 Keiichi Tomishige
Keiichi Tomishige Yu Gu
Yu Gu- School of Engineering, Tohoku University, Sendai, Japan
Reaction of CO2 with alcohols to organic carbonates is one of non-reductive CO2 conversion methods. The catalysts are needed for this reaction, at the same time, effective H2O removal methods are also needed because the yield of organic carbonates is strongly limited by the equilibrium. The development of heterogeneous catalysts for the synthesis of dimethyl carbonate from CO2 and methanol, which is a model and typical reaction, is described. This is because heterogeneous catalysts are more suitable to the practical process than homogeneous catalysts from the viewpoint of the separation of catalysts from the products and the reusability of the catalysts. One of the reported heterogeneous catalysts is CeO2, and it has been also reported that the combination of dimethyl carbonate synthesis from CO2 and methanol with the hydration of nitriles such as 2-cyanopyridine, where both reactions are catalyzed by CeO2, enabled high yield of the carbonate. In addition, the combination of CeO2 catalyst + nitriles can be applied to the synthesis of a variety of linear-, cyclic (five- and six-membered ring)-, and poly-carbonates from CO2 and corresponding alcohols.
Introduction
Much attention has been recently paid to chemical utilization of CO2, although the chemicals produced from CO2 are so limited at present. Generally speaking, it is not easy to decrease the net CO2 emission in the production of a chemical from CO2 because of the CO2 emission due to the separation and purification of CO2. On the other hand, it is possible that the CO2 emission from the CO2-based chemical production becomes smaller than that from conventional production methods. This can be connected to the replacement of the conventional method by CO2-based one.
Chemical utilization of CO2 can be divided into two categories: one is reductive conversion of CO2 and the other is non-reductive conversion of CO2. One of typical methods for reductive conversion of CO2 is the hydrogenation of CO2. In most cases of the CO2 hydrogenation, the production of renewable hydrogen is required for the decrease in the CO2 emission and it is thought that the development of the feasible production method for hydrogen from renewable resources needs more time. In contrast, non-reductive conversion of CO2 does not need the renewable hydrogen.
One of the non-reductive conversions of CO2 includes the synthesis of organic carbonates, carbamates, and ureas from CO2, and alcohols, and/or amines. The synthesis of organic carbonates, carbamates, and ureas is possible from phosgene, and alcohols, and/or amines. Much higher reactivity of phosgene than CO2 enables no equilibrium limitation of these reactions and also connected to the non-catalytic reactions. In contrast, the synthesis of organic carbonates, carbamates, and ureas from CO2, and alcohols, and/or amines is commonly reversible reactions and can be limited by the equilibrium. At the same time, much lower reactivity of CO2 than phosgene requires effective catalysts. It has been reported that CeO2-based catalysts are effective to the synthesis of organic carbonates, carbamates, and ureas from CO2, and alcohols, and/or amines (Tamura et al., 2014; Tomishige et al., 2019, 2020). The product yield at the equilibrium is strongly influenced by the substrates such as alcohols and amines. As is known, the equilibrium limitation of the synthesis of organic carbonates from CO2 and alcohols is usually much more serious than that of the synthesis of carbamates and ureas. Therefore, the shift of the equilibrium is very important for high product yield in the synthesis of organic carbonates from CO2 and alcohols. Recently, it is found that the combination of the synthesis of organic carbonates from CO2 and alcohols with the hydration of nitriles to amides is effective to the shift of the equilibrium to the product side (Honda et al., 2014a). The present review mainly mentions the synthesis of the organic carbonates such as linear-, cyclic, and poly-carbonates from CO2, and corresponding alcohols combined with the hydration of the nitriles over CeO2 as a heterogeneous catalyst comparing to other related catalysts, organic dehydrants, and so on. First, various synthesis routes of organic carbonates are introduced, and challenging subjects such as the low equilibrium level, and the catalyst development are mentioned. Next, we mention the method for the removal of H2O in the synthesis of organic carbonates from CO2 and alcohols to shift the equilibrium since the production of target organic carbonates in higher yield is important and can enhance the impact. Then, various examples of catalyst development for the reaction are shown and compared, indicating that CeO2 can be regarded as one of promising heterogeneous catalysts in terms of the catalytic activity such as the formation rate of organic carbonates. Another superiority of CeO2 is that the combination of the synthesis of DMC synthesis from CO2 and methanol with the hydration of nitriles over CeO2 enhanced the DMC yield remarkably, and the applicability of this system is also mentioned in this review.
Synthesis Routes of Organic Carbonates
A variety of synthesis routes of organic carbonates have been considered, and we would like to compare the routes from the thermodynamic viewpoint. This can clarify the thermodynamic difficulty level of the routes, and this is a fundamental aspect before the development of catalysts for each reaction. In this chapter, synthesis routes of organic carbonates are divided into three categories: non-CO2-based route, indirect CO2-based route, and direct CO2-based route.
Table 1 lists the energy change in carbonate formation reactions and their related reactions calculated by DFT for the evaluation of the severity of the equilibrium limitation and part of calculation was carried out for this article and some calculation has been described in our previous review (Tomishige et al., 2019). We adopted the DFT calculations because it is generally difficult to collect all the thermodynamic data such as formation enthalpy and so on regarding various substrates and products. The experimental data of the heat of reaction are also shown in Table 1 for representative entries. Although the obtained values can be different from the experimentally determined ones, the comparison of the obtained values can be suggestive. In particular, we have to be careful that all the substrate and product molecules are present in the gas phase. The negative and large energy change means that the reaction is highly exothermic and the positive and large one means that the reaction is highly endothermic. Since these values are almost the same to enthalpy change, the equilibrium is also affected by entropy change, especially when the number of molecules is changed in the reaction. Comparison between reactions with different changes in the number of molecules should be careful because the entropy term is much different between such reactions. Since the standard entropy (S0) of liquid alcohols is about 150 J mol−1 K−1, the difference in the number of reactant alcohol molecules by one corresponds to about 60 kJ mol−1 difference in free energy at 400 K.

Table 1. Summary of energy changes for related reactions for the synthesis of organic carbonates by DFT calculation (calculated at B3LYP/6-311++G(d,p) level theory with Gaussian 16 program package (Gaussian, Inc.) and the reference experimental data.
Non-CO2-Based Synthesis Route of Linear Organic Carbonates
Phosgene method is the most typical one for the synthesis of the linear organic carbonates from alcohols (Equation 1).
The problem of this method is the toxicity of phosgene and large amount of byproduct NaCl. The formation of DMC from phosgene and methanol is highly exothermic even when the product HCl is not neutralized (Table 1, Entry 1). This behavior is explained by the high energy state of phosgene itself, which is also connected to high reactivity of phosgene. It should be noted that the exothermicity becomes much larger considering the reaction of HCl with NaOH for the neutralization (Table 1, Entry 2). Totally, the high energy state and high reactivity of phosgene enables the practical production of organic carbonates by the reaction with alcohols, and this reaction can proceed without any catalysts.
Transesterification of linear organic carbonates with alcohols is applied to the synthesis of other organic carbonates, for example, DMC is the simplest linear organic carbonate and transesterification of DMC with other alcohols gives various organic carbonates (Equation 2) (Ono, 1997). High yield of the target carbonates will be obtained by large excess of DMC. This reaction has been also applied to the production of diphenyl carbonate, which can be rather endothermic due to the high energy of diphenyl carbonate (Table 1, Entry 7) (Ono, 1997).
If DMC can be produced by CO2-based routes, this route can be also categorized to indirect CO2-based ones.
Indirect CO2-Based Synthesis Route of Organic Carbonates
The most typical CO2-based chemical in the industry is the production of urea from CO2 and NH3 (Equation 3).
As listed in Table 1, Entry 3, the reaction seems to be endothermic, however, the reaction can be exothermic if (NH2)2CO is obtained as solid. Methanolysis of urea (Table 1, Entry 4) can give DMC as below (Equation 4). The overall reaction including Equations 2, 3 is regarded as indirect CO2-based DMC synthesis.
An example of the reports on alcoholysis of urea in supercritical methanol shows that 98% urea-based yield of DMC was obtained at 538 K, the molar ratio of methanol/urea of 14, and 9.2 MPa (Hou et al., 2014). Although the large excess of methanol is necessary for high DMC yield, the equilibrium of Equation 4 is not serious judging from high DMC yield. One of the merits of this process is that ammonia evolved in this synthesis route (Equation 4) can be recycled back to urea by reacting it with CO2 (Equation 3). This method has been also applied to the synthesis of diethyl carbonate (DEC), glycerol carbonate, propylene carbonate, ethylene carbonate and so on (Shukla and Srivastava, 2017).
Another approach for the DMC synthesis is the transesterification of ethylene carbonate with methanol. Ethylene carbonate can be produced by the reaction of CO2 with ethylene oxide (Table 1, Entry 5). The energy change of Equation 5 represents the exothermic reaction, which can be due to the high energy of ethylene oxide.
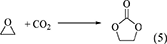
DMC can be synthesized by the transesterification of ethylene carbonate with methanol (Equation 6) (Table 1, Entry 7).
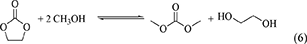
High DMC yield (93%) has been reported in Equation 6, for example, at 333 K and the molar ratio of methanol/ethylene carbonate of 16 (Liu et al., 2013). There have been a lot of reports on the transesterification of ethylene carbonate as summarized in a previous review (Santos et al., 2014). These reactions have been applied to non-phosgene route for the production of polycarbonate via diphenyl carbonate (Table 1, Entry 6) developed by Asahi Kasei Corporation (Fukuoka et al., 2003). The overall reaction including Equations 4, 5 is also regarded as indirect CO2-based DMC synthesis. This process can be very effective when ethylene oxide is available and ethylene glycol can be consumed by other processes such as the production of polyethylene terephthalate.
Direct CO2-based Synthesis Route of Linear Organic Carbonates
The direct synthesis of linear organic carbonates from CO2 and corresponding alcohols is described in Equation 7.
It has been well-known that this reaction is strongly limited by the equilibrium. Another important point is the selectivity to organic carbonates. A possible side reaction is the formation of ethers (Equation 8).
The energy changes of DMC synthesis from CO2 and methanol (Entry 8) and dimethyl ether (DME) formation from methanol (Entry 9) are also listed in Table 1. The comparison between the energy changes of DMC synthesis and DME formation indicates that the equilibrium limitation of DMC synthesis is much more serious than that of DME and the formation of DME is more favorable than the DMC synthesis from the thermodynamic viewpoint. The formation of H2O with DME can suppress the DMC synthesis. Therefore, very high selectivity to DMC is required in the catalytic performance.
Regarding the catalysts for the DMC synthesis from CO2 and methanol, homogeneous Sn alkoxide was firstly reported to be effective as mentioned in our previous review (Honda et al., 2014a). Regarding the heterogeneous catalysts for direct DMC synthesis from CO2 and methanol, it has been firstly reported that ZrO2-based oxides are effective catalysts (Tomishige et al., 1999, 2000; Ikeda et al., 2000, 2001), and then CeO2-based oxides have been found to be effective catalysts giving almost 100% selectivity to DMC (Tomishige et al., 2001; Yoshida et al., 2006). In particular, heterogeneous CeO2 catalysts have been utilized to the direct CO2-based synthesis route in various organic carbonates, therefore, the experimental determination of the equilibrium level of the reaction over CeO2 is mentioned here. The equilibrium limitation of DMC and diethyl carbonate (DEC) was investigated on CeO2 catalyst (Yoshida et al., 2006). The amount of DMC and DEC was 0.66 and 0.41 mmol, respectively, using 0.1 g CeO2 at 403 K under CH3OH or C2H5OH: CO2 = 200: 200 mmol, where the equilibrium yield of DMC and DEC is calculated to be as low as 0.7 and 0.4%, respectively. This tendency is also supported by the data in Table 1, Entries 8 and 10, and suggests that higher primary alcohols give lower equilibrium yield of corresponding organic carbonates, although the difference between methanol, and ethanol can be rather smaller than that between ethanol and n-propyl alcohol. The serious equilibrium limitation of the direct synthesis of organic carbonates from CO2 and corresponding alcohols has been also supported by other reports. The thermodynamic calculation on the effect of the CO2 pressure showed that very high pressure of CO2 such as more than 2.41 × 104 MPa is required in order to progress the reaction spontaneously (Leino et al., 2010). Prof. Urakawa described that methanol conversion increased with the reaction pressure but it reached the level of ca. 1% at even 40 MPa without dehydrating agents (Bansode and Urakawa, 2014). These indicate that the pressurized CO2 is not suitable to the equilibrium shift to the product side in the reaction of CO2 with methanol to DMC.
Another important behavior is the reaction of CO2 with secondary alcohols such as isopropanol and phenol (Table 1, Entries 12 and 13), suggesting higher level of difficulty in the synthesis of di-isopropyl carbonate and diphenyl carbonate. The formation of di-isopropyl carbonate using CeO2 was unfavorable in the reaction of CO2+aniline+2-propanol+2-cyanopyridine compared to the formation of DMC in the reaction of CO2+aniline+methanol+2-cyanopyridine (Tamura et al., 2018a; Gu et al., 2019a). In fact, the energy change between CO2+primary alcohols and CO2+2-propanol is not so different, however, the reactivity of 2-propanol seems to be much lower than that of primary alcohols over CeO2 catalyst, which may not be controlled by thermodynamics but by kinetics. Further investigation is necessary for low reactivity of secondary monoalcohol such as 2-propanol over CeO2 catalyst.
Direct CO2-Based Synthesis Route of Cyclic- and Poly-Carbonates
Cyclic carbonates and polycarbonates can be synthesized by the reaction of urea with diols, the transesterification of DMC with diols, and so on, which are similar reactions as described above and can be categorized to indirect CO2-based synthesis route of cyclic carbonates. In this section, direct synthesis of cyclic carbonates and polycarbonates from CO2 and corresponding diols.
Five membered ring carbonates can be synthesized by the reaction of CO2 and 1,2-diols such as ethylene glycol, propylene glycol and so on (Equation 9, Table 1, Entries 14-17).
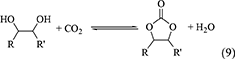
The selective formation of ethylene carbonate and propylene carbonate from CO2 and corresponding diols catalyzed by CeO2-based catalysts has been demonstrated (Tomishige et al., 2004a,b). An example of the equilibrium level of the synthesis of propylene carbonate from propylene glycol has been reported to be around 2% propylene glycol-based yield of propylene carbonate at 403 K and propylene glycol: CO2: CH3CN (solvent) = 100: 200: 120 mmol using CeO2-ZrO2 (Ce/(Ce+Zr)) = 0.33) catalyst (Tomishige et al., 2004a,b), which seems to be similar to or a little higher equilibrium level of DMC synthesis from CO2 and methanol. The level of difficulty in the synthesis of propylene carbonate may be comparable to that in the synthesis of DMC. The energy change of the synthesis of propylene carbonate is clearly higher than that of the synthesis of DMC. The inconsistency of the level of difficulty obtained from experimental results and the calculated energy changes may be explained by the difference in the number of molecules. In the case of DMC synthesis, the number of molecules is decreased from three as reactants to two as products, in contrast, in the case of propylene carbonate synthesis, the number of molecules is not changed during the reaction (two as reactants and two as products).
Therefore, it is not suitable to compare the energy change in the dialkyl carbonate synthesis (Table 1, Entries 8, 10–13) and the five membered ring carbonate synthesis (Table 1, Entries 14–17), on the other hand, the energy changes of five membered ring carbonate synthesis from CO2 and 1,2-diols can be compared in Table 1, Entries 14–17. The order of the energy change is as follows.
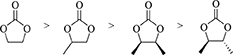
The comparison suggests that methyl group binding with five membered ring carbonate structure can decrease the energy change, probably by the electron donating properties of methyl group. The difference in the energy change of the carbonate from (R, R)-2,3-butanediol and (R, S)-2,3-butanediol can be explained by conformation of the two neighboring methyl groups.
Six membered ring carbonates can be synthesized by the reaction of CO2 and 1,3-diols such as 1,3-propanediol, 1,3-butanediol, and 2,4-pentanediol (Table 1, Entries 18–21) (Equation 10).
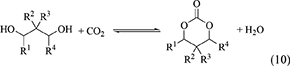
The energy change of the synthesis of six membered ring carbonates is clearly positive and larger than that that of the synthesis of five membered ring carbonates (Table 1, Entries 14–17). This can be interpreted by the difference in the stability of the ring structure. This tendency can also be related to high reactivity of six membered ring carbonates, for example, in the ring-opening polymerization (Honda and Abe, 2018; Tamura et al., 2018b). High reactivity in the ring-opening polymerization makes the synthesis of the reaction of CO2 and 1,3-diols more difficult. The energy change of the carbonate from (R, R)-2,4-pentanediol and (R, S)-2,4-pentanediol is relatively small (Table 1, Entries 20, 21), which can be explained by the longer distance between the two methyl groups. Without dehydrating agents, six membered ring carbonate synthesis is very difficult, however, it has been reported that six membered ring carbonates can be synthesized from 1,3-diols using CeO2 and 2-cyanopyridine as mentioned below (Honda et al., 2014b).
The energy change of seven membered ring carbonate formation from CO2 and 1,4-butanediol (Equation 11) is clearly positive and higher than that of five or six membered ring carbonates from CO2 and corresponding diols, which can be due to much lower stability of seven membered ring carbonates (Table 1, Entry 22).

The energy change of ring opening polymerization of five, six and seven membered ring carbonates is estimated by the reaction of the cyclic carbonates and its dimer, and the obtained results are also listed in Table 1. The values of the energy change are negative, and the order of the absolute values is as follows: five<six<seven. This tendency can be explained by the stability of the ring structure and it also corresponds to the reactivity of cyclic carbonates. The energy change of ring opening polymerization of five-, six-, and seven-membered ring carbonates are also listed in Table 1, Entries 23–25. The energy change is negative and larger and the order is five < six < seven, and this order can be connected to the order of the reactivity in the ring opening polymerization.
The energy change of the direct synthesis of polycarbonates from CO2 and α,ω-diols (Table 1, Entries 26–28) can be calculated from that of the carboxylation of the diol (Table 1, Entries 14, 18, 22) and the ring-opening polymerization of the ring carbonate (Table 1, Entries 23, 24, 25), and the results are also listed in Table 1. The copolymerization of CO2 and 1,4-butanediol is more preferable to that of CO2 and 1,3-propanediol or ethylene glycol, although the equilibrium level of the formation of polycarbonate is so serious and the removal of H2O is necessary for high yield of the polycarbonate.
Removal of H2O in the Synthesis of Carbonates From CO2 and Alcohols
One of the common problems in the reaction of CO2 and alcohols is the very low equilibrium yield of the target organic carbonates. The problem of the low yield of organic carbonates at the equilibrium cannot be solved by the catalyst development. Instead, the approach for the equilibrium shift to the product side is required, and one of the possible methods to shift the equilibrium is the removal of H2O, one of which is the utilization of organic dehydrants. As mentioned above, very low equilibrium yield of the carbonates means very low H2O concentration in the reactor. Therefore, the organic dehydrant can react with low concentration of H2O selectively under the reaction conditions like the presence of large amount of alcohol substrates. There have been various reports on the carbonate synthesis from CO2 and alcohols using organic dehydrants, for example, 2,2-dimethoxypropane (DMP) (Tomishige and Kunimori, 2002; Choi et al., 2008), 1,1,1-trimethoxymethane (TMM) (Zhang et al., 2011), butylene oxide (Leino et al., 2011, 2013), nitriles (Honda et al., 2009, 2010, 2011a, 2013, 2014c), and so on.
The reaction equations of organic dehydrants such as DMP (Equation 12), TMM (Equation 13), Butylene oxide (Equation 14), nitriles (Equation 15) are shown below.
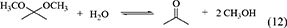
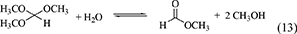
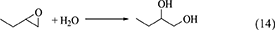
One of advantages of DMP and TMM in the synthesis of DMC from CO2 and methanol is that methanol is the hydration product. At the same time, this can be disadvantage in the synthesis of organic carbonates from CO2 and other alcohols than methanol. This is because it is not easy to get the acetal of acetone or methyl formate containing the corresponding alcohols. The energy change of DMP is positive (Table 1, Entry 29) and that of TMM is negative (Table 1, Entry 31), and this suggests that TMM can be a stronger dehydrant than DMP. The addition of DMP to the DMC synthesis from CO2 and methanol clearly enhanced the yield of DMC above the equilibrium level in the reaction of CO2+methanol, however, the yield of DMC is limited again to the equilibrium level in the reaction of CO2+methanol+DMP (Equations 16–18). In particular, in the reaction of CO2+methanol+DMP, the overall reaction (Equation 18) is the sum of the DMC synthesis from CO2 and methanol (Equation 16) and the hydration of DMP (Equation 17). Methanol is not consumed or formed in the overall reaction (DMP+CO2 to DMC and acetone; Equation 18), which is also reversible and limited by the equilibrium (Table 1, Entry 30) (Tomishige and Kunimori, 2002).
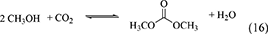
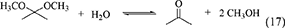
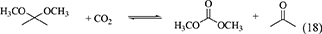
The overall reaction of the combination of DMC synthesis and TMM hydrolysis is similar to the case of DMP. An important point is that the hydrolysis of TMM is more thermodynamically favorable than that of DMP (Table 1, Entries 30 and 32). The yield of DMC using TMM can be clearly higher than that using DMP. The effect of TMM addition to the synthesis of DMC from CO2 and methanol over Ce0.5Zr0.5O2 has been reported (Zhang et al., 2011). Another possible dehydrant is 1,1,1-trimethoxyethane, where the hydrolysis of 1,1,1-trimethoxyethane gives methyl acetate and methanol (Table 1, Entry 33). 1,1,1-Trimethoxyethane can be a stronger dehydrant than TMM in terms of the energy change of the hydrolysis.
In the utilization of DMP, TMM, and 1,1,1-trimethoxyethane, acetone, methyl formate, and methyl acetate, which are produced from the hydrolysis, had better be regenerated to corresponding acetals. However, the regeneration to acetals with methanol is the reverse reaction of the hydrolysis acetals. This means that the stronger dehydrants can be more difficult to be regenerated. The energy balance between the hydrolysis and the acetalization can be important for the system construction including DMC synthesis and the regeneration of the dehydrants.
Recently, ester has been attempted to be used as a dehydrant, although the example is a little limited. The combination of DMC synthesis from CO2 and methanol with the hydrolysis of methyl trichloroacetate has been reported (Marciniak et al., 2019). The reaction formula of the hydrolysis of methyl trichloroacetate is shown below and the energy change of this reaction is also listed in Table 1, Entry 34.
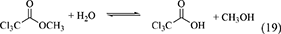
For the reference, the energy change of the hydrolysis of methyl acetate and methyl formate is also listed in Table 1, Entries 35 and 36. Judging from the energy change, it seems that methyl acetate or methyl formate can be more suitable dehydrants than methyl trichloroacetate. When the esters are used as a dehydrant, the produced carboxylic acids can decrease the catalytic activity of CeO2 because of the interaction between the carboxylic group and CeO2 is rather strong. It seems that it is not easy to obtain high alcohol-based carbonate yield because the amount of produced acids is very large at high methanol conversion.
Epoxides such as 1,2-epoxybutane (butylene oxide), styrene oxide (Equation 20) and cyclohexene oxide (Equation 21) have been used as a dehydrant in DMC synthesis from CO2 and methanol or ethanol (Leino et al., 2011, 2013, 2018; Tamboli et al., 2018), and hydration of epoxides above are shown below and the energy change is listed in Table 1, Entries 37, 40, 43. In the case of epoxides, the selective reaction of epoxides with H2O is not easy. In the combination of DMC synthesis with hydration of epoxides, the reaction of epoxides with methanol can proceed in a parallel way. Therefore, the energy change of epoxides with methanol is also listed in Table 1 (Entries 38, 39, 41, 42, 44). The difference in the energy change of the reaction of epoxides with H2O and methanol can be important because this can be connected to the selectivity of the H2O removal. However, the difference in the energy change is rather small, suggesting that the reaction of epoxides with methanol can proceed more preferably than the reaction of epoxide with H2O, in particular, considering much higher concentration of methanol than that of H2O in the combination of DMC synthesis from CO2 and methanol with hydration of epoxides. Therefore, the selectivity of the H2O removal is not so high, as a result, the yield of DMC is not so high and larger amount of epoxides than the stoichiometry of H2O removal is required.
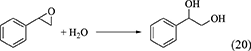

Nitriles such as acetonitrile (Equation 22), benzonitrile (Equation 23), 2-cyanopyridine (Equation 24), and 2-furonitrile (Equation 25) have been used as a dehydrant in DMC synthesis from CO2 and methanol (Honda et al., 2009, 2010, 2011a, 2014c), and the reaction formulae of nitrile hydration are shown below.
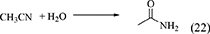
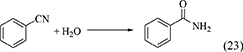
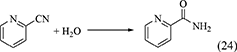
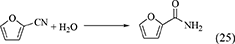
In the combination of DMC synthesis with hydration of nitriles, the reaction of nitriles with methanol may proceed in a parallel way. Here, the energy change of nitriles with H2O and methanol is listed in Table 1, Entries 45–52. The difference in the energy change of the reaction of nitrile with H2O and methanol is clearly larger than the case of the reaction of epoxides. This supports that the reaction of nitriles with H2O can proceed more preferably than the reaction of nitriles with methanol. The difference between epoxides and nitriles can be explained by the stabilization with keto–enol tautomerism after H2O addition to nitriles to give corresponding amides. In fact, the product in the reaction of nitriles with methanol was not detected in the combination of DMC synthesis from CO2 and methanol with hydration of nitriles. Therefore, the selectivity of the H2O removal can be very high, therefore, the yield of DMC is high and the stoichiometric amount of nitrile can be enough for H2O removal.
Another side reaction in the combination of DMC from CO2 and methanol with the hydration of nitriles is methanolysis of amides (R-CONH2 + CH3OH → RCOOCH3 + NH3). The energy change of the methanolysis of picolinamide is positive and that of acetamide and benzamide is negative (Table 1, Entries 53–55). This means that the loss of picolinamide by the methanolysis can be suppressed. In particular, if NH3 is formed by the methanolysis of amides, NH3 can react with DMC to give methyl carbamate causing the decrease of the DMC yield.
Heterogeneous Catalysts for the Synthesis of Organic Carbonates From CO2 and Alcohols
A variety of effective catalysts have been reported for the synthesis of organic carbonates from CO2 and alcohols, mainly DMC from CO2 and methanol. The catalysts can be divided into two categories: homogeneous and heterogeneous catalysts. Typical homogeneous catalysts are Sn alkoxides, Zn acetate, ionic liquids, inorganic, or organic base, and so on, which have been introduced in previous reviews (Honda et al., 2014a; Huang et al., 2015; Chaemchuen et al., 2019). The present review is focusing on the recent progress on heterogeneous catalysts. The most typical heterogeneous catalysts are CeO2-based materials, which have been mentioned in the very recent review (Tomishige et al., 2020). Therefore, the recent progress on the development of heterogeneous catalysts except for CeO2-based ones is focused in this review article, and the heterogeneous catalysts are divided into three categories: oxide catalysts, supported metal catalysts, and other catalysts including metal-organic framework (MOF). In addition, the recent reports on the catalyst development are summarized in Table 2.
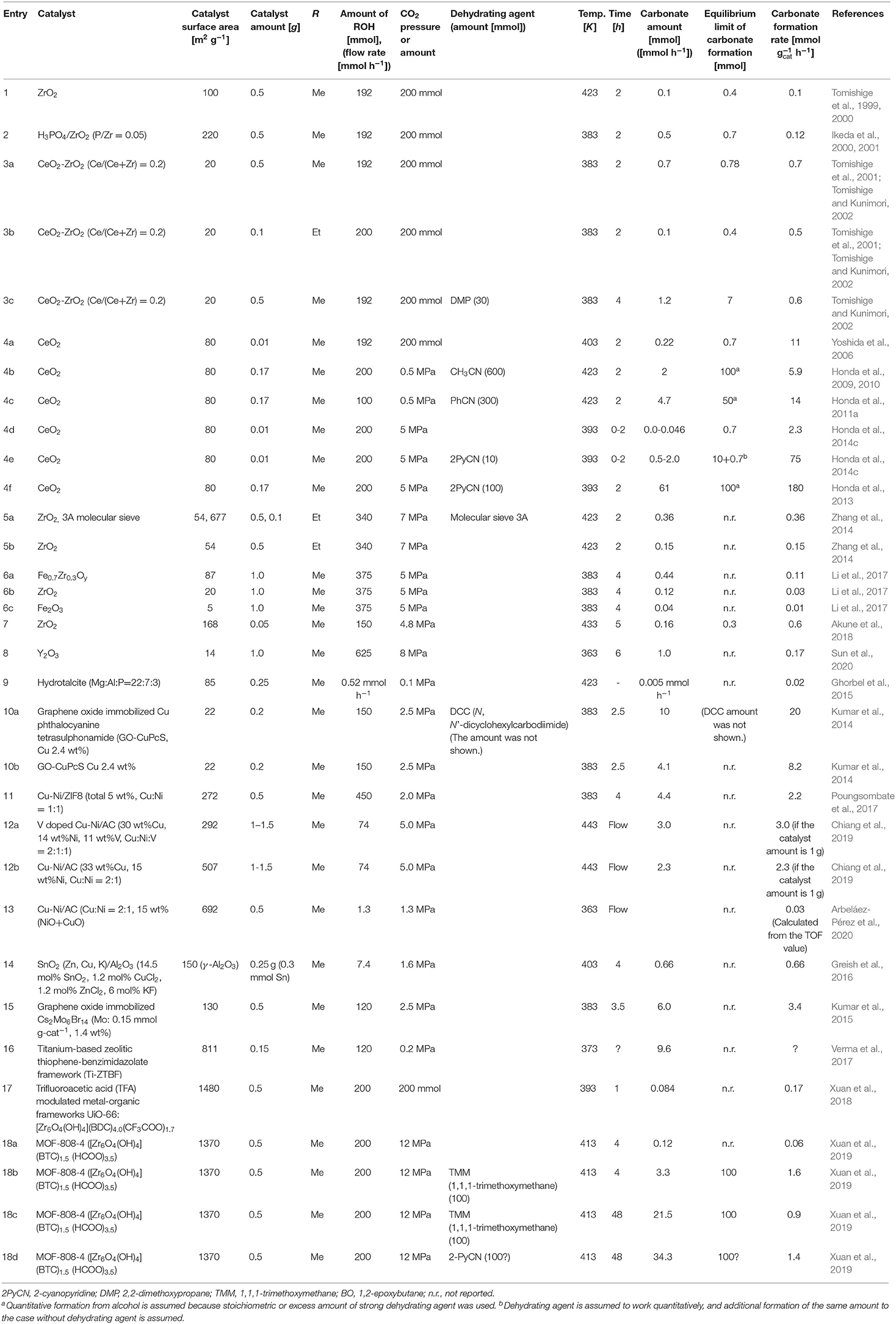
Table 2. Summary of dialkyl carbonate synthesis systems from ROH and CO2 using heterogeneous catalysts.
Oxide Catalysts
The development of oxide catalysts for the synthesis of DMC from CO2 and methanol probably started with the reports on the finding ZrO2 as an effective heterogeneous catalyst (Tomishige et al., 1999, 2000). It has been also reported that the modification of ZrO2 with H3PO4 enhanced the catalytic activity (Ikeda et al., 2000, 2001). Next, CeO2-ZrO2 solid solution catalysts were tested as a derivative of ZrO2-based materials, and they showed clearly higher catalytic activity than ZrO2 (Tomishige et al., 2001; Tomishige and Kunimori, 2002). At this time, we also tested pure CeO2 as one of the reference catalysts of CeO2-ZrO2 solid solution and recognized that CeO2 itself exhibited high catalytic activity in the DMC synthesis from CO2 and methanol as a heterogeneous catalyst (Yoshida et al., 2006). The catalytic activity per surface area of this pure CeO2 was almost comparable to that of CeO2-ZrO2. Based on these results, we have studied the application of pure CeO2 for the synthesis of organic carbonates, carbamates, and ureas from CO2 with alcohols, and/or amines (Honda et al., 2011b; Tamura et al., 2013a,b, 2016a, 2018a; Gu et al., 2019a).
Table 2 lists the catalytic performance of heterogeneous catalysts for the organic carbonate synthesis from CO2 and alcohols. The results of our reports on ZrO2- and CeO2-based catalysts are also listed (Table 2, Entries 1–4), which can be regarding as a kind of standards in the organic carbonate synthesis from CO2 and alcohols. As mentioned above, the reaction of CO2 with alcohols to the corresponding organic carbonates is limited by the equilibrium, therefore, one of the important factors regarding the catalytic performance is the catalytic activity, for example, per gram catalyst. Based on this policy, we selected the data, which can be far from the equilibrium level, from each report if possible. At the same time, we also list the BET surface area of the catalysts. In Table 2, the formation rates of the carbonates are described and this enables the estimation of the catalytic activity per surface area, which is a crucial index for the catalyst design of metal oxides.
Other ZrO2-based catalysts have been studied (Table 2, Entries 5–7) and the activity seems to be comparable to that in previous reports (Zhang et al., 2014; Li et al., 2017; Akune et al., 2018). It has been recently reported that Y2O3 was an effective catalyst for the DMC synthesis from CO2 and methanol (Table 2, Entry 8), and it is characteristic that Y2O3 has some activity at lower reaction temperature (363 K) than usual (for example, 383–423 K) (Sun et al., 2020). Unfortunately, the formation rate of DMC is not determined on Y2O3 precisely, and the formation rate can be clearly higher than the estimated value. however, considering the low BET surface area, Y2O3 may have high potential. The further development of Y2O3 may be promising and it is important to make clear the difference between Y2O3 and CeO2 catalysts. It has been also reported that DMC synthesis is catalyzed by phosphoric acid intercalated Mg-Al hydrotalcite-like compounds (Ghorbel et al., 2015), although the catalytic activity is not so high (Table 2, Entry 9).
Supported Metal Catalysts
The most typical supported metal catalysts for DMC synthesis are Cu-Ni bimetallic catalysts, and related catalysts have been reported to show the activity in the synthesis of DMC from CO2 and methanol. It has been reported that copper phthalocyanine tetrasulphonamide (Cu-PcS) showed catalytic activity as a homogeneous catalyst in the DMC synthesis from CO2 and methanol with high selectivity. The Cu-PcS complex was immobilized on graphene oxide (GO Cu-PcS) and this was demonstrated to be a heterogeneous and recyclable catalyst for the selective synthesis of DMC from CO2 and methanol, in particular, in the presence of N, N'-dicyclohexylcarbodiimide (DCC). The catalytic activity per g-cat of this heterogeneous catalyst is rather high (Table 2, Entry 10) (Kumar et al., 2014). Unfortunately, the amount of DCC is not shown, therefore, the highest yield of DMC in the presence of DCC cannot be calculated. It should be noted that the activity of heterogeneous GO Cu-PcS was comparable to that of homogeneous Cu-PcS.
ZIF-8 supported Cu-Ni catalyst showed the activity in the DMC synthesis from CO2 and methanol (Table 2, Entry 11) (Poungsombate et al., 2017). The reported methanol conversion is relatively high (12.8%), which may be higher than the equilibrium level of methanol conversion. ZIF-8 may have a role on the H2O removal and the shift of the equilibrium. V doped Cu-Ni/AC (30 wt% Cu, 14 wt% Ni, 11 wt% V, Cu:Ni:V = 2:1:1) was investigated in DMC synthesis from CO2 and methanol (Table 2, Entry 12) (Chiang et al., 2019). From the results of the reaction time profiles, it seems that the catalyst system has some induction period, although the reduction pretreatment was carried out before the reaction. The formation rate of DMC on V doped Cu-Ni/AC is rather high, and the effect of V is remarkable. However, the role of each catalyst component (Cu, Ni, V) is not clarified. Cu-Ni/AC (Cu:Ni = 2:1, 15 wt% (CuO+NiO) showed much higher activity than monometallic Cu/AC and Ni/AC catalysts in the DMC synthesis from CO2 and methanol, suggesting the strong synergy between Cu and Ni species (Table 2, Entry 13) (Arbeláez-Pérez et al., 2020). The activity is rather low due to the low reaction temperature (363 K). It is suggested that the Cu0/NiOx interface may be the catalytically active species. The selectivity of DMC formation is about 80%, and the byproducts were dimethyl ether and CO. It has been reported that SnO2 (Zn, Cu, K)/Al2O3 (14.5 mol% SnO2, 1.2 mol% CuCl2, 1.2 mol% ZnCl2, 6 mol% KF) catalyst gave high yield of DMC (17.8%) in high selectivity (about 99%) (Table 2, Entry 14) (Greish et al., 2016). This yield of DMC seems to be higher than the equilibrium level. The reaction time profiles of DMC on this catalyst is complicated: 3.2, 12.7, and 17.8% at 1, 2, and 4 h, respectively. At the initial stage, the activation of the catalyst may be necessary. Another important point is that the substrate to catalyst ratio is rather low (0.3 ml methanol to 0.25 g-catalyst), and high yield of DMC may be due to this low ratio. It has been reported that graphene oxide immobilized Cs2Mo6Br14 (Mo: 0.15 mmol g-cat−1, 1.4 wt%) exhibited rather high catalytic activity in the DMC synthesis from CO2 and methanol and the catalyst reusability was also demonstrated (Table 2, Entry 15) (Kumar et al., 2015). The yield of DMC seems to be higher than the equilibrium level, and the GO may have a role on the H2O removal and the shift of the equilibrium. An interesting point is the utilization of Mo species as a catalytically active species for the DMC synthesis. It is very important to elucidate the reaction mechanism of DMC formation on Mo species, which will be able to expand the active elements for the DMC synthesis.
Other Catalysts Including Metal-Organic Framework (MOF)
Titanium-based zeolitic thiophene-benzimidazolate framework (Ti-ZTBF) derived from Ti(IV) isobutoxide and 2-(thophen-2-yl)-1-((thiophen-2-yl)methyl)-1H-benzo[d]imidazole is an effective and recyclable heterogeneous catalyst for the DMC synthesis from CO2 and methanol (Table 2, Entry 16) (Verma et al., 2017). The 16% yield of DMC seems to be higher than the equilibrium, and ZTBT may have a role of H2O removal from the reaction system. Unfortunately, the activity cannot be calculated because of the lack of the reaction time.
UiO-66 is a class of Zr-based MOFs typically constructed with Zr6 cluster and terephthalic acid (BDC) linker, and a series of trifluoroacetic acid (TFA) modulated metal-organic frameworks UiO-66 catalysts were synthesized and applied to the DMC synthesis from CO2 and methanol (Table 2, Entry 17) (Xuan et al., 2018). This MOF has the catalytic activity of DMC formation, however, the activity is not high. The reusability of the catalyst was verified.
A series of metal-organic frameworks MOF-808-X (6-connected) were synthesized by regulating the ZrOCl2·8H2O/1,3,5-benzenetricarboxylic acid (BTC) molar ratio (X) and they were applied to DMC synthesis from CO2 and CH3OH with 1,1,1-trimethoxymethane (TMM) or 2-cyanopyridine as a dehydrating agent (Table 2, Entry 17) (Xuan et al., 2019). It was reported that MOF-808-4 ([Zr6O4(OH)4] (BTC)1.5 (HCOO)3.5) was an effective and recyclable heterogeneous catalyst, and the combination of the hydration of TMM and 2-cyanopyridine increased the DMC yield remarkably. The formation rate of DMC on this MOF is not so high as that on CeO2 in the presence of 2-cyanopyridine.
The comparison in Table 2 indicates that CeO2 showed higher catalytic activity (carbonate formation rate / mmol g-cat−1 h−1) in the DMC synthesis from CO2 and methanol without the addition of dehydrating agents than other catalysts (Table 2, Entry 4a) and that CeO2 showed higher catalytic activity in the reaction in the presence of dehydrating agents than other catalysts (Table 2, Entry 4f). In addition, the reports on the applicability of heterogeneous catalysts except for CeO2 catalysts to the direct CO2-based synthesis routes of organic carbonates except for DMC are so limited. Based on the superiority of CeO2 from the viewpoint of high formation rate and the broad scope, we focused on CeO2 in this review.
Unique Properties of CeO2 as a Heterogeneous Catalyst
As mentioned above, CeO2 is one of effective catalysts in the DMC synthesis from CO2 and methanol, and at the same time, broad applicability of CeO2 catalysts to the synthesis of other linear-, cyclic-, poly-carbonates have been reported than that of other catalysts. Here, we would like to mention the reasons for this uniqueness of CeO2. The formation of DMC and H2O from CO2 and methanol is thought to be acid-base-catalyzed reaction. When strongly acidic catalysts are used for the reaction of CO2 and methanol, the main product can be DME. It is clear that strongly acidic catalysts are not suitable to DMC synthesis from CO2 and methanol. Strongly basic catalysts such as MgO are not suitable to the DMC synthesis, which can be explained by the poisoning by CO2. It is characteristic that CeO2 has medium acidic and medium basic bifunctional properties. It has been known that ZrO2 has similar acidic and basic bifunctional properties in the field of heterogeneous catalysis, although the oxides with acid-base bifunctionality are so limited. The basicity of CeO2 is stronger than that of ZrO2, and the acidity of CeO2 is weaker than that of ZrO2 (Tomishige et al., 2004a). These acid-base bifunctionality can be explained by rather large ionic radius and high oxidation state (+4). In particular, cerium is unique in the lanthanoid elements from the viewpoint of the stability of Ce4+. As is known, cations with +3 of lanthanoid tends to be more stable, in contrast, Ce4+ is more stable than the 4+ cations of other lanthanoids. Higher oxidation state can be connected to higher Lewis acidity. As a result, CeO2 can have medium acidic and medium basic bifunctional properties. Modification of CeO2 with the additive such as Nb5+ and Al3+ has been also attempted for the improvement of the catalytic performance of CeO2 (Aresta et al., 2010; Dibenedetto et al., 2012).
The most important properties of CeO2 as a catalytic function is redox properties between Ce4+ and Ce3+. In most cases, CeO2 works as a redox catalyst at clearly higher temperature than that for the organic carbonate synthesis. In the case of DMC synthesis from CO2 and methanol, methanol is not oxidized with CeO2 at all, although benzyl alcohol can be oxidized with CeO2 at similar reaction temperature range (Tamura and Tomishige, 2015). The formation of Ce3+ is connected to the oxide ion defect on CeO2 surface, it has been reported that the oxide ion defects can have some function in the synthesis of DMC from methanol and CO2, however, which cannot be demonstrated clearly. If only the oxide ion defect can be the catalytically active site for DMC synthesis from CO2 and methanol, the catalytic activity is strongly dependent on the number of the oxide ion defect. Generally, the activity of CeO2 with more oxide ion defect may be a little higher compared to that of CeO2 with less oxide ion defect. This kind of behaviors suggests that the main active site on the CeO2 surface, although the oxide ion defect may have higher activity. At present, the structure of catalytically active site and the reaction mechanism are under debated. Further investigation by means of in-situ spectroscopic studies and theoretical approach is necessary. Various CeO2-based materials in the direct CO2-based synthesis routes of organic carbonates from corresponding alcohols have been compared in the recent review (Tomishige et al., 2020).
Hydration of Nitriles: Suitable to H2O Removal in DMC Synthesis From CO2 and Methanol Over Ceria
Development of heterogeneous catalysts for the reaction of CO2 with alcohols has been carried out mainly using DMC or DEC synthesis from CO2 and methanol or ethanol. The previous chapter shows that CeO2 is one of promising heterogeneous catalysts. In the above chapter of removal of H2O in the synthesis of carbonates from CO2 and alcohols, various reactions for H2O removal are listed. Among them, nitrile hydration is also catalyzed by CeO2 and it can be combined with DMC synthesis from CO2 and methanol. It has been found that the combination of DMC synthesis from CO2 and methanol with the hydration of nitriles, in particular, 2-cyanopyridine drastically shifted the equilibrium of the DMC formation (Table 2, Entry 4f, Honda et al., 2013). The effect of the nitrile addition from the viewpoint of H2O removal was strongly dependent on the type of nitriles. Table 3 lists the DMC synthesis from CO2 and methanol over CeO2 catalyst in the presence of various nitriles, and the energy change of hydration of nitriles, the addition of methanol to nitriles, and the methanolysis of amides are also listed in Table 3 (Honda et al., 2014c). Energy changes of the hydration of nitriles are more negative than that of methanol addition to nitriles in the case of all the nitriles. This indicates that the hydration of nitriles proceeds more preferably than methanol addition to nitriles in all the cases. It seems that the combination of DMC synthesis from CO2 and methanol with nitrile hydration can be effective to the equilibrium shift of DMC synthesis from the thermodynamic viewpoint. Energy change of the methanolysis is strongly dependent on the amides in the range of positive values to negative ones. From the viewpoint of the regeneration of nitriles by the dehydration of amides, the methanolysis of amides, which is regarded as a side reaction to esters and NH3, should be suppressed. Therefore, positive and larger energy change of the methanolysis of amides is better. It should be noted that three nitriles, 2-cyanopyridine, cyanopyradine, and 2-cyano-1,3-pyrimidine, promoted the formation of DMC remarkably (Table 3, Entries 1-3). The effect of other nitriles than these three nitriles on the formation rate of DMC is not so significant. The energy change of hydration of nitriles is not so different, which cannot explain the reactivity tendency. The explanation is as follows. These three nitriles have cyano group at the 2nd position of pyridine ring as a common structure. The investigation of the interaction between 2-cyanopyridine (Table 3, Entry 1) and CeO2 surface suggests that the nitrogen on the pyridine ring is coordinated to surface Ce4+ and the lattice oxide ion is coordinated to C atom in cyano group, resulting in the strongly basic nitrogen species in cyano group as illustrated in Figure 1A (Tamura et al., 2015, 2017). It is interpreted that this strongly basic species can activate alcohols to enhance the catalytic activity in the presence of 2-cyanopyridine on CeO2.
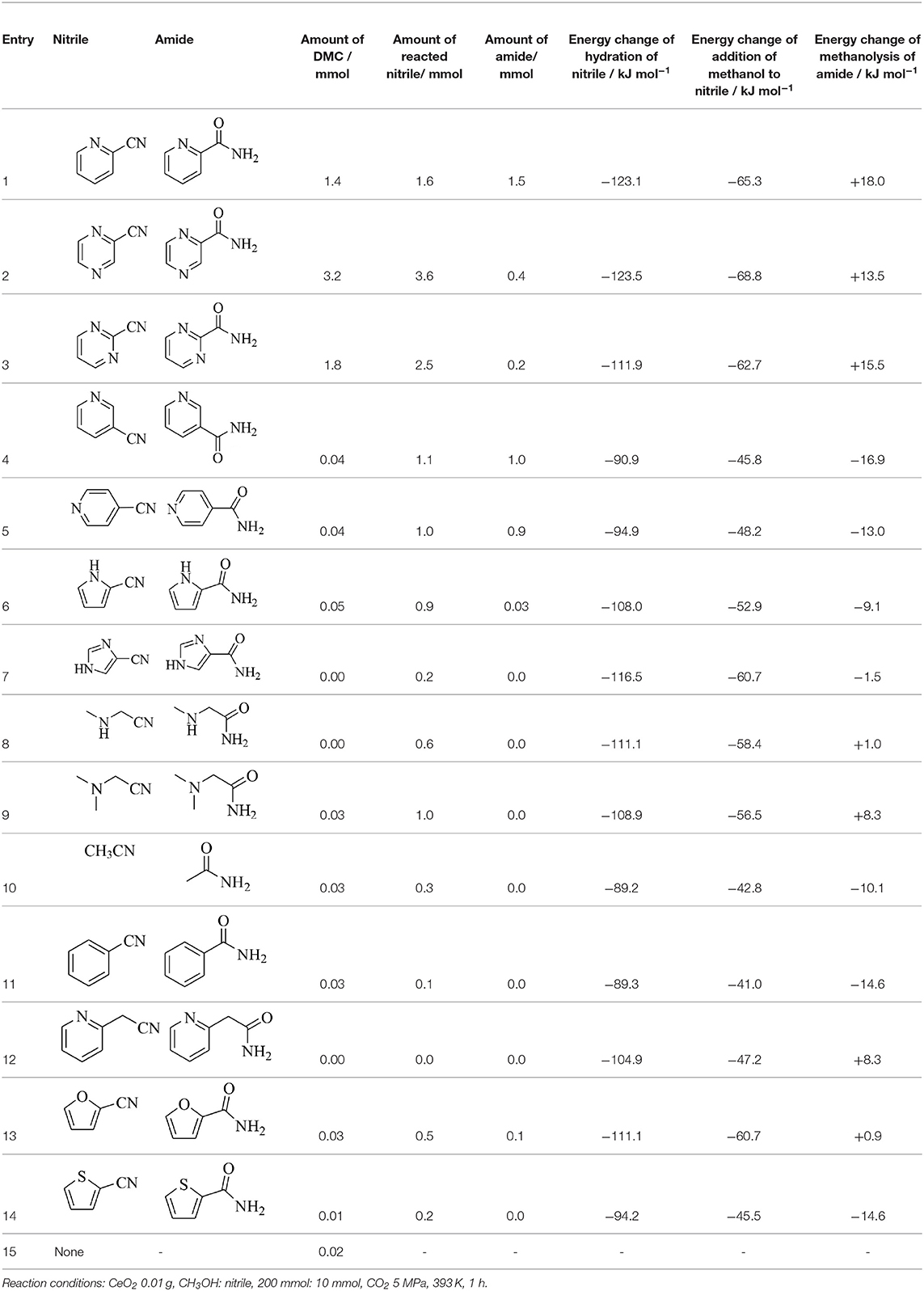
Table 3. DMC synthesis from CO2 and methanol over CeO2 with various nitriles (Honda et al., 2014c).
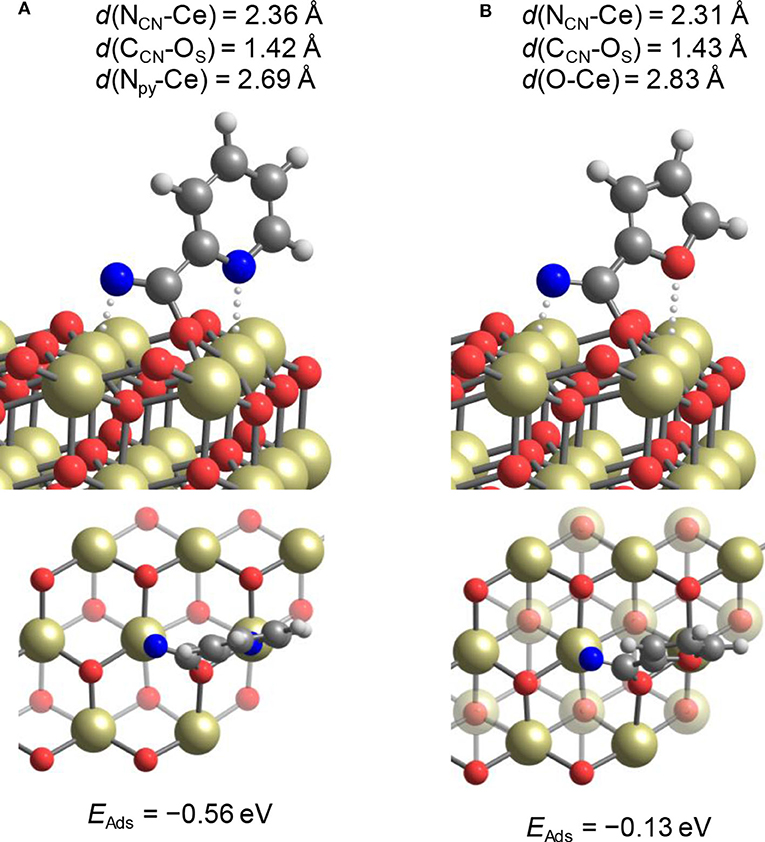
Figure 1. Adsorption structures of (A) 2-cyanopyridine and (B) 2-furonitrile on the CeO2(111) surface (Gu et al., 2019b). (Reprint with permission by American Chemical Society). The atoms are color coded as follows: gray, H; dark gray, C; blue, N; red O; dark yellow, Ce.
On the other hand, it has been also reported that the hydration of acetonitrile and benzonitrile (Entries 10, 11) can shift the equilibrium of DMC synthesis from CO2 and methanol, although the formation rate of DMC was not promoted so significantly (Table 3, Entries 10,11) (Honda et al., 2010, 2011a). In addition, it has been recently reported that CeO2 + 2-furonitrile was effective for the direct synthesis of alternating polycarbonates from CO2 and α,ω-diols (Gu et al., 2019b). The comparison between 2-cyanopyridine and 2-furonitrile in the DMC synthesis from CO2 and methanol over CeO2 is listed in Table 4 (Gu et al., unpublished data). The addition of 2-furonitrile also enhanced the yield of DMC with high selectivity, which was comparable to the case of 2-cyanopyridine, although the formation rate of DMC in the presence of 2-cyanopyridine is clearly higher than that in the presence of 2-furonitrile as listed in Table 3, Entries 1 and 13. According to the DFT calculations, the adsorption of 2-cyanopyridine on CeO2 (111) surface is stronger than that of 2-furonitrile (Figure 1B), which can be connected to higher formation rate of DMC in the presence of 2-cyanopyridine. Another possible demerit of 2-furonitrile is the methanolysis of the amide, which is related to lower energy change of methanolysis of 2-furamide than that of picolinamide. However, the methanolysis of 2-furamide was suppressed sufficiently.
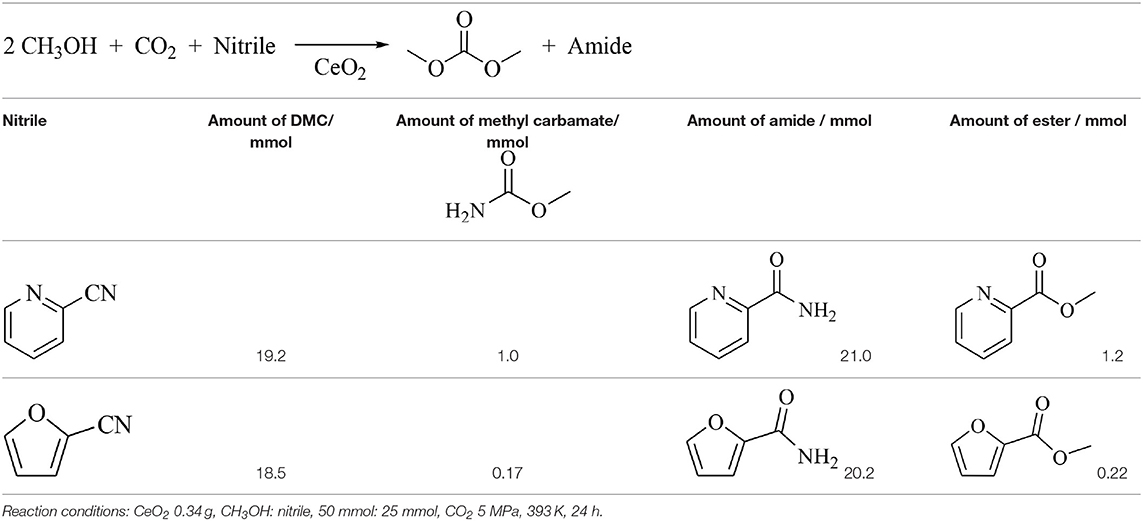
Table 4. Comparison between 2-cyanopyridine and 2-furonitrile in DMC synthesis from CO2 and methanol on CeO2 (Gu et al., unpublished data).
Synthesis of Various Organic Carbonates From CO2 and Corresponding Alcohols by Using CeO2 and Hydration of Nitriles
Various kinds of organic carbonates like cyclic- and poly-carbonates are recognized as value-added chemicals. The above chapters show that DMC can be synthesized from CO2 and methanol in high yield beyond the equilibrium level by the combination with the hydration of nitriles, in other words, the finding of CeO2 as the effective catalyst and nitriles as the effective dehydrating agent enables the direct synthesis of DMC from CO2 and methanol in high yield. As introduced above, various kinds of catalysts and dehydrating methods have been developed, however, the methods whose broad applicability has been demonstrated are so limited. Therefore, in this chapter, the applicability of the method using CeO2 catalyst and nitrile hydration to the direct synthesis of linear-, cyclic-, and poly-carbonates from CO2 and corresponding alcohols is mentioned.
Table 5 lists the synthesis of linear carbonates from CO2 and corresponding monoalcohols using CeO2 and 2-cyanopyridine. Various linear primary alcohols can be converted to corresponding dialkyl carbonates in high yield (around 90%) and the stoichiometric amount of picolinamide was detected (Entries 1–8) (Honda et al., 2014c).
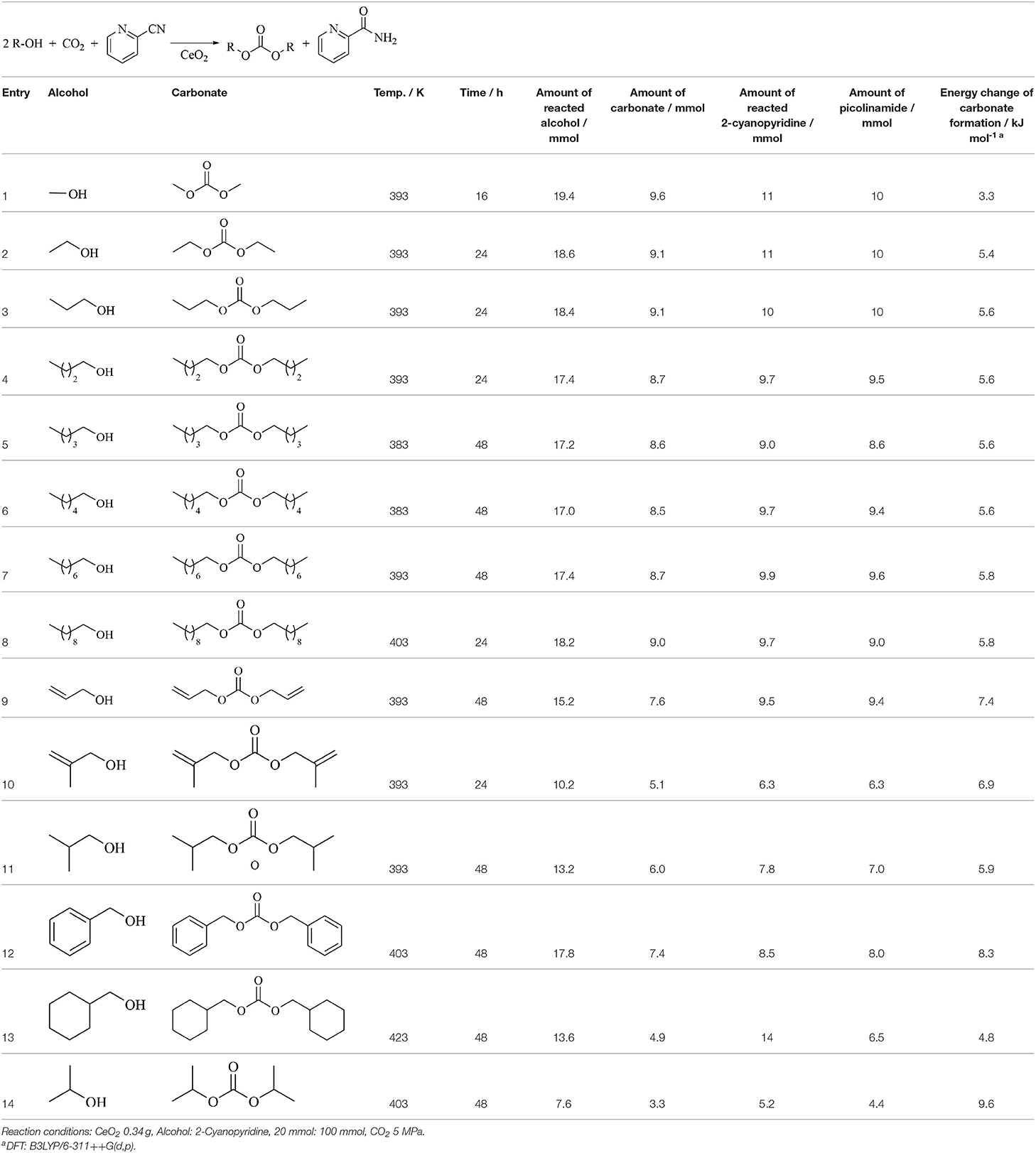
Table 5. Synthesis of linear carbonates from CO2 and corresponding monoalcohols using CeO2 and 2-cyanopyridine (Honda et al., 2014c).
The synthesis of linear carbonates from unsaturated alcohols was also possible to some extent (Table 5, Entries 9, 10). The yield of the carbonates from branched primary alcohol (Table 5, Entries 11, 13), benzyl alcohol (Table 5, Entry 12), and secondary alcohol (Table 5, Entry 14) is not so high. Energy change of the carbonate formation calculated by DFT is also listed in Table 5. The energy change is not so dependent on the alcohols and its absolute values are clearly smaller than that of hydration of 2-cyanopyridine. Therefore, the difficulty of the reaction is not controlled by the thermodynamics, and CeO2 does not seem to be good at converting the alcohols with large steric hindrance. Low reactivity of secondary alcohol such as 2-propanol (Table 5, Entry 14) is utilized to the carbamate synthesis from aniline, CO2, and 2-propanol using CeO2 + 2-cyanopyridine (Gu et al., 2019a). Here, it is very important to suppress the carbonate formation of alcohols, which can be connected to excess consumption of alcohols and 2-cyanopyridine. Actually, the carbamate synthesis from aniline, CO2, and methanol gave a large amount of DMC and excess picolinamide (Tamura et al., 2018a).
Table 6 lists the synthesis of cyclic carbonates from CO2 and corresponding diols using CeO2 and 2-cyanopyridine (Honda et al., 2014b). 1,2-Diols (Table 6, Entries 1–6) gave the corresponding five-membered ring carbonates in high yield. 1,2-Cyclopentanediol and 1,4-anhydroerythirtiol showed rather different reactivity, however, the corresponding five-membered ring carbonates were obtained in high yield (Entries 7, 8). The yield of the six-membered ring carbonates from CO2 and 1,3-diols (Table 6, Entries 9–12) tends to be clearly lower than the case of the synthesis of five-membered ring carbonates. For example, in the case of 1,3-propanediol (Table 6, Entry 9), the formation of cooligomers of CO2 and 1,3-propanediol was detected and this can explain the difference between the amount of the reacted 1,3-propanediol and that of the six-membered ring carbonate (Tomishige et al., 2019). The six-member ring carbonates in the case of Entries 13 and 14 (Table 6) were obtained in high yield, probably because of the suppression of the formation of cooligomers of CO2 and the diols. In this study, the synthesis of glycerol carbonate from CO2 and glycerol using CeO2 and 2-cyanopyridine was attempted, however, our yield of glycerol carbonate under similar reaction conditions was much lower than the case of other five- and six-membered ring carbonates as listed in Table 6, therefore, the result was not reported and we thought that the synthesis of glycerol carbonate is not so easy. On the other hand, this problem was solved to some extent. In the following study after our report, high yield (78.9%) of glycerol carbonate was reported using large amount of CeO2 (1.72 g, Ce 10 mmol) and dimethyl formamide as a solvent as summarized in Scheme 1 (Liu et al., 2016). In Table 6, the amount of substrates is clearly higher than that of CeO2 catalyst, meaning the formation of the carbonates proceeds catalytically. On the other hand, in the synthesis of glycerol carbonate, large amount of CeO2 is very crucial for high yield. The amount of glycerol carbonate was comparable to the amount of CeO2, and this reaction may proceed non-catalytically, suggesting that CeO2 can be deactivated rapidly. The reason for the necessity of large amount of CeO2 should be elucidated for further development of the glycerol carbonate synthesis.
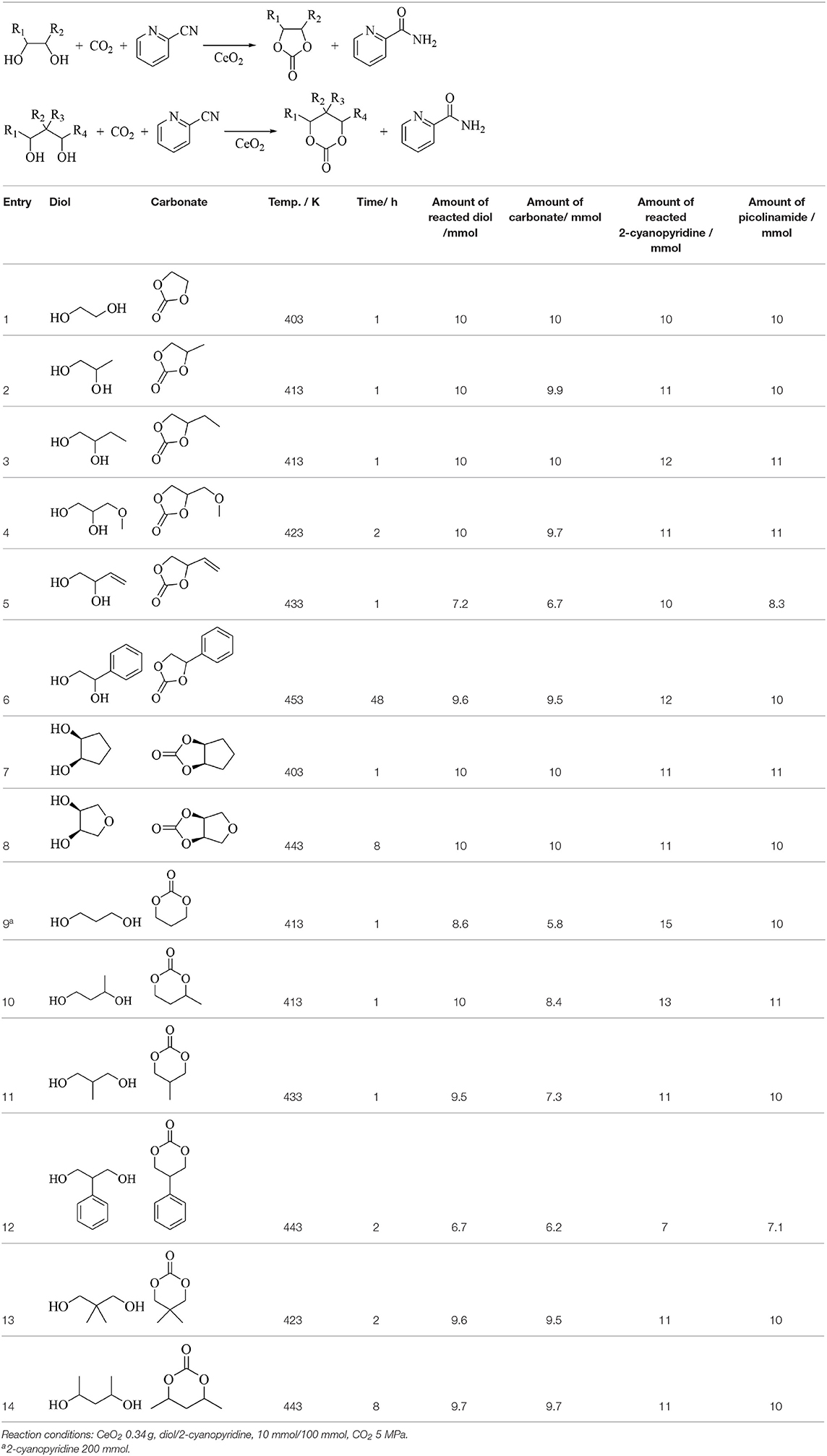
Table 6. Synthesis of cyclic carbonates from CO2 and corresponding diols using CeO2 and 2-cyanopyridine (Honda et al., 2014b).
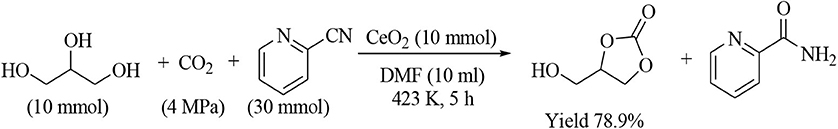
Scheme 1. Glycerol carbonate synthesis from CO2 and glycerol using CeO2 and 2-cyanopyridine (Liu et al., 2016).
The system of CeO2 + nitriles has been also applied to the direct copolymerization of CO2 and diols. It has been recently demonstrated that the copolymerization of CO2 and diols using CeO2 catalyst and 2-cyanopyridine promotor, providing the alternating cooligomers in high diol-based yield (up to 99%) and selectivity (up to >99%). This catalyst system is applicable to various diols including linear C4-C10 α,ω-diols to provide high yields of the corresponding cooligomers, which cannot be obtained by conventional methods such as copolymerization of CO2 and cyclic ethers and ring-opening polymerization of cyclic carbonates (Tamura et al., 2016b). In addition, the reaction of CO2 with 1,4-butanediol in the presence of 2-cyanopyridine was studied by using CeO2 with different morphologies such as nanorods (79 m2 g−1), nanocubes (18 m2 g−1), nanoparticles (59 m2 g−1), and submicroparticles (5 m2 g−1) (Gong et al., 2020). CeO2 nanorods catalyst was more effective than CeO2 with other different morphologies. The catalytic performance of CeO2 nanorods from the viewpoint of activity, selectivity, and polymerization degree was actually comparable to that in our previous report (Tamura et al., 2016b). In contrast, the effect of nitrile was more remarkable. The synthesis of alternating polycarbonates with higher molecular weight from CO2 and diols by using the catalyst system of CeO2 + 2-furonitrile than the case of CeO2 + 2-cyanopyridine (Gu et al., 2019b). Table 7 lists the comparison between 2-cyanopyridine and 2-furonitrile in the copolymerization of CO2 and diols over CeO2 catalyst under the similar reaction conditions. It is clear that 2-furonitrile is more effective than 2-cyanopyridine, from the viewpoint of the polymerization, although the reaction rate on CeO2 + 2-furonitrile is clearly lower than that of CeO2 + 2-cyanopyridine as shown above in the case of DMC synthesis. The effectiveness of 2-furonitrile is explained by two factors. First, the reactivity of 2-furamide, which is produced by the hydration of 2-furonitrile, with OH groups of polycarbonate diols leads to low selectivity of the ester-capped polycarbonates and this can increase the polymerization degree. The other is the weak adsorption of 2-furonitrile on CeO2, decreasing the steric hindrance at the active sites of CeO2 and enabling the reaction of longer diols, such as polycarbonate diols (Gu et al., 2019b).
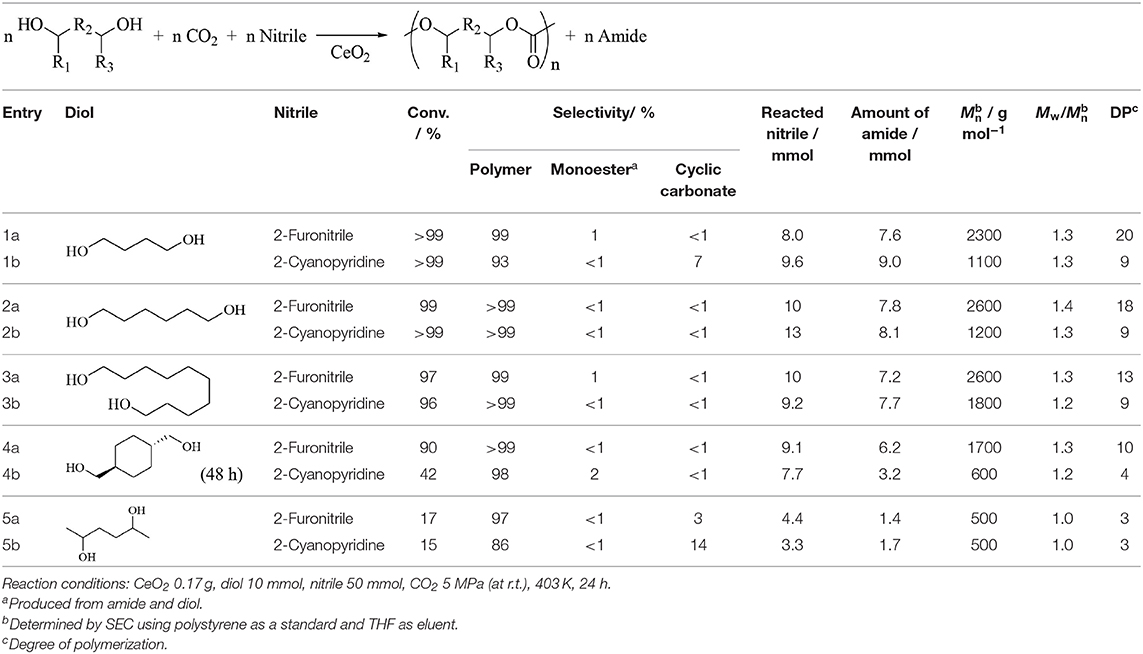
Table 7. Comparison between 2-cyanopyridine and 2-furonitrile in copolymerization of CO2 and diols on CeO2 (Gu et al., 2019b).
Summary and Outlook
The direct synthesis of organic carbonates from CO2 and alcohols is a challenging subject, which includes two key issues: development of catalysts and H2O removal. This article is dealing with CeO2 catalyst and the combination with the nitrile hydration mainly. A variety of heterogeneous catalysts have been developed for the organic carbonates from CO2 and alcohols. In these studies, methanol has been used as an alcohol and the application of each heterogeneous catalyst to wide scope of alcohols is hardly demonstrated. In contrast, it is demonstrated that CeO2 is applicable to the carbamate synthesis from CO2, alcohols, amines, and the urea synthesis from CO2 and amines as well as linear-, cyclic- and poly-carbonates from CO2 and alcohols. It is recommended that the applicability and the limitation of the catalysts should be demonstrated in each study. These results can be very useful for further development of catalysts and H2O removal methods. At present, the system of CeO2 + nitriles has limitation in the synthesis of organic carbonates from CO2 and alcohols: examples of reactions not achieved include the linear carbonate formation from CO2 and secondary alcohols, the synthesis of diphenyl carbonate from CO2 and phenol, the synthesis of seven-membered ring carbonate from CO2 and diols, the synthesis of polycarbonate with higher molecular weight, and so on. Moreover, the dehydration of picolinamide to 2-cyanopyridine is very important from the practical viewpoint. At present, the researches on the dehydration of picolinamide is so limited, and the catalyst development is essential. It is may be possible to replace 2-cyanopyridine by other nitriles or organic dehydrating agents with easier regeneration ability. As mentioned above, 2-furonitrile is one of the candidates, however, the dehydration of 2-furamide is not investigated.
The evaluation of the catalytic performance in the organic carbonate synthesis from CO2 and alcohols using fixed bed flow reactors is more important from the practical viewpoint. This can give the information on the catalyst deactivation profiles, which can be also connected to the further development of catalysts. This kind of studies have been conducted recently (Stoian et al., 2017, 2018), where the visual inspection was combined with IR and Raman spectroscopic studies to identify the origin of the catalyst deactivation and establish an efficient catalyst reactivation protocol.
Process design for the synthesis of carbonates from CO2 and alcohols and the evaluation of the process by techno-economic and CO2 emission assessment are necessary. We feel that this kind of study on non-reductive CO2 conversion including the synthesis of organic carbonates is increased steadily. It will be more important to develop the catalytic system on the basis of the suggestions from the studies on the process design and techno-economic analysis.
Author Contributions
KT made manuscript design and wrote the paper. YG summarized the literature. YN carried out DFT calculation and made discussion in theoretical view. MT made discussion in view of materials chemistry. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Akune, T., Morita, Y., Shirakawa, S., Katagiri, K., and Inumaru, K. (2018). ZrO2 nanocrystals as catalyst for synthesis of dimethyl carbonate from methanol and carbon dioxide: catalytic activity and elucidation of active sites. Langmuir 34, 23–29. doi: 10.1021/acs.langmuir.7b01294
Arbeláez-Pérez, O. F., Domínguez-Cardozo, S., Orrego-Romero, A. F., Villa-Holguín, A. L., and Bustamante, F. (2020). Gas phase synthesis of dimethyl carbonate from CO2 and CH3OH over Cu-Ni/AC. A kinetic study. Revista Facultad de Ingeniería Universidad de Antioquia. 95, 88–99. doi: 10.17533/udea.redin.20190941
Aresta, M., Dibenedetto, A., Pastore, C., Angelini, A., Aresta, B., and Papai, I. (2010). Influence of Al2O3 on the performance of CeO2 used as catalyst in the direct carboxylation of methanol to dimethyl carbonate and the elucidation of the reaction mechanism. J. Catal. 269, 44–52. doi: 10.1016/j.jcat.2009.10.014
Bansode, A., and Urakawa, A. (2014). Continuous DMC synthesis from CO2 and methanol over a CeO2 catalyst in a fixed bed reactor in the presence of a dehydrating agent. ACS Catal. 4, 3877–3880. doi: 10.1021/cs501221q
Chaemchuen, S., Semyonov, O. V., Dingemans, J., Xu, W., Zhuiykov, S., Khan, A., et al. (2019). Progress on catalyst development for direct synthesis of dimethyl carbonate from CO2 and methanol. Chem. Africa 2, 533–549. doi: 10.1007/s42250-019-00082-x
Chiang, C.-L., Lin, K.-S., and Yu, S.-H. (2019). Improvement of dimethyl carbonate formation via methanol carbonation over vanadium-doped Cu–Ni/AC catalyst. J. Taiwan Inst. Chem. Eng. 98, 132–149. doi: 10.1016/j.jtice.2018.08.001
Choi, J. C., Kohno, K., Ohshima, Y., Yasuda, H., and Sakakura, T. (2008). Tin- or titanium-catalyzed dimethyl carbonate synthesis from carbon dioxide and methanol: large promotion by a small amount of triflate salts. Catal. Commun. 9, 1630–1633. doi: 10.1016/j.catcom.2008.01.013
Dibenedetto, A., Aresta, M., Angelini, A., Ethiraj, J., and Aresta, B. M. (2012). Synthesis, characterization, and use of NbV/CeIV-mixed oxides in the direct carboxylation of ethanol by using pervaporation membranes for water removal. Chem. Eur. J. 18, 10324–10334. doi: 10.1002/chem.201201561
Fukuoka, S., Kawamura, M., Komiya, K., Tojo, M., Hachiya, H., Hasegawa, K., et al. (2003). A novel non-phosgene polycarbonate production process using by-product CO2 as starting material. Green Chem. 5, 497–507. doi: 10.1039/B304963A
Ghorbel, S. B., Medina, F., Ghorbel, A., and Segarra, A. M. (2015). Phosphoric acid intercalated Mg–Al hydrotalcite-like compounds for catalytic carboxylation reaction of methanol in a continuous system. Appl. Catal. A. 493, 142–148. doi: 10.1016/j.apcata.2015.01.004
Gong, Z.-J., Li, Y.-R., Wu, H.-L., Lin, S. D., and Yu, W.-Y. (2020). Direct copolymerization of carbon dioxide and 1,4-butanediol enhanced by ceria nanorod catalyst. Appl. Catal. B 265:118524. doi: 10.1016/j.apcatb.2019.118524
Greish, A. A., Finashina, E. D., Tkachenko, O. P., Shuvalova, E. V., and Kustov, L. M. (2016). Synthesis of dimethyl carbonate from methanol and CO2 on the SnO2/Al2O3-based catalyst. Mendeleev Commun. 26, 497–499. doi: 10.1016/j.mencom.2016.11.012
Gu, Y., Matsuda, K., Nakayama, A., Tamura, M., Nakagawa, Y., and Tomishige, K. (2019b). Direct synthesis of alternating polycarbonates from CO2 and diols by using a catalyst system of CeO2 and 2-furonitrile. ACS Sustain. Chem. Eng. 7, 6304–6315. doi: 10.1021/acssuschemeng.8b06870
Gu, Y., Miura, A., Tamura, M., Nakagawa, Y., and Tomishige, K. (2019a). Highly efficient synthesis of alkyl N-arylcarbamates from CO2, anilines, and branched alcohols with a catalyst system of CeO2 and 2-cyanopyridine. ACS Sustain. Chem. Eng. 7, 16795–16802. doi: 10.1021/acssuschemeng.9b04318
Honda, M., and Abe, H. (2018). Development of a H3PW12O40/CeO2 catalyst for bulk ring-opening polymerization of a cyclic carbonate. Green Chem. 20, 4995–5006. doi: 10.1039/C8GC01909F
Honda, M., Kuno, S., Begum, N., Fujimoto, K., Suzuki, K., Nakagawa, Y., et al. (2010). Catalytic synthesis of dialkyl carbonate from low pressure CO2 and alcohols combined with acetonitrile hydration catalyzed by CeO2. Appl. Catal. A 384, 165–170. doi: 10.1016/j.apcata.2010.06.033
Honda, M., Kuno, S., Sonehara, S., Fujimoto, K., Suzuki, K., Nakagawa, Y., et al. (2011a). Tandem carboxylation-hydration reaction system from methanol, CO2 and benzonitrile to dimethyl carbonate and benzamide catalyzed by CeO2. ChemCatChem 3, 365–370. doi: 10.1002/cctc.201000339
Honda, M., Sonehara, S., Yasuda, H., Nakagawa, Y., and Tomishige, K. (2011b). Heterogeneous CeO2 catalyst for the one-pot synthesis of organic carbamates from amines, CO2 and alcohols. Green Chem. 13, 3406–3413. doi: 10.1039/c1gc15646b
Honda, M., Suzuki, A., Noorjahan, B., Fujimoto, K., Suzuki, K., and Tomishige, K. (2009). Low pressure CO2 to dimethyl carbonate by the reaction with methanol promoted by acetonitrile hydration. Chem. Commun. 4596–4598. doi: 10.1039/b909610h
Honda, M., Tamura, M., Nakagawa, Y., Nakao, K., Suzuki, K., and Tomishige, K. (2014c). Organic carbonate synthesis from CO2 and alcohol over CeO2 with 2-cyanopyridine: scope and mechanistic studies. J. Catal. 318, 95–107. doi: 10.1016/j.jcat.2014.07.022
Honda, M., Tamura, M., Nakagawa, Y., Sonehara, S., Suzuki, K., Fujimoto, K., et al. (2013). Ceria-catalyzed conversion of carbon dioxide into dimethyl carbonate with 2-cyanopyridine. ChemSusChem 6, 1341–1344. doi: 10.1002/cssc.201300229
Honda, M., Tamura, M., Nakagawa, Y., and Tomishige, K. (2014a). Catalytic CO2 conversion to organic carbonates with alcohols in combination with dehydration system. Catal. Sci. Technol. 4, 2830–2845. doi: 10.1039/C4CY00557K
Honda, M., Tamura, M., Nakao, K., Suzuki, K., Nakagawa, Y., and Tomishige, K. (2014b). Direct cyclic carbonate synthesis from CO2 and diol over carboxylation/hydration cascade catalyst of CeO2 with 2-cyanopyridine. ACS Catal. 4, 1893–1896. doi: 10.1021/cs500301d
Hou, Z. Q., Luo, L. G., Liu, K., Liu, C. Z., Wang, Y. Y., and Dai, L. Y. (2014). High-yield synthesis of dimethyl carbonate from the direct alcoholysis of urea in supercritical methanol. Chem. Eng. J. 236, 415–418. doi: 10.1016/j.cej.2013.09.024
Huang, S. Y., Yan, B., Wang, S. P., and Ma, X. B. (2015). Recent advances in dialkyl carbonates synthesis and applications. Chem. Soc. Rev. 44, 3079–3116. doi: 10.1039/C4CS00374H
Ikeda, Y., Asadullah, M., Fujimoto, K., and Tomishige, K. (2001). Structure of the active sites on H3PO4/ZrO2 catalysts for dimethyl carbonate synthesis from methanol and carbon dioxide. J. Phys. Chem. B 105, 10653–10658. doi: 10.1021/jp0121522
Ikeda, Y., Sakaihori, T., Tomishige, K., and Fujimoto, K. (2000). Promoting effect of phosphoric acid on zirconia catalysts in selective synthesis of dimethyl carbonate from methanol and carbon dioxide. Catal. Lett. 66, 59–62. doi: 10.1023/A:1019043422050
Kumar, S., Khatri, O. P., Cordier, S., Boukherroub, R., and Jain, S. L. (2015). Graphene oxide supported molybdenum cluster: first heterogenized homogeneous catalyst for the synthesis of dimethyl carbonate from CO2 and methanol. Chem. Eur. J. 21, 3488–3494. doi: 10.1002/chem.201404949
Kumar, S., Kumar, P., and Jain, S. L. (2014). Graphene oxide immobilized copper phthalocyanine tetrasulphonamide: the first heterogenized homogeneous catalyst for dimethyl carbonate synthesis from CO2 and methanol. J. Mater. Chem. A 2, 18861–18866. doi: 10.1039/C4TA03420A
Leino, E., Kumar, N., Mäki-Arvela, P., Rautio, A.-R., Dahl, J., Roine, J., et al. (2018). Synthesis and characterization of ceria-supported catalysts for carbon dioxide transformation to diethyl carbonate. Catal. Today 306, 128–137. doi: 10.1016/j.cattod.2017.01.016
Leino, E., Mäki-Arvela, P., Eränen, K., Tenho, M., Murzin, D. Y., et al. (2011). Enhanced yields of diethyl carbonate via one-pot synthesis from ethanol, carbon dioxide and butylene oxide over cerium (IV) oxide. Chem. Eng. J. 176–177, 124–133. doi: 10.1016/j.cej.2011.07.054
Leino, E., Mäki-Arvela, P., Eta, V., Kumar, N., Demoisson, F., Samikannu, A., et al. (2013). The influence of various synthesis methods on the catalytic activity of cerium oxide in one-pot synthesis of diethyl carbonate starting from CO2, ethanol and butylene oxide. Catal. Today 210, 47–54. doi: 10.1016/j.cattod.2013.02.011
Leino, E., Mäki-Arvela, P., Eta, V., Murzin, D. Y., Salmi, T., and Mikkola, J.-P. (2010). Conventional synthesis methods of short-chain dialkylcarbonates and novel production technology via direct route from alcohol and waste CO2. Appl. Catal. A 383, 1–13. doi: 10.1016/j.apcata.2010.05.046
Li, A.-X., Pu, Y.-F., Li, F., Luo, J., Zhao, N., and Xiao, F.-K. (2017). Synthesis of dimethyl carbonate from methanol and CO2 over Fe–Zr mixed oxides. J. CO2 Util. 19, 33–39. doi: 10.1016/j.jcou.2017.02.016
Liu, J., Guo, H. T., Zhou, Q. B., Wang, J. Y., Lin, B. K., Zhang, H. B., et al. (2013). Highly efficient enzymatic preparation for dimethyl carbonate catalyzed by lipase from penicillium expansum immobilized on CMC–PVA film. J. Mol. Catal. B 96, 96–102. doi: 10.1016/j.molcatb.2013.06.013
Liu, J. X., Li, Y. M., Zhang, J., and He, D. H. (2016). Glycerol carbonylation with CO2 to glycerol carbonate over CeO2 catalyst and the influence of CeO2 preparation methods and reaction parameters. Appl. Catal. A 513, 9–18. doi: 10.1016/j.apcata.2015.12.030
Marciniak, A. A., Alves, O. C., Appel, L. G., and Mota, C. J. A. (2019). Synthesis of dimethyl carbonate from CO2 and methanol over CeO2: role of copper as dopant and the use of methyl trichloroacetate as dehydrating agent. J. Catal. 371, 88–95. doi: 10.1016/j.jcat.2019.01.035
Ono, Y. (1997). Catalysis in the production and reactions of dimethyl carbonate, an environmentally benign building block. Appl. Catal. A. 155, 133–166. doi: 10.1016/S0926-860X(96)00402-4
Poungsombate, A., Imyen, T., Dittanet, P., Embley, B., and Kongkachuichay, P. (2017). Direct synthesis of dimethyl carbonate from CO2 and methanol by supported bimetallic Cu–Ni/ZIF-8 MOF catalysts. J. Taiwan Inst. Chem. Eng. 80, 16–24. doi: 10.1016/j.jtice.2017.07.019
Santos, B. A. V., Silva, V. M. T. M., Loureiro, J. M., and Rodrigues, A. E. (2014). Review for the direct synthesis of dimethyl carbonate. ChemBioEng Rev. 1, 214–229. doi: 10.1002/cben.201400020
Shukla, K., and Srivastava, V. C. (2017). Synthesis of organic carbonates from alcoholysis of urea: a review. Catal. Rev. Sci. Eng. 59, 1–43. doi: 10.1080/01614940.2016.1263088
Stoian, D., Bansode, A., Medina, F., and Urakawa, A. (2017). Catalysis under microscope: Unraveling the mechanism of catalyst de- and re-activation in the continuous dimethyl carbonate synthesis from CO2 and methanol in the presence of a dehydrating agent. Catal. Today 283, 2–10. doi: 10.1016/j.cattod.2016.03.038
Stoian, D., Medina, F., and Urakawa, A. (2018). Improving the stability of CeO2 catalyst by rare earth metal promotion and molecular insights in the dimethyl carbonate synthesis from CO2 and methanol with 2-cyanopyridine. ACS Catal. 8, 3181–3193. doi: 10.1021/acscatal.7b04198
Sun, W., Zheng, L., Wang, Y.-Q., Li, D.-D., Liu, Z.-R., Wu, L., et al. (2020). A study for thermodynamics and experiment on direct synthesis of dimethyl carbonate from carbon dioxide and methanol over yttrium oxide. Ind. Eng. Chem. Res. 59, 4281–4290. doi: 10.1021/acs.iecr.9b06092
Tamboli, A. H., Chaugule, A. A., Gosavi, S. W., and Kim, H. (2018). CexZr1−−xO2 solid solutions for catalytic synthesis of dimethyl carbonate from CO2: reaction mechanism and the effect of catalyst morphology on catalytic activity. Fuel. 216, 245–254. doi: 10.1016/j.fuel.2017.12.008
Tamura, M., Honda, M., Nakagawa, Y., and Tomishige, K. (2014). Direct conversion of CO2 with diols, aminoalcohols and diamines to cyclic carbonates, cyclic carbamates and cyclic ureas using heterogeneous catalysts. J. Chem. Technol. Biotechnol. 89, 19–33. doi: 10.1002/jctb.4209
Tamura, M., Honda, M., Noro, K., Nakagawa, Y., and Tomishige, K. (2013a). Heterogeneous CeO2-catalyzed selective synthesis of cyclic carbamates from CO2 and aminoalcohols in acetonitrile solvent. J. Catal. 305, 191–203. doi: 10.1016/j.jcat.2013.05.013
Tamura, M., Ito, K., Honda, M., Nakagawa, Y., Sugimoto, H., and Tomishige, K. (2016b). Direct copolymerization of CO2 and diols. Sci. Rep. 6:24038. doi: 10.1038/srep24038
Tamura, M., Ito, K., Nakagawa, Y., and Tomishige, K. (2016a). CeO2-catalyzed direct synthesis of dialkylureas from CO2 and amines. J. Catal. 343, 75–85. doi: 10.1016/j.jcat.2015.11.015
Tamura, M., Kishi, R., Nakagawa, Y., and Tomishige, K. (2015). Self-assembled hybrid metal oxide base catalysts prepared by simply mixing with organic modifiers. Nat. Commun. 6:8580. doi: 10.1038/ncomms9580
Tamura, M., Kishi, R., Nakayama, A., Nakagawa, Y., Hasegawa, J.-,y., and Tomishige, K. (2017). Formation of a new, strongly basic nitrogen anion by metal oxide modification. J. Am. Chem. Soc. 139, 11857–11867. doi: 10.1021/jacs.7b05227
Tamura, M., Matsuda, K., Nakagawa, Y., and Tomishige, K. (2018b). Ring-opening polymerization of trimethylene carbonate to poly(trimethylene carbonate) diol over a heterogeneous high-temperature calcined CeO2 catalyst. Chem. Commun. 54, 14017–14020. doi: 10.1039/C8CC08405J
Tamura, M., Miura, A., Honda, M., Gu, Y., Nakagawa, Y., and Tomishige, K. (2018a). Direct catalytic synthesis of N-arylcarbamates from CO2, anilines and alcohols. ChemCatChem. 10, 4821–4825. doi: 10.1002/cctc.201801443
Tamura, M., Noro, K., Honda, M., Nakagawa, Y., and Tomishige, K. (2013b). Highly efficient synthesis of cyclic ureas from CO2 and diamines by a pure CeO2 catalyst using a 2-propanol solvent. Green Chem. 15, 1567–1577. doi: 10.1039/c3gc40495a
Tamura, M., and Tomishige, K. (2015). Redox properties of CeO2 at low temperature: the direct synthesis of imines from alcohol and amine. Angew. Chem. Int. Ed. 54, 864–867. doi: 10.1002/anie.201409601
Tomishige, K., Furusawa, Y., Ikeda, Y., Asadullah, M., and Fujimoto, K. (2001). CeO2-ZrO2 solid solution catalyst for selective synthesis of dimethyl carbonate from methanol and carbon dioxide. Catal. Lett. 76, 71–74. doi: 10.1023/A:1016711722721
Tomishige, K., Gu, Y., Chang, T., Tamura, M., and Nakagawa, Y. (2020). Catalytic function of CeO2 in non-reductive conversion of CO2 with alcohols. Mater. Today Sustain. 9:100035. doi: 10.1016/j.mtsust.2020.100035
Tomishige, K., Ikeda, Y., Sakaihori, T., and Fujimoto, K. (2000). Catalytic properties and structure of zirconia catalysts for direct synthesis of dimethyl carbonate from methanol and carbon dioxide. J. Catal. 192, 355–362. doi: 10.1006/jcat.2000.2854
Tomishige, K., and Kunimori, K. (2002). Catalytic and direct synthesis of dimethyl carbonate starting from carbon dioxide using CeO2-ZrO2 solid solution heterogeneous catalyst: effect of H2O removal from the reaction system. Appl. Catal. A 237, 103–109. doi: 10.1016/S0926-860X(02)00322-8
Tomishige, K., Sakaihori, T., Ikeda, Y., and Fujimoto, K. (1999). A novel method of direct synthesis of dimethyl carbonate from methanol and carbon dioxide catalyzed by zirconia. Catal. Lett. 58, 225–229. doi: 10.1023/A:1019098405444
Tomishige, K., Tamura, M., and Nakagawa, Y. (2019). CO2 Conversion with alcohols and amines into carbonates, ureas, and carbamates over CeO2 catalyst in the presence and absence of 2-cyanopyridine. Chem. Rec. 19, 1354–1379. doi: 10.1002/tcr.201800117
Tomishige, K., Yasuda, H., Yoshida, Y., Nurunnabi, M., Li, B. T., and Kunimori, K. (2004a). Catalytic performance and properties of ceria based catalysts for cyclic carbonate synthesis from glycol and carbon dioxide. Green Chem. 6, 206–214. doi: 10.1039/b401215a
Tomishige, K., Yasuda, H., Yoshida, Y., Nurunnabi, M., Li, B. T., and Kunimori, K. (2004b). Novel route to propylene carbonate: selective synthesis from propylene glycol and carbon dioxide. Catal. Lett. 95, 45–49. doi: 10.1023/B:CATL.0000023720.39110.4e
Verma, S., Baig, R. B. N., Nadagouda, M. N., and Varma, R. S. (2017). Fixation of carbon dioxide into dimethyl carbonate over titanium-based zeolitic thiophenebenzimidazolate framework. Sci. Rep. 7:655. doi: 10.1038/s41598-017-00736-1
Xuan, K., Pu, Y.-F., Li, F., Li, A.-X., Luo, J., Li, L., et al. (2018). Direct synthesis of dimethyl carbonate from CO2 and methanol over trifluoroacetic acid modulated UiO-66. J. CO2 Util. 27, 272–282. doi: 10.1016/j.jcou.2018.08.002
Xuan, K., Pu, Y.-F., Li, F., Luo, J., Zhao, N., and Xiao, F.-K. (2019). Metal-organic frameworks MOF-808-X as highly efficient catalysts for direct synthesis of dimethyl carbonate from CO2 and methanol. Chin. J. Catal. 40, 553–566. doi: 10.1016/S1872-2067(19)63291-2
Yoshida, Y., Arai, Y., Kado, S., Kunimori, K., and Tomishige, K. (2006). Direct synthesis of organic carbonates from the reaction of CO2 with methanol and ethanol over CeO2 catalysts. Catal. Today 115, 95–101. doi: 10.1016/j.cattod.2006.02.027
Zhang, X.-Z., Jia, D.-D, Zhang, J., and Sun, Y.-Y. (2014). Direct synthesis of diethyl carbonate from CO2 and ethanol catalyzed by ZrO2/molecular sieve. Catal. Lett. 144, 2144–2150. doi: 10.1007/s10562-014-1403-5
Keywords: carbon dioxide, ceria, alcohol, carbonate, equilibrium shift, nitrile hydration
Citation: Tomishige K, Gu Y, Nakagawa Y and Tamura M (2020) Reaction of CO2 With Alcohols to Linear-, Cyclic-, and Poly-Carbonates Using CeO2-Based Catalysts. Front. Energy Res. 8:117. doi: 10.3389/fenrg.2020.00117
Received: 20 March 2020; Accepted: 18 May 2020;
Published: 23 June 2020.
Edited by:
Michele Aresta, IC2R srl, ItalyReviewed by:
Kenichi Shimizu, Hokkaido University, JapanJordi Llorca, Universitat Politecnica de Catalunya, Spain
Copyright © 2020 Tomishige, Gu, Nakagawa and Tamura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Keiichi Tomishige, dG9taUBlcmVjLmNoZS50b2hva3UuYWMuanA=