
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 10 March 2025
Sec. Reproduction
Volume 16 - 2025 | https://doi.org/10.3389/fendo.2025.1558271
This article is part of the Research Topic Traditional Chinese Medicine Strategies for Preventing and Treating Reproductive Endocrine Disorders Caused by Various Factors View all 5 articles
 Qing Qi1†
Qing Qi1† Yaonan Li1†Ziqin Chen2Zhihui Luo3Ting Zhou3
Yaonan Li1†Ziqin Chen2Zhihui Luo3Ting Zhou3 Jing Zhou4Yanlin Zhang5Song Chen5*
Jing Zhou4Yanlin Zhang5Song Chen5* Ling Wang6,7,8*
Ling Wang6,7,8*Endometriosis, the most prevalent cause of infertility, is associated with anatomical distortion leading to adhesions and fibrosis, as well as endocrine abnormalities and immune disorders. This review discusses the mechanisms underlying endometriosis-related infertility. Firstly, alterations in the hypothalamic-pituitary-ovarian axis lead to the secretion of gonadotropins and steroid hormones, with adverse effects on ovulation and implantation, leading to fertility decline. Secondly, dysregulation of the hypothalamic-pituitary-adrenal axis induces elevated serum cortisol and prolactin levels in patients with endometriosis, accounting for its regulation of stress, depression, and anxiety. Abnormal interactions between endometrial cells and the immune system change the local microenvironment, resulting in epithelial-mesenchymal transition and inflammation. Activated epithelial cells, stromal cells, and immunocytes produce various chemokines, cytokines, or autoantibodies, creating an unfavorable environment for embryo implantation. These findings suggest that alterations in the immune spectrum play a crucial role in endometriosis-related infertility. Thirdly, oxidative stress has adverse effects on the ovarian reserve and subsequent embryonic development, predicting another promising strategy for endometriosis-related infertility. An unbalanced redox state, including impaired mitochondrial function, dysregulated lipid metabolism, and iron-induced oxidative stress, generates a pro-oxidative microenvironment, which negatively impacts oocyte quality and sperm and embryo viability. Thus, an updated understanding of the mechanisms involved in this disease will help to develop effective strategies to manage endometriosis-related infertility.
Endometriosis is an estrogen-dependent disease characterized by the implantation of endometrial glands and extrauterine stroma, affecting 10% of women of reproductive age (1). Approximately 30–50% of women with endometriosis experience infertility, contributing to anxiety and depression in these patients and their partners (2). Mechanical damage caused by pelvic adhesions and ectopic ovarian cysts in women with advanced endometriosis can hinder oocyte release, obstruct the fallopian tubes, and impair the uterine environment for embryo implantation, resulting in female infertility (3). However, even in cases where there is no distortion of the pelvic anatomy, infertility is considered a possible consequence of early-stage endometriosis. The complex interactions among the endocrine, immune systems, and oxidative stress, collectively contribute to infertility.
There are still many questionable aspects concerning the pathogenesis of endometriosis-related infertility, making it a hot topic in current research. The pathological mechanism of subfertility in patients with endometriosis is complicated, including mechanical distortion caused by pelvic adhesion, reduced oocyte quality, gametal and embryonic toxicity, dysfunctional uterotubal motility, and altered endometrial receptivity. This review discusses the potential mechanisms underlying endometriosis-related infertility based on current evidence. This includes the involvement of the neuro-endocrine axis and sex hormones, the interaction between endometrial cells and the immune system, and oxidative stress. An updated understanding of the mechanisms involved in this condition will help to develop effective management strategies for endometriosis-related infertility.
Endometriosis is associated with alterations in the HPO axis (4), which tightly regulates the cyclic secretion of gonadotropins and steroid hormones, and the ovulation of the dominant follicle, preparing for subsequent fertilization and implantation. The development and maintenance of endometriosis are usually accompanied by abnormal fluctuations in these hormones and their receptors. For example, changes in gonadotropin ratios, estrogen dominance, and progesterone resistance caused by abnormal regulation of steroidogenesis are the main features of endometriosis-related infertility (5) (Figure 1).
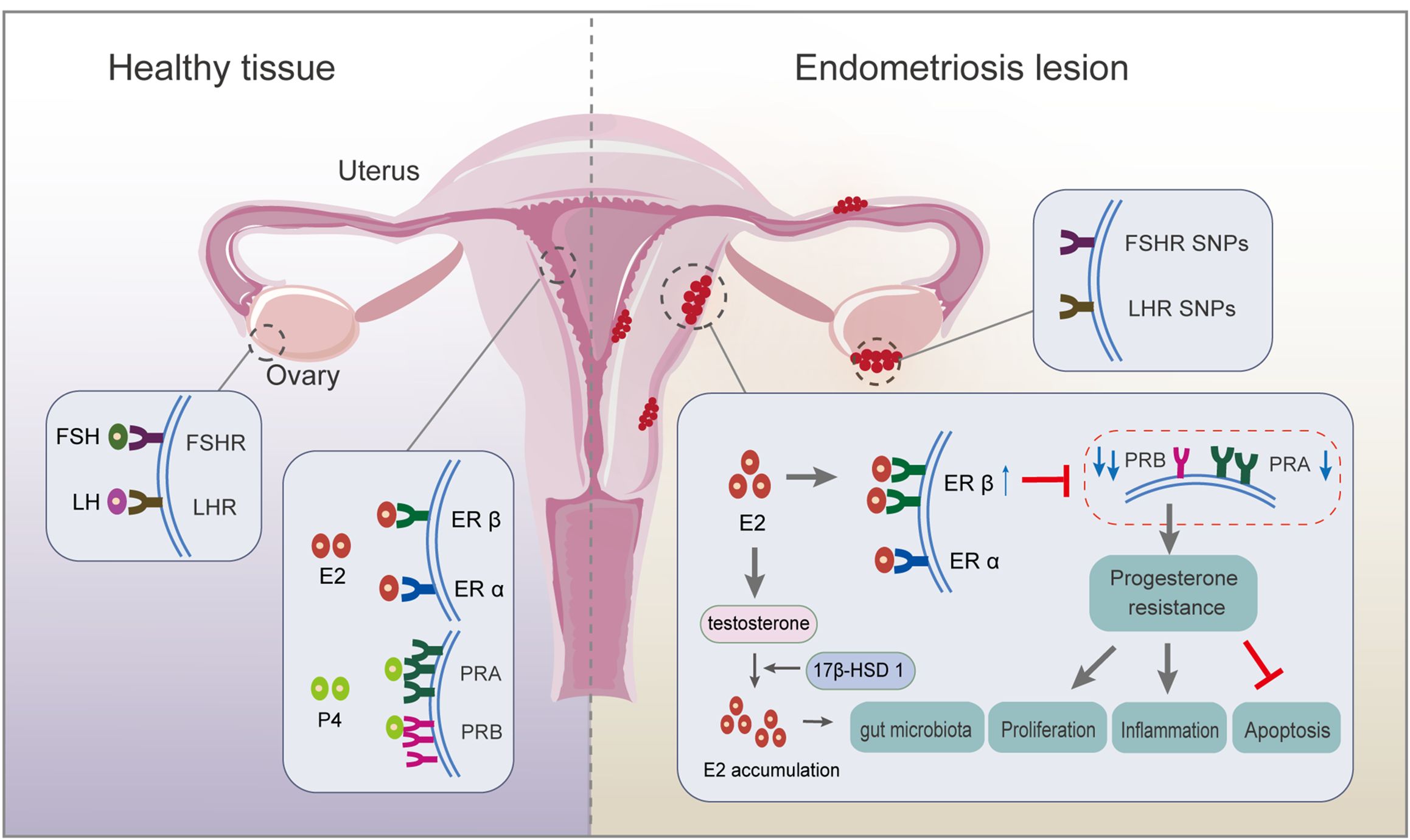
Figure 1. Respective roles of endocrine-related receptors in normal endometrial and endometriotic lesions. Firstly, SNP in the FSHR gene and LHR gene are associated with ovulation disorders. Deletion of FSHR, LHR, or both impedes granulosa cell survival and follicle development. Secondly, circulating E2 acts mainly on ERα; ERβ expression is upregulated, and ERα expression is weakened in endometriosis lesions. There is local accumulation of E2, mainly because of the ability of endometriotic lesions to synthesize E2, resulting from higher expression of 17β-hydroxysteroid dehydrogenase 1, a key enzyme for E2 production. Thirdly, PRA and PRB regulate the progesterone response. An imbalanced PRA: PRB ratio alters the progesterone pathway, inducing progesterone resistance, which finally inhibits apoptosis and promotes proliferation and inflammation. ER, estrogen receptor; FSH, follicle-stimulating hormone; FSH receptor, FSHR; LH, luteinizing hormone; LH receptor, LHR; PR, progesterone receptor; SNP, single nucleotide polymorphism.
The pituitary gland secretes FSH and LH, which travel via the bloodstream to the ovaries, where they synergistically induce folliculogenesis, oocyte maturation, ovulation, granulosa cell growth, and aromatase synthesis during reproduction (6). In patients with endometriosis combined with infertility, although the patients retain the ability to ovulate, there are significant disturbances in the rhythm and secretion of serum gonadotropic hormones, which severely affects their reproductive function. Evidence demonstrates reduced ovarian reserve function in endometriosis (7, 8): i) elevated serum concentrations of FSH and low concentrations of LH relative to FSH levels are observed in endometriosis, while lower FSH levels are potential indexes for successful postoperative pregnancy; and ii) single nucleotide polymorphisms (SNPs) in the FSH receptor (FSHR) gene and LH receptor (LHR) gene might be associated with ovulation disorders. The deletion of FSHR, LHR, or both, impedes granulosa cell survival and follicle development, ultimately altering oocyte quality through failure of cell maturation (4). A high incidence of mutations is observed in LHR in patients with endometriosis and infertility, suggesting impaired LH function in patients with endometriosis/infertility (9).
Endometriosis is an estrogen-dependent disease in which estrogen accumulation affects the progression of endometriotic lesions by binding to and activating ERs, including ERα and ERβ (10). Endometriotic tissue expresses aromatase and shows a high aromatase activity, partly accounting for the increased levels of E2 in endometrial tissue (11). Upregulated expression of 17β-hydroxysteroid dehydrogenase 1, a key enzyme for E2 production, is observed in the in situ endometrium, accompanied by high estrogen levels, which are responsible for a high rate of implantation failure in patients with infertile ovarian endometriosis (5). Overproduction of E2 drives the ERβ signaling pathway, leading to survival and inflammation of endometriotic tissue (11). Further study revealed that E2-induced estrogen in breast cancer 1, as an early prototypical estrogen-responsive gene, encodes a protein that modulates downstream gene expression to promote endometriosis development (12). The relative ratio of ERα to ERβ (ERα/ERβ) in endometriotic tissues is significantly lower than in the eutopic endometrium, accompanied by enhanced ERβ activity (13, 14). This results in evasion of endogenous immune surveillance for cell survival, inhibition of tumor necrosis factor (TNF)-α-induced apoptosis, an increase in interleukin (IL)-1β to enhance its cellular adhesion and proliferation properties, and strengthening of epithelial-mesenchymal transition (EMT) signaling (14). The close association of estrogen with inflammation is strongly supported by the estrogen-mediated expression of cytokines, such as IL-8, IL-6, IL-1β, and TNF-α, partly from macrophages (15). Thus, estrogen-macrophage interplay might be involved in endometriosis and infertility because of the remarkable ability of macrophages to respond to estrogens in vitro, probably creating a permissive environment for oocyte movement, fertilization, and implantation (16). Hence, estrogen pathways lead to an inflammatory state and disturbances in the immune environment, which affect morphological and biochemical changes associated with endometrial tolerance, resulting in pregnancy outcomes.
The interactions between high estrogen levels and the gut microbiota (GM) detrimentally affect embryo implantation and fertility by altering the cervical vaginal microenvironment, such as increased pH, bacterial overgrowth, and increased levels of inflammatory factors. The two major gut bacteria with significantly increased abundance, Blautia and Dorea, were shown to correlate positively with estrogen levels in patients with stage 3/4 endometriosis (17). The GM influences estrogen levels by regulating the enzymes responsible for estrogen metabolism (18). Estrogome, an estrogen-metabolizing enzyme, dissociates conjugated estrogen from bile acids by enhancing the activity of β-glucuronase, glucosidase, and hydroxysteroid dehydrogenase, resulting in increased levels of free estrogen in the peripheral blood (19). Uterine microbiota changes might affect the endometrium’s receptivity-related pathways during endometriosis (20). Metabolites produced by the GM might also modulate neurohormone release through the gut-brain axis. The neuroactive metabolites, such as short-chain fatty acids (21) and 5-hydroxytryptamine (22), resulting from GM metabolism can affect the brain’s neural signaling process through the circulatory system and influence downstream estrogen generation through the HPO axis (23). These metabolites activate gonadotropin-releasing hormone (GnRH)-producing neurons in the hypothalamus. The secreted GnRH signals to the pituitary gland increase the production of LH and FSH and stimulate the ovary to produce estrogen. Accordingly, when the intestine is in a biological imbalance, it is hypothesized to aggravate endometriosis-related infertility through the gut-brain axis (24). However, a causal relationship between microbiota and endometriosis cannot be established because of the insufficiency of current evidence and lack of further in-depth exploration.
Ovarian-derived progesterone regulates the periodic decidualization of endometrial stromal cells and facilitates the formation of an endometrial microenvironment that promotes blastocyst receptivity and embryo implantation. Excessive progesterone production during the follicular phase is involved in the abnormal signaling that impairs oocyte quality in endometriosis-associated infertility (25). Overexpression of progesterone is activated by beclin-1-induced autophagy in granulosa cells in patients with endometriosis (26). There are two functionally distinct isoforms of PR, PRA and PRB, which regulate the progesterone response. Regarding altered ER activity, undermethylation of the ERB promoter inhibits estrogen-induced PR expression, thereby reducing progesterone sensitivity (27). Abnormal overexpression of PRA and high DNA methylation level in the PRB promoter region in the endometriotic tissue of ovarian endometriosis patients increases the PRA: PRB ratio. Then, PRB, a strong transactivator in response to progesterone, loses its dominant position in the secretory nuclei, thus inducing progesterone resistance and leading to repeated implantation failure (5, 10). The imbalance of the PRA: PRB ratio alters the activity of progesterone and promotes the overexpression of specific response target genes, such as that encoding matrix metalloproteinase (MMP)-9, leading to extensive reconstruction of the endometrial layer and extracellular matrix, which directly affects the follicular microenvironment and the quality of oocytes and embryos. Mechanistically, dual inhibition of protein kinase B (AKT) and extracellular-signal-regulated kinase (ERK)1/2 pathways can reduce E2 biosynthesis and restore specific PRB signaling of endometrial cells (28). Moreover, how the interaction occurs between progesterone and E2 is still not fully understood. Further study demonstrated that mitogen inducible gene 6 mediates progesterone inhibition of E2 signaling by suppressing the Erb-B2 receptor tyrosine kinase 2 (ERBB2)-ERK signaling (29). However, the in-depth mechanism still requires further investigation.
Dysfunction of the HPA axis has been identified in patients with endometriosis, being associated with the regulation of stress, depression, and anxiety (30). A significant correlation was found between cortisol levels and infertility and difficulties with sexual intercourse, with elevated serum levels of cortisol and prolactin being identified in patients with endometriosis-related infertility (31). This might explain why anxiety and depression are common in infertile patients and their partners (2). There are a number of possible explanations: i) neurons that secrete corticotropin-releasing hormone (CRH) are activated during stressful states, resulting in elevated plasma cortisol levels and prolactin release (30, 32); ii) neuroendocrine regulation and CRH release affected by chronic stress activate an inflammatory response, affecting the growth of an ectopic endometrium and the physiopathology of the pelvic organs, and leading to exacerbation of symptoms in endometriosis (33, 34); and iii) pleiotropy, such as SNP rs12666606 in DGKB (encoding diacylglycerol kinase beta), likely contributes to endometriosis-associated mental health (2).
A two-sample Mendelian randomization method identified specific immune cells involved in different types of endometrioses, uniquely establishing causal relationships between immune cells and endometriosis (35). High-resolution single-cell reference atlas profiled dysregulation in endometrial cells and inflammatory conditions in women with and without endometriosis (36, 37). Local or systemic inflammatory response dominates the pathological progression of endometriosis. It compromises the reproductive capacity by affecting folliculogenesis and oocyte maturation (38), suggesting the critical role of immune profile alteration in endometriosis-related infertility (Figure 2).
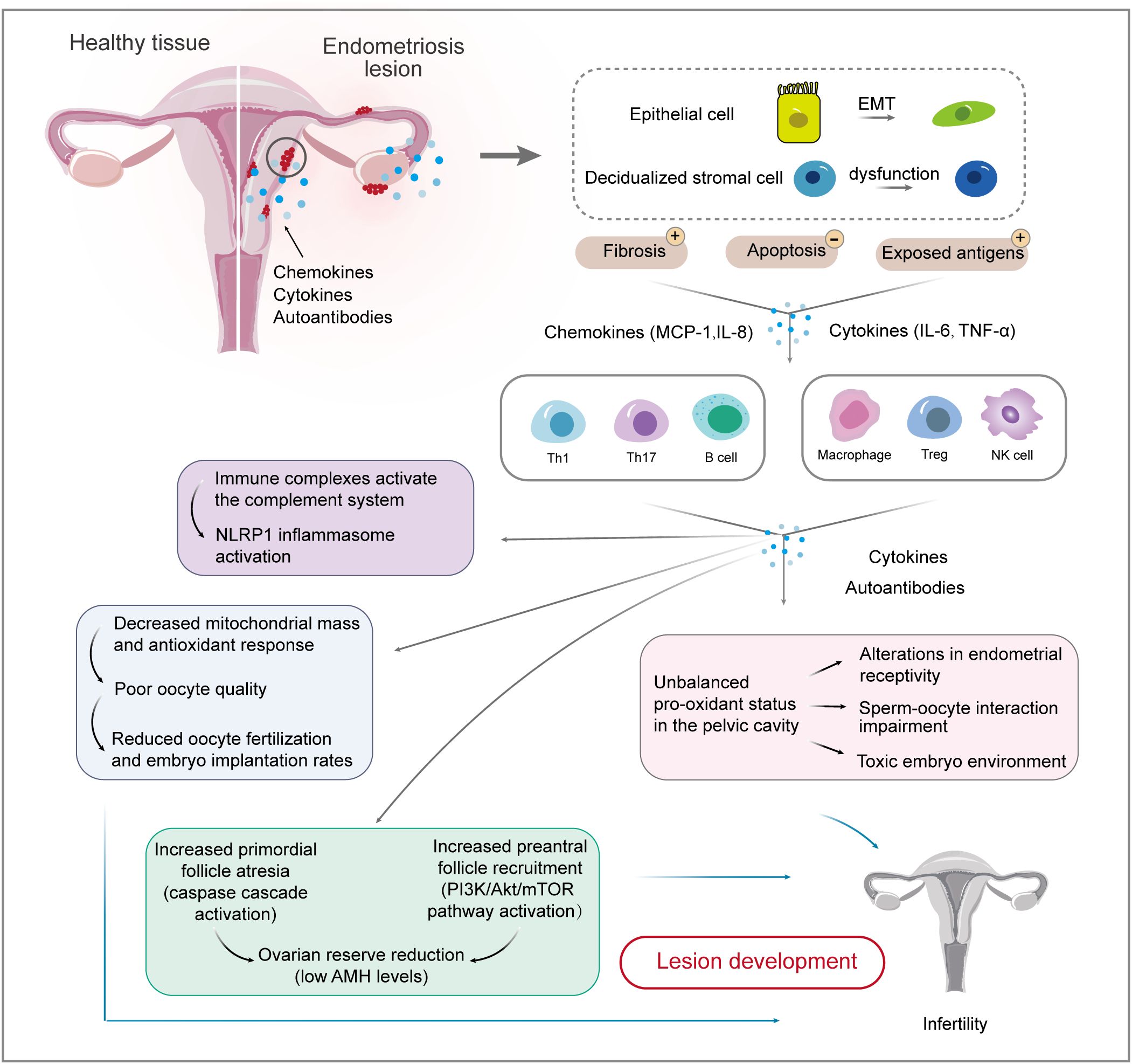
Figure 2. Interactions between endometrial cells and the immune system involved in endometriosis-related infertility. Firstly, causal relationships between endometrial cells and immune cells are well-established. EMT biological processes and stromal cell dysfunction account for fibrosis and exposed antigens, which cause ongoing inflammation and impaired immune surveillance. Immune cell populations, including T cells, macrophages, and NK cells, are involved in the development and maintenance of endometriotic lesions. The inflammatory environment of endometriosis induces immune complexes and an unbalanced pro-oxidant status, which affects ovarian reserve and oocyte quality, thereby reducing the chance of fertilization and implantation. EMT, epithelial-mesenchymal transition; IL, interleukin; MCP, monocyte chemoattractant protein; TNF, tumor necrosis factor.
The pathophysiology of endometriosis involves EMT, a biological process in which polarized epithelial cells transform to highly motile mesenchymal cells (39). Globally, plastic consumption has been rapidly increasing, and persistent pollutant exposure, such as plasticizers, has increased endometriosis-related EMT development, contributing to disease onset and progression (40). Mesothelial-to-mesenchymal transition (MMT), a type II EMT process, occurs in mesothelial cells that lose their polarity and take on a fibroblast-like phenotype in a pathological state (41). It was observed that MMT can trigger peritoneal fibrosis, angiogenesis, and implantation of an ectopic endometrium, in which peritoneal mesothelial cells undergoing MMT can lead to cell adhesions (41). Mechanistically, EMT production is associated with the pathophysiology of fibrosis through the IL-33-suppression of tumorigenicity 2 (ST2) axis, the transforming growth factor (TGF)-β signaling pathway, and the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)/AKT/glycogen synthase kinase 3 beta (GSK-3β)/β-catenin axis, which promote the migration and invasion of endometrial mesenchymal cells and mediate peritoneal adhesions (39–42). Targeting the IL-33-ST2 axis might be a feasible method to control fibrosis in endometriosis (39). Epithelial cells enhance ectopic endometrial cell survival and apoptotic resistance, decrease natural killer (NK) cell activity, and induce macrophage recruitment by overexpressing TGF-β1. This promotes fibrosis and adhesion formation by upregulating tissue inhibitor of metalloproteinase 1 expression and inducing myofibroblast differentiation (43). TGF-β-dependent signaling, activated by soluble endoglin and growth differentiation factor 15, promotes inflammatory chemokines in the ovary and pelvic adhesions, contributing to reduced fertility (44). In conclusion, the EMT theory provides a rationale for the involvement of endometrial cell transformation in the development of endometriosis-associated infertility.
Decidualized stromal cells are highlighted in endometriosis (36). Endometrial cells account for the increased number of peritoneal macrophages by producing monocyte chemoattractant protein (MCP)-1 and IL-6 (45). MCP-1 has a critical function in the pathogenesis of endometriosis-associated infertility, especially in the early stages, being implicated in the regulation of follicular growth, ovulation, luteal development, and the induction of intrafollicular inflammatory state, as well as being a potential predictor of worse prognosis (46). However, distinguishable changes in MCP-1 levels were also detected between patients with endometriosis and those without, which was independent of cycle and fertility status (47).
In addition, endometrial stromal cells account for IL-6 and IL-8 production in endometriotic tissue (48), affecting blastocyst adhesion and inhibiting embryo outgrowth to a great extent (49). High levels of IL-6 are detected in follicular fluid from naturally mature follicles in patients with moderate and severe endometriosis, which is associated with lower pregnancy rates (50). The reasons may be that, i) high levels of IL-6 might affect follicular biology and reduce oocyte quality (50); and ii) the combination of IL-6 and its soluble receptor significantly reduces sperm motility (51). Therefore, it is hypothesized that combining IL-6 and IL-8 with serum carcinogenic antigen-125 levels, a major biomarker of endometriosis, could predict infertility in patients with endometriosis (52). Moreover, IL-8 blockade is hypothesized to reduce fibrosis in endometriosis (53). The data related to these biomarkers need to be validated in future studies, followed by testing of their clinical utility for routine screening, as non-surgical diagnostic markers, and as targets for immunomodulatory therapy, with the aim of identifying immunomodulatory strategies for this disease.
Endometrial stromal cells serve as progenitors of adenomyosis, in which multiple WNT proteins originating from fibroblasts, epithelial cells, and endothelial cells constitute a critical paracrine network (54). The hippo/yes-associated protein 1 (YAP1) signaling pathway plays an important role in endometriosis. YAP1 reduces PR expression and inhibits endometrial stromal cell decidualization by upregulating the expression of the microRNA miR-21-5p (54). In addition, methyltransferase-like 3-regulated m6A modification impairs the decidualization of endometrial stromal cells by regulating degradation of the mRNA encoding Forkhead box O1, a core decidualization marker in endometrial stromal cells (55, 56). Activation of the AKT/mechanistic target of rapamycin kinase (mTOR) pathway promotes endometrial stromal cells to secret stromal cell-derived factor-1 (SDF-1), which enhances the migration and recruitment of endothelial progenitor cells in the SDF-1-C-X-C motif chemokine receptor 4 (CXCR4) axis (57). Moreover, endometrial fibroblasts exhibit pro-inflammatory characteristics, with activated IL-8, cellular communication network factor 2, and transgelin expression, which organize cell migration and the extracellular matrix (58). The coculture with endometrial stromal cells induced depressed cytotoxicity and aberrant metabolism of CD8+ T cells via signal transducer and activator of transcription (STAT)1/programmed cell death (PDCD)1 pathway; correspondingly, CD8+ T cells depressed the proliferation of ectopic stromal cells (ESCs) through inhibiting cyclin-dependent kinase (CDK)1/cyclin B (CCNB)1 pathway to arrest the cell cycle and triggered inflammation (59). Hence, in endometriosis, pro-inflammatory pathways are dysregulated in epithelial and stromal cells (60).
The presence of ectopic endometrial implants may cause ongoing inflammation and impaired immune surveillance, which might be responsible for maintaining the dysregulated state and impaired reproductive function (53). Immunocytes account for cytokine activation in developing endometriosis-related infertility (47). Aberrant distribution of immune cell populations, including macrophages, NK cells, T cells, and B cells, is often observed in the peritoneal fluid and eutopic endometrium of patients with endometriosis, resulting in chronic inflammation and an inhospitable environment for embryo implantation, thus contributing to endometriosis-associated infertility (61).
An integrated single-cell reference atlas highlighted the critical function of macrophages in endometriosis (36). The pro-inflammatory M1 phenotype and a propensity for M2 to M1 polarization were observed in the endometrium of patients with endometriosis (62). However, as the disease progresses, macrophages polarize toward the M2 phenotype (63), evidenced by M2 macrophage accumulation in adenomyosis, which enhances the invasion capacity of adenomyotic cells (64). Moreover, M0 to M2 transition caused by gut dysbiosis-derived β-glucuronidase promoted the proliferation and migration of endometrial stromal cells (65). In accordance with M2 polarization, the inverse correlation between aggravating indicators of endometriosis and the expression of pattern recognition receptors, toll-like receptor 4 (TLR4), and receptor of advanced glycation end-products on macrophages indicates the presence of immunosuppression in endometriosis (66). Subsequently, downstream events are activated. Vascular endothelial growth factor (VEGF)-A release and blood vessel formation both increase, facilitating Schwann cell migration and peripheral axon regeneration (67). Interestingly, repolarization of M2 to M1 can inhibit ectopic endometrial cell growth (68).
Infertile patients with endometriosis showed altered expression of macrophages in the endometrium and increased numbers of uterine NK (uNK) cells or plasma cells at higher concentrations of macrophages (69). Activation of insulin like growth factor binding protein 5 (IGFBP5)+ macrophages with pro-inflammatory properties is notably observed in ovarian endometrioma (58). Increased serum levels of TNF-α, IL-6, and IL-1β, released from disease-associated macrophages, were observed in patients with endometriosis-induced infertility (70, 71), creating a feedforward loop to aggravate endothelial cell activation. IL-1β inhibits ERα and PRs in primary human endometrial stromal cells via activation of the ERK1/2 pathway, ultimately inhibiting decidualization (72). Administration of an anti-IL-1 receptor 1 (IL-1R1) antibody or IL-1R1-associated kinase 4 inhibitor limited endometrial lesions or suppressed lesion formation (73). Anakinra, an IL-1 inhibitor and an FDA-approved injectable medication to treat rheumatoid arthritis, was proposed to reduce pelvic pain caused by endometriosis in a human pilot study; however, no results were posted after the study (74). TNF-α facilitates endometrial cell proliferation in ectopic environments (75). Peri-implantation treatment with a TNF-α inhibitor increased the implantation and clinical pregnancy rates, while the ongoing pregnancy and live birth rates were not significantly different (76). However, the safety of TNF-α inhibitors on embryos and fetuses has not been fully clarified.
In addition, macrophages regulate ectopic lesion growth and angiogenesis in an autocrine and paracrine manner via extracellular vesicles (77). The endothelial cell-macrophage interaction facilitates epithelial cell proliferation and results in sterile endometrial inflammation (78). Hence, therapeutic strategies, such as suppression of ‘inflammation,’ might dysregulate the cross-regulation of ‘pro- and anti-inflammatory mediators,’ leading to detrimental effects in patients with endometriosis (43).
In the uterus, resident uNK cells play a crucial role in successful pregnancy by generating and secreting angiogenic factors, such as VEGF and angiotensin 2, and promoting revascularization, vasodilation, and neovascularization in the maternal endometrium and trophoblast cell invasion into the endometrium. Thus, defects in uNK cell activity or dysfunction of uNK cells might affect embryo implantation and development, resulting in decreased fertility and adverse pregnancy outcomes. NK cell exhaustion with aberrant upregulation of apoptosis-related genes has been identified in ovarian endometrioma (58). However, the increased number of CD16+ cytotoxic uNK cells observed in the eutopic endometrium of patients with endometriosis exhibit higher toxic activity and produce cytotoxic factors in response to trophoblast cells, possibly leading to infertility (79). Similarly, a concomitant increase of endometrial CD56+ NK cells was found in patients with endometriosis (58). Based on these studies, an altered proportion of uNK cells might also be responsible for an excessive inflammatory and hypoxic environment during embryo implantation or decidualization, thereby increasing the risk of infertility; however, more studies are needed to understand the exact mechanisms by which uNK cells affect fertility.
Elevated levels of IL-17A, an inflammatory cytokine produced by T helper (Th)17 cells, are found in serum and follicular fluid to varying degrees when endometriosis and infertility co-exist (80). As a stimulator of recruitment and activation of immune cells, such as monocytes and neutrophils, upregulation of IL-17 could further activate many downstream cytokines, such as IL-1, IL-6, IL-8, and MCP-1, and participate in the occurrence of endometriosis-related infertility by aggravating inflammation, enhancing toxic effects, and affecting the function of the female reproductive system. Peritoneal fluid-derived IL-17 induces monocytes to differentiate into M2 macrophages (81), contributing to an immunosuppressive environment in the pelvic cavity and promoting the proliferation, growth, invasion, and immune escape of ectopic lesions. The involvement of IL-17A and Th17 cells in the preservation of endometrial cell viability is achieved through the suppression of apoptosis and the inhibition of NK cell-induced cytotoxicity toward endometrial cells, mediated by the ERK1/2 pathway (82). Extracellular vesicles from individuals with endometriosis-associated infertility significantly inhibited the total motility, progressive motility, linear velocity, and the acrosome reaction rate of human sperm by inducing the production of inflammatory cytokines leading to a Th17/regulatory T cell (Treg) imbalance (83).
Dysregulation of T lymphocyte homeostasis, especially deficiency in the number and activity of Tregs, might be one of the causes of endometriosis predisposing to the development of infertility (84). Tregs might induce type 2 immunity in the endometriotic microenvironment, likely suppressing the recruitment of immune cells, which can recognize and target endometrial antigens, contributing to the progression and fibrogenesis of endometriosis (85). Consistently, Th2-skewing occurs in endometriotic lesions, whereas Th1-skewing occurs in peripheral blood and peritoneal fluid (86). Treg dysfunction might exaggerate local inflammation and stimulate angiogenesis by interacting with effector T cells and macrophages, thereby facilitating the attachment and growth of endometrial implants and increasing the risk of reproductive and perinatal complications.
Studies have shown an association between endometriosis and autoimmune diseases (87). Various autoantibodies, including anti-histone H1.2 and anti-alpha 2-HS glycoprotein, were found in the peritoneal fluid or serum in infertile patients with endometriosis, affecting 60% of women with endometriosis-associated infertility (88, 89). Women with recurrent miscarriages have higher levels of anti-laminin-1 antibodies compared with those in healthy controls in the early stages of pregnancy (90). Anti-β2-Glycoprotein I/HLA-DR antibodies might be associated with the pathophysiology of infertility and endometriosis (91). In addition, immune complexes activate the complement system and induce NLR family pyrin domain containing 1 (NLRP1) inflammasome activation to release IL-1β, thus dysregulating pro-inflammatory pathways (60, 92). Complement C1q is the first recognition molecule of the complement classical pathway. High expression of C1q in endometriotic lesions induces a robust proangiogenic effect on endothelial cells that overexpress the receptor for the globular heads of C1q (93). The mechanisms involve linker for activation of T cells family member 2 (LAT2, also known as NTAL and LAB)-mediated calcium mobilization, TGF-β, integrin-mediated cell adhesion, platelet-derived growth factor, and Rac family small GTPase 1 (RAC1)/P21 (RAC1) activated kinase 1 (PAK1)/p38/MMP2 signaling (89). Enriching the LAT2 pathway in activated B cells and monocytes supports the involvement of B cell-mediated autoimmunity in endometriosis-associated infertility (89). It is likely that Th2 polarization at ectopic sites and the consequent generation of IL-4 are also triggering factors for the appearance of these autoantibodies (94). In light of these findings, serum IgA and IgG anti-endometrial antibody levels might predict the pregnancy outcome for patients with endometriosis and tubal factor infertility (95).
Pyroptosis, a novel form of inflammatory cell death, was revealed to have a significant but neglected role in endometriosis via bioinformatic analysis of published data from human and animal models. Pyroptosis exacerbates immune dysfunction by recruiting activated immune cells, including macrophages, neutrophils, central memory CD8+ T cells, and Tregs with unregulated chemokines (96). It is worth noting that the complex inflammatory milieu results from the synergistic activity among different immune cell populations rather than a single role. The question of whether immune dysfunction serves as a pathogenetic mechanism leading to endometriosis-associated infertility or is a consequence of the disease remains unknown.
Various immune cells and the inflammatory cascade are involved in the development and maintenance of endometriosis; therefore, more work is required to determine the mechanisms of endometriosis-related signaling pathways. Several endometriosis-related signaling pathways have been discovered, including nuclear factor kappa B (NF-κB), mitogen-activated protein kinase (MAPK), fibroblast growth factor receptor (FGFR), PI3K/AKT/mTOR, YAP, Wnt/β‐catenin, Rho/Rho associated coiled-coil containing protein kinase (ROCK), and TGF‐β (97). NF-κB plays a crucial role in the immune response as a node in the main inflammation-activated signaling pathway, which facilitates signaling between IL-1 and TNF-α receptors and the production of leukemia inhibitory factor and IL-6 (98). Inhibition of NF-κB signaling reduced endometrial stromal cell proliferation, migration, invasion, EMT, and inflammation in endometriosis (99). The MAPK signaling pathway regulates cell proliferation and invasion, and the Ras-related protein Rap-1 (RAP1) pathway interacts with the MAPK pathway to promote endometriosis development (97). FGFR2 promotes the proliferation, migration, and invasion of ESCs via activation of the ERK signaling pathway in endometriosis: an ERK1/2 inhibitor could counteract the effects of FGFR2 on ESC proliferation and invasion (100). Signaling pathways have been confirmed to be involved in endometriosis; therefore, new strategies should be developed to target these pathways.
Chronic low-grade inflammation can induce oxidative stress. Reactive oxygen species (ROS) derived from cytokines and immunocytes in the follicular fluid microenvironment play a dual role in female reproduction. Altered oxidative stress was observed in patients with endometriosis (101). In addition, differentially expressed genes related to oxidative phosphorylation and mitochondrial function were highlighted for oocyte competence acquisition in cumulus cells of endometriosis (102). Oxidative stress, resulting from the duration and severity of exposure to pro-oxidant molecules and antioxidant defenses, can damage the ovarian reserve and adversely affect oocyte and subsequent embryo development. Elevated systemic oxidative stress can lead to notable fibrinogen oxidation and structural alterations, which are associated with impaired fibrin polymerization and enhanced resistance to plasmin-induced lysis. These changes might increase the thrombotic risk in women with endometriosis, which is detrimental to pregnancy (103).
Oxidative stress plays a key role in the pathogenesis of endometriosis-related infertility. High levels of oxidative stress-sensitive markers, such as advanced oxidation protein products and myeloperoxidase, found in endometriosis patients’ follicular fluid, severely impair mitochondrial metabolism and spindle formation during oocyte meiosis, thereby accelerating follicular depletion, reducing oocyte quality, and lowering fertilization rates (104, 105). High-quality oocytes, the prerequisite for successful pregnancy, are closely related to mitochondrial function and an enzymatic environment that balances intracellular ROS levels (106). However, abnormal mitochondrial accumulation, such as swelling and cristae disruption, is observed in oocytes from women with endometriosis, which would significantly affect energy supply and lead to oocyte stagnation and degeneration during fertilization (107). One explanation might be the elevated expression of prohibitin 1, a highly conserved protein related to mitochondrial function, which leads to increased glucose consumption and lactic acid production, as well as aberrant expression of glycolysis-related enzymes, in the granulosa cells of patients with endometriosis (108). In contrast, ectopic endometrial stromal cells maintain high-energy mitochondrial metabolism through the antioxidant effect of superoxide dismutase overexpression, and promote proliferation and migration in patients with ovarian endometriosis (107). The local microenvironment comprises key constituents to which gametes and preimplantation embryos are exposed (109). High ROS activities and associated oxidative stress in the follicular fluid and serum from patients with endometriosis have the ability to damage gametes and embryos (109). Ovarian endometrioma, one of the major subtypes of endometriosis, leads to a loss of the ovarian cortex-specific stroma and decreased follicle density through occupying compression and local inflammatory invasion, which was confirmed by the presence of early follicular atresia and increased expression of apoptosis-related proteins in ovarian cortical biopsies (110).
A multi-omics study using a mouse model and women with endometriosis revealed the dysregulation of lipid metabolism, identifying 55 upregulated and 67 downregulated metabolites (111). The mechanism involves cellular reactive oxidative stress, cell proliferation inhibition, senescence, apoptosis, and regulation of MAPK-ERK1/2 signaling, leading to a reduced number of mature oocytes (111). Upregulated expression of senescence-associated β-galactosidase can aggravate endoplasmic reticulum stress and differentially affect senescence-associated secretory phenotype factors (IL-1β, MMP-9, and keratinocyte growth factor), which are useful indexes to evaluate the oocyte retrieval number and mature oocyte number. The changes further promote the senescence of ovarian granulosa cells (112). Sphingosine 1-phosphate (S1P) is a metabolite of sphingolipid and an inflammatory mediator. The S1P concentration significantly increased during the non-menstrual phase in patients with endometriosis compared with that in the controls. Further experiments showed that S1P administration increased the size of the endometriotic-like lesion via biasing peritoneal macrophages toward M2 polarization and increasing the expression of IL-6 and mitochondrially encoded cytochrome C oxidase II (113).
Lipid rafts, an acceptor of TLR4, are micro-structure areas on the membrane that are rich in cholesterol. Lipid raft levels increased markedly in uterine tissues after lipopolysaccharide (LPS) treatment, accompanied by the production of inflammatory factors (114). Activation of the liver X receptor-alpha (LxRα)-ATP binding cassette subfamily A member 1 (ABCA1) pathway might account for the outflow of cholesterol (114). Moreover, lipophagy, a special form of autophagy, can deliver the lipid cargo within the lipid droplets to lysosomes for degradation to release free cholesterol (115). In addition, density lipoprotein receptor (LDLR) genes are overexpressed in the foci of deep bowel endometriosis, according to an exploratory case-control study (116), supporting the feasibility of LDLR-targeted therapy in deep endometriosis. Meanwhile, an apolipoprotein E mimetic decreased the lesion size and modified the macrophage phenotype in gain of function experiments (117). Lipid-soluble statins, exemplified by simvastatin, lovastatin, and atorvastatin, effectively inhibit the growth and invasiveness of primary human endometrial stromal cells compared with that of pravastatin, a water-soluble statin (118). Five mg/kg atorvastatin has been shown to regress the ectopic lesions without discernible harm to the ovary in endometriosis mice (119). However, further study is needed to verify the preclinical results.
Extremely high levels of iron are found in peritoneal fluid, ectopic cyst fluid, and ovarian follicles of patients with endometriosis because of numerous lysed erythrocytes in accumulated refluxed menstrual blood (120). In the presence of transferrin insufficiency and iron overload, iron toxicity induces oxidative stress through the Fenton reaction, the most biologically potent ROS reaction, to produce superoxide anions and hydroxyl radicals, resulting in direct damage to follicles, oocytes, sperm, and embryos (121, 122). The iron overload-induced oocyte damage and embryo toxicity might be related to ferroptosis mediated by multiple signaling pathways, including nuclear receptor coactivator 4-dependent ferritinophagy (123), glutathione peroxidase 4-dependent downregulation (124), and heme oxygenase 1 (HMOX1) overexpression (120), in which mitochondrial dysfunction and structural damage play a central role. HMOX1 regulates the delicate balance of iron-induced oxidative stress in endometriotic cyst fluid (125). However, when ROS production exceeds the capacity of cellular antioxidant defenses to remove these toxic agents, oxidative stress occurs, as confirmed by elevated specific biomarkers of oxidative damage in cyst fluid, including lactose dehydrogenase, lipid peroxidation, and 8-hydroxy-2’-deoxyguanosine (126). This explains why the severity of endometriosis-related infertility can be predicted to some extent by measuring iron levels in ectopic cysts. Moreover, iron overload-induced gasdermin E (GSDME)-mediated pyroptosis can trigger the activation and release of IL-16, which drives inflammation in ovarian endometriosis through CD4+ T cells. Indeed, compound Z30702029 inhibited GSDME-N-terminal domain-mediated pyroptosis in preclinical experiments (127).
As mentioned above, it is reasonable to conclude that a pro-oxidant microenvironment in the local vicinity of the follicle adversely affects the quality of oocytes, sperm, and embryos. The mobilization of potent antioxidants might protect them from oxidative damage. Coenzyme Q10 improved the in vitro maturation of oocytes exposed to follicular fluid from patients with endometriosis (128). The administration of vitamins E and C significantly decreased the myeloperoxidase levels in follicular fluid, but not plasma, in moderate/severe endometriosis (105). Rapamycin, used as an anti-senescence drug, could elevate in vitro fertilization (IVF) outcomes, including embryo transfer success and fertility rates via IVF (129). Thus, antioxidant stress might be a promising strategy to target oxidative stress to inhibit endometriotic lesion progression and alleviate endometriosis-associated symptoms, including infertility.
Although the onset of endometriosis-associated symptoms often occurs during adolescence and adulthood, the disease occurs across the life course. Early assessment of risk factors associated with endometriosis could help clinicians to make a diagnosis and reduce diagnostic delay.
An updated understanding of the pathogenesis of endometriosis is needed to develop medical approaches. In recent years, researchers have progressively elucidated the mechanisms underlying endometriosis-related infertility, summarizing multiple factors that influence patient fertility. Firstly, estrogen dominance and progesterone resistance, along with abnormal steroidogenesis, are hallmark features of endometriosis-associated infertility. Additionally, dysregulation of the HPA axis contributes to elevated serum cortisol and prolactin levels in patients with endometriosis. Secondly, ectopic lesions facilitate chronic inflammation, creating an unfavorable environment that is associated with the release of various inflammatory factors. The latter stimulates angiogenesis and induces the further spread of the lesions. Furthermore, interactions among endocrine functions and immune responses play a crucial role. Therefore, maintaining the balance among these systems is essential for optimal endometrial function. The GM, as a central regulator of inflammatory and proliferative conditions, is involved crucially in the onset and progression of endometriosis. The disease involves a multi-system disorder and hormone dysregulation, which might open the door for inter-organ crosstalk mediated by the GM-endocrine system for endometriosis (130, 131). A genetic correlation between endometriosis and gastrointestinal disorders was reported for the co-existence of endometriosis and gastrointestinal symptoms. Based on the shared pathogenesis, including estrogen regulation and inflammation, potential therapeutic drug targets (cholecystokinin B receptor and phosphodiesterase 4B) were identified, providing evidence of shared disease etiology and implicating clinical diagnostic and treatment decisions for both diseases (131). More studies are required to further explore the connection between endometriosis and gastrointestinal disorders and the novel view of the pathogenesis of endometriosis. Thirdly, an unbalanced redox state generates a pro-oxidative microenvironment around follicles, negatively impacting oocyte quality, and sperm and embryo viability. Iron-induced oxidative stress and ferroptosis within granulosa cells lead to suboptimal oocyte development—a focus area for current research endeavors.
Endometriosis is a heterogeneous disease with a complex pathogenesis; therefore, a single-molecular model is insufficient to explain endometriosis-related infertility. A complex ex vivo endometrial model was developed to recapitulate the organization and properties of the human endometrium using innovative tissue bioengineering methods (132). This preclinical approach is hypothesized to predict individual responses to different types of hormonal treatment, providing a better understanding of their effects. Furthermore, much work has been done to develop biomarkers in blood and endometrial tissue, such as microRNAs, B-cell lymphoma 6 protein, and sirtuin 1 protein immunostaining (133). However, most of the non-surgical diagnostic techniques remain to be developed or at least fully validated.
With improving insights into endometriosis pathophysiology, further advances in treatment strategies will emerge. Can diet or supplements have an impact on endometriosis-associated symptoms? Studies have reported generally positive responses to a range of dietary modifications. Changes in diet might affect symptoms through anti-inflammatory effects and a contribution to a more favorable GM. Selenium intake alleviates LPS-induced endometritis by reducing cholesterol levels (114). Copper’s chelation with ammonium tetrathiomolybdate showed a highly effective antiproliferative and antiangiogenic effect by modulating the oxidative imbalance in a murine model of endometriosis. The results indicate an anti-endometriotic potential as a possible non-hormonal treatment (134). However, most studies were conducted for a short time on small groups of individuals, often without a control group (135). Novel delivery systems, such as mifepristone-loaded nanostructured lipid carriers (NLCs), have been developed as an effective strategy for endometriosis-targeted therapy (136, 137). Furthermore, NLC will benefit certain drugs that are difficult to dissolve (137). A targeted nano delivery system as the carrier of mifepristone could selectively target M2 macrophages highly enriched in ectopic endometrial tissue via the secreted protein acidic and cysteine rich receptor. The targeting strategy reduces side effects by increasing the local drug concentration (68).
Endometriosis is associated with a reduced quality of life, and its heterogeneous presentation impedes diagnosis and treatment. Moreover, current medical management, including surgical and medical methods, is ineffective for a sizable proportion of women (138). Women often endure symptoms without good health treatments because of their complex pathogenesis. Global public health policies are urgently needed to promote awareness and implement multidisciplinary care for patients with endometriosis (139). In particular, self-management strategies should be investigated to improve patients’ overall quality of life.
QQ: Conceptualization, Funding acquisition, Methodology, Project administration, Writing – original draft, Writing – review & editing. YL: Conceptualization, Methodology, Project administration, Writing – original draft, Writing – review & editing. ZC: Conceptualization, Writing – review & editing. ZL: Conceptualization, Writing – review & editing. TZ: Writing – review & editing. JZ: Formal Analysis, Writing – review & editing. YZ: Writing – review & editing. SC: Conceptualization, Project administration, Writing – review & editing. LW: Conceptualization, Funding acquisition, Project administration, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The present study was supported by the National Natural Science Foundation of China (grant no. 82374243) and the Wuhan Business University project (grant no. 2024KB021).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Havers-Borgersen E, Hartwell D, Ekelund C, Butt JH, Østergaard L, Holgersson C, et al. Endometriosis and long-term cardiovascular risk: a nationwide danish study. Eur Heart J. (2024) 45:4734–43. doi: 10.1093/eurheartj/ehae563
2. Koller D, Pathak GA, Wendt FR, Tylee DS, Levey DF, Overstreet C, et al. Epidemiologic and genetic associations of endometriosis with depression, anxiety, and eating disorders. JAMA Netw Open. (2023) 6:e225121. doi: 10.1001/jamanetworkopen.2022.51214
3. Vercellini P, Vigano P, Bandini V, Buggio L, Berlanda N, Somigliana E. Association of endometriosis and adenomyosis with pregnancy and infertility. Fertil Steril. (2023) 119:727–40. doi: 10.1016/j.fertnstert.2023.03.018
4. Vannuccini S, Clemenza S, Rossi M, Petraglia F. Hormonal treatments for endometriosis: the endocrine background. Rev Endocr Metab Disord. (2022) 23:333–55. doi: 10.1007/s11154-021-09666-w
5. Anupa G, Sharma JB, Roy KK, Sengupta J, Ghosh D. An assessment of the multifactorial profile of steroid-metabolizing enzymes and steroid receptors in the eutopic endometrium during moderate to severe ovarian endometriosis. Reprod Biol Endocrinol. (2019) 17:111. doi: 10.1186/s12958-019-0553-0
6. Kumar TR. Rerouting of follicle-stimulating hormone secretion and gonadal function. Fertil Steril. (2023) 119:180–3. doi: 10.1016/j.fertnstert.2022.12.005
7. Sanchez AM, Vanni VS, Bartiromo L, Papaleo E, Zilberberg E, Candiani M, et al. Is the oocyte quality affected by endometriosis? A review of the literature. J Ovarian Res. (2017) 10:43. doi: 10.1186/s13048-017-0341-4
8. Wang Z, Guo S, Xie Y, Tong Y, Qi W, Wang Z. Endometrial expression of errβ and errγ: prognostic significance and cinical correlations in severe endometriosis. Front Endocrinol (Lausanne). (2024) 15:1489097. doi: 10.3389/fendo.2024.1489097
9. Schmitz CR, Souza CA, Genro VK, Matte U, Conto E, Cunha-Filho JS. Lh (trp8arg/ile15thr), lhr (inslq) and fshr (asn680ser) polymorphisms genotypic prevalence in women with endometriosis and infertility. J Assist Reprod Genet. (2015) 32:991–7. doi: 10.1007/s10815-015-0477-3
10. Taylor HS, Kotlyar AM, Flores VA. Endometriosis is a chronic systemic disease: clinical challenges and novel innovations. Lancet. (2021) 397:839–52. doi: 10.1016/s0140-6736(21)00389-5
11. Bulun SE, Yilmaz BD, Sison C, Miyazaki K, Bernardi L, Liu S, et al. Endometriosis. Endocr Rev. (2019) 40:1048–79. doi: 10.1210/er.2018-00242
12. Chadchan SB, Popli P, Liao Z, Andreas E, Dias M, Wang T, et al. A greb1-steroid receptor feedforward mechanism governs differential greb1 action in endometrial function and endometriosis. Nat Commun. (2024) 15:1947. doi: 10.1038/s41467-024-46180-4
13. Matsuzaki S, Murakami T, Uehara S, Canis M, Sasano H, Okamura K. Expression of estrogen receptor alpha and beta in peritoneal and ovarian endometriosis. Fertil Steril. (2001) 75:1198–205. doi: 10.1016/s0015-0282(01)01783-6
14. Han SJ, Jung SY, Wu SP, Hawkins SM, Park MJ, Kyo S, et al. Estrogen receptor B modulates apoptosis complexes and the inflammasome to drive the pathogenesis of endometriosis. Cell. (2015) 163:960–74. doi: 10.1016/j.cell.2015.10.034
15. Fan D, Wang X, Shi Z, Jiang Y, Zheng B, Xu L, et al. Understanding endometriosis from an immunomicroenvironmental perspective. Chin Med J (Engl). (2023) 136:1897–909. doi: 10.1097/CM9.0000000000002649
16. Pepe G, Locati M, Della Torre S, Mornata F, Cignarella A, Maggi A, et al. The estrogen-macrophage interplay in the homeostasis of the female reproductive tract. Hum Reprod Update. (2018) 24:652–72. doi: 10.1093/humupd/dmy026
17. Shan J, Ni Z, Cheng W, Zhou L, Zhai D, Sun S, et al. Gut microbiota imbalance and its correlations with hormone and inflammatory factors in patients with stage 3/4 endometriosis. Arch Gynecol Obstet. (2021) 304:1363–73. doi: 10.1007/s00404-021-06057-z
18. García-Peñarrubia P, Ruiz-Alcaraz AJ, Martínez-Esparza M, Marín P, MaChado-Linde F. Hypothetical roadmap towards endometriosis: prenatal endocrine-disrupting chemical pollutant exposure, anogenital distance, gut-genital microbiota and subclinical infections. Hum Reprod Update. (2020) 26:214–46. doi: 10.1093/humupd/dmz044
19. Ervin SM, Li H, Lim L, Roberts LR, Liang X, Mani S, et al. Gut microbial B-glucuronidases reactivate estrogens as components of the estrobolome that reactivate estrogens. J Biol Chem. (2019) 294:18586–99. doi: 10.1074/jbc.RA119.010950
20. Zhu J, Chen Y, Chen H, Sun Y, Yan L, Zhu M, et al. Comparison of microbial abundance and diversity in uterine and peritoneal fluid in infertile patients with or without endometriosis. BMC Womens Health. (2024) 24:148. doi: 10.1186/s12905-024-02985-5
21. Majumdar A, Siva Venkatesh IP, Basu A. Short-chain fatty acids in the microbiota-gut-brain axis: role in neurodegenerative disorders and viral infections. ACS Chem Neurosci. (2023) 14:1045–62. doi: 10.1021/acschemneuro.2c00803
22. Jones LA, Sun EW, Martin AM, Keating DJ. The ever-changing roles of serotonin. Int J Biochem Cell Biol. (2020) 125:105776. doi: 10.1016/j.biocel.2020.105776
23. Martinez-Ibarra A, Cerbon M, Martinez-Razo LD, Morales-Pacheco M, Torre-Villalvazo I, Kawa S, et al. Impact of dehp exposure on female reproductive health: insights into uterine effects. Environ Toxicol Pharmacol. (2024) 107:104391. doi: 10.1016/j.etap.2024.104391
24. Salliss ME, Farland LV, Mahnert ND, Herbst-Kralovetz MM. The role of gut and genital microbiota and the estrobolome in endometriosis, infertility and chronic pelvic pain. Hum Reprod Update. (2021) 28:92–131. doi: 10.1093/humupd/dmab035
25. Reis FM, Coutinho LM, Vannuccini S, Batteux F, Chapron C, Petraglia F. Progesterone receptor ligands for the treatment of endometriosis: the mechanisms behind therapeutic success and failure. Hum Reprod Update. (2020) 26:565–85. doi: 10.1093/humupd/dmaa009
26. Ding Y, Zhu Q, He Y, Lu Y, Wang Y, Qi J, et al. Induction of autophagy by beclin-1 in granulosa cells contributes to follicular progesterone elevation in ovarian endometriosis. Transl Res. (2021) 227:15–29. doi: 10.1016/j.trsl.2020.06.013
27. Lu R, Zhu J, Li X, Zeng C, Huang Y, Peng C, et al. Erβ-activated linc01018 promotes endometriosis development by regulating the Cdc25c/Cdk1/Cyclinb1 pathway. J Genet Genomics. (2024) 51:617–29. doi: 10.1016/j.jgg.2023.12.012
28. Arosh JA, Lee J, Banu SK. Effects of dual inhibition of Akt and Erk1/2 pathways on endometrial pro-inflammatory, hormonal, and epigenetic microenvironment in endometriosis. Mol Cell Endocrinol. (2022) 539:111446. doi: 10.1016/j.mce.2021.111446
29. Yoo JY, Kim TH, Shin JH, Marquardt RM, Müller U, Fazleabas AT, et al. Loss of mig-6 results in endometrial progesterone resistance via erbb2. Nat Commun. (2022) 13:1101. doi: 10.1038/s41467-022-28608-x
30. Mokhtari T, Irandoost E, Sheikhbahaei F. Stress, pain, anxiety, and depression in endometriosis-targeting glial activation and inflammation. Int Immunopharmacol. (2024) 132:111942. doi: 10.1016/j.intimp.2024.111942
31. Giudice LC. Clinical practice. Endometr N Engl J Med. (2010) 362:2389–98. doi: 10.1056/NEJMcp1000274
32. Inda C, Armando NG, Dos Santos Claro PA, Silberstein S. Endocrinology and the brain: corticotropin-releasing hormone signaling. Endocr Connect. (2017) 6:R99–r120. doi: 10.1530/ec-17-0111
33. Lai TT, Liou CW, Tsai YH, Lin YY, Wu WL. Butterflies in the gut: the interplay between intestinal microbiota and stress. J BioMed Sci. (2023) 30:92. doi: 10.1186/s12929-023-00984-6
34. De Hoyos G, Ramos-Sostre D, Torres-Reveron A, Barros-Cartagena B, Lopez-Rodriguez V, Nieves-Vazquez C, et al. Efficacy of an environmental enrichment intervention for endometriosis: a pilot study. Front Psychol. (2023) 14:1225790. doi: 10.3389/fpsyg.2023.1225790
35. Li W, Wang X, Zhang X, Sun S, Feng J, Aziz AUR. A Mendelian randomization approach to study causal association between four types of endometriosis and immune cells: experimental studies. Int J Surg. (2024) 111:1461–5. doi: 10.1097/js9.0000000000001909
36. Marečková M, Garcia-Alonso L, Moullet M, Lorenzi V, Petryszak R, Sancho-Serra C, et al. An integrated single-cell reference atlas of the human endometrium. Nat Genet. (2024) 56:1925–37. doi: 10.1038/s41588-024-01873-w
37. Rahmioglu N, Mortlock S, Ghiasi M, Møller PL, Stefansdottir L, Galarneau G, et al. The genetic basis of endometriosis and comorbidity with other pain and inflammatory conditions. Nat Genet. (2023) 55:423–36. doi: 10.1038/s41588-023-01323-z
38. Yang S, Wang H, Li D, Li M. An estrogen-NK cells regulatory axis in endometriosis, related infertility, and miscarriage. Int J Mol Sci. (2024) 25:3362. doi: 10.3390/ijms25063362
39. Ruan J, Tian Q, Li S, Zhou X, Sun Q, Wang Y, et al. The il-33-st2 axis plays a vital role in endometriosis via promoting epithelial-mesenchymal transition by phosphorylating B-catenin. Cell Commun Signal. (2024) 22:318. doi: 10.1186/s12964-024-01683-x
40. Kim HG, Lim YS, Hwang S, Kim HY, Moon Y, Song YJ, et al. Di-(2-Ethylhexyl) phthalate triggers proliferation, migration, stemness, and epithelial-mesenchymal transition in human endometrial and endometriotic epithelial cells via the transforming growth factor-B/smad signaling pathway. Int J Mol Sci. (2022) 23:3938. doi: 10.3390/ijms23073938
41. Sandoval P, Jimenez-Heffernan JA, Guerra-Azcona G, Perez-Lozano ML, Rynne-Vidal A, Albar-Vizcaino P, et al. Mesothelial-to-mesenchymal transition in the pathogenesis of post-surgical peritoneal adhesions. J Pathol. (2016) 239:48–59. doi: 10.1002/path.4695
42. Kabil SL, Rashed HE, Mohamed NM, Elwany NE. Parthenolide repressed endometriosis induced surgically in rats: role of Pten/Pi3kinase/Akt/Gsk-3β/B-Catenin signaling in inhibition of epithelial mesenchymal transition. Life Sci. (2023) 331:122037. doi: 10.1016/j.lfs.2023.122037
43. Matsuzaki S, Pouly JL, Canis M. Il-10 is not anti-fibrotic but pro-fibrotic in endometriosis: il-10 treatment of endometriotic stromal cells in vitro promotes myofibroblast proliferation and collagen type I protein expression. Hum Reprod. (2023) 38:14–29. doi: 10.1093/humrep/deac248
44. Santoso B, Rahmawati NY, Sa’adi A, Dwiningsih SR, Annas JY, Tunjungseto A, et al. Elevated peritoneal soluble endoglin and gdf-15 in infertile women with severe endometriosis and pelvic adhesion. J Reprod Immunol. (2021) 146:103343. doi: 10.1016/j.jri.2021.103343
45. Cao X, Yang D, Song M, Murphy A, Parthasarathy S. The pesence of endometrial cells in the peritoneal cavity enhances monocyte recruitment and induces inflammatory cytokines in mice: implications for endometriosis. Fertil Steril. (2004) 82 Suppl 3:999–1007. doi: 10.1016/j.fertnstert.2004.04.040
46. Bouet PE, Chao de la Barca JM, Boucret L, Descamps P, Legendre G, Hachem HE, et al. Elevated levels of monocyte chemotactic protein-1 in the follicular fluid reveals different populations among women with severe endometriosis. J Clin Med. (2020) 9:1306. doi: 10.3390/jcm9051306
47. Jørgensen H, Fedorcsak P, Isaacson K, Tevonian E, Xiao A, Beste M, et al. Endometrial cytokines in patients with and without endometriosis evaluated for infertility. Fertil Steril. (2022) 117:629–40. doi: 10.1016/j.fertnstert.2021.11.024
48. Bergqvist A, Bruse C, Carlberg M, Carlstrom K. Interleukin 1beta, interleukin-6, and tumor necrosis factor-alpha in endometriotic tissue and in endometrium. Fertil Steril. (2001) 75:489–95. doi: 10.1016/s0015-0282(00)01752-0
49. Paktinat S, Hashemi SM, Novin MG, Mohammadi-Yeganeh S, Salehpour S, Karamian A, et al. Seminal exosomes induce interleukin-6 and interleukin-8 secretion by human endometrial stromal cells. Eur J Obstet Gyn R B. (2019) 235:71–6. doi: 10.1016/j.ejogrb.2019.02.010
50. Ali R, Ahmed Khan T, Gul H, Rehman R. An interplay of progesterone, leukemia inhibitor factor and interleukin-6 in the window of implantation; impact on fertility. Cytokine. (2023) 170:156332. doi: 10.1016/j.cyto.2023.156332
51. Borojerdi ASD, Welchowski T, Peng WM, Buchen A, Novak N, Haidl G, et al. Human spermatozoa of male patients with subfertility express the interleukin-6 receptor. Andrologia. (2020) 52:e13511. doi: 10.1111/and.13511
52. Gica N, Panaitescu AM, Iancu G, Botezatu R, Peltecu G, Gica C. The role of biological markers in predicting infertility associated with non-obstructive endometriosis. Ginekol Pol. (2020) 91:189–92. doi: 10.5603/gp.2020.0039
53. Bordon Y. Cxcl8 blockade reduces fibrosis in endometriosis. Nat Rev Immunol. (2023) 23:203. doi: 10.1038/s41577-023-00861-1
54. Lin SC, Li WN, Lin SC, Hou HT, Tsai YC, Lin TC, et al. Targeting Yap1 ameliorates progesterone resistance in endometriosis. Hum Reprod. (2023) 38:1124–34. doi: 10.1093/humrep/dead071
55. Adiguzel D, Celik-Ozenci C. Foxo1 is a cell-specific core transcription factor for endometrial remodeling and homeostasis during menstrual cycle and early pregnancy. Hum Reprod Update. (2021) 27:570–83. doi: 10.1093/humupd/dmaa060
56. Li X, Jin J, Long X, Weng R, Xiong W, Liang J, et al. Mettl3-regulated m6a modification impairs the decidualization of endometrial stromal cells by regulating ythdf2-mediated degradation of foxo1 mrna in endometriosis-related infertility. Reprod Biol Endocrinol. (2023) 21:99. doi: 10.1186/s12958-023-01151-0
57. Zhao R, Feng D, Zhuang G, Liu Y, Chi S, Zhang J, et al. Protein kinase ck2 participates in estrogen-mediated endothelial progenitor cell homing to endometriotic lesions through stromal cells in a stromal cell-derived factor-1- cxcr4-dependent manner. Fertil Steril. (2020) 113:1067–79.e5. doi: 10.1016/j.fertnstert.2019.12.035
58. Wu J, Xia S, Ye W, Sun Y, Cai J, Yu F, et al. Dissecting the cell microenvironment of ovarian endometrioma through single-cell rna sequencing. Sci China Life Sci. (2024) 68:116–29. doi: 10.1007/s11427-024-2638-9
59. Huang ZX, Lin DC, Zhang HY, Yang MJ, Chen JH, Ding XY, et al. The dysfunction of cd8(+) T cells triggered by endometriotic stromal cells promotes the immune survival of endometriosis. Immunology. (2024) 172:469–85. doi: 10.1111/imm.13786
60. Fonseca MAS, Haro M, Wright KN, Lin X, Abbasi F, Sun J, et al. Single-cell transcriptomic analysis of endometriosis. Nat Genet. (2023) 55:255–67. doi: 10.1038/s41588-022-01254-1
61. Huang X, Wu L, Pei T, Liu D, Liu C, Luo B, et al. Single-cell transcriptome analysis reveals endometrial immune microenvironment in minimal/mild endometriosis. Clin Exp Immunol. (2023) 212:285–95. doi: 10.1093/cei/uxad029
62. Vallve-Juanico J, Santamaria X, Vo KC, Houshdaran S, Giudice LC. Macrophages display proinflammatory phenotypes in the eutopic endometrium of women with endometriosis with relevance to an infectious etiology of the disease. Fertil Steril. (2019) 112:1118–28. doi: 10.1016/j.fertnstert.2019.08.060
63. Laganà AS, Salmeri FM, Ban Frangež H, Ghezzi F, Vrtačnik-Bokal E, Granese R. Evaluation of M1 and M2 macrophages in ovarian endometriomas from women affected by endometriosis at different stages of the disease. Gynecol Endocrinol. (2020) 36:441–4. doi: 10.1080/09513590.2019.1683821
64. Stratopoulou CA, Cussac S, d’Argent M, Donnez J, Dolmans MM. M2 Macrophages enhance endometrial cell invasiveness by promoting collective cell migration in uterine adenomyosis. Reprod BioMed Online. (2023) 46:729–38. doi: 10.1016/j.rbmo.2023.01.001
65. Wei Y, Tan H, Yang R, Yang F, Liu D, Huang B, et al. Gut dysbiosis-derived B-glucuronidase promotes the development of endometriosis. Fertil Steril. (2023) 120:682–94. doi: 10.1016/j.fertnstert.2023.03.032
66. Shiraishi T, Ikeda M, Watanabe T, Negishi Y, Ichikawa G, Kaseki H, et al. Downregulation of pattern recognition receptors on macrophages involved in aggravation of endometriosis. Am J Reprod Immunol. (2024) 91:e13812. doi: 10.1111/aji.13812
67. Tao Y, Wang F, Xu Z, Lu X, Yang Y, Wu J, et al. Gasdermin D in peripheral nerves: the pyroptotic microenvironment inhibits nerve regeneration. Cell Death Discovery. (2021) 7:144. doi: 10.1038/s41420-021-00529-6
68. Zhang M, Ye Y, Chen Z, Wu X, Chen Y, Zhao P, et al. Targeting delivery of mifepristone to endometrial dysfunctional macrophages for endometriosis therapy. Acta Biomater. (2024) 189:505–18. doi: 10.1016/j.actbio.2024.09.037
69. Freitag N, Baston-Buest DM, Kruessel JS, Markert UR, Fehm TN, Bielfeld AP. Eutopic endometrial immune profile of infertility-patients with and without endometriosis. J Reprod Immunol. (2022) 150:103489. doi: 10.1016/j.jri.2022.103489
70. Rostami S, Alyasin A, Saedi M, Nekoonam S, Khodarahmian M, Moeini A, et al. Astaxanthin ameliorates inflammation, oxidative stress, and reproductive outcomes in endometriosis patients undergoing assisted reproduction: a randomized, triple-blind placebo-controlled clinical trial. Front Endocrinol (Lausanne). (2023) 14:1144323. doi: 10.3389/fendo.2023.1144323
71. Lv H, Liu B, Dai Y, Li F, Bellone S, Zhou Y, et al. Tet3-overexpressing macrophages promote endometriosis. J Clin Invest. (2024) 134:e181839. doi: 10.1172/jci181839
72. Yu J, Berga SL, Zou W, Taylor RN. Interleukin-1β inhibits estrogen receptor-A, progesterone receptors A and B and biomarkers of human endometrial stromal cell differentiation: implications for endometriosis. Mol Hum Reprod. (2019) 25:625–37. doi: 10.1093/molehr/gaz045
73. Kato T, Yasuda K, Matsushita K, Ishii KJ, Hirota S, Yoshimoto T, et al. Interleukin-1/-33 signaling pathways as therapeutic targets for endometriosis. Front Immunol. (2019) 10:2021. doi: 10.3389/fimmu.2019.02021
74. Agarwal S. Pilot study of the il-1 antagonist anakinra for the treatment of endometriosis related symptoms (2021-5-28) [2025-01-23. Available online at: https://clinicaltrials.gov/study/NCT03991520 (Accessed January 23, 2025).
75. Braun DP, Ding J, Dmowski WP. Peritoneal fluid-mediated enhancement of eutopic and ectopic endometrial cell proliferation is dependent on tumor necrosis factor-alpha in women with endometriosis. Fertil Steril. (2002) 78:727–32. doi: 10.1016/s0015-0282(02)03318-6
76. Liu M, Li Y, Yuan Y, Jiang M, Yin P, Yang D. Peri-implantation treatment with tnf-a inhibitor for endometriosis and/or adenomyosis women undergoing frozen-thawed embryo transfer: a retrospective cohort study. J Reprod Immunol. (2024) 167:104415. doi: 10.1016/j.jri.2024.104415
77. Gao X, Gao H, Shao W, Wang J, Li M, Liu S. The extracellular vesicle-macrophage regulatory axis: a novel pathogenesis for endometriosis. Biomolecules. (2023) 13:1376. doi: 10.3390/biom13091376
78. Pan J, Qu J, Fang W, Zhao L, Zheng W, Zhai L, et al. Shp2-triggered endothelial cell activation fuels estradiol-independent endometrial sterile inflammation. Adv Sci (Weinh). (2024) 11:e2403038. doi: 10.1002/advs.202403038
79. Quan Q, Gu H, Wang Y, Yu M. Immune micro-environment analysis and drug screening for ovarian endometriosis. Genes Genomics. (2024) 46:803–15. doi: 10.1007/s13258-024-01497-8
80. Jiang Y, Wang L, Peng Y, Qin J, Tan A, Wang S. Interleukin 17 receptor E identifies heterogeneous T helper 17 cells in peritoneal fluid of moderate and severe endometriosis patients. Clin Exp Immunol. (2022) 207:360–9. doi: 10.1093/cei/uxac004
81. Miller JE, Ahn SH, Marks RM, Monsanto SP, Fazleabas AT, Koti M, et al. Il-17a modulates peritoneal macrophage recruitment and M2 polarization in endometriosis. Front Immunol. (2020) 11:108. doi: 10.3389/fimmu.2020.00108
82. Kang YJ, Cho HJ, Lee Y, Park A, Kim MJ, Jeung IC, et al. Il-17a and Th17 cells contribute to endometrial cell survival by inhibiting apoptosis and NK cell mediated cytotoxicity of endometrial cells via erk1/2 pathway. Immune Netw. (2023) 23:e14. doi: 10.4110/in.2023.23.e14
83. Zhang Z, Xiong Y, Jiang H, Wang Q, Hu X, Wei X, et al. Vaginal extracellular vesicles impair fertility in endometriosis by favoring Th17/Treg imbalance and inhibiting sperm activity. J Cell Physiol. (2024) 239:e31188. doi: 10.1002/jcp.31188
84. Knez J, Kovačič B, Goropevšek A. The role of regulatory T-cells in the development of endometriosis. Hum Reprod. (2024) 39:1367–80. doi: 10.1093/humrep/deae103
85. Xiao F, Liu X, Guo SW. Platelets and regulatory T cells may induce a type 2 immunity that is conducive to the progression and fibrogenesis of endometriosis. Front Immunol. (2020) 11:610963. doi: 10.3389/fimmu.2020.610963
86. Chang LY, Shan J, Hou XX, Li DJ, Wang XQ. Synergy between Th1 and Th2 responses during endometriosis: A Review of Current Understanding. J Reprod Immunol. (2023) 158:103975. doi: 10.1016/j.jri.2023.103975
87. Shigesi N, Kvaskoff M, Kirtley S, Feng Q, Fang H, Knight JC, et al. The association between endometriosis and autoimmune diseases: a systematic review and meta-analysis. Hum Reprod Update. (2019) 25:486–503. doi: 10.1093/humupd/dmz014
88. Kolanska K, Alijotas-Reig J, Cohen J, Cheloufi M, Selleret L, d’Argent E, et al. Endometriosis with infertility: a comprehensive review on the role of immune deregulation and immunomodulation therapy. Am J Reprod Immunol. (2021) 85:e13384. doi: 10.1111/aji.13384
89. Harden S, Tan TY, Ku CW, Zhou J, Chen Q, Chan JKY, et al. Peritoneal autoantibody profiling identifies P53 as an autoantibody target in endometriosis. Fertil Steril. (2023) 120:176–87. doi: 10.1016/j.fertnstert.2023.02.025
90. Caccavo D, Pellegrino NM, Nardelli C, Vergine S, Leone L, Marolla A, et al. Anti-laminin-1 antibodies in serum and follicular fluid of women with Hashimoto’s thyroiditis undergoing in vitro fertilization. Int J Immunopathol Pharmacol. (2016) 29:280–7. doi: 10.1177/0394632015627281
91. Ono Y, Wada S, Ota H, Fukushi Y, Tanimura K, Yoshino O, et al. Anti-beta2-glycoprotein I/hla-dr antibody in infertility. J Reprod Immunol. (2023) 158:103955. doi: 10.1016/j.jri.2023.103955
92. Titiz M, Landini L, Souza Monteiro de Araujo D, Marini M, Seravalli V, Chieca M, et al. Schwann cell C5ar1 co-opts inflammasome nlrp1 to sustain pain in a mouse model of endometriosis. Nat Commun. (2024) 15:10142. doi: 10.1038/s41467-024-54486-6
93. Agostinis C, Toffoli M, Zito G, Balduit A, Pegoraro S, Spazzapan M, et al. Proangiogenic properties of complement protein c1q can contribute to endometriosis. Front Immunol. (2024) 15:1405597. doi: 10.3389/fimmu.2024.1405597
94. Agostinis C, Balduit A, Mangogna A, Zito G, Romano F, Ricci G, et al. Immunological basis of the endometriosis: the complement system as a potential therapeutic target. Front Immunol. (2020) 11:599117. doi: 10.3389/fimmu.2020.599117
95. Sarapik A, Haller-Kikkatalo K, Utt M, Teesalu K, Salumets A, Uibo R. Serum anti-endometrial antibodies in infertile women - potential risk factor for implantation failure. Am J Reprod Immunol. (2010) 63:349–57. doi: 10.1111/j.1600-0897.2010.00808.x
96. Guo Q, Zhou C, Xiang Y, Liang X. Pyroptosis orchestrates immune responses in endometriosis. Int Immunopharmacol. (2023) 118:110141. doi: 10.1016/j.intimp.2023.110141
97. Zhang M, Xu T, Tong D, Li S, Yu X, Liu B, et al. Research advances in endometriosis-related signaling pathways: a review. BioMed Pharmacother. (2023) 164:114909. doi: 10.1016/j.biopha.2023.114909
98. Laird SM, Tuckerman EM, Cork BA, Li TC. Expression of nuclear factor kappa B in human endometrium; role in the control of interleukin 6 and leukaemia inhibitory factor production. Mol Hum Reprod. (2000) 6:34–40. doi: 10.1093/molehr/6.1.34
99. Wu M, Zhang Y. Mir-182 inhibits proliferation, migration, invasion and inflammation of endometrial stromal cells through deactivation of nf-κb signaling pathway in endometriosis. Mol Cell Biochem. (2021) 476:1575–88. doi: 10.1007/s11010-020-03986-2
100. Xu Y, Gao F, Zhang J, Cai P, Xu D. Fibroblast growth factor receptor 2 promotes the proliferation, migration, and invasion of ectopic stromal cells via activation of extracellular-signal-regulated kinase signaling pathway in endometriosis. Bioengineered. (2022) 13:8360–71. doi: 10.1080/21655979.2022.2054207
101. Da Broi MG, Ferreira EM, Andrade AZ, Jordão AA, Ferriani RA, Navarro PA. The impact of controlled ovarian stimulation on serum oxidative stress markers in infertile women with endometriosis undergoing icsi. Antioxid (Basel). (2022) 11:1161. doi: 10.3390/antiox11061161
102. Da Luz CM, Da Broi MG, Plaça JR, Silva WA Jr., Meola J, Navarro PA. Altered transcriptome in cumulus cells of infertile women with advanced endometriosis with and without endometrioma. Reprod BioMed Online. (2021) 42:952–62. doi: 10.1016/j.rbmo.2021.01.024
103. Fini E, Argento FR, Borghi S, Giurranna E, Nencini F, Cirillo M, et al. Fibrinogen structural changes and their potential role in endometriosis-related thrombosis. Antioxid (Basel). (2024) 13:1456. doi: 10.3390/antiox13121456
104. Song Y, Liu J, Qiu Z, Chen D, Luo C, Liu X, et al. Advanced oxidation protein products from the follicular microenvironment and their role in infertile women with endometriosis. Exp Ther Med. (2018) 15:479–86. doi: 10.3892/etm.2017.5390
105. Santanam N, Zoneraich N, Parthasarathy S. Myeloperoxidase as a potential target in women with endometriosis undergoing IVF. Reprod Sci. (2017) 24:619–26. doi: 10.1177/1933719116667225
106. Cacciottola L, Donnez J, Dolmans MM. Oxidative stress, mitochondria, and infertility: is the relationship fully established? Fertil Steril. (2021) 116:306–8. doi: 10.1016/j.fertnstert.2021.04.026
107. Chen C, Zhou Y, Hu C, Wang Y, Yan Z, Li Z, et al. Mitochondria and oxidative stress in ovarian endometriosis. Free Radic Biol Med. (2019) 136:22–34. doi: 10.1016/j.freeradbiomed.2019.03.027
108. Mao J, Zhang J, Cai L, Cui Y, Liu J, Mao Y. Elevated prohibitin 1 expression mitigates glucose metabolism defects in granulosa cells of infertile patients with endometriosis. Mol Hum Reprod. (2022) 28:gaac018. doi: 10.1093/molehr/gaac018
109. Prieto L, Quesada JF, Cambero O, Pacheco A, Pellicer A, Codoceo R, et al. Analysis of follicular fluid and serum markers of oxidative stress in women with infertility related to endometriosis. Fertil Steril. (2012) 98:126–30. doi: 10.1016/j.fertnstert.2012.03.052
110. Kitajima M, Dolmans MM, Donnez O, Masuzaki H, Soares M, Donnez J. Enhanced follicular recruitment and atresia in cortex derived from ovaries with endometriomas. Fertil Steril. (2014) 101:1031–7. doi: 10.1016/j.fertnstert.2013.12.049
111. Dai Y, Lin X, Liu N, Shi L, Zhuo F, Huang Q, et al. Integrative analysis of transcriptomic and metabolomic profiles reveals abnormal phosphatidylinositol metabolism in follicles from endometriosis-associated infertility patients. J Pathol. (2023) 260:248–60. doi: 10.1002/path.6079
112. Lin X, Dai Y, Tong X, Xu W, Huang Q, Jin X, et al. Excessive oxidative stress in cumulus granulosa cells induced cell senescence contributes to endometriosis-associated infertility. Redox Biol. (2020) 30:101431. doi: 10.1016/j.redox.2020.101431
113. Ono Y, Kawakita T, Yoshino O, Sato E, Kano K, Ohba M, et al. Sphingosine 1-phosphate (S1p) in the peritoneal fluid skews M2 macrophage and contributes to the development of endometriosis. Biomedicines. (2021) 9:1519. doi: 10.3390/biomedicines9111519
114. Chen Y, Zhao YF, Yang J, Jing HY, Liang W, Chen MY, et al. Selenium alleviates lipopolysaccharide-induced endometritis via regulating the recruitment of tlr4 into lipid rafts in mice. Food Funct. (2020) 11:200–10. doi: 10.1039/c9fo02415h
115. Esmaeilian Y, Hela F, Bildik G, İltumur E, Yusufoglu S, Yildiz CS, et al. Autophagy regulates sex steroid hormone synthesis through lysosomal degradation of lipid droplets in human ovary and testis. Cell Death Dis. (2023) 14:342. doi: 10.1038/s41419-023-05864-3
116. Gibran L, Maranhão RC, Tavares ER, Carvalho PO, Abrão MS, Podgaec S. Mrna levels of low-density lipoprotein receptors are overexpressed in the foci of deep bowel endometriosis. Hum Reprod. (2017) 32:332–9. doi: 10.1093/humrep/dew303
117. Henlon Y, Panir K, McIntyre I, Hogg C, Dhami P, Cuff AO, et al. Single-cell analysis identifies distinct macrophage phenotypes associated with prodisease and proresolving functions in the endometriotic niche. Proc Natl Acad Sci U.S.A. (2024) 121:e2405474121. doi: 10.1073/pnas.2405474121
118. Sokalska A, Hawkins AB, Yamaguchi T, Duleba AJ. Lipophilic statins inhibit growth and reduce invasiveness of human endometrial sromal cells. J Assist Reprod Genet. (2019) 36:535–41. doi: 10.1007/s10815-018-1352-9
119. Qin X, Wang Q, Xu D, Sun Y, Xu W, Wang B, et al. Atorvastatin exerts dual effects of lesion regression and ovarian protection in the prevention and treatment of endometriosis. Eur J Pharmacol. (2024) 964:176261. doi: 10.1016/j.ejphar.2023.176261
120. Li S, Zhou Y, Huang Q, Fu X, Zhang L, Gao F, et al. Iron overload in endometriosis peritoneal fluid induces early embryo ferroptosis mediated by hmox1. Cell Death Discovery. (2021) 7:355. doi: 10.1038/s41420-021-00751-2
121. Wu Y, Yang R, Lan J, Wu Y, Huang J, Fan Q, et al. Iron overload modulates follicular microenvironment via ros/hif-1alpha/fshr signaling. Free Radic Biol Med. (2023) 196:37–52. doi: 10.1016/j.freeradbiomed.2022.12.105
122. Hayashi S, Nakamura T, Motooka Y, Ito F, Jiang L, Akatsuka S, et al. Novel ovarian endometriosis model causes infertility via iron-mediated oxidative stress in mice. Redox Biol. (2020) 37:101726. doi: 10.1016/j.redox.2020.101726
123. Ni Z, Li Y, Song D, Ding J, Mei S, Sun S, et al. Iron-overloaded follicular fluid increases the risk of endometriosis-related infertility by triggering granulosa cell ferroptosis and oocyte dysmaturity. Cell Death Dis. (2022) 13:579. doi: 10.1038/s41419-022-05037-8
124. Chen X, Zhou Y, Wu D, Shu C, Wu R, Li S, et al. Iron overload compromises preimplantation mouse embryo development. Reprod Toxicol. (2021) 105:156–65. doi: 10.1016/j.reprotox.2021.08.010
125. Imanaka S, Yamada Y, Kawahara N, Kobayashi H. A delicate redox balance between iron and heme oxygenase-1 as an essential biological feature of endometriosis. Arch Med Res. (2021) 52:641–7. doi: 10.1016/j.arcmed.2021.03.006
126. Wyatt J, Fernando SM, Powell SG, Hill CJ, Arshad I, Probert C, et al. The role of iron in the pathogenesis of endometriosis: a systematic review. Hum Reprod Open. (2023) 2023:hoad033. doi: 10.1093/hropen/hoad033
127. Zhang J, Zhao W, Zhou Y, Xi S, Xu X, Du X, et al. Pyroptotic T cell-derived active il-16 has a driving function in ovarian endometriosis development. Cell Rep Med. (2024) 5:101476. doi: 10.1016/j.xcrm.2024.101476
128. Romero S, Pella R, Zorrilla I, Berrío P, Escudero F, Pérez Y, et al. Coenzyme q10 improves the in vitro maturation of oocytes exposed to the intrafollicular environment of patients on fertility treatment. JBRA Assist Reprod. (2020) 24:283–8. doi: 10.5935/1518-0557.20200003
129. Fan J, Chen C, Zhong Y. A cohort study on ivf outcomes in infertile endometriosis patients: the effects of rapamycin treatment. Reprod BioMed Online. (2024) 48:103319. doi: 10.1016/j.rbmo.2023.103319
130. Bany Bakar R, Reimann F, Gribble FM. The intestine as an endocrine organ and the role of gut hormones in metabolic regulation. Nat Rev Gastroenterol Hepatol. (2023) 20:784–96. doi: 10.1038/s41575-023-00830-y
131. Yang F, Wu Y, Hockey R, Doust J, Mishra GD, Montgomery GW, et al. Evidence of shared genetic factors in the etiology of gastrointestinal disorders and endometriosis and clinical implications for disease management. Cell Rep Med. (2023) 4:101250. doi: 10.1016/j.xcrm.2023.101250
132. Lyon H. Developing a complex ex vivo endometrial tissue model to improve endometriosis care (Otendo) (2024-07-15) [2025-01-27. Available online at: https://clinicaltrials.gov/study/NCT06331676?cond=Endometriosis&aggFilters=sex:f&term=IL&rank=2 (Accessed January 27, 2025).
133. Young SL. Nonsurgical approaches to the diagnosis and evaluation of endometriosis. Fertil Steril. (2024) 121:140–4. doi: 10.1016/j.fertnstert.2023.12.020
134. Delsouc MB, Conforti RA, Vitale DL, Alaniz L, Pacheco P, Andujar S, et al. Antiproliferative and antiangiogenic effects of ammonium tetrathiomolybdate in a model of endometriosis. Life Sci. (2021) 287:120099. doi: 10.1016/j.lfs.2021.120099
135. Griffiths MJ, Horne AW, Gibson DA, Roberts N, Saunders PTK. Endometriosis: recent advances that could accelerate diagnosis and improve care. Trends Mol Med. (2024) 30:875–89. doi: 10.1016/j.molmed.2024.06.008
136. Yuan M, Ding S, Meng T, Lu B, Shao S, Zhang X, et al. Effect of a-317491 delivered by glycolipid-like polymer micelles on endometriosis pain. Int J Nanomed. (2017) 12:8171–83. doi: 10.2147/ijn.S146569
137. Yan J, Peng Y, Huang S, Fei W, Chen Y, Zheng C, et al. Treatment of endometriosis with mifepristone mediated by nanostructured lipid carriers. Drug Delivery Transl Res. (2025) 15:1181–92. doi: 10.1007/s13346-024-01661-3
138. Zondervan KT, Becker CM, Missmer SA. Endometriosis. N Engl J Med. (2020) 382:1244–56. doi: 10.1056/NEJMra1810764
Keywords: endometriosis, infertility, endocrine, immune dysfunction, oxidative stress
Citation: Qi Q, Li Y, Chen Z, Luo Z, Zhou T, Zhou J, Zhang Y, Chen S and Wang L (2025) Update on the pathogenesis of endometriosis-related infertility based on contemporary evidence. Front. Endocrinol. 16:1558271. doi: 10.3389/fendo.2025.1558271
Received: 10 January 2025; Accepted: 19 February 2025;
Published: 10 March 2025.
Edited by:
Lihua Yang, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Zhenzhong Deng, University of Southern California, United StatesCopyright © 2025 Qi, Li, Chen, Luo, Zhou, Zhou, Zhang, Chen and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ling Wang, ZHIud2FuZ2xpbmdAdmlwLjE2My5jb20=; Song Chen, Y2hlbnNvbmdAaGJ6eXlkeC53ZWNvbS53b3Jr
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.