 M. Hassan Beyzavi1†
M. Hassan Beyzavi1† Casey J. Stephenson1†
Casey J. Stephenson1† Yangyang Liu1†
Yangyang Liu1† Olga Karagiaridi1
Olga Karagiaridi1 Joseph T. Hupp1*
Joseph T. Hupp1* Omar K. Farha1,2*
Omar K. Farha1,2*- 1Department of Chemistry, International Institute for Nanotechnology (IIN), Northwestern University, Evanston, IL, USA
- 2Department of Chemistry, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
As a C1 feedstock, CO2 has the potential to be uniquely highly economical in both a chemical and a financial sense. In particular, the highly atom-economical acid-catalyzed cycloaddition of CO2 to epoxides to yield cyclic organic carbonates (OCs), a functionality having many important industrial applications, is an attractive reaction for the utilization of CO2 as a chemical feedstock. Metal–organic frameworks (MOFs) are promising candidates in catalysis as they are a class of crystalline, porous, and functional materials with remarkable properties including great surface area, high stability, open channels, and permanent porosity. MOFs structure tunability and their affinity for CO2, makes them great catalysts for the formation of OCs using CO2 and epoxides. In this review, we examine MOF-based catalytic materials for the cycloaddition of carbon dioxide to epoxides. Catalysts are grouped based on the location of catalytic sites, i.e., at the struts, nodes, defect sites, or some combination thereof. Additionally, important features of each catalyst system are critically discussed.
Introduction
Humankind is highly dependent on fossil fuels as the primary source of energy. However, consumption of fossil fuels through combustion releases the greenhouse gas CO2 into the atmosphere, which is believed to contribute adversely to climate changes globally. Currently, carbon capture and storage/sequestration (CCS) from point sources is one of the most promising strategies being implemented to reduce carbon emissions. An ancillary benefit of the capture of CO2 in concentrated form may be its ready availability for use as a chemical reagent. While the volume of CO2 that can be consumed with chemical production, and especially, fine chemicals, is small in comparison to the amount generated by fossil fuel combustion, its diversion for the former nevertheless would be favorable from a green chemistry perspective. If done efficiently, using CO2 as a reagent would likely constitute a net positive contribution toward sustainability (Aresta and Dibenedetto, 2007; Schrag, 2007; Haszeldine, 2009; Markewitz et al., 2012).
Cycloaddition of CO2 to Epoxide Leading to Cyclic Organic Carbonates
The cycloaddition reaction of CO2 to epoxides is a 100% atom-economic reaction, and thus constitutes one of the most efficient examples of artificial CO2 fixation. This chemistry is advantageous compared to traditional syntheses, which usually involve highly toxic and corrosive phosgene (Shaikh and Sivaram, 1996; Sakakura and Kohno, 2009). Because of these advantages, cycloaddition of CO2 to epoxides has been studied extensively (Zevenhoven et al., 2006; Mikkelsen et al., 2010; North et al., 2010).
The mechanism for the cycloaddition of CO2 to epoxides typically involves an acid catalyst (e.g., a metal ion or a proton) that coordinates to the epoxide thereby activating it toward nucleophilic attack by the co-catalyst (typically a tetraalkylammonium halide) to form a halo-alkoxide. This halo-alkoxide intermediate can then react with carbon dioxide through cycloaddition to yield the cyclic carbonate with regeneration of the tetraalkylammonium halide co-catalyst (Figure 1) (North and Pasquale, 2009).
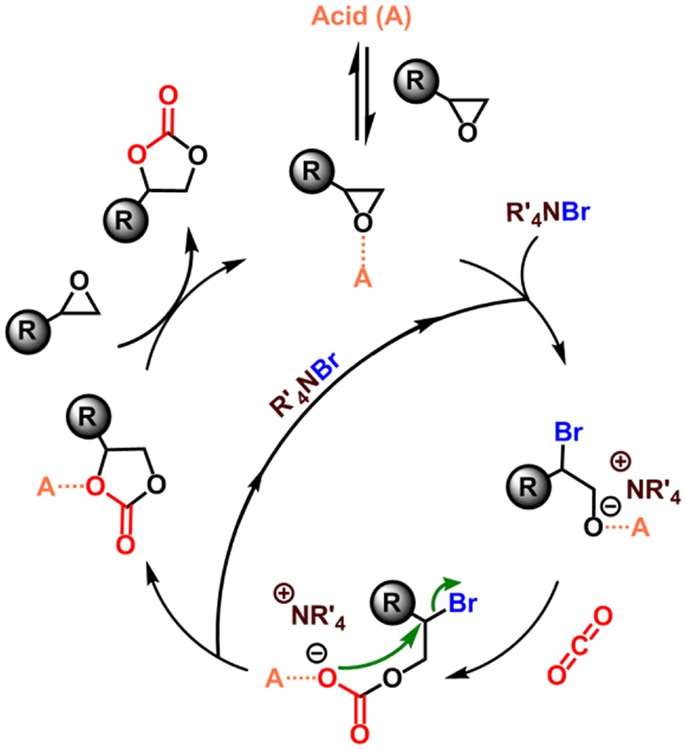
Figure 1. Proposed mechanism for the acid (A)-catalyzed carbon dioxide insertion into epoxide in the presence of a tetraalkylammonium halide (bromide) (North and Pasquale, 2009).
The rate law for the cycloaddition of CO2 to epoxide using a homogeneous aluminum complex as the Lewis acid has been determined (North and Pasquale, 2009). By varying the concentrations of catalyst, co-catalyst, and carbon dioxide, the rate equation is ultimately described by Eq. 1 indicating that the reaction is first-order for epoxide, carbon dioxide, and catalyst concentration and second-order for the co-catalyst concentration.
Scope of This Review
Several types of heterogeneous catalysts, including metal oxides (Yano et al., 1997; Yamaguchi et al., 1999; Yasuda et al., 2002, 2005), zeolites (Doskocil et al., 1999), silica-supported salts (Wang et al., 2006), titanosilicates (Srivastava et al., 2005), an organic network (Chun et al., 2013), and a microporous polymer (Xie et al., 2013) have been applied for the CO2 fixation reaction. However, they mostly require elevated temperatures and/or pressures and/or separation and purification steps. Additionally, product yields from these reactions are often low, thus increasing energy and cost due to the increased number of separation and recycling steps in a given chemical process. Therefore, the development of highly efficient catalysts for the synthesis of cyclic carbonates from CO2 is critical.
Promising candidates that could fulfill the abovementioned requirements for an efficient catalyst (Farrusseng et al., 2009; Lee et al., 2009; Ma et al., 2010; Jiang and Xu, 2011; Dhakshinamoorthy et al., 2012; Yoon et al., 2012; Gu et al., 2014; Zhao et al., 2014) are a novel class of hybrid, crystalline, highly porous materials called metal-organic frameworks (MOFs) (Long and Yaghi, 2009; Farha and Hupp, 2010; Serre et al., 2012; Xuan et al., 2012; Zhou et al., 2012a; Furukawa et al., 2013). MOFs can be functionalized with acidic catalytic sites (present as coordinatively unsaturated metal centers either at the nodes or at metalloligand struts) and nucleophilic functional groups (e.g., amines) that can fulfill the role of the co-catalyst in the cycloaddition of CO2 to an epoxide. Moreover, because they are crystalline solids, MOFs are easily separated from reaction streams and recycled. Whereas many supported catalysts suffer from an inhomogeneous distribution of environments and coordination modes that reduce product selectivity, the regularity of the catalytic sites in MOFs results in product selectivities that are comparable with homogeneous catalysts. As a result of these promising features, MOF materials have been developed for a myriad of intriguing catalytic applications such as the insertion of carbon dioxide into epoxide leading to organic carbonates (OCs).
In this review, we focus on the cycloaddition of CO2 to epoxides catalyzed by MOFs, forming OCs as the products. To date, 16 MOFs and their derivatives have been reported to catalyze this type of reactions using 11 different epoxides shown in Figure 2 (Kleist et al., 2009; Song et al., 2009; Cho et al., 2012; Lescouet et al., 2012; Macias et al., 2012; Miralda et al., 2012; Yang et al., 2012, 2014; Zhou et al., 2012b; Feng et al., 2013; Kim and Park, 2013; Kim et al., 2013; Ren et al., 2013; Zalomaeva et al., 2013a,b; Zhu et al., 2013; Beyzavi et al., 2014; Gao et al., 2014a,b; Guillerm et al., 2014). In our discussion, we categorized the MOFs based on the composition of the catalytic sites [the nodes/secondary building units (SBU), the functional ligand, or both] and reviewed each individual case in detail. As summarized in Table 1, MOFs that engage in the so-called “opportunistic catalysis” of CO2 insertion (performed by defect sites, rather than by deliberately introduced functionalities) include ZIF-8 (Miralda et al., 2012; Kim et al., 2013; Zhu et al., 2013), ZIF-68 (Yang et al., 2014), and MOF-5 (Song et al., 2009). In these MOFs, structural defects as well as the surface of MOF crystals play an important role in their catalytic activity. Co- and Mg-MOF-74 (Cho et al., 2012; Yang et al., 2012; Kim et al., 2013), MOF-505 (Gao et al., 2014a), HKUST-1 (Macias et al., 2012; Kim et al., 2013; Gao et al., 2014a), Cr- and Fe-MIL-101 (Kim et al., 2013; Zalomaeva et al., 2013a,b), gea-MOF-1 (Guillerm et al., 2014), and most recently Hf-NU-1000 are MOFs that catalyze the CO2 insertion reaction by catalytically active metal nodes (Beyzavi et al., 2014). As an extension to the former category, there are also MOFs in which ligands incorporating a Lewis acid center as well as nodes with open metal sites are present to catalyze the cycloaddition reaction. These MOFs with double Lewis acid character include PCN-224(Co) (Feng et al., 2013), MMPF-9 (Gao et al., 2014b), Ni(salphen)-MOF (Ren et al., 2013), and MMCF-2 (Gao et al., 2014a). The last category includes MOFs that are constructed from linkers with functional groups, but saturated metal nodes, such as MIL-68(In)-NH2 (Lescouet et al., 2012), UiO-66-(NH2) (Kim et al., 2013), ZIF-68 (Yang et al., 2014), F-IRMOF-3 (Zhou et al., 2012b; Kim and Park, 2013; Kim et al., 2013), and Zn4O(BDC)x(ABDC)3– x based MOF-5 (Kleist et al., 2009). In Tables 1 and 2, we list all the MOF examples and references from the literature, and we discuss selected reports in the following sections.
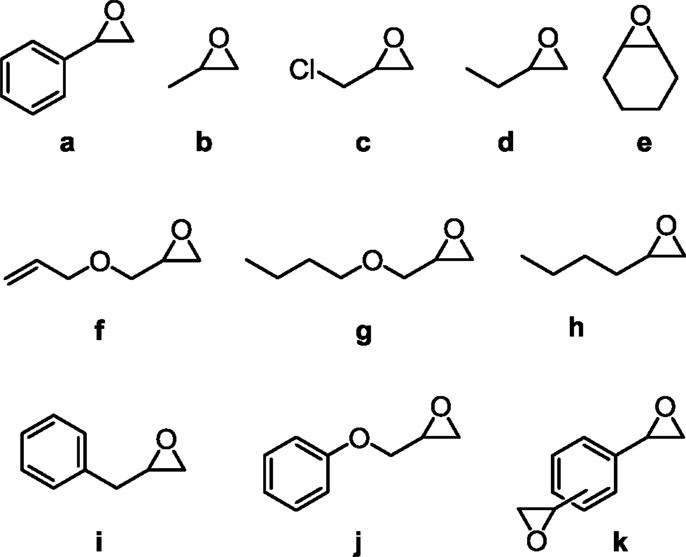
Figure 2. Epoxides utilized for the cycloaddition reaction of CO2 catalyzed by MOFs and covered in this review.
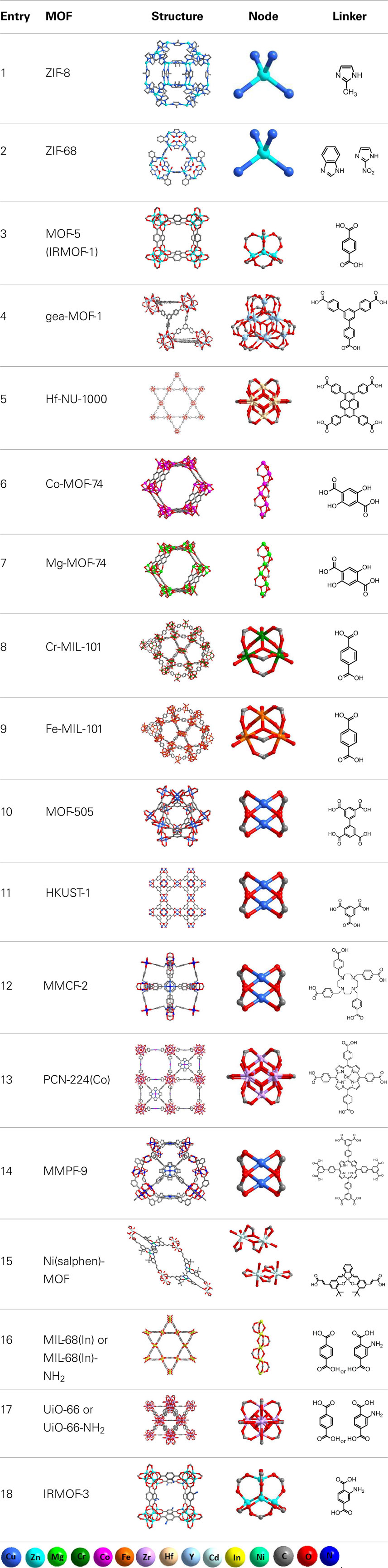
Table 1. Structural details of the MOFs, which have been used as catalysts for the cycloaddition of CO2 to epoxides leading to five-membered ring OCs.
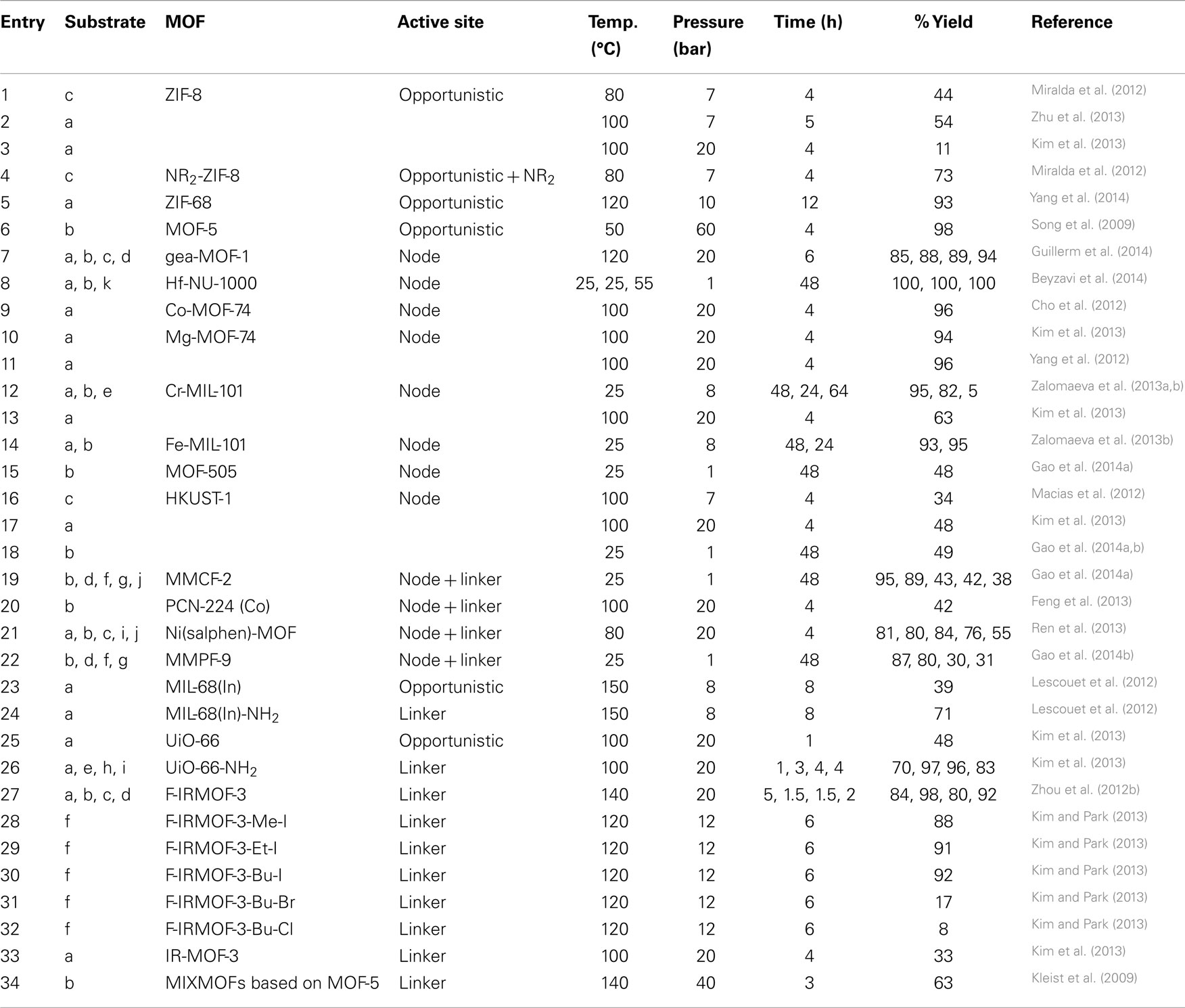
Table 2. Conditions utilized for the CO2 insertion into epoxides leading to OCs for each MOF.
Reported MOFs as Catalysts for the Cycloaddition of CO2 to Epoxides Leading to OCs
MOFs with Opportunistic Active Catalytic Sites
In this section, we present three examples in which the linkers are not designed to be catalytically active, and the metal centers are coordinatively saturated. Therefore, the transformation of epoxide to OCs is catalyzed either by the defects in the MOF structure and/or the defects on the surface of the MOF. These examples are ZIF-8 (Miralda et al., 2012; Kim et al., 2013; Zhu et al., 2013), ZIF-68, and MOF-5 (Song et al., 2009), all of which are Zn2+-containing materials. Some function as single component catalysts (that is they have both Lewis/Brønsted basic and Lewis acidic defect sites and can function without the addition of a co-catalyst). Others have Lewis acidic defect sites exclusively and need a co-catalyst.
Zeolitic imidazole frameworks or ZIFs are composed of imidazolate-based linkers and a divalent metal node, most commonly Zn2+ (Banerjee et al., 2008; Phan et al., 2009). An archetypal member of this class of materials, ZIF-8, has a major pore of 11.6 Å and apertures of 3.4 Å (Table 1, Entry 1). The narrow aperture of ZIF-8 restricts what substrates can enter the cavity, as only substrates with the right kinetic diameter will be able to enter the ZIF. As a result, catalysis generally occurs on the exterior of the crystallites where Lewis or Brønsted sites that terminate the surface of the ZIF are found (Chizallet et al., 2010).
ZIF-8 was the first ZIF used as a catalyst in organic carbonate synthesis (Miralda et al., 2012). It was used as a catalyst for the cycloaddition of CO2 to c to form chloropropene carbonate in the absence of solvent or co-catalyst. The reaction was carried out at 70–100°C for 4 h at 7 bar of CO2 with poor to moderate yields (Table 2, Entry 1). As amines have high affinity for CO2 and are used in CO2 scrubbers, the surface sites of ZIF-8 were functionalized with ethylene diamine to improve CO2 affinity and increase the carbonate yields. Epoxide conversion and carbonate yield were improved from a maximum of 44% yield in ZIF-8 to 73% in functionalized ZIF-8 (Table 2, Entry 4). Control experiments that tested whether zinc nitrate hexahydrate, ethylene diamine, and 2-methylimidazole were capable of catalyzing the reaction were carried out at 80°C for 4 h at 7 bar CO2 and yielded only diol, confirming that the ZIF-8 topology is a crucial component of the reaction process. Catalytic trials were performed using zeolite beta, zeolite TS-1, zeolite HY, and SBA-15 as well; neither of the materials’ performance matched that of ZIF-8. Zeolites, as determined by numerous vibrational spectroscopy studies of adsorbed Lewis bases such as NH3, have a preponderance of Lewis/Brønsted acid sites and few Lewis/Brønsted base sites. Likewise, silicates have very weakly acidic Si–OH bonds while titanosilicate-1 has extremely Lewis acidic Ti4+ sites but few basic sites (Srivastava et al., 2005). In contrast, ethylene diamine-functionalized ZIF-8 possesses both types of sites on its surface and thus outperforms these catalysts.
In a follow-up report (Zhu et al., 2013), ZIF-8 was used as a catalyst for the synthesis of styrene carbonate (SC) from CO2 and a. Reactions were conducted at 50–100°C under 7 bar of CO2 for 4 h. Styrene oxide is too large to enter the pore of ZIF-8 and catalysis is expected to occur at defect sites at the surface of the crystallites. A maximum yield of ~54% was obtained at 100°C (Table 2, Entry 2). When the catalyst was recycled, there was no significant drop in the product yield despite a reduction in the specific surface area by ~10%. Ammonia and pyridine binding studies were used to assay the nature of the acid sites of ZIF-8 in both fresh and recycled samples. DRIFTS of adsorbed pyridine and ammonia revealed the presence of both Brønsted and Lewis acid sites in the fresh samples, but the almost complete disappearance of Brønsted acid sites in recycled samples. A comparison of fresh and recycled samples for CO2 temperature programed desorption (TPD) reveals a loss of ~15% of basic sites, which explains the slight loss in carbonate yield during catalyst recycling trials.
Building on the strategy of using ZIFs with high CO2 affinity as catalysts for cyclic carbonate synthesis, Yang et al. (2014) explored the catalytic behavior of ZIF-68 for the synthesis of SC. ZIF-68, which contains 2-nitroimidazolate and benzimidazolate linkers, has one of the highest calculated adsorption capacities for CO2 (Phan et al., 2009). It has a GME topology and larger pores (10.3 and 7.5 Å) than ZIF-8 and can act as a more active catalyst for the cycloaddition of CO2 than ZIF-8 (Table 1, Entry 2) due to its ability to capture CO2 (and thus essentially create high substrate concentrations at the catalytic sites). Catalytic trials were performed between 80 and 130°C in a pressure range of 2–30 bar with a reaction time of 12 h. Under optimized conditions, total conversion to SC could be achieved after 10 h at 120°C (Table 2, Entry 5). Recyclability studies with ZIF-68 revealed a loss in product yield with each additional time that the catalyst was reused.
In a separate study, Song et al. (2009) utilized a different zinc-based MOF, MOF-5, which has coordinatively saturated metal centers in the node with unfunctionalized linkers (Table 1, Entry 3). MOF-5 is composed of Zn4O nodes with benzene-1,4-dicarboxylate linkers and has an aperture of 8 and a 12 Å pore diameter. Due to the lack of basic sites in MOF-5, catalysis is expected to occur at the node and tetraalkylammonium halide co-catalysts are required for the cycloaddition of CO2 to epoxides. Cycloaddition reactions were carried out for a variety of epoxides under mild conditions at 50°C, under 60 bar CO2 with nearly 100% yield of propylene carbonate after 4 h (Table 2, Entry 6). As MOF-5 does not have any catalytic functionality built into the linkers, all catalysis must occur at defect sites. Indeed, studies by Ravon et al. (2008, 2010) have shown that it is acidic –OH groups at defect sites that are responsible for the catalysis. Ammonia and CO2 TPD was performed on MOF-5 as part of a separate study to quantify the number and strength of acidic and basic sites. CO2 TPD indicated that there were no basic sites in MOF-5 (which explains the requirement for using a co-catalyst when MOF-5 is employed as a catalyst for the cycloaddition of CO2). Me4NCl, Me4NBr, Et4NBr, Pr4NBr, and Bu4NBr were all investigated as co-catalysts in catalytic trials. Less bulky quaternary ammonium salts gave little to no yield of propylene carbonate while bulkier ones like Pr4NBr and Bu4NBr gave the best results. These results indicated that the halide ion was more tightly bound to the less bulky quaternary ammonium cation and thus less available for catalysis. Epoxides with electron withdrawing and electron donating substituents were investigated and the reactions proceeded to completion in all cases.
MOFs with Active Catalytic Nodes
In this section, we discuss six examples of utilization of MOF nodes with catalytically active coordinately unsaturated sites for the transformation of epoxides into OCs. The nodes function as Lewis acid sites for the activation of the epoxide but lack any Lewis basic functionality that would open the epoxide ring, thus requiring the use of a co-catalyst. The catalytically active nodes are the building blocks of M2(DHTP)-based MOFs (CPO-27-M/M-MOF-74) (Cho et al., 2012; Yang et al., 2012; Kim et al., 2013), MOF-505 (Gao et al., 2014a), HKUST (Cu3(BTC)2) (Macias et al., 2012; Kim et al., 2013; Gao et al., 2014a), Cr- and Fe-MIL-101 (Kim et al., 2013; Zalomaeva et al., 2013a,b), gea-MOF-1 (Guillerm et al., 2014), and Hf-NU-1000 (Beyzavi et al., 2014). Based on Lewis acidity alone, one would expect the most active catalysts to have nodes based on early transition metal catalysts and lanthanides (Y3+, Zr4+, and Hf4+) followed by mid group transition metals in high oxidation states (Cr3+, Fe3+, and Co3+) and finally late transition metals (Cu2+ and Zn2+) should have the lowest catalytic activity. It must be noted, however, that the nodes of these MOF are not mononuclear species but multinuclear complexes, which leads to additional factors that determine the catalytic activity.
The highly Lewis acidic Y3+-based MOF using a nonanuclear carboxylate-based cluster reported by Guillerm et al. (2014) is one of the most recent catalysts for carbonate synthesis. This cluster serves as an 18-connected MBB for the formation of MOFs with (3,18)-connected gea topology. Single crystals of gea-MOF-1 were prepared from the solvothermal reaction between H3BTB and Y(NO3)3⋅6H2O in DMF and water. Structural analysis reveals that the anionic metal cluster [Y9(μ3 − OH)8(μ2 − OH)3(O2C−)18]2− contains nine metal ions, which form a tricapped trigonal prism. The yttrium cluster is linked to 18 carboxylate groups from 18 BTB linkers to construct a three-dimensional framework (Table 1, Entry 4). Considering its high porosity and great thermal stability, gea-MOF-1 was explored as a heterogeneous catalyst for the cycloaddition of CO2 to epoxides. The reactions were carried out using 200 mmol substrate to 0.15 mmol yttrium (60 mg catalyst) and 0.15 mmol tetrabutylammonium bromide (TBAB) at 120°C and 20 bar CO2 for 6 h (Table 2, Entry 7) in neat epoxide. The catalyst converted 86% of the propylene oxide. However, under similar reaction conditions the homogeneous YCl3/TBAB was more than 10× more active than gea-MOF-1, indicating mass-transport limitations and/or less Lewis acidic (Y3+) sites in the node in the gea-MOF-1 framework. A heterogeneous control, Y2O3/TBAB, was also studied for comparison and, as expected, conversion of propylene oxide was much lower than in the case of gea-MOF-1/TBAB. The highly accessible Lewis acid (Y3+) sites of gea-MOF-1 are the key for its superior catalytic activity, as they are important for the epoxide activation that is the first step in the reaction.
Recently, we reported an Hf-based MOF, Hf-NU-1000, which is an excellent catalyst for the cycloaddition reaction of CO2 to epoxides (Beyzavi et al., 2014). The MOF was synthesized via the solvothermal reaction of HfOCl4⋅8H2O and 1,3,6,8-tetrakis(p-benzoic acid)pyrene (H4TBAPyr) affording yellow microcrystalline product. As shown in Table 1, Entry 5, Hf-NU-1000 has triangular and hexagonal channels with a diameter of 1.4 and 2.9 nm. The node consists of an Hf6 cluster capped by eight bridging carboxylate-containing linkers. Vibrational spectroscopy was used to assign the coordination geometry of the Hf-OH and Hf-OH2 groups bound to the node, which was found to be [Hf6(μ3 − O)4(μ3 − OH)4(OH)4(OH2)4(TBAPyr)2]. Hf-NU-1000 converted a quantitatively at room temperature under 1 bar CO2 (Table 2, Entry 8) after 55 h. By increasing the temperature to 55°C, the reaction proceeded to completion in 12 h. We proposed that catalysis occurs at the –OH groups at the nodes via a Brønsted acid pathway, as it was calculated that the hydroxyl protons of Hf-NU-1000 are slightly more acidic than those of the Zr4+ analog NU-1000. As a result, when NU-1000 was used in the cycloaddition reaction, it yielded only 46% conversion under identical conditions as Hf-NU-1000.
Metal-organic frameworks based on hard acid centers like Mg2+ are expected to excel as Lewis acid catalysts for epoxide activation. In 2012, Cho et al. (2012) and Yang et al. (2012) reported catalysis of the cycloaddition of CO2 to epoxides by M2(DHTP)(H2O)2⋅8H2O (CPO-27-M/M-MOF-74 where M = Co2+ or Mg2+). The crystal structures of the Co- and Mg-MOF-74 (CPO-27-Co and -Mg) are shown in Table 1, Entries 6 and 7, respectively. Larger substrates can enter the pores of MOF-74 as it has pore diameters of ~1.1 nm. The metal nodes are coordinated in a square pyramidal fashion (to five oxygen atoms from the carboxylate and hydroxy groups on the organic linker), with a water molecule (that can be easily removed by heating in vacuo, yielding a coordinatively unsaturated metal site) filling out the coordination sphere. Performing the reaction in neat epoxide or in non-coordinating solvents is ideal as then catalysis rates will not be retarded by competitive binding at the metal sites.
Both Co- and Mg-MOF-74 (CPO-27-Co and -Mg) were used as catalysts for the insertion of CO2 into a and they show high catalytic activities under 20 bar and 100°C for 4 h (Table 2, Entries 9–11) (Cho et al., 2012; Yang et al., 2012; Kim et al., 2013). The catalytic trials were run under identical conditions using a non-coordinating solvent that would not compete with the epoxide for open coordination sites. An interesting feature of this system is that catalysis occurs in the absence of the ammonium salt co-catalyst. It is proposed, but not confirmed that the oxygen atoms of the carboxylate linkers act as the base. Conversions were comparable, however, because catalyst mass loadings were identical (20 mg catalyst). On the basis of hard and soft acid–base theory, Mg2+ is a hard acid while Co2+ is more intermediate, so one would expect more of a difference in catalysis. However, the two systems had very similar performance, thus implying the importance of the MOF topology in enhancing the catalytic activity. Indeed, both the Co2+ and the Mg2+ analogs have very high CO2 affinities, which results in a higher local concentration of CO2, thus increasing catalyst performance.
As expected Cr-MIL-101 (Ferey et al., 2005) has all of the properties required from a good catalyst: large mesopores (two types of mesoporous cages with diameters of 2.9 and 3.4 nm), thermal stability, and coordination sites available for epoxide binding (see Table 1, Entry 8). However, as a result of crystal field effects, Cr3+ in an octahedral coordination environment tends to be more inert (water exchange rate constant of solvated Cr3+ has been measured as ~10−6), thus impacting its ability to be an efficient catalyst. The catalytic properties of Cr-MIL-101 for CO2 insertion reactions were studied by Zalomaeva et al. (2013a,b). In their studies, the reaction was performed under mild solvent free conditions, in which neat epoxide a, Cr-MIL-101, and co-catalyst (TBAB) were reacted under 8 bar CO2. The SC yield reaches 95% under 25°C after 48 h. Under the same conditions, the yield of propylene carbonate (PC) from b is 82% (Table 2, Entry 12). For comparison, the catalytic performance of Fe-MIL-101 (structural analog of Cr-MIL-101, see Table 1, Entry 9) loaded with the same amount of TBAB was also studied. Under the same pressure, temperature, and catalyst loading, Fe-MIL-101 catalyst gives the yield of 93% for SO and 95% for PO, respectively (Table 2, Entry 14). Although MIL-101 (Cr/Fe) together with TBAB (co-catalyst) are effective heterogeneous catalysts for the reaction of a with CO2 under mild conditions (8 bar CO2 and 25°C), the catalytic activity of MIL-101 tends to decrease after a few cycles due to the structural loss or blockage of the pores.
Metal-organic frameworks based on “soft” Cu2+ nodes have also been used for epoxide activation. The advantage of using a soft metal like Cu2+ to activate epoxides is that the epoxide will not bind so strongly to the metal as to slow down catalysis. MOF-505 (Chen et al., 2005), a Cu2(BPTC) based MOF, and Cu3(BTC)2 [also known as HKUST-1 (Macias et al., 2012)] both share similar Cu2-paddlewheel SBUs, which are based on the dimeric cupric tetracarboxylate units. The linkers and copper paddlewheel nodes form cuboctahedral cages in the MOF structure (Table 1, Entry 10 and vide infra). Open coordination sites can be generated by dissociation or removal of the water molecules that occupy the terminus of the Cu2+ atoms. HKUST-1 also has high CO2 affinity, which should improve its efficiency as a catalyst. Both MOF-505 and HKUST-1 were used for cycloaddition reactions of CO2 to b (Table 2, Entry 15 and 18) (Gao et al., 2014a). The reactions were carried out under 1 bar CO2 at room temperature for 48 h yielding 48 and 49% of corresponding product, respectively. In a separate study (Macias et al., 2012), HKUST-1 was used for the cycloaddition of CO2 to epichlorohydrin. However, HKUST-1 was found not to be catalytically selective for the formation of chloropropene carbonate below 100°C as the corresponding diol and dimers of epichlorohydrin were observed. At 100°C and under 7 atm of CO2, HKUST-1 in the absence of solvent and co-catalyst converts CO2 and c to chloropropene carbonate with the co-occurrence of the corresponding diol (34%) and dimer (14%) (Table 2, Entry 16).
MOFs with Catalytically Active Nodes and Linkers
Another interesting approach to construct suitable MOFs for the CO2 fixation reaction is to incorporate and utilize Lewis acid containing linkers for preparing MOF catalysts. This method could be considered as a dual acidic activation approach; besides the nodes with acidic character, the organometallic linkers also would participate in the catalysis, thus enhancing the catalytic efficiency. This approach requires designing new MOF topologies allowing incorporation of the functional linkers into the structure and results in higher concentration of acid sites compared to those MOFs, which only have catalytic sites in their nodes as discussed in the previous section.
In order to investigate whether increasing the number of Lewis acid sites could result in higher efficiency of the catalytic performance, a NbO MOF platform was constructed by solvothermally reacting an azamacrocycle ligand (H4TACTMB) with Cu(NO3)2 (Gao et al., 2014a) The Cu2+ is not only applied to form the Cu2-paddlewheel nodes but also to in situ metalate the azamacrocycle. As shown in Table 1 Entry 12, the resulting MOF, MMCF-2, has cuboctahedral cages with six square faces, which are occupied by the azamacrocycle metalated with Cu2+. Since the presented MOF has high concentration of Cu2+ centers (18 Cu2+ sites in the cuboctahedral cage) it could be a suitable candidate for the CO2 insertion into epoxides and a superior catalyst to its counterpart, MOF-505 (Chen et al., 2005) (Table 1, Entry 10), which has only 12 Cu2+ sites in its cuboctahedral cages. From the point of view of the topology, both MMCF-2 and MOF-505 are made up of cuboctahedral cages through 12 Cu2-paddlewheel nodes. However, in MOF-505 the BPTC linkers cannot incorporate Cu2+ ions and are therefore not catalytically active. Moreover, since in MOF-505 the Lewis acid Cu2+ sites are located at the corners of the octahedral cages, they are not pointing to the center of the cage, thus their accessibility by the reagents (and consequently the catalytic activity) is limited. This limitation is addressed in the custom-designed MMCF-2, as additional active Cu2+ sites are introduced by decorating the six square faces of the cage with Cu2+ Lewis acid sites, which are positioned toward the cage center, thus improving the interactions between the catalytic sites and substrates. The resulting overall catalytic efficiency is very pronounced. MOF-505 or homogeneous Cu(TACTMB) convert b in 48 and 47% yield, respectively, at room temperature and 1 bar CO2 after 48 h, while MMCF-2 almost doubled the yield under the same reaction conditions (Table 2, Entries 15 and 19). Besides substrate b, substrates d, f, g, and j have been utilized for the CO2 insertion reaction using MMCF-2; however, the yields for f, g, and j are below 50%, which can be justified by the restricted diffusion of large epoxides into the octahedral cage of the catalyst. This example clearly shows that by incorporating additional accessible Lewis acid centers, the catalytic activity of the MOF could be remarkably enhanced if the substrate can access these sites.
Other interesting metal complexes with Lewis acid character are metalloporphyrins and metallosalens, which both have been incorporated into MOF structures and their catalytic activities have been investigated for the cycloaddition reaction of CO2 with epoxides. The metalloporphyrin complex example was reported (Feng et al., 2013) by preparing a series of wide-range pH stable porous metalloporphyrinic zirconium-based MOFs denoted PCN-224, which are constructed from a Zr6 cluster and MTCPP ligand, resulting in 3D channels as large as 19 Å (Table 1, Entry 13). It is known that metalloporphyrins are catalytically active for the CO2 fixation reaction due to their Lewis acid character, therefore, it was expected that PCN-224(Co), which has porphyrin linkers functionalized with Co2+ would be catalytically active for the cycloaddition reaction of CO2 with an epoxide. As an example, b was reacted with CO2 in the presence of PCN-224(Co) and after 4 h under 20 bars CO2 at 100°C, 42% conversion was observed by the authors (Table 2, Entry 20).
Similarly, metallosalen complexes were used as linkers exemplifying again a double Lewis catalytic effect MOF design. This report (Ren et al., 2013) describes a Ni(salphen)-based MOF material which features the dicarboxylfunctionalized nickel salphen complex (Ni–H2L) and shows catalytic activity for the cycloaddition of CO2 to epoxides. As shown in Table 1 Entry 15, there are two types of dinuclear Cd2 nodes which form clusters by two bidentate and two tridentate carboxylate groups. One of the Cd2+ ions in both Cd2 clusters is coordinated by five oxygen atoms and two aqua ligands. Therefore, each cluster has four water molecules, which could simply be replaced by the epoxide oxygen to enable epoxide activation as the first step for CO2 insertion. Besides the Cd2+ open sites, the coordinatively unsaturated Ni2+ ions in the functional ligands are accessible to epoxide molecules for double-Lewis acid activation. The catalytic activity of the presented MOF was tested by using a, b, c, i, and j under 20 bars CO2 and 80°C and the highest yield was obtained for c in 84% (Table 2, Entry 21) in the presence of NBu4Br. Although Br− is less nucleophilic than I−, NBu4Br shows slightly higher reactivity than NBu4I, which could be a result of the less hindered diffusion of smaller Br− into the microporous MOF. In order to examine if the cycloaddition reactions are catalyzed by the coordinatively unsaturated Ni–L units or the Cd2+ active sites, Ni–H2L and [Cd(bpdc)]n, were tested separately as catalysts under the same conditions and it was found that the yield of the carbonate product was significantly lower compared to the reaction catalyzed by the MOF. This clearly indicates that the Ni2+ and Cd2+ centers operate synergistically to activate the epoxide.
MOFs with Functional Linkers
As discussed, the open metal nodes in MOFs, which act as Lewis acids, are catalytically active for the CO2 cycloaddition reaction to epoxides. In most cases, however, use of a co-catalyst such as tetraalkylammonium halide is required in the reaction to obtain a higher SC yield under relatively mild conditions (Zalomaeva et al., 2013a). As it was mentioned above, the role of the co-catalyst in the acid-catalyzed CO2 insertion reaction is to attack the epoxides to form a halo-alkoxide (Figure 1). More importantly, it was also discovered that the co-existence of the Lewis acid sites and the co-catalyst could activate both epoxides and CO2 to form an intermediate that facilitates the intramolecular reaction (North and Pasquale, 2009). As MOFs are built from two components, metals nodes and linkers, if the organic linkers can be employed as “co-catalysts” to activate CO2 while the Lewis acidic metal nodes are used to activate the epoxides, a MOF itself would be an efficient catalyst. Encouraged by this hypothesis and the diversity of MOF structures, the MOF community started to explore MOFs with functional linkers as potential catalysts.
To date, several MOFs with functional linkers have been studied for their catalytic activity toward the CO2 cycloaddition reaction (Kleist et al., 2009; Lescouet et al., 2012; Zhou et al., 2012b; Kim and Park, 2013; Kim et al., 2013). Among them, the amine group is one of the most studied functional groups in these MOF catalysts. As the open metal nodes in MOFs are Lewis acidic, the introduction of Lewis basic functional groups such as amine (-NR2) into the organic linkers would result in a catalyst with acid-base pairs. Previous research has shown that traditional catalysts (zeolites, mesoporous silica) with acid-base pairs are effective in CO2 cycloaddition, in which CO2 is activated on the basic sites forming a carbamate, which reacts with epoxides that are adsorbed on the adjacent acidic sites. This concerted action of acidic and basic sites in one catalyst could promote the production of OCs, with the proposed mechanism shown in Figure 2 (Tu and Davis, 2001; Lescouet et al., 2012; Kim et al., 2013). Compared to traditional catalysts, the well-defined yet versatile structures of MOFs provide an excellent platform for heterogeneous catalysis. More specifically, in MOFs bearing acid-base pairs, the base functionalities embedded in the organic linkers are separated from the Lewis acidic open metal nodes due to the well-defined 3D structure, thus preventing catalyst poisoning. Meanwhile, the connections between organic linkers and metal nodes ensure the acid site and adjacent base site are within a certain distance so that the intramolecular reaction could occur between the two species that are adsorbed to these two sites (Figure 3).
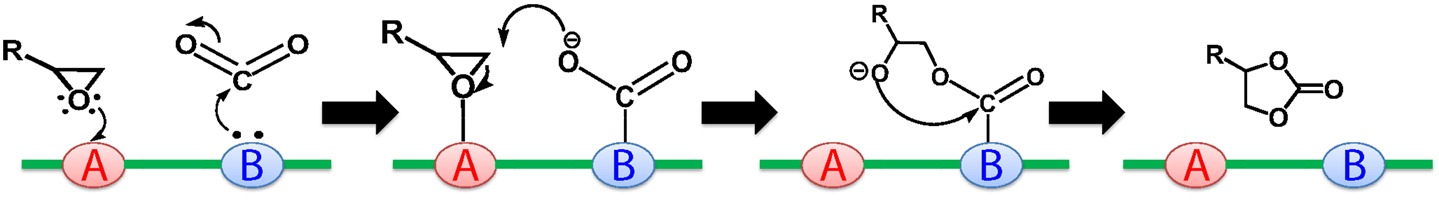
Figure 3. Proposed reaction mechanism for CO2 cycloaddition to an epoxide by a catalyst with acid–base pairs (A, acid; B, base) (Kim et al., 2013).
In general, no co-catalysts are needed when using these MOFs with functional linkers as catalysts, and they usually exhibit better catalytic activities compared to their parent MOFs that do not possess the basic functional linkers. For example, the catalytic performance of amine-functionalized MIL-68(In)-NH2, a MOF containing acid (indium nodes)-base (amine) pairs (Table 1, Entry 16), was found to be better than that of MIL-68 (In) in the CO2 cycloaddition reaction (Lescouet et al., 2012). Under 150°C and 8 bar CO2, the conversions of a after 8 h are 71 and 39% for MIL-68(In)-NH2 and MIL-68(In), respectively (Table 2, Entries 23 and 24). Another example is UiO-66-NH2, which gives higher conversion than UiO-66 for CO2 cycloaddition to a. After 1 h, 70 and 48% conversions of a were found for UiO-66-NH2 and UiO-66 (Table 1, Entry 17), respectively (under 100°C and 20 bar, Table 2, Entry 25 and 26), although this difference becomes less significant when extending the reaction time (Kim et al., 2013). It is worth noting that in both UiO-66-NH2 and UiO-66 the active Lewis acid sites are exposed Zr4+ nodes resulting from the defects in the structures. Kim et al. (2013) also carried an interesting study on the correlation between the catalytic activities of MOFs and their acid–base characteristics using NH3- and CO2-TPD. They discovered that UiO-66-NH2 has the highest catalytic activity among all the MOFs investigated (UiO-66, Mg-MOF-74, MIL-101, HKUST-1, IRMOF-3, ZIF-8, and MOF-5), mainly because it contains more weak basic sites (-NH2) than the other MOFs as well as a comparable number of acid sites. On the other hand, strong basic sites were found not as beneficial for the catalysis presumably due to the slow desorption of the reaction products.
Besides using predesigned functionalized linkers, various catalytically active groups can also be introduced into MOFs by post-synthetic modification techniques or mixed-ligand approaches (Kleist et al., 2009; Zhou et al., 2012b; Kim and Park, 2013). For instance, quaternary ammonium salt functional groups were successfully introduced into IRMOF-3 (Table 1, Entry 18) by post-synthetic functionalization entailing the reaction of IRMOF-3 with various alkyl halides (MeI, EtI, BuI, BuBr, and BuCl) (Kim and Park, 2013). In this study F-IRMOF-3 was prepared via a fast precipitation method that favors the formation of structural defects, which in turn open up Lewis acidic sites on node-based Zn2+ ions. The co-existence of Lewis acidic sites and linker-based Lewis basic sites (quaternary ammonium salts) enhances the catalytic activity of F-IRMOF-3. Analogs of F-IRMOF-3 featuring a diverse range of tetraalkylammonium species were tested for transforming substrate f to the corresponding carbonate. The catalyst activity was paralleled with the size of the tetraalkylammonium group and the size of the anion, both of which may affect the nucleophilicity of the anions. The nucleophilic attack to the epoxide is an important intermediate step in this reaction (Sun et al., 2005). The best catalyst was a tetrabutylammonium iodide-functionalized MOF, which gave the carbonate in 92% yield (under 120°C, 12 bar CO2 for 6 h, Table 2, Entries 27–32). Free quaternary ammonium salts are well known homogeneous catalysts for the synthesis of OCs, but they are characterized by poor catalytic recyclability (Kihara et al., 1993; Tu and Davis, 2001; Yasuda et al., 2005; Kim and Park, 2013). In contrast, the heterogeneous catalyst F-IRMOF-3 can be easily separated from reaction mixtures by filtration, and then redeployed. Recyclability tests with F-IRMOF-3 show that little loss of activity accompanies its reuse with two additional reaction mixtures.
Conclusion and Outlook
Metal-organic framework-based catalysts blend some of the best features of homogeneous and heterogeneous catalysts. Most notably, they offer an ease of modification that has traditionally been available only for homogeneous catalysts. Taking advantage of this tunability, one can synthesize nodes with Lewis acidic character so that they can rapidly activate epoxides. The density of catalytic sites can be increased by having catalytically active linkers in addition to catalytically active nodes. The incorporation of functional groups such as amino groups into the organic linkers may increase the CO2 affinity of a MOF thereby increasing the local concentration of CO2 near the catalytic sites. In addition, organic linkers with Lewis basic functional groups could act as co-catalysts to activate CO2, enabling the catalysis to occur without additional co-catalyst.
Taking into account the lessons learned from developing MOFs capable of catalyzing the cycloaddition of carbon dioxide to epoxides, we can expect interesting future work aimed at producing MOFs that effectively catalyze a broader spectrum of CO2 consuming reactions, including reactions that require the MOF additionally to function as a receptor and/or conduit for capture and delivery of photonic or other external energy. The catalytic properties of MOFs that show high isosteric heats of adsorption for CO2 are worth exploring, as their CO2 capture properties can lead to enhanced substrate concentration and enhanced reaction kinetics. Depending on the binding mode of CO2, such materials might also open up additional ways of activating CO2. Optimizing the acid–base properties of the MOF materials could have a profound effect on catalyst performance, and perhaps efficient catalysts that operate under at or near room temperature and atmospheric pressure could be realized. Finally, predictive modeling to optimize these properties and propose MOF candidates will expedite the search for the ideal MOF catalyst.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
CCS, carbon capture and sequestration; CPO, coordination polymer of Oslo; DHTP, 2,5-dihydroxyterephthalate; DRIFTS, diffuse reflectance infrared Fourier-transformed spectroscopy; F-IRMOF, functionalized isoreticular metal-organic framework; H2ABDC, 2-aminobenzene-1,4-dicarboxylic acid; H2BPTC, 4,4′-biphenyldicarboxylic acid; H3BTB, 1,3,5-tris(4-carboxyphenyl)benzene; H3BTC, benzene-1,3,5-tricarboxylic acid; H4BPTC, 3,3′,5,5′-biphenyltetracarboxylic acid; H4TBAPyr, 1,3,6,8-tetrakis(p-benzoic acid)pyrene; HKUST, Hong Kong University of Science and Technology; H4TACTMB, 1,4,7,10-tetrazazcyclododecane-N,N′,N″,N′″-tetra-p-methylbenzoic acid; MBB, molecular building blocks; MIL, Materials Institut Lavoisier; MMCF, metal-macrocyclic framework; MMPF, metal-metalloporphyrin framework; MOF, metal-organic framework; MTCPP, tetrakis(p-carboxyphenyl)-porphyrin metalated with various metals, M); NU, Northwestern University; OC, organic carbonate; PCN, porous coordination network; RE, rare earth; SC, styrene carbonate; TBAB, tetrabutylammonium bromide; TPD, temperature programed desorption; UiO, Universitetet i Oslo; ZIF, zeolitic imidazolate framework.
References
Aresta, M., and Dibenedetto, A. (2007). Utilisation of CO2 as a chemical feedstock: opportunities and challenges. Dalton Trans. 2975–2992. doi:10.1039/b700658f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Banerjee, R., Phan, A., Wang, B., Knobler, C., Furukawa, H., O’Keeffe, M., et al. (2008). High-throughput synthesis of zeolitic imidazolate frameworks and application to CO2 capture. Science 319, 939–943. doi:10.1126/science.1152516
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Beyzavi, M. H., Klet, R. C., Tussupbayev, S., Borycz, J., Vermeulen, N. A., Cramer, C. J., et al. (2014). A hafnium-based metal-organic framework as an efficient and multifunctional catalyst for facile CO2 fixation and regioselective and enantioretentive epoxide activation. J. Am. Chem. Soc. 136, 15861–15864. doi:10.1021/ja508626n
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, B. L., Ockwig, N. W., Millward, A. R., Contreras, D. S., and Yaghi, O. M. (2005). High H2 adsorption in a microporous metal-organic framework with open metal sites. Angew. Chem. 44, 4745–4749. doi:10.1002/anie.200462787
Chizallet, C., Lazare, S., Bazer-Bachi, D., Bonnier, F., Lecocq, V., Soyer, E., et al. (2010). Catalysis of transesterification by a nonfunctionalized metal-organic framework: acido-basicity at the external surface of ZIF-8 probed by ftir and ab initio calculations. J. Am. Chem. Soc. 132, 12365–12377. doi:10.1021/ja103365s
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cho, H. Y., Yang, D. A., Kim, J., Jeong, S. Y., and Ahn, W. S. (2012). CO2 adsorption and catalytic application of Co-MOF-74 synthesized by microwave heating. Catal. Today 185, 35–40. doi:10.1016/j.cattod.2011.08.019
Chun, J., Kang, S., Kang, N., Lee, S. M., Kim, H. J., and Son, S. U. (2013). Microporous organic networks bearing metal-salen species for mild CO2 fixation to cyclic carbonates. J. Mater. Chem. A 1, 5517–5523. doi:10.1039/c3ta10477j
Dhakshinamoorthy, A., Alvaro, M., and Garcia, H. (2012). Commercial metal-organic frameworks as heterogeneous catalysts. Chem. Commun. 48, 11275–11288. doi:10.1039/c2cc34329k
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Doskocil, E. J., Bordawekar, S. V., Kaye, B. G., and Davis, R. J. (1999). UV-vis spectroscopy of iodine adsorbed on alkali-metal-modified zeolite catalysts for addition of carbon dioxide to ethylene oxide. J. Phys. Chem. B 103, 6277–6282. doi:10.1021/jp991091t
Farha, O. K., and Hupp, J. T. (2010). Rational design, synthesis, purification, and activation of metal-organic framework materials. Acc. Chem. Res. 43, 1166–1175. doi:10.1021/ar1000617
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Farrusseng, D., Aguado, S., and Pinel, C. (2009). Metal-organic frameworks: opportunities for catalysis. Angew. Chem. 48, 7502–7513. doi:10.1002/anie.200806063
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Feng, D. W., Chung, W. C., Wei, Z. W., Gu, Z. Y., Jiang, H. L., Chen, Y. P., et al. (2013). Construction of ultrastable porphyrin Zr metal-organic frameworks through linker elimination. J. Am. Chem. Soc. 135, 17105–17110. doi:10.1021/ja408084j
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ferey, G., Mellot-Draznieks, C., Serre, C., Millange, F., Dutour, J., Surble, S., et al. (2005). A chromium terephthalate-based solid with unusually large pore volumes and surface area. Science 309, 2040–2042. doi:10.1126/science.1116275
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Furukawa, H., Cordova, K. E., O’Keeffe, M., and Yaghi, O. M. (2013). The chemistry and applications of metal-organic frameworks. Science 341, 974. doi:10.1126/science.1230444
Gao, W.-Y., Chen, Y., Niu, Y., Williams, K., Cash, L., Perez, P. J., et al. (2014a). Crystal engineering of an nbo topology metal-organic framework for chemical fixation of CO2 under ambient conditions. Angew. Chem. Int. Ed. 53, 2615–2619. doi:10.1002/anie.201309778
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gao, W.-Y., Wojtas, L., and Ma, S. (2014b). A porous metal-metalloporphyrin framework featuring high-density active sites for chemical fixation of CO2 under ambient conditions. Chem. Commun. 50, 5316–5318. doi:10.1039/c3cc47542e
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gu, Z.-Y., Park, J., Raiff, A., Wei, Z. W., and Zhou, H. C. (2014). Metal-organic frameworks as biomimetic catalysts. ChemCatChem 6, 67–75. doi:10.1002/cctc.201300493
Guillerm, V., Weselinski, L. J., Belmabkhout, Y., Cairns, A. J., D’Elia, V., Wojtas, L., et al. (2014). Discovery and introduction of a (3,18)-connected net as an ideal blueprint for the design of metal-organic frameworks. Nat. Chem. 6, 673–680. doi:10.1038/nchem.1982
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haszeldine, R. S. (2009). Carbon capture and storage: how green can black be? Science 325, 1647–1652. doi:10.1126/science.1172246
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jiang, H. L., and Xu, Q. (2011). Porous metal-organic frameworks as platforms for functional applications. Chem. Commun. 47, 3351–3370. doi:10.1039/c0cc05419d
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kihara, N., Hara, N., and Endo, T. (1993). Catalytic activity of various salts in the reaction of 2,3-epoxypropyl phenyl ether and carbon dioxide under atmospheric pressure. J. Org. Chem. 58, 6198–6202. doi:10.1021/jo00075a011
Kim, J., Kim, S. N., Jang, H. G., Seo, G., and Ahn, W. S. (2013). CO2 cycloaddition of styrene oxide over MOF catalysts. Appl. Catal. A Gen. 453, 175–180. doi:10.1016/j.apcata.2012.12.018
Kim, Y. J., and Park, D. W. (2013). Functionalized IRMOF-3: an efficient heterogeneous catalyst for the cycloaddition of allyl glycidyl ether and CO2. J. Nanosci. Nanotechnol. 13, 2307–2312. doi:10.1166/jnn.2013.6878
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kleist, W., Jutz, F., Maciejewski, M., and Baiker, A. (2009). Mixed-linker metal-organic frameworks as catalysts for the synthesis of propylene carbonate from propylene oxide and CO2. Eur. J. Inorg. Chem. 3552–3561. doi:10.1002/ejic.200900509
Lee, J., Farha, O. K., Roberts, J., Scheidt, K. A., Nguyen, S. T., and Hupp, J. T. (2009). Metal-organic framework materials as catalysts. Chem. Soc. Rev. 38, 1450–1459. doi:10.1039/b807080f
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lescouet, T., Chizallet, C., and Farrusseng, D. (2012). The origin of the activity of amine-functionalized metal-organic frameworks in the catalytic synthesis of cyclic carbonates from epoxide and CO2. ChemCatChem 4, 1725–1728. doi:10.1002/cctc.201200288
Long, J. R., and Yaghi, O. M. (2009). The pervasive chemistry of metal-organic frameworks. Chem. Soc. Rev. 38, 1213–1214. doi:10.1039/b903811f
Ma, L. Q., Falkowski, J. M., Abney, C., and Lin, W. B. (2010). A series of isoreticular chiral metal-organic frameworks as a tunable platform for asymmetric catalysis. Nat. Chem. 2, 838–846. doi:10.1038/nchem.738
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Macias, E. E., Ratnasamy, P., and Carreon, M. A. (2012). Catalytic activity of metal organic framework Cu3(BTC)2 in the cycloaddition of CO2 to epichlorohydrin reaction. Catal. Today 198, 215–218. doi:10.1016/j.cattod.2012.03.034
Markewitz, P., Kuckshinrichs, W., Leitner, W., Linssen, J., Zapp, P., Bongartz, R., et al. (2012). Worldwide innovations in the development of carbon capture technologies and the utilization of CO2. Energy Environ. Sci. 5, 7281–7305. doi:10.1039/c2ee03403d
Mikkelsen, M., Jorgensen, M., and Krebs, F. C. (2010). The teraton challenge. A review of fixation and transformation of carbon dioxide. Energy Environ. Sci. 3, 43–81. doi:10.1039/b912904a
Miralda, C. M., Macias, E. E., Zhu, M. Q., Ratnasamy, P., and Carreon, M. A. (2012). Zeolitic imidazole framework-8 catalysts in the conversion of CO2 to chloropropene carbonate. ACS Catal. 2, 180–183. doi:10.1021/cs200638h
North, M., and Pasquale, R. (2009). Mechanism of cyclic carbonate synthesis from epoxides and CO2. Angew. Chem. 48, 2946–2948. doi:10.1002/anie.200805451
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
North, M., Pasquale, R., and Young, C. (2010). Synthesis of cyclic carbonates from epoxides and CO2. Green Chem. 12, 1514–1539. doi:10.1039/c0gc00065e
Phan, A., Doonan, C. J., Uribe-Romo, F. J., Knobler, C. B., O’Keeffe, M., and Yaghi, O. M. (2009). Synthesis, structure, and carbon dioxide capture properties of zeolitic imidazolate frameworks. Acc. Chem. Res. 43, 58–67. doi:10.1021/ar900116g
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ravon, U., Domine, M. E., Gaudillere, C., Desmartin-Chomel, A., and Farrusseng, D. (2008). MOFs as acid catalysts with shape selectivity properties. New J. Chem. 32, 937–940. doi:10.1039/b803953b
Ravon, U., Savonnet, M., Aguado, S., Domine, M. E., Janneau, E., and Farrusseng, D. (2010). Engineering of coordination polymers for shape selective alkylation of large aromatics and the role of defects. Microporous Mesoporous Mater. 129, 319–329. doi:10.1016/j.micromeso.2009.06.008
Ren, Y. W., Shi, Y. C., Chen, J. X., Yang, S. R., Qi, C. R., and Jiang, H. F. (2013). Ni(salphen)-based metal-organic framework for the synthesis of cyclic carbonates by cycloaddition of CO2 to epoxides. RSC Adv. 3, 2167–2170. doi:10.1039/c2ra22550f
Sakakura, T., and Kohno, K. (2009). The synthesis of organic carbonates from carbon dioxide. Chem. Commun. 1312–1330. doi:10.1039/b819997c
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Schrag, D. P. (2007). Preparing to capture carbon. Science 315, 812–813. doi:10.1126/science.1137632
Serre, C., Kitagawa, S., and Dietzel, P. D. C. (2012). Introduction to special issue: metal organic frameworks. Microporous Mesoporous Mater. 157, 1–2. doi:10.1016/j.micromeso.2012.04.044
Shaikh, A. A. G., and Sivaram, S. (1996). Organic carbonates. Chem. Rev. 96, 951–976. doi:10.1021/cr950067i
Song, J. L., Zhang, Z. F., Hu, S. Q., Wu, T. B., Jiang, T., and Han, B. X. (2009). MOF-5/n-Bu4NBr: an efficient catalyst system for the synthesis of cyclic carbonates from epoxides and CO2 under mild conditions. Green Chem. 11, 1031–1036. doi:10.1039/b902550b
Srivastava, R., Srinivas, D., and Ratnasamy, P. (2005). CO2 activation and synthesis of cyclic carbonates and alkyl/aryl carbamates over adenine-modified Ti-SBA-15 solid catalysts. J. Catal. 233, 1–15. doi:10.1016/j.jcat.2005.03.023
Sun, J., Fujita, S.-I., Zhao, F., and Arai, M. (2005). A highly efficient catalyst system of ZnBr2/n-Bu4NI for the synthesis of styrene carbonate from styrene oxide and supercritical carbon dioxide. Appl. Catal. A Gen. 287, 221–226. doi:10.1016/j.apcata.2005.03.035
Tu, M., and Davis, R. J. (2001). Cycloaddition of CO2 to epoxides over solid base catalysts. J. Catal. 199, 85–91. doi:10.1006/jcat.2000.3145
Wang, J. Q., Kong, D. L., Chen, J. Y., Cai, F., and He, L. N. (2006). Synthesis of cyclic carbonates from epoxides and carbon dioxide over silica-supported quaternary ammonium salts under supercritical conditions. J. Mol. Catal. A Chem. 249, 143–148. doi:10.1016/j.molcata.2006.01.008
Xie, Y., Wang, T. T., Liu, X. H., Zou, K., and Deng, W. Q. (2013). Capture and conversion of CO2 at ambient conditions by a conjugated microporous polymer. Nat. Commun. 4, 1960. doi:10.1038/ncomms2960
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xuan, W. M., Zhu, C. F., Liu, Y., and Cui, Y. (2012). Mesoporous metal-organic framework materials. Chem. Soc. Rev. 41, 1677–1695. doi:10.1039/c1cs15196g
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yamaguchi, K., Ebitani, K., Yoshida, T., Yoshida, H., and Kaneda, K. (1999). Mg-Al mixed oxides as highly active acid-base catalysts for cycloaddition of carbon dioxide to epoxides. J. Am. Chem. Soc. 121, 4526–4527. doi:10.1021/ja9902165
Yang, D. A., Cho, H. Y., Kim, J., Yang, S. T., and Ahn, W. S. (2012). CO2 capture and conversion using Mg-MOF-74 prepared by a sonochemical method. Energy Environ. Sci. 5, 6465–6473. doi:10.1039/c1ee02234b
Yang, L., Yu, L., Diao, G., Sun, M., Cheng, G., and Chen, S. (2014). Zeolitic imidazolate framework-68 as an efficient heterogeneous catalyst for chemical fixation of carbon dioxide. J. Mol. Catal. A Chem. 392, 278–283. doi:10.1016/j.molcata.2014.05.033
Yano, T., Matsui, H., Koike, T., Ishiguro, H., Fujihara, H., Yoshihara, M., et al. (1997). Magnesium oxide-catalysed reaction of carbon dioxide with an epoxide with retention of stereochemistry. Chem. Commun. 1129–1130. doi:10.1039/a608102i
Yasuda, H., He, L. N., and Sakakura, T. (2002). Cyclic carbonate synthesis from supercritical carbon dioxide and epoxide over lanthanide oxychloride. J. Catal. 209, 547–550. doi:10.1006/jcat.2002.3662
Yasuda, H., He, L. N., Sakakura, T., and Hu, C. W. (2005). Efficient synthesis of cyclic carbonate from carbon dioxide catalyzed by polyoxometalate: the remarkable effects of metal substitution. J. Catal. 233, 119–122. doi:10.1016/j.jcat.2005.04.030
Yoon, M., Srirambalaji, R., and Kim, K. (2012). Homochiral metal-organic frameworks for asymmetric heterogeneous catalysis. Chem. Rev. 112, 1196–1231. doi:10.1021/cr2003147
Zalomaeva, O. V., Chibiryaev, A. M., Kovalenko, K. A., Kholdeeva, O. A., Balzhinimaev, B. S., and Fedin, V. P. (2013a). Cyclic carbonates synthesis from epoxides and CO2 over metal-organic framework Cr-MIL-101. J. Catal. 298, 179–185. doi:10.1016/j.jcat.2012.11.029
Zalomaeva, O. V., Maksimchuk, N. V., Chibiryaev, A. M., Kovalenko, K. A., Fedin, V. P., and Balzhinimaev, B. S. (2013b). Synthesis of cyclic carbonates from epoxides or olefins and CO2 catalyzed by metal-organic frameworks and quaternary ammonium salts. J. Energy Chem. 22, 130–135. doi:10.1016/S2095-4956(13)60017-0
Zevenhoven, R., Eloneva, S., and Teir, S. (2006). Chemical fixation of CO2 in carbonates: routes to valuable products and long-term storage. Catal. Today 115, 73–79. doi:10.1016/j.cattod.2006.02.020
Zhao, M., Ou, S., and Wu, C.-D. (2014). Porous metal-organic frameworks for heterogeneous biomimetic catalysis. Acc. Chem. Res. 47, 1199–1207. doi:10.1021/ar400265x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhou, H. C., Long, J. R., and Yaghi, O. M. (2012a). Introduction to metal-organic frameworks. Chem. Rev. 112, 673–674. doi:10.1021/cr300014x
Zhou, X., Zhang, Y., Yang, X. G., Zhao, L. Z., and Wang, G. Y. (2012b). Functionalized IRMOF-3 as efficient heterogeneous catalyst for the synthesis of cyclic carbonates. J. Mol. Catal. A Chem. 361, 12–16. doi:10.1016/j.molcata.2012.04.008
Keywords: metal-organic frameworks, carbon dioxide utilization, carbon dioxide fixation, cycloaddition reactions, heterogeneous catalysts, cyclic carbonates
Citation: Beyzavi MH, Stephenson CJ, Liu Y, Karagiaridi O, Hupp JT and Farha OK (2015) Metal–organic framework-based catalysts: Chemical fixation of CO2 with epoxides leading to cyclic organic carbonates. Front. Energy Res. 2:63. doi: 10.3389/fenrg.2014.00063
Received: 28 November 2014; Accepted: 18 December 2014;
Published online: 21 January 2015.
Edited by:
Camille Petit, Imperial College London, UKReviewed by:
Banglin Chen, The University of Texas at San Antonio, USAAntoine Buchard, University of Bath, UK
Copyright: © 2015 Beyzavi, Stephenson, Liu, Karagiaridi, Hupp and Farha. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph T. Hupp and Omar K. Farha, Department of Chemistry, International Institute for Nanotechnology (IIN), Northwestern University, Evanston, IL, USA e-mail:ai1odXBwQG5vcnRod2VzdGVybi5lZHU=;by1mYXJoYUBub3J0aHdlc3Rlcm4uZWR1
†M. Hassan Beyzavi, Casey J. Stephenson and Yangyang Liu have contributed equally to this work.