
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Energy Res. , 30 March 2016
Sec. Electrochemical Energy Storage
Volume 4 - 2016 | https://doi.org/10.3389/fenrg.2016.00010
This article is part of the Research Topic Fast ionic conductors and solid-solid interfaces designed for next generation solid-state batteries View all 15 articles
 Kazunori Takada1,2*
Kazunori Takada1,2* Takahisa Ohno2,3
Takahisa Ohno2,3
Solid-state batteries with inorganic solid electrolytes are expected to be an efficient solution to the issues of current lithium-ion batteries that are originated from their organic-solvent electrolytes. Although solid-state batteries had been suffering from low rate capability due to low ionic conductivities of solid electrolytes, some sulfide solid electrolytes exhibiting high ionic conductivity of the order of 10−2 S cm−1 have been recently developed. Since the conductivity is comparable to or even higher than that of liquid electrolytes, when taking the transport number of unity into account, ion transport in solid electrolytes has ceased from rate determination; however, it has been replaced by that across interfaces. The sulfide electrolytes show high interfacial resistance to the high-voltage cathodes. Our previous studies have demonstrated that oxide solid electrolytes interposed at the interface reduce the resistance, and they also suggest that the high resistance is attributable to a lithium-depleted layer formed at the interface. This study employs the first-principles calculation in order to gain insight into the interface. The interface structure between an oxide cathode/sulfide electrolyte simulated by the first-principles molecular dynamics has disclosed the presence of lithium-depleted layer at the interface, and the electronic structure calculated on the basis of density functional theory strongly suggests that the charge current preferentially removes lithium ions from the sulfide electrolyte side of the interface to deplete the lithium ion there. These calculation results are consistent with the transport mechanism proposed from the experimental results.
Lithium-ion batteries predominate in today’s rechargeable battery systems because of their high energy densities. They have been used portable electronics devices including cellular phones and notebook PC’s and serving as a key component in this information society for more than 20 years. Moreover, they start to be installed in vehicles and smart grids for realizing a low-carbon society; however, some issues including safety originating from the thermal and electrochemical instability of their organic-solvent electrolytes have not gone out completely. Because inorganic solid electrolytes exhibit higher thermal and electrochemical stability, use of the solid electrolytes is expected to be an efficient solution (Tarascon and Armand, 2001), even if the electrochemical stability is attributed to the kinetic stabilization as suggested in Zhu et al. (2015). However, solid-state lithium batteries had been suffering from low power densities.
Essential materials to improve the power densities are, of course, solid electrolytes with high ionic conductivities. Since ions generally diffuse faster in liquids than in solids, the use of solid electrolytes lowers the power densities. However, high ionic conductivities that are comparable to that in liquid systems have been achieved among sulfide solid electrolytes. Sulfide ions are larger in size and higher in polarizability than oxide counterparts. The large ionic radius leaves wide opening for ionic conduction, and the higher polarizability weakens attraction between lithium and sulfide ions to make the lithium ions mobile (Zheng et al., 2003). Therefore, high ionic conductivities have been often observed in sulfides, as reviewed in Minami et al. (2006) and Takada (2013b), and the highest conductivities in sulfide electrolyte systems have recently reached 10−2 S cm−1 (Kamaya et al., 2011; Seino et al., 2014).
In addition, sulfide solid electrolytes exhibit high processability. They are relatively soft materials (Sakuda et al., 2013) and show low grain boundary resistance only by compression. Therefore, sintering process is not essential to connect the particles, whereas high temperature sintering is inevitable to connect ionic conduction between particles in oxide solid electrolytes, for instance. Besides the high ionic conductivity, this feature is beneficial for developing solid-state batteries, because high temperature sintering promotes mutual diffusion that forms impurity phases at the interface. However, even such solid electrolytes had not improve the power density to be high enough for practical application by themselves.
As reported in Takada et al. (2008), when a sulfide electrolyte is combined with graphite and LiCoO2, which are typical anode and cathode materials, respectively, to assemble a lithium-ion-type solid-state battery, high resistance is found in its cathode. Because the resistance is the highest in the battery to be rate-determining in the battery reaction, power density will not be improved without reducing the cathode resistance. Ohta et al. (2006) integrates a novel interface structure between the sulfide electrolyte and LiCoO2 into the cathode in order to improve the power density to a practical level. In the interface structure, surface of the LiCoO2 particles is coated with Li4Ti5O12, which act as an oxide solid electrolyte on the surface. This surface coating has reduced the cathode resistance by 1/20 and increased the power density to be as high as that of commercial lithium-ion batteries with liquid electrolytes. The interface is designed on the basis of “nanoionics” (Maier, 1995).
Ionic conductors often exhibit anomalous ionic conduction at their surface or interface. It is explained that the anomalous ionic conduction is induced by the changing concentration of mobile ions to form space-charge layers at the interface. Nanoionics has been often reported as enhanced ionic conduction at interfaces, as reviewed in Maier (2014). On the other hand, Takada (2013a) suggests that the space-charge layer appears as a lithium-depleted layer at the cathode interface in solid-state batteries with sulfide electrolytes. Noble potential of the cathode material depletes lithium ions of the sulfide electrolyte at the interface to the cathode material and makes the interface highly resistive. The surface coating, which interposes the oxide electrolyte between the high-voltage cathode and sulfide electrolyte materials, suppresses the lithium depletion to reduce the interfacial resistance.
Although the changes in the electrode properties upon the surface coating are well explainable by the interface model based on nanoionics, origin of the high interfacial resistance and its reduction are still under debate, because changes in the interfacial structure upon the coating, which are predicted by the interface model, have not been visualized yet. Since the space-charge layer lies between solids with only several nanometers in thickness, it has never shown up even on advanced analyses. This study employs computational science using the first-principle calculations in order to get a clear picture of the interfacial phenomena. Results from density functional theory (DFT) calculation for the interfaces to Li1−xFePO4 cathode materials are combined with experimental results from the interfaces to Li1−xCoO2 and Li1−xMn2O4 in Ohta et al. (2007) and Takada et al. (2012) to reveal the validity of the space-charge layer model.
The materials presented in this paper have been already reported in Ohta et al. (2007) and Takada et al. (2012). Thin layer of LiNbO3 was formed on the particle surface of LiCoO2 and LiMn2O4 from an ethanol solution of alkoxides of Li and Nb by a rolling fluidized coating method. The LiCoO2 and LiMn2O4 powders are commercially available (D10 and M811, respectively; Toda Kogyo). Average particle size and Brunauer–Emmett–Teller (BET) surface area of the LiCoO2 powder are 11.2 μm and 0.26 m2 g−1, respectively, while those of the LiMn2O4 powder are 5 μm and 0.40 m2 g−1, respectively. Alkoxides used to form LiNbO3 layer are lithium ethoxide and niobium pentaethoxide. The amount of LiNbO3 coated on the surface varies from 0 (uncoated) to 94 mg m−2. The maximum amount corresponds to LiNbO3 layer of 20 nm in thickness, if the surface of active material particles is uniformly covered with the LiNbO3.
The sulfide solid electrolyte to make the interface to the cathode materials in this study is Li3.25Ge0.25P0.75S4 (Kanno and Murayama, 2001). It is mixed with the LiCoO2 and the LiMn2O4 at weight ratios of 3:7 and 1:1, respectively, to form the interfaces in the composite electrodes. Solid-state cells for the electrochemical measurements are assembled from the mixtures (12.7 mg for the LiCoO2 and 15 mg for the LiMn2O4) as the working electrodes, In–Li alloy as the counter electrodes and Li3.25Ge0.25P0.75S4 as the separators. Because electrode potential of the In–Li electrode is 0.62 V vs. Li+/Li, cell voltage is presented by adding 0.62 V in this paper as if the electrode properties were investigated in electrochemical cells with lithium metal anodes. The LiCoO2 cathodes are charged to 4.2 V vs. Li+/Li, that is, 3.58 V in the cell voltage at a constant current of 127.4 μA cm−2, while the LiMn2O4 are charged to 4.3 V vs. Li+/Li at 20 μA cm−2, before the electrochemical impedance spectroscopy to evaluate the electrode impedance.
The cathode material used in the computations is LiFePO4. Li3PO4 and Li3PS4 are selected as oxide and sulfide-based solid electrolytes, respectively, in order to compare cathode/oxide electrolyte and cathode/sulfide electrolyte interfaces. The LiFePO4/Li3PO4 interface is constructed between LiFePO4 (010) and γ-Li3PO4 (100) planes by the computation method reported in Sumita et al. (2015) and Sumita et al. (2016).
On the other hand, because lattice mismatch between LiFePO4 and Li3PS4 is too large to form a coherent interface, Li3PS4 is amorphized on the LiFePO4 (010) plane by first principles molecular dynamics simulation (FP-MD) to form the interface as follows. The amorphous Li3PS4 is constructed from 16 Li3PS4 units stacked on (010) plane of a 1 × 3 × 2 supercell of LiFePO4. The interface is stabilized by FP-MF calculation with a constant-temperature, constant-pressure (NPT) ensemble at the temperature of 400 K, and the pressure of 1.0 atom with the valuable cell dimension perpendicular to the interface. The interface structure is equilibrated under the temperature control by canonical sampling through velocity rescaling thermostat (Bussi et al., 2007) for 40 ps.
In this study, cathode interfacial structures during the cell operation are also investigated. Fully charged LiFePO4, i.e., FePO4, is stacked onto Li3PO4 and Li3PS4, respectively, and the interface structures are relaxed by FP-MD simulations with an NPT ensemble at 400 K under 1.0 atom for ca. 200 ps. These interfaces are connected to lithium metal in order to calculate energies relative to its Fermi level.
Computational analyses for these interfaces are according with the procedures in Sumita et al. (2015) and Sumita et al. (2016). Total energies are calculated at the Γ point in the super cell approach within DFT implemented in CP2K at www.cp2k.org. The Goedecker, Teter, and Hutter (GTH) pseudopotentials are selected for PBE functional (Perdew et al., 1996) with the local spin density approximation in this study. Valence pseudowave functions are expanded in Gaussian-type orbitals, and the density is represented in a plane wave auxiliary basis in the hybrid Gaussian (MOLOPT DZVP) and plane-wave (240 Ry for cutoff energy) basis set. The +U strategy is applied to include the electronic correlation within the d orbital of Fe, in which the effective U is set to 4.3 eV as in Wang et al. (2007).
The surface coating reduces the cathode impedance, as demonstrated in Figure 1. Each Nyquist plot consists of an arc followed by a tail in the low frequency region. The impedance at the high frequency limit agrees with the resistance of the solid electrolyte layer calculated from the cross section and the thickness of the electrolyte layer and the ionic conductivity of the solid electrolyte. It is, in general, very difficult to derive intrinsic interfacial resistance from impedance data of composite electrodes; however, it is clear that the surface coating decreases the interfacial resistance. The impedance including the interfacial resistance, which draws an arc in the Nyquist plot, is decreased from 670 to 10 Ω cm2 for the Li1−xCoO2 cathode and from 8000 to 150 Ω cm2 for the Li1−xMn2O4 cathode, that is, by almost two orders of magnitude.
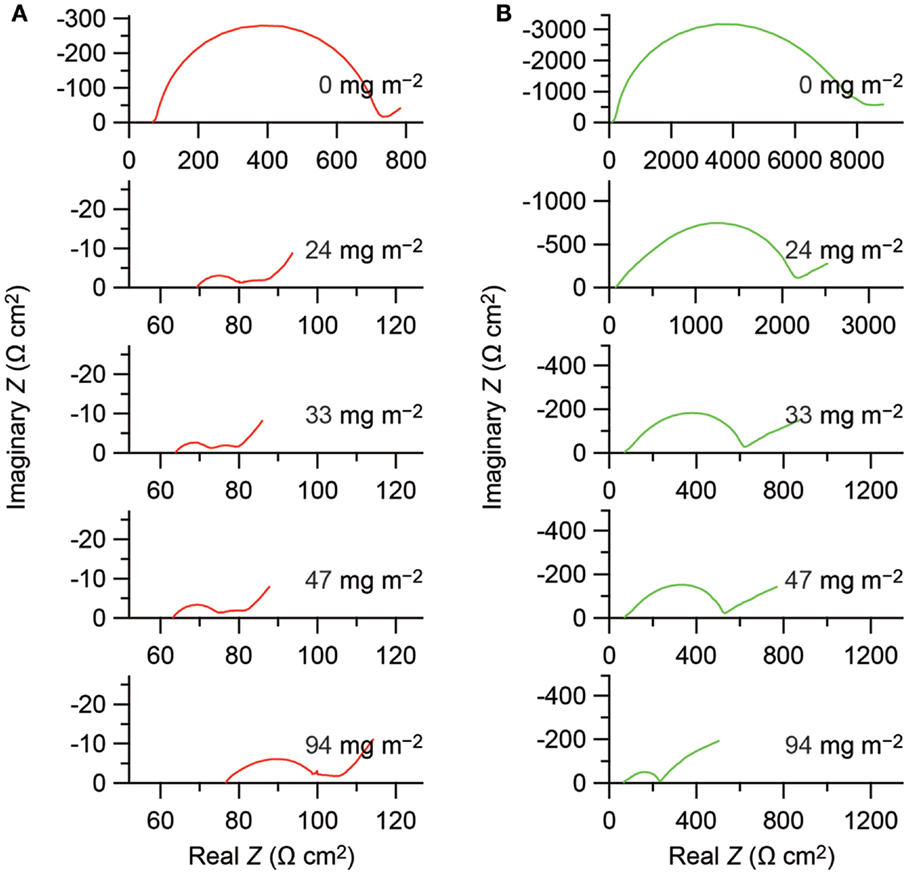
Figure 1. Nyquist plots obtained from the solid-state electrochemical cells with configuration of In–Lix/Li1−xCoO2 (A) and In–Lix/Li1−xMn2O4 (B). Horizontal and vertical axes indicate real and imaginary parts of the complex impedance based on the projection areas of the cathodes, respectively. The amounts of the LiNbO3 coating on the active materials are indicated on the basis of the BET surface areas of the active materials in the figures. Impedance date reported in Ohta et al. (2007) and Takada et al. (2012) are replotted in this figure, copyright © 2007 and 2012, with permission from Elsevier.
The surface coating has another effect on the electrode properties. Lithium ions are deintercalated from LiCoO2 and LiMn2O4 in potential plateaus at 4 V vs. Li+/Li. However, they exhibit additional potential slopes prior to the 4 V plateaus in the sulfide solid electrolyte during the charging, as shown in the top panels of Figure 2. The surface coating changes the length of the slopes. Before surface coating, both of the LiCoO2 and LiMn2O4 show potential slopes with almost the same length of 0.2 μAh cm−2. The increasing amount of LiNbO3 gradually shortens the potential slopes, and the slopes finally disappear at the LiNbO3 amount of 94 mg m−2.
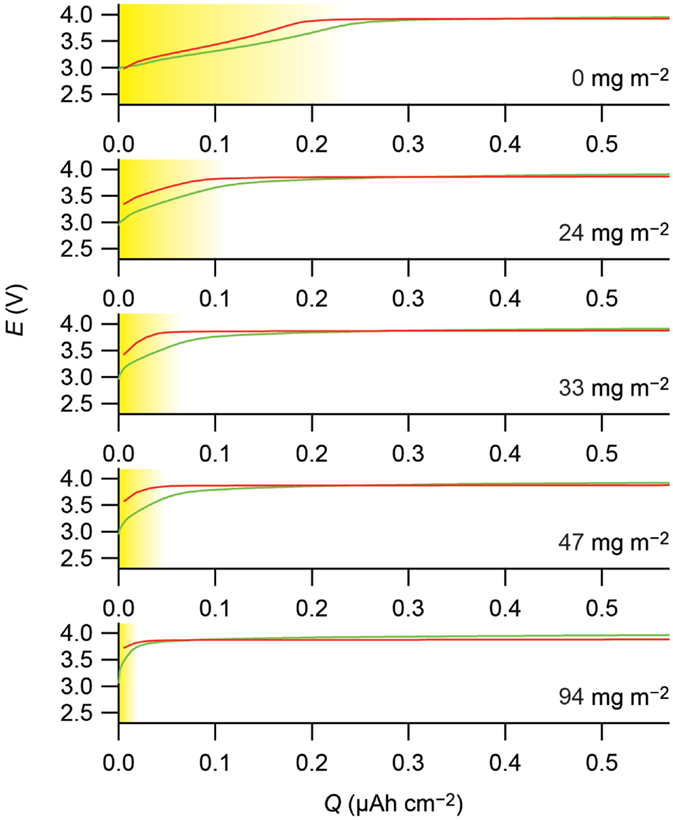
Figure 2. The beginning parts of the charge curves of the cathodes. The curves for the In–Lix/Li1−xCoO2 and In–Lix/Li1−xMn2O4 are shown in red and green, respectively. The horizontal axes indicate the charge capacities normalized by the BET surface areas of the active materials, and the vertical axes indicate the cathode potential calculated by adding 0.62 V, which is the electrode potential vs. Li+/Li of the In–Lix counter electrodes, to the cell voltage. The amounts of the LiNbO3 coated on the active materials are indicated on the basis of the BET surface areas of the active materials in the figures. The potential slopes described in the text are shaded in yellow. Charge curves reported in Ohta et al. (2007) and Takada et al. (2012) are replotted in this figure, copyright © 2007 and 2012, with permission from Elsevier.
The capacities in the potential slopes are very small. For example, the capacity of 0.2 μAh cm−2 based on the BET surface area corresponds only to 0.5 mAh g−1 for the LiCoO2 and 0.8 mAh g−1 for the LiMn2O4. However, the slopes are not artifact. They appear both in LiCoO2 and LiMn2O4 with almost the same capacity based on the BET surface areas of the materials and fade to the same degree with increasing amounts of LiNbO3.
Figure 3 plots density of states (DOS) in LiFePO4, Li3PO4, and Li3PS4 against energy relative to Fermi level of lithium metal. They are semiconductors with band gap energies of 3.7, 5.9, and 2.4 eV, respectively. The valence band maximum (VBM) of the LiFePO4, which mainly consists of Fe 3d orbital, is located at −3.5 V, indicating that charge reaction of LiFePO4 extracts electrons from the 3d orbitals at 3.5 V to give the electromotive force in a Li/Li1−xFeO4 cell. The VBM’s in Li3PO4, and Li3PS4, where contribution of O 2p and S 3p orbitals is predominant, are located at −4.0 and −2.0 V, respectively.
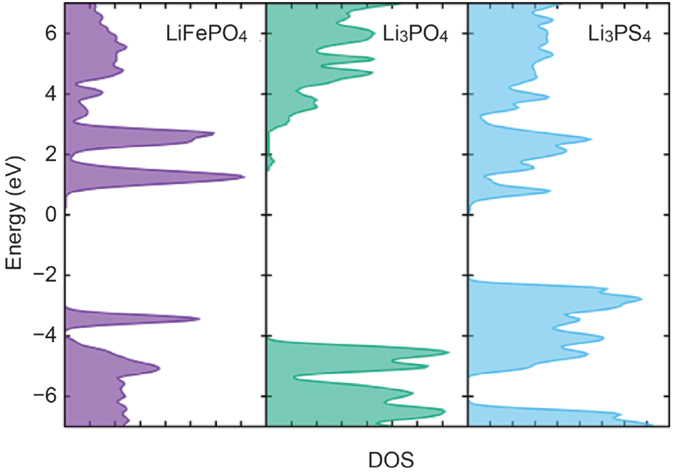
Figure 3. Density of states calculated for LiFePO4, Li3PO4, and Li3PS4. Zero in energy is set to the Fermi level of Li metal.
Figures 4B and 5B show the crystallographic structures of the interfaces during the cell operation, which are computed by relaxing FePO4/Li3PO4 and FePO4/Li3PS4 stacks displayed in Figures 4A and 5A, respectively, by the FP-MD. It shows a clear difference between the interfaces. The structure relaxation leads to little change in the FePO4/Li3PO4 interface, while it transfers a significant amount of lithium ions residing in the interface region of Li3PS4 to the FePO4. As a result, FePO4 has become Li1−xFePO4. At the same time, the interface region of the Li3PS4 becomes low in the lithium-ion concentration, while that in the bulk region is not changed significantly, as can be seen in Figure 5B.
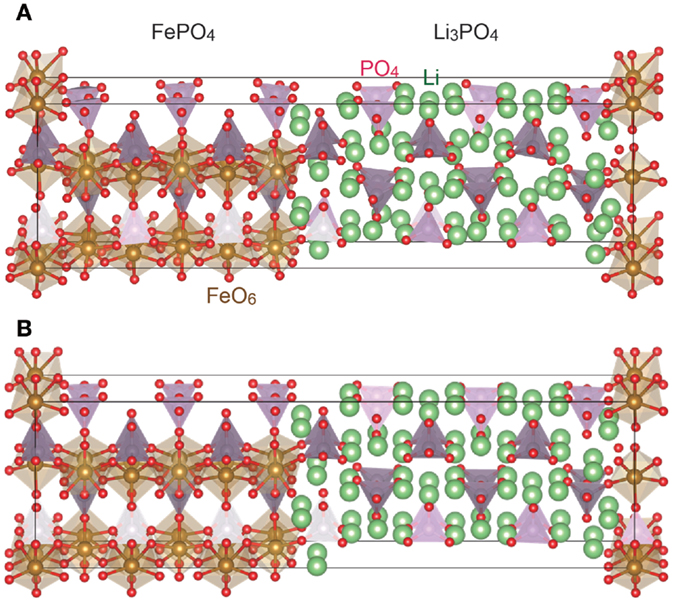
Figure 4. Interface structure between FePO4 and Li3PO4 before (A) and after (B) the FP-MD. The crystal structures are drawn by a computer program, VESTA (Momma and Izumi, 2011).
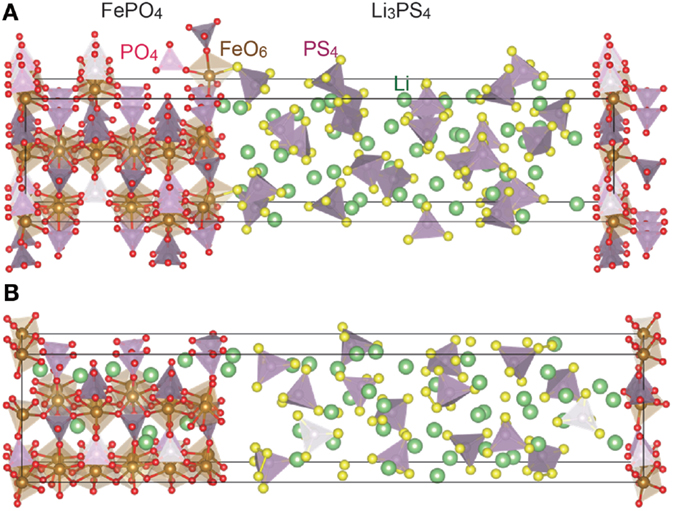
Figure 5. Interface structure between FePO4 and Li3PS4 before (A) and after (B) the FP-MD. The crystal structures are drawn by a computer program, VESTA (Momma and Izumi, 2011).
Origin of the high interface resistance at oxide cathodes/sulfide electrolytes can be found in Figure 5B. The FP-MD, which leads the interface to the equilibrium, transfers lithium ions from Li3PS4 to FePO4 and depletes lithium ions on the Li3PS4 side of the interface. This result reveals that the space-charge layer appears as a lithium-depleted layer at the Li1−xFePO4/Li3PS4 interface, which should make the interface highly resistive owing to the absence of charge-carrying lithium ions.
This computational results demonstrate that the ion concentration on the electrode surface will be a critical factor governing the interfacial resistance. It is needless to mention that the ion concentration is related to the electrode potential through a Nernst equation.
In the Nernst equation, electrode potential, E is connected to activities of oxidant and reductant, aOx and aRed, respectively, with the standard electrode potential, E0; the gas constant, R; the absolute temperature, T; the number of transferred electrons, z; and the Faraday constant, F. Because activities are related to ion concentration through activity coefficient, the concentrations of oxidant and reductant determine the E and vice versa. In solid electrolytes, only lithium ions are mobile to change the concentration; therefore, presence and absence of lithium should be regarded as the reductant and oxidant, respectively. That is, lithium-ion concentration at the interface is governed by the electrode potential, and noble potential of the oxide cathodes will decrease it.
E0 in sulfide will be much lower than that in oxides, because lithium ions are much more loosely bonded to the lattice framework in sulfides. Therefore, aOx/aRed, which corresponds to concentration of lithium vacancies, should become very large in order to make the E reach the redox potential of the cathode and start the charge reaction. In other word, the oxidation current in the beginning of charging is spent to remove lithium ions from the sulfide solid electrolyte side of the cathode/sulfide electrolyte interface to increase the aOx/aRed in equation (1) before E reaches the redox potential. After the E becomes equal to the redox potential of the cathode, lithium deintercalation from the cathode material begins.
This classical picture may be rough but seems reasonable. The preferential removal of lithium ions from the sulfide electrolyte side can be seen as potential slopes appearing for the uncoated materials in Figure 2, which are distinguishable from the following potential plateaus originated from lithium deintercalation from the cathode materials. Moreover, because E determines the lithium-ion concentrations on the electrode surface in equation (1), the potential slopes should have the same capacity between the LiCoO2 and LiMn2O4 due to the same electrode potential of 4 V, which is also evidenced by the top panels in Figure 2.
In addition, equation (1) suggests that change in the electrode potential changes lithium-ion concentration exponentially; that is, increasing E results in the drastic decrease of lithium ions. A buffer layer in the first report on the reduced interfacial resistance (Ohta et al., 2006) is Li4Ti5O12. Although it decreases the interfacial resistance at Li1−xCoO2/Li3.25Ge0.25P0.75S4 by 1/20, its ionic conductivity is only 10−13–10−10 S cm−1 (Wilkening et al., 2007; Fehr et al., 2010), while ionic conductivity of the sulfide electrolyte, Li3.25Ge0.25P0.75S4, is as high as 2.2 × 10−3 S cm–1 (Kanno and Murayama, 2001). A reasonable explanation for the reduced resistance inferred from the Nernst equation is that the high bulk conductivity of Li3.25Ge0.25P0.75S4 becomes so low at the interface due to the exponential decrease in the carrier concentration that even the Li4Ti5O12 with low ionic conductivity reduces the resistance.
The DFT calculation results shown in Figure 3 gives a clear explanation for the preferential removal of lithium ions from the Li3PS4 side upon charging. Charge reaction in LiFePO4 is lithium deintercalation accompanied by the extraction of electrons in 3d orbitals of LiFePO4 located around −3.5 V. On the other hand, VBM of the Li3PS4 is located at −2.0 eV, which is higher than the 3d orbital of LiFePO4. Therefore, electrons will be extracted not from the LiFePO4 but from the Li3PS4 at the beginning of the charge reaction, and simultaneously lithium ions will be extracted from the Li3PS4 to keep charge neutrality.
High interfacial resistance at oxide cathodes/sulfide electrolytes is considered to come from lithium-depleted layer formed at the interface, as discussed above. Therefore, reduction of the interfacial resistance by interposing oxide electrolytes observed in the experiment should be attributed to the suppression of lithium depletion; that is, lithium-depleted layer is not formed at oxide cathode/oxide solid electrolyte interfaces.
In fact, the FP-MD to relax the interfacial structure does not change distribution of lithium ions around the FePO4/Li3PO4 interface, as demonstrated in Figure 4. The Li3PO4 keeps its original structure at the interface to the FePO4, and lithium depletion is not recognized there. Moreover, the DOS’s calculated for LiFePO4 and Li3PO4 indicate that lithium ions are not preferentially removed from the solid electrolyte. VBM of the Li3PO4 is located above 3d orbital of LiFePO4 as shown in Figure 3, and thus charging current should be electrons removed from LiFePO4 even at the beginning. That is, lithium deintercalation from the LiFePO4 takes place at the interface to Li3PO4 from the beginning of charging.
Because lithium ions are not preferentially removed from the oxide electrolyte side of the interface, cathode/oxide electrolyte interfaces do not provide a potential slope to the charging curves. In fact, Figure 2 indicates fading of the potential slopes for the LiCoO2 and LiMn2O4 observed in the Li3.25Ge0.25P0.75S4 interfaces upon the LiNbO3 coating. The increasing amount of LiNbO3 coated on the cathode materials gradually shortens the potential slope, which is attributable to the increasing coverage by the coating layer. It decreases the interface area, where the cathode materials are directly contacted to the sulfide solid electrolyte, to eliminate the potential slope. Simultaneously, it increases the cathode/oxide electrolyte interface area, where lithium-depleted layer is not formed, to decrease the interfacial resistance.
The above discussions have revealed that the high interfacial resistance and the effects of surface coating on the electrode properties are explainable by the space-charge model. On the other hand, Sakuda et al. (2010) has proposed another mechanism for the interfacial resistance and its reduction. It reports that transition metals and anions, as well as lithium ions, diffuse across a Li1−xCoO2/Li2S–P2S5 to form an impurity phase. The impurity phase is concluded to be the reason for the high interfacial resistance, and the surface coating suppresses the mutual diffusion and reduces the interfacial resistance. On the other hand, such mutual diffusion is not observed in the present computations. Figure 5B exhibits that the structure relaxation changes the lithium-ion distribution around the interface without changing the flamework structure. However, it is premature to deny the mutual diffusion, because the relaxation time in the FP-MD simulations is only 200 ps.
It should be noted that Li1−xFePO4 is used as the cathode material in the computations instead of Li1−xCoO2 in the experiments in this study. LiFePO4 forms a coherent interface to Li3PO4, as reported in Sumita et al. (2015). In addition, the surface spin-state of LiCoO2 is complicated, although the manifold spin-states may not affect the band alignment at the interface significantly (Sumita and Ohno, 2016). That is, Li1−xFePO4 is better than Li1−xCoO2 to eliminate ambiguities in the computation models, and thus is used in this study. Although computations using Li1−xCoO2 is preferable to provide an accurate picture on the interfacial phenomena observed in the experiments, coincidences in the electrode properties between the LiCoO2 and LiMn2O4 in the Li3.25Ge0.25P0.75S4 and the effects of the surface coating to the interfaces, which are displayed in Figures 1 and 2, and the above discussions strongly suggest that electrode potential of cathode materials dominates the lithium-ion concentration at the interface and the interfacial resistance. Of course, electrode potential of Li1−xFePO4 is 3.5 V and lower than 4.0 V for Li1−xCoO2 to a certain extent; however, both of them are far enough from the VBM of Li3PS4 not to give significant differences. Therefore, the picture given from the Li1−xFePO4 will be valid also for the interface to Li1−xCoO2. Computations on Li1−xCoO2 are in progress, and the results will appear in near future.
However, some differences may be found between the interfaces to Li1−xFePO4 and Li1−xCoO2. These cathode materials are quite different in the electronic conduction: Li1−xCoO2 exhibits metallic conduction, while Li1−xFePO4 is poor in electronic conduction and moreover insulating at x = 0. When they come into contact with a sulfide electrolyte, part of lithium ions in the electrolyte move into the cathode materials, as displayed in Figure 5B. By the way, Li1−xFePO4 is a two-phase system. Roughly speaking, Li1−xFePO4 is a mixture of FePO4 and LiFePO4. Therefore, the movement of lithium ions from the electrolyte into the material may form a LiFePO4 layer at the electrode surface. Consequently, Li1−xFePO4 particles become to have a core–shell structure in sulfide electrolytes, where the core is a mixture of FePO4 and LiFePO4, while the shell mainly consists of LiFePO4. Because the LiFePO4 is electronically insulating, the lithium depletion may be suppressed at the surface of Li1−xFePO4, and thus the Li1−xFePO4 may work relatively well even without surface coating in sulfide electrolytes. That is, Li1−xFePO4 may have a self-origanized core–shell structure for high power, which is similar to that proposed in Xu et al. (2011), where the coating layer is formed only by compositional change in the cathode material.
KT and TO are responsible for the experimental and computational studies, respectively.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Dr. Narumi Ohta and Dr. Masato Sumita for their collaboration in the experiments and computations, respectively. This work was partially funded by the Ministry of Economy, Trade and Industry (METI), New Energy and Industrial Technology Development Organization (NEDO), ALCA-SPRING project of Japan Science and Technology Agency, and Ministry of Education, Culture, Sports, Science and Technology (MEXT).
Bussi, G., Donadio, D., and Parrinello, M. (2007). Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101. doi:10.1063/1.2408420
Fehr, K., Holzapfel, M., Laumann, A., and Schmidbauer, E. (2010). DC and AC conductivity of Li4/3Ti5/3O4 spinel. Solid State Ionics. 181, 1111–1118. doi:10.1016/j.ssi.2010.05.026
Kamaya, N., Homma, K., Yamakawa, Y., Hirayama, M., Kanno, R., Yonemura, M., et al. (2011). A lithium superionic conductor. Nat. Mater. 10, 682–686. doi:10.1038/nmat3066
Kanno, R., and Murayama, M. (2001). Lithium ionic conductor thio-lisicon: the Li2S–GeS2–P2S5 system. J. Electrochem. Soc. 148, A742–A746. doi:10.1149/1.1379028
Maier, J. (1995). Ionic conduction in space charge regions. Prog. Solid State Chem. 23, 171–263. doi:10.1016/0079-6786(95)00004-E
Maier, J. (2014). Pushing nanoionics to the limits: charge carrier chemistry in extremely small systems. Chem. Mater. 26, 348–360. doi:10.1021/cm4021657
Minami, T., Hayashi, A., and Tatsumisago, M. (2006). Recent progress of glass and glass-ceramics as solid electrolytes for lithium secondary batteries. Solid State Ionics. 177, 2715–2720. doi:10.1016/j.ssi.2006.07.017
Momma, K., and Izumi, F. (2011). VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276. doi:10.1107/S0021889811038970
Ohta, N., Takada, K., Sakaguchi, I., Zhang, L., Ma, R., Fukuda, K., et al. (2007). LiNbO3-coated LiCoO2 as cathode material for all solid-state lithium secondary batteries. Electrochem. commun. 9, 1486–1490. doi:10.1016/j.elecom.2007.02.008
Ohta, N., Takada, K., Zhang, L., Ma, R., Osada, M., and Sasaki, T. (2006). Enhancement of the high-rate capability of solid-state lithium batteries by nanoscale interfacial modification. Adv. Mater. 18, 2226–2229. doi:10.1002/adma.200502604
Perdew, J. P., Burke, K., and Ernzerhof, M. (1996). Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868. doi:10.1103/PhysRevLett.77.3865
Sakuda, A., Hayashi, A., and Tatsumisago, M. (2010). Interfacial observation between LiCoO2 electrode and Li2S–P2S5 solid electrolytes of all-solid state lithium secondary batteries using transmission electron microscopy. Chem. Mater. 22, 949–956. doi:10.1021/cm901819c
Sakuda, A., Hayashi, A., and Tatsumisago, M. (2013). Sulfide solid electrolyte with favorable mechanical property for all-solid state lithium batteries. Sci. Rep. 3, 2261. doi:10.1038/srep02261
Seino, Y., Ota, T., Takada, K., Hayashi, A., and Tatsumisago, M. (2014). A sulphide lithium super ion conductor is superior to liquid ion conductors for use in rechargeable batteries. Energy Environ. Sci. 7, 627–631. doi:10.1039/C3EE41655K
Sumita, M., and Ohno, T. (2016). Multi-spin-state at Li3PO4/LiCoO2 (104) interface. Phys. Chem. Chem. Phys. 18, 4316–4319. doi:10.1039/C5CP07735D
Sumita, M., Tanaka, Y., Ikeda, M., and Ohno, T. (2015). Theoretically designed Li3PO4 (100)/LiFePO4 (010) coherent electrolyte/cathode interface for all solid-state Li ion secondary batteries. J. Phys. Chem. C 119, 14–22. doi:10.1021/jp5060342
Sumita, M., Tanaka, Y., Ikeda, M., and Ohno, T. (2016). Theoretical insight into charging process in a Li3PO4 (100)/LiFePO4 (010) coherent interface system. Solid State Ionics. 285, 59–65. doi:10.1016/j.ssi.2015.09.029
Takada, K. (2013a). Interfacial nanoarchitectonics for solid-state lithium batteries. Langmuir 29, 7538–7541. doi:10.1021/la3045253
Takada, K. (2013b). Progress and prospective of solid-state lithium batteries. Acta Mater. 61, 759–770. doi:10.1016/j.actamat.2012.10.034
Takada, K., Ohta, N., Zhang, L., Cu, X., Hang, B. T., Ohnishi, T., et al. (2012). Interfacial phenomena in solid-state lithium battery with sulfide solid electrolyte. Solid State Ionics. 225, 594–597. doi:10.1016/j.ssi.2012.01.009
Takada, K., Ohta, N., Zhang, L., Fukuda, K., Sakaguchi, I., Ma, R., et al. (2008). Interfacial modification for high-power solid-state lithium batteries. Solid State Ionics. 179, 1333–1337. doi:10.1016/j.ssi.2008.02.017
Tarascon, J.-M., and Armand, M. (2001). Issues and challenges facing rechargeable lithium batteries. Nature 414, 359–367. doi:10.1038/35104644
Wang, L., Zhou, F., Meng, Y. S., and Ceder, G. (2007). First-principles study of surface properties of LiFePO4: surface energy, structure, wulff shape, and surface redox potential. Phys. Rev. B 76, 165435. doi:10.1103/PhysRevB.76.165435
Wilkening, M., Amade, R., Iwaniak, W., and Heitjans, P. (2007). Ultraslow Li diffusion in spinel-type structured Li4Ti5O12 – a comparison of results from solid state nmr and impedance spectroscopy. Phys. Chem. Chem. Phys. 9, 1239–1246. doi:10.1039/B616269J
Xu, X., Takada, K., Watanabe, K., Sakaguchi, I., Akatsuka, K., Hang, B. T., et al. (2011). Self-organized core–shell structure for high-power electrode in solid-state lithium batteries. Chem. Mater. 23, 3798–3804. doi:10.1021/cm103665w
Zheng, N., Bu, X., and Feng, P. (2003). Synthetic design of crystalline inorganic chalcogenides exhibiting fast-ion conductivity. Nature 426, 428–432. doi:10.1038/nature02159
Keywords: nanoionics, space-charge layer, electrode/electrolyte interface, solid-state battery, solid electrolyte, first-principles calculation
Citation: Takada K and Ohno T (2016) Experimental and Computational Approaches to Interfacial Resistance in Solid-State Batteries. Front. Energy Res. 4:10. doi: 10.3389/fenrg.2016.00010
Received: 01 February 2016; Accepted: 14 March 2016;
Published: 30 March 2016
Edited by:
Jeff Sakamoto, University of Michigan, USAReviewed by:
Mehmet Kadri Aydinol, Middle East Technical University, TurkeyCopyright: © 2016 Takada and Ohno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazunori Takada, dGFrYWRhLmthenVub3JpQG5pbXMuZ28uanA=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.