 Michele Aresta1*
Michele Aresta1* Angela Dibenedetto1,2*
Angela Dibenedetto1,2*- 1Interuniversity Consortium on Chemical Reactivity and Catalysis – CIRCC, University of Bari, Bari, Italy
- 2Department of Chemistry, University of Bari, Bari, Italy
The conversion of CO2 into added value chemicals, materials and fuels is a case of transition from the linear to the cyclic-C economy, a necessary change for stopping the putative negative effect of CO2 on climate and the environment. Several strategies can be implemented for CO2 conversion and their potential and timeframe is discussed in this perspective paper. The overall amount of avoided CO2 is evaluated in the short-, medium-, and long-term. The distinct contribution of Catalysis, Solar Chemistry and integrated Chemocatalysis-Biosystems is discussed.
Introduction
The continuous immission of anthropogenic-, nonbiogenic-CO2 in the atmosphere is hold liable of climate-change and extreme events we observe with higher frequency these days. In our opinion, such negative impact must be more correctly ascribed to the general scarce efficiency (Aresta and Dibenedetto, 2020) in the use of the fossil-C chemical energy we do [only an average 32% of the fossil energy is converted into usable energy (electrical-, mechanical-, thermal-energy), the rest ending to heat the atmosphere and our planet]. In fact, direct heating of the atmosphere is causing an increase of water transfer from soil to air (Dai, 2006) with water vapor having a stronger Green House Effect-GHE (at least twice) than CO2 (Dessler et al., 2008). The continuous increase of CO2 atmospheric level since the beginning of the industrial-age, is the witness and alarm light of our inefficient use of fossil-C and of our lavishness in the use of any form of energy (electric, thermal, mechanical). Nevertheless, whether the atmospheric level of CO2 is the protagonist of climate change or has a lower level play-role, whichever is(are) the real responsible for the impact on climate, the reduction of use of fossil-C is a necessity and an opportunity. A necessity for preserving natural resources that are not illimited (Table 1), an opportunity for making the best use (direct and indirect) of perennial (solar, wind, hydro, geothermal-SWHG) and renewable energy (biomass) in conjunction with CO2 use and C-recycling, stepping toward the Circular-C economy.
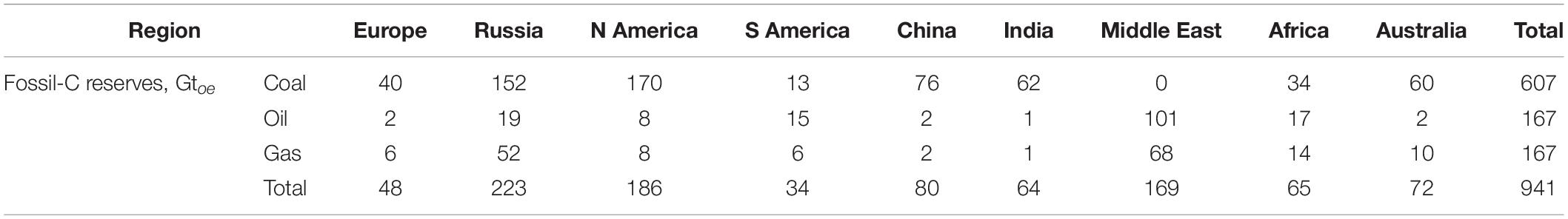
Table 1. Estimated worldwide fossil-C availability (Gtoe, toe = tons oil equivalent).
C-recycling means building a man-made C-cycle that can integrate the natural one and make available at a higher rate, than natural processes, and with a higher selectivity toward a single product, goods that are in our common use and will continue to be used for next generations. The exploitation of such ability will ensure longer usability of the limited natural resources (Table 1). The direct use of perennial SWHG energies is possible in a number of cases, but is not so general as fossil-C derivatives are. Fossil-C and their derivatives can be shipped all around the world and used, perennial energies are not (solar-, wind-, hydro, geothermal-power are not exportable) and their key derivative (electricity) has a limited distribution potential. Biomass has limitations to the direct utilization (emissions of particulate and noxious molecules such as N-aromatics and dioxins) and requires upgrading for the best use: anyway alone it cannot solve the problem also because its rate of combustion is much faster (1000–10,000 times) than the rate of formation and this will not solve the problem of CO2 accumulation in the atmosphere. Fossil-C derivatives (liquid and gaseous fuels) are ideal energy vectors (diesel and gasoline are the most concentrated form of solar energy, see Table 2) and, furthermore, are suited for being used in actual infrastructures, without requiring unviable worldwide economic investments.

Table 2. Energy density of some energy sources/vectors.
Do we have the possibility for converting CO2 on a large scale? Which energy sources we can exploit? Within which time-frame? What kind of products we can target?
This paper will make a survey of opportunities and possibilities, trying to put a timeframe to plausible targets and wrapping an all-inclusive conversion budget of CO2. Next paragraphs will each make the analysis of specific fields, highlighting barriers to overcome for a transition to a large-scale conversion of CO2, well beyond actual 220 MtCO2/y.
Noteworthy, in this Introduction we have used two different terms relevant to CO2 applications: utilization and conversion. They are not synonyms: conversion infers the chemical transformation of CO2 into other products such as polymers, materials, fine-chemicals, and fuels; utilization includes the conversion and other applications of CO2, such as the use of CO2 as a technical fluid without chemical conversion: additive to beverages, packaging including food conservation, low temperature generation, cleaning of surfaces, fire extinguisher, basic-waters treatment, water-wells regeneration, etc. These aspects will not be treated in this paper, interested readers can eventually look at Chapter 8 in Ref 1.
We recall here, that reactions of CO2 can be categorized in three main Classes which link the status of the CO2 molecule to the energetics of reaction, namely: (i) Processes (either exoergonic or moderately endoergonic), in which the entire CO2 molecule is incorporated into chemicals (carbonates, both organic and inorganic; polycarbonates; carbamates and polyurethanes). The oxidation state of C remains equal to 4; (ii) Average energy processes, with formation of C-C bonds without hydrogen consumption. The oxidation state of C is reduced to 3; (iii) Processes requiring both energy and hydrogen. The oxidation state of C goes down until to reach -4 in CH4.
In a “energy frame” based on “fossil-C” only the exploitation of Class (i) reactions makes sense, in a “energy frame” that includes substantial contributions from perennial energies, Class (iii) processes are feasible (Aresta et al., 2013a). And this is the great change, which revolutionizes the CO2 problem. CO2 is a resource not a waste (Aresta and Forti, 1987; Aresta, 2003, 2010). We have been firm supporters of such strategy and already in 1989 we elaborated a strategic approach to CO2 utilization, based on the use of solar energy (Figure 1). The conversion of large volumes of CO2 had no sense in the past (the perennial and renewable energies contribution to the entire energy budget was very limited), but it is now at the center of the scientific challenge for the future because perennial energies are entering the energy market with growing weight. Using solar energy for coprocessing water and CO2 allows to convert two very stable molecules into useful products, such as energy-rich hydrocarbons or their derivatives, plus oxygen, that can be separately collected and marketed. This is the future, that is fully mimic of natural processes.
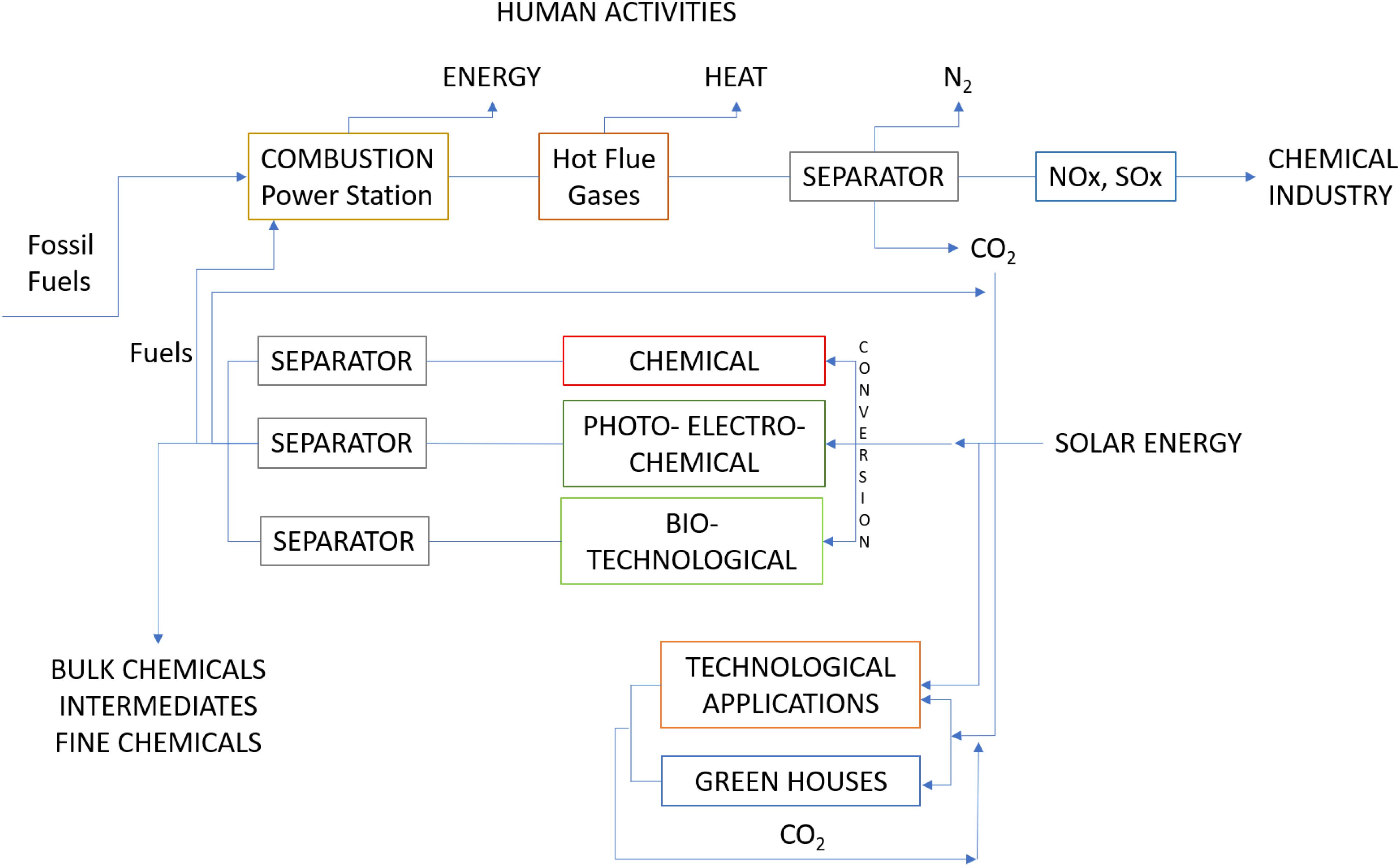
Figure 1. Utilization of CO2 under solar energy powering of processes (Aresta and Forti, 1987).
Low Energy Processes for CO2 Conversion
The utilization of CO2 for the synthesis of urea from ammonia (NH3) and CO2 (Woehler, 1828), and salicylic acid from phenol (Kolbe, 1860) are the oldest industrial processes known. But the process based on CO2 utilization with longer life is the plaster lime, made from quicklime and water, used to cover walls. By exposition to the atmosphere, calcium hydroxide [Ca(OH)2] reacts with CO2 and makes Ca(HCO3)2, then converted into CaCO3. This technique is very old, used most likely first by Jordans, then by Egyptians, Romans up to our times, passing through the exceptional uses Venetians made of it during Renaissance.
All just mentioned processes maintain the intact CO2 moiety in the final product, as also do carbamates (RHN-CO2R’) easily formed from amines (aliphatic, primary and secondary), and polymers (polycarbonates and polyurethanes). Such uses have been moderately exploited during last decades, in an energy frame based on fossil-C, as they are characterized by a negative Gibbs free-energy (inorganic carbonates, carbamates or polymers), or slightly positive (organic carbonates) in the circle in Figure 2.
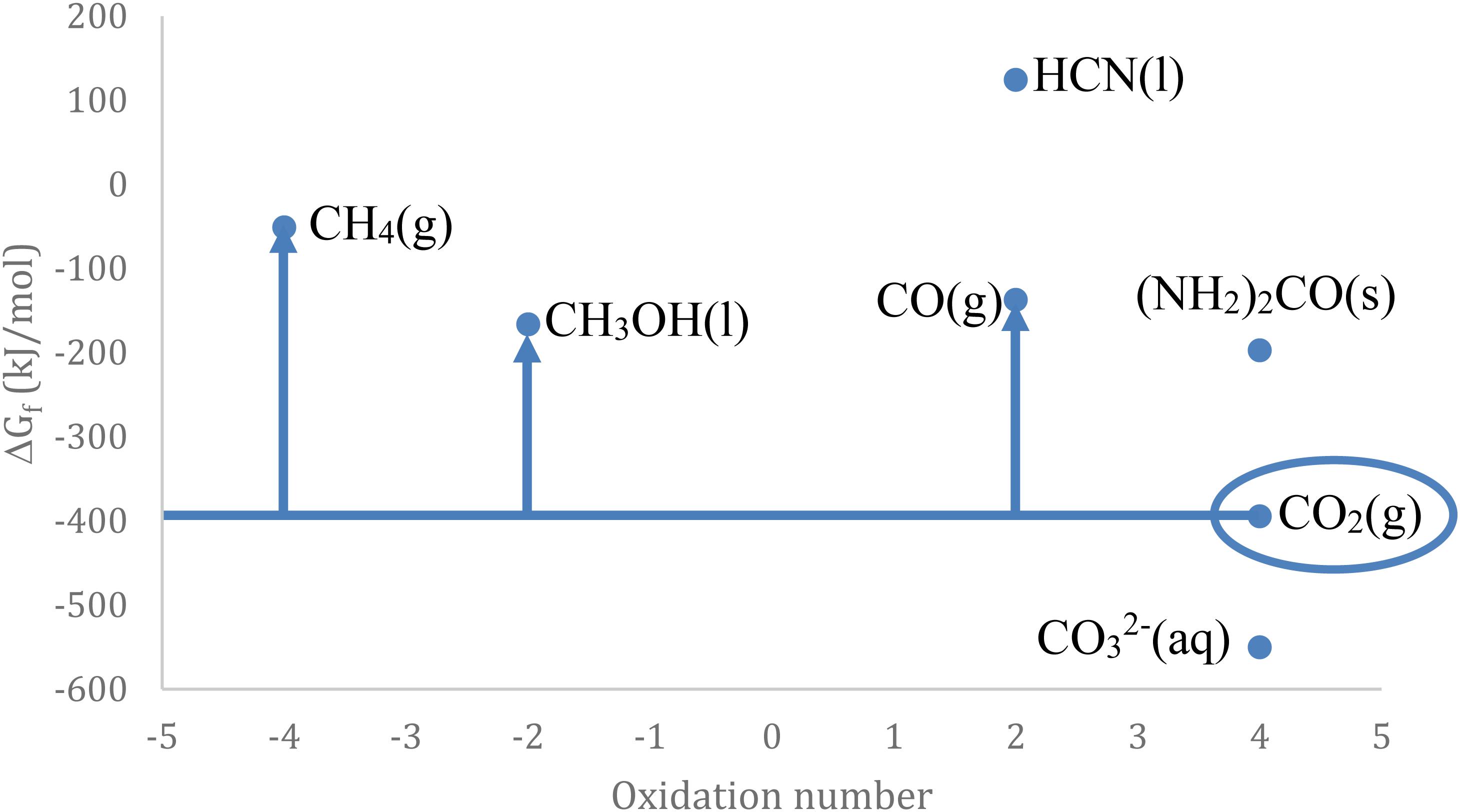
Figure 2. Gibbs free energy for some C1 species.
The total amount of CO2 used today is around 220 Mt/y, most of it used in the synthesis of urea (over 137 Mt/y), followed by inorganic carbonates and left with CO in the synthesis of methanol. The urea world market has reached 187.8 Mt in 2019 with a foreseen CAGR of 2% until 2025.1 To such amount, some 20 Mt/y must be added as technical fluid in applications such as: metal cleaning in cuttings and soldering, electronic industry, dry-cleaning, cooling agent, food industry (preservation during transport or stockage and modified packaging atmosphere-MPA), fire extinguisher, water-wells restauration, water treatment, green houses (for rising the atmospheric CO2 concentration up to 600 ppm) and Enhanced Oil Recovery-EOR. In the latter applications, even if CO2 is vented at the end of use, there is a large benefit because it substitutes products that would have a much larger Climate Change Power-CCP (for example, ChloroFluoroCarbons-CFCs have a CCP some > 10,000 times higher than CO2) (Aresta and Dibenedetto, 2020, Ch4). However, some 240 MtCO2/y are used today, mainly (92%) in the chemical industry. (The Catalyst Group Resources [TGCR], 2012) Per se it is a considerable amount, but it represents a drop in the ocean of the emitted CO2 (35 000+ Mt/y).
In the short term (5–7 years) this amount may grow to 350 MtCO2/y if CO2-based synthetic technologies will be promoted. In the long term (2040) the market of CO2 for chemicals may grow to some 1000 Mt/y, making a difference (Aresta et al., 2018). It is worth to recall, at this point, that what really matters is not the amount of CO2 converted but the amount of “avoided” CO2, e.g., of CO2 “not emitted”. In fact, the overall objective is to reduce the CO2 emission, a target that can be reached by reducing the “Carbon Footprint-CF” of a product or a process. The total reduction is given by the sum of the amount converted plus the amount not emitted because of the process improvement in terms or energy and waste production reduction. This amount represents the “avoided CO2” and is calculated by using the Life Cycle Assessment-LCA methodology, (Aresta and Galatola) by comparing a process on stream to an innovative one based on CO2 utilization. The avoided CO2 is in the range of 2,3.5, with a good average around 2.8, for a CO2 based process with respect to a process on stream not based on CO2. Therefore, if we apply such coefficient to the estimated amount of CO2 used in the synthesis of chemicals at 2040, considering those chemicals today not made from CO2 [1000 – (urea+carbonates)] it comes out that to 1000 MtCO2 converted correspond some 2500 MtCO2 avoided, that is 8% of the amount today emitted. This makes sense and suggests that the use of CO2 may help to considerably reduce its emissions and make more sustainable the chemical industry.
There are some CO2-based processes which are highly wished at the industrial level because they would substitute either processes which use toxic raw materials (COCl2, used in the synthesis of carbonates and carbamates) or implement direct syntheses, avoiding large waste production (synthesis of acrylates or carboxylates). We shall appraise briefly synthetic methodologies relevant to chemicals with a market over 1 Mt/y, namely: acrylates, carbamates and polyurethanes, carbonates and polycarbonates, and specialty chemicals, highlighting the benefits of using CO2.
Acrylates
Acrylates (RCH = CHCO2R’; R, R’ = H, alkyl moiety) are compounds with a market of some 6 Mt/y; they find utilization in a variety of different fields, from polymers, to varnishes and disposable napkins. Actually, acrylic acid is prepared by stepwise propane oxidation (Eq. 1, left) while could be prepared by a one-step coupling of ethene and CO2 (Eq 1, right):
The use of Pd (Aresta et al., 2007; Limbach et al., 2012; Li et al., 2018) instead of historical Mo (Carmona et al., 1985) or Ni (Hoberg et al., 1987; Papai et al., 2004) has allowed to rise the TON to 10 (Aresta et al., 2007; Limbach et al., 2012)-100 (Li et al., 2018) from 1 (Carmona et al., 1985): a lot there is to do for rising the one-step process to application level that would bring a substantial change in this field with a large atom economy and waste reduction considered that the oxidation process has a large C-footprint due to its low selectivity. This case can be used for highlighting the difference between “used” and “avoided” CO2. The used CO2 is one mol per mol of acrylate (Eq. 1 right), while the avoided CO2 is the sum of such amount plus the non-emitted CO2 because the CF of the synthetic methodology is neatly improved stepping from the old process (high temperature and low selectivity in oxidation) to the new one (low temperature and CUF = 1).
Carbamates and Polyurethanes
Both title compounds are characterized by bearing the moiety -NH-CO2−. Monomeric carbamates are made by reacting amines with phosgene and alcohols (Eq. 2, left), while could be made by substituting the more reactive and toxic phosgene with CO2 (Eq. 2, right) (Aresta and Quaranta, 1997, Aresta et al., 2016b).
Even organic carbonates can be used for making carbamates by reaction with amines (Eq. 3)
Polyurethanes are made by copolymerization of di-isocyanates, made from dicarbamates, (Eq. 4) with diols (Eq. 5)
All such products can be made by using CO2. Due to its inertness, reactions based on CO2 require active and selective catalysts, so to work at the lowest temperature while lowering the waste burden of synthetic processes.
Carbonates and Polycarbonates
These compounds are discussed elsewhere in this SI (Tomishighe et al., this SI). Here we wish to emphasize that the use of CO2 in the synthesis of both linear (Eq. 6) or cyclic (Eq. 7) is greatly beneficial as reduces the environmental impact on a number of categories (Aresta and Galatola, 1999). Kinetic and thermodynamic issues have to be yet resolved for a practical utilization of Eqs. 6 and 7.
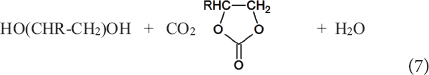
Water is the only co-product in both Eqs. 6 and 7. It may affect the catalyst and shifts the equilibrium to the left. Water elimination has been achieved by using organic [Aresta et al., 2005 and Tomishige et al., 2020, this (SI)] or inorganic water traps (Lercher et al., 2018). The use of cyanides (RCN >> RC(O)NH2) as water trap (Tomishige et al., 2020) requires energy for post-process product separation by distillation; while the use of carbodiimides (RN = C = NR) (Aresta et al., 2005) has the advantage of a spontaneous separation from the reaction medium of the hydrated form urea (RHN-C(O)-NHR). All three processes above demand energy for recovering the water trap for re-use. The use of pervaporation membranes for water separation is a promising technique (Aresta et al., 2012) that has a lower energy demand (Aresta et al., 2017a).
Another approach to cyclic carbonates is the oxidative carboxylation (Aresta et al., 2000; Aresta and Dibenedetto, 2002; Hutchings et al., 2018; Wang et al., 2020) of olefins that has to win the drawback of olefin loss by radical oxidation.
Specialty Chemicals
The direct carboxylation of organic substrates (Aresta et al., 2018) may allow making complex structures shifting from “several steps-several pots” to “one step-one pot” processes with a lot of simplifications and substantially lowering the Carbon Footprint-CF and waste production of such processes. In general, such reactions imply a C–H activation, a non-simple process to achieve by thermal catalysis but more feasible under photochemical conditions (Aresta et al., 2015, 2017b). Actually, the formation of a carboxylic moiety passes through the oxidation of organic moieties (-CH3, Eq. 8) or partial oxidative destruction of cyclic structures (naphthalene to dicarboxylic acids, Eq. 9) or even hydrolysis of cyanides (Eq. 10) or use of Grignard reagents (Eq. 11) or carbonylation processes using CO.
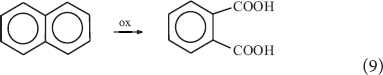
All such processes are not C-effective and have a high environmental impact. The direct carboxylation of organic substrates would be a winning option (Eq. 12) as a “one step-one pot” reaction.
The direct carboxylation of organic substrates can find application in many fields for the production of large-scale chemicals, such as long-chain carboxylic acids (potential market >10 Mt/y as surfactants and additives to product formulations), and specialty chemicals used in the pharmaceutical industry. Noteworthy, more complex if the structure of a chemical higher is the E-factor associated to its production (the E- factor is the ratio twaste produced/tuseful product that grows with the complexity of a chemical and the number of steps necessary for its production). Therefore, introducing a carboxylic moiety through a multistep process into a complex organic molecule such as an active principle may cause the generation of large volumes of waste-C. Direct syntheses can be more effective, reduce the amount of waste, lower the CF and the overall environmental impact.
High Energy Processes for CO2 Conversion
However, it will be the use of CO2 as source of carbon for making fuels that will produce a big change because the fuels market is 15 times that of chemicals. In an energy frame based on fossil fuels, does not make sense to produce fuels from CO2 because the production of the necessary hydrogen will emit more CO2 than is fixed. For making a CnH2n+2 hydrocarbon from CO2, nCO2 will be converted using (n+1)H2 that will emit (n+1)CO2 assuming that H2 is made in a Syngas production frame, according to reaction 13, the known Water Gas Shift Reaction-WGSR:
To such amount, the CO2 emitted for producing the necessary thermal energy for driving the synthetic and separation processes must be added, making the whole conversion a non-sense. In the medium-long term a new paradigm will be possible (Aresta et al., 2014a; Aresta et al., 2013a), as H2 necessary for CO2 reduction would be generated from water by electrolysis using PV. By 2040, the amount of PV power installed will grow to an estimated 3500-4500 GW, with respect to ca. 300 GW today. This will make the difference. The proof of concept is given by the already existing pilot or mini-demo plants2 that use PV-H2 for the methanation of CO2. Today the cost of PV-H2 is some 2.5,3 times higher than that of H2 from Methane Reforming-MR. Such difference can be leveled in time by the increased efficiency of future solar cells (from actual 20 to 40+% efficiency), their lower cost (ca. −50%) and longer resistance: the recent history of PV supports such trend. By 2040, PV-H2 and MR-H2 might have the same or very close price, allowing the large-scale industrial exploitation of the methanation reaction for which the development of large scale and robust electrolyzers is necessary. Having the correct infrastructure and non-fossil hydrogen at low cost, it will be possible to reach a significant capacity of conversion of solar energy into fuels (StF), with a net conversion of fossil-CO2. Are we getting closer to a “Man-made C-Cycle”? Sure, we are.
In fact, we can take CO2 directly from the atmosphere (DAC, Direct Atmosphere Capture, [Wang and Song, 2019]) and convert it into fuels using solar energy: this is mimic of Nature, with the advantage that the Man-Made cycle may be faster than the natural one and even more efficient as light use efficiency toward a single product, even if the efficient use of energy may not be an issue considered the large availability of solar energy. In such approach, it is compulsory that the energy necessary must come from perennial sources: it does not make sense use fossil-C. As for today, it is possible to carry out the conversions categorized below with the specific reported efficiencies:
| Solar energy to electricity (StE) | 20% |
| Electricity to H2 (electrolysis of water) (EtH) | 90% |
| Hydrogen to fuels (CO2 hydrogenation) (HtF) | 70% |
| or an overall Solar to Fuels (StF) | 12.6% |
That is much higher than plants (1.8–2.2%) or even microalgae (6–10%), the best solar light utilizers.
Therefore, the Man-Made C-cycle targeting fuels such as methanol and methane is at our hand: it could be exploited in short term even if not yet at costs competitive with fossil-C based technologies, and in the medium term at close to market costs. The conversion of CO2 into long-chain hydrocarbons is also an important objective that is under deep investigation and may give a strong contribution to C-recycling through CO2 conversion. Parameters such as selectivity toward target products and chain elongation are still at low TRL (2–3). They need to be improved so to bring the process to a marketable level.
What we have seen so far is based on pure chemo-catalysis and thermal processes, even if combined with PV-hydrogen production. Such approach has a limitation in the fact that it still has some issues for what concerns the storage and use of H2. Electrolytic hydrogen is already produced today, amounting to a few units per cent of the total amount: most of H2 comes from MR (85%). The real innovation discussed above is in the use (not yet economically viable, as already specified) of PV in electrolysis. Electrolytic hydrogen is produced at ambient pressure, it needs to be stored (as liquid H2) and distributed to the utilizers (industrial processes) which need pressurized (3.0-6.0 MPa) H2. The discontinuity of PV generates a mismatch between the production and utilization phases of H2 and storage of H2 is necessary (unless electrolyzers are integrated into the electric grid and use fossil-C generated electricity during dark). We do not consider here the transport of H2, because in the ideal case production of hydrogen, capture of CO2, hydrogenation of CO2 should be clustered on the same site and transport would be internal to the site. The storage of H2 is not a trivial operation and it requires that high safety standards are met: such safety issues play a fundamental role in moving from a pilot plant to a full-scale plant, and even more when passing to a generalized, disseminated use of the option. The fact that H2 is produced, rises the comments of some people about: “Why should we lose 30% efficiency in the conversion to C-based fuels? Let us use H2 directly”. This legitim comment finds an answer in the fact that the loss of efficiency is more than compensated by the fact that we can continue to use the same infrastructures, avoiding huge investment costs and reducing risk.
Are there alternative options to H2-production? Yes: we can avoid producing hydrogen. This specific aspect, that is really innovative, will be discussed in next section.
Integration of Photo-Catalysis and Biotechnology
Avoiding the production, storage and distribution of hydrogen means to move from the use of solar energy for “water splitting” (typical approach of the hydrogen scientific community, so far) to the use of solar energy “mimicking nature”, i.e., “co-processing CO2 and water” affording directly reduced forms of CO2, without H2 production. This shift requires a high ability to implement the “proton coupled to electron transfer-PCET” (“H+ + e–” transfer to CO2 with parallel O2 formation) in water utilization as reducing agent of CO2. The direct use of solar energy in driving chemical processes was foreseen by the Italian Chemist Giacomo Ciamician at the beginning of 1900 (Ciamician, 1912). A new paradigm, that changes the rules of the play and opens the way to a completely different approach to problem solving, leaving apart the old schemes. Nature teaches that for reducing CO2 it is not necessary to generate hydrogen (Dibenedetto, 2016). However, it is PCET that must operate more than water splitting. The use of engineered semiconductors, coupling n-p-type materials for avoiding exciton collapse is the future. Having stable and efficient photocatalysts will allow CO2 and water co-processing to energy products. Such approach is under investigation since long time, with alternate fortune. In the 1980s the light efficiency was around 0.1 % max (Mackor et al., 1987). Today, the literature shows best cases with > 10% light efficiency, (Hagfeldt and Graetzel, 1995; Esswein and Nocera, 2007; Aresta et al., 2016a) paving the way to a possible application.
A singular approach is the integration of photocatalysis with biotechnology, that means with the use of biosystems, either enzymes or whole microorganisms, for CO2 reduction in water driven by the injection of electrons generated by using solar energy.
Hybrid bioinspired systems have been used for the enzymatic reduction of CO2 to CH3OH in water at ambient conditions (Aresta et al., 2014b). The 6e–-reduction of CO2 to CH3OH is carried out by three NADH-dependent enzymes (Formate dehydrogenase-FateDH, Formaldehyde dehydogenase- FaldDH and Alcohol dehydrogenase-ADH) (Figure 3) co-encapsulated into an alginate matrix. As Figure 3 shows each step uses one NADH as donor of 2e– and protons: in total 3 mol of NADH are used per each mol of CH3OH. Unless NADH is regenerated, this dream reaction is not economically viable.
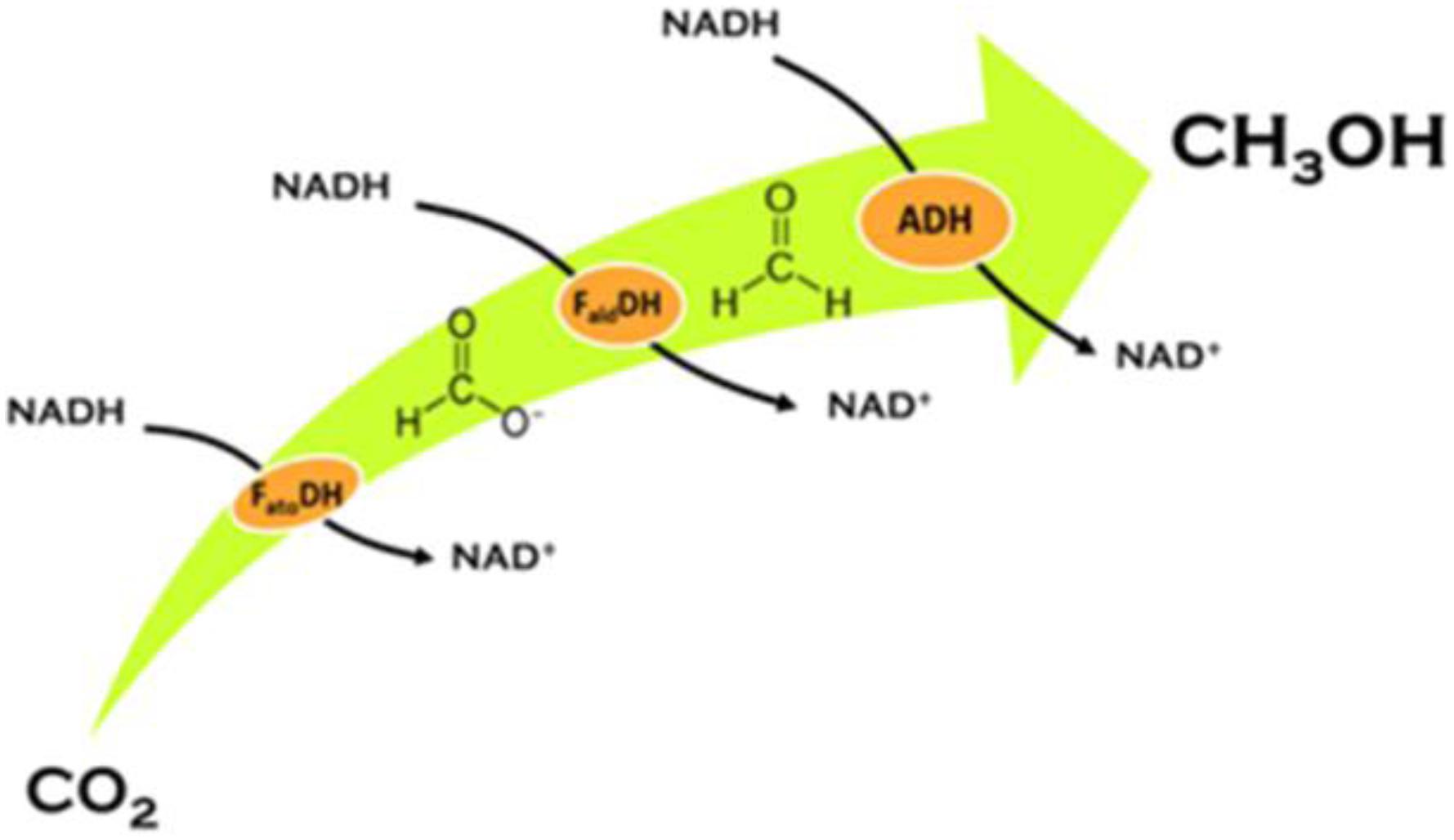
Figure 3. CO2 reduction to methanol by three dehydrogenase enzymes (Aresta et al., 2014b).
An in situ photocatalytic reduction of NAD+ to afford NADH, using semiconductors such as Cu2O, InVO4, TiO2 modified either with the organic compound rutin, or with the inorganic complex [CrF5(H2O)]2–) (Aresta et al., 2013b) and the doped sulfide Fe/ZnS has been carried out. The efficiency of the regeneration process is enhanced by using a RhIII-complex for facilitating the electron and hydride transfer from the H-donor (water or a water-glycerol solution) to NAD+. In this way one mol of NADH was used for producing over 100 mol of CH3OH, opening the way to a practical application (Aresta et al., 2016b). The bottleneck in this process is the gap between the rate of consumption and that of regeneration of NADH, an issue still under investigation in our Lab. Following such idea and using catalytic electrodes for heterogeneous electrochemical CO2 reduction, avoiding the use of NADH, co-encapsulated enzymes and co-factors were immobilized (Figure 4) on a carbon felt electrode (Schlager et al., 2017) with direct injection of electrons into them.
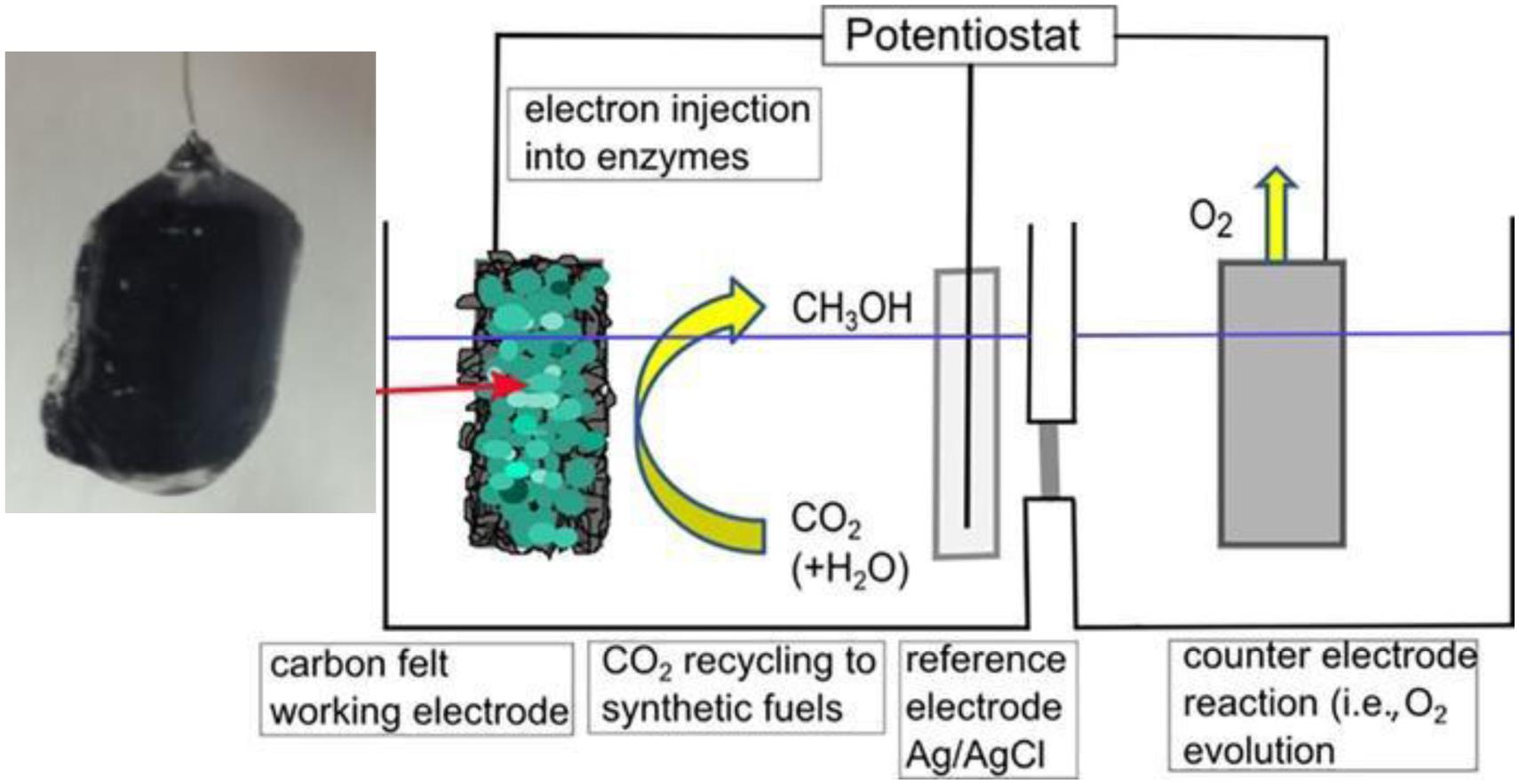
Figure 4. Electrochemical cell for electroenzymatic CO2 reduction.
CO2 is reduced to methanol at the working electrode (Figure 4) while oxidation reactions take place at the counter electrode. The bottleneck of this approach is the life of enzymes.
Other approaches have been followed by other research groups based on either the use of enzymes for NADH regeneration or on electrochemical NADH regeneration, with alternate fortune (Cazelles et al., 2013; Ullah et al., 2015).
Coupling of electrodes as source of electrons and more robust (than enzymes) whole microorganisms has been attempted in the last decade with the intent of reducing CO2 to energy products. So, electrodes and hydrogenophilic methanogenic cultures have been used for the direct and indirect extracellular electron transfer for CO2 reduction to methane (Villano et al., 2010). Such systems are known as BioElectrochemical Systems-BES (see the paper by Pant, this SI) that are rising great expectations as a new and sustainable technology for generation of energy and useful products from waste (Blankenship et al., 2011; Lovley and Nevin, 2013). In such devices, bacteria interact with solid-state electrodes (covered with semiconductor materials so that electricity is generated by shining light on the electrode surface) ad exchange electrons with them, either directly or via redox mediators. Microbial fuel cells (MFCs) and microbial electrolysis cells (MECs) are examples of a BES.
Combining the advantages of solar chemistry and BES could be the way to solve problems related to the use of metal enzymes, trying to make profit of their high conversion rate and selectivity. A “two-step” approach for CO2 reduction to methane has been recently proposed, that electrochemically converts CO2 into formate, used as substrate for methanogen Methanococcus maripaludis (Huang and Hu, 2018).
BES can be integrated into several processes. For example, biomass is converted into ethanol with less than 50% yield, co-producing liquid and solid waste rich in organics (including lignin) that could be advantageously used. Residual C6 and C5 sugars can be used as substrate in anodic oxidation in BES coupled to other reduction systems, or can be converted to higher energy products.
Even the large-scale production of biodiesel from palm-oil can be integrated with BES. In fact, the production of biodiesel from oils and fats is energy intensive. For example, for 100 kg of biodiesel produced, 5.3 kg of oily waste and 10.7 kg of glycerol are co-produced, while 466 MJ of energy are consumed. BES could be used for the conversion of watery glycerol either into propanediol (used as source of monomers for polymers (Aresta et al., 2013c) or ethanol, (Ito et al., 2005) or even hydrogen (Aresta and Dibenedetto, 2009).
A further step toward hi-tech applications is the building of “hybrid” systems integrating chemo- and-enzymatic catalysis (Dumeignil et al., 2018). Such approach aims at extreme simplification of reactive systems by developing one single device that incorporates chemo-catalysts and enzymes for a single step-single pot reaction targeting high selectivity and yield. This is not a easy-to-build system, as enzymes often suffer (can be deactivated) the presence of chemicals: in order to have effective systems, high complexity must be win in building both the integrated catalytic system and the reactor.
Conclusion
Innovative technologies are pushing the shift from the linear to C-circular economy (Dibenedetto and Nocito, 2020). Such trend is being more and more integrated with Bio-economy. The “Linear economy” is based on a non-natural attitude: the consumerism (take natural resources-convert into goods-use-throw away), that generates plentiful waste that impacts natural systems and our life. The “Cyclic economy” is close to Nature: like Nature, that does not produce waste, the cyclic-economy recycles used goods, lowering both the extraction of natural resources and the environmental burden of human activities. Process integration will favor this trend.
Processes based on hybrid systems may in the future take the place of pure chemocatalytic-thermal processes, with saving of large amounts of fossil-C and reduction of CO2 emission. “Cheap and clean” electrons generated by the use of perennial SWHG energies will supplant “dirty” thermal-electrons. CO2 is renewable carbon, as Nature teaches. The conversion of CO2 into chemical reactors is faster than both the natural conversion into added value products and the fossilization of C through biomass capture in soil. The integration of chemo-catalysis with bio-systems is happening with increasing intensity. Circular-Economy and Bio-economy are two faces of the same medal, are the strategies that will drive the world economy in future years and both aim at C-recycle through CO2 conversion. The implementation of all tools we have in our hands, and of the new ones that will be discovered, will allow the conversion of ca. 9-10 GtCO2/y, as depicted in Figure 5 and this is a great target.
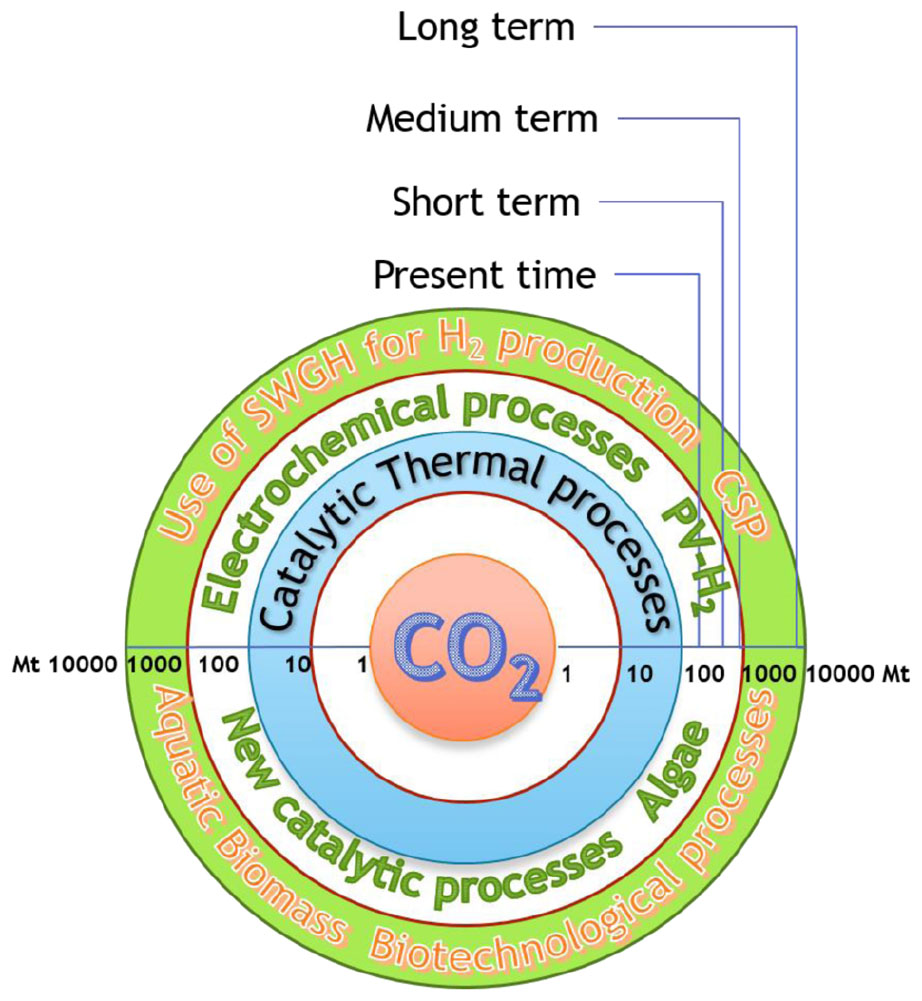
Figure 5. Stepwise implementation of strategic technologies for CO2 conversion, innovative processes and amount of CO2 converted into added value products, fuels and materials.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Author Contributions
MA and AD have contributed on the conceptualization and writing of the manuscript. AD has the responsibility on data collection while MA analyzed the data and provided feedback on the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was carried out with the financial support of MiUR - PRIN 2017, CO2Only, 2017WR2LRS.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
IC2R, METEA, University of Bari and CIRCC are acknowledged for funding.
Footnotes
- ^ https://www.expertmarketresearch.com/reports/urea-market
- ^ https://www.caranddriver.com/news/a15370096/audi-opens-first-e-gas-synthetic-fuel-production-facility/
References
Aresta, M., and Dibenedetto, A. (2002). Carbon dioxide as building block for the synthesis of organic carbonates: behavior of homogeneous and heterogeneous catalysts in the oxidative carboxylation of olefins. J. Mol. Catal. A 182–183, 399–409. doi: 10.1016/s1381-1169(01)00514-3
Aresta, M., and Dibenedetto, A. (2009). “Energy from organic waste: influence of the process parameters on the production of methane and hydrogen,” in Catalysis for Sustainable Energy Production, eds P. Barbaro and C. Bianchini (Hoboken, NJ: Wiley).
Aresta, M., Dibenedetto, A., and Angelini, A. (2013a). The changing paradigm in CO2 utilization. J. CO2 Util. 3–4, 65–73. doi: 10.1016/j.jcou.2013.08.001
Aresta, M., Dibenedetto, A., Di Bitonto, L., and Dubois, J. L. (2013b). Synthetic Process of Trimethylene Carbonate From 1,3-Propanediol and Urea by Heterogeneous Catalysts Patent No: EP 13192912.7.
Aresta, M., Dibenedetto, A., Macyk, W., and Baran, T. (2013c). Photocatalysts Working in the Visible Region for the Reduction of NAD+ to NADH within an Hybrid Chemo-enzymatic Process of CO2 Reduction to Methanol Patent No: MI2013A001135.
Aresta, M., Dibenedetto, A., and Angelini, A. (2014a). From CO2 to chemicals and fuels. Chem. Rev. 114:1709.
Aresta, M., Dibenedetto, A., Baran, T., Angelini, A., Łabuz, P., et al. (2014b). An integrated photocatalytic-enzymatic system for the reduction of co2 to methanol in bio-glycerol-water Beilstein. J. Org. Chem. 10, 2556–2565. doi: 10.3762/bjoc.10.267
Aresta, M., Dibenedetto, A., Angelini, A., and Ethiraj, J. (2012). Synthesis, characterization and use of Nb(V)/Ce(IV)-mixed oxides in the direct carboxylation of ethanol with water removal with pervaporation membranes. Chem. A Eur. J. 18, 10324–10334. doi: 10.1002/chem.201201561
Aresta, M., Dibenedetto, A., Baran, T., Wojtyła, S., and Macyk, W. (2015). Solar energy utilization in the direct photocarboxylation of 2,3-dihydrofuran using CO2. Faraday Discuss. 183, 413–427. doi: 10.1039/c5fd00040h
Aresta, M., Dibenedetto, A., and Dutta, A. (2017a). Energy issues in the utilization of CO2 in the synthesis of chemicals: the case of the direct carboxylation of alcohols to dialkyl-carbonates. Catal. Today 281, 345–351. doi: 10.1016/j.cattod.2016.02.046
Aresta, M., Dibenedetto, A., Zhang, J., Trochowski, M., Angelini, A., et al. (2017b). Photocatalytic carboxylation of CH bonds promoted by popped graphene oxide (PGO) either bare or loaded with CuO. J. CO2 Util. 20, 97–104. doi: 10.1016/j.jcou.2017.05.010
Aresta, M., Dibenedetto, A., Fracchiolla, E., Giannoccaro, P., Pastore, C., Papai, I., et al. (2005). Mechanism of formation of organic carbonates from aliphatic alcohols and carbon dioxide under mild conditions promoted by carbodiimides. DFT calculation and experimental study. J. Org. Chem. 70, 6177–6186. doi: 10.1021/jo050392y
Aresta, M., Dibenedetto, A., and Macyk, W. (2016a). Photocatalytic carbon dioxide reduction at p-type copper(I) iodide. ChemSusChem 9, 2933–2938. doi: 10.1002/cssc.201600289
Aresta, M., Dibenedetto, A., and Quaranta, E. (2016b). Reaction Mechanisms in Carbon Dioxide Conversion. Berlin: Springer.
Aresta, M., Dibenedetto, A., and Tommasi, I. (2000). Direct synthesis of organic carbonates by oxidative carboxylation of olefins catalysed by metal oxides: developing green chemistry based on carbon dioxide. Appl. Organomet. Chem. 14, 799–802. doi: 10.1002/1099-0739(200012)14:12<799::aid-aoc82>3.0.co;2-8
Aresta, M., and Forti, G. (eds) (1987). Carbon Dioxide as Carbon Source, NATO ASI Series C206. Dordrecht: Reidel Publ.
Aresta, M., and Galatola, M. (1999). Life cycle analysis applied to the assessment of the environmental impact of alternative synthetic processes. The dimethylcarbonate case: part 1. J. Clean. Product. 7, 181–190.
Aresta, M., Nocito, F., and Dibenedetto, A. (2018). What catalysis can do for boosting CO2 utilization. Adv. Catal. 63, 49–111. doi: 10.1016/bs.acat.2018.08.002
Aresta, M., Pastore, C., Giannoccaro, P., Kovacs, G., Dibenedetto, A., et al. (2007). Evidence for spontaneous release of acrylates from a transition metal complex upon coupling ethene or propene with a carboxylic moiety or CO2. Chem. A Eur. J. 13, 9028–9034. doi: 10.1002/chem.200700532
Blankenship, R. E., Tiede, D. M., Barber, J., Brudvig, G. W., Fleming, G., Ghirardi, M., et al. (2011). Comparing photosynthetic and photovoltaic efficiencies and recognizing the potential for improvement. Science 332, 805–809. doi: 10.1126/science.1200165
Carmona, E., Alvarez, R., Cole-Hamilton, D. J., Galindo, A., Gutiérrez-Puebla, E., Monge, A., et al. (1985). Formation of acrylic acid derivatives from the reaction of carbon dioxide with ethylene complexes of molybdenum and tungsten. J. Am. Chem. Soc. 107, 5529–5531. doi: 10.1021/ja00305a037
Cazelles, R., Drone, J., Fajula, F., Ersen, O., Moldovan, S., et al. (2013). Reduction of CO2 to methanol by a polyenzymatic system encapsulated in phospholipids-silica nanocapsules. New J. Chem. 37, 3721–3730.
Ciamician, G. (1912). “La fotochimica dell’avvenire (The photochemistry of the future),” in Proceedings of the 8th International Congress on Applied Chemistry, (New York, NY), 135–150.
Dai, A. (2006). Recent climatology, variability, and trends in global surface humidity. J. Clim. 19, 3589–3606. doi: 10.1175/jcli3816.1
Dessler, A. E., Zhang, Z., and Yang, P. (2008). Water-vapor climate feedback inferred from climate fluctuations. Geophys. Res. Lett. 35, 2003–2008.
Dibenedetto, A., and Nocito, F. (2020). Atmospheric CO2 mitigation technologies: carbon capture utilization and storage. Curr. Op. Green Sust. Chem. 21, 34–43. doi: 10.1016/j.cogsc.2019.10.002
Dumeignil, F., Guehl, M., Gimbernat, A., Capron, M., Ferreira, N. L., Froidevaux, R., et al. (2018). From sequential chemoenzymatic synthesis to integrated hybrid catalysis: taking the best of both worlds to open up the scope of possibilities for a sustainable future. Catal. Sci. Technol. 8, 5708–5734. doi: 10.1039/c8cy01190g
Esswein, A. J., and Nocera, D. G. (2007). Hydrogen production by molecular photocatalysis. Chem. Rev. 107, 4022–4047. doi: 10.1021/cr050193e
Hagfeldt, A., and Graetzel, M. (1995). Light-induced redox reactions in nanocrystalline systems. Chem. Rev. 95, 49–68. doi: 10.1021/cr00033a003
Hoberg, H., Jenni, K., Angermund, K., and Kruger, C. (1987). CC-linkages of ethene with CO2 on an iron (0) complex—synthesis and crystal structure analysis of [(PEt3) 2Fe (C2H4) 2]. Angew. Chem. 26, 153–155. doi: 10.1002/anie.198701531
Huang, Y. X., and Hu, Z. (2018). An integrated electrochemical and biochemical system for sequential reduction of CO2 to methane. Fuel 220, 8–13. doi: 10.1016/j.fuel.2018.01.141
Hutchings, G. J., Engel, R. V., Alsaiari, R., Nowicka, E., Pattisson, S., Miedziak, P. J., et al. (2018). Oxidative carboxylation of 1-Decene to 1, 2-Decylene carbonate. Top. Catal. 61, 509–518. doi: 10.1007/s11244-018-0900-y
Ito, T., Nakashimada, Y., Senba, K., Matsui, T., and Nishio, N. (2005). Hydrogen and ethanol production from glycerol-containing wastes discharged after biodiesel manufacturing process. J. Biosci. Bioeng. 100, 260–265. doi: 10.1263/jbb.100.260
Kolbe, H. (1860). Ueber Synthese der Salicylsäure. Justus Liebigs Ann. Chem. 113, 125–127. doi: 10.1002/jlac.18601130120
Lercher, J. A., Peng, B., Dou, H., Shi, H., and Ember, E. E. (2018). Overcoming thermodynamic limitations in dimethyl carbonate synthesis from methanol and CO2. Catal. Lett. 148, 1914–1919. doi: 10.1007/s10562-018-2402-8
Li, Y., Liu, Z., Cheng, R., and Liu, B. (2018). Mechanistic aspects of acrylic acid formation from CO2–ethylene coupling over palladium-and nickel-based catalysts. ChemCatChem 10, 1420–1430. doi: 10.1002/cctc.201701763
Limbach, M., Lejkowski, M. L., Lindner, R., Kageyama, T., Bódizs, G. E., Plessow, P. N., et al. (2012). The first catalytic synthesis of an acrylate from CO2 and an alkene—a rational approach. Chem. A Eur. J. 18, 14017–14025. doi: 10.1002/chem.201201757
Lovley, D. R., and Nevin, K. P. (2013). Electrobiocommodities: powering microbial production of fuels and commodity chemicals from carbon dioxide with electricity. Curr. Opin. Biotechnol. 24, 385–390. doi: 10.1016/j.copbio.2013.02.012
Mackor, A., Tinnemans, A. H. A., and Koster, T. P. M. (1987). “Reduction of CO2 at titanate powders in sunligth and at electrodes in the light or dark,” in Carbon Dioxide as Carbon Source, NATO ASI Series C206, eds M. Aresta and G. Forti (Dordrecht: Reidel Publ.), 393.
Papai, I., Schubert, G., Mayer, I., Besennyei, G., and Aresta, M. (2004). Mechanistic details of nickel (0)-assisted oxidative coupling of CO2 with C2H4. Organomet 23, 5252–5259. doi: 10.1021/om049496%2B
Schlager, S., Dibenedetto, A., Aresta, M., Apaydin, D. H., Dumitru, L. M., Neugebauer, H., et al. (2017). Biocatalytic and bioelectrocatalytic approaches for the reduction of carbon dioxide using enzymes. Energy Technol. 5, 812–821. doi: 10.1002/ente.201600610
The Catalyst Group Resources [TGCR] (2012). Analysis of Demand for Captured CO2 and Products from CO2 Conversion. Bethlehem, PA: The Catalyst Group Resources [TGCR].
Tomishige, K., Gu, Y., Nakagawa, Y., and Tamura, M. (2020). Reaction of CO2 with alcohols to linear-, cyclic-, and poly-carbonates using CeO2-based catalysts. Front. Energy Res. 8:117. doi: 10.3389/fenrg.2020.00117
Ullah, N., Ali, I., and Omanovic, S. M. (2015). Direct electrocatalytic reduction of coenzyme NAD(+) to enzymatically-active 1,4-NADH employing an iridium/ruthenium-oxide electrode. Chem. Phys. 149–150, 413–417. doi: 10.1016/j.matchemphys.2014.10.038
Villano, M., Aulenta, F., Ciucci, C., Ferri, T., Giuliano, A., et al. (2010). Bioelectrochemical reduction of CO2 to CH4 via direct and indirect extracellular electron transfer by a hydrogenophilic methanogenic culture. Biores. Technol. 101, 3085–3090. doi: 10.1016/j.biortech.2009.12.077
Wang, L., Que, S., Ding, Z., and Vessally, E. (2020). Oxidative carboxylation of olefins with CO2: environmentally benign access to five-membered cyclic carbonates. RSC Adv. 10, 9103–9115. doi: 10.1039/c9ra10755j
Wang, X., and Song, C. (2019). “Capture of CO2 from concentrated sources and the atmosphere,” in An Economy based on Carbon Dioxide and Water, eds M. Aresta, I. Karimi, and S. Kawi (Cham: Springer).
Keywords: CO2 conversion, cyclic-C economy, catalysis, biotechnological processes, integrated systems
Citation: Aresta M and Dibenedetto A (2020) Carbon Recycling Through CO2-Conversion for Stepping Toward a Cyclic-C Economy. A Perspective. Front. Energy Res. 8:159. doi: 10.3389/fenrg.2020.00159
Received: 21 May 2020; Accepted: 23 June 2020;
Published: 16 July 2020.
Edited by:
Youngjune Park, Gwangju Institute of Science and Technology, South KoreaReviewed by:
Guanhe Rim, Columbia University, United StatesMenglian Zheng, Zhejiang University, China
Copyright © 2020 Aresta and Dibenedetto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michele Aresta, bWljaGVsZS5hcmVzdGFAdW5pYmEuaXQ=; bWljaGVsZS5hcmVzdGFAaWMyci5jb20=; Angela Dibenedetto, YW5nZWxhLmRpYmVuZWRldHRvQHVuaWJhLml0; YS5kaWJlbmVkZXR0b0BjaGltaWNhLnVuaWJhLml0