 Siyuan Liu1†
Siyuan Liu1† Deju Zhang
Deju Zhang Xiaoping Yin
Xiaoping Yin Peng Yu
Peng Yu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 03 April 2025
Sec. Diabetes: Molecular Mechanisms
Volume 16 - 2025 | https://doi.org/10.3389/fendo.2025.1541809
Diabetes is a growing health concern, accompanied by significant complications like cardiovascular disease, kidney disease, and retinopathy. Metal ions, including iron, zinc, and copper, play a crucial role in maintaining human health through their balance within the body. Disruptions in metal ion balance can intensify diabetic conditions. For instance, iron overload induces oxidative stress, which harms islet β cells and impacts vascular complications of diabetes. Abnormal copper levels heighten insulin resistance, and zinc deficiency has a strong connection with type 1 diabetes. Future in - depth exploration of the association between metal metabolism and diabetes holds the potential to uncover novel treatment avenues, enhancing both the quality of life and health prognosis for patients.
The increasing global prevalence of obesity and unhealthy lifestyles has contributed to the rising incidence and prevalence of diabetes-related diseases. The International Diabetes Federation (IDF) reported that, in 2021, 10.5% of adults aged 20 to 79 had diabetes. Among all diabetic individuals, type 1 diabetes mellitus (T1DM) accounts for between 5-15%, and type 2 diabetes accounts for the majority. It is currently estimated that the number of people living with diabetes will increase from 536.6 million to 783.2 million by 2045 (1).
Diabetes can be divided into type 1 diabetes mellitus (T1DM) and type 2 diabetes mellitus (T2DM). The main difference between the two is the different inactivation pathways of pancreatic β cells. T1DM is an autoimmune destruction of beta cells, which usually leads to insulin deficiency, while T2DM is a frequent progressive decrease in beta cell insulin secretion and insulin resistance (2). Based on current research, metal elements play a crucial role in the human body, and several essential metals are required for many enzymes, transcription factors, and proteins to play important roles in various biochemical pathways. In diabetes, metal ions play an indispensable role. For example, chromium can enhance the action of insulin and help lower blood glucose levels (3). Zinc is involved in the synthesis and release of insulin and affects blood glucose regulation (4). Copper plays a role in redox reactions and may affect insulin function (5). The imbalance of these metal ions may lead to the development or aggravation of diabetes. Additionally, the plasma levels of trace metal elements such as magnesium, copper, selenium, and zinc differ between patients with T1DM and T2DM. In T1DM, the plasma magnesium concentration changes most and decreases. Besides, in T2DM, plasma levels of selenium and copper were significantly affected, and the plasma selenium and copper concentrations decreased (6).
Diabetes is associated with high rates of microvascular (neuropathy, nephropathy, and retinopathy) and macrovascular (coronary artery disease, cerebrovascular disease, and peripheral artery disease) complications. There is a close relationship between metal ions and diabetic complications. For example, chromium levels may decrease in hyperglycemic states, which may affect insulin sensitivity and increase cardiovascular disease risk (7). Zinc deficiency may lead to impaired insulin synthesis and secretion, affecting glycemic control (8). The role of copper in diabetic complications is still under investigation, but its involvement in REDOX reactions may affect vascular health and neurological function (9).
Investigating the connection between diabetic complications and metal ions could help us to understand the pathogenesis and offer new targets and strategies for treatment and prevention, thereby enhancing patients’ quality of life and decreasing the complication incidence.
Essential metals in the human body include Na, K, Ca, Mg, Zn, Cu, Fe, Mn, Co, and Mo (10). Metal elements play a vital role in the human body (11) (Table 1). For example, iron ions contribute to the construction of hemoglobin and participate in oxygen transport in the body. Copper ions serve as cofactors for various enzymes, participate in iron transport, and promote angiogenesis. Zinc ions are components of zinc-containing metalloenzymes and zinc finger proteins, and are essential for insulin synthesis. Calcium ions are involved in building and maintaining bone health, maintaining neuromuscular excitability, and regulating the activity of many enzymes.
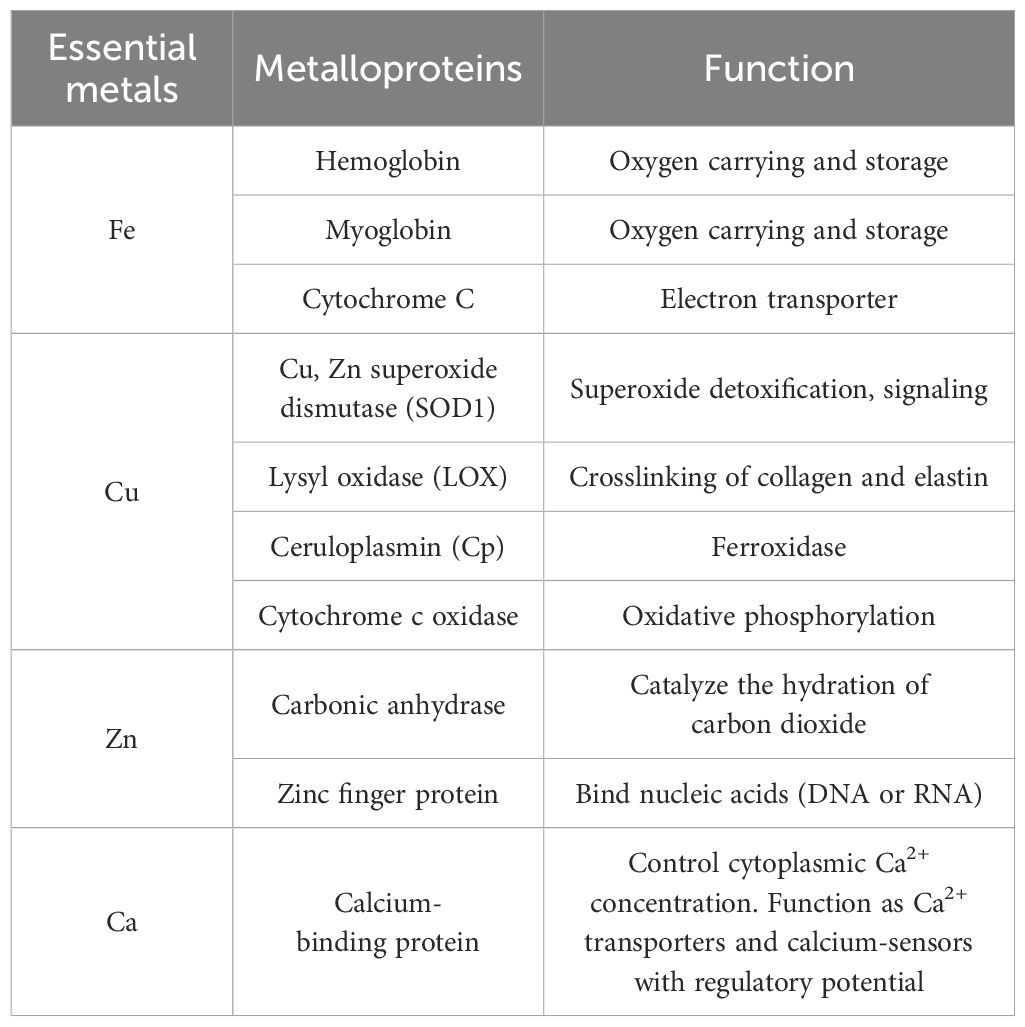
Table 1. Metalloprotein species of essential metals and their functions.
Metal ion metabolism refers to the regulation of the absorption, distribution, storage, and removal of metal ions in cells (12). Intracellular metal ion balance refers to the maintenance process of intracellular metal ion levels within a certain range. The steady-state regulation of metal ions is mainly through accurate absorption and adaptation (such as Zn, Fe, Cu, and Mn). Studies have shown that a critical absorption pathway is mediated by transporters located at the brush border of the intestine, which can selectively collect metal ions. The homeostatic regulation of absorption appears to depend on the expression of these proteins and/or molecules in the following transport line from the cell lumen to the bloodstream. However, renal excretion is less adaptive (Zn), and usually cannot quantitatively promote metabolic regulation (Fe, Cu, Mn) (13). For example, the regulation of iron ion homeostasis, which regulates the binding and dissociation of iron-regulated sequences (IREs) and iron-regulated proteins (IRPs) at the protein synthesis level to regulate the production of ferritin, which in turn maintains iron ion homeostasis (14). During the absorption stage, iron ion homeostasis is also regulated by the intestinal mucosa. Ferritin is an essential mechanism for regulating iron homeostasis in absorbing epithelial cells. When iron overload occurs, newly formed epithelial cells contain ferritin, which binds iron to ferritin. Iron is lost when cells are shed at the end of their life cycle. The iron that bypasses ferritin will be intercepted by macrophages in the villi. When iron is lacking, the absorption cells will lack ferritin, and the entry of dietary iron into the body is relatively unimpeded (15). The precise regulation of copper ions is achieved by metal chaperones. (see 4.1) (the mechanism is shown in Figure 1).
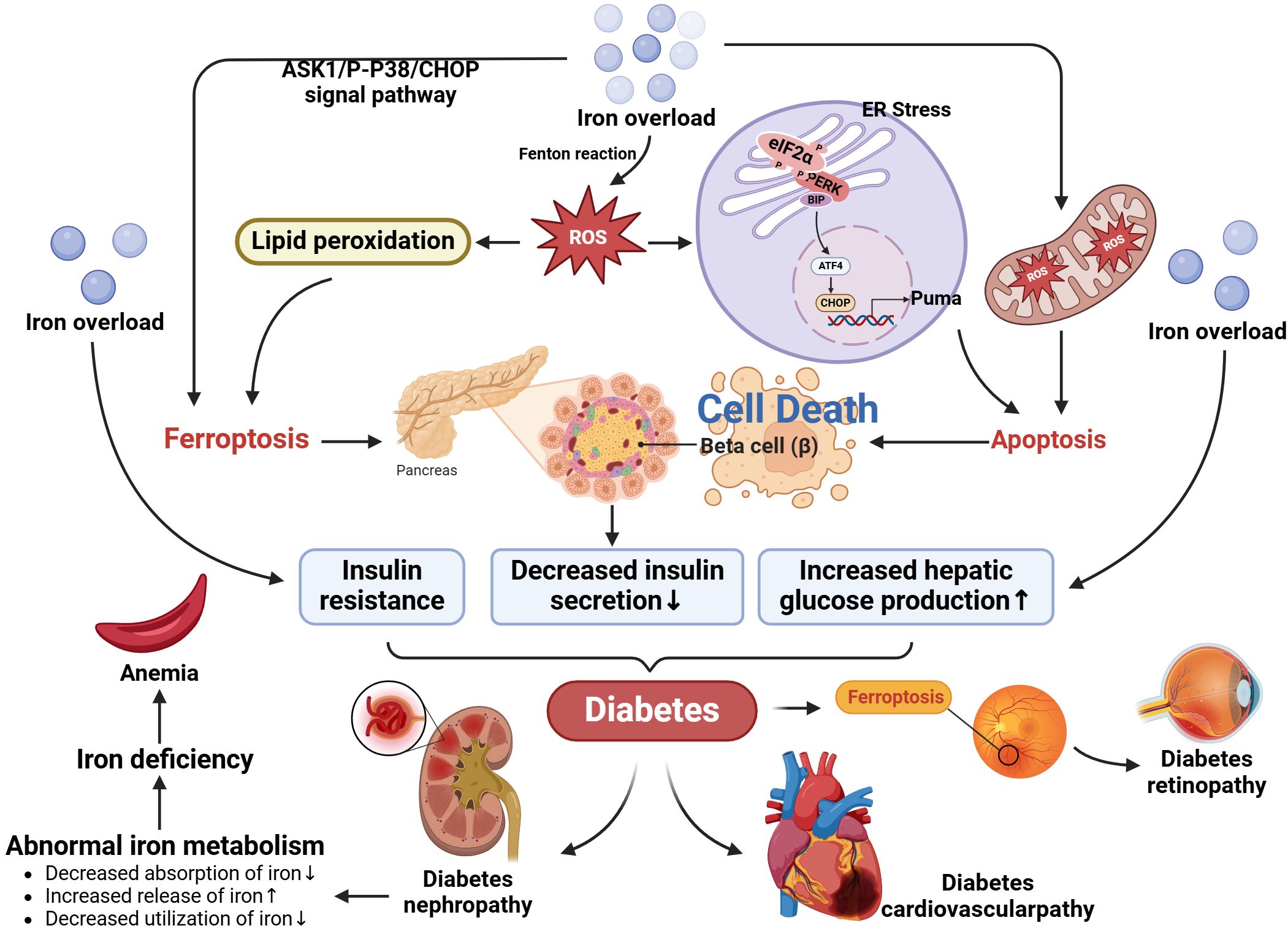
Figure 1. Metal ion metabolism. The main components involved in metal ion metabolism and homeostatic regulation include transporters, metal-blinding chaperones, and metalloproteins.
In addition, the absorbed metal ions will bind to the transporter and be transported to the target site. For example, iron is exported to the circulation through the basolateral membrane of intestinal epithelial cells, bound to transferrin and transported to the sites of utilization and storage. The iron bound with transferrin enters the target cells, primarily red blood cells, through receptor-mediated endocytosis, and iron is stored in hemoglobin and myoglobin (14). At the same time, copper is bound to albumin after intestinal absorption and transported to the liver, and then released into the blood and other tissues. Metallothionein is bound to copper in the liver (16).
If metal ion metabolism is out of balance, it can have a variety of adverse effects on human health, including:
Iron deficiency leads to iron-deficiency anemia, while iron overload can lead to iron deposition in organs such as the heart, liver, and brain, increasing the risk of diabetes, cirrhosis, liver cancer, arrhythmia, heart failure, retinal degeneration, and neurodegenerative diseases (17). Copper deficiency leads to anemia, neutropenia, osteoporosis, and neurological problems, while copper overload leads to gastrointestinal symptoms, cirrhosis, and neurological diseases (18). Zinc deficiency can lead to growth retardation, glossitis, slow wound healing, hair loss, and decreased immunity (19). Calcium deficiency can lead to rickets, osteomalacia, hypertension, atherosclerosis, diabetes, neurodegenerative diseases, degenerative joint disease, etc. Calcium overload can lead to hypercalcemia and urinary calculi (20).
Iron overload (IO) is a hazard factor for diabetes. In the pathogenesis of diabetes, iron has a direct and causal role, which is mediated through β-cell failure and insulin resistance (21). It impacts most of its essential features: decreased insulin resistance, insulin secretion, and increased hepatic glucose production (22). The liver maintains iron homeostasis through heparin (23). Heparin is involved in the storage, transport and release of iron ions in the body by regulating the expression of ferritin and transporter proteins in the liver (24). In addition, heparin also regulates the absorption of iron in the intestine and the recycling of iron in the liver to maintain iron balance in the body (25). According to clinical trials, patients with T2DM have reduced heparin levels and increased plasma iron and ferritin levels (26). On the other hand, recent research has revealed that the accumulation of lipid peroxides and free iron will induce a type of cell death called ferroptosis (27). The mechanism is that free iron catalyzes the Fenton reaction, induces cellular oxidative stress, generates a large quantity of free radicals, and triggers a sequence of cell damage reactions (28). Free radicals will attack beta cells, leading to impaired blood glucose regulation in the human body, and then develop diabetes. According to recent studies, CHOP is one of the ferroptosis inducers related to the activation of the PERK-eIF2α-ATF4 pathway that mediates endoplasmic reticulum stress (29). Iron overload may activate the ASK1/P-P38/CHOP/COX-2 pathway, thereby inducing severe oxidative stress, resulting in the accumulation of numerous lipid peroxides and the imbalance of REDOX status in cells, which in turn induces ferroptosis of β cells (30). Secondly, iron accumulates in hepatocytes and leads to insulin resistance through oxidative stress (31, 32). Its mechanism mainly involves two aspects: oxidative stress and lipid metabolism disorder. Oxidative stress interferes with the structure and function of the insulin receptor and reduces the activity of the insulin receptor, thereby weakening the action of insulin (33). In addition, oxidative stress may also damage the intracellular molecules of the insulin signal transduction pathway, such as insulin receptor substrate, insulin receptor substrate binding protein, insulin receptor kinase, and so on, leading to the blockage of insulin signal transmission (22). Oxidative stress is one of the essential mechanisms of insulin resistance by damaging all aspects of insulin signal transduction pathway. Excess iron ions can also lead to disorders of lipid metabolism, including lipid peroxidation, fat accumulation, and abnormal lipid metabolism (34).
Iron causes damage to cells through producing reactive oxygen species (ROS) and free radicals from peroxides. Iron is a critical participant in redox reactions, so excessive iron ions can promote the progress of redox reactions, leading to the production of large amounts of ROS. Secondly, excessive iron ions may undergo a Fenton reaction with oxidizing substances such as hydrogen peroxide in the cell to produce more active oxygen radicals, such as peroxyl radicals and hydroxyl radicals (35). These ROS are able to oxidize cell membrane lipids, proteins, and nucleic acids, triggering oxidative stress responses and damaging cell structure and function (36). In addition, excessive iron ions may also affect mitochondrial function, interfere with the normal operation of the electron transport chain, and increase the production of ROS within mitochondria. Islet beta cells are particularly sensitive to ROS (22). Since the expression rate of antioxidants such as SOD2 and catalase is low, experimental data show that antioxidant enzymes are usually ±50% in the liver, while gene expression is significantly reduced in pancreatic islets. In the liver, the gene expression levels of mitochondrial Mn-SOD and cytoplasmic Cu, Zn superoxide dismutase (Cu, Zn-SOD) were in the range of 30 - 40%. In islets, the gene expression of glutathione peroxidase was 15%, while catalase gene expression was not detectable (37, 38).
Iron metabolism disorders are highly relevant to a variety of diabetic complications. In diabetic retinopathy (DR), retinal damage has a close connection with oxidative stress, in which iron overload is not only a catalyst but also a cofactor to coordinate oxidative upheaval. Retinal iron content accelerates with age, and diabetes exacerbates pathological iron deposition in the retinal domain (39). The iron influx in the retina, through the Fenton/Haber-Weiss reaction, induces the generation of hydroxyl radicals, aggravates oxidative stress, disseminates lipid peroxidation, and causes damage to neurons, retinal endothelial cells (REC) and retinal pigment epithelial cells (RPEC), which may catalyze the induction of ferroptosis (40). Iron deficiency plays a vital role in chronic kidney disease (CKD). Iron is a component of red cell hemoglobin, and iron deficiency can lead to decreased red cell production and aggravate anemia. Patients with CKD are often accompanied by chronic inflammation and kidney damage. These pathophysiological processes lead to abnormal iron metabolism, including decreased absorption, increased release, and decreased utilization of iron, which aggravates iron deficiency (41). According to experiments, the prevalence of iron deficiency anemia is very high among patients with T2DM, and most patients with diabetes also show iron deficiency (42). Chronic kidney disease (CKD) is often accompanied by anemia, and the mechanism by which iron deficiency leads to impaired erythropoiesis in the context of decreased renal function is remarkable. This may be due to a true or relative deficiency of iron stores, which are absolute and functional iron deficiencies, respectively (41, 43). In addition, the present data strongly suggest that peripheral blood iron levels are related to coronary atherosclerosis, and iron concentration is significantly reduced in acute myocardial infarction (AMI) and coronary artery disease (SCAD) (44). Iron also plays a crucial role in cardiac mitochondrial and cardiomyocyte function. Because iron deficiency may severely perturb mitochondrial energy, and iron overload may react with iron in mitochondria via ROS to generate harmful hydroxyl radicals (45), resulting in the opening of the mitochondrial permeability overpore (46) and the mitochondrial membrane potential depolarization and eventually mitochondrial swelling (47). Mitochondrial dysregulation is associated with various pathological conditions of the heart, ranging from advanced heart failure (caused by iron shortage) to cardiac ischemia (due to iron overload) (48). These deleterious effects are partly due to the increase in oxidative stress. (the mechanism is shown in Figure 2).
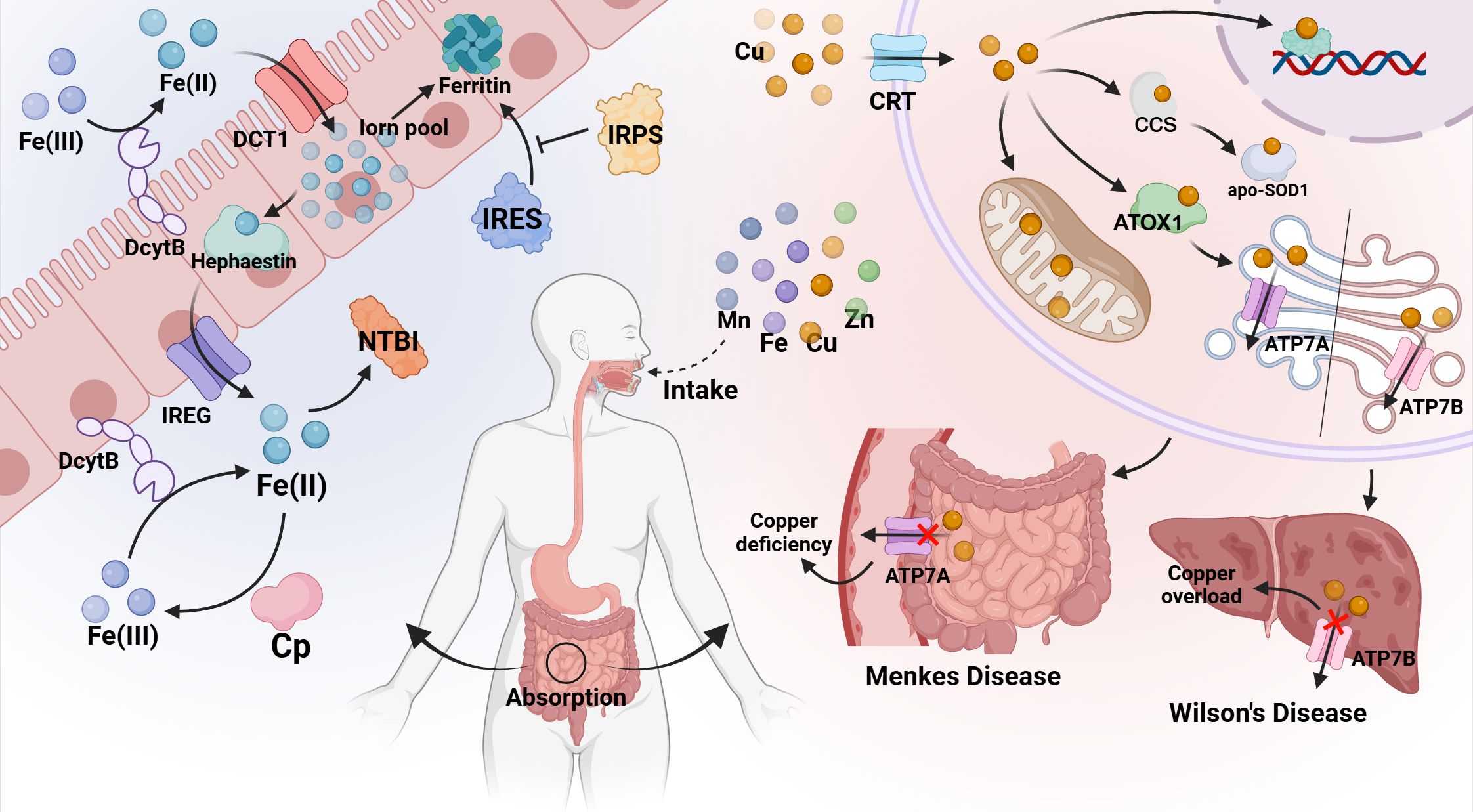
Figure 2. Relationship between iron metabolism disorder and diabetes and its complications. Plasma iron and ferritin levels increase in T2DM patients. Iron accumulates in liver cells and leads to insulin resistance and reduced secretion through oxidative stress pathways. In addition, free iron can induce iron death of beta cells by activating the ASK1/P-P38/CHOP signaling pathway. Besides, an iron influx in the retina, through the Fenton/Haber-Weiss reaction, causes the generation of a series of hydroxyl radicals, aggravates oxidative stress, propagates lipid peroxidation, and damages neurons, retinal endothelial cells (REC) and retinal pigment epithelium (RPEC), which may catalyze the induction of ferroptosis, and lead to diabetic retinopathy (DR). Renal damage in patients with diabetes nephropathy leads to abnormal iron metabolism, including decreased absorption, increased release and decreased utilization of iron, which aggravates iron deficiency and leads to anemia.
Iron chelators are a class of drugs used to treat conditions of iron overload or iron toxicity. By binding to free iron in the body, these drugs form stable complexes, thereby reducing the concentration of free iron and preventing its damage to cells and tissues. The main effects of iron chelators include promoting the excretion of excess iron from the body, reducing iron absorption, and blocking iron recycling in the body (49). Common iron chelators include deferoxamine and aminoglycolic acid, which play an essential role in the treatment of iron-related diseases, such as β-thalassemia major and iron poisoning. Iron chelation therapy can decrease the incidence of diabetes, and strengthening iron chelation and the appropriate combination of chelating agents can reduce insulin resistance (IR) and improve glucose tolerance (50). Iron chelators improve β-cell function and insulin sensitivity by reducing the concentration of free intracellular iron. Firstly, iron chelators can reduce the level of intracellular free iron and reduce the degree of oxidative stress, thereby protecting the structure and function of islet beta cells and helping to maintain the normal secretion of insulin. Secondly, iron chelators can reduce the presence of free iron, reduce the release of inflammatory factors, and alleviate the inflammatory response, which is beneficial to the protection of β cells. In addition, iron chelators can reduce the interference of intracellular iron overload on the insulin signaling pathway, which helps to improve insulin signaling and improve insulin sensitivity (51).
As a component or essential cofactor of many enzymes, copper is involved in numerous physiological pathways, including preventing ROS damage (such as Cu, Zn-SOD), participating in the mitochondrial respiratory chain (such as cytochrome C oxidase), and iron metabolism (such as ceruloplasmin) (52). Precise regulation of copper is achieved by a protein known as a metal chaperone. Copper enters cells through cell surface transport proteins (CTRs), and upon entry, it binds to molecular chaperones and is transported to the Golgi, apo-SOD1 or to the mitochondria. After arriving at the destination, copper is transported to the copper transport P-type ATPase molecules on the Golgi apparatus, including Wilson’s disease protein (ATP7B) and Menkes disease protein (ATP7A), and released. Among them, ATP7B is mainly present in the liver, and Wilson’s disease can damage this protein, leading to the accumulation of Cu. ATP7A is present in extrahepatic tissues, which can be damaged by Menkes disease and hinder the absorption of Cu in the intestinal mucosa, leading to systemic copper deficiency (53). Copper ion homeostasis must be tightly controlled. Copper imbalances can lead to a wide variety of diseases, chief among them are those caused by copper overload. Copper overload includes excessive copper intake and hereditary copper overload. Excessive copper intake is mostly acute poisoning, which is related to copper pollution in tableware and eating foods with excessive copper content. Chronic poisoning has a low incidence and is likely to present a genetic predisposition. Studies have shown that it is usually autosomal recessive transmission, and the genetic origin is not yet clear (54). Hereditary copper overload is further divided into copper-associated infantile cirrhosis and Wilson’s disease.
Wilson’s Disease (WD), also referred to as Hepatolenticular Degeneration (HLD), is an autosomal recessive copper metabolism disorder. WD is caused by a mutation in the copper-transporting ATPase(ATP 7B), which is in charge of copper biliary excretion, resulting in a decrease in the transport of copper from the liver to the bile, and the deposition of excess copper in the liver leading to hepatocellular damage. Copper is released into the blood and deposited in extrahepatic tissues such as the kidneys, cornea, and brain, resulting in clinical symptoms in the corresponding organs (18).
Under physiological conditions, the body has an antioxidant defense system that clears oxidative products such as ROS, and oxidative stress refers to the disruption of this balance, damaging tissues and biological macromolecules. Due to the copper absorption process (conversion between the Cu+ and Cu2+), positive ions can be released to generate reactive oxygen species (ROS) (55). Oxidative stress can result in peripheral insulin resistance and the impairment of islet β-cell function, which can trigger diabetes and even serious complications (e.g., diabetic cardiovascular disease, diabetic retinopathy, and diabetic neuropathy) (56). Thus, copper deposition due to Wilson’s disease mediates ROS production, leading to diabetes.
Cu and Zn are a pair of antagonistic microelements. During intestinal absorption, they compete for the common carrier protein, which is metallothionein. Cu increases have an impact on zinc absorption, leading to significant Zn loss from the body, and the reduction of Zn promotes diabetes (see 5) (56).
Diabetes predisposes to Cu overload, leading to oxidative stress, which ultimately in turn promotes diabetic complications, primarily copper ions in cardiovascular, renal and retinal damage (55). Diabetic complications can be categorized into large and small vascular complications, which are the effects of the chronic hyperglycemic state of diabetes on the body’s vascular system, leading to vascular damage and disease progression. Large vascular complications are diseases of the cardiovascular. Small vessel complications include diabetic neuropathy, diabetic retinopathy and diabetic nephropathy. The association between copper dysmetabolism and diabetic complications is discussed below in terms of large and small vascular classifications.
It has been suggested that copper is an antioxidant nutrient with beneficial effects on cardiovascular health. Cu serves as a component of many enzymes, some of which are important for cardiovascular health, such as lysyl oxidase, which is required for the cross-linking of arterial elastin and collagen, and arterial proteoglycan metabolism is also interrupted due to copper deficiency (57). Studies have shown that superoxide dismutase is decreased in patients with myocardial infarction. This is because copper-zinc superoxide dismutase (SOD1) can catalyze the dismutation of superoxide radicals into hydrogen peroxide, which is the key enzyme against ROS. When this enzyme is deficient, cardiomyocytes will be damaged through oxidative stress (58) (Table 2).
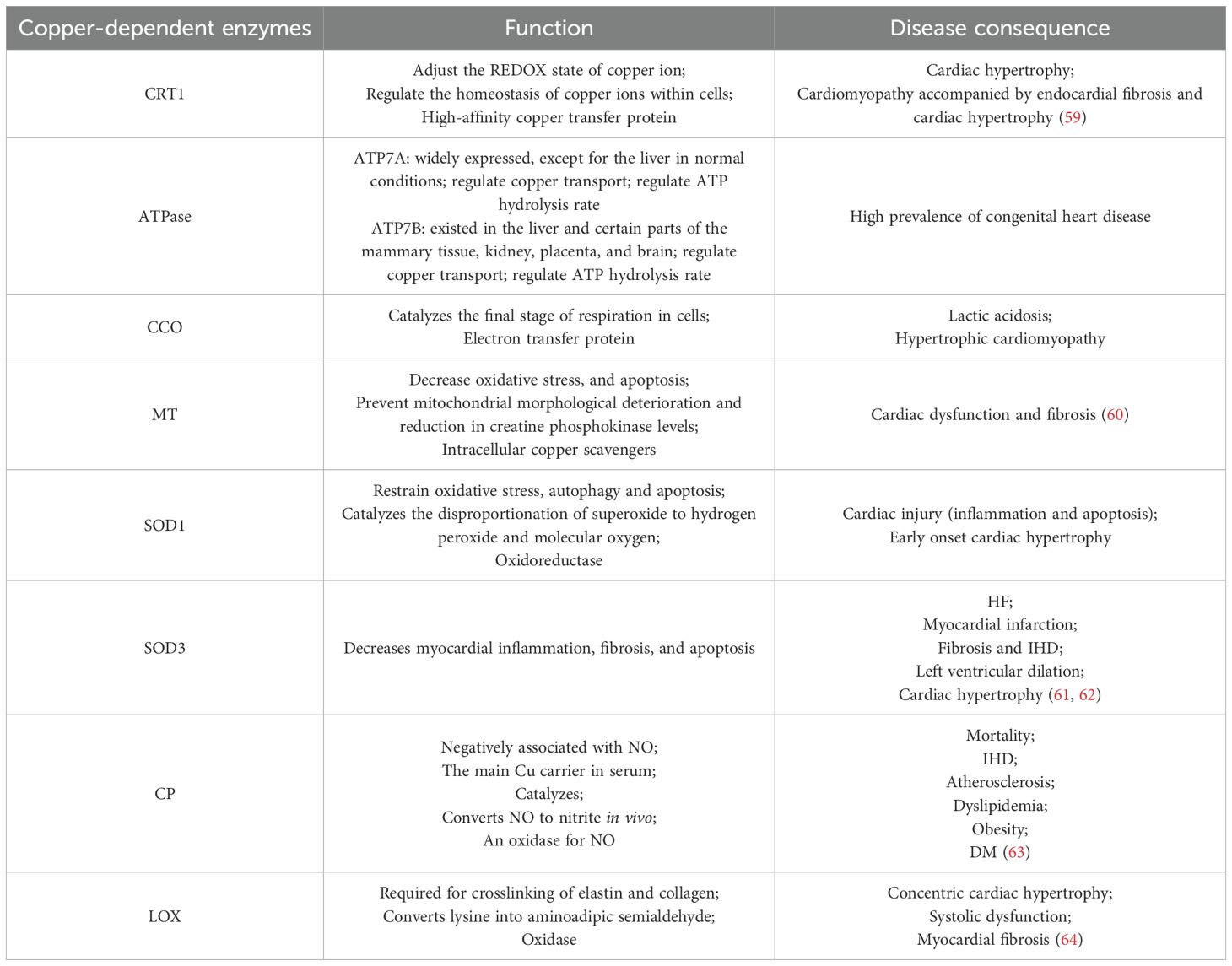
Table 2. Physiological effects of associated copper chaperone proteins and their mediation in cardiac diseases.
Studies have shown that Cu is involved with the EDRF-dependent arteriolar diastolic defects and impaired peripheral blood flow that occur in diabetes, leading to reduced intraneural blood flow and impaired peripheral nerve function. EDRF is thought to be a thiol adduct of nitric oxide or nitric oxide. In diabetic patients, the internal elastic layer of the arteries accumulates glycated proteins, which are able to bind Cu, and the abundance of Cu within the arterial wall will enhance hyperglycemia through metal catalysis, leading to ROS production via glucose autoxidation, disrupting NO or NO adducts, preventing normal EDRF-mediated diastole, and resulting in defective arterial relaxation and reduced peripheral blood flow (65). In addition to this, copper deficiency in the body may also lead to small vessel complications depending on the role of SOD. Superoxide dismutase (SOD), which is mammals’ main antioxidant defense system, requires the activation of metal catalysts (Cu or Mn). Whereas, in the presence of Cu deficiency, the ability of SOD to protect NO-mediated vasodilation is greatly reduced (66).
In addition, increased production of reactive oxygen species may itself impair EDRF-dependent vasodilation. Patients with diabetes have excessively high levels of reactive and exchangeable copper in their tissues, thus causing a variety of small vessel complications associated with peripheral blood reduction (65).
Diabetes leads to pathogenic copper overload, accompanied by enhanced copper-mediated oxidative stress, which in turn results in tissue damage in the nerves, retina, kidneys, heart and blood vessels, known as diabetic complications. For example, Renal tubular dysfunction in diabetes is likely a consequence of copper overload in the kidneys, in which copper-induced oxidative stress impairs EDRF-dependent vasodilation, resulting in reduced renal blood flow, which is the key to the molecular mechanism that causes diabetic nephropathy (67). (The above mechanism is shown in Figure 3).
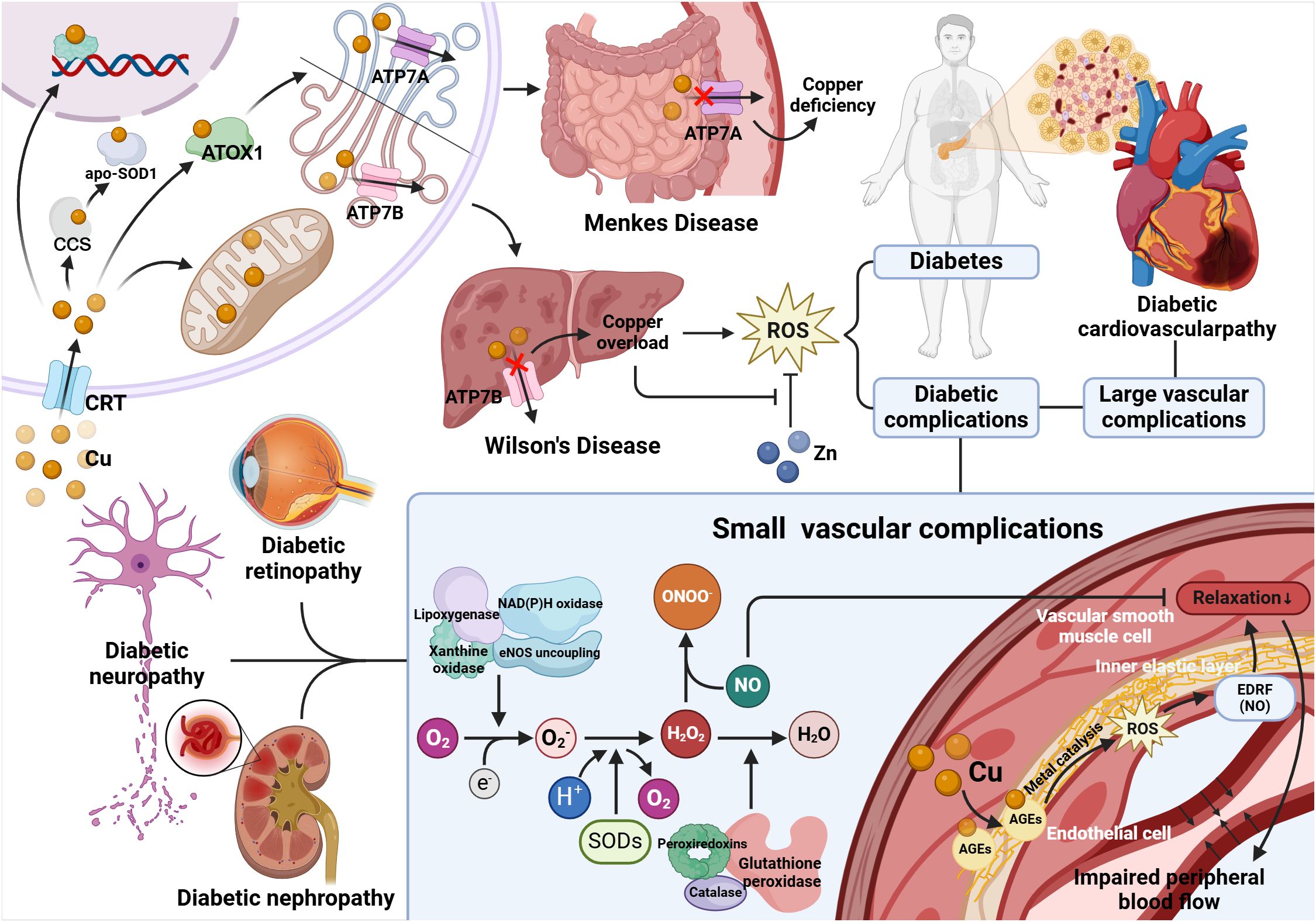
Figure 3. Relationship between copper metabolism disorder and diabetes and its complications. Wilson's disease protein (ATP7B) is mainly present in the liver, and Wilson's disease can damage this protein, leading to the accumulation of Cu. ATP7A is present in extrahepatic tissues, which can be damaged by Menkes disease and hinder the absorption of Cu in the intestinal mucosa, leading to systemic copper deficiency. Copper overload will lead to diabetes and diabetes complications through oxidative stress, in which diabetes small vessel disease is closely related to EDRF-dependent arterial relaxation. In diabetic patients, the internal elastic layer of the arteries (composed of elastin) accumulates glycated proteins that bind Cu, and the Cu enriched in the arterial wall catalyzes the destruction of NO or NO adducts, which is EDRF, by the metal catalysis, which prevents normal EDRF-mediated diastole, leading to arterial diastolic defects and impaired peripheral blood flow. And SOD can protect against NO-mediated vasodilation. Superoxide (O2-) is produced by mitochondrial enzymes, lipoxygenase, nitric oxide synthase (NOS), xanthine oxidase and NADPH oxidase. Superoxide dismutase (SOD) converts superoxide to H2O2. Then, peroxide reductase (Prx), glutathione peroxidase (GPx), and catalase reduce H2O2 to water. Nitric oxide (NO) is rapidly deactivated through reacting with O2-, resulting in the generation of the peroxynitrite (ONOO-), which is a strong oxidant. Therefore, SOD is the first line of defense against the toxicity of superoxide anion radicals. Moreover, this enzyme protects NO and its ability to mediate vasodilation.
Insufficient insulin production is the sole cause of T1DM. Zinc plays a variety of roles in the insulin signaling pathways, pancreatic storage and secretion, and structure and conformation of insulin (68). Therefore, zinc deficiency may contribute to the development of diabetes. According to the model study, the risk of diabetes will be significantly increased in drinking groundwater with low zinc content, and there is a close relationship between zinc and diabetes (69, 70). In most mammals, insulin is stored as zinc crystals in β cells of the pancreas (71). Moreover, adding zinc to insulin leads to a conformational change and strengthens the binding of insulin to its receptor (72). According to a series of animal experiments, zinc deficient rats show peripheral insulin resistance, and zinc supplementation improves insulin sensitivity to a certain extent (73).
It is well known that the beneficial impacts of zinc supplementation in diabetes include modulating oxidative stress, promoting glycemic control, and reducing insulin resistance. At the same time, zinc supplementation also has a beneficial effect on the secondary complications of diabetes. First, zinc supplementation may decrease the risk of dyslipidemia and cardiovascular disease associated with diabetes. A number of clinical studies have shown that zinc supplementation has significant benefits on blood lipids. Zinc ions can promote the synthesis and stability of HDL, increase its functional activity, and thus increase the level of HDL in the body. In addition, zinc ions can regulate the synthesis and metabolism of LDL and reduce its level in the blood, thereby reducing the risk of atherosclerosis. Zinc ions are involved in regulating the synthesis and metabolism of cholesterol in the body and help maintain the balance of cholesterol (74). It is able to reduce the levels of LDL cholesterol and total cholesterol while increasing the level of HDL cholesterol, thereby improving the lipid profile and reducing the risk of cardiovascular disease (75, 76). Second, zinc supplementation slowed the progression of diabetic nephropathy. Clinical trials have shown that zinc supplementation is beneficial in reducing urinary protein excretion and inflammation in patients with diabetic nephropathy (77, 78). According to another animal experiment, zinc deficiency leads to pathological changes in the renal cortex of rats, including enlargement of Bowman’s space, enlargement of mitochondria in the proximal distal flexural tubules, and fibrosis of the cortical interstitium, while zinc supplementation can minimize histopathological changes (79). In addition, zinc supplementation can improve diabetic cataract by regulating lens protein and polyol pathway, and can also reduce diabetic peripheral neuropathy (80).
Calcium is essential for bone health and many important physiologic functions. Most of the calcium present in the body is in the form of Ca2+ ions, complexes, and inorganic salts (52). Calcium ions play a key role in pancreatic islet cells and are involved in insulin secretion.
In islet beta cells, glucose-induced rapid increase of Ca2+ is a central signaling messenger that triggers insulin secretion (81). Insulin secreted by pancreatic β cells is stored in large dense core vesicles (LDCV), which are recruited to the plasma membrane and secreted insulin by regulating exocytosis. In β-cells, Glucose metabolism in β-cells stimulates mitochondria to produce ATP, which causes ATP-sensitive K+ channel (KATP) membranes to depolarize closure and voltage-dependent Ca2+ channels to open, promote Ca2+ influx, and bind cations to gelsolin and synaptotagmin, which are conducive to cortical actin (limiting but not preventing vesicles from entering the plasma membrane) and membrane fusion, respectively (82).
Diabetic patients have impaired renal function due to hypovolemia. In the case of renal failure, kidney’s phosphorus excreted is reduced, resulting in hyperphosphatemia. Hyperphosphatemia triggers hypocalcemia through interfering with the kidney’s phosphorus excretion (83). Diabetes also has hypomagnesemia, which leads to hypocalcemia. Hypomagnesemia in diabetic patients may be caused by poor oral intake, gastrointestinal malabsorption, and increased renal magnesium excretion. Mg2+ depletion impairs parathyroid hormone (PTH) secretion and renal tubules and bone are resistant to PTH action, resulting in hypocalcemia (83, 84). In addition, in patients with type 1 diabetes, a small decrease in PTH secretion will also reduce blood calcium levels (83).
In type 2 diabetes, reduced glucose transport through normal insulin stimulation leads to increased intracellular free calcium concentration, increasing the demand for insulin, resulting in excessive insulin production and secretion, leading to insulin resistance mediated by hyperparathyroidism. In addition, patients with diabetes who take thiazide diuretics are more likely to develop hypercalcemia (85).
As mentioned above, insulin secretion depends on calcium signaling channels, and calcium deficiency can lead to insulin resistance and decreased insulin secretion. Therefore, regulating calcium channels can improve insulin resistance and increase insulin secretion.
Polymorphisms in the calmodulin-dependent kinase subtype D (CaMK1D) gene region are linked to an increased incidence of diabetes (86). However, in a European population-based study, no significant correlation between T2D and CaMK1D SNP was identified. Therefore, the function of CaMK1D gene in the pathophysiology of T2D is still ambiguous (87). A study using mice as a model showed that highly selective calmodulin-dependent kinase inhibitors, such as CaMK1 selective inhibitors, can restore insulin sensitivity, confirming CaMK1D as a target for diabetes treatment (86).
The effect of metal ions on diabetes and its complications, mainly through oxidative stress, promotes the use of antioxidant compounds in the treatment of diabetes. Antioxidant therapy includes low molecular weight tissue-permeable compounds such as vitamin E, lipoic acid, or acetylcysteine (88). In addition to common antioxidants, C-peptide, as a natural antioxidant, is secreted together with insulin. Research has shown that C-peptides mainly exhibit antioxidant, anti-apoptotic, and anti-inflammatory effects by binding to cell surface signaling molecules to activate downstream pathways or regulate intracellular transcription processes. It can protect beta cells from oxidative stress and inflammation damage, and also has a protective effect on other cell types (89, 90). Therefore, C-peptide has potential therapeutic value in the prevention and treatment of diabetes and its complications (89).
In addition, the role of alpha-lipoic acid (ALA) as a powerful antioxidant in the treatment of diabetes has been studied. Alpha-lipoic acid and its reduced form, DHLA, are considered to be powerful natural antioxidants with the ability to scavenge many reactive oxygen species, which can inhibit oxidative stress-induced pancreatic β Cell damage has a potential protective effect. Antioxidants including ala can improve endothelial nitric oxide synthase (eNOS) to form NO, but not O2-, to improve vascular elasticity, which holds significant importance in the treatment of diabetic complications (91, 92).
Essential metals are critical in maintaining intracellular homeostasis, but overloading may increase the production of reactive oxygen species (ROS), thereby leading to metal-induced toxicity. Iron and copper are the two metals with the greatest potential for ROS production. They can both be chelated by ethylenediaminetetraacetate (93). Disodium ethylenediaminetetraacetate is a synthetic amino acid that has a high affinity for ions with +2 ~ +6 valence. It is able to trap metal ions and excrete them through the kidneys or bile, thus removing the metal from the body (94). Patients with chronic renal failure and ischemic heart disease were treated with ethylenediaminetetraacetic acid (EDTA) chelation therapy alone to promote the elimination of copper/iron ions from the body and reduce the damage to kidney cells and cardiomyocytes (95).
In addition, the transition metal chelator Trientine exerted a significant beneficial effect on endothelium-dependent diastole mediated by EDHF and NO in mesenteric vessels. The effects of Trientine, which are attributed to protection against the harmful effects of oxidative stress, may imply potential therapeutic approaches for diabetic microangiopathy and vasculopathy (96). It has been shown that diabetic patients show structural improvement in the heart after long-term Trientine treatment (97).
Metal ions also play a role in medications commonly employed in the treatment of diabetes, including angiotensin II receptor blocker (ARB), angiotensin-converting enzyme inhibitors (ACEIs), and metformin (98). ACEIs and ARB inhibit metal-catalyzed oxidation of ascorbic acid in vitro and reduce AGE formation. Metformin can play a role in AMP-activated protein kinase by interacting with mitochondrial copper, trapping α-dicarbonyl groups and leading to reduced gluconeogenesis (94).
There are many reports regarding the benefits of plant nutrients and extracts (e.g., resveratrol, quercetin, polyphenols, rutin) against diabetic complications. Among these compounds, some can inhibit the formation of AGE in diabetic animals. Part of their action mechanism might be related to limiting the absorption of metal ions or facilitating their excretion through chelating activity (99). For example, rutin can inhibit the small intestine’s absorption of glucose, as well as stimulate β Cells to secrete insulin and increase glucose uptake by tissues, thereby lowering blood glucose. In addition, rutin can reduce the formation of inflammatory cytokines by inhibiting the formation of age, as well as inhibit aldose reductase and reduce the concentration of sorbitol in erythrocytes, maintaining intracellular NADPH levels, and reduce oxidative stress, thereby protecting against dyslipidemia and hyperglycemia induced cardiovascular disease, liver injury, neuropathy, and nephropathy (100).
Metallothionein (MT) is a metal-binding protein that can be induced by numerous conditions, including zinc supplementation. It prevents or treats diabetes and its complications through anti-inflammatory effects, anti-apoptotic, and anti-OS, as well as reducing the acute toxic effects of heavy metals. Some studies have demonstrated successful treatment and prevention of type 2 diabetes, diabetic neuropathy, diabetic nephropathy, and diabetic complications using MT protein therapy. In addition, MT prevents diabetes-induced myocardial ER stress, thus contributing to the prevention and treatment of diabetic cardioprotection (101) (Table 3).
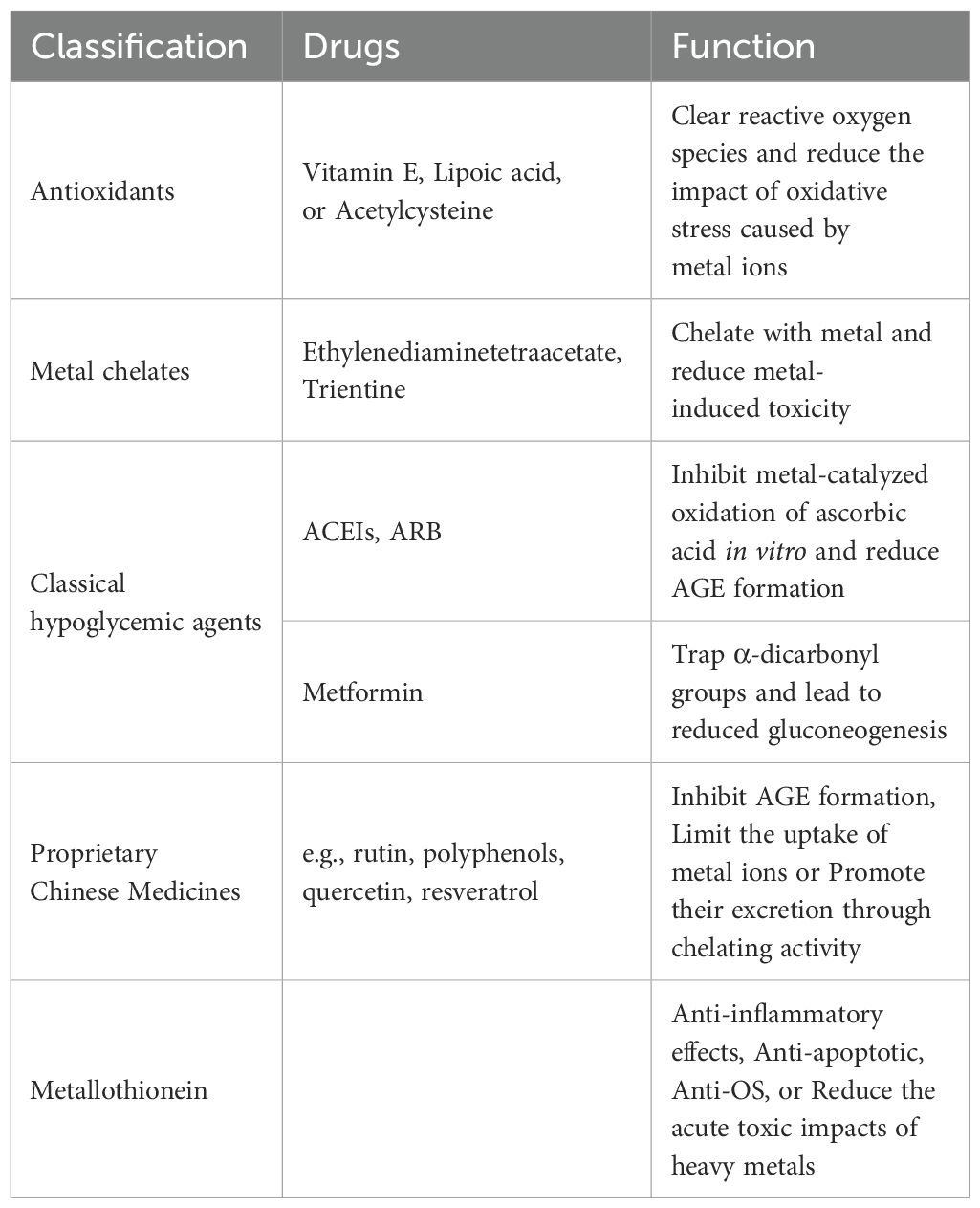
Table 3. Metal related drugs and treatment of diabetes.
In summary, the involvement of metallic ions in the etiology and progression of diabetes and its associated complications is multifaceted and pivotal. Dysregulations in the metabolism of minerals such as iron, copper, and zinc not only impact insulin secretion and cellular integrity but are also integrally linked to the onset and exacerbation of diabetic complications. Despite the promising therapeutic implications of iron-chelating agents and antioxidants in managing diabetes, comprehensive investigative efforts and empirical clinical validation are imperative. Future endeavors should aim at a profound comprehension and precise modulation of these processes.
SL: Formal Analysis, Investigation, Writing – original draft. XC: Formal Analysis, Investigation, Writing – original draft. XQ: Conceptualization, Investigation, Methodology, Writing – review & editing. JB: Writing – review & editing. BT: Investigation, Writing – review & editing. DZ: Writing – review & editing, Funding acquisition. XY: Writing – review & editing, Funding acquisition, Supervision. PY: Writing – review & editing, Conceptualization, Funding acquisition, Supervision.
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by the National Natural Science Foundation of China (No. 82360162 to PY); the Jiangxi Province Thousands of Plans (No. jxsq2023201105 to PY); and the Hengrui Diabetes Metabolism Research Fund (No. Z-2017-26-2202-4 to PY).
The figures were created with BioRender software (BioRender.com).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
T2DM, Type2 diabetes mellitus; SOD, Superoxide dismutase; ROS, reactive oxygen species; Cp, Ceruloplasmin; LOX, Lysyl oxidase; CCO, Cytochrome c oxidase; MT, Metallothionein; HDL, high-density lipoprotein; LDL, Low-Density Lipoprotein; AGE, advanced glycation end products.
1. Rosengren A, Dikaiou P. Cardiovascular outcomes in type 1 and type 2 diabetes. Diabetologia. (2023) 66(3):425–37. doi: 10.1007/s00125-022-05857-5
2. Eizirik DL, Pasquali L, Cnop M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: different pathways to failure. Nat Rev Endocrinol. (2020) 16(7):349–62. doi: 10.1038/s41574-020-0355-7
3. Vincent JB. Effects of chromium supplementation on body composition, human and animal health, and insulin and glucose metabolism. Curr Opin Clin Nutr Metab Care. (2019) 22(6):483–9. doi: 10.1097/MCO.0000000000000604
4. Li YV. Zinc and insulin in pancreatic beta-cells. Endocrine. (2014) 45(2):178–89. doi: 10.1007/s12020-013-0032-x
5. Bjørklund G, Dadar M, Pivina L, Doşa MD, Semenova Y, Aaseth J. The role of zinc and copper in insulin resistance and diabetes mellitus. Curr Med Chem. (2020) 27(39):6643–57. doi: 10.2174/0929867326666190902122155
6. Sobczak AIS, Stefanowicz F, Pitt SJ, Ajjan RA, Stewart AJ. Total plasma magnesium, zinc, copper and selenium concentrations in type-I and type-II diabetes. Biometals. (2019) 32(1):123–38. doi: 10.1007/s10534-018-00167-z
7. Hummel M, Standl E, Schnell O. Chromium in metabolic and cardiovascular disease. Horm Metab Res. (2007) 39(10):743–51. doi: 10.1055/s-2007-985847
8. Chausmer AB. Zinc, insulin and diabetes. J Am Coll Nutr. (1998) 17(2):109–15. doi: 10.1080/07315724.1998.10718735
9. Zheng Y, Li XK, Wang Y, Cai L. The role of zinc, copper and iron in the pathogenesis of diabetes and diabetic complications: therapeutic effects by chelators. Hemoglobin. (2008) 32(1-2):135–45. doi: 10.1080/03630260701727077
10. Zoroddu MA, Aaseth J, Crisponi G, Medici S, Peana M, Nurchi VM. The essential metals for humans: a brief overview. J Inorg Biochem. (2019) 195:120–9. doi: 10.1016/j.jinorgbio.2019.03.013
11. Khan AR, Awan FR. Metals in the pathogenesis of type 2 diabetes. J Diabetes Metab Disord. (2014) 13(1):16. doi: 10.1186/2251-6581-13-16
12. Zhang Y. Trace elements and healthcare: A bioinformatics perspective. Adv Exp Med Biol. (2017) 1005:63–98. doi: 10.1007/978-981-10-5717-5_4
13. Windisch W. Interaction of chemical species with biological regulation of the metabolism of essential trace elements. Anal Bioanal Chem. (2002) 372(3):421–5. doi: 10.1007/s00216-001-1117-6
14. Crichton RR, Ward RJ. An overview of iron metabolism: molecular and cellular criteria for the selection of iron chelators. Curr Med Chem. (2003) 10(12):997–1004. doi: 10.2174/0929867033457566
15. Crosby WH. The control of iron balance by the intestinal mucosa. Blood. (1963) 22:441–9. doi: 10.1182/blood.V22.4.441.441
16. Laurie SH. Transport and storage of metals. J Inherit Metab Dis. (1983) 6 Suppl 1:9–14. doi: 10.1007/BF01811317
17. Andrews NC. Iron metabolism: iron deficiency and iron overload. Annu Rev Genomics Hum Genet. (2000) 1:75–98. doi: 10.1146/annurev.genom.1.1.75
18. Scheiber I, Dringen R, Mercer JF. Copper: effects of deficiency and overload. Met Ions Life Sci. (2013) 13:359–87. doi: 10.1007/978-94-007-7500-8_11
20. Fujita T. Calcium paradox: consequences of calcium deficiency manifested by a wide variety of diseases. J Bone Miner Metab. (2000) 18(4):234–6. doi: 10.1007/PL00010637
21. Simcox JA, McClain DA. Iron and diabetes risk. Cell Metab. (2013) 17(3):329–41. doi: 10.1016/j.cmet.2013.02.007
22. Harrison AV, Lorenzo FR, McClain DA. Iron and the pathophysiology of diabetes. Annu Rev Physiol. (2023) 85:339–62. doi: 10.1146/annurev-physiol-022522-102832
23. Asperti M, Denardo A, Gryzik M, Arosio P, Poli M. The role of heparin, heparanase and heparan sulfates in hepcidin regulation. Vitam Horm. (2019) 110:157–88. doi: 10.1016/bs.vh.2019.01.008
24. Graham RM, Chua AC, Herbison CE, Olynyk JK, Trinder D. Liver iron transport. World J Gastroenterol. (2007) 13(35):4725–36. doi: 10.3748/wjg.v13.i35.4725
25. Doguer C, Ha JH, Collins JF. Intersection of iron and copper metabolism in the mammalian intestine and liver. Compr Physiol. (2018) 8(4):1433–61. doi: 10.1002/cphy.c170045
26. Altamura S, Kopf S, Schmidt J, Müdder K, da Silva AR, Nawroth P. Uncoupled iron homeostasis in type 2 diabetes mellitus. J Mol Med (Berl). (2017) 95(12):1387–98. doi: 10.1007/s00109-017-1596-3
27. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
28. Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev. (2011) 111(10):5944–72. doi: 10.1021/cr200084z
29. Lee YS, Lee DH, Choudry HA, Bartlett DL, Lee YJ. Ferroptosis-induced endoplasmic reticulum stress: Cross-talk between ferroptosis and apoptosis. Mol Cancer Res. (2018) 16(7):1073–6. doi: 10.1158/1541-7786.MCR-18-0055
30. Deng L, Mo MQ, Zhong J, Li Z, Li G, Liang Y. Iron overload induces islet β cell ferroptosis by activating ASK1/P-P38/CHOP signaling pathway. PeerJ. (2023) 11:e15206. doi: 10.7717/peerj.15206
31. Houstis N, Rosen ED, Lander ES. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature. (2006) 440(7086):944–8. doi: 10.1038/nature04634
32. Qiu R, Alikhanyan K, Volk N, Marques O, Mertens C, Agarvas AR. Repression of the iron exporter ferroportin may contribute to hepatocyte iron overload in individuals with type 2 diabetes. Mol Metab. (2022) 66:101644. doi: 10.1016/j.molmet.2022.101644
33. Andreadi A, Bellia A, Di Daniele N, Meloni M, Lauro R, Della-Morte D. The molecular link between oxidative stress, insulin resistance, and type 2 diabetes: A target for new therapies against cardiovascular diseases. Curr Opin Pharmacol. (2022) 62:85–96. doi: 10.1016/j.coph.2021.11.010
34. Ma J, Zhang H, Chen Y, Liu X, Tian J, Shen W. The role of macrophage iron overload and ferroptosis in atherosclerosis. Biomolecules. (2022) 12(11):1702. doi: 10.3390/biom12111702
35. Nakamura T, Naguro I, Ichijo H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim Biophys Acta Gen Subj. (2019) 1863(9):1398–409. doi: 10.1016/j.bbagen.2019.06.010
36. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
37. Beguin Y. Soluble transferrin receptor for the evaluation of erythropoiesis and iron status. Clin Chim Acta. (2003) 329(1-2):9–22. doi: 10.1016/S0009-8981(03)00005-6
38. Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med. (1996) 20(3):463–6. doi: 10.1016/0891-5849(96)02051-5
39. Zuber M. Diagnosis and significance of antinuclear antibodies. Dtsch Med Wochenschr. (1994) 119:392–8. doi: 10.1055/s-2008-1058707
40. Ouyang J, Zhou L, Wang Q. Spotlight on iron and ferroptosis: research progress in diabetic retinopathy. Front Endocrinol (Lausanne). (2023) 14:1234824. doi: 10.3389/fendo.2023.1234824
41. Batchelor EK, Kapitsinou P, Pergola PE, Kovesdy CP, Jalal DI. Iron deficiency in chronic kidney disease: Updates on pathophysiology, diagnosis, and treatment. J Am Soc Nephrol. (2020) 31(3):456–68. doi: 10.1681/ASN.2019020213
42. Praveen M, Jain N, Raizada N, Sharma S, Narang S, Madhu SV. Anaemia in patients with type 2 diabetes mellitus without nephropathy is related to iron deficiency. Diabetes Metab Syndr. (2020) 14(6):1837–40. doi: 10.1016/j.dsx.2020.09.020
43. Gafter-Gvili A, Schechter A, Rozen-Zvi B. Iron deficiency anemia in chronic kidney disease. Acta Haematol. (2019) 142(1):44–50. doi: 10.1159/000496492
44. Meng H, Wang Y, Ruan J, Chen Y, Wang X, Zhou F. Decreased iron ion concentrations in the peripheral blood correlate with coronary atherosclerosis. Nutrients. (2022) 14(2):319. doi: 10.3390/nu14020319
45. Aisen P, Enns C, Wessling-Resnick M. Chemistry and biology of eukaryotic iron metabolism. Int J Biochem Cell Biol. (2001) 33(10):940–59. doi: 10.1016/S1357-2725(01)00063-2
46. Chan S, Lian Q, Chen MP, Jiang D, Ho JTK, Cheung YF. Deferiprone inhibits iron overload-induced tissue factor bearing endothelial microparticle generation by inhibition oxidative stress induced mitochondrial injury, and apoptosis. Toxicol Appl Pharmacol. (2018) 338:148–58. doi: 10.1016/j.taap.2017.11.005
47. Sripetchwandee J, KenKnight SB, Sanit J, Chattipakorn S, Chattipakorn N. Blockade of mitochondrial calcium uniporter prevents cardiac mitochondrial dysfunction caused by iron overload. Acta Physiol (Oxf). (2014) 210(2):330–41. doi: 10.1111/apha.12162
48. Melenovsky V, Petrak J, Mracek T, Benes J, Borlaug BA, Nuskova H. Myocardial iron content and mitochondrial function in human heart failure: a direct tissue analysis. Eur J Heart Fail. (2017) 19(4):522–30. doi: 10.1002/ejhf.640
49. Malik A, Firke SD, Patil RR, Shirkhedkar AA, Surana SJ. Determination of iron chelating agents by analytical methods: A review. Crit Rev Anal Chem. (2020) 50(3):254–64. doi: 10.1080/10408347.2019.1620095
50. De Sanctis V, Soliman AT, Elsedfy H, Pepe A, Kattamis C, El Kholy M. Diabetes and glucose metabolism in thalassemia major: An update. Expert Rev Hematol. (2016) 9(4):401–8. doi: 10.1586/17474086.2016.1136209
51. Mechlovich D, Amit T, Bar-Am O, Mandel S, Youdim MB, Weinreb O. The novel multi-target iron chelator, M30 modulates HIF-1α-related glycolytic genes and insulin signaling pathway in the frontal cortex of APP/PS1 alzheimer's disease mice. Curr Alzheimer Res. (2014) 11(2):119–27. doi: 10.2174/1567205010666131212112529
52. Jomova K, Makova M, Alomar SY, Alwasel SH, Nepovimova E, Kuca K. Essential metals in health and disease. Chem Biol Interact. (2022) 367:110173. doi: 10.1016/j.cbi.2022.110173
53. Ba LA, Doering M, Burkholz T, Jacob C. Metal trafficking: from maintaining the metal homeostasis to future drug design. Metallomics. (2009) 1(4):292–311. doi: 10.1039/b904533c
54. Trocello JM, Chappuis P, El Balkhi S, Poupon J, Leyendecker A, Chaine P. [Abnormal copper metabolism in adult]. Rev Med Interne. (2010) 31(11):750–6. doi: 10.1016/j.revmed.2009.11.012
55. Lowe J, Taveira-da-Silva R, Hilário-Souza E. Dissecting copper homeostasis in diabetes mellitus. IUBMB Life. (2017) 69(4):255–62. doi: 10.1002/iub.1614
56. Liu Z, Wang M, Zhang C, Zhou S, Ji G. Molecular functions of ceruloplasmin in metabolic disease pathology. Diabetes Metab Syndr Obes. (2022) 15:695–711. doi: 10.2147/DMSO.S346648
57. Klevay LM. Cardiovascular disease from copper deficiency–a history. J Nutr. (2000) 130(2S Suppl):489s–92s. doi: 10.1093/jn/130.2.489S
58. Viktorínová A, Toserová E, Krizko M, Duracková Z. Altered metabolism of copper, zinc, and magnesium is associated with increased levels of glycated hemoglobin in patients with diabetes mellitus. Metabolism. (2009) 58(10):1477–82. doi: 10.1016/j.metabol.2009.04.035
59. Kim BE, Turski ML, Nose Y, Casad M, Rockman HA, Thiele DJ. Cardiac copper deficiency activates a systemic signaling mechanism that communicates with the copper acquisition and storage organs. Cell Metab. (2010) 11(5):353–63. doi: 10.1016/j.cmet.2010.04.003
60. Yin X, Zhou S, Zheng Y, Tan Y, Kong M, Wang B. Metallothionein as a compensatory component prevents intermittent hypoxia-induced cardiomyopathy in mice. Toxicol Appl Pharmacol. (2014) 277(1):58–66. doi: 10.1016/j.taap.2014.03.007
61. Kliment CR, Suliman HB, Tobolewski JM, Reynolds CM, Day BJ, Zhu X. Extracellular superoxide dismutase regulates cardiac function and fibrosis. J Mol Cell Cardiol. (2009) 47(5):730–42. doi: 10.1016/j.yjmcc.2009.08.010
62. Kobylecki CJ, Afzal S, Nordestgaard BG. Genetically low antioxidant protection and risk of cardiovascular disease and heart failure in diabetic subjects. EBioMedicine. (2015) 2(12):2010–5. doi: 10.1016/j.ebiom.2015.11.026
63. Shukla N, Maher J, Masters J, Angelini GD, Jeremy JY. Does oxidative stress change ceruloplasmin from a protective to a vasculopathic factor? Atherosclerosis. (2006) 187(2):238–50. doi: 10.1016/j.atherosclerosis.2005.11.035
64. Mujumdar VS, Tyagi SC. Temporal regulation of extracellular matrix components in transition from compensatory hypertrophy to decompensatory heart failure. J Hypertens. (1999) 17(2):261–70. doi: 10.1097/00004872-199917020-00011
65. Eaton JW, Qian M. Interactions of copper with glycated proteins: possible involvement in the etiology of diabetic neuropathy. Mol Cell Biochem. (2002) 234-235(1-2):135–42. doi: 10.1023/A:1015988817587
66. Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. (2011) 15(6):1583–606. doi: 10.1089/ars.2011.3999
67. Cooper GJ. Therapeutic potential of copper chelation with triethylenetetramine in managing diabetes mellitus and alzheimer's disease. Drugs. (2011) 71(10):1281–320. doi: 10.2165/11591370-000000000-00000
68. Li X, Cai L, Feng W. Diabetes and metallothionein. Mini Rev Med Chem. (2007) 7(7):761–8. doi: 10.2174/138955707781024490
69. Haglund B, Ryckenberg K, Selinus O, Dahlquist G. Evidence of a relationship between childhood-onset typediabetes and low groundwater concentration of zinc. Diabetes Care. (1996) 19(8):873–5. doi: 10.2337/diacare.19.8.873
70. Zhao HX, Mold MD, Stenhouse EA, Bird SC, Wright DE, Demaine AG. Drinking water composition and childhood-onset type 1 diabetes mellitus in devon and cornwall, england. Diabetes Med. (2001) 18(9):709–17. doi: 10.1046/j.1464-5491.2001.00554.x
71. Lazaris YA, Kuts LG, Bavel'skii ZE. Zinc and insulin content in pancreatic islets of rats after administration of hypoglycemic sulfonamides. Bull Exp Biol Med. (1974) 77(6):647–9. doi: 10.1007/BF00789991
72. Arquilla ER, Packer S, Tarmas W, Miyamoto S. The effect of zinc on insulin metabolism. Endocrinology. (1978) 103(4):1440–9. doi: 10.1210/endo-103-4-1440
73. Faure P, Roussel A, Coudray C, Richard MJ, Halimi S, Favier A. Zinc and insulin sensitivity. Biol Trace Elem Res. (1992) 32:305–10. doi: 10.1007/BF02784615
74. Heidari Seyedmahalleh M, Montazer M, Ebrahimpour-Koujan S, Azadbakht L. The effect of zinc supplementation on lipid profiles in patients with type 2 diabetes mellitus: A systematic review and dose-response meta-analysis of randomized clinical trials. Adv Nutr. (2023) 14(6):1374–88. doi: 10.1016/j.advnut.2023.08.006
75. Ranasinghe P, Wathurapatha WS, Galappatthy P, Katulanda P, Jayawardena R, Constantine GR. Zinc supplementation in prediabetes: A randomized double-blind placebo-controlled clinical trial. J Diabetes. (2018) 10(5):386–97. doi: 10.1111/1753-0407.12621
76. Ranasinghe P, Wathurapatha WS, Ishara MH, Jayawardana R, Galappatthy P, Katulanda P. Effects of zinc supplementation on serum lipids: a systematic review and meta-analysis. Nutr Metab (Lond). (2015) 12:26. doi: 10.1186/s12986-015-0023-4
77. Parham M, Amini M, Aminorroaya A, Heidarian E. Effect of zinc supplementation on microalbuminuria in patients with type 2 diabetes: a double blind, randomized, placebo-controlled, cross-over trial. Rev Diabetes Stud. (2008) 5(2):102–9. doi: 10.1900/RDS.2008.5.102
78. Khan MI, Siddique KU, Ashfaq F, Ali W, Reddy HD, Mishra A. Effect of high-dose zinc supplementation with oral hypoglycemic agents on glycemic control and inflammation in type-2 diabetic nephropathy patients. J Nat Sci Biol Med. (2013) 4(2):336–40. doi: 10.4103/0976-9668.117002
79. Elsaed WM, Mohamed HA. Dietary zinc modifies diabetic-induced renal pathology in rats. Ren Fail. (2017) 39(1):246–57. doi: 10.1080/0886022X.2016.1256321
80. Barman S, Srinivasan K. Diabetes and zinc dyshomeostasis: Can zinc supplementation mitigate diabetic complications? Crit Rev Food Sci Nutr. (2022) 62(4):1046–61. doi: 10.1080/10408398.2020.1833178
81. Draznin B. Intracellular calcium, insulin secretion, and action. Am J Med. (1988) 85:44–58. doi: 10.1016/0002-9343(88)90397-X
82. Gauthier BR, Wollheim CB. Synaptotagmins bind calcium to release insulin. Am J Physiol Endocrinol Metab. (2008) 295(6):E1279–86. doi: 10.1152/ajpendo.90568.2008
83. Ahn C, Kang JH, Jeung EB. Calcium homeostasis in diabetes mellitus. J Vet Sci. (2017) 18(3):261–6. doi: 10.4142/jvs.2017.18.3.261
84. Pham PC, Pham PM, Pham SV, Miller JM, Pham PT. Hypomagnesemia in patients with type 2 diabetes. Clin J Am Soc Nephrol. (2007) 2(2):366–73. doi: 10.2215/CJN.02960906
85. Liamis G, Liberopoulos E, Barkas F, Elisaf M. Diabetes mellitus and electrolyte disorders. World J Clin cases. (2014) 2(10):488–96. doi: 10.12998/wjcc.v2.i10.488
86. Fromont C, Atzori A, Kaur D, Hashmi L, Greco G, Cabanillas A. Discovery of highly selective inhibitors of calmodulin-dependent kinases that restore insulin sensitivity in the diet-induced obesity in vivo mouse model. J Med Chem. (2020) 63(13):6784–801. doi: 10.1021/acs.jmedchem.9b01803
87. Cheng J, Tang L, Hong Q, Ye H, Xu X, Xu L. Investigation into the promoter DNA methylation of three genes (CAMK1D, CRY2 and CALM2) in the peripheral blood of patients with type 2 diabetes. Exp Ther Med. (2014) 8(2):579–84. doi: 10.3892/etm.2014.1766
88. Cameron NE, Cotter MA. Effects of an extracellular metal chelator on neurovascular function in diabetic rats. Diabetologia. (2001) 44(5):621–8. doi: 10.1007/s001250051669
89. Chen J, Huang Y, Liu C, Chi J, Wang Y, Xu L. The role of c-peptide in diabetes and its complications: an updated review. Front Endocrinol (Lausanne). (2023) 14:1256093. doi: 10.3389/fendo.2023.1256093
90. Luppi P, Drain P. C-peptide antioxidant adaptive pathways in β cells and diabetes. J Intern Med. (2017) 281(1):7–24. doi: 10.1111/joim.12522
91. Jeffrey S, Isaac Samraj P, Sundara Raj B. Therapeutic benefits of alpha-lipoic acid supplementation in diabetes mellitus: A narrative review. J Diet Suppl. (2022) 19(4):566–86. doi: 10.1080/19390211.2021.2020387
92. Gomes MB, Negrato CA. Alpha-lipoic acid as a pleiotropic compound with potential therapeutic use in diabetes and other chronic diseases. Diabetol Metab Syndr. (2014) 6(1):80. doi: 10.1186/1758-5996-6-80
93. Diaz D, Fonseca V, Aude YW, Lamas GA. Chelation therapy to prevent diabetes-associated cardiovascular events. Curr Opin Endocrinol Diabetes Obes. (2018) 25(4):258–66. doi: 10.1097/MED.0000000000000419
94. Calderon Moreno R, Navas-Acien A, Escolar E, Nathan DM, Newman J, Schmedtje JF. Potential role of metal chelation to prevent the cardiovascular complications of diabetes. J Clin Endocrinol Metab. (2019) 104(7):2931–41. doi: 10.1210/jc.2018-01484
95. Glicklich D, Shin CT, Frishman WH. Heavy metal toxicity in chronic renal failure and cardiovascular disease: Possible role for chelation therapy. Cardiol Rev. (2020) 28(6):312–8. doi: 10.1097/CRD.0000000000000304
96. Inkster ME, Cotter MA, Cameron NE. Effects of trientine, a metal chelator, on defective endothelium-dependent relaxation in the mesenteric vasculature of diabetic rats. Free Radic Res. (2002) 36(10):1091–9. doi: 10.1080/1071576021000028325
97. Cooper GJ, Phillips AR, Choong SY, Leonard BL, Crossman DJ, Brunton DH. Regeneration of the heart in diabetes by selective copper chelation. Diabetes. (2004) 53(9):2501–8. doi: 10.2337/diabetes.53.9.2501
98. Frizzell N, Baynes JW. Chelation therapy for the management of diabetic complications: a hypothesis and a proposal for clinical laboratory assessment of metal ion homeostasis in plasma. Clin Chem Lab Med. (2014) 52(1):69–75. doi: 10.1515/cclm-2012-0881
99. Nagai R, Murray DB, Metz TO, Baynes JW. Chelation: a fundamental mechanism of action of AGE inhibitors, AGE breakers, and other inhibitors of diabetes complications. Diabetes. (2012) 61(3):549–59. doi: 10.2337/db11-1120
100. Ghorbani A. Mechanisms of antidiabetic effects of flavonoid rutin. BioMed Pharmacother. (2017) 96:305–12. doi: 10.1016/j.biopha.2017.10.001
Keywords: diabetes, metal ion, metabolism, iron ion, copper ion, complications
Citation: Liu S, Chen X, Qi X, Bai J, Tong B, Zhang D, Yin X and Yu P (2025) The role of metal ion metabolism in the pathogenesis of diabetes and associated complications. Front. Endocrinol. 16:1541809. doi: 10.3389/fendo.2025.1541809
Received: 08 December 2024; Accepted: 14 March 2025;
Published: 03 April 2025.
Edited by:
Marcia Hiriart, Universidad Nacional Autonoma de Mexico, MexicoReviewed by:
Peter Drain, University of Pittsburgh, United StatesCopyright © 2025 Liu, Chen, Qi, Bai, Tong, Zhang, Yin and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Peng Yu, eXU4MjIwMTgyQDE2My5jb20=; Xiaoping Yin, eGlhb3BpbmdidXhpYW9AMTI2LmNvbQ==; Deju Zhang, dTMwMDU3NTdAY29ubmVjdC5oa3UuaGs=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.