hydrogen atoms of iso-octane can be divided into four groups a, b, c, and d depending on their carbon sites, as depicted in Figure 6.
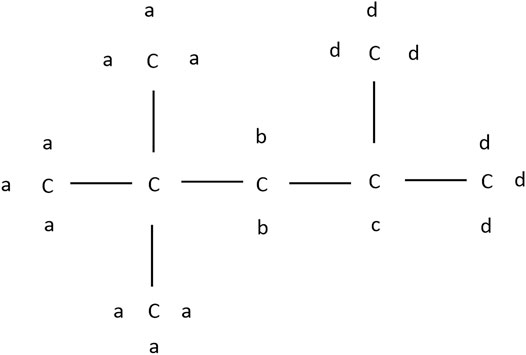
FIGURE 6. Molecular structure of iso-octane.
According to this structure, every abstraction of hydrogen within a group forms the same radical and each group forms a different radical. Each possible iso-octyl (C8H17) radical will lead to a different set of products. The different reaction mechanisms already yield different predictions for the mainly formed iso-octyl radical in the very first reaction step of the reaction scheme shown in Figures 7, 8. To analyze the main formation pathways of the experimentally observed intermediates in our experiment, we performed a sensitivity and reaction pathway analysis with respect to fuel conversion.
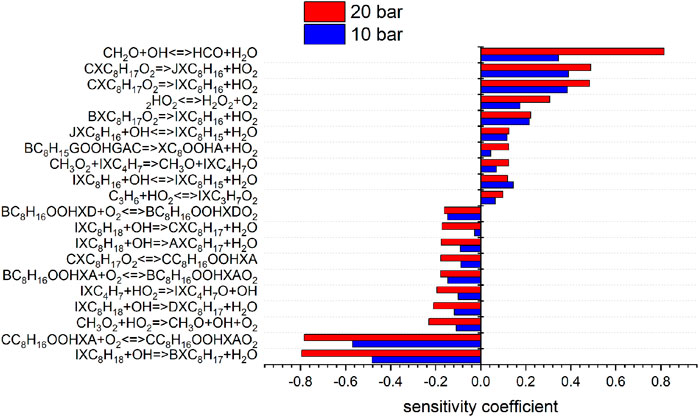
FIGURE 7. Sensitivity analysis of C8H18 on reaction rate coefficients performed with the model of Cai et al. at 10 and 20 bar, 623 K.
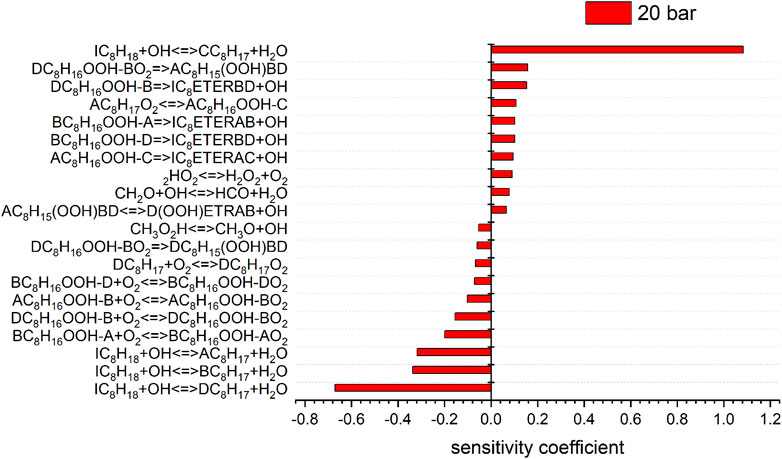
FIGURE 8. Sensitivity analysis of C8H18 on reaction rate coefficients performed with the model of Atef et al. at 10 (sensitivity coefficients = 0) and 20 bar, 623 K.
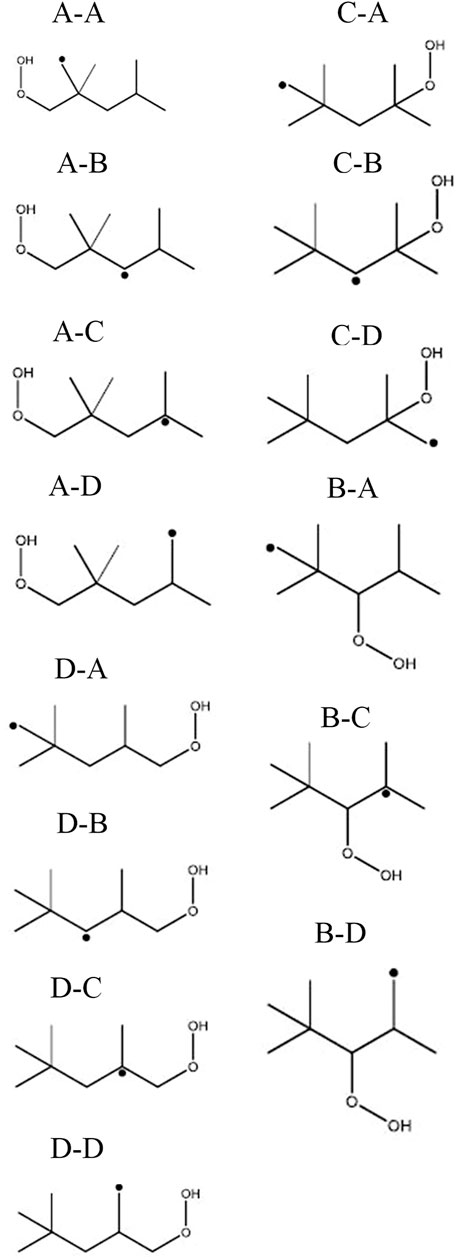
FIGURE 9. Species dictionary of QOOH radicals.
4.4 Sensitivity -and Reaction Path Analyses
To shed more light on the underlying kinetics governing the low-temperature oxidation of iso-octane, sensitivity analyses of the fuel mole fraction with respect to elementary reaction rate coefficients were performed using the mechanisms of Atef et al. (2017) and Cai et al. (2019) at a temperature of 623 K (corresponding to the region of low-temperature reactivity in the flow reactor measurements) and pressures of 10 and 20 bar. The results are shown in Figures 7, 8. The kinetic models of Atef et al. (2017) and Cai et al. (2019) are used. The model of Atef et al. (2017) contains new reaction rates related to the C8-submechanism and it is comparatively detailed for C8-combustion since it has the third O2 addition pathway considered. The mechanism of Cai et al. (2019) has been developed in a very systematic manner. The mechanism of Cai et al. (2019) relies on rate rules optimized based on experimental ignition data (Cai et al., 2016), while the mechanism of Atef et al. (2017) uses rate rules from Sivaramakrishnan and Michael (2009) and Badra and Farooq (2015) for the H-atom abstraction reactions. The percentage in the reaction flux analysis refers to the relative rates of production (ROP), which shows the main consumption path of a certain species. The reaction flux analysis corresponds to a reactor position, where around 25% of the maximum conversion of the fuel for the specific condition is reached. The influence of increasing pressure on the reaction configuration is evaluated and an initial oxidation scheme is presented.
The consumption of iso-octane is initiated by H-abstraction primarily by OH radicals. At temperatures higher than in the NTC region, reactivity is more controlled by high-temperature chemistry and the fuel consumption by HO2 radicals increases. With increasing pressure the contributions of H-abstraction reactions become higher. Considering the reaction flux analysis derived by the mechanism of Atef et al. (2017), the highest fluxes are observed for the primary-site radicals. However, abstraction from primary carbons is energetically more difficult than abstraction from secondary and tertiary carbons. The fact that 9 times more H-atoms are avaliable at the primary carbon site, compared to the tertiary carbon site, lead to the dominant route at the primary carbon site with the model of Atef et al. (2017). A comparison with the reaction fluxes derived by the model of Cai et al. (2019) indicates the highest flux in forming iso-octyl radicals by abstraction from secondary and tertiary carbons. Ning et al. (2015) calculated the C–C and C–H bond dissociation energies (BDE) within the iso-octane structure. It has been shown that the C–H BDEs for the primary carbons (101.3 and 100.4 kcal/mol) are higher than those for the secondary (96.0 kcal/mol) and tertiary carbons (92.9 kcal/mol). Increasing the pressure does not noticeably change the distribution in forming iso-octyl radicals. The sensitivity analysis shows that fuel H-abstraction reactions by OH radicals are the most sensitive reactions, as can be seen in Figure 7.
The first addition of alkyl radicals (Ṙ + O2 = RO2) to O2 initiates the low-temperature oxidation mechanism and the second addition of QOOH to O2 initiates low-temperature chain branching. Wang et al. (2016) and Wang et al. (2019) proposed additionally a third O2 addition and an alternative isomerization reaction path, where peroxy-hydroperoxy alkyls (ȮOQOOH) with a tertiary hydroperoxy group form alkyl-dihydroperoxides isomerization. After the first addition of iso-octyl radicals to O2, the formed alkylperoxy radicals can build diisobutylene isomers (2,4,4-trimethyl-1-pentene and 2,4,4-trimethyl-2-pentene) by concerted HȮ2 radical elimination reactions. The reaction flux analysis (Figure 10) reveals a large pressure dependence of the unimolecular decomposition reaction of alkylperoxy radicals to diisobutylene (DIB) isomers. While the formation of DIB isomers at 10 bar is negligible, the flux at 20 bar of this reaction channel is high.
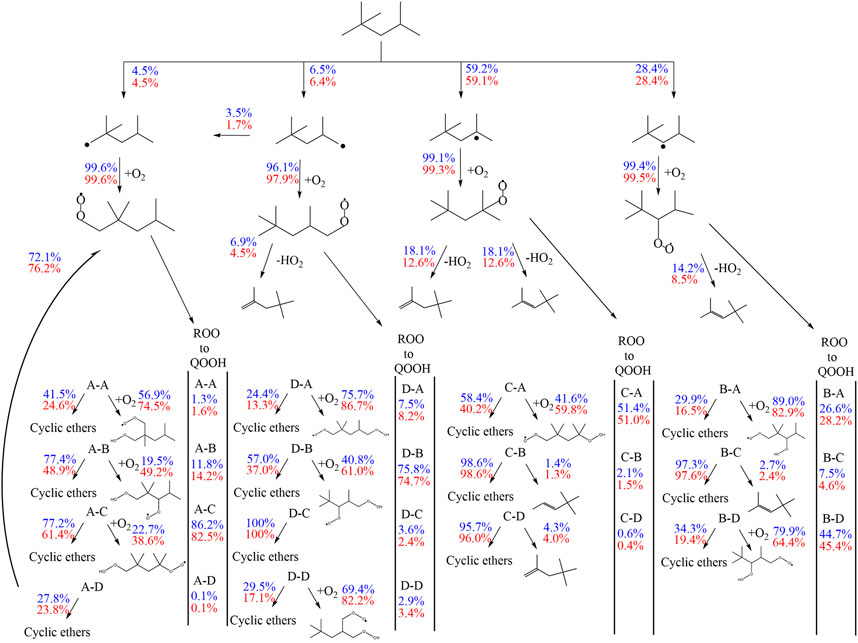
FIGURE 10. Flux analysis performed with the model of Cai et al. (2019) at 10 (blue) and 20 bar (red), 623 K at 25% of the maximum achieved conversion of iso-octane at the specific condition.
DIB isomers are important intermediates in the oxidation of iso-octane, but pressure dependent reactions are missing in current DIB sub-mechanisms. Further kinetic investigations of diisobutylene will improve the model predictions and benefit the understanding of iso-octane kinetics.
The most sensitive reactions differ significantly between the two mechanisms. Especially striking is the difference between the sensitivities for the H-atom abstraction reactions by OH radicals from the different carbon sites of the fuel. The mechanism of Atef et al. (2017) shows a strongly positive sensitivity for abstraction from the tertiary site of the fuel, while negative sensitivities are observed for the abstractions from other carbon sites. Conversely, the sensitivities on the rate coefficients of all H-atom abstraction reactions from the fuel by OH, including that on the tertiary site, are negative in the case of the mechanism of Cai et al. (2019). These differences are rooted in different rate coefficients assigned to these and subsequent reactions in the two mechanisms (as outlined earlier). In the mechanism of Atef et al. (2017), the H-abstraction reaction by OH from the fuel a-site exhibits the largest branching ratio at 623 K, while the H-abstractions from the other carbon sites show relatively similar rate coefficients at this condition, which is also manifested in the corresponding reaction fluxes shown in Figure 11. Since the produced radicals derived from the tertiary carbon site almost entirely add with O2 to form tertiary-peroxy radicals, which then largely undergo concerted HO2 elimination to form diisobutylene isomers, the tertiary-channel is by far the least reactive one in case of the mechanism of Atef et al. (2017).
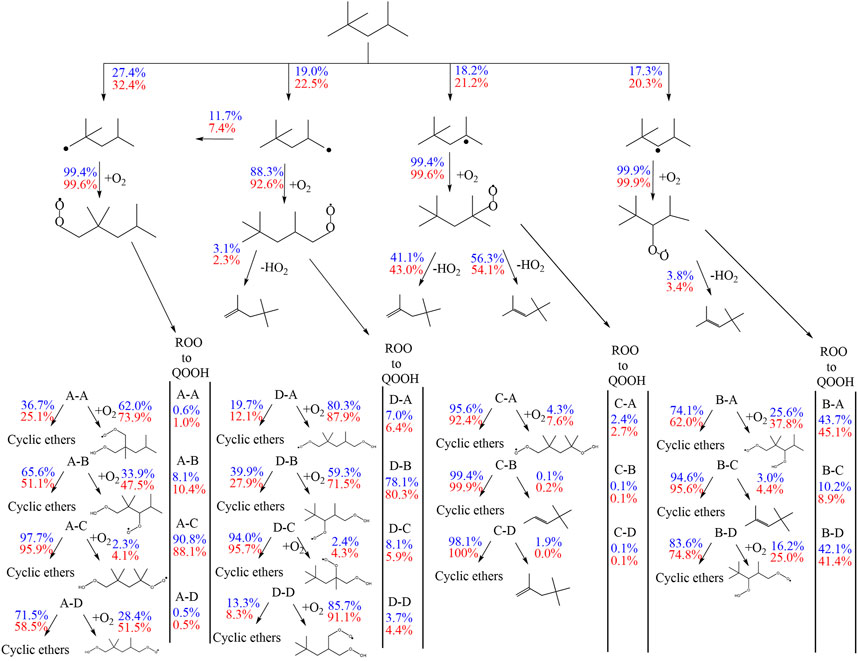
FIGURE 11. Flux analysis performed with the model of Atef et al. (2017) at 10 (blue) and 20 bar (red), 623 K at 25% of the maximum achieved conversion of iso-octane at the specific condition.
This observation explains the strongly negative sensitivity of the corresponding H-abstraction reaction by OH. On the other hand, in the mechanism of Cai et al. (2019), the H-abstraction reaction from the tertiary site of the fuel exhibits the highest rate coefficient at 623 K among all H-abstraction reactions by OH, which explains the high reaction flux through the tertiary-channel predicted by this mechanism (see Figure 11). The subsequently produced peroxy radicals derived from the tertiary carbon site mostly undergo isomerization to hydroperoxide radicals, which partly react into peroxy hydroperoxide radicals through the second O2 addition. While no ketohydroperoxides can ultimately be formed through this channel as the hydroperoxide moiety is attached to a tertiary carbon site, this reaction sequence can still lead to chain branching through the alternative isomerization channel (Bugler et al., 2015) which was considered in the mechanism (Cai et al., 2019). This higher importance of the chain branching pathway relative to the concerted HO2 elimination pathway of the peroxy radicals derived from the tertiary carbon site in the mechanism of Cai et al. (2019) explains the moderately negative sensitivities of the H-atom abstraction reaction by OH on the tertiary carbon site, in contrast to the strongly positive corresponding sensitivity predicted by the mechanism of Atef et al. (2017). The importance of the chain branching also explains the strongly negative sensitivity of the corresponding second O2 addition reaction CC8H16OOHXA + O2 = CC8H16OOHXAO2 (where the hydroperoxide radical originates from the tertiary carbon site channel) in the mechanism of Cai et al. (2019), which competes here with the less reactive formation of cyclic ethers. In contrast, the negligible reaction fluxes through the isomerization channels of the peroxy radical of the tertiary carbon site in the mechanism of Atef et al. (2017) enforce that only the second addition of hydroperoxide radicals to O2 originating from the a-, b-, and d-channels, as well as their competing cyclic ether formation reactions, show significant negative and positive sensitivities, respectively, in the model of Atef et al. (2017).
Besides, high sensitivities in the model of Cai et al. (2019) are also observed for the concerted HO2 elimination reactions of b- and c-peroxy radicals. These peroxy radicals are predicted to be the two most abundant isomers by this mechanism. As mentioned earlier, the peroxide formation serves as main competing pathway to the isomerization reactions. Other reactions involving HO2 radicals are observed to be sensitive in the mechanism of Cai et al. (2019) as well, due to the relative importance of these HO2-producing reactions. In contrast, the concerted elimination reactions are not particularly sensitive in the mechanism of Atef et al. (2017), where their competition with the O2 addition reactions is significantly less pronounced.
These results demonstrate that the results from the two mechanisms assessed here can lead to partly different conclusions regarding the underlying kinetics. In particular, the varying importance of the different fuel radicals due to different branching ratios in the two mechanisms deserves further investigations for a more consistent treatment. While the high rate coefficient for H-atom abstraction from the fuel a-site in the mechanism of Atef et al. (2017) is partly supported by the fact that nine H-atoms are available on this site compared to fewer H-atoms on other sites (despite the fact that primary radicals exhibit the highest C-H bond dissociation energies), the large rate coefficient for H-abstraction from the c-site in the mechanism of Cai et al. (2019) is partly supported by the significantly lower C-H bond dissociation energy at tertiary carbon sites (despite the fact that only one H-atom is available here for abstraction).
Finally, it can be noted that the main effect of pressure on the low-temperature reactivity results largely from the competition between the second O2 addition reactions of hydroperoxide radicals and their less reactive reactions into cyclic ethers and OH. While the forward rates of the unimolecular reactions into cyclic ethers and OH scale linearly with pressure, the forward rates of the bimolecular O2 addition reactions scale quadratically. Consequently, the relative importance of the latter increases with pressure, as seen in the reaction flux analysis in Figure 11, and so does thus the fuel consumption at low temperatures, as seen in Figure 1. The model of Atef et al. (2017) tends to overpredict fuel mole fractions at low temperatures regardless of pressure, while the model of Cai et al. (2019) rather tends to predict lower fuel mole fractions than experimentally observed (see Figure 1). More refined rate coefficients for these reactions as well as for those discussed earlier may further improve the model predictions for the present experimental data.
4.4.1 TMTHF
2,2,4,4-tetramethyl-tetrahydrofuran (TMTHF, C8H16O) is the most abundant species among the cyclic ethers and could be unambiguously identified in this work. It can be formed as a cyclic ether from either a- or c-iso-octyl radicals. Figure 12 compares the temperature dependence of the TMTHF mole fractions at different pressures to the mole fractions from the simulations.
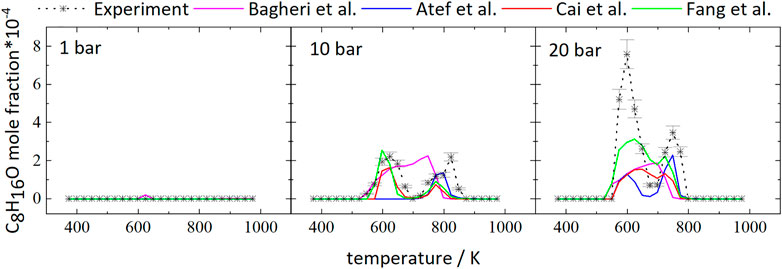
FIGURE 12. Mole fractions of 2,2,4,4-tetramethyl-tetrahydrofuran vs. reactor-temperature at 1, 10, and 20 bar. Dashed lines with symbols and solid lines illustrates the experimental and modeled results, respectively.
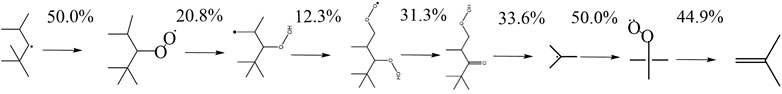
FIGURE 13. Reaction pathway of b-iso-octyl-radical to iso-butene derived by the mechanism of Atef et al. (2017) at 623 K and 20 bar.
TMTHF was not detected at 1 bar because here the iso-octane consumption only starts at temperatures above 898 K and low-temperature reactions leading to C8H16O are not expected to contribute significantly to the fuel consumption. In agreement with the low signal intensities in the experiment, the absolute mole fraction of TMTHF in the simulation was very small. At 20 bar, 3 times more TMTHF was detected experimentally compared to the measurement at 10 bar. The model of Atef et al. (2017) predicts mole fractions of TMTHF of ×(C8H16O) = 1.36‧10−4 and ×(C8H16O) = 2.29‧10−4 at 10 and 20 bar, respectively, and does not capture the experimentally observed mole fraction ratio of 3.5 (xEXP,max (C8H16O) = 2.19‧10−4 at 10 bar and xEXP,max (C8H16O) = 7.58‧10−4 at 20 bar).
Differences can also be observed in the simulated and measured profile shapes of the curves at 10 and 20 bar. The experimental curve at 20 bar displays two peaks at 598 and 748 K, respectively. The temperature of the larger peak at 598 K matches the temperature of maximum mole fraction predicted by all models, but the second peak is not predicted well by any model, beside the model of Bagheri et al. (2020). Instead, formation of TMTHF over a temperature range of 523–773 K is predicted by all models except by the model of Atef et al. (2017), which predicts two pronounced peaks. At 10 bar the C8H16O curve reaches a peak at 623 and 823 K in the experiment, while the model of Atef et al. predicts only one peak at 798 K. The model of Cai et al. (2019) predicts a small peak at 773 K and the mole fraction maximum at 623 K, which both match the prediction of Fang et al. (2020). The model of Bagheri et al. (2020) predicts formation of C8H16O over a temperature range of 523–798 K.
Rate-of-production analyses performed with the model of Atef et al. (2017) at 623, 673, and 773 K for pressures of 10 and 20 bar at a position in the reactor where 25% of the maximum conversion of the fuel for the specific condition is reached show that for all these conditions, TMTHF is produced via the a-iso-octyl radical, which is formed in almost constant fraction from the fuel. Two of the main decomposition reactions of TMTHF are:
In the mechanism, the rate constants of these reactions are barrierless and only depend on the A-factor. An explicit pressure dependence of the rate constants is not included in the reaction mechanism. Potentially, the missing pressure dependence of the decomposition/formation reactions leads to the deviations between simulation and experiment, especially since bimolecular reactions are favored with increasing pressure, which would lead to more a-iso-octyl radicals. Another reason for this discrepancy could be a poor description of the production of TMTHF via the decomposition of iso-octylhydroperoxy radicals. Unfortunately, radicals cannot be detected in our experiment. Nevertheless, the uncertainty in rate constants for the formation of cyclic ethers and their consumption chemistry need to be considered as well to further optimize reaction models of iso-octane combustion.
4.4.2 Formation of iso-C4H8 and C3H6O
The branched structure of iso-octane easily enables the formation pathway to iso-butene. Iso-octane decomposes to iso-butyl radicals and other branched alkyl radicals, whose decomposition reactions to iso-butene and iso-butyl radicals lead to high formation of iso-butene. Figure 14 displays the measured and simulated mole fraction profiles of iso-C4H8 for 1, 10, and 20 bar and compares the measured and simulated profiles of acetone. The simulations underpredict the absolute mole fractions of iso-butene for all conditions. Rate-of-production analyses have been performed at specific temperatures to identify important reaction pathways which lead to the formation of iso-butene and acetone. At 1 bar, almost quantitative agreement is observed. With increasing pressure, the deviations between experimental values and simulations increase. Higher pressure favors the formation of acetone, which can be seen in the decreasing and increasing mole fractions of iso-butene and acetone, respectively. At 1 bar, iso-butene is only formed at high temperatures. According to a reaction path analysis performed with the model of Atef et al. (2017), ß-scission reactions of the a- and c-iso-octyl radicals as well as C7H15 are responsible for the iso-butene formation.
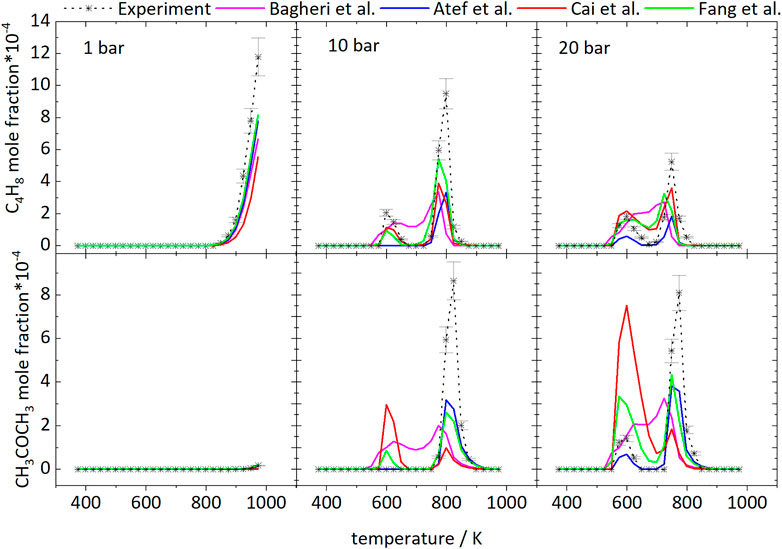
FIGURE 14. Mole fractions of iso-butene and acetone vs. reactor-temperature at 1, 10, and 20 bar. Dashed lines with symbols and solid lines illustrates the experimental and modeled results, respectively.
At 10 bar, the iso-butene formation has two maxima. One occurs in the LTC region and the other at 798 K for the measurements. At 20 bar the highest concentration is reached at 748 K. In the LTC region at 10 bar the iso-butene is formed by ß-scission reactions of the c-iso-octyl radical, which is predominately formed at these low temperatures. At 20 bar, iso-butene formation is dominated by decomposition of the dihydroperoxide formed from the b-iso-octyl radical.
At 798 K at 10 bar and 748 K at 20 bar, the same reaction pathways are active as at 1 bar at high temperatures. Consequently, the higher pressure leads to a shift of the high-temperature chemistry to lower temperatures.
Acetone was detected in the experiments with a maximum mole fraction of ×(C3H6O) = 8‧10−4 at both 10 and 20 bar. At 1 bar, it was below the detection limit in agreement with very small mole fractions predicted in the simulations. Acetone is mainly formed from iso-butene according to the Waddington mechanism shown in Figure 15 reaction that is implemented in all mechanisms used for the simulations shown here, beside the mechanism of Cai et al. (2019). Regarding the models of Cai et al. (2019), Bagheri et al. (2020), and Fang et al. (2020) a comparison of the experimental and simulated mole fraction profiles of acetone at 10 and 20 bar shown in Figure 14 reveals high deviations at all temperatures. Better agreement can be achieved with the models of Atef et al. (2017) and Fang et al. (2020). In the experiments acetone is formed in detectable quantities only at high temperatures at 10 bar. At 20 bar it is formed in the LTC region and at high temperatures. The appearance in the LTC region at 598 and 748 K at 20 bar and at 823 K at 10 bar is qualitatively captured by the model of Atef et al. (2017) with quantitative deviations. The formation can be traced in a reaction path analysis to the same route that leads to the formation of iso-butene from the b-iso-octyl radical.
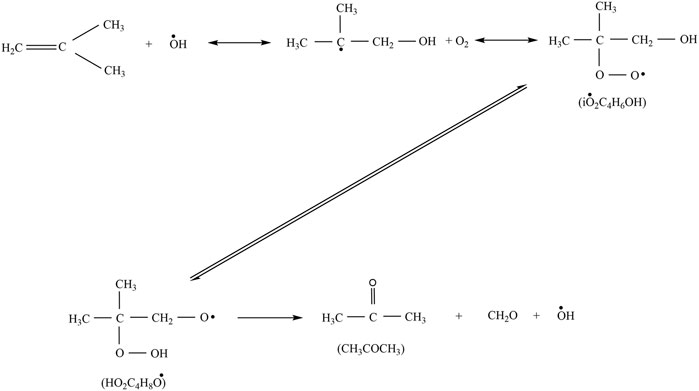
FIGURE 15. Formation of acetone via the Waddington mechanism (Chen et al., 2000).
At these temperatures acetone is not formed from iso-butene according to the Waddington mechanism but from oxidation of tert-butyl radicals that are produced in the oxidation of the b-iso-octyl radical. The same pathway is active at 10 bar in the simulation with the model of Atef et al. (2017). The fact that no acetone was found in the experiment at 10 bar at low temperature indicates that the reaction rates within this pathway are overestimated at lower pressure. At higher temperatures (>700 K) the Waddington mechanism contributes to the acetone formation and converts iso-butene to acetone. As a result, the acetone mole fractions peak at 25 K larger temperatures than the iso-butene mole fractions at 10 and 20 bar. This observation corroborates the conclusion that higher pressures shift the occurrence of high-temperature reaction pathways to lower temperature. Also, addition of OH to a double bond could become less important. Thus, reactions with HO2 become more important with increasing temperature.
5 Conclusion
The oxidation of iso-octane is investigated in a flow reactor at 1, 10, and 20 bar, 473 ≤ T ≤ 973 K, and ɸ = 1. These conditions include the region of low- and intermediate-temperatures including the NTC region in a wide pressure range. Previous studies mostly focused either on the low- or high-temperature oxidation at atmospheric pressure. The mole fraction profiles of reactants, products, and several intermediates as function of temperature have been determined with GC analysis and the data sets are provided for reaction mechanism development. The concentration profiles at each pressure have been compared to simulation results obtained with the recent mechanisms of Atef et al. (2017), Cai et al. (2019), Bagheri et al. (2020), and Fang et al. (2020). In general, a shift of the NTC region to higher temperatures is seen with increasing pressure. While appreciable reactivity is only observed at higher temperature at atmospheric pressure, low-temperature reactivity starts being observed with higher pressures. The pressure-dependent mole fraction changes of important experimentally detected intermediates, e.g., 2,2,4,4-tetramethyl-tetrahydrofuran, iso-butene, and acetone, are discussed in detail with respect to the formation pathways implemented in the reaction mechanisms.
The new data set complements existing validation data for reaction mechanism development in a pressure and temperature range that is of particular interest to control strategies of low-temperature combustion in engines.
Data Availability Statement
The original contribution presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Author Contributions
SS: investigation, methodology, formal analysis, visualization, writing—original draft. FvL: formal analysis, writing—review and editing. JB: supervision, formal analysis—review and editing. HP: supervision—review and editing. TK: supervision, funding acquisition, project administration, writing—review and editing.
Funding
Financial support by the Deutsche Forschungsgemeinschaft within the framework of the DFG research unit FOR2401 “Optimierungsbasierte Multiskalenregelung motorischer Niedertemperatur-Brennverfahren” under projects 277012063 is gratefully acknowledged.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenrg.2022.859112/full#supplementary-material
References
Atef, N., Kukkadapu, G., Mohamed, S. Y., Rashidi, M. A., Banyon, C., Mehl, M., et al. (2017). A Comprehensive Iso-Octane Combustion Model with Improved Thermochemistry and Chemical Kinetics. Combustion and Flame 178, 111–134. doi:10.1016/j.combustflame.2016.12.029
Badra, J., and Farooq, A. (2015). Site-Specific Reaction Rate Constant Measurements for Various Secondary and Tertiary H-Abstraction by OH Radicals. Combustion and Flame 162, 2034–2044. doi:10.1016/j.combustflame.2015.01.001
Bagheri, G., Ranzi, E., Pelucchi, M., Parente, A., Frassoldati, A., and Faravelli, T. (2020). Comprehensive Kinetic Study of Combustion Technologies for Low Environmental Impact: MILD and OXY-Fuel Combustion of Methane. Combustion and Flame 212, 142–155. doi:10.1016/j.combustflame.2019.10.014
Buda, F., Bounaceur, R., Warth, V., Glaude, P. A., Fournet, R., and Battin-Leclerc, F. (2005). Progress toward a Unified Detailed Kinetic Model for the Autoignition of Alkanes from C4 to C10 between 600 and 1200 K. Combustion and Flame 142, 170–186. doi:10.1016/j.combustflame.2005.03.005
Bugler, J., Somers, K. P., Silke, E. J., and Curran, H. J. (2015). Revisiting the Kinetics and Thermodynamics of the Low-Temperature Oxidation Pathways of Alkanes: A Case Study of the Three Pentane Isomers. J. Phys. Chem. AA 119, 7510–7527. doi:10.1021/acs.jpca.5b00837
Burke, S. M., Simmie, J. M., and Curran, H. J. (2015). Critical Evaluation of Thermochemical Properties of C 1 –C 4 Species: Updated Group-Contributions to Estimate Thermochemical Properties. J. Phys. Chem. Reference Data 44, 13101. doi:10.1063/1.4902535
Cai, L., Pitsch, H., Mohamed, S. Y., Raman, V., Bugler, J., Curran, H., et al. (2016). Optimized Reaction Mechanism Rate Rules for Ignition of Normal Alkanes. Combustion and Flame 173, 468–482. doi:10.1016/j.combustflame.2016.04.022
Cai, L., Ramalingam, A., Minwegen, H., Alexander Heufer, K., and Pitsch, H. (2019). Impact of Exhaust Gas Recirculation on Ignition Delay Times of Gasoline Fuel: An Experimental and Modeling Study. Proc. Combustion Inst. 37, 639–647. doi:10.1016/j.proci.2018.05.032
Callahan, C. V., Held, T. J., Dryer, F. L., Minetti, R., Ribaucour, M., Sochet, L. R., et al. (1996). Experimental Data and Kinetic Modeling of Primary Reference Fuel Mixtures. Symp. (International) Combustion 26, 739–746. doi:10.1016/s0082-0784(96)80282-7
Chen, J.-S., Litzinger, T. A., and Curran, H. J. (2000). The Lean Oxidation of Iso-Octane in the Intermediate Temperature Regime at Elevated Pressures. Combustion Sci. Techn. 156, 49–79. doi:10.1080/00102200008947296
Ciajolo, A., D'Anna, A., and Mercogijano, R. (1993). Slow-Combustion of N-Heptane, Iso-Octane and a Toluene/n-Heptane Mixture. Gcst 90, 357–371. doi:10.1080/00102209308907622
Cox, A., Griffiths, J. F., Mohamed, C., Curran, H. J., Pitz, W. J., and Westbrook, C. K. (1996). Extents of Alkane Combustion During Rapid Compression Leading to Single-And Two-Stage Ignition. Symp. (International) Combustion 26, 2685–2692. doi:10.1016/s0082-0784(96)80104-4
Cox, R. A., and Cole, J. A. (1985). Chemical Aspects of the Autoignition of Hydrocarbonair Mixtures. Combustion and Flame 60, 109–123. doi:10.1016/0010-2180(85)90001-x
Curran, H. (2002). A Comprehensive Modeling Study of Iso-Octane Oxidation. Combustion and Flame 129, 253–280. doi:10.1016/s0010-2180(01)00373-x
D'Anna, A., Mercoguano, R., Barbella, R., and Ciajolo, A. (1992). Low Temperature Oxidation Chemistry of Iso-Octane Under High Pressure Conditions. Combustion Sci. Techn. 83, 217–232. doi:10.1080/00102209208951833
Dagaut, P., Reuillon, M., and Cathonnet, M. (1993). High Pressure Oxidation of Liquid Fuels from Low to High Temperature. 1. N-Heptane and Iso-Octane. Combustion Sci. Techn. 95, 233–260. doi:10.1080/00102209408935336
Dagaut, P., Reuillon, M., and Cathonnet, M. (1994). High Pressure Oxidation of Liquid Fuels from Low to High Temperature. 2. Mixtures of N-Heptane and Iso-Octane. Combustion Sci. Techn. 103, 315–336. doi:10.1080/00102209408907701
Davidson, D. F., Gauthier, B. M., and Hanson, R. K. (2005). Shock Tube Ignition Measurements of Iso-Octane/Air and Toluene/Air at High Pressures. Proc. Combustion Inst. 30, 1175–1182. doi:10.1016/j.proci.2004.08.004
Davidson, D. F., Oehlschlaeger, M. A., Herbon, J. T., and Hanson, R. K. (2002). Shock Tube Measurements of Iso-Octane Ignition Times and OH Concentration Time Histories. Proc. Combustion Inst. 29, 1295–1301. doi:10.1016/s1540-7489(02)80159-6
Davis, S. G., and Law, C. K. (1998). Laminar Flame Speeds and Oxidation Kinetics of Iso-Octane-Air and N-Heptane-Air Flames. Symp. (International) Combustion 27, 521–527. doi:10.1016/s0082-0784(98)80442-6
Dryer, F. L., and Brezinsky, K. (1986). A Flow Reactor Study of the Oxidation of N-Octane and Iso-Octane. Combustion Sci. Techn. 45, 199–212. doi:10.1080/00102208608923850
Fang, R., Kukkadapu, G., Wang, M., Wagnon, S. W., Zhang, K., Mehl, M., et al. (2020). Fuel Molecular Structure Effect on Autoignition of Highly Branched Iso-Alkanes at Low-To-Intermediate Temperatures: Iso-Octane versus Iso-Dodecane. Combustion and Flame 214, 152–166. doi:10.1016/j.combustflame.2019.12.037
Fieweger, K., Blumenthal, R., and Adomeit, G. (1997). Self-Ignition of S.I. Engine Model Fuels: A Shock Tube Investigation at High Pressure. Combustion and Flame 109, 599–619. doi:10.1016/s0010-2180(97)00049-7
Glaude, P. A., Conraud, V., Fournet, R., Battin-Leclerc, F., Côme, G. M., Scacchi, G., et al. (2002). Modeling the Oxidation of Mixtures of Primary Reference Automobile Fuels. Energy Fuels 16, 1186–1195. doi:10.1021/ef020025e
Griffiths, J. F., Halford-Maw, P. A., and Rose, D. J. (1993). Fundamental Features of Hydrocarbon Autoignition in a Rapid Compression Machine. Combustion and Flame 95, 291–306. doi:10.1016/0010-2180(93)90133-n
Halstead, M. P., Kirsch, L. J., and Quinn, C. P. (1977). The Autoignition of Hydrocarbon Fuels at High Temperatures and Pressures-Fitting of a Mathematical Model. Combustion and Flame 30, 45–60. doi:10.1016/0010-2180(77)90050-5
Hartmann, M., Gushterova, I., Fikri, M., Schulz, C., Schießl, R., and Maas, U. (2011). Auto-Ignition of Toluene-Doped N-Heptane and Iso-Octane/Air Mixtures: High-Pressure Shock-Tube Experiments and Kinetics Modeling. Combustion and Flame 158, 172–178. doi:10.1016/j.combustflame.2010.08.005
He, X., Donovan, M. T., Zigler, B. T., Palmer, T. R., Walton, S. M., Wooldridge, M. S., et al. (2005). An Experimental and Modeling Study of Iso-Octane Ignition Delay Times Under Homogeneous Charge Compression Ignition Conditions. Combustion and Flame 142, 266–275. doi:10.1016/j.combustflame.2005.02.014
He, X., Walton, S. M., Zigler, B. T., Wooldridge, M. S., and Atreya, A. (2007). Experimental Investigation of the Intermediates of Isooctane During Ignition. Int. J. Chem. Kinet. 39, 498–517. doi:10.1002/kin.20254
Hu, H., and Keck, J. (1987). Autoignition of Adiabatically Compressed Combustible Gas Mixtures. SAE Technical Paper 872110.
Jia, M., and Xie, M. (2006). A Chemical Kinetics Model of Iso-Octane Oxidation for HCCI Engines. Fuel 85, 2593–2604. doi:10.1016/j.fuel.2006.02.018
Kaczmarek, D., Atakan, B., and Kasper, T. (2019). Investigation of the Partial Oxidation of Methane/n-Heptane-Mixtures and the Interaction of Methane and N-Heptane under Ultra-Rich Conditions. Combustion and Flame 205, 345–357. doi:10.1016/j.combustflame.2019.04.005
Kaczmarek, D., Atakan, B., and Kasper, T. (2019). Plug-Flow Reactor Study of the Partial Oxidation of Methane and Natural Gas at Ultra-Rich Conditions. Combustion Sci. Techn. 191, 1571–1584. doi:10.1080/00102202.2019.1577829
Korkmaz, M., Zweigel, R., Jochim, B., Beeckmann, J., Abel, D., and Pitsch, H. (2018). Triple-Injection Strategy for Model-Based Control of Premixed Charge Compression Ignition Diesel Engine Combustion. Int. J. Engine Res. 19, 230–240. doi:10.1177/1468087417730485
Kukkadapu, G., Kumar, K., Sung, C.-J., Mehl, M., and Pitz, W. J. (2013). Autoignition of Gasoline and its Surrogates in a Rapid Compression Machine. Proc. Combustion Inst. 34, 345–352. doi:10.1016/j.proci.2012.06.135
Kukkadapu, G., Kumar, K., Sung, C.-J., Mehl, M., and Pitz, W. J. (2012). Experimental and Surrogate Modeling Study of Gasoline Ignition in a Rapid Compression Machine. Combustion and Flame 159, 3066–3078. doi:10.1016/j.combustflame.2012.05.008
Leppard, W. R. (1992). The Autoignition Chemistries of Primary Reference Fuels, Olefin/Paraffin Binary Mixtures, and Non-Linear Octane Blending. SAE Technical Paper 922325.
Li, H., Prabhu, S. K., Miller, D. L., and Cernansky, N. P. (1994). Autoignition Chemistry Studies on Primary Reference Fuels in a Motored Engine. SAE Technical Paper 940478.
Li, Y., Zhou, C.-W., Somers, K. P., Zhang, K., and Curran, H. J. (2017). The Oxidation of 2-Butene: A High Pressure Ignition Delay, Kinetic Modeling Study and Reactivity Comparison with Isobutene and 1-Butene. Proc. Combustion Inst. 36, 403–411. doi:10.1016/j.proci.2016.05.052
Lignola, P. G., Di Maio, F. P., Marzocchella, A., Mercogliano, R., and Reverchon, E. (1989). JSFR Combustion Processes of N-Heptane and Isooctane. Symp. (International) Combustion 22, 1625–1633. doi:10.1016/s0082-0784(89)80174-2
Lü, X.-C., Chen, W., and Huang, Z. (2005). A Fundamental Study on the Control of the HCCI Combustion and Emissions by Fuel Design Concept Combined with Controllable EGR. Part 1. The Basic Characteristics of HCCI Combustion. Fuel 84, 1074–1083.
Lu, Z., Yang, Y., and Brear, M. J. (2019). Oxidation of PRFs and Ethanol/Iso-Octane Mixtures in a Flow Reactor and the Implication for Their Octane Blending. Proc. Combustion Inst. 37, 649–656. doi:10.1016/j.proci.2018.05.134
Masurier, J.-B., Foucher, F., Dayma, G., and Dagaut, P. (2015). Investigation of Iso-Octane Combustion in a Homogeneous Charge Compression Ignition Engine Seeded by Ozone, Nitric Oxide and Nitrogen Dioxide. Proc. Combustion Inst. 35, 3125–3132. doi:10.1016/j.proci.2014.05.060
Maynard, J. B., Legate, C. E., and Graiff, L. B. (1967). Pre-Flame Reaction Products of Isooctane Formed in a Motored Engine. Combustion and Flame 11, 155–166. doi:10.1016/0010-2180(67)90119-8
Mehl, M., Pitz, W. J., Westbrook, C. K., and Curran, H. J. (2011). Kinetic Modeling of Gasoline Surrogate Components and Mixtures Under Engine Conditions. Proc. Combustion Inst. 33, 193–200. doi:10.1016/j.proci.2010.05.027
Metcalfe, W. K., Pitz, W. J., Curran, H. J., Simmie, J. M., and Westbrook, C. K. (2007). The Development of a Detailed Chemical Kinetic Mechanism for Diisobutylene and Comparison to Shock Tube Ignition Times. Proc. Combustion Inst. 31, 377–384. doi:10.1016/j.proci.2006.07.207
Minetti, R., Ribaucour, M., Carlier, M., and Sochet, L. R. (1996). Autoignition Delays of a Series of Linear and Branched Chain Alkanes in the Intermediate Range of Temperature. Combustion Sci. Techn. 113, 179–192. doi:10.1080/00102209608935493
Mittal, G., and Sung, C.-J. (2007). A Rapid Compression Machine for Chemical Kinetics Studies at Elevated Pressures and Temperatures. Combustion Sci. Techn. 179, 497–530. doi:10.1080/00102200600671898
Musculus, M. P. B., Miles, P. C., and Pickett, L. M. (2013). Conceptual Models for Partially Premixed Low-Temperature Diesel Combustion. Prog. Energ. Combustion Sci. 39, 246–283. doi:10.1016/j.pecs.2012.09.001
Ning, H., Gong, C., Li, Z., and Li, X. (2015). Pressure-Dependent Kinetics of Initial Reactions in Iso-Octane Pyrolysis. J. Phys. Chem. AA 119, 4093–4107. doi:10.1021/acs.jpca.5b02013
Nuss, E., Wick, M., Andert, J., de Schutter, J., Diehl, M., Abel, D., et al. (2019). Nonlinear Model Predictive Control of a Discrete-Cycle Gasoline-Controlled Auto Ignition Engine Model: Simulative Analysis. Int. J. Engine Res. 20, 1025–1036. doi:10.1177/1468087418824915
Ogink, R., and Golovitchev, V. (2001). Gasoline HCCI Modeling: Computer Program Combining Detailed Chemistry and Gas Exchange Processes. SAE Technical Paper 973614.
Ranzi, E., Faravelli, T., Gaffuri, P., Sogaro, A., D'Anna, A., and Ciajolo, A. (1997). A Wide-Range Modeling Study of Iso-Octane Oxidation. Combustion and Flame 108, 24–42. doi:10.1016/s0010-2180(95)00274-x
Scanlon, J. T., and Willis, D. E. (1985). Calculation of Flame Ionization Detector Relative Response Factors Using the Effective Carbon Number Concept. J. Chromatogr. Sci. 23, 333–340. doi:10.1093/chromsci/23.8.333
Sen, F., Kasper, T., Bergmann, U., Hegner, R., and Atakan, B. (2015). Partial Oxidation of Methane at Elevated Pressures and Effects of Propene and Ethane as Additive: Experiment and Simulation. Z. für Physikalische Chem. 229. doi:10.1515/zpch-2014-0576
Shen, H.-P. S., Vanderover, J., and Oehlschlaeger, M. A. (2008). A Shock Tube Study of Iso-Octane Ignition at Elevated Pressures: The Influence of Diluent Gases. Combustion and Flame 155, 739–755. doi:10.1016/j.combustflame.2008.06.001
Sivaramakrishnan, R., and Michael, J. V. (2009). Rate Constants for OH with Selected Large Alkanes: Shock-Tube Measurements and an Improved Group Scheme. J. Phys. Chem. AA 113, 5047–5060. doi:10.1021/jp810987u
Tanaka, S., Ayala, F., and Keck, J. C. (2003). A Reduced Chemical Kinetic Model for HCCI Combustion of Primary Reference Fuels in a Rapid Compression Machine. Combustion and Flame 133, 467–481. doi:10.1016/s0010-2180(03)00057-9
Tanaka, S. (2003). Two-Stage Ignition in HCCI Combustion and HCCI Control by Fuels and Additives. Combustion and Flame 132, 219–239. doi:10.1016/s0010-2180(02)00457-1
Vermeer, D. J., Meyer, J. W., and Oppenheim, A. K. (1972). Auto-Ignition of Hydrocarbons Behind Reflected Shock Waves. Combustion and Flame 18, 327–336. doi:10.1016/s0010-2180(72)80183-4
Wang, Q., Tang, X., Yang, J., Zhai, Y., Zhang, Y., Cao, C., et al. (2019). Investigations on Pyrolysis of Isooctane at Low and Atmospheric Pressures. Energy Fuels 33, 3518–3528. doi:10.1021/acs.energyfuels.8b04029
Wang, Z., and Sarathy, S. M. (2016). Third O2 Addition Reactions Promote the Low-Temperature Auto-Ignition of N-Alkanes. Combustion and Flame 165, 364–372. doi:10.1016/j.combustflame.2015.12.020
Wang, Z., Zhang, L., Moshammer, K., Popolan-Vaida, D. M., Shankar, V. S. B., Lucassen, A., et al. (2016). Additional Chain-Branching Pathways in the Low-Temperature Oxidation of Branched Alkanes. Combustion and Flame 164, 386–396. doi:10.1016/j.combustflame.2015.11.035
Westbrook, C. K., Pitz, W. J., and Leppard, W. R. (1991). The Autoignition Chemistry of Paraffinic Fuels and Pro-Knock and Anti-Knock Additives: A Detailed Chemical Kinetic Study. SAE Technical Paper 912314.
Yao, M., Zheng, Z., and Liu, H. (2009). Progress and Recent Trends in Homogeneous Charge Compression Ignition (HCCI) Engines. Prog. Energ. Combustion Sci. 35, 398–437. doi:10.1016/j.pecs.2009.05.001
Zhang, K., Banyon, C., Burke, U., Kukkadapu, G., Wagnon, S. W., Mehl, M., et al. (2019). An Experimental and Kinetic Modeling Study of the Oxidation of Hexane Isomers: Developing Consistent Reaction Rate Rules for Alkanes. Combustion and Flame 206, 123–137. doi:10.1016/j.combustflame.2019.04.011
Zheng, J., Yang, W., Miller, D. L., and Cernansky, N. P. (2002). A Skeletal Chemical Kinetic Model for the HCCI Combustion Process. SAE Technical Paper 983115.
Keywords: iso-octane, reaction kinetics, flow reactor, mechanism, validation
Citation: Shaqiri S, Kaczmarek D, vom Lehn F, Beeckmann J, Pitsch H and Kasper T (2022) Experimental Investigation of the Pressure Dependence of Iso-Octane Combustion. Front. Energy Res. 10:859112. doi: 10.3389/fenrg.2022.859112
Received: 20 January 2022; Accepted: 07 April 2022;
Published: 18 May 2022.
Edited by:
Maria Abian, University of Zaragoza, SpainReviewed by:
Goutham Kukkadapu, Lawrence Livermore National Laboratory (DOE), United StatesGuillaume Dayma, Université d’Orléans, France
Lars Seidel, LOGE Deutschland GmbH, Germany
Copyright © 2022 Shaqiri, Kaczmarek, vom Lehn, Beeckmann, Pitsch and Kasper. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: S. Shaqiri, c2hrZWxxaW0uc2hhcWlyaUB1bmktZHVlLmRl