 Fengxia Yue
Fengxia Yue Wu Lan1
Wu Lan1 John Ralph
John Ralph
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Energy Res. , 30 March 2021
Sec. Bioenergy and Biofuels
Volume 9 - 2021 | https://doi.org/10.3389/fenrg.2021.640337
This article is part of the Research Topic Advances in the Structural Elucidation and Utilization of Lignins View all 10 articles
Pinoresinol is a high-value monolignol-derived lignan used in plant defense and with human health-supporting effects. The synthetic yield and isolation efficiency of racemic pinoresinol from coniferyl alcohol by conventional radical coupling methods is sub-optimal. In this work, a facile and efficient synthetic approach was developed to synthesize pinoresinol with much higher yield. By using 5-bromoconiferyl alcohol, which was synthesized in high yield from 5-bromovanillin, to make 5,5′-bromopinoresinol via a peroxidase-mediated radical coupling reaction takes advantage of the smaller variety of radical coupling products from the 5-substituted monolignol, producing simpler product mixtures from which 5,5′-bromopinoresinol may be readily crystalized with good yield (total yield of 44.1% by NMR; isolated crystalline yield of 24.6%). Hydro-debromination of the crystalline 5,5′-bromopinoresinol to pinoresinol was essentially quantitative. Gram quantities of pinoresinol were conveniently synthesized by using this approach. This simple alternative pathway to make pinoresinol will impact pinoresinol-related research including structural characterization and modification of lignins, as well as clinical applications of pinoresinol and its derivatives.
Pinoresinol structures with a β-β′-linkage between two monolignols are important structures in the lignins from softwoods, dicots, and monocots, especially in some technical lignins from hardwood (Yue et al., 2012b; Li et al., 2020). Not only do they affect the lignification pathway, but also have implications for the biorefinery. Pinoresinol is also a vital lignan, a usually optically-active dimerization product of two coniferyl alcohol monolignols, that displays important physiological functions in planta, as well as in human nutrition and medicine (Lewis et al., 1995; Pellegrini et al., 2010). Lignans have drawn enormous attention because of their abundance in nature and their various biological activities including antibacterial, antifungal, antiviral, antioxidative, and/or cytotoxic properties (Paska et al., 2002; Min et al., 2003; Umezawa, 2003; Milder et al., 2005; Suzuki and Umezawa, 2007; Lin et al., 2016). Such lignans are commonly found as optically active β-β′-linked dimers (Davin et al., 1992; Pare et al., 1994), such as pinoresinol and syringaresinol as shown in Figure 1. Some lignans are structurally similar to dilignols, or units in oligolignols, that are metabolites involved in lignification (Eberhardt et al., 1993; Pare et al., 1994; Morreel et al., 2004). Chiral (+)-pinoresinol and (–)-pinoresinol are formed through a dirigent-protein-mediated radical coupling of coniferyl alcohol (Halls et al., 2004). However, racemic pinoresinol and oligomers containing pinoresinol are often found in plant metabolites destined for lignin, a racemic polymer that contains significant amounts of pinoresinol substructures, especially for softwood lignins or plants that are down-regulated in syringyl-specific genes (Eberhardt et al., 1993; Ralph et al., 1999).
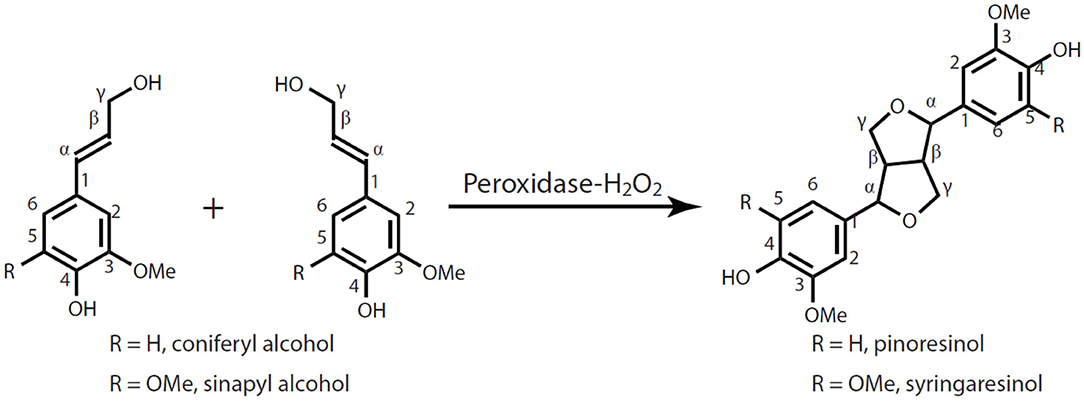
Figure 1. Traditional pathways for synthesis of pinoresinol and syringaresinol.
As one of the structurally simplest lignin dimers, pinoresinol is an important nucleation site formed by the self-coupling of coniferyl alcohol during lignification (Umezawa, 2003; Zhang et al., 2003; Ralph J. et al., 2004; Schroeder et al., 2006; Ralph et al., 2008; Yue et al., 2012a). The structure is frequently present in the lignin of woody and fibrous plants, but the amount varies significantly in different plants. Because of its wide range of bioactivities and its chemical structure related to lignin, pinoresinol and its lignan analog play very important roles in pharmaceutical research, lignin biosynthesis, and other related studies (Umezawa, 2003; Zhang et al., 2003; Ralph J. et al., 2004; Jung et al., 2006; Ralph et al., 2008; Yue et al., 2012a; Tamura et al., 2014). Moreover, substantial amounts of resinol (β-β′) structures, including pinoresinol and syringaresinol, remained and can be identified in several technical lignins, which is crucial to the phenolation of alkaline lignin, for example (Li et al., 2020; Zhao et al., 2020). Studies on the biosynthesis of pinoresinol structures in lignin, technical lignin modifications, pinoresinol-related lignans, and pinoresinol contributions to a healthy human diet or for disease prevention require easy access to pinoresinol. Normally, commercially available pinoresinol is isolated from natural sources, such as seeds of flax, sesame, whole-grain cereals, legumes, fruits, and some vegetables. Although pinoresinol is widely distributed in vascular plants, the content is at low level (Umezawa, 2003; Ralph et al., 2008; Li et al., 2019). The isolation processes, which involve a series of time-consuming and costly separation/purification steps, are not efficient and at very low yields (such as, 2.6 mg pinoresinol isolated from 8 kg of dried cinnamon) (Milder et al., 2005; Li et al., 2019). It is difficult to isolate sufficient pure pinoresinol and its derivatives from plant materials due to its low content and the complexity of plant extractives. Therefore, current commercial pinoresinol that is isolated from natural sources is expensive [$281 for 10 mg (>95%), Sigma-Aldrich]. Although many synthetic routes have been proposed to make optically active pinoresinol (Davin et al., 1992; Pare et al., 1994; Umezawa, 2003; Feng et al., 2006, 2007; Kim et al., 2010), racemic pinoresinol has been more often prepared from radical coupling of coniferyl alcohol catalyzed by various oxidants including silver oxide, ferric chloride, manganese acetate, or copper acetate, as well as oxidative enzymes (peroxidase or laccase) (Brezny and Alfoldi, 1982; Vermes et al., 1991; Quideau and Ralph, 1994; Zhang et al., 2003; Saliu et al., 2011; Yue et al., 2012a; Tamura et al., 2014). The yields of syringaresinol (analog of pinoresinol) from radical coupling reactions has always been much higher than those of pinoresinol because the occupied C5-position on sinapyl alcohol prevents the production of dimers from other coupling modes (Zhang et al., 2003). Therefore, the difficult access to pinoresinol limits its further exploration in research and clinical applications, and therefore a low-cost, environmentally friendly method is required to address this issue.
Herein we report an efficient synthetic strategy for pinoresinol. In this method, we used 5-bromoconiferyl alcohol for the radical coupling reaction to produce the β-β′-coupled compound as the main product. It is readily crystallized and subsequently hydrodebrominated to produce clean racemic pinoresinol.
5-Bromovanillin (98%) was purchased from Acros Organics (New Jersey, USA). Horseradish peroxidase (HRP) (EC 1.11.1.7, 181 purpurogallin units per mg solid, type II) was purchased from Sigma Aldrich. All other chemicals and solvents used in this study were purchased from Aldrich (Milwaukee, WI, USA) and used as supplied.
Flash-chromatography was performed on a Biotage® Isolera One (Biotage, Charlottesville, VA) flash-chromatography instrument, using pre-packed (or re-packed) SNAP cartridges (50 or 100 g of silica-gel). Thin-layer chromatography separation was performed on 1 mm normal-phase silica-gel plates, UniplateTM (UV 254, 20 × 20 cm). All synthesized compounds were characterized by the usual array of NMR. NMR spectra were acquired on a Bruker Biospin (Billerica, MA, USA) AVANCE 500 (500 MHz) spectrometer fitted with a cryogenically cooled 5 mm TCI gradient probe with inverse geometry (proton coils closest to the sample). Spectra were processed using Bruker's Topspin 3.5 (Mac) software. Standard Bruker implementations of one- and two-dimensional (gradient-selected COSY, HSQC and HMBC) NMR experiments were used for routine structural assignments of all synthesized compounds. Typically, 5–10 mg of sample was dissolved in about 0.5 mL deuterated solvent (acetone-d6), and the central solvent peak (δH/δC 2.04/29.80) was used as the internal reference.
As shown in Figures 2, 3, pinoresinol was synthesized via a simple multi-step route.
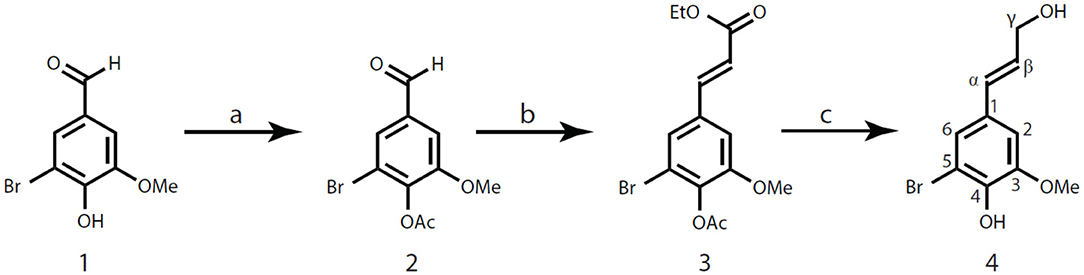
Figure 2. Synthesis of 5-bromoconiferyl alcohol 4. (a) pyridine, acetic anhydride; (b) NaH, triethyl phosphonoacetate, THF; (c) diisobutylaluminum hydride (DIBAL-H), cyclohexane.
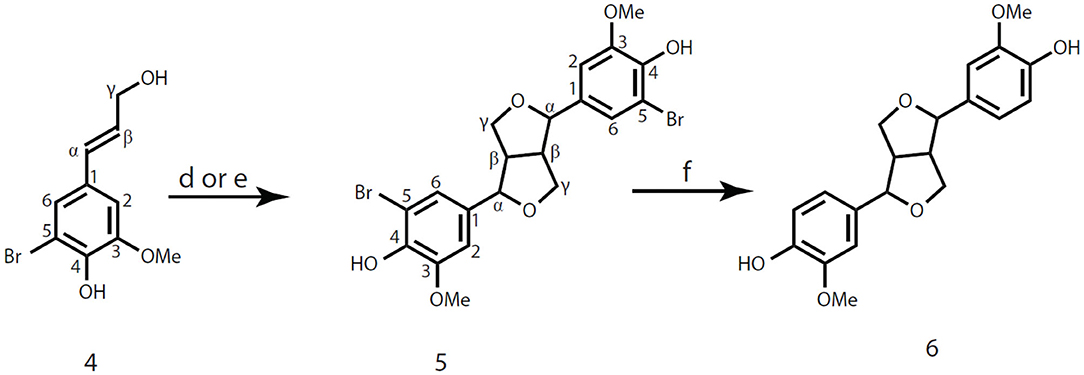
Figure 3. Synthesis of pinoresinol from 5-bromoconiferyl alcohol 4. (d) H2O2, peroxidase; acetone-buffer; (e) FeCl3, acetone-buffer; (f) Et3N, Pd/C, H2, methanol.
5-Bromovanillin acetate 2 was prepared by acetylation of 5-bromovanillin 1 with pyridine and acetic anhydride (1:1, v/v), which was accomplished in the traditional way using acetic anhydride in pyridine (~100% yield). Compound 2, NMR (acetone-d6, Supplementary Figure 1) δH: 2.35 (3H, s, OAc), 3.95 (3H, s, OMe), 7.60 (1H, d, J = 1.61, A2), 7.80 (1H, d, J = 1.61, A6), 9.97 (1H, s, Hα); δC: 20.19 (OAc), 56.99 (A-OMe), 111.73 (A2), 118.45 (A5), 127.21 (A6), 136.71 (A1), 143.48 (A4), 154.33 (A3), 167.60 (OAc), 190.87 (α).
Compound 3 was synthesized via a Horner-Wadsworth-Emmons reaction (HWE reaction) from compound 2 (Ralph et al., 1992; Brandt et al., 1998; Touchard, 2004). Briefly, NaH (sodium hydride 60% dispersion in mineral oil, 3.62 g, 90.42 mmol) was washed with 50 mL cyclohexane by stirring more than 10 min in the reaction flask and the supernatant was carefully removed, to which 100 mL THF (tetrahydrofuran anhydrous, 99.9%) was added. The resultant slurry was continuously stirred for more than 5 min at 0°C (ice-water bath) before triethyl phosphonoacetate (10.14 g, 45.22 mmol) was added dropwise. After the addition of triethyl phosphonoacetate, the resulting solution was well-stirred at 0°C for 20 min until gas evolution ceased. 5-Bromovanillin acetate 2 (11.23 g, 41.11 mmol) dissolved in 10 mL THF was slowly added to the yellow solution in ice-water bath. The reaction mixture was kept stirring at 0°C for 1 h and monitored by TLC (n-hexanes/EtOAc, 5:1, v/v). After the reaction was completed, the excess agent was quenched by adding 1 M aqueous HCl solution to a pH value <3. Then, the reaction mixture was evaporated to remove THF. The resultant product was re-dissolved in EtOAc and water, and extracted with EtOAc (300 mL × 2). The combined organic phase was washed with distilled water and saturated NH4Cl solution, dried over anhydrous MgSO4, filtered, and concentrated under reduced pressure at 40°C. The total recovery of crude product was 13.45 g (39.33 mmol, 95.7%). The crude product was dissolved and recrystallized in n-hexanes/EtOAc (5:1, v/v) to obtain crystalline compound 3 with a yield of 70.9%. Compound 3, NMR (acetone-d6, Supplementary Figure 2) δH: 1.27 (3H, t, J = 7.09, Aγ-OCH2Me), 2.31 (3H, s, OAc), 3.92 (3H, s, OMe), 4.20 (2H, q, J = 7.13, Aγ-OCH2Me), 6.62 (1H, d, J = 15.96, β), 7.49 (1H, d, J = 1.81, A2), 7.55 (1H, d, J = 1.81, A6), 7.60 (1H, d, J = 15.96, α); δC: 14.53 (Aγ-OCH2Me), 20.21 (OAc), 56.87 (OMe), 60.90 (Aγ-OCH2Me), 111.64 (A2), 118.02 (A5), 120.81 (β), 125.08 (A6), 135.21 (A1), 140.10 (A4), 143.08 (α), 153.84 (A3), 166.68 (Aγ), 167.87 (OAc). Melting point, 104.5–106.0°C.
Compound 4 was produced via DIBAL-H (di-iso-butyl-aluminum hydride) reduction of ethyl 5-bromoferulate 3 in n-hexanes as previously described (Quideau and Ralph, 1992). Ethyl 5-bromoferulate acetate 3 (6 g, 17.54 mmol) was added into a 500 mL reaction vessel and dissolved in 50 mL cyclohexane after which it was kept stirring for 5 min in an ice-water bath (to make sure reaction mixture at 0 °C). DIBAL-H (105 mL, 1.0 M solution in n-hexanes) was slowly added into the vigorously stirring solution via syringe, reaction vessel was sealed immediately with plastic cap after the addition. The reaction mixture turned clear in about 10 min, and was kept stirring at 0°C (ice-water bath) for 1 h. After the consumption of starting material 3 (monitored by TLC), the reaction was carefully quenched by with ethanol (5 mL) added dropwise at 0°C and released the gas generated at the same time. Subsequent addition of 1 M aqueous HCl solution until the mixture turned clear and colorless. The final pH value of the solution was <3, and then extracted with EtOAc (200 mL × 2). Combined extracts were washed with saturated brine, dried over anhydrous MgSO4, filtered, and evaporated under reduced pressure at 40°C to result in compound 4 as light yellow oil with the isolated yield over 90%. Compound 4, NMR (acetone-d6, Supplementary Figure 3) δH: 3.88 (3H, s, OMe), 4.20 (2H, dd, J = 5.35, 1.68, γ), 6.29 (1H, dt, J = 15.90, 5.27; β), 6.47 (1H, dt, J = 15.90, 1.55; α), 7.06 (1H, d, J = 1.88, A2), 7.11 (1H, d, J = 1.88, A6); δC: 56.56 (OMe), 63.07 (γ), 108.86 (A2), 109.27 (A5), 123.58 (A6), 128.77 (α), 129.80 (β), 130.96 (A1), 144.29 (A4), 149.04 (A3).
5,5′-Bromopinoresinol 5 was then prepared via two different coupling reactions catalyzed by peroxidase or FeCl3 from 5-bromoconiferyl alcohol 4 (Figure 3).
5-Bromoconiferyl alcohol 4 (207 mg, 0.80 mmol) was dissolved in acetone (20 mL), to which phosphate buffer (200 mL, pH 5.0) was slowly added while kept stirring. (Note: We recognize that the buffering capacity is low for this system, but it is non-nucleophilic, so doesn't produce undesired quinone-methide-trapping products, and has been used reproducibly over many decades for radical coupling reactions, including for the preparation of synthetic lignins.) Then, H2O2-urea complex (38.2 mg, 1.02 eq. relative to the phenols) dissolved in buffer (~2 mL) was added into the acetone-buffer system and followed by the addition of peroxidase (0.1 mg in 1 mL buffer). The solution turned light yellow and then became cloudy immediately after the addition of peroxidase. The reaction mixture was continuously stirring at room temperature for 45 min until the starting material 4 disappeared (monitored by TLC, n-hexanes/EtOAc, 1:1, v/v). The reaction was quenched by adding 1 M aqueous HCl solution (2 mL) to pH <3. 4,4′-Ethylenebisphenol (20.9 mg, 0.098 mmol) was then added as internal standard for quantification, and the resultant mixture was extracted with EtOAc (80 mL × 3). The combined organic phase was washed with saturated brine, dried over anhydrous MgSO4, and concentrated under reduced pressure at 40°C. One part of the crude product mixture (20–30 mg dissolved in 0.6 mL acetone-d6) was used for NMR directly. The calculated yield of product 5,5′-bromopinoresinol 5 was 44.1%, based on 1H NMR integration. Another fraction of the products (about 55 mg) was loaded onto a 1 mm silica-gel plate and developed twice with EtOAc/n-hexanes (1:1). Each isolated product was characterized by NMR. Internal standard (5 mg) and 15 mg of 5,5′-bromopinoresinol 5 were isolated from the TLC plate. The yield of 5,5′-bromopinoresinol 5 was 30.4% after TLC purification.
5-Bromoconiferyl alcohol 4 (104 mg, 0.4 mmol) was dissolved in acetone (12.5 mL) to which phosphate buffer (125 mL, pH 5.02) was added. Then, iron (III) chloride (FeCl3, 65 mg, 1.0 eq. relative to the phenols) dissolved in buffer (1–2 mL) was added into the acetone-buffer system. The reaction system was kept stirring at room temperature for 60 min until TLC (n-hexanes/EtOAc, 1:1, v/v) showed no starting material 4 remaining. Then, 4,4′-ethylenebisphenol (10.51 mg, 0.049 mmol) was added into the reaction mixture as an internal standard. Following the same workup procedure as for the peroxidase-catalyzed reaction, the crude product was used for NMR measurement and TLC purification. The calculated yield of product 5,5′-bromopinoresinol 5 was 30.1% (based on 1H NMR integration) whereas the isolated yield was 20.4% (by TLC plate separation).
Scaled-up coupling of 5-bromoconiferyl alcohol 4 was carried out under the peroxidase-catalyzed condition as abovementioned. Briefly, 5-bromoconiferyl alcohol 4 (4.08 g, 15.75 mmol) was dissolved in acetone-phosphate buffer system (500 mL/2 L, v/v), followed by the addition of H2O2-urea complex (814 mg, 8.66 mmol) and peroxidase (10 mg) dissolved in the buffer, respectively. After the addition of peroxidase, the solution turned bright yellow immediately and then became a yellow slurry in 2 min. The reaction mixture was kept stirring at room temperature and monitored by TLC (n-hexanes/EtOAc, 1:1, v/v). The reaction was quenched when it was completed by adding 1 M aqueous HCl to pH <3 and then the acetone was evaporated. The resulting mixture was extracted with EtOAc (400 mL × 3). The combined organic phase was washed with brine, dried over anhydrous MgSO4, and concentrated under reduced pressure. The crude product was purified by flash-chromatography (Biotage, 100 g silica gel column × 2) using n-hexanes/EtOAc (1:1, v/v) to obtain compound 5,5′-bromopinoresinol 5. Compound 5 was then re-dissolved and crystallized from ethanol, with an isolated crystalline yield of 24.6%. Compound 5, NMR (acetone-d6, Supplementary Figure 4) δH: 3.10 (2H, m, β), 3.84 (2H, d, J = 3.68, γ1), 3.85 (6H, s, OMe), 4.22 (2H, m, γ2), 4.69 (2H, d, J = 4.10, α), 6.99 (2H, d, J = 1.86, A2), 7.11 (2H, dd, J = 1.88, 0.51; A6); δC: 55.03 (β), 56.60 (OMe), 72.29 (γ), 85.77 (α), 108.99 (A5), 109.38 (A2), 122.64 (A6), 134.99 (A1), 144.05 (A4), 148.91 (A3). Melting point, 210.2–211.5°C.
Pinoresinol 6 was produced via debromination of compound 5 under catalytic hydrogenation conditions at the presence of palladium on activated carbon (Pd/C, 10 wt% loading) and Et3N in methanol (Sajiki et al., 2002). 5,5′-Bromopinoresinol 5 (810 mg, 1.57 mmol) was dissolved in 80 mL methanol (1% solution of compound 5) with stirring, followed by the addition of Et3N (381.3 mg, 3.77 mmol, 1.2 eq. vs. bromine) and Pd/C (24.3 mg, 10% Palladium on activated carbon, 3% of the weight of the aromatic bromide). The resulting mixture was kept stirring under a hydrogen-filled balloon for 2 h until TLC (CH2Cl2/MeOH, 20:1, v/v) showed that all compound 5 had been consumed. The solid catalyst was filtered off using a polyamide membrane (Whatman, 0.2 μm). The resulting filtrate was evaporated to remove methanol, then extracted with distilled water and EtOAc (100 mL × 2). The combined organic phase was washed with saturated NH4Cl, dried over anhydrous MgSO4, and concentrated under reduced pressure at 40°C to obtain pinoresinol 6 (560 mg, 1.57 mmol) as a light-yellow oil with the isolated yield of ~100%. The obtained pinoresinol could be crystalized from the oily state (kept in refrigerator for a long time) or recrystallized in ethanol/n-hexanes and gave white crystals. Compound 6, NMR (acetone-d6, Supplementary Figure 5) δH: 3.07 (2H, m, β), 3.79 (2H, dd, J = 9.05, 3.80, γ1), 3.83 (6H, s, OMe), 4.19 (2H, m, γ2), 4.66 (2H, d, J = 4.32, α), 6.78 (2H, d, J = 8.01, A5), 6.83 (2H, dd, J = 8.15, 1.94; A6), 6.98 (2H, d, J = 1.93, A2), 7.53 (Ar-OH); δC: 55.16 (β), 56.14 (OMe), 72.12 (γ), 86.56 (α), 110.47 (A2), 115.46 (A5), 119.54 (A6), 134.08 (A1), 146.76 (A4), 148.21 (A3). The NMR data of compound 6 are consistent with the previously published data (Ralph S. et al., 2004). Melting point, 113.5–114.5°C.
As described above, pinoresinol can be produced by dehydrodimerization of coniferyl alcohol in lignin biosynthesis. Many synthetic routes have been proposed to make optically active or racemic pinoresinol via chemical or enzymatic syntheses by radical coupling of coniferyl alcohol (Brezny and Alfoldi, 1982; Vermes et al., 1991; Davin et al., 1992; Pare et al., 1994; Umezawa, 2003; Zhang et al., 2003; Kim et al., 2006; Saliu et al., 2011; Yue et al., 2012a; Tamura et al., 2014; Ricklefs et al., 2015). Currently, the most commonly used approaches for the synthesis of pinoresinol are using oxidative enzymes (peroxidase or laccase) to catalyze coniferyl alcohol dimerization, though with poor selectivity and yield. The highest yield of pinoresinol that can be achieved by coupling of coniferyl alcohol is reported to be no more than 13% (Ricklefs et al., 2015). The low yield and selectivity resulted in difficult isolation and purification of pinoresinol from the crude mixtures (Sih et al., 1976; Vermes et al., 1991; Davin et al., 1992; Fukuhara et al., 2013). Another reason for the high cost of pinoresinol is that the starting material (coniferyl alcohol) of these reactions is also expensive. On the other hand, syringaresinol that results from homo-coupling of sinapyl alcohol was at much better selectivity and yield. The logical reason is that the substitution by the methoxyl group on the aromatic C-5 position prevents coupling reactions at this site and therefore reduces the number of possible coupling routes, leading to higher selectivity (Vermes et al., 1991; Zhang et al., 2003). This indicates that an ideal way to improve the synthesis of pinoresinol is to suppress the undesired side-reactions by substitution at the C-5 position. We therefore decided to use the 5-bromo analog as a starting substrate anticipating the subsequent efficient elimination of aryl bromine.
Accordingly, an efficient synthetic approach was designed to synthesize pinoresinol with higher yield than that of the conventional radical coupling pathway from coniferyl alcohol (Figures 2, 3). 5-Bromoconiferyl alcohol 4 was used to make 5,5′-bromopinoresinol 5 via a peroxidase-mediated radical coupling reaction for the purpose of producing less complex product mixtures. In this study, we used 5-bromovanillin 1 as starting material as the bromine protects the aromatic C-5 position. Acetylation of 5-bromovanillin 1 using pyridine and acetic anhydride gave a quantitative conversation yield of bromovanillin acetate 2. Next, an HWE reaction (using triethyl phosphonoacetate) was carried out to produce ethyl 5-bromoferulate 3 from compound 2. The HWE reaction is a variation of the Wittig olefination reactions (Brandt et al., 1998). It is a chemical reaction of stabilized phosphonate carbanions with aldehydes (or ketones) to produce predominantly (E)-alkenes, and is most commonly applied to triesters of phosphono-acetic acid that leads to α,β-unsaturated esters. In contrast to phosphonium ylides used in the Wittig reaction, phosphonate-stabilized carbanions are more nucleophilic and more basic. Likewise, phosphonate-stabilized carbanions can be alkylated, unlike phosphonium ylides the dialkylphosphate salt by-product is easily removed by aqueous extraction (Maryanoff and Reitz, 1989). In this approach, excess triethyl phosphonoacetate was added to the reaction solution to ensure the complete conversion of compound 2 as the phosphonate can be easily removed during a workup procedure. After the reaction, the obtained product was pure enough for the next step without requiring further purification. The total recovery yield of ethyl 5-bromoferulate 3 achieved 95.7%, and the yield for crystalline products was 70.9%. The following step was DIBAL-H reduction of ethyl 5-bromoferulate 3 to produce 5-bromoconiferyl alcohol 4 with a yield of over 90%. DIBAL-H, being a liquid and miscible in numerous solvents, is a unique and versatile organometallic hydride that has been widely used as reduction reagent for the preparation of fine organic chemicals and pharmaceuticals (Self et al., 1990; Quideau and Ralph, 1992; Yue et al., 2012a). For the reduction of compound 3, DIBAL-H in n-hexanes (1.0 M solution in n-hexanes) solution was used instead of DIBAL-H in toluene. Although no significant difference on reaction activity between DIBAL-H hexane solution and toluene solution, using the n-hexanes solution simplifies the work-up procedure since it can easily form phase separation from water during extraction (Quideau and Ralph, 1992). It is important to note that the reaction should be slowly quenched with ethanol first, and then fully quenched with dilute HCl (1 M). Both ethanol and dilute HCl were added carefully and slowly while the reaction mixture was placed in ice-water bath and stirred vigorously. Ethanol was first added to degrade excess DIBAL-H and cleave the aluminum-oxygen bond. No precipitates can be observed during the ethanol quenching stage. Aqueous 1 M HCl was then slowly added and white precipitates quickly generated, and continued until the precipitates disappeared (normally when pH <3). In particular, the reaction during quenching process, to some extent, was delayed when adding ethanol and dilute HCl, Slow additions of ethanol and dilute HCl are therefore necessary to avoid the rapid generation of gas. Concentrated HCl is not recommended for the quenching process as it will destroy the product. This step provided a high yield and purity of 5-bromoconiferyl alcohol 4 in 90% of yield with almost 100% for purity. As the 5-bromoconiferyl alcohol 4 is not stable to long-term storage, it should be used immediately for the next coupling reaction.
For the synthesis of pinoresinol and syringaresinol, radical coupling of coniferyl alcohol and sinapyl alcohol catalyzed by peroxidase(s) in the presence of H2O2 are the traditional methods (Vermes et al., 1991; Davin et al., 1992; Zhang et al., 2003). In this study, for a better comparison purpose, coupling reactions of 5-bromoconiferyl alcohol 4 catalyzed by two different catalysts, peroxidase and FeCl3, were studied. Quantitative 1H NMR characterization showed that the yield of 5,5′-bromopinoresinol 5 catalyzed by peroxidase was 44.1%, which was much higher than that of coupling of coniferyl alcohol (at most 10–12%) (Vermes et al., 1991; Quideau and Ralph, 1994; Ricklefs et al., 2015). The 5,5′-bromopinoresinol 5 can be purified by TLC separation to give 30.4% isolation yield and by recrystallization in hexane/ethanol to give 24.6% crystal yield. For the radical coupling catalyzed by FeCl3, the yield of 5,5′-bromopinoresinol was 30.1% from 1H NMR, not 58% as reported previously (Brezny and Alfoldi, 1982). In this case, the isolated yield from TLC purification was 20%, accounting for about 66% of the total obtained 5,5′-bromopinoresinol, which was consistent with the case using peroxidase as catalyst.
By comparison, the yield of 5,5′-bromopinoresinol obtained via peroxidase-catalyzed radical coupling was higher, about 1.5 times, than that from FeCl3 catalyzed coupling. Nevertheless, in contrast to sinapyl alcohol, the β-β′ bond formation was not the dominant radical coupling pathway for the coupling reaction of 5-bromoconiferyl alcohol. More side-reactions than expected still occurred even with the aromatic C-5 position blocked by bromine, such as β-O-4 dimer, etc. (see Supplementary Figure 6). Additionally, it is worth pointing out that, the molar equivalents of H2O2 used and the reaction time are two key variables in the radical coupling reactions. There will be no pinoresinol formed if H2O2 was omitted, whereas the product will be polymerized (deeply colored precipitates) with sharply decreased pinoresinol yields (lower than 10%) as reaction time increased once the H2O2 was overcharged. In this study, 1.1 equivalents (relative to the phenols) of H2O2 were used to ensure full conversion of 5-bromoconiferyl alcohol 4 but to avoid further coupling reactions beyond dimerization. The reaction was quenched after 45 min to minimize formation of oligomers. After the normal workup procedure, crystalline product 5,5′-bromopinoresinol 5 was easily obtained after column purification.
In the last step, a mild and efficient one-pot method was modified and used for the hydrodebromination of 5,5′-bromopinoresinol 5 (Sajiki et al., 2002). The reaction proceeded at room temperature under catalytic hydrogenation conditions, which gave a nearly 100% yield of the required product. It is worth pointing out that the pinoresinol obtained after hydro-debromination was pure enough to crystalize without initial purification. No side-reactions occurred during the debromination, and the NMR spectra of the final product confirmed the near 100% purity of the pinoresinol.
In summary, we have developed a feasible method for the synthesis of pinoresinol with higher yield than that of previous studies. The formation of the β-β′ bond during radical coupling was greatly improved by using 5-bromoconiferyl alcohol, an aromatic compound in which the C-5 position is protected by bromine, over the conventional use of coniferyl alcohol. The yield of 5,5′-bromopinoresinol 5 (which can be quantitatively converted into pinoresinol) obtained via peroxidase-catalyzed radical coupling was much higher than that via an FeCl3-catalyzed reaction. With the relatively higher yield, it provides a feasible way to obtain pinoresinol on a large scale, which will benefit the various research activities on pinoresinol structural characterization in lignins, in human nutrition, and for pharmacological applications.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding authors.
FY designed, performed the synthesis and analysis, wrote the paper, and obtained funding in China. WL aided in the analysis and synthesized some starting compounds. FL designed the project and obtained funding in China. LZ and RS aided in the analysis. JR advised on experiments, aided in analysis, and obtained the funding in the US. All authors were involved in the writing and revisions.
The authors are grateful to the financial support for this work by the National Natural Science Foundation of China (31870560 and 21908072), Natural Science Foundation of Guangdong Province (2018A030313840), Guangxi Key Laboratory of Clean Pulp and papermaking and pollution control (KF201805-5), and the DOE Great Lakes Bioenergy Research Center (DOE BER Office of Science DE-SC0018409).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fenrg.2021.640337/full#supplementary-material
Brandt, P., Norrby, P. O., Martin, I., and Rein, T. (1998). A quantum chemical exploration of the Horner-Wadsworth-Emmons reaction. J. Org. Chem. 63, 1280–1289. doi: 10.1021/jo971973t
Brezny, R., and Alfoldi, J. (1982). Prins reaction in the synthesis of lignin model compounds: 3. Alternative synthesis of pinoresinol, coniferyl aldehyde and guaiacyl vinyl ketone. Chem. Zvesti 36, 267–276.
Davin, L. B., Bedgar, D. L., Katayama, T., and Lewis, N. G. (1992). On the stereoselective synthesis of (+)-pinoresinol in Forsythia Suspensa from its achiral precursor, coniferyl alcohol. Phytochemistry 31, 3869–3874. doi: 10.1016/S0031-9422(00)97544-7
Eberhardt, T. L., Bernards, M. A., He, L., Davin, L. B., Wooten, J. B., and Lewis, N. G. (1993). Lignification in cell suspension cultures of Pinus taeda: in situ characterization of a gymnosperm lignin. J. Biol. Chem. 268, 21088–21096. doi: 10.1016/S0021-9258(19)36897-8
Feng, S. M., Gan, Z. J., Zhai, X. F., Fu, P. F., and Sun, W. J. (2006). Content comparison of pinoresinol diglucoside in original and reborn bark of Eucommia ulmoides. Zhong Yao Cai 29, 792–794. doi: 10.3321/j.issn:1001-4454.2006.08.017
Feng, S. M., Ni, S. F., and Sun, W. J. (2007). Preparative isolation and purification of the lignan pinoresinol diglucoside and liriodendrin from the bark of Eucommia ulmoides Oliv. by high speed countercurrent chromatography. J. Liq. Chromatogr. Relat. Technol. 30, 135–145. doi: 10.1080/10826070601036324
Fukuhara, Y., Kamimura, N., Nakajima, M., Hishiyama, S., Hara, H., Kasai, D., et al. (2013). Discovery of pinoresinol reductase genes in sphingomonads. Enzyme Microb. Technol. 52, 38–43. doi: 10.1016/j.enzmictec.2012.10.004
Halls, S. C., Davin, L. B., Kramer, D. M., and Lewis, N. G. (2004). Kinetic study of coniferyl alcohol radical binding to the (+)-pinoresinol forming dirigent protein. Biochemistry 43, 2587–2595. doi: 10.1021/bi035959o
Jung, J. C., Kim, J. C., Moon, H. I., and Park, O. S. (2006). Stereoselective total synthesis of furofuran lignans through dianion aldol condensation. Tetrahedron Lett. 47, 6433–6437. doi: 10.1016/j.tetlet.2006.06.127
Kim, H. Y., Kim, J. K., Choi, J. H., Jung, J. Y., Oh, W. Y., Kim, D. C., et al. (2010). Hepatoprotective effect of pinoresinol on carbon tetrachloride-induced hepatic damage in mice. J. Pharmacol. Sci. 112, 105–112. doi: 10.1254/jphs.09234FP
Kim, J. C., Kim, K. H., Jung, J. C., and Park, O. S. (2006). An efficient asymmetric synthesis of furofuran lignans: (+)-sesamin and (-)-sesamin. Tetrahedron Asymmetry 17, 3–6. doi: 10.1016/j.tetasy.2005.11.007
Lewis, N. G., Kato, M. J., Lopes, N., and Davin, L. B. (1995). Lignans -diversity, biosynthesis, and function. Chem. Amazon 588, 135–167. doi: 10.1021/bk-1995-0588.ch013
Li, A. L., Li, G. H., Li, Y. R., Wu, X. Y., Ren, D. M., Lou, H. X., et al. (2019). Lignan and flavonoid support the prevention of cinnamon against oxidative stress related diseases. Phytomedicine 53, 143–153. doi: 10.1016/j.phymed.2018.09.022
Li, S. X., Shi, L. L., Wang, C., Yue, F. X., and Lu, F. C. (2020). Naphthalene structures derived from lignins during phenolation. Chemsuschem 13, 5549–5555. doi: 10.1002/cssc.202001693
Lin, B., Sun, L. N., Xin, H. L., Nian, H., Song, H. T., Jiang, Y. P., et al. (2016). Anti-inflammatory constituents from the root of Litsea cubeba in LPS-induced RAW 264.7 macrophages. Pharm. Biol. 54, 1741–1747. doi: 10.3109/13880209.2015.1126619
Maryanoff, B. E., and Reitz, A. B. (1989). The wittig olefination reaction and modifications involving phosphoryl-stabilized carbanions - stereochemistry, mechanism, and selected synthetic aspects. Chem. Rev. 89, 863–927. doi: 10.1021/cr00094a007
Milder, I. E., Arts, I. C., Van De Putte, B., Venema, D. P., and Hollman, P. C. (2005). Lignan contents of Dutch plant foods: a database including lariciresinol, pinoresinol, secoisolariciresinol, and matairesinol. Br. J. Nutr. 93, 393–402. doi: 10.1079/BJN20051371
Min, T. P., Kasahara, H., Bedgar, D. L., Youn, B. Y., Lawrence, P. K., Gang, D. R., et al. (2003). Crystal structures of pinoresinol-lariciresinol and phenylcoumaran benzylic ether reductases and their relationship to isoflavone reductases. J. Biol. Chem. 278, 50714–50723. doi: 10.1074/jbc.M308493200
Morreel, K., Ralph, J., Kim, H., Lu, F., Goeminne, G., Ralph, S. A., et al. (2004). Profiling of oligolignols reveals monolignol coupling conditions in lignifying poplar xylem. Plant Physiol. 136, 3537–3549. doi: 10.1104/pp.104.049304
Pare, P. W., Wang, H. B., Davin, L. B., and Lewis, N. G. (1994). (+)-Pinoresinol synthase - a stereoselective oxidase catalyzing 8,8′-lignan formation in Forsythia-Intermedia. Tetrahedron Lett. 35, 4731–4734. doi: 10.1016/S0040-4039(00)76953-X
Paska, C., Innocenti, G., Ferlin, M., Kunvari, M., and Laszlo, M. (2002). Pinoresinol from Ipomoea cairica cell cultures. Nat. Prod. Lett. 16, 359–363. doi: 10.1080/1057530290033123
Pellegrini, N., Valtuena, S., Ardigo, D., Brighenti, F., Franzini, L., Del Rio, D., et al. (2010). Intake of the plant lignans matairesinol, secoisolariciresinol, pinoresinol, and lariciresinol in relation to vascular inflammation and endothelial dysfunction in middle age-elderly men and post-menopausal women living in Northern Italy. Nutr. Metab. Cardiovasc. Dis. 20, 64–71. doi: 10.1016/j.numecd.2009.02.003
Quideau, S., and Ralph, J. (1992). Facile large-scale synthesis of coniferyl, sinapyl, and p-coumaryl alcohol. J. Agr. Food Chem. 40, 1108–1110. doi: 10.1021/jf00019a003
Quideau, S., and Ralph, J. (1994). A biomimetic route to lignin model compounds via silver (I) oxide oxidation. 1. Synthesis of dilignols and non-cyclic benzyl aryl ethers. Holzforschung 48, 12–22. doi: 10.1515/hfsg.1994.48.1.12
Ralph, J., Brunow, G., Harris, P. J., Dixon, R. A., Schatz, P. F., and Boerjan, W. (2008). “Lignification: Are lignins biosynthesized via simple combinatorial chemistry or via proteinaceous control and template replication?,” in Recent Advances in Polyphenol Research, eds. F. Daayf, A. El Hadrami, L. Adam, and G.M. Ballance (Oxford: Wiley-Blackwell Publishing), 36–66.
Ralph, J., Helm, R. F., Quideau, S., and Hatfield, R. D. (1992). Lignin-feruloyl ester cross-links in grasses. Part 1. Incorporation of feruloyl esters into coniferyl alcohol dehydrogenation polymers. J. Chem. Soc. Perkin Trans. 1, 2961–2969. doi: 10.1039/P19920002961
Ralph, J., Lundquist, K., Brunow, G., Lu, F., Kim, H., Schatz, P. F., et al. (2004). Lignins: natural polymers from oxidative coupling of 4-hydroxyphenylpropanoids. Phytochem. Rev. 3, 29–60. doi: 10.1023/B:PHYT.0000047809.65444.a4
Ralph, J., Peng, J. P., Lu, F. C., Hatfield, R. D., and Helm, R. F. (1999). Are lignins optically active? J. Agr. Food Chem. 47, 2991–2996. doi: 10.1021/jf9901136
Ralph, S., Landucci, L., and Ralph, J. (2004). NMR Database of Lignin and Cell Wall Model Compounds. Available online at: https://www.glbrc.org/databases_and_software/nmrdatabase/
Ricklefs, E., Girhard, M., Koschorreck, K., Smit, M. S., and Urlacher, V. B. (2015). Two-step one-pot synthesis of pinoresinol from eugenol in an enzymatic cascade. Chemcatchem 7, 1857–1864. doi: 10.1002/cctc.201500182
Sajiki, H., Kume, A., Hattori, K., and Hirota, K. (2002). Mild and general procedure for Pd/C-catalyzed hydrodechlorination of aromatic chlorides. Tetrahedron Lett. 43, 7247–7250. doi: 10.1016/S0040-4039(02)01622-2
Saliu, F., Tolppa, E. L., Zoia, L., and Orlandi, M. (2011). Horseradish peroxidase catalyzed oxidative cross-coupling reactions: the synthesis of “unnatural” dihydrobenzofuran lignans. Tetrahedron Lett. 52, 3856–3860. doi: 10.1016/j.tetlet.2011.05.072
Schroeder, F. C., Del Campo, M. L., Grant, J. B., Weibel, D. B., Smedley, S. R., Bolton, K. L., et al. (2006). Pinoresinol: a lignol of plant origin serving for defense in a caterpillar. Proc. Natl. Acad. Sci. U.S.A. 103, 15497–15501. doi: 10.1073/pnas.0605921103
Self, M. F., Pennington, W. T., and Robinson, G. H. (1990). Reaction of diisobutylaluminum hydride with a macrocyclic tetradentate secondary amine–synthesis and molecular-structure of [Al(I-Bu)]2[C10h20n4][Al(I-Bu)3]2–evidence of an unusual disproportionation of (I-Bu)2alh. Inorg. Chim. Acta 175, 151–153. doi: 10.1016/S0020-1693(00)84819-7
Sih, C. J., Ravikumar, P. R., Huang, F. C., Buckner, C., and Whitlock, H. Jr. (1976). Letter: Isolation and synthesis of pinoresinol diglucoside, a major antihypertensive principle of Tu-Chung (Eucommia ulmoides, Oliver). J. Am. Chem. Soc. 98, 5412–5413. doi: 10.1021/ja00433a070
Suzuki, S., and Umezawa, T. (2007). Biosynthesis of lignans and norlignans. J. Wood Sci. 53, 273–284. doi: 10.1007/s10086-007-0892-x
Tamura, M., Tsuji, Y., Kusunose, T., Okazawa, A., Kamimura, N., Mori, T., et al. (2014). Successful expression of a novel bacterial gene for pinoresinol reductase and its effect on lignan biosynthesis in transgenic Arabidopsis thaliana. Appl. Microbiol. Biotechnol. 98, 8165–8177. doi: 10.1007/s00253-014-5934-x
Touchard, F. P. (2004). New and efficient conditions for the Z-selective synthesis of unsaturated esters by the Horner-Wadsworth-Emmons olefination. Tetrahedron Lett. 45, 5519–5523. doi: 10.1016/j.tetlet.2004.05.026
Umezawa, T. (2003). Diversity in lignan biosynthesis. Phytochem. Rev. 2, 371–390. doi: 10.1023/B:PHYT.0000045487.02836.32
Vermes, B., Seligmann, O., and Wagner, H. (1991). Synthesis of biologically-active tetrahydro-furofuranlignan-(syringin, pinoresinol)-monoglucosides and bisglucosides. Phytochemistry 30, 3087–3089. doi: 10.1016/S0031-9422(00)98258-X
Yue, F., Lu, F., Sun, R., and Ralph, J. (2012b). Synthesis and characterization of new 5-linked pinoresinol lignin models. Chem. Eur. J. 18, 16402–16410. doi: 10.1002/chem.201201506
Yue, F., Lu, F., Sun, R.-C., and Ralph, J. (2012a). Syntheses of lignin-derived thioacidolysis monomers and their uses as quantitation standards. J. Agr. Food Chem. 60, 922–928. doi: 10.1021/jf204481x
Zhang, L., Henriksson, G., and Gellerstedt, G. (2003). The formation of β-β structures in lignin biosynthesis—are there two different pathways? Org. Biomol. Chem. 1, 3621–3624. doi: 10.1039/B306434D
Keywords: 5-bromoconiferyl alcohol, bromopinoresinol, coniferyl alcohol, radical coupling, lignin
Citation: Yue F, Lan W, Zhang L, Lu F, Sun R and Ralph J (2021) Efficient Synthesis of Pinoresinol, an Important Lignin Dimeric Model Compound. Front. Energy Res. 9:640337. doi: 10.3389/fenrg.2021.640337
Received: 11 December 2020; Accepted: 05 March 2021;
Published: 30 March 2021.
Edited by:
Liandong Zhu, Wuhan University, ChinaReviewed by:
Anna Kärkönen, Natural Resources Institute Finland (Luke), FinlandCopyright © 2021 Yue, Lan, Zhang, Lu, Sun and Ralph. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fengxia Yue, eXVlZnhAc2N1dC5lZHUuY24=; Fachuang Lu, ZmFjaHVhbmdsdUB3aXNjLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.