 Drace Penley
Drace Penley Stephen P. Vicchio
Stephen P. Vicchio Rachel B. Getman
Rachel B. Getman Burcu Gurkan
Burcu Gurkan- 1Chemical and Biomolecular Engineering, Case Western Reserve University, Cleveland, OH, United States
- 2Chemical and Biomolecular Engineering, Clemson University, Clemson, SC, United States
The energetics, coordination, and Raman vibrations of Li solvates in ionic liquid (IL) electrolytes are studied with density functional theory (DFT). Li+ coordination with asymmetric anions of cyano(trifluoromethanesulfonyl)imide ([CTFSI]) and (fluorosulfonyl)(trifluoro-methanesulfonyl)imide ([FTFSI]) is examined in contrast to their symmetric analogs of bis(trifluoromethanesulfonyl)imide ([TFSI]), bis(fluorosulfonyl)imide ([FSI]), and dicyanamide ([DCA]). The dissociation energies that can be used to describe the solvation strength of Li+ are calculated on the basis of the energetics of the individual components and the Li solvate. The calculated dissociation energies are found to be similar for Li+-[FTFSI], Li+-[TFSI], and Li+-[FSI] where only Li+-O coordination exists. Increase in asymmetry and anion size by fluorination on one side of the [TFSI] anion does not result in significant differences in the dissociation energies. On the other hand, with [CTFSI], both Li+-O and Li+-N coordination are present, and the Li solvate has smaller dissociation energy than the solvation by [DCA] alone, [TFSI] alone, or a 1:1 mixture of [DCA]/[TFSI] anions. This finding suggests that the Li+ solvation can be weakened by asymmetric anions that promote competing coordination environments through enthalpic effects. Among the possible Li solvates of (Li[CTFSI]n)−(n−1), where n = 1, 2, 3, or 4, (Li[CTFSI]2)−1 is found to be the most stable with both monodentate and bidentate bonding possibilities. Based on this study, we hypothesize that the partial solvation and weakened solvation energetics by asymmetric anions may increase structural heterogeneity and fluctuations in Li solvates in IL electrolytes. These effects may further promote the Li+ hopping transport mechanism in concentrated and multicomponent IL electrolytes that is relevant to Li-ion batteries.
Introduction
The structure of Li+ solvates in concentrated electrolytes has a significant impact on Li+ transport and is influential in the rate capability of rechargeable lithium ion batteries (LIBs) (Borodin et al., 2018; Yamada et al., 2019; Krachkovskiy et al., 2020; Pham et al., 2021). ILs present an extreme case of a concentrated electrolyte where the electrolyte is made entirely of discrete ions and lacks neutral solvent molecules. Generally, ILs are known to have high ionic conductivity, large electrochemical windows, and negligible volatility (Bae et al., 2013; Navarra, 2013). These properties are desirable for lithium battery systems and thus are of great interest for safe, high-density energy storage devices (Galinski et al., 2006; Bae et al., 2013; Navarra, 2013; Eftekhari et al., 2016). However, Li+ transport in ILs is complex and hindered by the high viscosity, mainly due to Coulombic interactions and specific Li+–anion interactions, which leads to the formation of clusters that decrease the mobility of Li+ in the bulk liquid (Lesch et al., 2014; Pham et al., 2021). Li+ transport in ILs has been studied mostly by Raman spectroscopy, nuclear magnetic resonance spectroscopy, transference measurements, density functional theory (DFT), and classical molecular dynamic (MD) simulations (Borodin et al., 2006; Umebayashi et al., 2007; Duluard et al., 2008; Lassègues et al., 2009; Umebayashi et al., 2010; Castiglione et al., 2011; Fujii et al., 2013; Haskins et al., 2014; Borodin et al., 2018). In this study, the DFT approach is employed to understand the impact of anion asymmetry and the presence of binary anions on the structure and energetics of the first solvation shell of Li+ in IL electrolytes.
The bis(trifluoromethanesulfonyl)imide, [TFSI], anion and its respective ILs have been widely studied as potential electrolytes for lithium metal batteries and LIBs, and were shown to improve battery performance over the state-of-the-art organic carbonate electrolytes with Li[PF6] (Sakaebe and Matsumoto, 2003; Garcia et al., 2004; Armand et al., 2009). Fundamental studies of IL electrolytes with [TFSI] anion showed that Li+ coordinates with anionic oxygen atoms where on average slightly less than four oxygen atoms are involved (Borodin et al., 2006), leading to two anions in the first solvation shell where cis conformation is favored with increased Li salt doping (Umebayashi et al., 2010). These studies were focused on Li salt mole fraction of
Decreasing the solvation strength and the lifetime of coordinated species in LIB electrolytes have been suggested to play an important role in improving Li+ mobility. Borodin et al. (2017) demonstrated the promotion of Li+ transport in concentrated electrolytes where nano-heterogeneity decouples cations from anion cages. In other examples where heterogeneity was achieved by simple mixtures, Li+ solvation was shown to weaken in binary organic carbonate mixtures (Matsuda et al., 2002), and the solvation shell was found to be more rigid in cyclic carbonates than in their linear counterparts (Fulfer and Kuroda, 2016). Shim (2018) studied the solvation structure and dynamics of a mixture of ethylmethyl carbonate (EMC), dimethyl carbonate (DMC), and ethylene carbonate (EC) with the Li[PF6] salt by MD simulations. They reported that as the EC concentration increased, the solvent fluctuations in Li solvates decreased due to the preferred coordination of Li+ with EC and increased local density by the ring structure of EC, which was consistent with the reports by Yang et al. (2010). The decreased solvent fluctuations resulted in increased viscosity and reduced Li+ diffusion. Similar to organic mixture electrolytes, mixtures of ILs have been studied to increase conductivity by Li solvate structural fluctuations and reduced solvation strength (Yang et al., 2010; Kerner et al., 2015; Huang et al., 2019a; Huang et al., 2019b; Chen and Forsyth, 2019).
IL mixtures with [TFSI]/[FSI] anions have been studied the most to date due to the superior electrochemical properties of ILs paired with these anions. The [DCA] anion has also garnered attention due to its lower viscosity and higher conductivity than [TFSI]- and [FSI]-based ILs. In a study by Yoon et al. (2013), the [DCA] anion paired with a pyrrolidinium cation in a Li|LiFePO4 cell showed stable cycling at elevated temperatures. Huang et al. (2019b) showed by a combined Raman spectroscopy and MD simulations study that at
Very recently, Nurnberg et al. (2020) studied the Li+ solvation and transport in IL electrolytes (
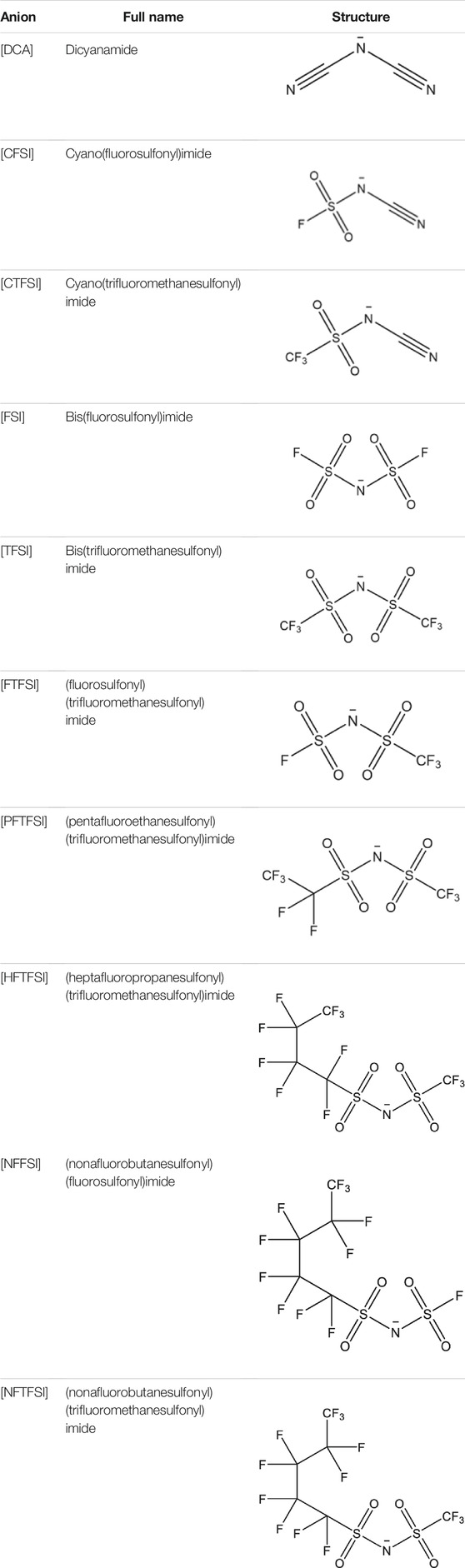
TABLE 1. Anions of interest with full name as well as an abbreviation used within the study.
The bulk properties and electrochemical applications of the three asymmetric anions such as [NFTFSI], [CTFSI], and [FTFSI] are relatively unexplored when compared to their parent anions. The [CTFSI] anion in an IL electrolyte has shown promise in LIBs, with improved thermal and anodic stability, and exhibited higher room temperature conductivity than [DCA] and [TFSI] parent anions (Hoffknecht et al., 2017). However, the structure of the Li solvate was not studied, and it is not clear what the role of the liquid structure is on the improved performance. An investigation on the [FTFSI]-based IL electrolyte showed that the vehicular motion dominates due to the formation of Li+ aggregates until 0.4 mole fraction of the Li salt after which the transport of Li+ via hopping increases (Brinkkötter et al., 2018). [NFTFSI] is unique in that rather than combining the functionality of two parent IL anions, the fluoromethane group on [TFSI] is replaced with a fluorobutane. This provides the ability to quantify how the increased fluorination impacts the structure and transport properties. There has been little investigation of the [NFTFSI] and [NFFSI] anions in the past. Palumbo et al. (2017) studied the conformer structures of [NFTFSI] in a pyrrolidinium IL. Han et al. (2011) utilized 1M Li[NFFSI] as the conducting salt in organic carbonates (EC/EMC 3:7 v/v) to improve high temperature performance of LIBs. Li[NFFSI] showed similar conductivity as Li[ClO4] and slightly lower than Li[PF6]; however, Li[NFFSI] did not corrode the aluminum current collector of the graphite/LiCoO2 cell with 63% capacity retention over 100 cycles at 60°C, while the cell with Li[PF6] failed rapidly. Therefore, how the length of fluorination impacts the stability of LIBs is important to understand for extreme temperature conditions. In order to do so, we need a better understanding of how these fluorinated anions solvate and interact with Li+ in the bulk electrolyte. The [CFSI], [PFTFSI], and [HFTFSI] anions were studied here to explore how the degree of fluorination of the asymmetric anions impacts solvation strength. Previously, it was found that the binding energies of Li solvates with organic ligands decreased with increased fluorination (Bauschlicher, 2018). It was also shown that with reduced dipole moment and an increased ligand size, the ligand–ligand repulsion was increased. This was discussed to possibly lead to different structures than the non-fluorinated analogs. In this study, we show how the strength of solvation is impacted by the asymmetry of the coordinating anions and how the anion mixtures compare to the analogous asymmetric anion that has binary coordination with Li+. We report the strength of the first solvation shell of Li+ in ILs without the impacts of the IL cation through DFT calculations and suggest which anions may help promote Li+ transport based on this first principles study.
Methods
Computational Details
Dissociation energies (ED) were used to assess the thermodynamic favorabilities of the different Li+-anion systems. ED represents the amount of energy it takes to break Li+-anion clusters into their constituents (i.e., the difference in energy going from the Li+-anion cluster to the individual components of Li+ and the anion at infinite separation). They were calculated using quantum mechanics in the absence of solvation, similar to a prior study on Li+ transport in cross-linked polymer electrolytes with ILs (Elmore et al., 2018), according to the equation:
where ELi+ is the electronic energy of the lithium ion, Eanion is the electronic energy of the isolated anion i, y is the number of anion i in the solvate structure, and ELi-solvate is the electronic energy of the Li solvate. In this study, 1 ≤ y ≤ 4. The isolated Li+ is modeled with a +1 charge, and each anion is modeled with a net charge of −1. The net charge of the Li solvate can hence be 0, −1, −2, and −3, depending on the number of coordinating anions. This analysis is limited to a maximum number of coordinating anions of four due to the repulsive forces between anions when the first solvation shell includes five anions or greater, resulting in concentrated environments of 0.20 ≤ xLi+ ≤ 0.50. According to Eq. 1, positive values of ED are favorable, with increasing values of ED indicating stronger Li+ ion bonds. Thus, a more positive ED value is interpreted as a more stable Li solvate, and a more negative ED value is interpreted as less stable, which would allow for faster breakdown of the first solvation shell and thus quicker exchange of anions within this shell.
Quantum mechanics calculations were carried out in the Gaussian 16 program (Frisch et al., 2016) using the three-parameter Becke model with the Lee–Yang–Par modification (B3LYP) (Lee et al., 1988) functional for exchange and correlation and the 6-311G(d,p) (Ditchfield et al., 1971) basis set. Structures of the isolated anions and Li+ solvate systems and their associated electronic energies were obtained using geometry optimizations with default convergence criteria; as the Li+ system does not require geometry optimization, its electronic energy was calculated as a “single point.” Frequency calculations were carried out at the same level of theory in order to obtain thermodynamic and Raman spectroscopic data. Raman frequencies were scaled by a factor of 0.964 (Palafox, 2018). An example Gaussian input file for a geometry optimization and frequency calculation is provided in the supplementary.
Coordination Number and Li Solvate Structures
It is important to note the distinction between the number of anions coordinating with Li+ and the coordination number (CN) associated with this first solvation shell. An anion can have more than one bonding site for Li+ in which case CN will be a greater number than the number of coordinating anions. For example, the cis and trans conformation of [TFSI] can yield bidentate and monodentate bonds which can result in CN of 1–4 as [TFSI] has four oxygen atoms that can coordinate with Li+. Figure 1 demonstrates the possible monodentate and bidentate binding with two coordinating anions. The represented anions are asymmetric as illustrated by the color code (blue and red). Thus, with two coordinating asymmetric anions, a CN of two, three, or four is possible.
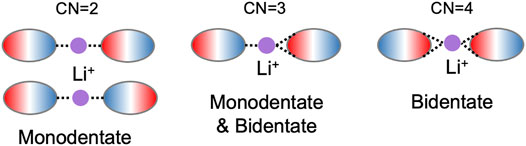
FIGURE 1. Representative coordination environments for Li+ with two asymmetric solvating anions forming bidentate and/or monodentate bonds. Li+ is represented by the purple circle and the asymmetric anion is represented by the ovals, with red and blue color codes corresponding to the different coordination sites.
Differential vs. Integral Analysis
Two methods were used to determine the energy difference associated with adding a new anion to the first solvation shell of Li+. These methods can be visualized in Figure 2 for a symmetric anion such as [TFSI] coordination with Li+. The integral binding energy, which is equal to −1 × ED, is shown via the green diagonal line. It refers to the energy to add n anions to Li+, for example, Li+ + 2[anion]− → Li[anion]2−. In contrast, the differential binding energy is the energy to add one anion at a time, for example, Li[anion] + [anion]− → Li[anion]2−. The differential binding energy is useful for assessing the maximum number of anions that will coordinate to Li+, whereas the integral binding energy is useful for assessing relative stabilities. A detailed example of the asymmetric [CTFSI] and [FTFSI] differential vs. integral analysis can be seen in Supplementary Tables S1, S2.
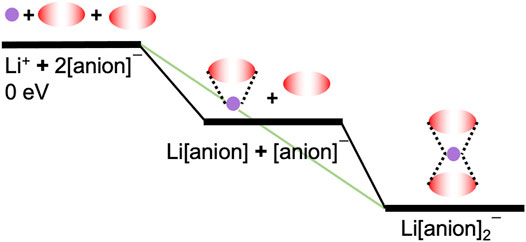
FIGURE 2. Comparison of differential and integral analyses for Li solvates with a symmetrical anion. Bidentate binding is illustrated for one and two coordinating anions. Energy levels are indicated by the black horizontal bars where the starting individual components have 0 eV as a reference and the Li solvates have <0 eV. The differential energies are represented by the solid black lines and the integral energy is represented by the diagonal green line that connects the 0 eV state with the Li[anion]2−1 solvate at the lowest energy state.
Ab Initio Thermodynamic Analysis
The thermodynamic stabilities of structurally and compositionally different Li solvates were calculated using an ab initio thermodynamic analysis (Soon et al., 2007; Getman et al., 2008; Grundner et al., 2015; Paolucci et al., 2016). This analysis was performed specifically for [TFSI]- and [DCA]-containing solvates. The ab initio thermodynamic analysis calculates the free energy of formation for different Li solvates at different mole fractions of [DCA] and [TFSI] in solution according to the following equation:
where
where
where
Results and Discussion
Thermodynamically Favored Structure
The Li+ solvation in the electrolyte is dynamic, and different populations of multiple solvate structures maybe present. In order to determine the most likely configurations, dissociation energies were calculated for all of the possible monodentate and bidentate coordination environments while incrementally increasing the number of anions around Li+. The list of calculated ED’s for all of the Li solvates studied is in Supplementary Table S3. For the asymmetric [CTFSI], it is determined that Li+ can coordinate with up to four anions where CN varies from one to four, as shown in Figure 3. Both the oxygen on the sulfonyl (Li+-O) and the nitrogen on the cyano group (Li+-N) can coordinate with Li+. Therefore, when there is one [CFTSI] coordinating with Li+, two configurations are possible, as seen in Figures 3A,B. With two coordinating anions, there are four possible configurations, as seen in Figures 3C–F. With three [CTFSI] anions, there are only two possible structures: all cyano coordination with monodentate bonds or two cyano and one sulfonyl oxygen coordination each with monodentate bonds, as shown in Figures 3G,H. Finally, moving to four coordinating anions, the only possible structure is seen in Figure 3I via monodentate Li+-N bonds. The calculated ED’s for these Li solvate structures at differing numbers of coordinating anions are shown in Figure 4. It is interpreted that the structure with the highest ED is the most stable since it requires more energy to break the coordination. Therefore, Li solvates with the highest ED (strongest) among the multiple possible configurations for a specific coordination are said to be thermodynamically favored. According to the energies in Figure 4, for both Li[CTFSI] and (Li[CTFSI]2)−1, Li+ prefers to coordinate with [CTFSI] via the cyano group (Li+-N) over the sulfonyl oxygen atoms (Li+-O). The most optimal Li solvate is (Li[CTFSI]2)−1 where Li+ coordinates with two cyano groups each through monodentate conformation as in Figure 3C and/or one monodentate with cyano and another monodentate with sulfonyl, as in Figure 3D. Based on the three and four anion systems relying solely on Li+-N coordination, all Li+-N coordinated (Li[CTFSI]2)−1 are used for further analysis. This methodology for selecting the thermodynamically favored structures was implemented for all of the anions investigated in this study. To better understand the change in solvation energy with the asymmetric anion in comparison to the symmetric anions, the Li+-[CTFSI] system was compared with the Li+-[TFSI] and Li+-[DCA] which are more widely studied experimentally (Sakaebe and Matsumoto, 2003; Garcia et al., 2004; Borgel et al., 2009; Yoon et al., 2013; Shen et al., 2015).
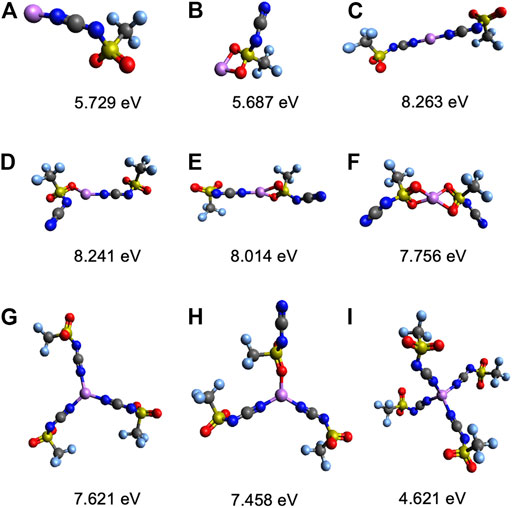
FIGURE 3. Geometry optimized structures and calculated integral energies, ED, of Li solvates with [CTFSI] for the number of coordinating anions of 1 (A,B), 2 (C–F), 3 (G,H), and 4 (I). The atom color code is as follows: red (oxygen), blue (nitrogen), yellow (sulfur), gray (carbon), purple (lithium), and light blue (fluorine).
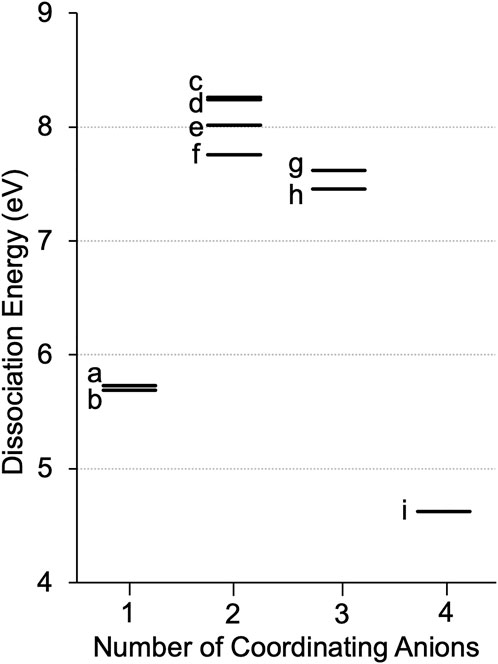
FIGURE 4. Integral dissociation energies, ED, for Li solvates with [CTFSI] according to the numbers of coordinating anions. Labels a–i correspond to structures shown in Figure 3.
Further, the Raman frequencies of [TFSI], [DCA], and [CTFSI] with and without Li+ coordination were calculated (spectra shown in Supplementary Figures S1–S3). The prominent vibrations were of the S-N-S and S=O bonds of [TFSI], and the C-N-C bond of [DCA]. For [CTFSI], the vibrations of interest are S-N-CCN, S=O, and C≡N. With Li+ coordination, the symmetric S-N-S stretch of cis-[TFSI] at 735 cm−1 experiences a blue shift by 10 cm−1, which is consistent with previously reported experiments (Huang et al., 2019a; Huang et al., 2019b; Nurnberg et al., 2020). The S=O symmetric stretch appears at 1,132 and 1,148 cm−1. With Li+ coordination (structure in Figure 5 with single [TFSI] coordinated to Li+), the S=O peaks shift to 1,100 and 1,111 cm−1. These vibrations have similar trends with [CTFSI]. Specifically, the S-N-CCN peak in [CTFSI] at 762 cm−1 shifts to 779 cm−1 with Li+ coordination (structure in Figure 3B). The S=O vibration of [CTFSI] at 1,118 cm−1 shifts to 1,099 cm−1. Similarly, the C-N-C asymmetric and symmetric stretches of [DCA] at 2,183 and 2,199 cm−1 show blue shifts by 15 and 45 cm−1 (structure in Figure 6 with single [DCA] coordinated to Li+), respectively, which are also consistent with previous experimental findings (Huang et al., 2019b). [CTFSI] shows a similar peak location as [DCA] for the C≡N symmetric stretch at 2,195 cm−1, with a blue shift of 42 cm−1 with Li+ coordination (structure in Figure 3A), which is similar to the previous experimental findings (Nurnberg et al., 2020). The overall consistency of the peak locations and shifts when moving from the pure anion to the Li solvate provide confidence that our converged structures closely match with the known experimental reports.
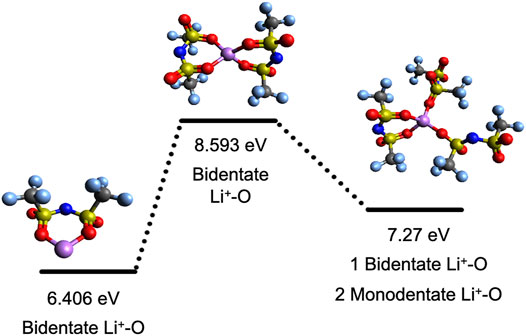
FIGURE 5. Integral disassociation energies, ED, and representative structures for Li+-[TFSI] solvates with the respective coordination type. The color code for atoms is as follows: red (oxygen), blue (nitrogen), yellow (sulfur), gray (carbon), purple (lithium), and light blue (fluorine).
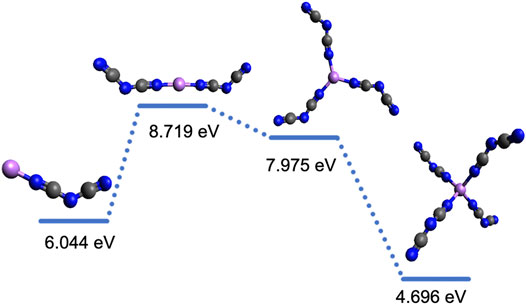
FIGURE 6. Integral dissociation energies, ED, and representative structures for Li+-[DCA] systems with the respective coordination type (all monodentate Li+-N). The color code for the atoms is as follows: blue (nitrogen), gray (carbon), and purple (lithium).
Li+ Solvation by [TFSI], [DCA], and [CTFSI]
Figures 5, 6 show the dissociation energies and structures for Li solvates with the [TFSI] and [DCA] anions, respectively. Li+ has a CN of four with both [TFSI] and [DCA], according to the number of coordination sites. However, the number of anions in the first solvation shell for [TFSI] is three, while it is four for [DCA]. This is a result of [TFSI] coordination via the oxygen atoms on the same anion where the two Li+-O coordination via bidentate bonds with two [TFSI] anions yield a CN of four, as seen in Figure 5. Previously, the integral binding energy for Li solvates with two [TFSI] anions calculated at the DFT/6-311+(d) level of theory was reported as −792.5 kJ/mol (8.2 eV) (Umebayashi et al., 2007), which is in reasonable agreement with our calculated Li[TFSI]2−1 (middle structure in Figure 5) ED of 8.59 eV (828.8 kJ/mol). For [DCA], there is Li+-N interaction via monodentate bonds; therefore, four [DCA] anions yield a CN of four, as seen in Figure 6. Based on these results, Li+-N coordination is stronger than Li+-O, especially when solvates with 2–3 anions are compared, which is consistent with previously reported results (Huang et al., 2019b). Therefore, one can suggest that Li solvates with a [DCA] anion will be more rigid, and this can hinder Li+ hopping in comparison to Li solvates with [TFSI]. This is counterintuitive to the low viscosities that are promoted with [DCA] anion in ILs. It should be recalled that while smaller and more rigid Li solvates may have higher vehicular motion, the hopping mechanism can prevail at high Li salt concentrations with weaker solvate structures.
To further understand the relationship of how the differing anions affect the strength of the interaction between Li+ and the [TFSI] and [DCA] anions, the ED of the mixture system, [TFSI]/[DCA], as well as the individual anions are compared, as in Figure 7.
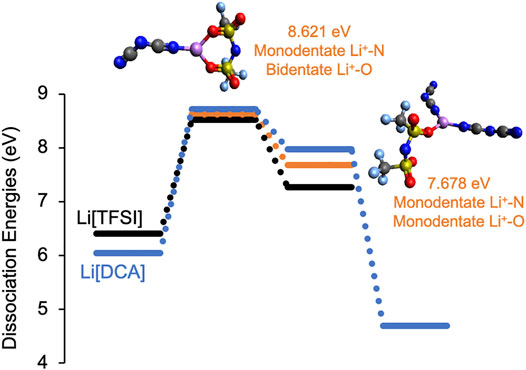
FIGURE 7. Integral dissociation energies, ED, and representative structures for Li solvates with [DCA]/[TFSI] anions (orange bars) in comparison to Li-[TFSI] (black bars) and Li-[DCA] (blue bars). The color code for the atoms is as follows: red (oxygen), blue (nitrogen), yellow (sulfur), gray (carbon), purple (lithium), and light blue (fluorine).
It should be noted that the minimum number of solvating anions for the [DCA]/[TFSI] system studied is two since a single anion would not capture the mixture behavior. Additionally, Li+ solvation by three anions can either contain two [DCA] and one [TFSI] or vice versa; however, Li+ coordination with two [DCA] anions provided a more thermodynamically favored structure (as shown in Figure 7). Comparing the Li+ solvation energies of the [DCA]/[TFSI] mixture with those of [DCA]-only and [TFSI]-only systems, it is seen that with a CN of three, the mix-anion system (represented by orange bars in Figure 7) has a weaker ED than Li-[DCA] (blue bars) and a stronger ED than Li-[TFSI] (black) systems. For a perspective on how the mix-anion system of [DCA]/[TFSI] compares with the asymmetric analog of [CTFSI], the calculated dissociation energies are shown in Figure 8.
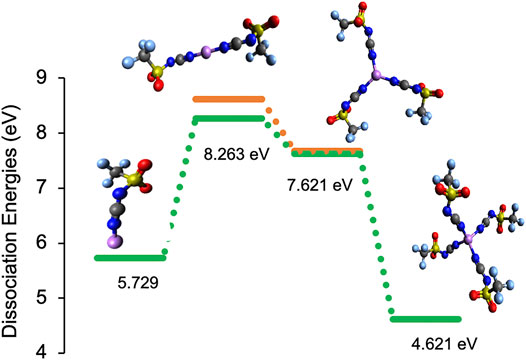
FIGURE 8. Integral dissociation energies, ED, and representative structures for Li-[CTFSI] solvates (green bars) in comparison to the Li-[DCA]/[TFSI] mixture (orange bars). All of the coordination environments are based on monodentate Li-N bonds. The color code for the atoms is as follows: red (oxygen), blue (nitrogen), yellow (sulfur), gray (carbon), purple (lithium), and light blue (fluorine).
Based on the lower dissociation energies of Li solvates with the [CTFSI] anion, it can be said that the solvation cage is weakened by the anion asymmetry and the presence of competing coordination sites for Li+. For example, comparing systems with two total solvation anions around Li+, (Li[CTFSI]2)−1 has the most facile ED of 8.26 eV in comparison to 8.59 eV for (Li[TFSI]2)−1, 8.62 eV for (Li[DCA][TFSI])−1, and 8.72 eV for (Li[DCA]2)−1. When [CTFSI] was compared with a slightly different asymmetric anion [CFSI], similar results were obtained, where Li-[CFSI] has more facile ED than the solvates with [FSI] or [DCA] alone. Further, Li-[CFSI] solvates had slightly reduced ED when compared to [CTFSI] (see Supplementary Figure S4). To understand the influence of solution composition on the Li+ solvate, we performed an ab initio thermodynamic analysis of the mix-anion [DCA]/[TFSI] system. The thermodynamic stability for the anionic [DCA]/[TFSI] system as a function of [DCA] and [TFSI] mole fraction is presented in Figure 9. Indeed, these results indicate that (Li[DCA]2)−1 is thermodynamically most preferred except for very low mole fractions of [DCA], where (Li[TFSI]2)−1 is preferred. None of the mix-anion structures are thermodynamically preferred according to the ab initio thermodynamic analysis.
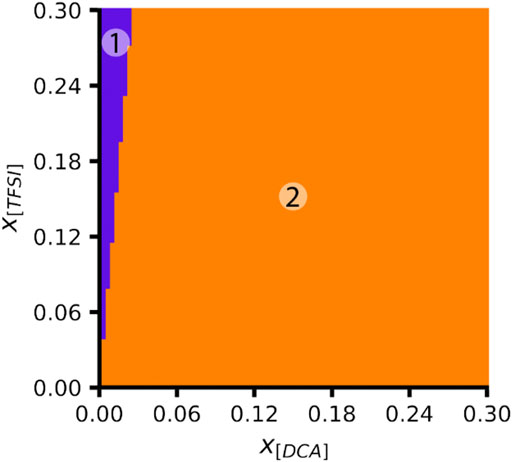
FIGURE 9. Phase diagram for Li+ solvates calculated in the ab initio thermodynamic analysis as a function of the mole fractions of [DCA] and [TFSI]. Color key: the purple region (1) = (Li[TFSI]2)−1 and the orange region (2) = (Li[DCA]2)−1.
Taken together, the above analysis suggests that assestment of ion pair lifetimes and measurement of the Li+ transport when solvated by asymmetric anions would be interesting and necessary to understand the transport mechanism as these studies may provide a method to tune Li+ hopping in concentrated electrolytes.
Li+ solvation by [FSI], [TFSI], and [FTFSI]
Li solvates with the asymmetric anion [FTSFI] were also studied in comparison to [TFSI], [FSI], and the [FSI]/[TFSI] mixture. It has been shown that Li+ transport is enhanced in ILs with [FSI]/[TFSI] mix-anions, resulting in improved battery cycling over the Li-IL binary mixtures with either the [FSI] or the [TFSI] anion (Lux et al., 2009; Bayley et al., 2011; Huang et al., 2019a). In order to understand whether the improvements in LIBs with these electrolytes originated from a change in solvation energies, we calculated the dissociation energies for this system, similar to the analysis performed for Li+-[CTFSI]. Figure 10 shows that the asymmetric [FTFSI] behaves similarly to [FSI] and the [FSI]/[TFSI] mixture. It should be noted that the Li solvate with the [FSI]/[TFSI] mixture and three coordinating anions consists of two [TFSI] and one [FSI], as this produced the more thermodynamically favored structure. The other configuration with two [FSI] and one [TFSI] system resulted in a dissociation energy of 6.907 eV, which is slightly lower than coordination with three [FTFSI] anions for (Li+-[FTFSI]3)−2 at 7.045 eV. Unlike the [CTFSI] asymmetric anion, Li solvates with [FTFSI] have higher ED than that of its parent anion [FSI], and the ED of all configurations is similar. This is due to the single coordination chemistry through the sulfonyl oxygen in all of the cases considered in Figure 10. The most significant difference among Li solvates with [TFSI], [FSI], and [FTFSI] is that with three coordinating anions, [FSI] has all monodentate Li+-O(=S) coordination, leading to a CN of three, while [FTFSI] and [TFSI] have one bidentate and two monodentate bonds, respectively, leading to a CN of four, as seen in Supplementary Figure S5. All other structures demonstrated bidentate configurations where CN was two for a single solvating anion and four for two solvating anions. Due to the similarities in the solvation structure, the only impact on the solvation strength comes from the fluorinated groups, either F-S ([FSI]) or CF3-S ([TFSI] and [FTFSI]).
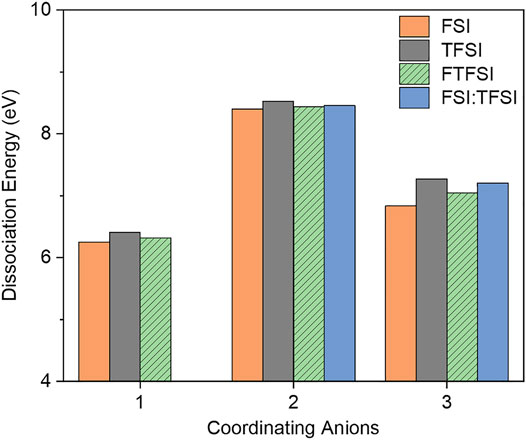
FIGURE 10. Integral dissociation energies, ED, of Li-[FSTFI] solvates in comparison to Li-[TFSI], Li-[FSI], and Li-[FSI]/[TFSI] mixture.
Fluorination Effects
The impacts of the length of fluorination and the degree of asymmetry on the calculated dissociation energies for Li solvates are captured in Figure 11. The asymmetry of the anions represented by the patterned bars in Figure 11 is dictated by the length of the fluorinated alkyl chain on either the [FSI] or the [TFSI] parent anion. Specifically, the PF, NF, and HF nomenclatures in front of TFSI stand for C2F5, C3F7, and C4F9 chains, respectively, on the one side of the derivatized [TFSI] anion. As seen, there are no significant differences in ED. The lowest dissociation energy is with [NFFSI] which has the longest fluorinated alkyl chains (C4F9) on the one side, with the other side being F-S.
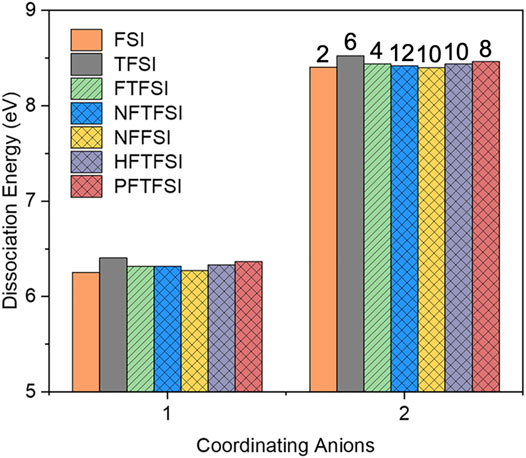
FIGURE 11. Integral dissociation energies with respect to the number of coordinating asymmetric anions. [FTFSI] anion is highlighted with slashes, and all other asymmetric fluorinated anions are highlighted with crosshatches. PF, NF, and HF nomenclatures in front of TFSI or FSI stand for C2F5, C3F7, and C4F9 chains, respectively. The numbers on top of the bars indicate the number of total fluorines in the anion.
While these results cannot point to a specific advantage of the longer fluorinated anions since they have the same enthalpic effects on Li+ coordination, we expect to find entropic effects in solution which are not present in gas phase energy evaluations. In the case of [NFTFSI] and [NFFSI], the long fluorinated butane chains of each anion align with each other when the Li solvate has two anions with a CN of four via two bidentate bonds (Supplementary Figure S6). This alignment of the fluorinated alkyl chains may have a significant impact on the bulk liquid structure that cannot be evaluated by gas phase DFT calculations. We expect that the increase in the fluorination length will improve the viscosity and the solvation dynamics more than the enthalpic contributions to the dissociation energies, due to the delocalization of charge along the alkyl chain and inability of fluorochains solvating Li+. In particular, the treatment of the entire system with IL cation and other cosolvents, if present, can provide more realistic solvate structures when longer chain fluorinated asymmetric anions are solvating Li+. By studying these systems with MD simulations and experimental spectroscopic studies (Raman and NMR), the impact of fluorination on the bulk solvation and transport properties can be probed to a greater extent.
Conclusion
The solvation structure and energies of Li+ with solvating anions of [TFSI], [DAC], [CTFSI], and [FTFSI] and a number of longer fluorinated anions with Li+-O and Li+-N coordination capability were calculated. The anions studied produced Li solvate structures with CNs of 2–4 depending on the size and functionality of the anion. Li solvates with a CN of two resulted in the highest dissociation energies (most stable conformers). Li solvates with a CN of one resulted in the lowest ED with the exception of [DCA] and [CTFSI]. [DCA] and [CTFSI] were able to coordinate with four anions, and these Li solvates produced the lowest ED (weakest). Through the ED analysis, we found that the asymmetric [CTFSI] anion coordination with Li+ is weaker than [TFSI] and [DCA] anions as well as the [TFSI]/[DCA] mixture. There are a larger number of solvate structures for Li-[CTFSI] solvate than those for [TFSI] and [DCA]. We suspect that this heterogeneity in the first solvation shell may lead to increased fluidity and structural diffusion (hopping) of Li+ compared to solvates with [TFSI] and [DCA]. The [FTFSI] anion showed very similar results in terms of ED as the [TFSI] and [FSI] parent anions due to the similar coordination environments between the three anions. However, [FTFSI] yielded slightly lower ED than that of the Li+ solvate with the parent anion mixture of [TFSI]/[FSI]. Given the weakened solvation energies, it would be interesting to study the impact of IL electrolytes with asymmetric anion on the Li+ transference and performance of LIBs. The impact of fluorination length seems to have limited impact on ED, with the longer fluorinated chains having slightly lower ED than [TFSI]. However, we anticipate that these long fluorinated alkyl chains will have more significant entropic effects, which are not captured in gas phase DFT calculations. By studying these systems with MD simulations and experimental spectroscopic studies (Raman and NMR), the impact of fluorination on the bulk solvation and transport properties can be probed to a greater extent. It will also be crucial for future studies to evaluate the impact of IL cations as well as the electric field present in Li batteries on the solvation structure and dynamics of Li+.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Author Contributions
DP performed the DFT calculations for geometries, vibrations, and dissociation energies. SV performed the ab initio thermodynamic analysis. RG supervised the ab initio thermodynamic analysis and contributed to the discussions. BG developed the plan, supervised DFT calculations, and contributed to the discussions. All authors contributed to the writing of the manuscript.
Funding
BG would like to acknowledge funding from the National Science Foundation (CBET, Award No. 1903259). BG and DP would also like to acknowledge the High Performance Computing Resource in the Core Facility for Advanced Research Computing at Case Western Reserve University. SV is supported by a GAANN Fellowship (Award Number: P200A180076) from the United States Department of Education.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenrg.2021.725010/full#supplementary-material
References
Armand, M., Endres, F., MacFarlane, D. R., Ohno, H., and Scrosati, B. (2009). Ionic-Liquid Materials for the Electrochemical Challenges of the Future. Nat. Mater. 8 (8), 621–629. doi:10.1038/nmat2448
Bae, S.-Y., Shim, E.-G., and Kim, D.-W. (2013). Effect of Ionic Liquid as a Flame-Retarding Additive on the Cycling Performance and thermal Stability of Lithium-Ion Batteries. J. Power Sourc. 244, 266–271. doi:10.1016/j.jpowsour.2013.01.100
Bauschlicher, C. W. (2018). Li+-Ligand Binding Energies and the Effect of Ligand Fluorination on the Binding Energies. Chem. Phys. Lett. 694, 86–92. doi:10.1016/j.cplett.2018.01.047
Bayley, P. M., Best, A. S., MacFarlane, D. R., and Forsyth, M. (2011). Transport Properties and Phase Behaviour in Binary and Ternary Ionic Liquid Electrolyte Systems of Interest in Lithium Batteries. ChemPhysChem 12 (4), 823–827. doi:10.1002/cphc.201000909
Borgel, V., Markevich, E., Aurbach, D., Semrau, G., and Schmidt, M. (2009). On the Application of Ionic Liquids for Rechargeable Li Batteries: High Voltage Systems. J. Power Sourc. 189 (1), 331–336. doi:10.1016/j.jpowsour.2008.08.099
Borodin, O., Giffin, G. A., Moretti, A., Haskins, J. B., Lawson, J. W., Henderson, W. A., et al. (2018). Insights into the Structure and Transport of the Lithium, Sodium, Magnesium, and Zinc Bis(trifluoromethansulfonyl)imide Salts in Ionic Liquids. J. Phys. Chem. C. 122 (35), 20108–20121. doi:10.1021/acs.jpcc.8b05573
Borodin, O., Smith, G. D., and Henderson, W. (2006). Li+ Cation Environment, Transport, and Mechanical Properties of the LiTFSI Doped N-Methyl-N-alkylpyrrolidinium+TFSI- Ionic Liquids. J. Phys. Chem. B. 110 (34), 16879–16886. doi:10.1021/jp061930t
Borodin, O., Suo, L., Gobet, M., Ren, X., Wang, F., Faraone, A., et al. (2017). Liquid Structure with Nano-Heterogeneity Promotes Cationic Transport in Concentrated Electrolytes. ACS Nano 11 (10), 10462–10471. doi:10.1021/acsnano.7b05664
Brinkkötter, M., Giffin, G. A., Moretti, A., Jeong, S., Passerini, S., and Schönhoff, M. (2018). Relevance of Ion Clusters for Li Transport at Elevated Salt Concentrations in [Pyr12O1][FTFSI] Ionic Liquid-Based Electrolytes. Chem. Commun. 54, 4278–4281. doi:10.1039/C8CC01416G
Castiglione, F., Ragg, E., Mele, A., Appetecchi, G. B., Montanino, M., and Passerini, S. (2011). Molecular Environment and Enhanced Diffusivity of Li+ Ions in Lithium-Salt-Doped Ionic Liquid Electrolytes. J. Phys. Chem. Lett. 2 (3), 153–157. doi:10.1021/jz101516c
Chen, F., and Forsyth, M. (2019). Computational Investigation of Mixed Anion Effect on Lithium Coordination and Transport in Salt Concentrated Ionic Liquid Electrolytes. J. Phys. Chem. Lett. 10 (23), 7414–7420. doi:10.1021/acs.jpclett.9b02416
Ditchfield, R., Hehre, W. J., and Pople, J. A. (1971). Self‐Consistent Molecular‐Orbital Methods. IX. An Extended Gaussian‐Type Basis for Molecular‐Orbital Studies of Organic Molecules. J. Chem. Phys. 54 (2), 724. doi:10.1063/1.1674902
Duluard, S., Grondin, J., Bruneel, J.-L., Pianet, I., Grélard, A., Campet, G., et al. (2008). Lithium Solvation and Diffusion in the 1-Butyl-3-Methylimidazolium Bis(trifluoromethanesulfonyl)imide Ionic Liquid. J. Raman Spectrosc. 39 (5), 627–632. doi:10.1002/jrs.1896
Eftekhari, A., Liu, Y., and Chen, P. (2016). Different Roles of Ionic Liquids in Lithium Batteries. J. Power Sourc. 334, 221–239. doi:10.1016/j.jpowsour.2016.10.025
Elmore, C., Seidler, M., Ford, H., Merrill, L., Upadhyay, S., Schneider, W., et al. (2018). Ion Transport in Solvent-free, Crosslinked, Single-Ion Conducting Polymer Electrolytes for Post-Lithium Ion Batteries. Batteries 4 (2), 28. doi:10.3390/batteries4020028
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., et al. (2016). Gaussian 16 Rev. C.01. Wallingford, CT.
Fujii, K., Hamano, H., Doi, H., Song, X., Tsuzuki, S., Hayamizu, K., et al. (2013). Unusual Li+Ion Solvation Structure in Bis(fluorosulfonyl)amide Based Ionic Liquid. J. Phys. Chem. C. 117 (38), 19314–19324. doi:10.1021/jp4053264
Fulfer, K. D., and Kuroda, D. G. (2016). Solvation Structure and Dynamics of the Lithium Ion in Organic Carbonate-Based Electrolytes: A Time-dependent Infrared Spectroscopy Study. J. Phys. Chem. C. 120 (42), 24011–24022. doi:10.1021/acs.jpcc.6b08607
Galiński, M., Lewandowski, A., and Stępniak, I. (2006). Ionic Liquids as Electrolytes. Electrochimica Acta. 51 (26), 5567–5580. doi:10.1016/j.electacta.2006.03.016
Garcia, B., Lavallée, S., Perron, G., Michot, C., and Armand, M. (2004). Room Temperature Molten Salts as Lithium Battery Electrolyte. Electrochim. Acta. 49 (26), 4583–4588. doi:10.1016/j.electacta.2004.04.041
Getman, R. B., Xu, Y., and Schneider, W. F. (2008). Thermodynamics of Environment-dependent Oxygen Chemisorption on Pt(111). J. Phys. Chem. C. 112 (26), 9559–9572. doi:10.1021/jp800905a
Grundner, S., Markovits, M. A. C., Li, G., Tromp, M., Pidko, E. A., Hensen, E. J. M., et al. (2015). Single-site Trinuclear Copper Oxygen Clusters in Mordenite for Selective Conversion of Methane to Methanol. Nat. Commun. 6 (1), 7546. doi:10.1038/ncomms8546
Han, H., Guo, J., Zhang, D., Feng, S., Feng, W., Nie, J., et al. (2011). Lithium (Fluorosulfonyl)(nonafluorobutanesulfonyl)imide (LiFNFSI) as Conducting Salt to Improve the High-Temperature Resilience of Lithium-Ion Cells. Electrochem. Commun. 13 (3), 265–268. doi:10.1016/j.elecom.2010.12.030
Haskins, J. B., Bennett, W. R., Wu, J. J., Hernández, D. M., Borodin, O., Monk, J. D., et al. (2014). Computational and Experimental Investigation of Li-Doped Ionic Liquid Electrolytes: [pyr14][TFSI], [pyr13][FSI], and [EMIM][BF4]. J. Phys. Chem. B. 118 (38), 11295–11309. doi:10.1021/jp5061705
Hoffknecht, J.-P., Drews, M., He, X., and Paillard, E. (2017). Investigation of the N -Butyl- N -Methyl Pyrrolidinium Trifluoromethanesulfonyl- N -cyanoamide (PYR 14 TFSAM) Ionic Liquid as Electrolyte for Li-Ion Battery. Electrochim. Acta. 250, 25–34. doi:10.1016/j.electacta.2017.08.020
Huang, Q., Lee, Y.-Y., and Gurkan, B. (2019a). Pyrrolidinium Ionic Liquid Electrolyte with Bis(trifluoromethylsulfonyl)imide and Bis(fluorosulfonyl)imide Anions: Lithium Solvation and Mobility, and Performance in Lithium Metal-Lithium Iron Phosphate Batteries. Ind. Eng. Chem. Res. 58, 22587–22597. doi:10.1021/acs.iecr.9b03202
Huang, Q., Lourenço, T. C., Costa, L. T., Zhang, Y., Maginn, E. J., and Gurkan, B. (2019b). Solvation Structure and Dynamics of Li+ in Ternary Ionic Liquid-Lithium Salt Electrolytes. J. Phys. Chem. B. 123 (2), 516–527. doi:10.1021/acs.jpcb.8b08859
Imai, M., Tanabe, I., Ikehata, A., Ozaki, Y., and Fukui, K.-i. (2020). Attenuated Total Reflectance Far-Ultraviolet and Deep-Ultraviolet Spectroscopy Analysis of the Electronic Structure of a Dicyanamide-Based Ionic Liquid with Li+. Phys. Chem. Chem. Phys. 22 (38), 21768–21775. doi:10.1039/D0CP03865B
Kerner, M., Plylahan, N., Scheers, J., and Johansson, P. (2015). Ionic Liquid Based Lithium Battery Electrolytes: Fundamental Benefits of Utilising Both TFSI and FSI Anions?. Phys. Chem. Chem. Phys. 17 (29), 19569–19581. doi:10.1039/C5CP01891A
Krachkovskiy, S., Dontigny, M., Rochon, S., Kim, C., Trudeau, M. L., and Zaghib, K. (2020). Determination of Binary Diffusivities in Concentrated Lithium Battery Electrolytes via NMR and Conductivity Measurements. J. Phys. Chem. C. 124 (45), 24624–24630. doi:10.1021/acs.jpcc.0c07383
Lassègues, J.-C., Grondin, J., Aupetit, C., and Johansson, P. (2009). Spectroscopic Identification of the Lithium Ion Transporting Species in LiTFSI-Doped Ionic Liquids. J. Phys. Chem. A. 113 (1), 305–314. doi:10.1021/jp806124w
Lee, C., Yang, W., and Parr, R. G. (1988). Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B. 37 (2), 785–789. doi:10.1103/physrevb.37.785
Lesch, V., Jeremias, S., Moretti, A., Passerini, S., Heuer, A., and Borodin, O. (2014). A Combined Theoretical and Experimental Study of the Influence of Different Anion Ratios on Lithium Ion Dynamics in Ionic Liquids. J. Phys. Chem. B. 118 (26), 7367–7375. doi:10.1021/jp501075g
Lesch, V., Li, Z., Bedrov, D., Borodin, O., and Heuer, A. (2016). The Influence of Cations on Lithium Ion Coordination and Transport in Ionic Liquid Electrolytes: a MD Simulation Study. Phys. Chem. Chem. Phys. 18 (1), 382–392. doi:10.1039/C5CP05111H
Li, Z., Smith, G. D., and Bedrov, D. (2012). Li+ Solvation and Transport Properties in Ionic Liquid/Lithium Salt Mixtures: A Molecular Dynamics Simulation Study. J. Phys. Chem. B. 116 (42), 12801–12809. doi:10.1021/jp3052246
Lux, S. F., Schmuck, M., Appetecchi, G. B., Passerini, S., Winter, M., and Balducci, A. (2009). Lithium Insertion in Graphite from Ternary Ionic Liquid-Lithium Salt Electrolytes: II. Evaluation of Specific Capacity and Cycling Efficiency and Stability at Room Temperature. J. Power Sourc. 192 (2), 606–611. doi:10.1016/j.jpowsour.2009.02.066
Matsuda, Y., Fukushima, T., Hashimoto, H., and Arakawa, R. (2002). Solvation of Lithium Ions in Mixed Organic Electrolyte Solutions by Electrospray Ionization Mass Spectroscopy. J. Electrochem. Soc. 149 (8), A1045. doi:10.1149/1.1489687
McEldrew, M., Goodwin, Z. A. H., Bi, S., Kornyshev, A. A., and Bazant, M. Z. (2021). Ion Clusters and Networks in Water-In-Salt Electrolytes. J. Electrochem. Soc. 168 (5), 050514. doi:10.1149/1945-7111/abf975
Moučka, F., Nezbeda, I., and Smith, W. R. (2015). Chemical Potentials, Activity Coefficients, and Solubility in Aqueous NaCl Solutions: Prediction by Polarizable Force Fields. J. Chem. Theor. Comput. 11 (4), 1756–1764. doi:10.1021/acs.jctc.5b00018
Navarra, M. A. (2013). Ionic Liquids as Safe Electrolyte Components for Li-Metal and Li-Ion Batteries. MRS Bull. 38 (7), 548–553. doi:10.1557/mrs.2013.152
Noda, A., Hayamizu, K., and Watanabe, M. (2001). Pulsed-Gradient Spin−Echo1H and19F NMR Ionic Diffusion Coefficient, Viscosity, and Ionic Conductivity of Non-chloroaluminate Room-Temperature Ionic Liquids. J. Phys. Chem. B. 105 (20), 4603–4610. doi:10.1021/jp004132q
Nürnberg, P., Lozinskaya, E. I., Shaplov, A. S., and Schönhoff, M. (2020). Li Coordination of a Novel Asymmetric Anion in Ionic Liquid-In-Li Salt Electrolytes. J. Phys. Chem. B. 124 (5), 861–870. doi:10.1021/acs.jpcb.9b11051
Palafox, M. A. (2018). DFT Computations on Vibrational Spectra: Scaling Procedures to Improve the Wavenumbers. Phys. Sci. Rev. 3 (6), 1–30. doi:10.1515/psr-2017-0184
Palumbo, O., Trequattrini, F., Appetecchi, G., and Paolone, A. (2017). A Study of the Conformers of the (Nonafluorobutanesulfonyl)imide Ion by Means of Infrared Spectroscopy and Density Functional Theory (DFT) Calculations. Challenges 8 (1), 7. doi:10.3390/challe8010007
Paolucci, C., Parekh, A. A., Khurana, I., Di Iorio, J. R., Li, H., Albarracin Caballero, J. D., et al. (2016). Catalysis in a Cage: Condition-dependent Speciation and Dynamics of Exchanged Cu Cations in SSZ-13 Zeolites. J. Am. Chem. Soc. 138 (18), 6028–6048. doi:10.1021/jacs.6b02651
Pham, T. D., Bin Faheem, A., Chun, S. Y., Rho, J. R., Kwak, K., and Lee, K. K. (2021). Synergistic Effects on Lithium Metal Batteries by Preferential Ionic Interactions in Concentrated Bisalt Electrolytes. Adv. Energ. Mater. 11 (11), 2003520. doi:10.1002/aenm.202003520
Sakaebe, H., and Matsumoto, H. (2003). N-Methyl-N-propylpiperidinium Bis(trifluoromethanesulfonyl)imide (PP13-TFSI) - Novel Electrolyte Base for Li Battery. Electrochem. Commun. 5 (7), 594–598. doi:10.1016/s1388-2481(03)00137-1
Shen, S., Fang, S., Qu, L., Luo, D., Yang, L., and Hirano, S.-i. (2015). Low-viscosity Ether-Functionalized Pyrazolium Ionic Liquids Based on Dicyanamide Anions: Properties and Application as Electrolytes for Lithium Metal Batteries. RSC Adv. 5 (114), 93888–93899. doi:10.1039/C5RA17539A
Shim, Y. (2018). Computer Simulation Study of the Solvation of Lithium Ions in Ternary Mixed Carbonate Electrolytes: Free Energetics, Dynamics, and Ion Transport. Phys. Chem. Chem. Phys. 20 (45), 28649–28657. doi:10.1039/C8CP05190A
Soon, A., Todorova, M., Delley, B., and Stampfl, C. (2007). Surface Oxides of the Oxygen-Copper System: Precursors to the Bulk Oxide Phase?. Surf. Sci. 601 (24), 5809–5813. doi:10.1016/j.susc.2007.06.062
Sun, Y., Xin, N., and Prausnitz, J. M. (2018). Solubilities of Five Lithium Salts in 1-Butyl-3-Methylimidazolium Dicyanamide and in 1-Butyl-3-Methylimidazolium Tetrafluoroborate from 298.15 to 343.15 K. J. Chem. Eng. Data. 63 (12), 4524–4531. doi:10.1021/acs.jced.8b00618
Umebayashi, Y., Mitsugi, T., Fukuda, S., Fujimori, T., Fujii, K., Kanzaki, R., et al. (2007). Lithium Ion Solvation in Room-Temperature Ionic Liquids Involving Bis(trifluoromethanesulfonyl) Imide Anion Studied by Raman Spectroscopy and DFT Calculations. J. Phys. Chem. B. 111 (45), 13028–13032. doi:10.1021/jp076869m
Umebayashi, Y., Mori, S., Fujii, K., Tsuzuki, S., Seki, S., Hayamizu, K., et al. (2010). Raman Spectroscopic Studies and Ab Initio Calculations on Conformational Isomerism of 1-Butyl-3-Methylimidazolium Bis-(trifluoromethanesulfonyl)amide Solvated to a Lithium Ion in Ionic Liquids: Effects of the Second Solvation Sphere of the Lithium Ion. J. Phys. Chem. B. 114 (19), 6513–6521. doi:10.1021/jp100898h
Yamada, Y., Wang, J., Ko, S., Watanabe, E., and Yamada, A. (2019). Advances and Issues in Developing Salt-Concentrated Battery Electrolytes. Nat. Energ. 4 (4), 269–280. doi:10.1038/s41560-019-0336-z
Yang, L., Xiao, A., and Lucht, B. L. (2010). Investigation of Solvation in Lithium Ion Battery Electrolytes by NMR Spectroscopy. J. Mol. Liquids. 154 (2), 131–133. doi:10.1016/j.molliq.2010.04.025
Keywords: ionic liquid electrolytes, lithium solvation, concentrated electrolytes, lithium-ion battery, ab initio thermodynamics
Citation: Penley D, Vicchio SP, Getman RB and Gurkan B (2021) Energetics of Li+ Coordination with Asymmetric Anions in Ionic Liquids by Density Functional Theory. Front. Energy Res. 9:725010. doi: 10.3389/fenrg.2021.725010
Received: 14 June 2021; Accepted: 10 September 2021;
Published: 15 October 2021.
Edited by:
Ah-Hyung Alissa Park, Columbia University, United StatesReviewed by:
Hailei Zhao, University of Science and Technology Beijing, ChinaShrihari Sankarasubramanian, University of Texas at San Antonio, United States
Copyright © 2021 Penley, Vicchio, Getman and Gurkan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Burcu Gurkan, YmVnMjNAY2FzZS5lZHU=