 Alicia Wong
Alicia Wong Emilyn U. Alejandro
Emilyn U. Alejandro
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 11 March 2025
Sec. Diabetes: Molecular Mechanisms
Volume 16 - 2025 | https://doi.org/10.3389/fendo.2025.1562646
This article is part of the Research TopicAdvances in β-cell Development & RegenerationView all 5 articles
Dysfunction of the insulin-secreting β-cells is a key hallmark of Type 2 diabetes (T2D). In the natural history of the progression of T2D, factors such as genetics, early life exposures, lifestyle, and obesity dictate an individual’s susceptibility risk to disease. Obesity is associated with insulin resistance and increased demand for insulin to maintain glucose homeostasis. Studies in both mouse and human islets have implicated the β-cell’s ability to compensate through proliferation and survival (increasing functional β-cell mass) as a tipping point toward the development of disease. A growing body of evidence suggests the reduction of β-cell mass in T2D is driven majorly by loss of β-cell identity, rather than by apoptosis alone. The development and maintenance of pancreatic β-cell identity, function, and adaptation to stress is governed, in part, by the spatiotemporal expression of transcription factors (TFs), whose activity is regulated by signal-dependent post-translational modifications (PTM). In this review, we examine the role of these TFs in the developing pancreas and in the mature β-cell. We discuss functional implications of post-translational modifications on these transcription factors’ activities and how an understanding of the pathways they regulate can inform therapies to promoteβ-cell regeneration, proliferation, and survival in diabetes.
Diabetes is a growing public health concern that affects an estimated 537 million individuals worldwide (1), a number that is projected to increase to 783 million by 2045 (2). 90% of diabetes cases are classified as Type 2 (T2D), which is characterized by insulin resistance (3), hyperglycemia, and loss of functional pancreatic β-cell mass (4). While conventionally thought mainly to be associated with ER stress mediated apoptosis, dedifferentiation, defined here as the loss of pancreatic β-cell identity as an insulin-producing cell, has increasingly been identified as another major driver of progressive β-cell failure in diabetes (5–9), a concept that has been extensively reviewed (10–17).
Developmentally, lineage determination, differentiation, and maturation, in the pancreas is controlled, in part, by activation of major transcription factors (TFs) and their interaction with gene regulatory networks (18). In the islets of Langerhans, Pdx1 (19) (Pancreatic Duodenal Homeobox 1) and Pax6 (20) (Paired Box 6), for example, both maintain pancreatic β-cell identity by suppressing genes that specify other islet cell types (19, 20). Functionally, as a nutrient-sensitive cell, it has been posited that signal-secretion coupling, where β-cells secrete insulin in response to flux in the nutrient milieu, relies on the collaboration between lineage-dependent TFs (ex. Pdx1) and signal-dependent TFs (ex. MafA) (21). Many of these TFs also undergo nutrient-dependent post-translational modifications (PTM), such as O-GlcNAcylation, which can alter their conformation, subcellular localization, stability, and activity (22).
In this review, we examine the role of PTM on major transcription factors governing pancreas and islet development and their role in maintaining the identity and function of pancreatic β-cells. We also examine their roles in β-cell identity loss in T2D, as well as how transcriptional activation of pathways during adaptive β-cell mass expansion events such as in obesity and pregnancy can be targeted in future β-cell regeneration therapies.
Pancreatic development is governed by a hierarchy of transcriptional activation. In the mouse, the pancreatic bud forms at embryonic day (e) 9.5 (23). At this stage, expression of master regulator Pdx1 is detectable on the foregut wall and specifies the pancreatic lineage (24, 25) (Figure 1). Deletion of Pdx1 at this point in development results in pancreatic agenesis in mice (26) due to uncoupling of the developing pancreatic epithelium from mesenchyme-derived morphogenesis signals (27). In humans, a homozygous point deletion that renders Pdx1 truncated and nonfunctional also causes pancreatic agenesis (28). By e15.5, the number of Ngn3+ endocrine progenitor cells peaks (29) and, together with expression of Isl1 (30), defines an endocrine islet cell fate (Figure 1) (25, 31, 32). Expression of NeuroD1 in these cells is required for differentiation into glucagon-secreting α-cells and insulin-producing β-cells (33). Later, the expression of Pdx1 and MafA are restricted to the mature β-cell (Figure 1).
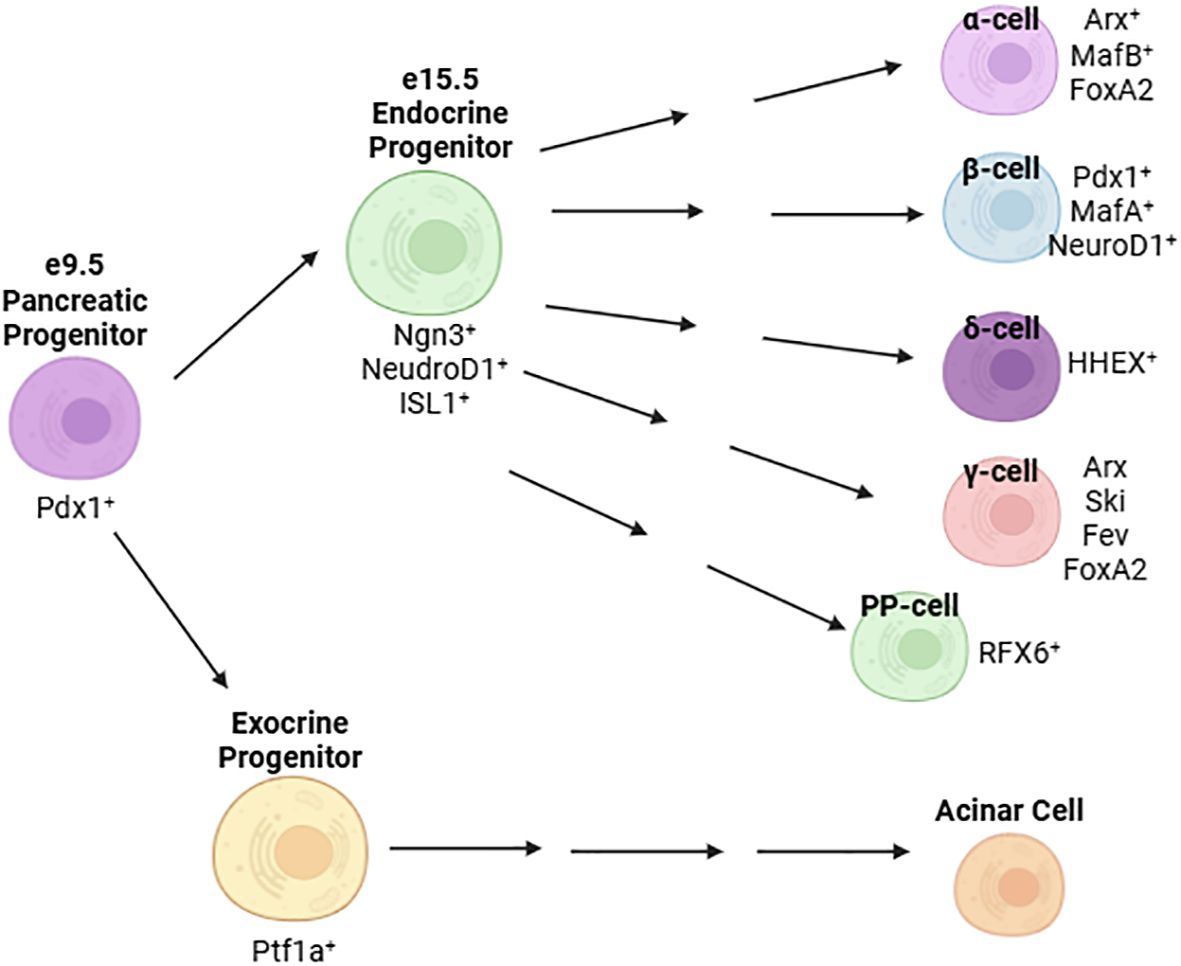
Figure 1. Major transcription factors defining the development and maturation of islet cells.
Strong evidence from human and murine studies have tied the loss of β-cell mass in T2D to, in part, altered β-cell identity and function in response to a high glucose, high lipid environment rather than apoptosis alone (6, 34–37). Here, the term “de-differentiation” broadly refers to a β-cell that has lost its identity as an insulin-producing cell, rather than explicit reversion to a less mature stage of development.
Obesity is associated with insulin resistance and is a risk factor for the development of T2D (38–41). In both human obesity and high fat diet-fed rodent models, the increased demand for insulin is compensated for by an increase in β-cell proliferation and functional β-cell mass (42–44). In primary islets and in MIN6 cells, there exists a heterogeneous population of β-cells consisting of mostly “mature” cells expressing both the TF Pdx1 and a high level of insulin transcription (Pdx1+/Inshigh) and a smaller population of Pdx1+/InsLow cells (45). In these Pdx1+/InsLow β-cells, genes characteristic of early β-cell development, namely MafB and Nkx2.2 are enriched (45). While these cells have a lower secretory capacity than the Pdx1+/Inshigh population, they have modestly improved proliferative capacity; a subset of them go on to become Pdx1+/Inshigh cells, suggesting Pdx1+/InsLow exist in a less mature developmental state more consistent with embryonic β-cell progenitors (45). Given these findings, it is possible that physiologically, this population of less mature but more proliferative β-cells is maintained to promote compensatory β-cell mass expansion in response to conditions such as obesity or pregnancy. Supporting this notion, chronic exposure to glucose in vivo, as demonstrated in rats, results in a compensatory expansion of β-cell mass to maintain euglycemia and also gives rise to a population of Pdx1+/InsLow cells (46). Given reduced transcriptional activity of Pdx1 in a diabetic milieu (47), it is feasible that this population of Pdx1+/InsLow cells is more susceptible to de-differentiation and cannot mount an effective compensatory response to a glucotoxic environment.
Other mechanisms have been proposed for β-cell de-differentiation in T2D. The forkhead TF, FoxO1, has been shown to be upregulated in islets in response to high fat diet feeding and orchestrates β-cell compensation in response to high-fat diet induced insulin resistance through expansion of β-cell mass (48). Knockdown of FoxO1 in murine β-cells results in defective compensation to physiological stressors such as successive pregnancies and aging (34). These mice experience reduced β-cell mass resulting from either the reversion of differentiated β-cells to a progenitor-like state or the trans-differentiation of β-cells to an α-like cell (34). More broadly, glucotoxicity and oxidative stress brought on by exposure to a diabetic milieu destabilizes the expression of TFs governing mature β-cell identity, including Pdx1 (49–51), MafA, and Nkx6.1 (51). As discussed earlier, these TFs have demonstrated roles in suppressing other islet cell programs.
There is evidence, however, that de-differentiation of β-cells is a potentially reversible process. In a mouse model of defective insulin secretion, lineage tracing studies demonstrate that β-cells revert to a Ngn3+/Insulin- state under hyperglycemia but that these cells can re-differentiate back to a mature β-cell identity following normalization of blood glucose levels (37), suggesting de-differentiated cells retain significant plasticity. Wang and colleagues propose re-differentiation as a potential mechanism through which some T2D patients partially recover β-cell mass and function following long-term treatment with insulin (37). Further supporting the notion of islet-cell plasticity, severe loss of β-cells following ablation by diphtheria toxin is compensated, in part, by trans-differentiation of α-cells to augment β-cell mass (52). Additionally, there is evidence to suggest the conversion of α-cells to β-cells in mothers following pregnancy parturition in a murine model (53). Taken together, β-cell plasticity and differentiation capacity, as well as the TFs that govern this process, prove to be an attractive therapeutic target for preservation or reconstitution of functional β-cell mass in diabetes.
Mature Onset Diabetes of the Young (MODY) describes a series of rare, heritable diabetic conditions that occur in young individuals and involves a dominant mutation of a single gene (54). Currently, 14 subtypes of MODY have been defined (54), half of which involve mutations in genes encoding transcription factors (Table 1). The causative link between loss of function mutations on TFs and the subsequent development of diabetes warrants further study on their regulatory role in pancreas/islet development and maintenance of glucose homeostasis in response to nutrient changes. Given that many of these TFs also undergo nutrient-dependent PTM (22), more efforts should be made in elucidating the molecular and biochemical effect of these PTM and the subsequence impact on β-cell survival and function.

Table 1. A summary of MODY subtypes involving mutations in transcription factors.
Post-translational modifications (PTM) are enzyme-catalyzed modifications onto the backbones or sidechains of translated proteins, often at specific amino acid residues (69). These PTM include phosphorylation (addition of phosphate group to Ser, Thr, Tyr), glycosylation (addition of sugar to Asn in N-linked, to Ser, Thr in O-linked), ubiquitylation (addition of ubiquitin to Lys), and acetylation (addition of acetyl group to Lys), among others (69). PTMs are a mechanism through which diversity of the proteome can be greatly increased apart from variations in amino acid sequence alone (69). These modifications can affect localization, function, and stability of target proteins (69), including many TFs in the pancreatic β-cell. Among them, Nkx2.2 (70), Nkx6.1 (71), and Pdx1 (19) are major players that govern islet development and play explicit roles in maintaining mature pancreatic β-cell identity through repression of genes conferring other islet cell fates. Other transcription factors, such as FoxM1 (72–75), play a role in maintaining or expanding functional β-cell mass. We examined these selected transcription factors and their post-translational modifications in detail. Other transcription factors involved in β-cell function and adaptation are summarized in Table 2.
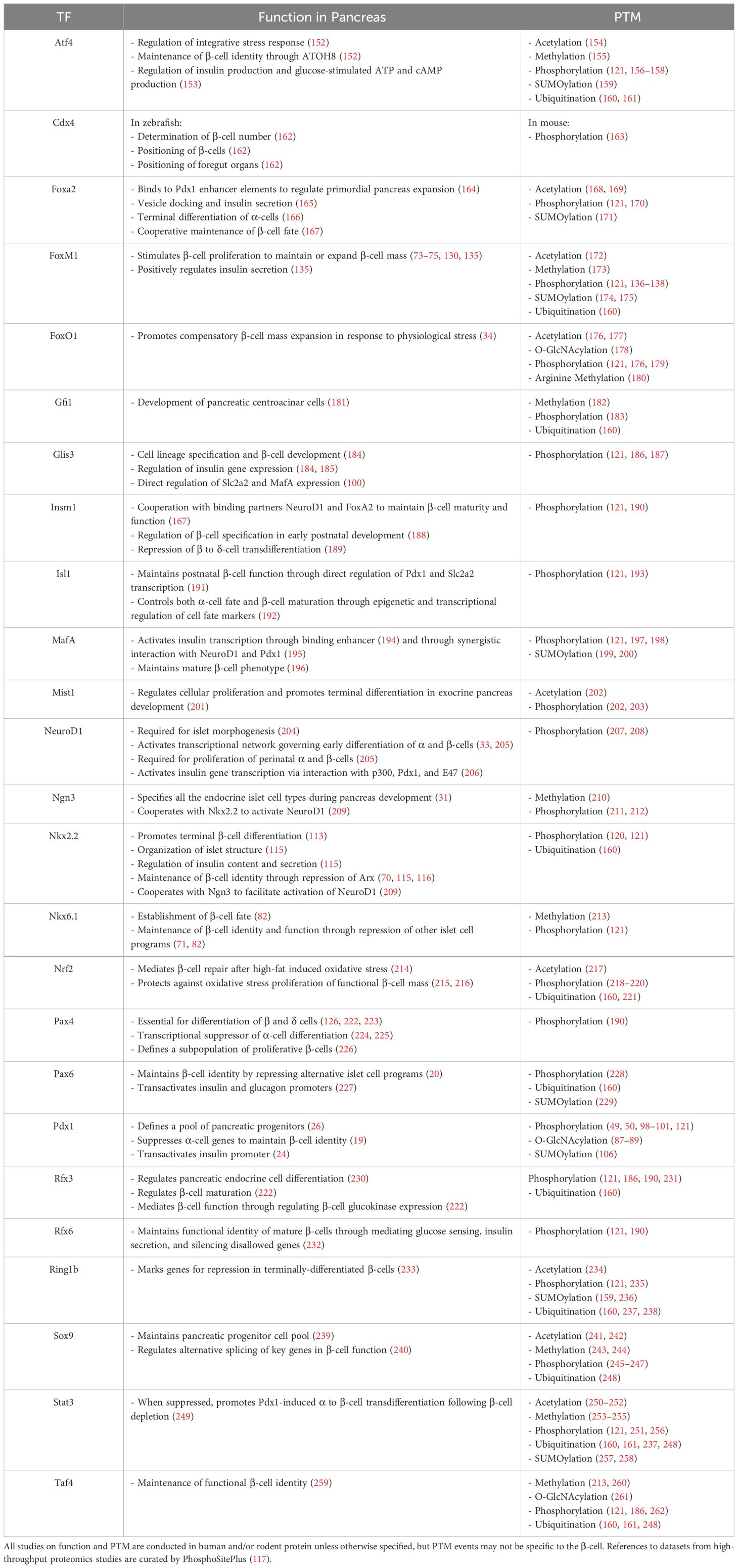
Table 2. PTMs of transcription factors governing β-cell development, identity, and function.
Pdx1 is a master regulator of pancreas development, as well as β-cell function, identity, and survival. Originally coined Insulin Promoter Factor 1 (IPF1), Pdx1 was first identified as a novel insulin promoter binding protein expressed solely in the β-cell (76) and was found to transactivate both insulin (24) and somatostatin (77) gene transcription. Autoantibodies against Pdx1 have been detected in Type 1 diabetes (78), and mutations in Pdx1 are associated with increased Type 2 diabetes (T2D) risk (79). A dominant loss of function mutation in Pdx1 causes Mature Onset Diabetes of the Young (MODY) Type 4 (60). Later genetic studies confirmed that Pdx1 is indispensable for pancreas organogenesis (26, 27, 80). Genetic ablation of Pdx1 results in pancreatic agenesis by blocking outgrowth of the pancreatic bud and uncouples mesenchymal and epithelial pancreas development (26, 27, 80). β-cell-specific knockdown of Pdx1 disrupts glucose homeostasis and causes mature-onset diabetes in mice (81). Islets of mice with Pdx1-deficient β-cells have disrupted islet architecture and impaired Glut2 expression, accompanied by increased glucagon-expressing cells and insulin/glucagon co-expressing cells (81). Pdx1-deficient β-cells exhibit α-cell like ultrastructure, along with an α-cell-like electrophysiology and transcriptomic profile, including increased MafB and glucagon expression (19). In MIN6 cells, Pdx1 is found to bind upstream to the MafB coding region, and in Ins1 cells, depletion of MafB in the absence of Pdx1 is sufficient to prevent induction of glucagon (19), suggesting Pdx1 maintains β-cell identity by blocking an α-cell program through repression of MafB. Furthermore, the reduction of Nkx6.1 expression in Pdx1-deficient β-cells (81) suggests another mechanism by which Pdx1 maintains β-cell identity may be through the stabilization of Nkx6.1, which has been shown to repress α-cell factor, Arx (82).
Pdx1 is highly conserved among species. Its expression has been mapped to chromosome 13 (83) in humans and chromosome 5 in mice (84). Pdx1 is composed of two exons separated by a single intronic region, with no reported splice variants (85). Exon 1 encodes the amino terminus (85), which houses the transactivation domain (residues 13-73 in both mice and humans) (86), and Exon 2 encodes the carboxyl terminus (85), which includes the nuclear localization signal (residues 198-204 in mice, 197-203 in mice) (86). In MIN6 cells, Pdx1 has been shown to undergo the nutrient-sensitive O-GlcNAc modification (87–89), which has been shown to increase DNA binding affinity (87). In O-GlcNAcylation, the enzyme O-GlcNAc transferase (Ogt) catalyzes the addition of a single GlcNAc sugar molecule onto Ser and Thr residues of nuclear, cytoplasmic, and mitochondrial proteins (90, 91), and this modification is removed by O-GlcNAcase (Oga) (92). YinOYang, a server that generates neural network predictions for O-GlcNAc sites in protein sequences, has computationally predicted Pdx1 to be O-GlcNAc modified at T11, S273, and S274 (93). However, no studies to date have confirmed these findings. Much like the deletion of Pdx1, loss of Ogt in the β-cells results in progressive diabetes and reduced β-cell mass, accompanied by significant reductions in islet Pdx1 protein levels (94, 95). This was recapitulated in a mouse model of Ogt loss in the endocrine progenitors, where immunoreactivity to Pdx1 was reduced (96). Additionally, genetic ablation of Ogt in the pancreatic epithelial progenitors results in pancreatic aplasia (89), phenocopying pancreatic Pdx1 knockdown (26). Interestingly, in the absence of Ogt, overexpression of Pdx1 in the β-cells improves mitochondrial morphology and function (95), while normalization of Pdx1 levels in the pancreatic epithelium can partially restore pancreas weight and β-cell mass (97). Together, these studies provide indirect evidence for the positive regulatory role of O-GlcNAcylation on Pdx1. However, given the many O-GlcNAc modified proteins in the β-cell, further molecular studies are warranted to elucidate whether this regulation occurs because of a direct O-GlcNAcylation on Pdx1 or due to factors upstream.
In contrast, numerous studies have examined the effects of various phosphorylation sites on Pdx1 (49, 50, 98–101). Ser61 was found to be the principal site of phosphorylation by nanofluidic proteomic assays in both endogenous and overexpressed mouse Pdx1 (98). In vitro, phosphorylation at this site was found to be unchanged under both high and low glucose conditions, and despite existing in a phosphorylated state during embryonic development, expression of a phospho-dead mutant, Pdx1 S61A, had no adverse effect on pancreas development in vivo (98), demonstrating the remarkable stability of this site under non-disease conditions. In contrast, under oxidative stress, which is associated with pathogenesis of T2D (102), increased phosphorylation of Pdx1 at several different residues targets it for degradation. Phosphorylation at S61 and/or S66 occurs in a glycogen synthase kinase 3 (GSK3)-dependent manner (49), while T11 is directly phosphorylated by Mammalian Sterile-20-like kinase (Mst1) (50), an amplifier of caspase-mediated apoptosis that is upregulated in a diabetic milieu (50, 103). In contrast, phosphorylation at T230 and S231 by CK2 increases Pdx1 transcriptional activity (104) through increasing Pdx1 stability (101). Pdx1 binds to E3-ubuiquitin ligase adaptor protein, SPOP, where it is targeted for ubiquitin-mediate proteasomal degradation (101). However, phosphorylation at T230 and S231 greatly decreases Pdx1 affinity for SPOP, allowing Pdx1 to maintain its function as a transcription factor (101). Taken together, these data implicate different phosphorylation events in the regulation of Pdx1 stability. In addition to protein levels, the localization Pdx1 in response to glucose is an important regulator of β-cell function. When the β-cell is exposed to high glucose, Pdx1 moves from the periphery of the nucleus to the nucleoplasm, where it can transactivate insulin transcription (105). In vitro, when S269 is phosphorylated by Homeodomain interacting protein kinase 2 (Hipk2), Pdx1 remains localized in the nuclear periphery (100), but further studies in vivo are necessary to characterize any effect on Pdx1 transactivation potential. Another mechanism governing Pdx1 localization is SUMOylation, the addition of small ubiquitin-like modifiers by Small Ubiquitin-related Modifier 1 (SUMO-1) (106). SUMOylation promotes both Pdx1 stability and nuclear localization; inhibition of SUMO-1 is associated with reduced transactivation of the insulin gene (106). Given the importance of Pdx1 in β-cell function and survival, targeted manipulation of Pdx1 PTM may inform therapies in maintaining β-cell function in diabetes.
As a homeobox gene, Nkx2.2 plays a pivotal role in the development of the central nervous system (107–110). In mice, expression of Nkx2.2 is detectable in the developing forebrain beginning 9 days post-coitum (dpc) (111). Originally thought to be brain-specific, Rudnick and colleagues detected expression of Nkx2.2 and other homeobox genes in murine β-cell lines (112). This was later confirmed in vivo by Sussel and colleagues, who additionally found Nkx2.2 expression in both α and PP cells (113). Mice carrying a homozygous null mutation of Nkx2.2 lack β-cells and have reduced α and PP cells, resulting in severe hyperglycemia and neonatal mortality (113). Interestingly, there is a large population of partially-differentiated “β-like” cells that express Isl1 and Pdx1 but neither secrete insulin nor express other canonical β-cell markers such as Glut2 and Nkx6.1, suggesting Nkx2.2 is required for terminal differentiation of β-cells (113). Supporting this notion, when given a series of developmental transcription factors in a timed manner, ending with Nkx2.2, human fibroblasts can be differentiated into β-cells with functional glucose-stimulated insulin secretion both in vitro and when transplanted into immunodeficient mice (114). In the mature islet, Nkx2.2 plays a functional role in regulating both β-cell function and islet architecture. When Nkx2.2 is repressed in the β-cells in mice, there is a downregulation of MafA, a downstream target of Nkx2.2 and a key TF in β-cell maturation and glucose response (115). Furthermore, these mice are glucose intolerant, accompanied by impairments in insulin content and secretion, as well as disruptions in islet structure during islet assembly at e18.5 and persisting into adulthood (115). In addition to function, Nkx2.2 also plays a critical role in the maintenance of β-cell identity. RNA sequencing of islets from mice with Nkx2.2 deficient β-cells indicated repression of factors governing β-cell function, such as Glut2 (70, 115) and Nkx6.1 (70), and lineage tracing of these β-cells confirmed the co-expression of hormones associated with other islet cell types such as glucagon, somatostatin, and pancreatic polypeptide (70). Mechanistically, Papizan and colleagues demonstrate that Nkx2.2 directly binds the promoter of the canonical α-cell gene, Arx, where it is proposed to recruit its binding partner, co-repressor protein Grg3, to repress Arx expression (116). Furthermore, Nkx2.2/Grg3 also complexes with HDAC1 and Dnmt3a at the Arx promoter in β-cells, lending credence to the notion Nkx2.2 and Dnmt3a work together to repress Arx expression in the β-cell (116). Taken together, these data suggest that Nkx2.2 plays a pivotal role in maintaining β-cell identity as an insulin-secreting cell by repressing other pancreatic endocrine cell programs.
Most of what is known about post-translational modifications on Nkx2.2 are from large-scale proteomics datasets in human and mouse tissues. In both mice and humans, the DNA-binding domain of Nkx2.2 is located on amino acid positions 128-187 (86). While PTM in this region have not been explicitly studied in the pancreatic β-cell, Akimov et al. demonstrated in the Hep2 and Jurkat human cell lines that K137 is ubiquitinated (116, 117). The specific effect of ubiquitination at this residue on Nkx2.2 has not been examined. However, the homeostatic balance of ubiquitination and de-ubiquitination is generally considered important in protein turnover and quality control. Protein degradation is regulated through the ubiquitin-proteasome system (118), allowing clearance of dysfunctional or misfolded proteins (119). In addition, a proteomic study in human ischemic breast and ovarian cancer samples indicated phosphorylation sites on Y152 and S163, residues within the homeobox region (117, 120). The effect of these PTM on the Nkx2.2, particularly on DNA-binding activity, warrants further study, including whether these same sites are modified in the β-cell. In the islet-specific context, Sacco and colleagues conducted a phospho-proteomic study of MIN6 cells and, combining stimulated and unstimulated conditions, found amino acid residues S27, S63, S103, S107, S199 to be phosphorylated (117, 121). To understand the biological impact of these PTMs, site-directed mutagenesis and immunoprecipitation studies should be performed to confirm the proteomics results and to elucidate the effect of phosphorylation at these residues.
Nkx6.1, a member of the NK homeobox family, is involved in β-cell formation and differentiation (122) and plays a role in suppressing acinar cell fate during pancreatic development through antagonism of Ptf1a (123). In humans, Nkx6.1 expression is detectable in the neural tube at Carnegie Stage (CS) 12 (29-31 dpc) and in the dorsal bud of the developing pancreas at CS 13 (30-33 dpc) (124). In mice, at e10.5, Nkx6.1 expression can be detected across the entire developing pancreatic epithelium (122). However, starting at the secondary transition of pancreatic development at e12.5, expression becomes restricted, ultimately becoming detectable only in the insulin-positive cells by e15.5, corresponding to the peak in β-cell formation (122). Overexpression of Nkx6.1 in Ngn3+ endocrine progenitors results in a reduction of non-insulin producing islet cell types (α, δ, ϵ, PP) with no differences in overall proliferation rates, suggesting expression of Nkx6.1 favors the establishment of a β-cell fate (82). Conversely, Nkx6.1 loss in Ngn3+ cells upregulates the expression of α-cell-associated TF, Arx, in insulin+ cells in neonates, but not in e15.5 embryos (82), suggesting a role for Nkx6.1 in the maintenance, but not formation, of β-cells. This is further supported by data showing the requirement of Nkx6.1 in postnatal β-cell function. Conditional inactivation of Nkx6.1 in β-cells of adult mice is associated rapid-onset glucose intolerance, hyperglycemia, and reduced circulating insulin; accompanied by reductions in genes associated with insulin-secretion and β-cell proliferation (71). Both constitutive (82) and conditional (71) inactivation of Nkx6.1 cause β-cells to adopt a δ-cell-like identity (71, 82). This, along with the increased Arx expression in insulin+ cells during Nkx6.1 deficiency (82), lends credence to the notion that Nkx6.1 regulates β-cell identity, in part, through repression of other islet cell programs.
There is evidence to support that the regulatory role of Nkx6.1 is carried out in a spaciotemporal manner (71, 125). Full-body ablation of Nkx6.1 in mice results in deficiency of insulin-producing cells when examined after the secondary transition in β-cell development at e13 but not prior (122). In contrast, the deletion of NK homeobox family member, Nkx2.2, yields a lack of insulin-producing cells through the entirety of pancreas development (122). Furthermore, concomitant loss of Nkx6.1 in the absence of Nkx2.2 phenocopies Nkx2.2 loss alone, suggesting the regulatory role of Nkx6.1 occurs hierarchically downstream of Nkx2.2 (122). In Nkx6.1-deficient mice, reconstitution of Nkx6.1 in the Pdx1+ domain, but not in Ngn3+ domain, can rescue β-cell development, suggesting that in the specification of β-cell fate, Nkx6.1 expression is required prior to Ngn3+ endocrine progenitor cell commitment (56). This sets Nkx6.1 regulation of β-cell fate apart from Ngn3-dependent, lineage-specifying TFs, such as Pax4 (125, 126). Later studies would also indicate substantial redundancy between Nkx6.1 and its paralog, Nkx6.2, with equivalent biochemical activities governing β-cell specification. In the absence of Nkx6.1, ectopic overexpression of Nkx6.2 in Pdx1+ cells can rescue the formation and maturation of β-cells, including restoring the expression of key β-cell maturity markers MafA and Glut2, as well as endocrine differentiation co-factor, Myt1, which is normally reduced in the absence of Nkx6.1 (56, 127). These results suggest the differential regulatory role of Nkx6.1 and Nkx6.2 are primarily due to the time in which they become expressed during pancreas development.
Through a phospho-proteomics study in stimulated and unstimulated MIN6 cells, Sacco and colleagues revealed phosphorylation of several Serine residues on Nkx6.1: S228, S335, S353, S359, S364, and S365 all within the C terminus with the exception of S228, located upstream of the homeodomain within the repressor region (86, 117, 121). Interestingly, S335, S353, S359, S364, and S365 were found to be significantly regulated by drug or glucose stimulation. However, to date, no studies have specifically confirmed these phosphorylation events. The C terminus of Nkx6.1 houses a binding interference domain, which in the mouse, is located from residues 306-364, which greatly decreases the DNA binding affinity of its own homeodomain (128). Interestingly, all but one of the purported phosphorylation sites identified by proteomics falls within this binding interference domain, warranting further studies of Nkx6.1 phosphorylation on DNA-binding affinity of its target genes. Furthermore, the S228 residue is located within the repressor domain spanning residues 102-269 (86). Assessing repression efficiency of gene targets using phospho-mimetic or phospho-dead Nkx6.1 mutants at this residue would provide valuable insights on potential mechanisms governing Nkx6.1 function.
Transcription factor FoxM1 is associated with cellular proliferation and growth of various cancers (129), and in the β-cells, is required for maintenance-level (73) and compensatory proliferation in response to partial pancreatectomy (130). In the murine embryonic and neonatal pancreas, FoxM1 is expressed in the endocrine cells (73). While genetic ablation of FoxM1 in the embryonic pancreatic endoderm results in normal β-cell mass at birth, these mice experience a decline in β-cell mass over time due to defective β-cell replication, suggesting FoxM1 is indispensable for postnatal β-cell proliferation (73). Expansion of postnatal β-cell mass results from replication of existing β-cells (131), and turnover significantly declines with age (132–134). In murine islets, this is accompanied by reduced islet FoxM1 gene expression (135). Activating FoxM1 expression in aged islets induces β-cell mass expansion through increased proliferation (135). Additionally, in young mice, a lack of FoxM1 expression in the β-cells results in reduced glucose-stimulated insulin secretion and induction of FoxM1 expression improves glucose homeostasis (135), suggesting that in addition to regulating proliferation, FoxM1 also regulates β-cell function. Phosphorylation of FoxM1 has been extensively studied in non-β-cells. Generally, it is thought that phosphorylation of FoxM1 controls its stability, nuclear entry, relief of its N-terminal repressor domain, and recruitment of co-factors. For example, in fibroblasts, phosphorylation by Raf/MEK/MAPK signaling allows for the nuclear translocation of FoxM1 during the G2/M phase of the cell cycle (136). In various human cell lines, Pololike kinase 1 (Plk1) directly phosphorylates FoxM1 at C terminus residues S715 and S725 (137, 138), which are located within the disordered but highly conserved αβα region of the transactivation domain (TAD) (138). During G1 of the cell cycle, FoxM1 forms an auto-repressive homodimer, wherein the αβα region of the TAD interacts with the ββαβ motif on the N terminal repressive domain (NRD) (138). Conversely, phosphorylation at S715 and S725 disrupts these interactions, allowing the αβα motif to, instead, interact with the intrinsically disordered region (spanning residues 328 – 583 in humans) (138). This super-activated homodimer conformation, common in the S – G2/M phases of the cell cycle, promotes cellular division (138). In 293FT cells and mouse embryonic fibroblasts, FoxM1 is shown to be phosphorylated by ABL1 at Y575, which stabilizes FoxM1 half-life through inhibition of ubiquitin-proteasomal degradation (139).
In MIN6 cells, Sacco and colleagues’ phosphoproteomic approach defined phosphorylation sites at S329, S332, and S635, none of which were significantly modulated by secretion-stimulating drug treatments (121). While biochemical approaches are warranted to validate the phosphorylation of the aforementioned residues, given the role of FoxM1 in regulating cellular proliferation, it may be necessary to assess FoxM1 phosphorylation using β-cells treated with proliferation-promoting agents in order to elucidate regulatory sites.
Expansion of functional β-cell mass through proliferation or regeneration has been a long-standing goal in the treatment of diabetes. Physiologically, β-cell mass has been known to expand in response to increased metabolic demand, such as in pregnancy (140–143) and obesity (4, 42, 144, 145). An understanding of the mechanisms governing this adaptive process can inform novel targets for future β-cell therapies. In the early stages of obesity, there is evidence for increased β-cell hyperplasia to compensate for increased insulin demand in non-diabetic individuals (4, 42, 144, 145). Using a non-diabetic mouse model of obesity, Leptinob/ob, Davis and colleagues identified upregulation of islet transcription factor, FoxM1, accompanied by higher circulating insulin levels and lower plasma glucose (74). Expression of FoxM1 can trigger proliferation in both murine and human donor islets through activation of the cell cycle, and like in mice, its expression is upregulated in islets of obese, non-diabetic human individuals (74). This, along with the lack of FoxM1 upregulation in diabetic Leptinob/ob islets, provides evidence that FoxM1-mediated β-cell proliferation is necessary for the compensatory regulation of glucose homeostasis under obesogenic stress (74).
Normal pregnancy is associated with maternal insulin resistance, necessitating greater insulin demand, and consequently, the upregulation of β-cell mass (140, 146, 147). This has been demonstrated extensively in rodent models (140–143). During pregnancy, β-cell FoxM1 expression is upregulated and has been shown to be a downstream effector of placental lactogen, though the exact mechanism is not entirely clear (75, 141). Lack of FoxM1 expression in the pancreas results in gestational diabetes, associated with inadequate compensatory β-cell proliferation (75). In mice, epidermal growth factor receptor (EGFR) signaling has been shown to orchestrate the pro-survival and proliferative effects of placental lactogen on β-cells during pregnancy (148), and serotonin has been shown molecularly and transcriptomically to act downstream of placental lactogen signaling to promote β-cell proliferation (149, 150). Furthermore, treatment of human immortalized β-cells with serum from pregnant human donors significantly increased rates of proliferation, indirectly supporting the role of pregnancy-specific circulating factors in the upregulation of β-cell expansion in humans (151). Unsurprisingly, a comparison of the islet transcriptome in pregnant and non-pregnant mice revealed an upregulation of genes regulating cell growth, proliferation, and apoptosis, as well as genes governing insulin secretion and secretory granule biosynthesis (150). Taken together, these data support highly orchestrated genetic and hormonal regulations of β-cell mass expansion during pregnancy. Continued research to isolate and modulate key factors during this process could inform therapies to promote β-cell proliferation while simultaneously preventing cell death in the treatment of diabetes.
In adults, new β-cells are formed by replication of existing β-cells rather than by differentiation of stem cells (263). However, the rate of proliferation to maintain basal β-cell mass is low (72). For this reason, targeting the health of existing β-cells or generating new functional β-cells in vitro have become two major focuses in the development of potential therapies for diabetes. TFs play an indispensable role in regulating pancreas and islet development and can serve as potential target in rescuing, preserving, or re-generating β-cells. Enhancing the expression of certain transcription factors via the addition of pharmaceutical compounds can have a protective effect on β-cells. For example, the flavonoid compound, tectorigenin, has been shown to enhance Pdx1 expression and protect β-cell viability under glucolipotoxic conditions (264). On the other hand, the successful generation of β-cells through differentiation of stem cells via the controlled addition of TFs has caused much excitement (114, 265, 266). Recently, islets derived from chemically induced pluripotent stem cells were successfully transplanted and engrafted into a human patient (267).
In the β-cell, many signal-dependent TFs cooperate with other proteins in response to stimuli, such as nutrient flux, to regulate adaptation responses (21). Many of these TFs also undergo nutrient-sensitive PTMs, which may influence their localization, stability, and function (22). However, while there are many loss-of-function studies examining the role of these TFs in the cell, few look at the stimulus-sensitive molecular mechanisms regulating the TFs themselves, such as PTM or epigenetics (not reviewed here). A deeper understanding of these elements can aid in developing more refined diabetes treatments targeting the pancreatic β-cell.
EA: Funding acquisition, Supervision, Writing – review & editing. AW: Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by NIH grant NIDDK (R56DK136293, R01DK115720, and R01DK136237 to EA; T32HL166142 to AW). This work was also supported by the Department of Integrative Biology and Physiology Accelerator Program and the McKnight Land-Grant Professorship and Presidential Fellows Program.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Atf4: Activating Transcription Factor 4
ATOH8: Atonal Basic Helix-Loop-Helix Transcription Factor 8
Cdx4: Caudal Type Homeobox 4
ER: Endoplasmic Reticulum
FoxA2: Forkhead Box Protein A2
FoxM1: Forkhead Box Protein M1
FoxO1: Forehead Box Protein O1
Gfi1: Growth Factor Independent 1 Transcriptional Repressor
Glis3: GLIS Family Zinc Finger 3
GSK3: Glycogen Synthase Kinase 3
Insm1: Insulinoma-Associated Protein 1
Isl1: Insulin Enhancer Protein Islet 1
MafA: MAF BZIP Transcriptional Factor A
Mist1: Basic Helix-Loop-Helix Family Member A15
MODY: Mature Onset Diabetes of the Young
NeuroD1: Neuronal Differentiation 1
Ngn3: Neurogenin-3
Nkx2.2: NK2 Homeobox 2
Nkx6.1: NK6 Homeobox 1
Nrf2: NFE2 Like BZIP Transcription Factor
Pax4: Paired Box 4
Pax6: Paired Box 6
Pdx1: Pancreatic Duodenal Homeobox 1
Plk1: Pololike Kinase 1
Rfx3: Regulatory Factor X3
Rfx6: Regulatory Factor X6
Ring1b: Ring Finger Protein 2
PTM: Posttranslational Modification
Sox9: SRY-Box Transcription Factor 9
Stat3: Signal Transducer and Activator of Transcription 3
T2D: Type 2 diabetes
Taf4: TATA-Box Binding Protein Associated Factor 4
TF: Transcription Factor
1. Federation ID. IDF Diabetes Atlas (2021). Available online at: https://www.diabetesatlas.org (Accessed November 30).
2. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
3. Ludvik B, Nolan JJ, Baloga J, Sacks D, Olefsky J. Effect of obesity on insulin resistance in normal subjects and patients with NIDDM. Diabetes. (1995) 44:1121–5. doi: 10.2337/diab.44.9.1121
4. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. (2003) 52:102–10. doi: 10.2337/diabetes.52.1.102
5. Groen N, Leenders F, Mahfouz A, Munoz-Garcia A, Muraro MJ, de Graaf N, et al. Single-cell transcriptomics links loss of human pancreatic β-cell identity to ER stress. Cells. (2021) 10:3585. doi: 10.3390/cells10123585
6. Spijker HS, Song H, Ellenbroek JH, Roefs MM, Engelse MA, Bos E, et al. Loss of β-cell identity occurs in type 2 diabetes and is associated with islet amyloid deposits. Diabetes. (2015) 64:2928–38. doi: 10.2337/db14-1752
7. White MG, Marshall HL, Rigby R, Huang GC, Amer A, Booth T, et al. Expression of mesenchymal and α-cell phenotypic markers in islet β-cells in recently diagnosed diabetes. Diabetes Care. (2013) 36:3818–20. doi: 10.2337/dc13-0705
8. Chhabra NF, Amend AL, Bastidas-Ponce A, Sabrautzki S, Tarquis-Medina M, Sachs S, et al. A point mutation in the Pdia6 gene results in loss of pancreatic β-cell identity causing overt diabetes. Mol Metab. (2021) 54:101334. doi: 10.1016/j.molmet.2021.101334
9. Dobosz AM, Janikiewicz J, Krogulec E, Dziewulska A, Ajduk A, Szpila M, et al. Inhibition of stearoyl-CoA desaturase 1 in the mouse impairs pancreatic islet morphogenesis and promotes loss of β-cell identity and α-cell expansion in the mature pancreas. Mol Metab. (2023) 67:101659. doi: 10.1016/j.molmet.2022.101659
10. Efrat S. Beta-cell dedifferentiation in type 2 diabetes: concise review. Stem Cells. (2019) 37:1267–72. doi: 10.1002/stem.3059
11. Patel S, Remedi MS. Loss of β-cell identity and dedifferentiation, not an irreversible process? Front Endocrinol (Lausanne). (2024) 15:1414447. doi: 10.3389/fendo.2024.1414447
12. Christensen AA, Gannon M. The beta cell in type 2 diabetes. Curr Diabetes Rep. (2019) 19:81. doi: 10.1007/s11892-019-1196-4
13. Alejandro EU, Gregg B, Blandino-Rosano M, Cras-Méneur C, Bernal-Mizrachi E. Natural history of β-cell adaptation and failure in type 2 diabetes. Mol Aspects Med. (2015) 42:19–41. doi: 10.1016/j.mam.2014.12.002
14. Weir GC, Aguayo-Mazzucato C, Bonner-Weir S. [amp]]beta;-cell dedifferentiation in diabetes is important, but what is it? Islets. (2013) 5:233–7. doi: 10.4161/isl.27494
15. Bensellam M, Jonas JC, Laybutt DR. Mechanisms of β-cell dedifferentiation in diabetes: recent findings and future research directions. J Endocrinol. (2018) 236:R109–43. doi: 10.1530/JOE-17-0516
16. Brereton MF, Rohm M, Ashcroft FM. [amp]]beta;-Cell dysfunction in diabetes: a crisis of identity? Diabetes Obes Metab. (2016) 18 Suppl 1:102–9. doi: 10.1111/dom.12732
17. Hunter CS, Stein RW. Evidence for loss in identity, de-differentiation, and. Front Genet. (2017) 8:35. doi: 10.3389/fgene.2017.00035
18. Zhu H, Wang G, Nguyen-Ngoc KV, Kim D, Miller M, Goss G, et al. Understanding cell fate acquisition in stem-cell-derived pancreatic islets using single-cell multiome-inferred regulomes. Dev Cell. (2023) 58:727–743.e11. doi: 10.1016/j.devcel.2023.03.011
19. Gao T, McKenna B, Li C, Reichert M, Nguyen J, Singh T, et al. Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab. (2014) 19:259–71. doi: 10.1016/j.cmet.2013.12.002
20. Swisa A, Avrahami D, Eden N, Zhang J, Feleke E, Dahan T, et al. PAX6 maintains β cell identity by repressing genes of alternative islet cell types. J Clin Invest. (2017) 127:230–43. doi: 10.1172/JCI88015
21. Wortham M, Sander M. Transcriptional mechanisms of pancreatic β-cell maturation and functional adaptation. Trends Endocrinol Metab. (2021) 32:474–87. doi: 10.1016/j.tem.2021.04.011
22. Hart GW. Nutrient regulation of signaling and transcription. J Biol Chem. (2019) 294:2211–31. doi: 10.1074/jbc.AW119.003226
23. Pictet RL, Clark WR, Williams RH, Rutter WJ. An ultrastructural analysis of the developing embryonic pancreas. Dev Biol. (1972) 29:436–67. doi: 10.1016/0012-1606(72)90083-8
24. Ohlsson H, Karlsson K, Edlund T. IPF1, a homeodomain-containing transactivator of the insulin gene. EMBO J. (1993) 12:4251–9. doi: 10.1002/j.1460-2075.1993.tb06109.x
25. Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. (2002) 129:2447–57. doi: 10.1242/dev.129.10.2447
26. Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. (1994) 371:606–9. doi: 10.1038/371606a0
27. Ahlgren U, Jonsson J, Edlund H. The morphogenesis of the pancreatic mesenchyme is uncoupled from that of the pancreatic epithelium in IPF1/PDX1-deficient mice. Development. (1996) 122:1409–16. doi: 10.1242/dev.122.5.1409
28. Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. (1997) 15:106–10. doi: 10.1038/ng0197-106
29. Apelqvist A, Li H, Sommer L, Beatus P, Anderson DJ, Honjo T, et al. Notch signalling controls pancreatic cell differentiation. Nature. (1999) 400:877–81. doi: 10.1038/23716
30. Du A, Hunter CS, Murray J, Noble D, Cai CL, Evans SM, et al. Islet-1 is required for the maturation, proliferation, and survival of the endocrine pancreas. Diabetes. (2009) 58:2059–69. doi: 10.2337/db08-0987
31. Gradwohl G, Dierich A, LeMeur M, Guillemot F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc Natl Acad Sci U.S.A. (2000) 97:1607–11. doi: 10.1073/pnas.97.4.1607
32. Lee CS, Perreault N, Brestelli JE, Kaestner KH. Neurogenin 3 is essential for the proper specification of gastric enteroendocrine cells and the maintenance of gastric epithelial cell identity. Genes Dev. (2002) 16:1488–97. doi: 10.1101/gad.985002
33. Bohuslavova R, Smolik O, Malfatti J, Berkova Z, Novakova Z, Saudek F, et al. NEUROD1 is required for the early α and β Endocrine differentiation in the pancreas. Int J Mol Sci. (2021) 22:13. doi: 10.3390/ijms22136713
34. Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell. (2012) 150:1223–34. doi: 10.1016/j.cell.2012.07.029
35. Marselli L, Suleiman M, Masini M, Campani D, Bugliani M, Syed F, et al. Are we overestimating the loss of beta cells in type 2 diabetes? Diabetologia. (2014) 57:362–5. doi: 10.1007/s00125-013-3098-3
36. Guo H, Guo D, An M, Zhang R, Wang C, He J. Angiotensin-(1-7) improves islet β-cell dedifferentiation by activating PI3K/akt/foxO1 pathway. Protein Pept Lett. (2023) 30:1009–19. doi: 10.2174/0109298665257646231020054036
37. Wang Z, York NW, Nichols CG, Remedi MS. Pancreatic β cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. (2014) 19:872–82. doi: 10.1016/j.cmet.2014.03.010
38. Szukiewicz D. Molecular mechanisms for the vicious cycle between insulin resistance and the inflammatory response in obesity. Int J Mol Sci. (2023) 24:12. doi: 10.3390/ijms24129818
39. Gallagher EJ, Leroith D, Karnieli E. Insulin resistance in obesity as the underlying cause for the metabolic syndrome. Mt Sinai J Med. (2010) 77:511–23. doi: 10.1002/msj.20212
40. Gobato AO, Vasques AC, Zambon MP, Barros Filho AeA, Hessel G. Metabolic syndrome and insulin resistance in obese adolescents. Rev Paul Pediatr. (2014) 32:55–62. doi: 10.1590/s0103-05822014000100010
41. Jiang J, Cai X, Pan Y, Du X, Zhu H, Yang X, et al. Relationship of obesity to adipose tissue insulin resistance. BMJ Open Diabetes Res Care. (2020) 8:e0000741. doi: 10.1136/bmjdrc-2019-000741
42. Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. β-cell mass and turnover in humans: effects of obesity and aging. Diabetes Care. (2013) 36:111–7. doi: 10.2337/dc12-0421
43. Stamateris RE, Sharma RB, Hollern DA, Alonso LC. Adaptive β-cell proliferation increases early in high-fat feeding in mice, concurrent with metabolic changes, with induction of islet cyclin D2 expression. Am J Physiol Endocrinol Metab. (2013) 305:E149–59. doi: 10.1152/ajpendo.00040.2013
44. Gupta D, Jetton TL, LaRock K, Monga N, Satish B, Lausier J, et al. Temporal characterization of β cell-adaptive and -maladaptive mechanisms during chronic high-fat feeding in C57BL/6NTac mice. J Biol Chem. (2017) 292:12449–59. doi: 10.1074/jbc.M117.781047
45. Szabat M, Luciani DS, Piret JM, Johnson JD. Maturation of adult beta-cells revealed using a Pdx1/insulin dual-reporter lentivirus. Endocrinology. (2009) 150:1627–35. doi: 10.1210/en.2008-1224
46. Jetton TL, Everill B, Lausier J, Roskens V, Habibovic A, LaRock K, et al. Enhanced beta-cell mass without increased proliferation following chronic mild glucose infusion. Am J Physiol Endocrinol Metab. (2008) 294:E679–87. doi: 10.1152/ajpendo.00569.2007
47. Ruiz L, Gurlo T, Ravier MA, Wojtusciszyn A, Mathieu J, Brown MR, et al. Proteasomal degradation of the histone acetyl transferase p300 contributes to beta-cell injury in a diabetes environment. Cell Death Dis. (2018) 9:600. doi: 10.1038/s41419-018-0603-0
48. Zhang T, Kim DH, Xiao X, Lee S, Gong Z, Muzumdar R, et al. FoxO1 plays an important role in regulating β-cell compensation for insulin resistance in male mice. Endocrinology. (2016) 157:1055–70. doi: 10.1210/en.2015-1852
49. Boucher MJ, Selander L, Carlsson L, Edlund H. Phosphorylation marks IPF1/PDX1 protein for degradation by glycogen synthase kinase 3-dependent mechanisms. J Biol Chem. (2006) 281:6395–403. doi: 10.1074/jbc.M511597200
50. Ardestani A, Paroni F, Azizi Z, Kaur S, Khobragade V, Yuan T, et al. MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat Med. (2014) 20:385–97. doi: 10.1038/nm.3482
51. Guo S, Dai C, Guo M, Taylor B, Harmon JS, Sander M, et al. Inactivation of specific β cell transcription factors in type 2 diabetes. J Clin Invest. (2013) 123:3305–16. doi: 10.1172/JCI65390
52. Thorel F, Népote V, Avril I, Kohno K, Desgraz R, Chera S, et al. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature. (2010) 464:1149–54. doi: 10.1038/nature08894
53. Rodriguez UA, Socorro M, Criscimanna A, Martins CP, Mohamed N, Hu J, et al. Conversion of α-cells to β-cells in the postpartum mouse pancreas involves lgr5 progeny. Diabetes. (2021) 70:1508–18. doi: 10.2337/db20-1059
54. Nkonge KM, Nkonge DK, Nkonge TN. The epidemiology, molecular pathogenesis, diagnosis, and treatment of maturity-onset diabetes of the young (MODY). Clin Diabetes Endocrinol. (2020) 6:20. doi: 10.1186/s40842-020-00112-5
55. Drewes T, Senkel S, Holewa B, Ryffel GU. Human hepatocyte nuclear factor 4 isoforms are encoded by distinct and differentially expressed genes. Mol Cell Biol. (1996) 16:925–31. doi: 10.1128/MCB.16.3.925
56. Yamagata K, Furuta H, Oda N, Kaisaki PJ, Menzel S, Cox NJ, et al. Mutations in the hepatocyte nuclear factor-4alpha gene in maturity-onset diabetes of the young (MODY1). Nature. (1996) 384:458–60. doi: 10.1038/384458a0
57. Shaw JT, Lovelock PK, Kesting JB, Cardinal J, Duffy D, Wainwright B, et al. Novel susceptibility gene for late-onset NIDDM is localized to human chromosome 12q. Diabetes. (1998) 47:1793–6. doi: 10.2337/diabetes.47.11.1793
58. Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, et al. Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3). Nature. (1996) 384:455–8. doi: 10.1038/384455a0
59. Chèvre JC, Hani EH, Boutin P, Vaxillaire M, Blanché H, Vionnet N, et al. Mutation screening in 18 Caucasian families suggest the existence of other MODY genes. Diabetologia. (1998) 41:1017–23. doi: 10.1007/s001250051025
60. Stoffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1. Nat Genet. (1997) 17:138–9. doi: 10.1038/ng1097-138
61. Abbott C, Piaggio G, Ammendola R, Solomon E, Povey S, Gounari F, et al. Mapping of the gene TCF2 coding for the transcription factor LFB3 to human chromosome 17 by polymerase chain reaction. Genomics. (1990) 8:165–7. doi: 10.1016/0888-7543(90)90239-q
62. Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, et al. Mutation in hepatocyte nuclear factor-1 beta gene (TCF2) associated with MODY. Nat Genet. (1997) 17:384–5. doi: 10.1038/ng1297-384
63. Tamimi R, Steingrimsson E, Copeland NG, Dyer-Montgomery K, Lee JE, Hernandez R, et al. The NEUROD gene maps to human chromosome 2q32 and mouse chromosome 2. Genomics. (1996) 34:418–21. doi: 10.1006/geno.1996.0306
64. Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, Orban T, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet. (1999) 23:323–8. doi: 10.1038/15500
65. Wang Z, Peters B, Klussmann S, Bender H, Herb A, Krieglstein K. Gene structure and evolution of Tieg3, a new member of the Tieg family of proteins. Gene. (2004) 325:25–34. doi: 10.1016/j.gene.2003.09.045
66. Neve B, Fernandez-Zapico ME, Ashkenazi-Katalan V, Dina C, Hamid YH, Joly E, et al. Role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc Natl Acad Sci U.S.A. (2005) 102:4807–12. doi: 10.1073/pnas.0409177102
67. Stapleton P, Weith A, Urbánek P, Kozmik Z, Busslinger M. Chromosomal localization of seven PAX genes and cloning of a novel family member, PAX-9. Nat Genet. (1993) 3:292–8. doi: 10.1038/ng0493-292
68. Sujjitjoon J, Kooptiwut S, Chongjaroen N, Tangjittipokin W, Plengvidhya N, Yenchitsomanus PT. Aberrant mRNA splicing of paired box 4 (PAX4) IVS7-1G>A mutation causing maturity-onset diabetes of the young, type 9. Acta Diabetol. (2016) 53:205–16. doi: 10.1007/s00592-015-0760-x
69. Walsh CT, Garneau-Tsodikova S, Gatto GJ. Protein posttranslational modifications: the chemistry of proteome diversifications. Angew Chem Int Ed Engl. (2005) 44:7342–72. doi: 10.1002/anie.200501023
70. Gutiérrez GD, Bender AS, Cirulli V, Mastracci TL, Kelly SM, Tsirigos A, et al. Pancreatic β cell identity requires continual repression of non-β cell programs. J Clin Invest. (2017) 127:244–59. doi: 10.1172/JCI88017
71. Taylor BL, Liu FF, Sander M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep. (2013) 4:1262–75. doi: 10.1016/j.celrep.2013.08.010
72. Kohata M, Imai J, Izumi T, Yamamoto J, Kawana Y, Endo A, et al. Roles of FoxM1-driven basal β-cell proliferation in maintenance of β-cell mass and glucose tolerance during adulthood. J Diabetes Investig. (2022) 13:1666–76. doi: 10.1111/jdi.13846
73. Zhang H, Ackermann AM, Gusarova GA, Lowe D, Feng X, Kopsombut UG, et al. The FoxM1 transcription factor is required to maintain pancreatic beta-cell mass. Mol Endocrinol. (2006) 20:1853–66. doi: 10.1210/me.2006-0056
74. Davis DB, Lavine JA, Suhonen JI, Krautkramer KA, Rabaglia ME, Sperger JM, et al. FoxM1 is up-regulated by obesity and stimulates beta-cell proliferation. Mol Endocrinol. (2010) 24:1822–34. doi: 10.1210/me.2010-0082
75. Zhang H, Zhang J, Pope CF, Crawford LA, Vasavada RC, Jagasia SM, et al. Gestational diabetes mellitus resulting from impaired beta-cell compensation in the absence of FoxM1, a novel downstream effector of placental lactogen. Diabetes. (2010) 59:143–52. doi: 10.2337/db09-0050
76. Ohlsson H, Thor S, Edlund T. Novel insulin promoter- and enhancer-binding proteins that discriminate between pancreatic alpha- and beta-cells. Mol Endocrinol. (1991) 5:897–904. doi: 10.1210/mend-5-7-897
77. Miller CP, McGehee RE, Habener JF. IDX-1: a new homeodomain transcription factor expressed in rat pancreatic islets and duodenum that transactivates the somatostatin gene. EMBO J. (1994) 13:1145–56. doi: 10.1002/j.1460-2075.1994.tb06363.x
78. Li SW, Koya V, Li Y, Donelan W, Lin P, Reeves WH, et al. Pancreatic duodenal homeobox 1 protein is a novel beta-cell-specific autoantigen for type I diabetes. Lab Invest. (2010) 90:31–9. doi: 10.1038/labinvest.2009.116
79. Hani EH, Stoffers DA, Chèvre JC, Durand E, Stanojevic V, Dina C, et al. Defective mutations in the insulin promoter factor-1 (IPF-1) gene in late-onset type 2 diabetes mellitus. J Clin Invest. (1999) 104:R41–8. doi: 10.1172/JCI7469
80. Offield MF, Jetton TL, Labosky PA, Ray M, Stein RW, Magnuson MA, et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. (1996) 122:983–95. doi: 10.1242/dev.122.3.983
81. Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev. (1998) 12:1763–8. doi: 10.1101/gad.12.12.1763
82. Schaffer AE, Taylor BL, Benthuysen JR, Liu J, Thorel F, Yuan W, et al. Nkx6.1 controls a gene regulatory network required for establishing and maintaining pancreatic Beta cell identity. PloS Genet. (2013) 9:e1003274. doi: 10.1371/journal.pgen.1003274
83. Stoffel M, Stein R, Wright CV, Espinosa R, Le Beau MM, Bell GI. Localization of human homeodomain transcription factor insulin promoter factor 1 (IPF1) to chromosome band 13q12. 1. Genomics. (1995) 28:125–6. doi: 10.1006/geno.1995.1120
84. Fiedorek FT, Kay ES. Mapping of the insulin promoter factor 1 gene (Ipf1) to distal mouse chromosome 5. Genomics. (1995) 28:581–4. doi: 10.1006/geno.1995.1193
85. Perez G, Barber GP, Benet-Pages A, Casper J, Clawson H, Diekhans M, et al. The UCSC Genome Browser database: 2025 update. Nucleic Acids Res. (2024) 52(D1):D1082–8. doi: 10.1093/nar/gkae974
86. Consortium U. UniProt: the universal protein knowledgebase in 2025. Nucleic Acids Res. (2024) 53(D1):D609–17. doi: 10.1093/nar/gkae1010
87. Gao Y, Miyazaki J, Hart GW. The transcription factor PDX-1 is post-translationally modified by O-linked N-acetylglucosamine and this modification is correlated with its DNA binding activity and insulin secretion in min6 beta-cells. Arch Biochem Biophys. (2003) 415:155–63. doi: 10.1016/s0003-9861(03)00234-0
88. Akimoto Y, Hart GW, Wells L, Vosseller K, Yamamoto K, Munetomo E, et al. Elevation of the post-translational modification of proteins by O-linked N-acetylglucosamine leads to deterioration of the glucose-stimulated insulin secretion in the pancreas of diabetic Goto-Kakizaki rats. Glycobiology. (2007) 17:127–40. doi: 10.1093/glycob/cwl067
89. Baumann D, Wong A, Akhaphong B, Jo S, Pritchard S, Mohan R, et al. Role of nutrient-driven O-GlcNAc-post-translational modification in pancreatic exocrine and endocrine islet development. Development. (2020) 147:dev186643. doi: 10.1242/dev.186643
90. Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. (2007) 446:1017–22. doi: 10.1038/nature05815
91. Love DC, Kochan J, Cathey RL, Shin SH, Hanover JA. Mitochondrial and nucleocytoplasmic targeting of O-linked GlcNAc transferase. J Cell Sci. (2003) 116:647–54. doi: 10.1242/jcs.00246
92. Dong DL, Hart GW. Purification and characterization of an O-GlcNAc selective N-acetyl-beta-D-glucosaminidase from rat spleen cytosol. J Biol Chem. (1994) 269:19321–30. doi: 10.1016/S0021-9258(17)32170-1
93. Gupta R, Brunak S. Prediction of glycosylation across the human proteome and the correlation to protein function. Pac Symp Biocomput. (2002), 310–22.
94. Alejandro EU, Bozadjieva N, Kumusoglu D, Abdulhamid S, Levine H, Haataja L, et al. Disruption of O-linked N-acetylglucosamine signaling induces ER stress and β Cell failure. Cell Rep. (2015) 13:2527–38. doi: 10.1016/j.celrep.2015.11.020
95. Mohan R, Jo S, Lockridge A, Ferrington DA, Murray K, Eschenlauer A, et al. OGT regulates mitochondrial biogenesis and function via diabetes susceptibility gene pdx1. Diabetes. (2021) 70:2608–25. doi: 10.2337/db21-0468
96. Wong A, Akhaphong B, Baumann D, Alejandro EU. Genetic ablation of the nutrient sensor ogt in endocrine progenitors is dispensable for β-cell development but essential for maintenance of β-cell mass. Biomedicines. (2022) 11:105. doi: 10.3390/biomedicines11010105
97. Wong SP A, Moore M, Akhaphong B, Avula N, Beetch M, Fujitani Y, et al. Overexpression of Pdx1, reduction of p53, or deletion of CHOP attenuates pancreas hypoplasia in mice with pancreas-specific O-GlcNAc Transferase deletion. J Biol Chem. (2023) 299(2):102878. doi: 10.1016/j.jbc.2023.102878
98. Frogne T, Sylvestersen KB, Kubicek S, Nielsen ML, Hecksher-Sørensen J. Pdx1 is post-translationally modified in vivo and serine 61 is the principal site of phosphorylation. PloS One. (2012) 7:e35233. doi: 10.1371/journal.pone.0035233
99. Lebrun P, Montminy MR, Van Obberghen E. Regulation of the pancreatic duodenal homeobox-1 protein by DNA-dependent protein kinase. J Biol Chem. (2005) 280:38203–10. doi: 10.1074/jbc.M504842200
100. An R, da Silva Xavier G, Semplici F, Vakhshouri S, Hao HX, Rutter J, et al. Pancreatic and duodenal homeobox 1 (PDX1) phosphorylation at serine-269 is HIPK2-dependent and affects PDX1 subnuclear localization. Biochem Biophys Res Commun. (2010) 399:155–61. doi: 10.1016/j.bbrc.2010.07.035
101. Ostertag MS, Messias AC, Sattler M, Popowicz GM. The structure of the SPOP-pdx1 interface reveals insights into the phosphorylation-dependent binding regulation. Structure. (2019) 27:327–334.e3. doi: 10.1016/j.str.2018.10.005
102. Caturano A, D’Angelo M, Mormone A, Russo V, Mollica MP, Salvatore T, et al. Oxidative stress in type 2 diabetes: impacts from pathogenesis to lifestyle modifications. Curr Issues Mol Biol. (2023) 45:6651–66. doi: 10.3390/cimb45080420
103. Lee KK, Murakawa M, Nishida E, Tsubuki S, Kawashima S, Sakamaki K, et al. Proteolytic activation of MST/Krs, STE20-related protein kinase, by caspase during apoptosis. Oncogene. (1998) 16:3029–37. doi: 10.1038/sj.onc.1201840
104. Meng R, Al-Quobaili F, Müller I, Götz C, Thiel G, Montenarh M. CK2 phosphorylation of Pdx-1 regulates its transcription factor activity. Cell Mol Life Sci. (2010) 67:2481–9. doi: 10.1007/s00018-010-0348-0
105. Rafiq I, Kennedy HJ, Rutter GA. Glucose-dependent translocation of insulin promoter factor-1 (IPF-1) between the nuclear periphery and the nucleoplasm of single MIN6 beta-cells. J Biol Chem. (1998) 273:23241–7. doi: 10.1074/jbc.273.36.23241
106. Kishi A, Nakamura T, Nishio Y, Maegawa H, Kashiwagi A. Sumoylation of Pdx1 is associated with its nuclear localization and insulin gene activation. Am J Physiol Endocrinol Metab. (2003) 284:E830–40. doi: 10.1152/ajpendo.00390.2002
107. Qi Y, Cai J, Wu Y, Wu R, Lee J, Fu H. Control of oligodendrocyte differentiation by the Nkx2.2 homeodomain transcription factor. Development. (2001) 128:2723–33. doi: 10.1242/dev.128.14.2723
108. Zhu Q, Zhao X, Zheng K, Li H, Huang H, Zhang Z, et al. Genetic evidence that Nkx2.2 and Pdgfra are major determinants of the timing of oligodendrocyte differentiation in the developing CNS. Development. (2014) 141:548–55. doi: 10.1242/dev.095323
109. Clark JK, O’keefe A, Mastracci TL, Sussel L, Matise MP, Kucenas S. Mammalian Nkx2.2+ perineurial glia are essential for motor nerve development. Dev Dyn. (2014) 243:1116–29. doi: 10.1002/dvdy.24158
110. Briscoe J, Sussel L, Serup P, Hartigan-O'Connor D, Jessell TM, Rubenstein JL, et al. Homeobox gene Nkx2.2 and specification of neuronal identity by graded Sonic hedgehog signalling. Nature. (1999) 398:622–7. doi: 10.1038/19315
111. Price M, Lazzaro D, Pohl T, Mattei MG, Rüther U, Olivo JC, et al. Regional expression of the homeobox gene Nkx-2.2 in the developing mammalian forebrain. Neuron. (1992) 8:241–55. doi: 10.1016/0896-6273(92)90291-k
112. Rudnick A, Ling TY, Odagiri H, Rutter WJ, German MS. Pancreatic beta cells express a diverse set of homeobox genes. Proc Natl Acad Sci U.S.A. (1994) 91:12203–7. doi: 10.1073/pnas.91.25.12203
113. Sussel L, Kalamaras J, Hartigan-O’Connor DJ, Meneses JJ, Pedersen RA, Rubenstein JL, et al. Mice lacking the homeodomain transcription factor Nkx2.2 have diabetes due to arrested differentiation of pancreatic beta cells. Development. (1998) 125:2213–21. doi: 10.1242/dev.125.12.2213
114. Fontcuberta-PiSunyer M, García-Alamán A, Prades È, Téllez N, Alves-Figueiredo H, Ramos-Rodríguez M, et al. Direct reprogramming of human fibroblasts into insulin-producing cells using transcription factors. Commun Biol. (2023) 6:256. doi: 10.1038/s42003-023-04627-2
115. Doyle MJ, Sussel L. Nkx2.2 regulates beta-cell function in the mature islet. Diabetes. (2007) 56:1999–2007. doi: 10.2337/db06-1766
116. Papizan JB, Singer RA, Tschen SI, Dhawan S, Friel JM, Hipkens SB, et al. Nkx2.2 repressor complex regulates islet β-cell specification and prevents β-to-α-cell reprogramming. Genes Dev. (2011) 25:2291–305. doi: 10.1101/gad.173039.111
117. Hornbeck PV, Kornhauser JM, Tkachev S, Zhang B, Skrzypek E, Murray B, et al. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. (2012) 40:D261–70. doi: 10.1093/nar/gkr1122
118. De Silva ARI, Page RC. Ubiquitination detection techniques. Exp Biol Med (Maywood). (2023) 248:1333–46. doi: 10.1177/15353702231191186
119. Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol. (2006) 17:1807–19. doi: 10.1681/ASN.2006010083
120. Mertins P, Yang F, Liu T, Mani DR, Petyuk VA, Gillette MA, et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics. (2014) 13:1690–704. doi: 10.1074/mcp.M113.036392
121. Sacco F, Humphrey SJ, Cox J, Mischnik M, Schulte A, Klabunde T, et al. Glucose-regulated and drug-perturbed phosphoproteome reveals molecular mechanisms controlling insulin secretion. Nat Commun. (2016) 7:13250. doi: 10.1038/ncomms13250
122. Sander M, Sussel L, Conners J, Scheel D, Kalamaras J, Dela Cruz F, et al. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development. (2000) 127:5533–40. doi: 10.1242/dev.127.24.5533
123. Schaffer AE, Freude KK, Nelson SB, Sander M. Nkx6 transcription factors and Ptf1a function as antagonistic lineage determinants in multipotent pancreatic progenitors. Dev Cell. (2010) 18:1022–9. doi: 10.1016/j.devcel.2010.05.015
124. Jennings RE, Berry AA, Kirkwood-Wilson R, Roberts NA, Hearn T, Salisbury RJ, et al. Development of the human pancreas from foregut to endocrine commitment. Diabetes. (2013) 62:3514–22. doi: 10.2337/db12-1479
125. Nelson SB, Schaffer AE, Sander M. The transcription factors Nkx6.1 and Nkx6.2 possess equivalent activities in promoting beta-cell fate specification in Pdx1+ pancreatic progenitor cells. Development. (2007) 134:2491–500. doi: 10.1242/dev.002691
126. Collombat P, Hecksher-Sørensen J, Broccoli V, Krull J, Ponte I, Mundiger T, et al. The simultaneous loss of Arx and Pax4 genes promotes a somatostatin-producing cell fate specification at the expense of the alpha- and beta-cell lineages in the mouse endocrine pancreas. Development. (2005) 132:2969–80. doi: 10.1242/dev.01870
127. Henseleit KD, Nelson SB, Kuhlbrodt K, Hennings JC, Ericson J, Sander M. NKX6 transcription factor activity is required for alpha- and beta-cell development in the pancreas. Development. (2005) 132:3139–49. doi: 10.1242/dev.01875
128. Mirmira RG, Watada H, German MS. Beta-cell differentiation factor Nkx6.1 contains distinct DNA binding interference and transcriptional repression domains. J Biol Chem. (2000) 275:14743–51. doi: 10.1074/jbc.275.19.14743
129. Liao GB, Li XZ, Zeng S, Liu C, Yang SM, Yang L, et al. Regulation of the master regulator FOXM1 in cancer. Cell Commun Signal. (2018) 16:57. doi: 10.1186/s12964-018-0266-6
130. Ackermann Misfeldt A, Costa RH, Gannon M. Beta-cell proliferation, but not neogenesis, following 60% partial pancreatectomy is impaired in the absence of FoxM1. Diabetes. (2008) 57:3069–77. doi: 10.2337/db08-0878
131. Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. (2008) 57:1584–94. doi: 10.2337/db07-1369
132. Stolovich-Rain M, Hija A, Grimsby J, Grimsby J, Glaser B, Dor Y. Pancreatic beta cells in very old mice retain capacity for compensatory proliferation. J Biol Chem. (2012) 287:27407–14. doi: 10.1074/jbc.M112.350736
133. Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. (2005) 54:2557–67. doi: 10.2337/diabetes.54.9.2557
134. Cnop M, Hughes SJ, Igoillo-Esteve M, Hoppa MB, Sayyed F, van de Laar L, et al. The long lifespan and low turnover of human islet beta cells estimated by mathematical modelling of lipofuscin accumulation. Diabetologia. (2010) 53:321–30. doi: 10.1007/s00125-009-1562-x
135. Golson ML, Dunn JC, Maulis MF, Dadi PK, Osipovich AB, Magnuson MA, et al. Activation of foxM1 revitalizes the replicative potential of aged β-cells in male mice and enhances insulin secretion. Diabetes. (2015) 64:3829–38. doi: 10.2337/db15-0465
136. Ma RY, Tong TH, Cheung AM, Tsang AC, Leung WY, Yao KM. Raf/MEK/MAPK signaling stimulates the nuclear translocation and transactivating activity of FOXM1c. J Cell Sci. (2005) 118:795–806. doi: 10.1242/jcs.01657
137. Fu Z, Malureanu L, Huang J, Wang W, Li H, van Deursen JM, et al. Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat Cell Biol. (2008) 10:1076–82. doi: 10.1038/ncb1767
138. Hsu CC, Yao X, Chen SY, Tsuo TC, Wang IC. The conformation of FOXM1 homodimers in vivo is crucial for regulating transcriptional activities. Nucleic Acids Res. (2024) 52:13625–43. doi: 10.1093/nar/gkae988
139. Dong Q, Wang D, Song C, Gong C, Liu Y, Zhou X, et al. ABL1-mediated phosphorylation promotes FOXM1-related tumorigenicity by Increasing FOXM1 stability. Cell Death Differ. (2024) 31:1285–301. doi: 10.1038/s41418-024-01339-w
140. Sorenson RL, Brelje TC. Adaptation of islets of Langerhans to pregnancy: beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm Metab Res. (1997) 29:301–7. doi: 10.1055/s-2007-979040
141. Parsons JA, Bartke A, Sorenson RL. Number and size of islets of Langerhans in pregnant, human growth hormone-expressing transgenic, and pituitary dwarf mice: effect of lactogenic hormones. Endocrinology. (1995) 136:2013–21. doi: 10.1210/endo.136.5.7720649
142. Green IC, Taylor KW. Effects of pregnancy in the rat on the size and insulin secretory response of the islets of Langerhans. J Endocrinol. (1972) 54:317–25. doi: 10.1677/joe.0.0540317
143. Van Assche FA. Quantitative morphologic and histoenzymatic study of the endocrine pancreas in nonpregnant and pregnant rats. Am J Obstet Gynecol. (1974) 118:39–41. doi: 10.1016/s0002-9378(16)33642-0
144. Klöppel G, Löhr M, Habich K, Oberholzer M, Heitz PU. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv Synth Pathol Res. (1985) 4:110–25. doi: 10.1159/000156969
145. Hanley SC, Austin E, Assouline-Thomas B, Kapeluto J, Blaichman J, Moosavi M, et al. {beta}-Cell mass dynamics and islet cell plasticity in human type 2 diabetes. Endocrinology. (2010) 151:1462–72. doi: 10.1210/en.2009-1277
146. Rieck S, Kaestner KH. Expansion of beta-cell mass in response to pregnancy. Trends Endocrinol Metab. (2010) 21:151–8. doi: 10.1016/j.tem.2009.11.001
147. Catalano PM, Roman-Drago NM, Amini SB, Amini SB, Sims EA. Longitudinal changes in body composition and energy balance in lean women with normal and abnormal glucose tolerance during pregnancy. Am J Obstet Gynecol. (1998) 179:156–65. doi: 10.1016/s0002-9378(98)70267-4
148. Hakonen E, Ustinov J, Palgi J, Miettinen PJ, Otonkoski T. EGFR signaling promotes β-cell proliferation and survivin expression during pregnancy. PloS One. (2014) 9:e93651. doi: 10.1371/journal.pone.0093651
149. Kim H, Toyofuku Y, Lynn FC, Chak E, Uchida T, Mizukami H, et al. Serotonin regulates pancreatic beta cell mass during pregnancy. Nat Med. (2010) 16:804–8. doi: 10.1038/nm.2173
150. Rieck S, White P, Schug J, Fox AJ, Smirnova O, Gao N, et al. The transcriptional response of the islet to pregnancy in mice. Mol Endocrinol. (2009) 23:1702–12. doi: 10.1210/me.2009-0144
151. Sylvester-Armstrong KR, Reeder CF, Powell A, Becker MW, Hagan DW, Chen J, et al. Serum from pregnant donors induces human beta cell proliferation. Islets. (2024) 16:2334044. doi: 10.1080/19382014.2024.2334044
152. Kitakaze K, Oyadomari M, Zhang J, Hamada Y, Takenouchi Y, Tsuboi K, et al. ATF4-mediated transcriptional regulation protects against β-cell loss during endoplasmic reticulum stress in a mouse model. Mol Metab. (2021) 54:101338. doi: 10.1016/j.molmet.2021.101338
153. Sobajima M, Miyake M, Hamada Y, Tsugawa K, Oyadomari M, Inoue R, et al. The multifaceted role of ATF4 in regulating glucose-stimulated insulin secretion. Biochem Biophys Res Commun. (2022) 611:165–71. doi: 10.1016/j.bbrc.2022.04.038
154. Lassot I, Estrabaud E, Emiliani S, Benkirane M, Benarous R, Margottin-Goguet F. p300 modulates ATF4 stability and transcriptional activity independently of its acetyltransferase domain. J Biol Chem. (2005) 280:41537–45. doi: 10.1074/jbc.M505294200
155. Jeong MH, Jeong HJ, Ahn BY, Pyun JH, Kwon I, Cho H, et al. PRMT1 suppresses ATF4-mediated endoplasmic reticulum response in cardiomyocytes. Cell Death Dis. (2019) 10:903. doi: 10.1038/s41419-019-2147-3
156. Lassot I, Ségéral E, Berlioz-Torrent C, Durand H, Groussin L, Hai T, et al. ATF4 degradation relies on a phosphorylation-dependent interaction with the SCF(betaTrCP) ubiquitin ligase. Mol Cell Biol. (2001) 21:2192–202. doi: 10.1128/MCB.21.6.2192-2202.2001
157. Wang A, Xu S, Zhang X, He J, Yan D, Yang Z, et al. Ribosomal protein RPL41 induces rapid degradation of ATF4, a transcription factor critical for tumour cell survival in stress. J Pathol. (2011) 225:285–92. doi: 10.1002/path.2918
158. Smith SG, Haynes KA, Hegde AN. Degradation of transcriptional repressor ATF4 during long-term synaptic plasticity. Int J Mol Sci. (2020) 21:8543. doi: 10.3390/ijms21228543
159. Hendriks IA, D’Souza RC, Yang B, Verlaan-de Vries M, Mann M, Vertegaal AC. Uncovering global SUMOylation signaling networks in a site-specific manner. Nat Struct Mol Biol. (2014) 21:927–36. doi: 10.1038/nsmb.2890
160. Akimov V, Barrio-Hernandez I, Hansen SVF, Hallenborg P, Pedersen AK, Bekker-Jensen DB, et al. UbiSite approach for comprehensive mapping of lysine and N-terminal ubiquitination sites. Nat Struct Mol Biol. (2018) 25:631–40. doi: 10.1038/s41594-018-0084-y
161. Udeshi ND, Svinkina T, Mertins P, Kuhn E, Mani DR, Qiao JW, et al. Refined preparation and use of anti-diglycine remnant (K-ϵ-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol Cell Proteomics. (2013) 12:825–31. doi: 10.1074/mcp.O112.027094
162. Kinkel MD, Eames SC, Alonzo MR, Prince VE. Cdx4 is required in the endoderm to localize the pancreas and limit beta-cell number. Development. (2008) 135:919–29. doi: 10.1242/dev.010660
163. Rinschen MM, Yu MJ, Wang G, Boja ES, Hoffert JD, Pisitkun T, et al. Quantitative phosphoproteomic analysis reveals vasopressin V2-receptor-dependent signaling pathways in renal collecting duct cells. Proc Natl Acad Sci U.S.A. (2010) 107:3882–7. doi: 10.1073/pnas.0910646107
164. Gao N, LeLay J, Vatamaniuk MZ, Rieck S, Friedman JR, Kaestner KH. Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev. (2008) 22:3435–48. doi: 10.1101/gad.1752608
165. Gao N, White P, Doliba N, Golson ML, Matschinsky FM, Kaestner KH. Foxa2 controls vesicle docking and insulin secretion in mature Beta cells. Cell Metab. (2007) 6:267–79. doi: 10.1016/j.cmet.2007.08.015
166. Lee CS, Sund NJ, Behr R, Herrera PL, Kaestner KH. Foxa2 is required for the differentiation of pancreatic alpha-cells. Dev Biol. (2005) 278:484–95. doi: 10.1016/j.ydbio.2004.10.012
167. Jia S, Ivanov A, Blasevic D, Müller T, Purfürst B, Sun W, et al. Insm1 cooperates with Neurod1 and Foxa2 to maintain mature pancreatic β-cell function. EMBO J. (2015) 34:1417–33. doi: 10.15252/embj.201490819
168. van Gent R, Di Sanza C, van den Broek NJ, Fleskens V, Veenstra A, Stout GJ, et al. SIRT1 mediates FOXA2 breakdown by deacetylation in a nutrient-dependent manner. PloS One. (2014) 9:e98438. doi: 10.1371/journal.pone.0098438
169. von Meyenn F, Porstmann T, Gasser E, Selevsek N, Schmidt A, Aebersold R, et al. Glucagon-induced acetylation of Foxa2 regulates hepatic lipid metabolism. Cell Metab. (2013) 17:436–47. doi: 10.1016/j.cmet.2013.01.014
170. Liu M, Lee DF, Chen CT, Yen CJ, Li LY, Lee HJ, et al. IKKα activation of NOTCH links tumorigenesis via FOXA2 suppression. Mol Cell. (2012) 45:171–84. doi: 10.1016/j.molcel.2011.11.018
171. Belaguli NS, Zhang M, Brunicardi FC, Berger DH. Forkhead box protein A2 (FOXA2) protein stability and activity are regulated by sumoylation. PloS One. (2012) 7:e48019. doi: 10.1371/journal.pone.0048019
172. Lv C, Zhao G, Sun X, Wang P, Xie N, Luo J, et al. Acetylation of FOXM1 is essential for its transactivation and tumor growth stimulation. Oncotarget. (2016) 7:60366–82. doi: 10.18632/oncotarget.11332
173. Cao XJ, Arnaudo AM, Garcia BA. Large-scale global identification of protein lysine methylation in vivo. Epigenetics. (2013) 8:477–85. doi: 10.4161/epi.24547
174. Zhang J, Yuan C, Wu J, Elsayed Z, Fu Z. Polo-like kinase 1-mediated phosphorylation of Forkhead box protein M1b antagonizes its SUMOylation and facilitates its mitotic function. J Biol Chem. (2015) 290:3708–19. doi: 10.1074/jbc.M114.634386
175. Myatt SS, Kongsema M, Man CW, Kelly DJ, Gomes AR, Khongkow P, et al. SUMOylation inhibits FOXM1 activity and delays mitotic transition. Oncogene. (2014) 33:4316–29. doi: 10.1038/onc.2013.546
176. Qiang L, Banks AS, Accili D. Uncoupling of acetylation from phosphorylation regulates FoxO1 function independent of its subcellular localization. J Biol Chem. (2010) 285:27396–401. doi: 10.1074/jbc.M110.140228
177. Masui K, Tanaka K, Akhavan D, Babic I, Gini B, Matsutani T, et al. mTOR complex 2 controls glycolytic metabolism in glioblastoma through FoxO acetylation and upregulation of c-Myc. Cell Metab. (2013) 18:726–39. doi: 10.1016/j.cmet.2013.09.013
178. Housley MP, Rodgers JT, Udeshi ND, Kelly TJ, Shabanowitz J, Hunt DF, et al. O-GlcNAc regulates FoxO activation in response to glucose. J Biol Chem. (2008) 283:16283–92. doi: 10.1074/jbc.M802240200
179. Saline M, Badertscher L, Wolter M, Lau R, Gunnarsson A, Jacso T, et al. AMPK and AKT protein kinases hierarchically phosphorylate the N-terminus of the FOXO1 transcription factor, modulating interactions with 14-3-3 proteins. J Biol Chem. (2019) 294:13106–16. doi: 10.1074/jbc.RA119.008649
180. Yamagata K, Daitoku H, Takahashi Y, Namiki K, Hisatake K, Kako K, et al. Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol Cell. (2008) 32:221–31. doi: 10.1016/j.molcel.2008.09.013
181. Qu X, Nyeng P, Xiao F, Dorantes J, Jensen J. Growth factor independence-1 (Gfi1) is required for pancreatic acinar unit formation and centroacinar cell differentiation. Cell Mol Gastroenterol Hepatol. (2015) 1:233–247.e1. doi: 10.1016/j.jcmgh.2014.12.004
182. Velinder M, Singer J, Bareyan D, Meznarich J, Tracy CM, Fulcher JM, et al. GFI1 functions in transcriptional control and cell fate determination require SNAG domain methylation to recruit LSD1. Biochem J. (2016) 473:3355–69. doi: 10.1042/BCJ20160558
183. Kuai X, Li L, Chen R, Wang K, Chen M, Cui B, et al. SCF. Cancer Res. (2019) 79:4387–98. doi: 10.1158/0008-5472.CAN-18-4032
184. Kang HS, Kim YS, ZeRuth G, Beak JY, Gerrish K, Kilic G, et al. Transcription factor Glis3, a novel critical player in the regulation of pancreatic beta-cell development and insulin gene expression. Mol Cell Biol. (2009) 29:6366–79. doi: 10.1128/MCB.01259-09
185. Scoville D, Lichti-Kaiser K, Grimm S, Jetten A. GLIS3 binds pancreatic beta cell regulatory regions alongside other islet transcription factors. J Endocrinol. (2019) 243(1):1–14. doi: 10.1530/JOE-19-0182
186. Sharma K, D’Souza RC, Tyanova S, Schaab C, Wiśniewski JR, Cox J, et al. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. (2014) 8:1583–94. doi: 10.1016/j.celrep.2014.07.036
187. Kettenbach AN, Schweppe DK, Faherty BK, Pechenick D, Pletnev AA, Gerber SA. Quantitative phosphoproteomics identifies substrates and functional modules of Aurora and Polo-like kinase activities in mitotic cells. Sci Signal. (2011) 4:rs5. doi: 10.1126/scisignal.2001497
188. Tao W, Zhang Y, Ma L, Deng C, Duan H, Liang X, et al. Haploinsufficiency of insm1 impairs postnatal baseline β-cell mass. Diabetes. (2018) 67:2615–25. doi: 10.2337/db17-1330
189. Liang X, Duan H, Mao Y, Koestner U, Wei Y, Deng F, et al. The SNAG domain of insm1 regulates pancreatic endocrine cell differentiation and represses β- to δ-cell transdifferentiation. Diabetes. (2021) 70:1084–97. doi: 10.2337/db20-0883
190. Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. (2016) 534:55–62. doi: 10.1038/nature18003
191. Ediger BN, Du A, Liu J, Hunter CS, Walp ER, Schug J, et al. Islet-1 Is essential for pancreatic β-cell function. Diabetes. (2014) 63:4206–17. doi: 10.2337/db14-0096
192. Bohuslavova R, Fabriciova V, Lebrón-Mora L, Malfatti J, Smolik O, Valihrach L, et al. ISL1 controls pancreatic alpha cell fate and beta cell maturation. Cell Biosci. (2023) 13:53. doi: 10.1186/s13578-023-01003-9
193. Shi Q, Ni X, Lei M, Xia Q, Dong Y, Zhang Q, et al. Phosphorylation of islet-1 serine 269 by CDK1 increases its transcriptional activity and promotes cell proliferation in gastric cancer. Mol Med. (2021) 27:47. doi: 10.1186/s10020-021-00302-6
194. Olbrot M, Rud J, Moss LG, Sharma A. Identification of beta-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc Natl Acad Sci U.S.A. (2002) 99:6737–42. doi: 10.1073/pnas.102168499
195. Aramata S, Han SI, Yasuda K, Kataoka K. Synergistic activation of the insulin gene promoter by the beta-cell enriched transcription factors MafA, Beta2, and Pdx1. Biochim Biophys Acta. (2005) 1730:41–6. doi: 10.1016/j.bbaexp.2005.05.009
196. Nishimura W, Takahashi S, Yasuda K. MafA is critical for maintenance of the mature beta cell phenotype in mice. Diabetologia. (2015) 58:566–74. doi: 10.1007/s00125-014-3464-9
197. Guo S, Vanderford NL, Stein R. Phosphorylation within the MafA N terminus regulates C-terminal dimerization and DNA binding. J Biol Chem. (2010) 285:12655–61. doi: 10.1074/jbc.M110.105759
198. Sii-Felice K, Pouponnot C, Gillet S, Lecoin L, Girault JA, Eychène A, et al. MafA transcription factor is phosphorylated by p38 MAP kinase. FEBS Lett. (2005) 579:3547–54. doi: 10.1016/j.febslet.2005.04.086
199. Kanai K, Reza HM, Kamitani A, Hamazaki Y, Han SI, Yasuda K, et al. SUMOylation negatively regulates transcriptional and oncogenic activities of MafA. Genes Cells. (2010) 15:971–82. doi: 10.1111/j.1365-2443.2010.01431.x
200. Shao C, Cobb MH. Sumoylation regulates the transcriptional activity of MafA in pancreatic beta cells. J Biol Chem. (2009) 284:3117–24. doi: 10.1074/jbc.M806286200
201. Jia D, Sun Y, Konieczny SF. Mist1 regulates pancreatic acinar cell proliferation through p21 CIP1/WAF1. Gastroenterology. (2008) 135:1687–97. doi: 10.1053/j.gastro.2008.07.026
202. Lundby A, Secher A, Lage K, Nordsborg NB, Dmytriyev A, Lundby C, et al. Quantitative maps of protein phosphorylation sites across 14 different rat organs and tissues. Nat Commun. (2012) 3:876. doi: 10.1038/ncomms1871
203. Huttlin EL, Jedrychowski MP, Elias JE, Goswami T, Rad R, Beausoleil SA, et al. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. (2010) 143:1174–89. doi: 10.1016/j.cell.2010.12.001
204. Naya FJ, Huang HP, Qiu Y, Mutoh H, DeMayo FJ, Leiter AB, et al. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. (1997) 11:2323–34. doi: 10.1101/gad.11.18.2323
205. Romer AI, Singer RA, Sui L, Egli D, Sussel L. Murine perinatal β-cell proliferation and the differentiation of human stem cell-derived insulin-expressing cells require NEUROD1. Diabetes. (2019) 68:2259–71. doi: 10.2337/db19-0117
206. Qiu Y, Guo M, Huang S, Stein R. Insulin gene transcription is mediated by interactions between the p300 coactivator and PDX-1, BETA2, and E47. Mol Cell Biol. (2002) 22:412–20. doi: 10.1128/MCB.22.2.412-420.2002
207. Khoo S, Griffen SC, Xia Y, Baer RJ, German MS, Cobb MH. Regulation of insulin gene transcription by ERK1 and ERK2 in pancreatic beta cells. J Biol Chem. (2003) 278:32969–77. doi: 10.1074/jbc.M301198200
208. Petersen HV, Jensen JN, Stein R, Serup P. Glucose induced MAPK signalling influences NeuroD1-mediated activation and nuclear localization. FEBS Lett. (2002) 528:241–5. doi: 10.1016/s0014-5793(02)03318-5
209. Anderson KR, Torres CA, Solomon K, Becker TC, Newgard CB, Wright CV, et al. Cooperative transcriptional regulation of the essential pancreatic islet gene NeuroD1 (beta2) by Nkx2.2 and neurogenin 3. J Biol Chem. (2009) 284:31236–48. doi: 10.1074/jbc.M109.048694
210. Cho G, Hyun K, Choi J, Shin E, Kim B, Kim H, et al. Arginine 65 methylation of Neurogenin 3 by PRMT1 is required for pancreatic endocrine development of hESCs. Exp Mol Med. (2023) 55:1506–19. doi: 10.1038/s12276-023-01035-8
211. Krentz NAJ, van Hoof D, Li Z, Watanabe A, Tang M, Nian C, et al. Phosphorylation of NEUROG3 links endocrine differentiation to the cell cycle in pancreatic progenitors. Dev Cell. (2017) 41:129–142.e6. doi: 10.1016/j.devcel.2017.02.006
212. Sancho R, Gruber R, Gu G, Behrens A. Loss of Fbw7 reprograms adult pancreatic ductal cells into α, δ, and β cells. Cell Stem Cell. (2014) 15:139–53. doi: 10.1016/j.stem.2014.06.019
213. Guo A, Gu H, Zhou J, Mulhern D, Wang Y, Lee KA, et al. Immunoaffinity enrichment and mass spectrometry analysis of protein methylation. Mol Cell Proteomics. (2014) 13:372–87. doi: 10.1074/mcp.O113.027870
214. Abebe T, Mahadevan J, Bogachus L, Hahn S, Black M, Oseid E, et al. Nrf2/antioxidant pathway mediates β cell self-repair after damage by high-fat diet-induced oxidative stress. JCI Insight. (2017) 2:e92854. doi: 10.1172/jci.insight.92854
215. Yagishita Y, Fukutomi T, Sugawara A, Kawamura H, Takahashi T, Pi J, et al. Nrf2 protects pancreatic β-cells from oxidative and nitrosative stress in diabetic model mice. Diabetes. (2014) 63:605–18. doi: 10.2337/db13-0909
216. Uruno A, Furusawa Y, Yagishita Y, Fukutomi T, Muramatsu H, Negishi T, et al. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol Cell Biol. (2013) 33:2996–3010. doi: 10.1128/MCB.00225-13
217. Sun Z, Chin YE, Zhang DD. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol Cell Biol. (2009) 29:2658–72. doi: 10.1128/MCB.01639-08
218. Niture SK, Jain AK, Jaiswal AK. Antioxidant-induced modification of INrf2 cysteine 151 and PKC-delta-mediated phosphorylation of Nrf2 serine 40 are both required for stabilization and nuclear translocation of Nrf2 and increased drug resistance. J Cell Sci. (2009) 122:4452–64. doi: 10.1242/jcs.058537
219. Huang HC, Nguyen T, Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem. (2002) 277:42769–74. doi: 10.1074/jbc.M206911200
220. Bloom DA, Jaiswal AK. Phosphorylation of Nrf2 at Ser40 by protein kinase C in response to antioxidants leads to the release of Nrf2 from INrf2, but is not required for Nrf2 stabilization/accumulation in the nucleus and transcriptional activation of antioxidant response element-mediated NAD(P)H:quinone oxidoreductase-1 gene expression. J Biol Chem. (2003) 278:44675–82. doi: 10.1074/jbc.M307633200
221. Tao S, Liu P, Luo G, Rojo de la Vega M, Chen H, Wu T, et al. p97 negatively regulates NRF2 by extracting ubiquitylated NRF2 from the KEAP1-CUL3 E3 complex. Mol Cell Biol. (2017) 37:e00660-16. doi: 10.1128/MCB.00660-16
222. Ait-Lounis A, Bonal C, Seguín-Estévez Q, Schmid CD, Bucher P, Herrera PL, et al. The transcription factor Rfx3 regulates beta-cell differentiation, function, and glucokinase expression. Diabetes. (2010) 59:1674–85. doi: 10.2337/db09-0986
223. Wang J, Elghazi L, Parker SE, Kizilocak H, Asano M, Sussel L, et al. The concerted activities of Pax4 and Nkx2.2 are essential to initiate pancreatic beta-cell differentiation. Dev Biol. (2004) 266:178–89. doi: 10.1016/j.ydbio.2003.10.018
224. Smith SB, Ee HC, Conners JR, German MS. Paired-homeodomain transcription factor PAX4 acts as a transcriptional repressor in early pancreatic development. Mol Cell Biol. (1999) 19:8272–80. doi: 10.1128/MCB.19.12.8272
225. Petersen HV, Jørgensen MC, Andersen FG, Jensen J, F-Nielsen T, Jørgensen R, et al. Pax4 represses pancreatic glucagon gene expression. Mol Cell Biol Res Commun. (2000) 3:249–54. doi: 10.1006/mcbr.2000.0220
226. Liew CG, Shah NN, Briston SJ, Shepherd RM, Khoo CP, Dunne MJ, et al. PAX4 enhances beta-cell differentiation of human embryonic stem cells. PloS One. (2008) 3:e1783. doi: 10.1371/journal.pone.0001783
227. Sander M, Neubüser A, Kalamaras J, Ee HC, Martin GR, German MS. Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes Dev. (1997) 11:1662–73. doi: 10.1101/gad.11.13.1662
228. Kim EA, Noh YT, Ryu MJ, Kim HT, Lee SE, Kim CH, et al. Phosphorylation and transactivation of Pax6 by homeodomain-interacting protein kinase 2. J Biol Chem. (2006) 281:7489–97. doi: 10.1074/jbc.M507227200
229. Yu F, Zhang W, Yan C, Yan D, Zhou M, Chen J, et al. PAX6, modified by SUMOylation, plays a protective role in corneal endothelial injury. Cell Death Dis. (2020) 11:683. doi: 10.1038/s41419-020-02848-5
230. Ait-Lounis A, Baas D, Barras E, Benadiba C, Charollais A, Nlend Nlend R, et al. Novel function of the ciliogenic transcription factor RFX3 in development of the endocrine pancreas. Diabetes. (2007) 56:950–9. doi: 10.2337/db06-1187
231. Rigbolt KT, Prokhorova TA, Akimov V, Henningsen J, Johansen PT, Kratchmarova I, et al. System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci Signal. (2011) 4:rs3. doi: 10.1126/scisignal.2001570
232. Piccand J, Strasser P, Hodson DJ, Meunier A, Ye T, Keime C, et al. Rfx6 maintains the functional identity of adult pancreatic β cells. Cell Rep. (2014) 9:2219–32. doi: 10.1016/j.celrep.2014.11.033
233. van Arensbergen J, García-Hurtado J, Maestro MA, Correa-Tapia M, Rutter GA, Vidal M, et al. Ring1b bookmarks genes in pancreatic embryonic progenitors for repression in adult β cells. Genes Dev. (2013) 27:52–63. doi: 10.1101/gad.206094.112
234. Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. (2010) 327:1000–4. doi: 10.1126/science.1179689
235. Rao PS, Satelli A, Zhang S, Srivastava SK, Srivenugopal KS, Rao US. RNF2 is the target for phosphorylation by the p38 MAPK and ERK signaling pathways. Proteomics. (2009) 9:2776–87. doi: 10.1002/pmic.200800847
236. Tammsalu T, Matic I, Jaffray EG, Ibrahim AFM, Tatham MH, Hay RT. Proteome-wide identification of SUMO2 modification sites. Sci Signal. (2014) 7:rs2. doi: 10.1126/scisignal.2005146
237. Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, et al. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics. (2011) 10:M111.013284. doi: 10.1074/mcp.M111.013284
238. Buchwald G, van der Stoop P, Weichenrieder O, Perrakis A, van Lohuizen M, Sixma TK. Structure and E3-ligase activity of the Ring-Ring complex of polycomb proteins Bmi1 and Ring1b. EMBO J. (2006) 25:2465–74. doi: 10.1038/sj.emboj.7601144
239. Seymour PA, Freude KK, Tran MN, Mayes EE, Jensen J, Kist R, et al. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc Natl Acad Sci U.S.A. (2007) 104:1865–70. doi: 10.1073/pnas.0609217104
240. Puri S, Maachi H, Nair G, Russ HA, Chen R, Pulimeno P, et al. Sox9 regulates alternative splicing and pancreatic beta cell function. Nat Commun. (2024) 15:588. doi: 10.1038/s41467-023-44384-8
241. Hattori T, Coustry F, Stephens S, Eberspaecher H, Takigawa M, Yasuda H, et al. Transcriptional regulation of chondrogenesis by coactivator Tip60 via chromatin association with Sox9 and Sox5. Nucleic Acids Res. (2008) 36:3011–24. doi: 10.1093/nar/gkn150
242. Liu L, Ge W, Zhang Z, Li Y, Xie M, Zhao C, et al. Sublytic C5b-9 triggers glomerular mesangial cell proliferation via enhancing FGF1 and PDGFα gene transcription mediated by GCN5-dependent SOX9 acetylation in rat Thy-1 nephritis. FASEB J. (2021) 35:e21751. doi: 10.1096/fj.202002814RR
243. Sun Q, Zhuang Z, Bai R, Deng J, Xin T, Zhang Y, et al. Lysine 68 methylation-dependent SOX9 stability control modulates chondrogenic differentiation in dental pulp stem cells. Adv Sci (Weinh). (2023) 10:e2206757. doi: 10.1002/advs.202206757
244. Ito T, Yadav N, Lee J, Furumatsu T, Yamashita S, Yoshida K, et al. Arginine methyltransferase CARM1/PRMT4 regulates endochondral ossification. BMC Dev Biol. (2009) 9:47. doi: 10.1186/1471-213X-9-47
245. Coricor G, Serra R. TGF-β regulates phosphorylation and stabilization of Sox9 protein in chondrocytes through p38 and Smad dependent mechanisms. Sci Rep. (2016) 6:38616. doi: 10.1038/srep38616
246. Al-Zahrani KN, Abou-Hamad J, Pascoal J, Labrèche C, Garland B, Sabourin LA. AKT-mediated phosphorylation of Sox9 induces Sox10 transcription in a murine model of HER2-positive breast cancer. Breast Cancer Res. (2021) 23:55. doi: 10.1186/s13058-021-01435-6
247. Huang W, Zhou X, Lefebvre V, de Crombrugghe B. Phosphorylation of SOX9 by cyclic AMP-dependent protein kinase A enhances SOX9’s ability to transactivate a Col2a1 chondrocyte-specific enhancer. Mol Cell Biol. (2000) 20:4149–58. doi: 10.1128/MCB.20.11.4149-4158.2000
248. Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. (2011) 44:325–40. doi: 10.1016/j.molcel.2011.08.025
249. Wakabayashi Y, Miyatsuka T, Miura M, Himuro M, Taguchi T, Iida H, et al. STAT3 suppression and β-cell ablation enhance α-to-β reprogramming mediated by Pdx1. Sci Rep. (2022) 12:21419. doi: 10.1038/s41598-022-25941-5
250. Ray S, Boldogh I, Brasier AR. STAT3 NH2-terminal acetylation is activated by the hepatic acute-phase response and required for IL-6 induction of angiotensinogen. Gastroenterology. (2005) 129:1616–32. doi: 10.1053/j.gastro.2005.07.055
251. Nie Y, Erion DM, Yuan Z, Dietrich M, Shulman GI, Horvath TL, et al. STAT3 inhibition of gluconeogenesis is downregulated by SirT1. Nat Cell Biol. (2009) 11:492–500. doi: 10.1038/ncb1857
252. Hou T, Ray S, Lee C, Brasier AR. The STAT3 NH2-terminal domain stabilizes enhanceosome assembly by interacting with the p300 bromodomain. J Biol Chem. (2008) 283:30725–34. doi: 10.1074/jbc.M805941200
253. Iwasaki H, Kovacic JC, Olive M, Beers JK, Yoshimoto T, Crook MF, et al. Disruption of protein arginine N-methyltransferase 2 regulates leptin signaling and produces leanness in vivo through loss of STAT3 methylation. Circ Res. (2010) 107:992–1001. doi: 10.1161/CIRCRESAHA.110.225326
254. Song D, Lan J, Chen Y, Liu A, Wu Q, Zhao C, et al. NSD2 promotes tumor angiogenesis through methylating and activating STAT3 protein. Oncogene. (2021) 40:2952–67. doi: 10.1038/s41388-021-01747-z
255. Dasgupta M, Dermawan JK, Willard B, Stark GR. STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc Natl Acad Sci U.S.A. (2015) 112:3985–90. doi: 10.1073/pnas.1503152112
256. Tang Q, Fang J, Lai W, Hu Y, Liu C, Hu X, et al. Hippo pathway monomerizes STAT3 to regulate prostate cancer growth. Cancer Sci. (2022) 113:2753–62. doi: 10.1111/cas.15463
257. Lumpkin RJ, Gu H, Zhu Y, Leonard M, Ahmad AS, Clauser KR, et al. Site-specific identification and quantitation of endogenous SUMO modifications under native conditions. Nat Commun. (2017) 8:1171. doi: 10.1038/s41467-017-01271-3
258. Zhou Z, Wang M, Li J, Xiao M, Chin YE, Cheng J, et al. SUMOylation and SENP3 regulate STAT3 activation in head and neck cancer. Oncogene. (2016) 35:5826–38. doi: 10.1038/onc.2016.124
259. Kleiber T, Davidson G, Mengus G, Martianov I, Davidson I. Single cell transcriptomics reveal trans-differentiation of pancreatic beta cells following inactivation of the TFIID subunit Taf4. Cell Death Dis. (2021) 12:790. doi: 10.1038/s41419-021-04067-y
260. Larsen SC, Sylvestersen KB, Mund A, Lyon D, Mullari M, Madsen MV, et al. Proteome-wide analysis of arginine monomethylation reveals widespread occurrence in human cells. Sci Signal. (2016) 9:rs9. doi: 10.1126/scisignal.aaf7329
261. Trinidad JC, Barkan DT, Gulledge BF, Thalhammer A, Sali A, Schoepfer R, et al. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol Cell Proteomics. (2012) 11:215–29. doi: 10.1074/mcp.O112.018366
262. Bai Y, Li J, Fang B, Edwards A, Zhang G, Bui M, et al. Phosphoproteomics identifies driver tyrosine kinases in sarcoma cell lines and tumors. Cancer Res. (2012) 72:2501–11. doi: 10.1158/0008-5472.CAN-11-3015
263. Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. (2004) 429:41–6. doi: 10.1038/nature02520
264. Yao X, Li K, Liang C, Zhou Z, Wang J, Wang S, et al. Tectorigenin enhances PDX1 expression and protects pancreatic β-cells by activating ERK and reducing ER stress. J Biol Chem. (2020) 295:12975–92. doi: 10.1074/jbc.RA120.012849
265. Pagliuca FW, Millman JR, Gürtler M, Segel M, Van Dervort A, Ryu JH, et al. Generation of functional human pancreatic β cells in vitro. Cell. (2014) 159:428–39. doi: 10.1016/j.cell.2014.09.040
266. Liang S, Zhao J, Baker RK, Tran E, Zhan L, Kieffer TJ. Differentiation of stem cell-derived pancreatic progenitors into insulin-secreting islet clusters in a multiwell-based static 3D culture system. Cell Rep Methods. (2023) 3:100466. doi: 10.1016/j.crmeth.2023.100466
Keywords: transcription factors, post-translational modification (PTM), pancreatic beta cells, pancreas development, beta cell proliferation, beta cell differentiation, diabetes, mature onset diabetes of the young (MODY)
Citation: Wong A and Alejandro EU (2025) Post translational modification regulation of transcription factors governing pancreatic β-cell identity and functional mass. Front. Endocrinol. 16:1562646. doi: 10.3389/fendo.2025.1562646
Received: 17 January 2025; Accepted: 17 February 2025;
Published: 11 March 2025.
Edited by:
Meritxell Rovira, Institut d’Investigacio Biomedica de Bellvitge (IDIBELL), SpainReviewed by:
Maria Golson, Johns Hopkins University, United StatesCopyright © 2025 Wong and Alejandro. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emilyn U. Alejandro, ZWFsZWphbmRAdW1uLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.