 Paula-Peace James-Okoro
Paula-Peace James-Okoro Jo Edward Lewis
Jo Edward Lewis Fiona Mary Gribble
Fiona Mary Gribble Frank Reimann
Frank Reimann- Institute of Metabolic-Science-Metabolic Research Laboratories and MRC-Metabolic Diseases Unit, University of Cambridge, Cambridge, United Kingdom
Glucose-dependent insulinotropic polypeptide (GIP) is one of two incretin hormones playing key roles in the control of food intake, nutrient assimilation, insulin secretion and whole-body metabolism. Recent pharmacological advances and clinical trials show that unimolecular co-agonists that target the receptors for the incretins – GIP and glucagon-like peptide 1 (GLP-1) – offer more effective treatment strategies for obesity and type 2 diabetes mellitus (T2D) compared with GLP-1 receptor (GLP1R) agonists alone, suggesting previously underappreciated roles of GIP in regulating food intake and body weight. The mechanisms by which GIP regulates energy balance remain controversial as both agonism and antagonism of the GIP receptor (GIPR) produce weight loss and improve metabolic outcomes in preclinical models. Recent studies have shown that GIPR signalling in the central nervous system (CNS), especially in regions of the brain that regulate energy balance, is essential for its action on appetite regulation. This finding has sparked interest in understanding the mechanisms by which GIP engages brain circuits to reduce food intake and body weight. In this review, we present key knowledge around the actions of GIP on food intake regulation and the potential mechanisms by which GIPR and GIPR/GLP1R agonists may regulate energy balance.
Introduction
Glucose-dependent insulinotropic polypeptide (previously known as gastric inhibitory peptide, GIP) is a gut-derived hormone produced and secreted from enteroendocrine K cells of the duodenum and upper jejunum upon meal ingestion (1). Along with glucagon-like peptide-1 (GLP-1), GIP plays a key role in regulating postprandial blood sugar levels through what is known as the incretin effect – where an estimated 50-70% of insulin release in response to a meal is mediated by GIP and GLP-1 (1). As the insulinotropic effect of GIP is diminished in patients with type 2 diabetes (T2D) (2), the therapeutic potential of the GIP axis has been relatively underexplored, whilst structurally optimised agonists of GLP-1 receptors (GLP1R) have been developed and introduced for the treatment of T2D and obesity. Focus is now turning to the therapeutic potential of targeting GIP receptors (GIPR), in light of the preclinical and clinical success of agents combining a GLP1R agonist with either a GIPR agonist or antagonist - strategies that deliver superior body weight reduction compared with GLP1R agonists alone (3–8).
Although research into the effects of GIP on appetite regulation is advancing, significant gaps remain in our understanding of the specific mechanisms underlying its anorectic action, particularly within the brain. Clarifying these central mechanisms is crucial, as the brain plays a vital role in regulating energy balance, and understanding how GIP affects central pathways to modulate food intake will be invaluable for improving GIP-based therapies for obesity and diabetes treatment. In this review, we present current knowledge on the role of GIPR signalling in the control of energy balance and examine emerging evidence regarding the potential mechanisms by which GIP affects food intake.
The physiology of GIP
The mature form of GIP, GIP(1-42), is a 42-amino acid hormone derived from posttranslational processing of the 153-amino acid precursor, pre-pro-GIP, through prohormone convertase (PC) 1/3-dependent cleavage after Arg-65 in the proGIP sequence (9). GIP belongs to the secretin/glucagon family of structurally related neuroregulatory peptides, which also includes pituitary adenylate cyclase-activating peptide (PACAP) and growth hormone-releasing hormone (GHRH). Its amino acid sequence is highly conserved across species, showing over 90% sequence identity across human, murine, porcine and bovine species (1). GIP(1-42) is susceptible to rapid degradation and inactivation by dipeptidyl peptidase 4 (DPP4), the same enzyme that inactivates GLP-1, resulting in a short plasma half-life of 5-7 minutes (10).
GIP is primarily produced by enteroendocrine K cells in the upper small intestinal epithelium (11, 12). A shorter form, GIP(1-30), has been reported in α-cells of pancreatic islets by immunohistochemistry, reverse-transcription polymerase chain reaction (RT-PCR), and in situ hybridisation (ISH) (13). In contrast, we were unable to detect GIP-peptides by mass spectrometry in human or mouse islets, or Gip-mRNA in FACS-purified islet cell types from mice raised in our facility (14, 15).
Expression of Gipr in peripheral tissues and the brain
The Gipr gene, while primarily known for its expression in pancreatic β-cells, is also found in other tissues, including adipose tissue, stomach, bone, adrenal cortex, heart, pituitary, endothelial cells, testis and several brain regions (1). Activation of GIPR thus exerts pleiotropic biological effects including neuroprotection (16), decreased bone resorption (17), and improved lipid metabolism and storage (18).
GIP was not historically considered to have any direct action on the brain, but intracerebroventricular (ICV) injection of high concentrations of GIP affected the secretion of anterior pituitary hormones including follicle-stimulating hormone (FSH) and growth hormone (GH), leading to the hypothesis that GIP could act on its receptors in hypothalamic regions near the third ventricle (19), and marking the first indication that GIP could have a regulatory role in the central nervous system (CNS). Subsequently, GIP receptors were identified in a rat cerebral cortex cDNA library and found to be similar to the receptors for glucagon and GLP-1, placing GIPR in the vasoactive intestinal polypeptide (VIP)/glucagon/secretin receptor superfamily (class B1) of seven transmembrane-domain G-protein coupled receptors (GPCRs) (20). GIP receptors primarily signal through Gαs/adenylyl cyclase activation, which increases intracellular cAMP levels; in pancreatic β-cells, this activates protein kinase A (PKA) and exchange protein activated by cAMP2 (EPAC2), resulting in a downstream increase in intracellular calcium levels and exocytosis of insulin (21–23).
Initial exploration of Gipr mRNA expression in the rat brain employing ISH and RT-qPCR revealed its wide distribution across many areas including the olfactory bulb, cerebral cortex, hippocampus, mammillary bodies, anterior and lateral septum, cortical amygdala, substantia nigra, thalamic nuclei, rostral raphe nuclei, choroid plexus, cuneate nucleus, cerebellum and brainstem (21, 24). In a study by Kaplan et al., autoradiographic localisation of saturable (25) GIP radioligand binding identified GIP binding sites in discrete areas of the rat brain, including the cortex, subiculum, anterior olfactory nucleus, inferior colliculus, lateral septal nucleus and the inferior olive (26). More recently, Adriaenssens et al. employed a Gipr-Cre knock-in mouse model, in which Gipr-expressing cells exhibit expression of a fluorescent EYFP reporter, to map Gipr expression in the mouse CNS (27). Immunostaining for EYFP highlighted Gipr expression in regions such as the medial preoptic area, subfornical organ, anterodorsal thalamic nucleus, magnocellular preoptic nucleus, suprachiasmatic nucleus and the interfascicular nucleus – alongside areas already identified in previous studies. Results from Cre-reporter lines must be interpreted with caution, as they can report cells expressing only very low levels of the receptor message, and might aberrantly report lineage tracing of cells that transiently expressed Cre-recombinase during development. Knock-in of Cre-recombinase into the native Gipr-locus, as in this model, should however reduce the risk of aberrant expression. Notably, Gipr mRNA expression was confirmed by qPCR in mouse hypothalamus and by ISH (RNAscope) specifically in the arcuate nucleus (ARH) and dorsomedial nucleus (DMH) of the hypothalamus of mice and several nuclei of the hypothalamus of humans (27). ISH (RNAscope) also confirmed expression of Gipr in the area postrema (AP) and nucleus tractus solitarius (NTS) of the brainstem in mice (28, 29) and Cynomolgus monkey (29). In the hindbrain, the signal in the AP was more pronounced compared to the NTS, suggesting that this area might be a particularly critical brain region for the regulation of energy balance and/or food intake regulation in response to GIPR-agonists.
Transcriptomic profiling using single-cell RNA sequencing of fluorescent cells isolated from Gipr-Cre mice further revealed heterogeneous expression of Gipr across neuronal and non-neuronal cell types in the hypothalamus. Based on the expression of cell-type-specific marker genes, Gipr was identified in mural cells, ependymocytes, pericytes, vascular and leptomeningeal cells (VLMCs), smooth muscle cells (SMCs), endothelial cells (ECs), oligodendrocytes (OLs), and neurons (27, 30). Analysis of the neuronal cluster showed Gipr expression in both glutamatergic and GABAergic neurons, along with co-expression of neurohormones involved in energy balance, including Sst, Avp, Pthlh, and fewer neurons expressing Cartpt and Tac1 (27). More recently active GIPR expression in mouse and human hypothalamus was confirmed by single-nucleus (sn) RNA sequencing and spatial transcriptomics (31). Gipr neurons expressed receptors for key gut peptides known to regulate energy homeostasis such as ghrelin and cholecystokinin (CCK), and calcium imaging analysis demonstrated that stimulating these receptors excites Gipr cells, suggesting that Gipr neurons can respond to food-related signals from the periphery.
Histological and snRNAseq have characterised Gipr expression in the brainstem, revealing differential expression of Gipr within the NTS and AP (27, 32–34). Gipr expression is abundant in inhibitory GABAergic neurons within the AP, but less so in the NTS and the nodose ganglia of the vagus nerve (28, 35). Projections from inhibitory GABAergic neurons are mostly confined within the AP itself with minimal projections to the proximal NTS (32). Some studies additionally identified Gipr in a small population of glutamatergic neurons in the dorsal vagal complex (DVC) of the mouse brainstem (28), potentially reflecting the sparse Gipr-positive cells in the NTS that were also identifiable by RNAscope. Both GABAergic and glutamatergic Gipr-positive neurons were identified in snRNAseq analyses of the combined AP and NTS from rats and mice (36, 37). Non-neuronal cells, particularly OLs and a few astrocytes express Gipr (33, 34, 37), revealing the multifaceted nature of Gipr distribution in the brainstem, as in hypothalamus, as outlined above.
Gip expression in the CNS
The expression of Gipr and identification of GIP binding sites in the brain, particularly in regions protected by the blood-brain barrier (BBB), suggests that GIP potentially plays a physiological role in the CNS. This raises important questions about whether central Gipr-expressing populations respond to circulating GIP from the periphery or if there is a central population of GIP-producing cells. Early studies, which attempted to detect Gip mRNA in the rat brain by northern blot hybridisation, ISH and RT-PCR were unable to detect any Gip mRNA (21, 38, 39). Given this, GIP was suggested to enter the brain through circumventricular organs (CVO) such as the AP and act on other brain regions (21). Alternatively, it was suggested that another ligand might activate GIPR within the brain (21). Some studies, however, have reported GIP mRNA and protein in rat retina (40), hippocampus (41), olfactory bulb, cerebellar Purkinje cells, cerebral cortex, substantia nigra (24, 42), and striatum (43), with moderate expression in the amygdala, lateral septal nucleus, pretectal nuclei, thalamic reticular nucleus as well as in several nuclei in the thalamus, hypothalamus, and brainstem (24, 42). In these studies, GIP immunoreactivity and mRNA colocalised with the neuronal marker NeuN, but not the glial marker GFAP (24, 42), suggesting that Gip-expressing cells may be neuronal. However, efforts by our research group using a Gip-Cre-reporter model that readily labels Gip-expressing cells in the duodenum (44) have been unable to detect Gip expression in the brain (45, 46). Given these findings, there is ongoing debate about whether GIP is truly produced in the brain or if its central effects are mediated by circulating GIP arriving from the periphery, or if an alternative ligand engages central GIPRs. It is plausible that GIP produced by the gut might affect brain function, as peripherally injected GIP was detected in cerebrospinal fluid (CSF) collected from mice cisterna magna (47). The questions of whether GIP is produced centrally and the physiological role of GIPR located behind the BBB continue to be subjects of active investigation.
Therapeutic GIPR agonism and GLP1R/GIPR dual agonism
Hormones released from the gut postprandially play key roles in regulating energy balance by modulating appetite and blood glucose levels (48), making them viable targets for the treatment of obesity and T2D. Among these, GLP-1 has gained significant attention for its ability to decrease body weight by inhibiting food intake, regulate glucose metabolism and improve renal and cardiovascular function (49, 50). This led to the development of GLP-1-based cardiometabolic medicines including liraglutide and semaglutide, which have shown clinical success in treating obesity (51–53). However, the use of GLP-1-based drugs comes with dose-dependent adverse effects, with up to 60% of patients reporting gastrointestinal (GI)-related issues. Furthermore, many patients struggle to reach their glycaemic and weight loss targets (54). This has spurred efforts to identify and develop agents that can enhance and complement GLP1R agonism.
An innovative approach in recent drug development is the design of unimolecular peptides that engage multiple receptors to improve therapeutic efficacy. Combining GLP1R agonists with activity against receptors for other hormones such as GIP, glucagon and amylin has shown promising results (3, 4, 55, 56). The approach stems from findings that co-treatment with GIPR and GLP1R agonists enhanced weight loss in diet-induced obese (DIO) mice (3, 4). Unimolecular GLP1R/GIPR dual agonists not only amplify the metabolic benefits of GLP-1 therapies but may also reduce common side effects, offering an effective strategy for managing obesity and T2D-related conditions (57, 58).
The first dual GLP1R/GIPR agonist, MAR709 (also known as NNC0090-2746) showed balanced in vitro activity at GIPR (EC50 = 3pM) and GLP1R (EC50 = 5pM) (3). In rodent models with genetic and diet-induced obesity, MAR709 produced greater weight loss and glycaemic improvements compared with pharmacokinetically matched GLP-1 treatments (3). In a phase 2b trial, the reductions in body weight and blood glucose in T2D patients treated with MAR709 at the single tested dose were similar to dose-titrated liraglutide (59). However, MAR709 was not further developed for commercial reasons (55).
A second GLP1R/GIPR coagonist, tirzepatide (previously called LY3298176, marketed as Mounjaro® for T2D and Zepbound® for obesity, Eli Lilly) is a 39-amino acid peptide acylated at the lysine 20 residue with a C20 fatty diacid. This acylation facilitates noncovalent binding to albumin, extending its half-life to approximately 160 hours in humans, compared with 19-25 hours for MAR709. Tirzepatide is designed as an imbalanced agonist, exhibiting greater affinity for human GIPR than for GLP1R. In signalling studies involving cell lines expressing GIPR or GLP1R, tirzepatide showed comparable potency to GIP in activating GIPR but was less potent than GLP-1 in activating GLP1R (4). In phase 3 clinical trials, tirzepatide demonstrated more effective reductions in body weight (approximately 20-22% weight loss with once-weekly 15 mg dosing) and glycated haemoglobin levels (up to 2.6%), along with overall greater improvements in lipid profiles compared with 1 mg semaglutide in T2D patients, although it should be noted that a higher dose of semaglutide (2.4 mg) is currently recommended for treating obesity (4, 51–53, 60, 61). It remains unclear whether the superior efficacy of tirzepatide in glucose and weight reduction, compared with the more balanced dual agonist MAR709, is due to its imbalanced pharmacology, its longer half-life or specific properties of tirzepatide such as biased GLP1R agonism (62). Collectively, these findings highlight the potential of GIPR agonism as a promising complementary target for achieving significant weight loss and blood glucose regulation, and there is growing interest in understanding how GIPR activation contributes to improved metabolic control and weight reduction (57).
As discussed in more detail below, a large body of preclinical research supports the idea that GIPR agonism plays a physiologically significant role in the mechanism of action of GLP1R/GIPR dual agonists. Mice fed a high-fat diet (HFD) showed improved body weight and glycaemic control when Gip was overexpressed (63). GIP analogues reduce appetite and lead to weight loss in DIO mice (64, 65), particularly when used in conjunction with GLP1R agonists. Importantly, cotreatment with agonists for GIPR and GLP1R reduced food intake and body weight to a greater extent than either agonist administered alone (3–6). These effects of GIP and GLP1R/GIPR dual agonists to reduce food intake suggest that GIP may act through central mechanisms and recruit neural networks that regulate energy balance and feeding behaviour.
GIPR antagonism
Despite the clear success of agonising GIPR in the treatment of obesity, there is evidence that taking the opposite approach and antagonising GIPR activity could also be an effective treatment strategy. Gipr knockout mice fed a HFD show protection against obesity and insulin resistance even on a hyperphagic leptin-deficient background (66). Some studies have shown that high-fat diets increase intestinal K cell density in ob/ob mice, or reported elevated circulating levels of GIP in obese animals and humans (67–71). It remains to be shown whether elevated GIP levels seen in these reports drive further fat accumulation or reflect a failing counterregulatory response; deleting GIP in the context of leptin deficiency, however, had little effect on the development of obesity in ob/ob mice (72). Various GIP or GIPR pharmacologic inhibition strategies in rodents, including genetic ablation of GIP-secreting K cells, infusions of neutralising antibodies against GIP, monoclonal antagonistic antibodies against GIPR, and vaccination against GIP, have all been shown to protect against HFD-induced weight gain without substantially deteriorating glucose homeostasis (47, 73–81). However, it is worth highlighting that chronic antagonism or knockout of GLP1R produces similar protection against diet-induced obesity in mice (82–84), implying that both incretin hormones exhibit paradoxical agonist/antagonist effects.
One of the strongest arguments for developing therapeutic GIPR antagonists is that in humans, common (E354Q) (85) and rare (R190Q, E288G) (86) coding variants of GIPR that are associated with decreased receptor signalling (87–89) have been associated with lower body mass index (BMI). A recent study found that GIPR missense mutations resulting in a loss of both Gs-coupled cAMP accumulation and β-arrestin coupling are associated with a lower BMI, whereas selective loss of function of Gs-coupling was not “protective” in a similar manner, pointing towards an important role of β-arrestin signalling in GIPR-function (90, 91). Several groups are developing GIPR antagonists for the treatment of obesity, as discussed in more detail below. These promote weight loss in preclinical and/or clinical studies (7, 8, 47, 78, 92–94) and demonstrate superior weight loss when combined with GLP1R-agonism compared to GLP1R-agonism alone, but it remains unclear what mechanisms these agents engage.
Some studies have suggested that increased energy expenditure is important for protection against DIO upon GIPR knockout or antagonism (73, 77, 81, 95, 96). Such studies consider effects on food intake to be secondary to weight loss, and emphasise the importance of altered glucose and lipid handling, possibly affected by altered postprandial insulin excursions (76). GIP mediates the uptake, storage and synthesis of fatty acids and triglycerides in adipocytes, especially under conditions of hyperinsulinemia (97–99) and it seems likely that GIPR-blocking agents and/or Gipr knock-out achieve some of their protection from diet-induced weight gain at the level of the adipose tissue. This lipogenic effect of GIP is thought to be mediated through mechanisms including increased secretion and activity of lipoprotein lipase (97, 100, 101), stimulation of insulin-dependent glucose uptake, increased adipocyte insulin receptor affinity, and conversion of glucose to lipids in the adipose tissue (98, 102, 103). Whilst the expression of GIPR in white adipocytes in vivo has been questioned (104), GIPR activation in combination with insulin does improve the lipid storage ability of adipose tissue by increasing responses to elevated insulin levels (105) whilst also improving lipid mobilisation when insulin levels are low (106) (Figure 1). It could be argued that inhibition of GIPR in adipose tissue interferes with its lipid buffering ability and that rodents, which as small animals are very sensitive to the insulating function of white adipose tissue, will under such circumstances increase their use of fatty acids to produce heat, e.g. increasing their energy expenditure. Consistent with this idea, Gipr knock-out mice have been reported to become obese on a HFD when kept at thermoneutrality (28°C) (107).
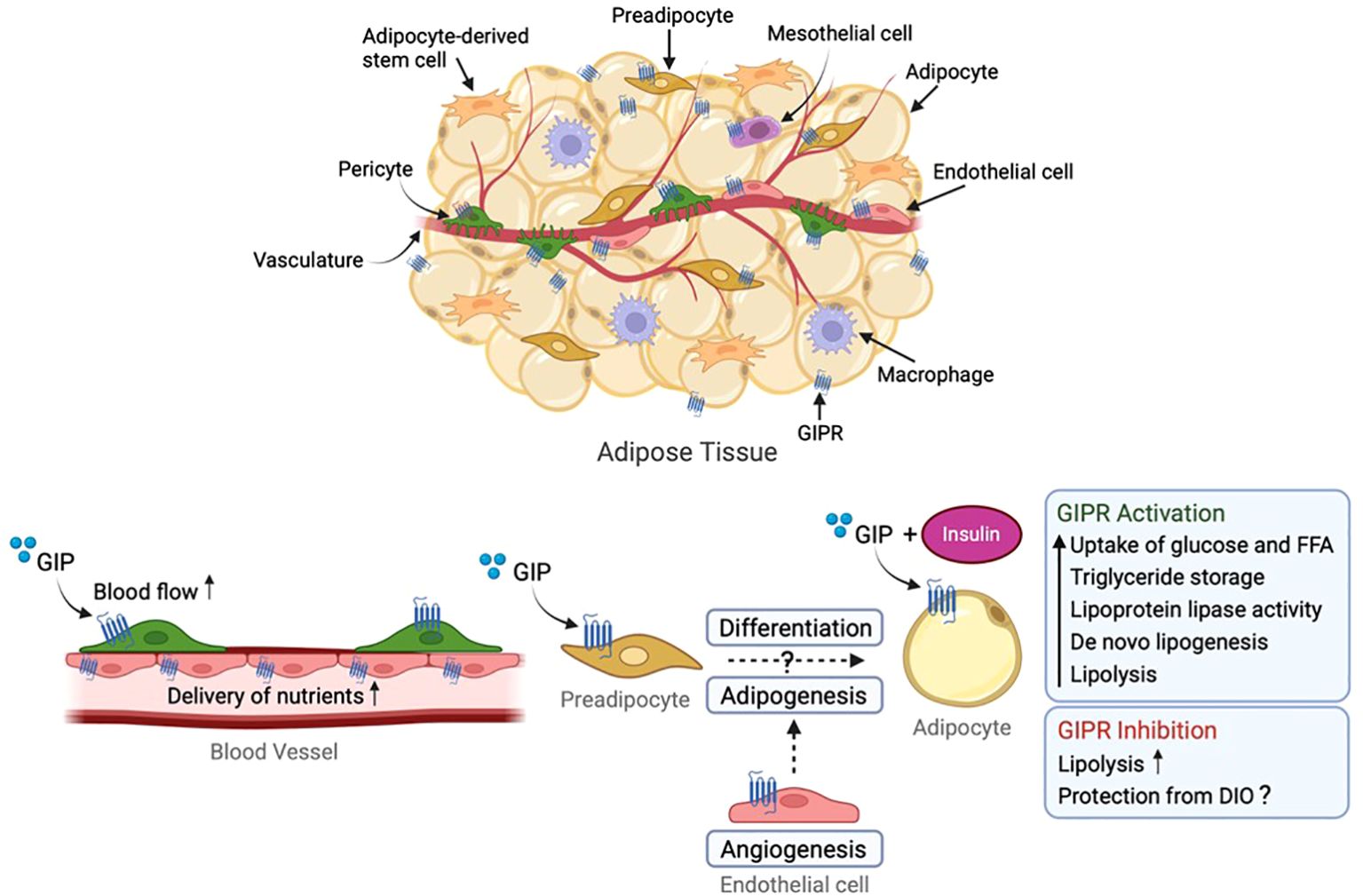
Figure 1. Role of GIP receptor in white adipose tissue (WAT). GIP receptor is expressed within adipose tissue in endothelial cells, macrophages, pericytes, mesothelial cells and adipocytes (the latter being discussed controversially). GIP stimulates blood flow and transport of nutrients to WAT, storage of triglycerides, lipoprotein lipase (LPL) activity, glucose and free fatty acid uptake, insulin sensitisation, de novo lipogenesis as well as lipolysis. Conversely, GIPR protects against HFD-induced weight gain presumably by inhibiting GIP-induced triglyceride storage and lipogenesis at the level of the adipose tissue. GIP, glucose-dependent insulinotropic polypeptide; GIPR, GIP receptor. Figure created with BioRender.com.
Other studies have pointed towards a direct effect of GIPR inhibition on food intake (7, 47). One study linked the anti-obesity effect of GIPR antagonism to leptin sensitivity in the hypothalamus, where genetically or pharmacologically blocking GIPR enhanced the anorexigenic properties of leptin in HFD-fed mice. Notably, Gipr knockout mice did not develop leptin resistance during HFD feeding (47). In the same study, it was also observed that central GIP could inhibit the appetite-suppressing effects of leptin. This GIP-induced leptin resistance occurred through activation of the small guanosine triphosphatase (GTPase) Ras-related protein 1 (Rap1), which inhibits the phosphorylation of signal transducer and activator of transcription 3 (STAT3) – a crucial mediator of leptin action (47, 108). This is, however, unlikely to be the only action of GIPR in the regulation of body weight, as whole-body Gipr knock-out also reduced body weight in the leptin-deficient ob/ob mouse model (66). It is also noteworthy that Rap1b activation downstream of the GIPR and Epac1 has recently been implicated in an anti-ageing action of GIP in the brain, reducing ferroptosis (109).
Emerging evidence suggests that the combination of antibody-based GIPR antagonism and GLP1R agonism leads to more significant weight loss in mice compared to the weight loss achieved through GLP1R agonism alone (7, 110). A bispecific molecule, maritide, in clinical trials comprises an antagonistic GIPR antibody coupled to two GLP-1 peptides. Phase 1 data on maritide, injected once monthly due to its long half-life, demonstrated effective body weight lowering in obese humans (8). The contribution of GIPR antagonism to the effectiveness of this molecule is not currently clear, as clinical dosing results in high circulating levels, raising the effective concentration of the GLP-1 component several orders of magnitude above the EC50 for GLP1R. As discussed later, GIP also acts to reduce nausea triggered by GLP-1, so antagonising GIPR could enhance the capability of the GLP-1 moiety to reduce food intake through the induction of nausea. An alternative approach of combining a GLP1R agonist with an antagonistic GIP peptide (AT-7687) is also under development, with some notable success in inducing weight loss in non-human primates (93).
Of note, whilst inhibition of food intake in response to peripheral treatment with a GIPR-blocking antibody was instantaneous in non-human primates, the effect took several days to develop in obese mice and might be secondary to weight loss (7). This group from Amgen also linked the effects of their GIPR-antagonistic antibody on body weight to adipose GIPR signalling and proposed that long-acting GIPR-agonism might act as functional antagonism due to receptor desensitisation/downregulation in adipocytes, but not other tissues such as pancreatic islets (92). This latter idea is, however, difficult to reconcile with reported effects of dual GLP1R/GIPR agonism which showed improved lipid flow into and out of adipose tissues, dependent on GIPR-agonism (106).
Effects of therapeutic GIPR antagonism should also be considered in the context of peripheral and central inflammation. Several studies suggest a pro-inflammatory role for GIP, contrasting with the well-documented anti-inflammatory action of GLP1R agonists (111, 112). Short term GIP infusion in human subjects increased IL-6 and monocyte chemoattractant protein-1 (MCP-1) in adipose tissue biopsies, likely reflecting enhanced macrophage-adipocyte crosstalk (113). Another study reported higher levels of inflammatory markers and differential expression of genes linked to leukocyte chemotaxis and tissue infiltration in obese adults with higher compared with lower fasting GIP levels, although participant numbers were relatively low and obesity parameters differed between the groups (114). In mouse models, central or peripheral administration of GIP promoted hypothalamic inflammation, with proinflammatory-related factors such as Il-6 and Socs3 elevated in the hypothalamus, which were reversed by an antagonistic GIPR antibody (115). By contrast, other studies reported anti-inflammatory effects of GIP, including a reduction in neuroinflammation surrounding amyloid plaques in a mouse model of Alzheimer’s disease after chronic treatment with [D-Ala2]GIP or a dual GLP1R/GIPR agonist. However, in these and other studies, it will be crucial to disentangle the extent to which inflammatory changes reflect direct effects of GIP on the inflammatory system rather than indirect effects arising from alterations in body weight and adiposity (116, 117).
Gut-brain circuits in the regulation of food intake
The gut-brain axis is a communication network of neuronal, biochemical, and hormonal signals connecting the GI tract to the brain. Following meal ingestion, changes in circulating nutrient levels, stimulation of GI mechanoreceptors, and the release of gut hormones work together to regulate subsequent food intake (118). Peripherally-derived adiposity signals such as leptin and insulin, alongside short-term satiety signals such as GLP-1, CCK and peptide YY (PYY), interact with brain circuits to modulate feeding behaviour and maintain energy balance (119, 120). Information from the gut is relayed to the brain by vagal and splanchnic afferent pathways; whilst these express receptors for gut hormones, discussion of these is beyond the scope of this review, given that Gipr expression within the CNS seems to be crucial for GIP’s action on food intake (discussed in next section). In the brain, neurons in the hypothalamus and the brainstem are essential for the control of energy homeostasis and are well placed anatomically to mediate the effects of gut peptides on feeding [reviewed in (121)]. The hypothalamus, particularly the ARH, is strategically located adjacent to the third ventricle and the median eminence (ME) – a CVO with fenestrated capillaries, lacking a BBB and allowing access of peripheral nutritional signals to these appetite-controlling regions of the CNS (122). Similarly, the DVC is located near the fourth ventricle in the medulla oblongata. It includes the AP, NTS and the dorsal motor nucleus of the vagus (DMV), with the AP being another CVO accessible to peripheral signals due to its leaky BBB. AP neurons and vagal sensory neurons project directly to the NTS, enabling the NTS to integrate and transmit gut-derived information to the brain (123, 124). Gastrointestinal, circulatory and central cues integrate in the DVC, which projects to the hypothalamus to regulate response to the body’s energy status and influence feeding behaviours, and vice versa (125, 126). This gut-brain communication forms a crucial feedback loop for maintaining energy balance and adapting feeding behaviour based on physiological needs.
Role of central GIPR signalling in appetite regulation
As apparent from the parallel development of drugs combining GLP1R agonism with either GIPR agonists or antagonists for the treatment of obesity, GIP appears to have several, sometimes opposing, actions on the brain. On the one hand, GIP signalling seems to favour weight gain, as evidenced by the protection of Gipr knockout mice from DIO, the lower BMI of humans with loss of function variants in GIPR, and a recently established ability of GIP to overcome nausea triggered by agents such as GLP1R agonists, discussed in more detail later. On the other hand, GIPR agonists have been demonstrated to have direct inhibitory effects on food intake in mice – a finding that is less evident in humans – and dual GLP1R/GIPR agonists in clinical practice trigger more weight loss than GLP1R agonism alone. These combined findings suggest that GIPR agonists have more than one action on the brain, potentially acting on different neuronal populations with different effects depending on the feeding status. Before embarking on a discussion of the different GIPR-containing neuronal circuitries identified in the literature, it should be acknowledged that a unifying consensus incorporating the multiple observations surrounding GIP biology has not yet been reached.
Owing to the rapid inactivation of endogenous GLP-1 and GIP by DPP4, it is currently debated whether endogenous gut-derived incretins reach their brain receptors at concentrations high enough to exert physiological actions. We recently demonstrated inhibition of food intake in response to activating intestinal K-cells that could be blocked by either systemic or central administration of a GIPR-blocking antibody (46). However, other studies failed to observe significant effects on food intake in mice when activating K-cells (25, 127). Degradation-resistant incretin receptor agonists undoubtedly have potential to act directly on the brain, likely through the CVOs, which are accessible to peripherally-administered fluorescent GIPR and GLP1R ligands (28, 128–130).
The expression of Gipr in feeding centres of the hypothalamus and brainstem suggests that GIP and GIP-based agonists could engage neuronal and/or non-neuronal circuitry in these regions to regulate food intake and body weight (27, 29, 30, 33, 64, 131). Emphasising the importance of brain GIPR signalling, deletion of Gipr from the CNS replicates the protection from DIO observed in global germline Gipr knockout models (64). Selective removal of Gipr from GABAergic cells using Vgat-Cre-dependent recombination further pointed to the importance of GABAergic neurons for both the protection against DIO and the reduction in food intake in response to systemically applied GIPR-agonists, especially when co-administered with a GLP1R-agonist (129, 132).
Both the brainstem and hypothalamus have been considered the primary central detectors of GIP and GIPR agonists in the peripheral circulation, since both areas contain neurons expressing Gipr within or adjacent to regions with a leaky BBB. Overlap of c-Fos staining and Gipr-Cre reporter-positive cells after administration of peripheral GIPR-agonist was more pronounced in the brainstem (particularly the AP) than the hypothalamus (46). Application of GIP also triggered cAMP elevation and increased neuronal activity as measured by electrophysiological recordings in hindbrain slices (133). A role for AP Gipr-positive neurons, the majority of which are GABAergic (32), does not, therefore, seem to be in any doubt, but whether this cell population underlies all the observed effects of GIPR agonists is less clear. A study by Adriaenssens et al., which characterised the distinct roles of Gipr-expressing neurons in the hypothalamus and brainstem, demonstrated that these neuronal populations engage different anorexigenic pathways depending on their neuroanatomical location (28). Whilst activation of Gipr-neurons in either region reduced dark-phase food intake, chemogenetic stimulation of DVC but not hypothalamic Gipr neurons induced conditioned taste aversion (CTA) (28). As evidence from other studies shows that AP Gipr neurons are anti-aversive, as discussed below, the researchers suggested that the CTA induced by DVC Gipr chemogenetic stimulation is mediated by direct activation of Gipr-neurons deeper in the NTS rather than the AP, and that the NTS is not a primary target of peripherally-administered GIPR agonists.
The weight loss and food intake suppressing effects of long-acting GIPR agonists and the GIP component of GLP1R/GIPR co-agonists have been attributed to inhibitory GABAergic neurons, likely reflecting Gipr-neurons in the AP (129) (Figure 2). In DIO WT mice, acyl-GIP and MAR709 decreased food intake, body weight and fat mass, concomitant with an increase in neuronal activation in the AP. Deletion of Gipr in Vgat-expressing GABAergic neurons blocked the effect of acyl GIP on AP neuronal activity, food intake, and body weight reduction, and abrogated the superior effectiveness of MAR709, suggesting that GIPR signalling via inhibitory GABAergic neurons is necessary for GIP’s anorectic effects (129). Further studies are necessary to clarify whether acyl-GIP and GLP1R/GIPR co-agonists act exclusively on brainstem GABAergic GIPR neurons or whether they also act on GABAergic GIPR neurons in other brain regions to reduce body weight and food intake.
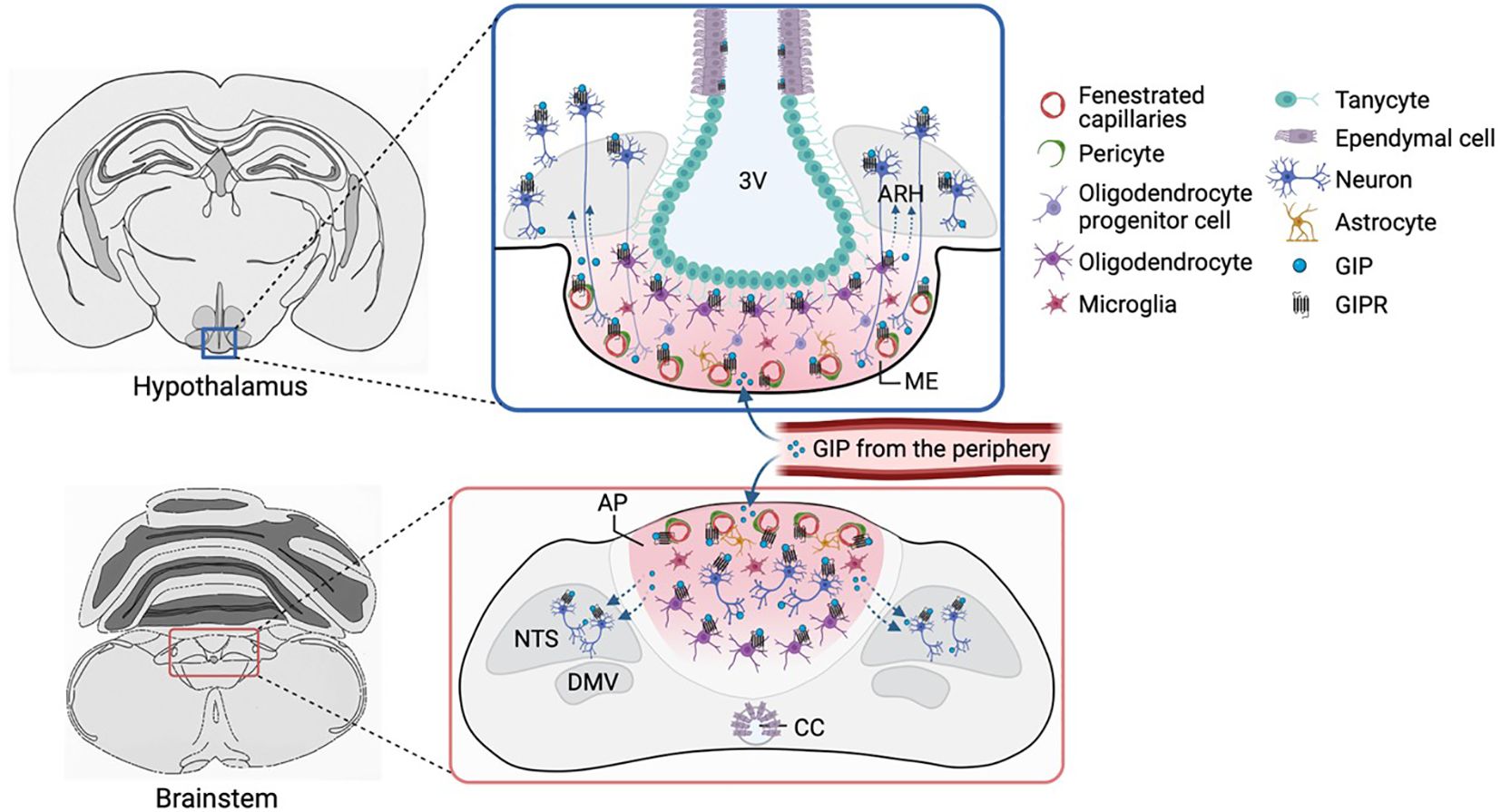
Figure 2. GIPR expression in the hypothalamus and brainstem. Gipr is expressed in oligodendrocytes, pericytes and other vascular and glial cells that control and regulate the transfer of circulating factors from CVOs (such as the ME and AP) to the brain parenchyma. Whilst GABAergic neurons in the AP have cell bodies, dendrites and axons outside the BBB, such neurons are scarce in the ME, although axons or dendrites from cells in adjacent areas (such as the ARH) might reach the ME, potentially allowing these neurons access to plasma-borne hormones. Alternatively, Gipr expression on non-neuronal cells might affect access to neuronal receptors behind the BBB. 3V, third ventricle; ARH, arcuate nucleus; ME, median eminence; CC, central canal; BBB, blood-brain barrier; CVO, circumventricular organs. Figure created with BioRender.com.
Of note, mice lacking Gipr in Vgat-expressing neurons were hypersensitive to GLP1R-agonist, losing more weight than wild-type animals in response to the same GLP1R-agonist dose (132). Similar GLP1R-agonist hypersensitivity was observed in a previous study when GIPRs were selectively removed by stereotactic injection of rAAV-Cre into the hypothalamus of Giprfxfx mice (28). However, this manipulation failed to prevent the additional food intake reduction that results from adding a GIPR-agonist on top of treatment with a GLP1R-agonist, suggesting that the hypothalamus is not the major site of GIPR-agonist action for food intake reduction, despite the reduction in food intake triggered by chemogenetic activation of hypothalamic Gipr-expressing neurons in the Gipr-Cre model (27).
In addition to the strong brainstem c-Fos signal triggered by administration of GIP analogues, this intervention also labels key hypothalamic regions implicated in feeding regulation, including the ARH, PVH, DMH, ventromedial (VMH), and lateral hypothalamus (LH) (29, 33, 46, 64, 129, 134). This suggests that hypothalamic engagement is involved in the overall neuronal circuitry important for and may underscore the action of GIP on food intake reduction. However, the overlap of c-Fos in response to peripheral GIPR-agonist administration and Gipr-Cre dependent reporter expression was limited in the hypothalamus (46),consistent with the idea that hypothalamic engagement occurs downstream of hindbrain activation by GIP. The relevance of the detected Gipr mRNA and the Gipr-positive cells reported by the Gipr-Cre model in the hypothalamus (Figure 2) therefore remains debated. Whilst it could be argued that these cells represent aberrant Gipr-Cre activity, the detection of Gipr mRNA in Hypomap argues against this (31). Alternatively, hypothalamic cells express Gipr at levels too low to elicit functional responses, or are not accessible to GIPR-agonists in the circulation; direct measurements of functional responses to GIP in Gipr-expressing neurons in brain slice preparations, which could potentially distinguish between these options, are currently lacking. However, it should be noted that cAMP or Ca2+ responses could be absent from a Gipr-labelled cell soma if the GIPR-containing compartment has been lost in the brain slice preparation or if the receptors are located in small, restricted compartments such as presynaptic terminals where they could play an important role in sensitising to other stimuli. Further studies will be crucial to disentangle this important area.
Regardless of exactly where GIP is primarily detected by the brain, the hypothalamus seems important in its downstream effects on food intake. Hunger-promoting AgRP neurons in the hypothalamus are necessary and sufficient to drive food intake and are key in maintaining energy balance (135–139), and although Gipr is not expressed in hypothalamic AgRP neurons themselves, GIPR and GLP1R agonism acutely inhibit AgRP neuronal activity in fasted mice, reducing their responses to food (140). In this context, it may be relevant that the ARH receives direct projections from the NTS to regulate feeding (141–143), and GABAergic projections from the DVC inhibit AgRP/NPY neurons in the ARH to decrease food intake and body weight (144). Studies by Kaneko and colleagues investigating the mechanism of action of intracranially-administered GIPR-antagonism alternatively implicated hypothalamic POMC neurons in underlying GIP-induced leptin resistance (47). However, another study involving systemic administration of the GIPR-agonist, GIPFA-085, suggested that increased plasma leptin levels recruit hypothalamic POMC-neurons in the ARH to underlie the reduced food intake (145). GIP’s interaction with leptin thus remains contentious, and additional research is needed to clarify the role of GIP in leptin signalling as well as to provide a more comprehensive understanding of the role of GIPR-expressing neurons in the hypothalamus in general.
GIP reduces nausea and emesis via the brainstem
The gut hormones, GLP-1 and PYY, reduce food intake by targeting overlapping neuronal circuits in the hypothalamus and brainstem that reduce appetite and trigger nausea and emesis (146–148). Despite their profound metabolic success, GLP-1-based medications cause nausea and vomiting in many patients (146–148). These GI side effects are considered a barrier to maximising the weight-loss profile of these treatments, suggesting a need to develop and test the efficacy of novel agents with improved tolerability. Interestingly, GIPR agonists have been shown to inhibit emetic responses caused by cisplatin, GLP-1 or PYY in dogs, ferrets and musk shrews, as well as nausea-associated behaviour in mice and rats (29, 33, 36, 134, 149). These findings have led to the concept that GIP acts as an anti-aversive hormone in response to GI-derived nauseating signals (134) and that GIPR agonism may improve the weight loss effectiveness of GLP1R agonists partly by reducing GLP1R agonist-induced nausea, allowing higher dosing of the GLP1R agonist.
The AP contains inhibitory neurons that counteract nausea and vomiting responses triggered by certain visceral stimuli (150–152), and as discussed above, Gipr is expressed in a subset of GABAergic inhibitory neurons in this region. GIP has been demonstrated to activate these inhibitory neurons, and thereby suppress the activity of nausea-promoting excitatory neurons, including those which express the growth/differentiation factor 15 (GDF15) receptor (Gfral), Glp1r (29) and the calcium-sensing receptor (Casr) (32, 133) (Figure 3). Diphtheria toxin-mediated ablation of AP Gipr neurons eliminated the anti-nausea effects of GIP (133). However, it is unknown whether these anti-aversive effects of GIP reflect its physiological as well as its pharmacological actions, and future studies will be required to determine whether intestinal GIP plays a physiological role in inhibiting avoidance responses to nauseating gastrointestinal signals.
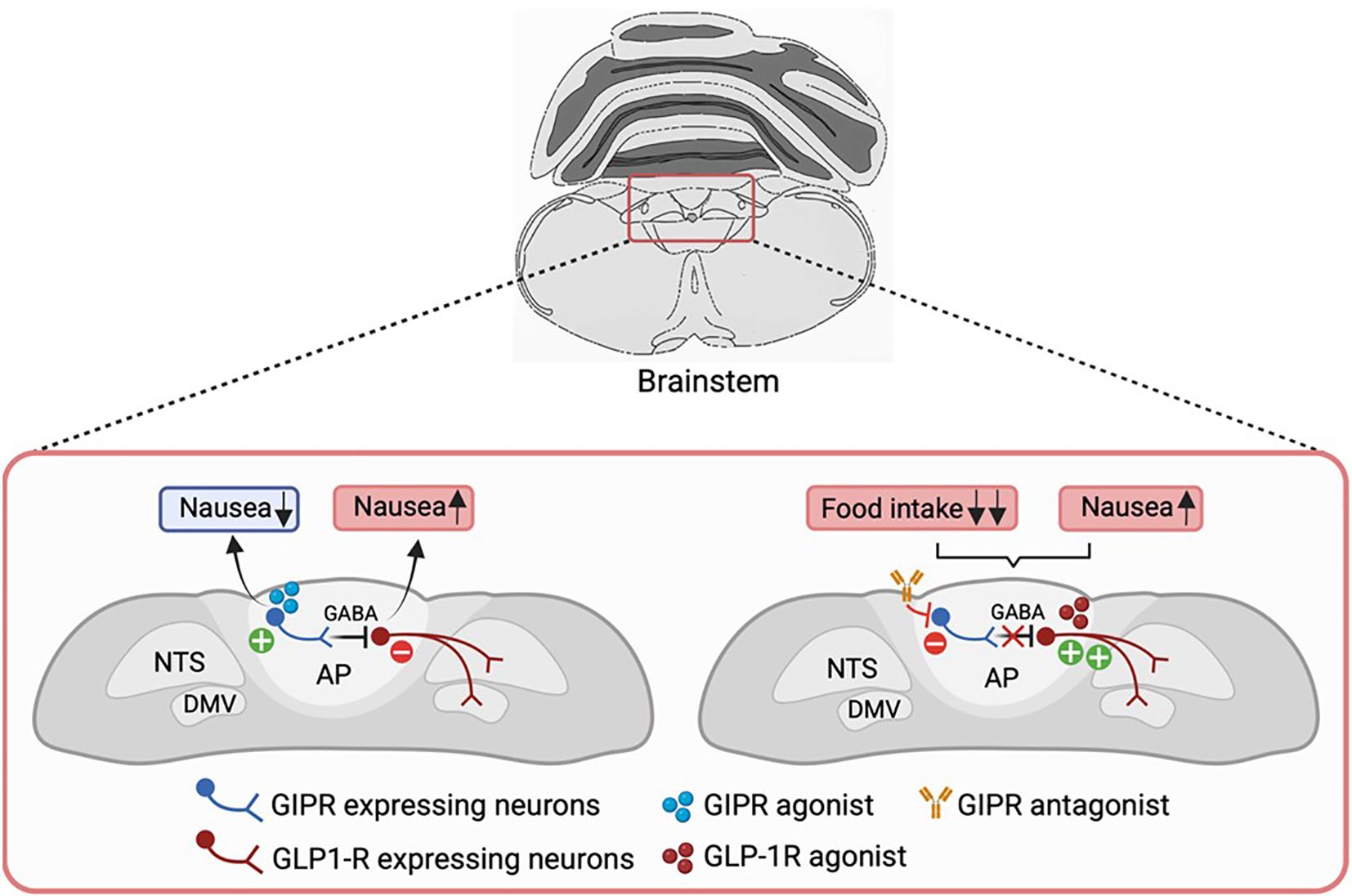
Figure 3. GIPR agonism in modulating GLP1R-induced nausea. Activation of GIPR might alleviate GLP-1-induced malaise by directly acting on the AP/NTS circuitry. Gipr is expressed in a subset of GABAergic inhibitory neurons in this region, through which GIP may act to suppress the activity of nausea-promoting GLP1R neurons, thereby reducing nausea and improving the therapeutic index of GLP-1-based medications. It remains to be seen if the same GABAergic GIPR-expressing neurons also underlie food intake inhibition by GIPR-agonists. Conversely, GIPR antagonism suppresses the activity of these inhibitory neurons, leading to enhanced activation of GLP1R-expressing neurons. This results in a greater reduction of food intake, albeit likely with increased nausea as a side effect of GLP1R activation. GABA, gamma-aminobutyric acid; AP, area postrema; NTS, nucleus of the solitary tract; DMV, dorsal motor nucleus of the vagus; GIPR, GIP receptor; GLP1R, GLP-1 receptor. Figure created with BioRender.com.
GIP may influence BBB permeability and brain access to peripheral factors
For GIP- and GLP-1-based therapies to exert their full anorectic effects, they must access their receptors located in CNS regions that modulate energy balance. In the majority of brain regions, the BBB carefully controls the entry of molecules from the bloodstream into the brain [reviewed in (153)]. Fluorescent GIPR and GLP1R agonists dosed peripherally can directly access brain areas that lie outside the BBB. This includes the choroid plexus and CVOs, which display a compromised BBB. CVOs with particular interest for the regulation of food intake include the ME of the hypothalamus and the AP of the brainstem (28, 128). Fluorescently labelled agonists were also detected, albeit at low levels, in brain regions supposed to be protected by the BBB, including the posterior ARH, NTS and DMV, which are adjacent to the ME and AP, respectively, and have been implicated in the regulation of food intake (28, 128). Radiolabelled GIP was additionally found to cross the BBB in a time-dependent saturable manner, which was inhibited by competition with native unlabelled GIP (26), suggesting involvement of a receptor-mediated mechanism, and GIP has been detected in the CSF of mice (47) and humans (154), which is interesting in the context of reported evidence of a neuroprotective effect of GIP in animal models for Alzheimer’s and Parkinson’s disease (16, 155).
While the majority of metabolic sensing neurons located in the ARH are separated from the ME by diffusion barriers, these neuronal populations may extend dendrites or axons to the ME or contact the ME through axonal terminals and directly access peripherally secreted hormones (122, 156–158). Tanycytes are thought to participate in the formation of CVO-parenchymal barriers (159, 160) and peripheral liraglutide has been shown to enter the hypothalamus via transcytosis across tanycytes (161). However, there is currently no similar evidence for GIP crossing into the brain through this mechanism; Gipr expression is especially high in ependymal cells surrounding the third ventricle (31), and oligodendrocytes but future work will need to address if these are involved in regulating access of hormones in the plasma to neurons behind the BBB. Enhancing the permeability and diffusion of these molecules to neuronal populations deeper in the brain that regulate energy balance may be a potential strategy to maximise the weight-loss effect of GLP1R/GIPR agonists (153, 162). Further research is needed to clarify the central mechanisms and routes through which these drugs reach their target sites in the brain, and the importance of brain penetration for induction of weight-loss.
Notably, in the hypothalamus and brainstem, Gipr is expressed by vascular and glial cells which affect perfusion and permeability, including pericytes, smooth muscle cells, vascular and leptomeningeal cells, endothelial cells and oligodendrocytes (27, 30, 37, 163). Oligodendrocytes are highly responsive to the nutritional state, as fasting triggers rapid proliferation and differentiation of OLs (74). Transcriptomic and FISH analyses of the murine and human hypothalamus have demonstrated that the Gipr is enriched in oligodendrocytes, particularly in the ME (27, 30, 31, 163–165). Supporting a functional role of GIPR signalling in the regulation of the oligodendrocyte lineage, a recent study demonstrated that oligodendrocyte-specific Gipr deletion in adult mice reduced oligodendrocyte survival and oligodendrogenesis, whilst treatment with GIPR agonists enhanced oligodendrocyte plasticity in the ME (165). GIPR signalling in oligodendrocytes increased access of GLP1R agonists to ME axons, and boosted the anorectic and weight-loss effects of GLP1R agonism (165), suggesting a novel mechanism by which GIPR agonism may enhance the weight loss profile of GLP1R agonists in incretin-based therapies.
Conclusion and future perspectives
The success of dual incretin receptor agonists as obesity pharmacotherapies has reignited interest in the previously overlooked role of GIPR signalling in energy balance. In addition to its insulinotropic effects, it is increasingly clear that GIP plays a role in modulating food intake and body weight, likely through central pathways that are not yet fully elucidated. GIP analogues act on regions of the brain that regulate energy balance, nausea and feeding behaviour to reduce food intake. Anti-emetic effects of GIP may enhance the therapeutic efficacy of GLP1R/GIPR dual agonists by mitigating the nauseating effects of GLP1R agonism – although this is yet to be proven in humans. Given the success of GLP1R/GIPR dual agonists in clinical trials, further understanding how GIPR signalling in the CNS affects energy balance is vital to fully harness the therapeutic benefits of GIP to combat obesity and T2D.
Author contributions
P-PJ-O: Conceptualization, Writing – original draft, Writing – review & editing. JL: Conceptualization, Writing – original draft, Writing – review & editing, Supervision. FG: Conceptualization, Supervision, Writing – original draft, Writing – review & editing, Funding acquisition. FR: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. Research in the Reimann/Gribble laboratory is funded by Wellcome (220271/Z/20/Z) and MRC-UK (MRC_MC_UU_12012/3). P-PJ-O is funded by the Cambridge School of Clinical Medicine’s Doctoral Training Programme in Medical Research and the Gates Cambridge Scholarship (#OPP1144).
Conflict of interest
PPJ-O is currently doing a placement at AZ. FMG has in the past consulted for Kallyope and Antag Therapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. (2007) 132:2131–57. doi: 10.1053/j.gastro.2007.03.054
2. Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest. (1993) 91:301–7. doi: 10.1172/JCI116186
3. Finan B, Ma T, Ottaway N, Müller TD, Habegger KM, Heppner KM, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med. (2013) 5(209):209ra151. doi: 10.1126/scitranslmed.3007218
4. Coskun T, Sloop KW, Loghin C, Alsina-Fernandez J, Urva S, Bokvist KB, et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept. Mol Metab. (2018) 18:3–14. doi: 10.1016/j.molmet.2018.09.009
5. Frias JP, Nauck MA, Van J, Kutner ME, Cui X, Benson C, et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet. (2018) 392:2180–93. doi: 10.1016/S0140-6736(18)32260-8
6. Wu Y, Ji T, Lv J, Wang Z. Rapid selection of a novel GLP-1/GIP dual receptor agonist with prolonged glycemic control and weight loss in rodent animals. Life Sci. (2020) 257:118025. doi: 10.1016/j.lfs.2020.118025
7. Killion EA, Wang J, Yie J, Shi SD-H, Bates D, Min X, et al. Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Sci Transl Med. (2018) 10:eaat3392. doi: 10.1126/scitranslmed.aat3392
8. Véniant MM, Lu S-C, Atangan L, Komorowski R, Stanislaus S, Cheng Y, et al. A GIPR antagonist conjugated to GLP-1 analogues promotes weight loss with improved metabolic parameters in preclinical and phase 1 settings. Nat Metab. (2024) 6:290–303. doi: 10.1038/s42255-023-00966-w
9. Ugleholdt R, Poulsen M-LH, Holst PJ, Irminger J-C, Orskov C, Pedersen J, et al. Prohormone convertase 1/3 is essential for processing of the glucose-dependent insulinotropic polypeptide precursor*. J Biol Chem. (2006) 281:11050–7. doi: 10.1074/jbc.M601203200
10. Kuhre RE, Wewer Albrechtsen NJ, Hartmann B, Deacon CF, Holst JJ. Measurement of the incretin hormones: glucagon-like peptide-1 and glucose-dependent insulinotropic peptide. J Diabetes Complications. (2015) 29:445–50. doi: 10.1016/j.jdiacomp.2014.12.006
11. Buchan AMJ, Polak JM, Capella C, Solcia E, Pearse AGE. Electronimmunocytochemical evidence for the K cell localization of gastric inhibitory polypeptide (GIP) im man. Histochemistry. (1978) 56:37–44. doi: 10.1007/BF00492251
12. Mortensen K, Christensen LL, Holst JJ, Orskov C. GLP-1 and GIP are colocalized in a subset of endocrine cells in the small intestine. Regul Pept. (2003) 114:189–96. doi: 10.1016/S0167-0115(03)00125-3
13. Fujita Y, Wideman RD, Asadi A, Yang GK, Baker R, Webber T, et al. Glucose-dependent insulinotropic polypeptide is expressed in pancreatic islet α-cells and promotes insulin secretion. Gastroenterology. (2010) 138:1966–1975.e1. doi: 10.1053/j.gastro.2010.01.049
14. Adriaenssens AE, Svendsen B, Lam BYH, Yeo GSH, Holst JJ, Reimann F, et al. Transcriptomic profiling of pancreatic alpha, beta and delta cell populations identifies delta cells as a principal target for ghrelin in mouse islets. Diabetologia. (2016) 59:2156–65. doi: 10.1007/s00125-016-4033-1
15. Galvin SG, Kay RG, Foreman R, Larraufie P, Meek CL, Biggs E, et al. The human and mouse islet peptidome: effects of obesity and type 2 diabetes, and assessment of intraislet production of glucagon-like peptide-1. J Proteome Res. (2021) 20:4507–17. doi: 10.1021/acs.jproteome.1c00463
16. Zhang ZQ, Hölscher C. GIP has neuroprotective effects in Alzheimer and Parkinson’s disease models. Glucose-Depend. Insul. Polypept. GIP. (2020) 125:170184. doi: 10.1016/j.peptides.2019.170184
17. Stensen S, Gasbjerg LS, Helsted MM, Hartmann B, Christensen MB, Knop FK. GIP and the gut-bone axis – Physiological, pathophysiological and potential therapeutic implications. Glucose-Depend. Insul. Polypept. GIP. (2020) 125:170197. doi: 10.1016/j.peptides.2019.170197
18. Hammoud R, Drucker DJ. Beyond the pancreas: contrasting cardiometabolic actions of GIP and GLP1. Nat Rev Endocrinol. (2023) 19:201–16. doi: 10.1038/s41574-022-00783-3
19. Ottlecz A, Samson WK, McCann SM. The effects of gastric inhibitory polypeptide (GIP) on the release of anterior pituitary hormones. Peptides. (1985) 6:115–9. doi: 10.1016/0196-9781(85)90086-5
20. Martin B, de Maturana RL, Brenneman R, Walent T, Mattson MP, Maudsley S. Class II G protein-coupled receptors and their ligands in neuronal function and protection. NeuroMolecular Med. (2005) 7:3–36. doi: 10.1385/NMM:7:1-2:003
21. Usdin TB, Mezey E, Button DC, Brownstein MJ, Bonner TI. Gastric inhibitory polypeptide receptor, a member of the secretin-vasoactive intestinal peptide receptor family, is widely distributed in peripheral organs and the brain. Endocrinology. (1993) 133:2861–70. doi: 10.1210/endo.133.6.8243312
22. Holst JJ. The incretin system in healthy humans: The role of GIP and GLP-1. Metabolism. (2019) 96:46–55. doi: 10.1016/j.metabol.2019.04.014
23. Mayo KE, Miller LJ, Bataille D, Dalle S, Göke B, Thorens B, et al. International union of pharmacology. XXXV. The glucagon receptor family. Pharmacol Rev. (2003) 55:167. doi: 10.1124/pr.55.1.6
24. Paratore S, Ciotti M, Basille M, Vaudry D, Gentile A, Parenti R, et al. Gastric inhibitory polypeptide and its receptor are expressed in the central nervous system and support neuronal survival. Cent Nerv Syst Agents Med Chem. (2011) 11:210–22. doi: 10.2174/187152411798047771
25. Hayashi M, Kaye JA, Douglas ER, Joshi NR, Gribble FM, Reimann F, et al. Enteroendocrine cell lineages that differentially control feeding and gut motility. eLife. (2023) 12:e78512. doi: 10.7554/eLife.78512
26. Kaplan AM, Vigna SR. Gastric inhibitory polypeptide (GIP) binding sites in rat brain. Peptides. (1994) 15:297–302. doi: 10.1016/0196-9781(94)90016-7
27. Adriaenssens AE, Biggs EK, Darwish T, Tadross J, Sukthankar T, Girish M, et al. Glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus regulate food intake. Cell Metab. (2019) 30:987–996.e6. doi: 10.1016/j.cmet.2019.07.013
28. Adriaenssens A, Broichhagen J, Bray A, Ast J, Hasib A, Jones B, et al. Hypothalamic and brainstem glucose-dependent insulinotropic polypeptide receptor neurons employ distinct mechanisms to affect feeding. JCI Insight. (2023) 8(10):e16492. doi: 10.1172/jci.insight.164921
29. Costa A, Ai M, Nunn N, Culotta I, Hunter J, Boudjadja MB, et al. Anorectic and aversive effects of GLP-1 receptor agonism are mediated by brainstem cholecystokinin neurons, and modulated by GIP receptor activation. Mol Metab. (2022) 55:101407. doi: 10.1016/j.molmet.2021.101407
30. Smith C, Patterson-Cross R, Woodward O, Lewis J, Chiarugi D, Merkle F, et al. A comparative transcriptomic analysis of glucagon-like peptide-1 receptor- and glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus. Appetite. (2022) 174:106022. doi: 10.1016/j.appet.2022.106022
31. Tadross JA, Steuernagel L, Dowsett GKC, Kentistou KA, Lundh S, Porniece M, et al. A comprehensive spatio-cellular map of the human hypothalamus. Nature. (2025). doi: 10.1038/s41586-024-08504-8
32. Zhang C, Kaye JA, Cai Z, Wang Y, Prescott SL, Liberles SD. Area postrema cell types that mediate nausea-associated behaviors. Neuron. (2021) 109:461–472.e5. doi: 10.1016/j.neuron.2020.11.010
33. Borner T, Geisler CE, Fortin SM, Cosgrove R, Alsina-Fernandez J, Dogra M, et al. GIP receptor agonism attenuates GLP-1 receptor agonist–induced nausea and emesis in preclinical models. Diabetes. (2021) 70:2545–53. doi: 10.2337/db21-0459
34. Ludwig MQ, Cheng W, Gordian D, Lee J, Paulsen SJ, Hansen SN, et al. A genetic map of the mouse dorsal vagal complex and its role in obesity. Nat Metab. (2021) 3:530–45. doi: 10.1038/s42255-021-00363-1
35. Nakagawa A, Satake H, Nakabayashi H, Nishizawa M, Furuya K, Nakano S, et al. Receptor gene expression of glucagon-like peptide-1, but not glucose-dependent insulinotropic polypeptide, in rat nodose ganglion cells. Auton. Neurosci. (2004) 110:36–43. doi: 10.1016/j.autneu.2003.11.001
36. Borner T, Reiner BC, Crist RC, Furst CD, Doebley SA, Halas JG, et al. GIP receptor agonism blocks chemotherapy-induced nausea and vomiting. Mol Metab. (2023) 73:101743. doi: 10.1016/j.molmet.2023.101743
37. Dowsett GKC, Lam BYH, Tadross JA, Cimino I, Rimmington D, Coll AP, et al. A survey of the mouse hindbrain in the fed and fasted states using single-nucleus RNA sequencing. Mol Metab. (2021) 53:101240. doi: 10.1016/j.molmet.2021.101240
38. Higashimoto Y, Simchock J, Liddle RA. Molecular cloning of rat glucose-dependent insulinotropic peptide (GIP). Biochim. Biophys. Acta BBA - Gene Struct Expr. (1992) 1132:72–4. doi: 10.1016/0167-4781(92)90054-4
39. Tseng C-C, Jarboe LA, Landau SB, Williams EK, Wolfe MM. Glucose-dependent insulinotropic peptide: structure of the precursor and tissue-specific expression in rat. Proc Natl Acad Sci U. S. A. (1993) 90:1992–6. doi: 10.1073/pnas.90.5.1992
40. Cho GJ, Ryu S, Kim YH, Kim YS, Cheon EW, Park JM, et al. Upregulation of glucose-dependent insulinotropic polypeptide and its receptor in the retina of streptozotocin-induced diabetic rats. Curr Eye Res. (2002) 25:381–8. doi: 10.1076/ceyr.25.6.381.14238
41. Nyberg J, Anderson MF, Meister B, Alborn AM, Ström AK, Brederlau A, et al. Glucose-dependent insulinotropic polypeptide is expressed in adult hippocampus and induces progenitor cell proliferation. J Neurosci. (2005) 25:1816. doi: 10.1523/JNEUROSCI.4920-04.2005
42. Nyberg J, Jacobsson C, Anderson MF, Eriksson PS. Immunohistochemical distribution of glucose-dependent insulinotropic polypeptide in the adult rat brain. J Neurosci Res. (2007) 85:2099–119. doi: 10.1002/jnr.21349
43. Sondhi S, Castellano J, Chong V, Rogoza R, Skoblenick K, Dyck B, et al. cDNA array reveals increased expression of glucose-dependent insulinotropic polypeptide following chronic clozapine treatment: role in atypical antipsychotic drug-induced adverse metabolic effects. Pharmacogenomics J. (2006) 6:131–40. doi: 10.1038/sj.tpj.6500346
44. Svendsen B, Pais R, Engelstoft MS, Milev NB, Richards P, Christiansen CB, et al. GLP1- and GIP-producing cells rarely overlap and differ by bombesin receptor-2 expression and responsiveness. J Endocrinol. (2016) 228:39–48. doi: 10.1530/joe-15-0247
45. Adriaenssens AE, Gribble FM, Reimann F. The glucose-dependent insulinotropic polypeptide signaling axis in the central nervous system. Glucose-Depend. Insul. Polypept. GIP. (2020) 125:170194. doi: 10.1016/j.peptides.2019.170194
46. Lewis JE, Nuzzaci D, James-Okoro P-P, Montaner M, O’Flaherty Rottenberger E, Darwish T, et al. Stimulating intestinal GIP release reduces food intake and body weight in mice. Mol Metab. (2024) 84:101945. doi: 10.1016/j.molmet.2024.101945
47. Kaneko K, Fu Y, Lin H-Y, Cordonier EL, Mo Q, Gao Y, et al. Gut-derived GIP activates central Rap1 to impair neural leptin sensitivity during overnutrition. J Clin Invest. (2019) 129:3786–91. doi: 10.1172/JCI126107
48. Murphy KG, Dhillo WS, Bloom SR. Gut peptides in the regulation of food intake and energy homeostasis. Endocr. Rev. (2006) 27:719–27. doi: 10.1210/er.2006-0028
49. Müller TD, Finan B, Bloom SR, D’Alessio D, Drucker DJ, Flatt PR, et al. Glucagon-like peptide 1 (GLP-1). Mol Metab. (2019) 30:72–130. doi: 10.1016/j.molmet.2019.09.010
50. Turton MD, O’Shea D, Gunn I, Beak SA, Edwards CMB, Meeran K, et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature. (1996) 379:69–72. doi: 10.1038/379069a0
51. Frías JP, Davies MJ, Rosenstock J, Pérez Manghi FC, Fernández Landó L, Bergman BK, et al. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med. (2021) 385:503–15. doi: 10.1056/NEJMoa2107519
52. Del Prato S, Kahn SE, Pavo I, Weerakkody GJ, Yang Z, Doupis J, et al. Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): a randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet. (2021) 398:1811–24. doi: 10.1016/S0140-6736(21)02188-7
53. Ludvik B, Giorgino F, Jódar E, Frias JP, Fernández Landó L, Brown K, et al. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): a randomised, open-label, parallel-group, phase 3 trial. Lancet. (2021) 398:583–98. doi: 10.1016/S0140-6736(21)01443-4
54. Sikirica M, Martin A, Wood R, Leith A, Piercy J, Higgins V, et al. Reasons for discontinuation of GLP1 receptor agonists: data from a real-world cross-sectional survey of physicians and their patients with type 2 diabetes. Diabetes Metab Syndr Obes. (2017) 29:403–12. doi: 10.2147/DMSO.S141235
55. Müller TD, Blüher M, Tschöp MH, DiMarchi RD. Anti-obesity drug discovery: advances and challenges. Nat Rev Drug Discovery. (2022) 21:201–23. doi: 10.1038/s41573-021-00337-8
56. Frias JP, Deenadayalan S, Erichsen L, Knop FK, Lingvay I, Macura S, et al. Efficacy and safety of co-administered once-weekly cagrilintide 2·4 mg with once-weekly semaglutide 2·4 mg in type 2 diabetes: a multicentre, randomised, double-blind, active-controlled, phase 2 trial. Lancet. (2023) 402:720–30. doi: 10.1016/S0140-6736(23)01163-7
57. Samms RJ, Coghlan MP, Sloop KW. How may GIP enhance the therapeutic efficacy of GLP-1? Trends Endocrinol Metab. (2020) 31:410–21. doi: 10.1016/j.tem.2020.02.006
58. Liu QK. Mechanisms of action and therapeutic applications of GLP-1 and dual GIP/GLP-1 receptor agonists. Front Endocrinol. (2024) 15. doi: 10.3389/fendo.2024.1431292
59. Frias JP, Bastyr EJ, Vignati L, Tschöp MH, Schmitt C, Owen K, et al. The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090-2746, in patients with type 2 diabetes. Cell Metab. (2017) 26:343–352.e2. doi: 10.1016/j.cmet.2017.07.011
60. Rosenstock J, Wysham C, Frías JP, Kaneko S, Lee CJ, Fernández Landó L, et al. Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet. (2021) 398:143–55. doi: 10.1016/S0140-6736(21)01324-6
61. Jastreboff AM, Aronne LJ, Ahmad NN, Wharton S, Connery L, Alves B, et al. Tirzepatide once weekly for the treatment of obesity. N Engl J Med. (2022) 387:205–16. doi: 10.1056/NEJMoa2206038
62. Willard FS, Douros JD, Gabe MBN, Showalter AD, Wainscott DB, Suter TM, et al. Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight. (2020) 5(17):e140532. doi: 10.1172/jci.insight.140532
63. Kim S-J, Nian C, Karunakaran S, Clee SM, Isales CM, McIntosh CHS. GIP-overexpressing mice demonstrate reduced diet-induced obesity and steatosis, and improved glucose homeostasis. PLoS One. (2012) 7:e40156. doi: 10.1371/journal.pone.0040156
64. Zhang Q, Delessa CT, Augustin R, Bakhti M, Colldén G, Drucker DJ, et al. The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling. Cell Metab. (2021) 33(4):833–844.e5. doi: 10.1016/j.cmet.2021.01.015
65. Mroz PA, Finan B, Gelfanov V, Yang B, Tschöp MH, DiMarchi RD, et al. Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism. Mol Metab. (2019) 20:51–62. doi: 10.1016/j.molmet.2018.12.001
66. Miyawaki K, Yamada Y, Ban N, Ihara Y, Tsukiyama K, Zhou H, et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med. (2002) 8:738–42. doi: 10.1038/nm727
67. Bailey CJ, Flatt PR, Kwasowski P, Powell CJ, Marks V. Immunoreactive gastric inhibitory polypeptide and K cell hyperplasia in obese hyperglycaemic (ob/ob) mice fed high fat and high carbohydrate cafeteria diets. Acta Endocrinol (Copenh.). (1986) 112:224–9. doi: 10.1530/acta.0.1120224
68. Flatt PR, Bailey CJ, Kwasowski P, Swanston-Flatt SK, Marks V. Abnormalities of GIP in spontaneous syndromes of obesity and diabetes in mice. Diabetes. (1983) 32:433–5. doi: 10.2337/diab.32.5.433
69. Brøns C, Jensen CB, Storgaard H, Hiscock NJ, White A, Appel JS, et al. Impact of short-term high-fat feeding on glucose and insulin metabolism in young healthy men. J Physiol. (2009) 587:2387–97. doi: 10.1113/jphysiol.2009.169078
70. Creutzfeldt W, Ebert R, Willms B, Frerichs H, Brown JC. Gastric inhibitory polypeptide (GIP) and insulin in obesity: Increased response to stimulation and defective feedback control of serum levels. Diabetologia. (1978) 14:15–24. doi: 10.1007/BF00429703
71. Ponter A, Salter D, Morgan L, Flatt P. The effect of energy source and feeding level on the hormones of the entero-insular axis and plasma glucose in the growing pig. Br J Nutr. (1991) 66:187–97. doi: 10.1079/bjn19910024
72. Shimazu-Kuwahara S, Harada N, Yamane S, Joo E, Sankoda A, Kieffer TJ, et al. Attenuated secretion of glucose-dependent insulinotropic polypeptide (GIP) does not alleviate hyperphagic obesity and insulin resistance in ob/ob mice. Mol Metab. (2017) 6:288–94. doi: 10.1016/j.molmet.2017.01.006
73. McClean PL, Irwin N, Cassidy RS, Holst JJ, Gault VA, Flatt PR. GIP receptor antagonism reverses obesity, insulin resistance, and associated metabolic disturbances induced in mice by prolonged consumption of high-fat diet. Am J Physiol.-Endocrinol. Metab. (2007) 293:E1746–55. doi: 10.1152/ajpendo.00460.2007
74. Ravn P, Madhurantakam C, Kunze S, Matthews E, Priest C, O’Brien S, et al. Structural and pharmacological characterization of novel potent and selective monoclonal antibody antagonists of glucose-dependent insulinotropic polypeptide receptor. J Biol Chem. (2013) 288:19760–72. doi: 10.1074/jbc.M112.426288
75. Fulurija A, Lutz TA, Sladko K, Osto M, Wielinga PY, Bachmann MF, et al. Vaccination against GIP for the treatment of obesity. PLoS One. (2008) 3:e3163. doi: 10.1371/journal.pone.0003163
76. Boylan MO, Glazebrook PA, Tatalovic M, Wolfe MM. Gastric inhibitory polypeptide immunoneutralization attenuates development of obesity in mice. Am J Physiol.-Endocrinol. Metab. (2015) 309:E1008–18. doi: 10.1152/ajpendo.00345.2015
77. Gault VA, McClean PL, Cassidy RS, Irwin N, Flatt PR. Chemical gastric inhibitory polypeptide receptor antagonism protects against obesity, insulin resistance, glucose intolerance and associated disturbances in mice fed high-fat and cafeteria diets. Diabetologia. (2007) 50:1752–62. doi: 10.1007/s00125-007-0710-4
78. Yang B, Gelfanov VM, El K, Chen A, Rohlfs R, DuBois B, et al. Discovery of a potent GIPR peptide antagonist that is effective in rodent and human systems. Mol Metab. (2022) 66:101638. doi: 10.1016/j.molmet.2022.101638
79. Wolfe MM, Apovian CM, Boylan MO. Glucose-dependent insulinotropic polypeptide monoclonal antibodies prevent and treat obesity in wild-type and hyperphagic mice. Obesity. (2023) 31:1499–504. doi: 10.1002/oby.23758
80. Min X, Yie J, Wang J, Chung BC, Huang C-S, Xu H, et al. Molecular mechanism of an antagonistic antibody against glucose-dependent insulinotropic polypeptide receptor. mAbs. (2020) 12:1710047. doi: 10.1080/19420862.2019.1710047
81. Althage MC, Ford EL, Wang S, Tso P, Polonsky KS, Wice BM. Targeted ablation of glucose-dependent insulinotropic polypeptide-producing cells in transgenic mice reduces obesity and insulin resistance induced by a high fat diet*. J Biol Chem. (2008) 283:18365–76. doi: 10.1074/jbc.M710466200
82. Hansotia T, Maida A, Flock G, Yamada Y, Tsukiyama K, Seino Y, et al. Extrapancreatic incretin receptors modulate glucose homeostasis, body weight, and energy expenditure. J Clin Invest. (2007) 117:143–52. doi: 10.1172/JCI25483
83. Svendsen B, Capozzi ME, Nui J, Hannou SA, Finan B, Naylor J, et al. Pharmacological antagonism of the incretin system protects against diet-induced obesity. Mol Metab. (2020) 32:44–55. doi: 10.1016/j.molmet.2019.11.018
84. Scrocchi LA, Drucker DJ. Effects of aging and a high fat diet on body weight and glucose tolerance in glucagon-like peptide-1 receptor(-/-) mice. Endocrinology. (1998) 139:3127–32. doi: 10.1210/endo.139.7.6092
85. Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. (2010) 42:937–48. doi: 10.1038/ng.686
86. Turcot V, Lu Y, Highland HM, Schurmann C, Justice AE, Fine RS, et al. Protein-altering variants associated with body mass index implicate pathways that control energy intake and expenditure in obesity. Nat Genet. (2018) 50:26–41. doi: 10.1038/s41588-017-0011-x
87. Mohammad S, Patel RT, Bruno J, Panhwar MS, Wen J, McGraw TE. A naturally occurring GIP receptor variant undergoes enhanced agonist-induced desensitization, which impairs GIP control of adipose insulin sensitivity. Mol Cell Biol. (2014) 34:3618–29. doi: 10.1128/MCB.00256-14
88. Gabe MBN, van der Velden WJC, Gadgaard S, Smit FX, Hartmann B, Bräuner-Osborne H, et al. Enhanced agonist residence time, internalization rate and signalling of the GIP receptor variant [E354Q] facilitate receptor desensitization and long-term impairment of the GIP system. Basic Clin Pharmacol Toxicol. (2020) 126:122–32. doi: 10.1111/bcpt.13289
89. Kizilkaya HS, Sørensen KV, Kibsgaard CJ, Gasbjerg LS, Hauser AS, Sparre-Ulrich AH, et al. Loss of function glucose-dependent insulinotropic polypeptide receptor variants are associated with alterations in BMI, bone strength and cardiovascular outcomes. Front Cell Dev Biol. (2021) 9:2296–634. doi: 10.3389/fcell.2021.749607
90. Kizilkaya HS, Sørensen KV, Madsen JS, Lindquist P, Douros JD, Bork-Jensen J, et al. Characterization of genetic variants of GIPR reveals a contribution of β-arrestin to metabolic phenotypes. Nat Metab. (2024) 6:1268–81. doi: 10.1038/s42255-024-01061-4
91. Holst JJ, Rosenkilde MM. GIP as a therapeutic target in diabetes and obesity: insight from incretin co-agonists. J Clin Endocrinol Metab. (2020) 105:e2710–6. doi: 10.1210/clinem/dgaa327
92. Killion EA, Chen M, Falsey JR, Sivits G, Hager T, Atangan L, et al. Chronic glucose-dependent insulinotropic polypeptide receptor (GIPR) agonism desensitizes adipocyte GIPR activity mimicking functional GIPR antagonism. Nat Commun. (2020) 11(1):4981. doi: 10.1038/s41467-020-18751-8
93. Jensen MH, Sanni SJ, Riber D, Holst JJ, Rosenkilde MM, Sparre-Ulrich AH. AT-7687, a novel GIPR peptide antagonist, combined with a GLP-1 agonist, leads to enhanced weight loss and metabolic improvements in cynomolgus monkeys. Mol Metab. (2024) 88:102006. doi: 10.1016/j.molmet.2024.102006
94. West JA, Tsakmaki A, Ghosh SS, Parkes DG, Grønlund RV, Pedersen PJ, et al. Chronic peptide-based GIP receptor inhibition exhibits modest glucose metabolic changes in mice when administered either alone or combined with GLP-1 agonism. PLoS One. (2021) 16:e0249239. doi: 10.1371/journal.pone.0249239
95. Irwin N, Hunter K, Flatt PR. Comparison of the metabolic effects of GIP receptor antagonism and PYY(3-36) receptor activation in high fat fed mice. Peptides. (2007) 28:2192–8. doi: 10.1016/j.peptides.2007.08.008
96. Irwin N, Hunter K, Flatt PR. Comparison of independent and combined chronic metabolic effects of GIP and CB1 receptor blockade in high-fat fed mice. Peptides. (2008) 29:1036–41. doi: 10.1016/j.peptides.2008.01.006
97. Eckel RH, Fujimoto WY, Brunzell JD. Gastric inhibitory polypeptide enhanced lipoprotein lipase activity in cultured preadipocytes. Diabetes. (1979) 28:1141–2. doi: 10.2337/diab.28.12.1141
98. Hauner H, Glatting G, Kaminska D, Pfeiffer EF. Effects of gastric inhibitory polypeptide on glucose and lipid metabolism of isolated rat adipocytes. Ann Nutr Metab. (1988) 32:282–8. doi: 10.1159/000177467
99. Beck B, Max J-P. Direct metabolic effects of gastric inhibitory polypeptide (GIP): dissociation at physiological levels of effects on insulin-stimulated fatty acid and glucose incorporation in rat adipose tissue. Diabetologia. (1986) 29:68. doi: 10.1007/BF02427284
100. Kim S-J, Nian C, McIntosh CHS. Activation of lipoprotein lipase by glucose-dependent insulinotropic polypeptide in adipocytes: A ROLE FOR A PROTEIN KINASE B, LKB1, AND AMP-ACTIVATED PROTEIN KINASE CASCADE*. J Biol Chem. (2007) 282:8557–67. doi: 10.1074/jbc.M609088200
101. Kim S-J, Nian C, McIntosh CHS. Resistin is a key mediator of glucose-dependent insulinotropic polypeptide (GIP) stimulation of lipoprotein lipase (LPL) activity in adipocytes. J Biol Chem. (2007) 282:34139–47. doi: 10.1074/jbc.M704896200
102. Starich GH, Bar RS, Mazzaferri EL. GIP increases insulin receptor affinity and cellular sensitivity in adipocytes. Am J Physiol.-Endocrinol. Metab. (1985) 249:E603–7. doi: 10.1152/ajpendo.1985.249.6.E603
103. Song DH, Getty–Kaushik L, Tseng E, Simon J, Corkey BE, Wolfe MM. Glucose-dependent insulinotropic polypeptide enhances adipocyte development and glucose uptake in part through akt activation. Gastroenterology. (2007) 133:1796–805. doi: 10.1053/j.gastro.2007.09.005
104. Campbell JE, Beaudry JL, Svendsen B, Baggio LL, Gordon AN, Ussher JR, et al. GIPR is predominantly localized to nonadipocyte cell types within white adipose tissue. Diabetes. (2022) 71:1115–27. doi: 10.2337/db21-1166
105. Samms RJ, Christe ME, Collins KAL, Pirro V, Droz BA, Holland AK, et al. GIPR agonism mediates weight-independent insulin sensitization by tirzepatide in obese mice. J Clin Invest. (2021) 131(12):e146353. doi: 10.1172/JCI146353
106. Regmi A, Aihara E, Christe ME, Varga G, Beyer TP, Ruan X, et al. Tirzepatide modulates the regulation of adipocyte nutrient metabolism through long-acting activation of the GIP receptor. Cell Metab. (2024) 36:1534–1549.e7. doi: 10.1016/j.cmet.2024.05.010
107. Furber EC, Hyatt K, Collins K, Yu X, Droz BA, Holland A, et al. GIPR agonism enhances TZD-induced insulin sensitivity in obese IR mice. Diabetes. (2023) 73:292–305. doi: 10.2337/db23-0172
108. Yue JTY, Lam TKT. Antiobesogenic effects of central GIPR antagonism. J Clin Invest. (2019) 129:3532–5. doi: 10.1172/JCI130755
109. Ko J, Jang S, Jang S, Park S, Yi J, Choi DK, et al. Glucose-dependent insulinotropic polypeptide (GIP) alleviates ferroptosis in aging-induced brain damage through the Epac/Rap1 signaling pathway. BMB Rep. (2024) 57:417–23. doi: 10.5483/BMBRep.2024-0067
110. Lu S-C, Chen M, Atangan L, Killion EA, Komorowski R, Cheng Y, et al. GIPR antagonist antibodies conjugated to GLP-1 peptide are bispecific molecules that decrease weight in obese mice and monkeys. Cell Rep Med. (2021) 2:100263. doi: 10.1016/j.xcrm.2021.100263
111. Wong CK, McLean BA, Baggio LL, Koehler JA, Hammoud R, Rittig N, et al. Central glucagon-like peptide 1 receptor activation inhibits Toll-like receptor agonist-induced inflammation. Cell Metab. (2024) 36:130–143.e5. doi: 10.1016/j.cmet.2023.11.009
112. Worthington JJ, Reimann F, Gribble FM. Enteroendocrine cells-sensory sentinels of the intestinal environment and orchestrators of mucosal immunity. Mucosal Immunol. (2018) 11:3–20. doi: 10.1038/mi.2017.73
113. Gögebakan Ö., Osterhoff MA, Schüler R, Pivovarova O, Kruse M, Seltmann A-C, et al. GIP increases adipose tissue expression and blood levels of MCP-1 in humans and links high energy diets to inflammation: a randomised trial. Diabetologia. (2015) 58:1759–68. doi: 10.1007/s00125-015-3618-4
114. Góralska J, Raźny U, Polus A, Stancel-Możwiłło J, Chojnacka M, Gruca A, et al. Pro-inflammatory gene expression profile in obese adults with high plasma GIP levels. Int J Obes. (2018) 42:826–34. doi: 10.1038/ijo.2017.305
115. Fu Y, Kaneko K, Lin H-Y, Mo Q, Xu Y, Suganami T, et al. Gut hormone GIP induces inflammation and insulin resistance in the hypothalamus. Endocrinology. (2020) 161:bqaa102. doi: 10.1210/endocr/bqaa102
116. Maskery M, Goulding EM, Gengler S, Melchiorsen JU, Rosenkilde MM, Hölscher C. The dual GLP-1/GIP receptor agonist DA4-JC shows superior protective properties compared to the GLP-1 analogue liraglutide in the APP/PS1 mouse model of alzheimer’s disease. Am J Alzheimers Dis Dementias®. (2020) 35:1533317520953041. doi: 10.1177/1533317520953041
117. Duffy AM, Hölscher C. The incretin analogue D-Ala2GIP reduces plaque load, astrogliosis and oxidative stress in an APP/PS1 mouse model of Alzheimer’s disease. Neuroscience. (2013) 228:294–300. doi: 10.1016/j.neuroscience.2012.10.045
118. Delzenne N, Blundell J, Brouns F, Cunningham K, De Graaf K, Erkner A, et al. Gastrointestinal targets of appetite regulation in humans. Obes Rev. (2010) 11:234–50. doi: 10.1111/j.1467-789X.2009.00707.x
119. Farooqi IS, Bullmore E, Keogh J, Gillard J, O’Rahilly S, Fletcher PC. Leptin regulates striatal regions and human eating behavior. Science. (2007) 317:1355–5. doi: 10.1126/science.1144599
120. Morris DL, Rui L. Recent advances in understanding leptin signaling and leptin resistance. Am J Physiol.-Endocrinol. Metab. (2009) 297:E1247–59. doi: 10.1152/ajpendo.00274.2009
121. Myers MG, Affinati AH, Richardson N, Schwartz MW. Central nervous system regulation of organismal energy and glucose homeostasis. Nat Metab. (2021) 3:737–50. doi: 10.1038/s42255-021-00408-5
122. Rodríguez E, Blázquez J, Guerra M. The design of barriers in the hypothalamus allows the median eminence and the arcuate nucleus to enjoy private milieus: The former opens to the portal blood and the latter to the cerebrospinal fluid. Peptides. (2010) 31:757–76. doi: 10.1016/j.peptides.2010.01.003
123. Grill HJ, Hayes MR. Hindbrain neurons as an essential hub in the neuroanatomically distributed control of energy balance. Cell Metab. (2012) 16:296–309. doi: 10.1016/j.cmet.2012.06.015
124. Cheng W, Gordian D, Ludwig MQ, Pers TH, Seeley RJ, Myers MG. Hindbrain circuits in the control of eating behaviour and energy balance. Nat Metab. (2022) 4:826–35. doi: 10.1038/s42255-022-00606-9
125. Morton GJ, Blevins JE, Williams DL, Niswender KD, Gelling RW, Rhodes CJ, et al. Leptin action in the forebrain regulates the hindbrain response to satiety signals. J Clin Invest. (2005) 115:703–10. doi: 10.1172/JCI22081
126. Schwartz GJ. Integrative capacity of the caudal brainstem in the control of food intake. Phil. Trans R Soc B. (2006) 361(1471):1275–80. doi: 10.1098/rstb.2006.1862
127. Oteng A-B, Liu L, Cui Y, Gavrilova O, Lu H, Chen M, et al. Activation of Gs signaling in mouse enteroendocrine K cells greatly improves obesity- and diabetes-related metabolic deficits. J Clin Invest. (2024) 134(24):e182325. doi: 10.1172/JCI182325
128. Gabery S, Salinas CG, Paulsen SJ, Ahnfelt-Rønne J, Alanentalo T, Baquero AF, et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight. (2021) 5(6):e133429. doi: 10.1172/jci.insight.133429
129. Liskiewicz A, Khalil A, Liskiewicz D, Novikoff A, Grandl G, Maity-Kumar G, et al. Glucose-dependent insulinotropic polypeptide regulates body weight and food intake via GABAergic neurons in mice. Nat Metab. (2023) 5(12):2075–85. doi: 10.1038/s42255-023-00931-7
130. Ast J, Arvaniti A, Fine NHF, Nasteska D, Ashford FB, Stamataki Z, et al. Super-resolution microscopy compatible fluorescent probes reveal endogenous glucagon-like peptide-1 receptor distribution and dynamics. Nat Commun. (2020) 11:467. doi: 10.1038/s41467-020-14309-w
131. Samms RJ, Sloop KW, Gribble FM, Reimann F, Adriaenssens AE. GIPR function in the central nervous system: implications and novel perspectives for GIP-based therapies in treating metabolic disorders. Diabetes. (2021) 70:1938–44. doi: 10.2337/dbi21-0002
132. Wean J, Kowalsky AH, Laker R, Will S, Drucker DJ, Rhodes CJ, et al. Specific loss of GIPR signaling in GABAergic neurons enhances GLP-1R agonist-induced body weight loss. Mol Metab. (2024) 26:102074. doi: 10.1016/j.molmet.2024.102074
133. Zhang C, Vincelette LK, Reimann F, Liberles SD. A brainstem circuit for nausea suppression. Cell Rep. (2022) 39:110953. doi: 10.1016/j.celrep.2022.110953
134. Samms RJ, Cosgrove R, Snider BM, Furber EC, Droz BA, Briere DA, et al. GIPR agonism inhibits PYY-induced nausea-like behavior. Diabetes. (2022) 71:1410–23. doi: 10.2337/db21-0848
135. Cai J, Chen J, Ortiz-Guzman J, Huang J, Arenkiel BR, Wang Y, et al. AgRP neurons are not indispensable for body weight maintenance in adult mice. Cell Rep. (2023) 42(7):112789. doi: 10.1016/j.celrep.2023.112789
136. Aponte Y, Atasoy D, Sternson SM. AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci. (2011) 14:351–5. doi: 10.1038/nn.2739
137. Gropp E, Shanabrough M, Borok E, Xu AW, Janoschek R, Buch T, et al. Agouti-related peptide–expressing neurons are mandatory for feeding. Nat Neurosci. (2005) 8:1289–91. doi: 10.1038/nn1548
138. Krashes MJ, Koda S, Ye CP, Rogan SC, Adams AC, Cusher DS, et al. Rapid, reversible activation of AgRP neurons drives feeding behavior in mice. J Clin Invest. (2011) 121:1424–8. doi: 10.1172/JCI46229
139. Luquet S, Perez FA, Hnasko TS, Palmiter RD. NPY/agRP neurons are essential for feeding in adult mice but can be ablated in neonates. Science. (2005) 310:683–5. doi: 10.1126/science.1115524
140. McMorrow HE, Lorch CM, Hayes NW, Fleps SW, Frydman JA, Xia JL, et al. Incretin hormones and pharmacomimetics rapidly inhibit AgRP neuron activity to suppress appetite. bioRxiv. (2024) 2024.03.18.585583. doi: 10.1101/2024.03.18.585583
141. Tsang AH, Nuzzaci D, Darwish T, Samudrala H, Blouet C. Nutrient sensing in the nucleus of the solitary tract mediates non-aversive suppression of feeding via inhibition of AgRP neurons. Mol Metab. (2020) 42:101070. doi: 10.1016/j.molmet.2020.101070
142. Aklan I, Sayar Atasoy N, Yavuz Y, Ates T, Coban I, Koksalar F, et al. NTS catecholamine neurons mediate hypoglycemic hunger via medial hypothalamic feeding pathways. Cell Metab. (2020) 31:313–26. doi: 10.1016/j.cmet.2019.11.016
143. D’Agostino G, Lyons DJ, Cristiano C, Burke LK, Madara JC, Campbell JN, et al. Appetite controlled by a cholecystokinin nucleus of the solitary tract to hypothalamus neurocircuit. eLife. (2016) 5:e12225. doi: 10.7554/eLife.12225
144. Martinez de Morentin PB, Gonzalez JA, Dowsett GKC, Martynova Y, Yeo GSH, Sylantyev S, et al. A brainstem to hypothalamic arcuate nucleus GABAergic circuit drives feeding. Curr Biol. (2024) 34:1646–1656.e4. doi: 10.1016/j.cub.2024.02.074
145. Han W, Wang L, Ohbayashi K, Takeuchi M, O’Farrell L, Coskun T, et al. Glucose-dependent insulinotropic polypeptide counteracts diet-induced obesity along with reduced feeding, elevated plasma leptin and activation of leptin-responsive and proopiomelanocortin neurons in the arcuate nucleus. Diabetes Obes Metab. (2023) 25:1534–46. doi: 10.1111/dom.15001
146. Kanoski SE, Rupprecht LE, Fortin SM, De Jonghe BC, Hayes MR. The role of nausea in food intake and body weight suppression by peripheral GLP-1 receptor agonists, exendin-4 and liraglutide. Neuropharmacology. (2012) 62:1916–27. doi: 10.1016/j.neuropharm.2011.12.022
147. Halatchev I, Cone R. Peripheral administration of PYY3–36 produces conditioned taste aversion in mice. Cell Metab. (2005) 1:159–68. doi: 10.1016/j.cmet.2005.02.003
148. Batterham RL, Cowley MA, Small CJ, Herzog H, Cohen MA, Dakin CL, et al. Gut hormone PYY3-36 physiologically inhibits food intake. Nature. (2002) 418:650–4. doi: 10.1038/nature00887
149. Asami T, Nishizawa N, Niida A. Gip receptor activating peptide. Int Pat. Appl. (2018). WO2018181864A1 Takeda Pharm. Co. Ltd. Available online at: https://patents.google.com/patent/WO2018181864A1/en.
150. Borison HL. Area Postrema: Chemoreceptor circumventricular organ of the medulla oblongata. Prog Neurobiol. (1989) 32:351–90. doi: 10.1016/0301-0082(89)90028-2
151. Miller AD, Leslie RA. The area postrema and vomiting. Front Neuroendocrinol. (1994) 15:301–20. doi: 10.1006/frne.1994.1012
152. Price CJ, Hoyda TD, Ferguson AV. The area postrema: A brain monitor and integrator of systemic autonomic state. Neuroscientist. (2008) 14:182–94. doi: 10.1177/1073858407311100
153. Neuwelt EA, Bauer B, Fahlke C, Fricker G, Iadecola C, Janigro D, et al. Engaging neuroscience to advance translational research in brain barrier biology. Nat Rev Neurosci. (2011) 12:169–82. doi: 10.1038/nrn2995
154. Chetram Deochand MT. CSF and brain indices of insulin resistance, oxidative stress and neuro-inflammation in early versus late alzheimer’s disease. J Alzheimer’s Dis Park. (2013) 03:128. doi: 10.4172/2161-0460.1000128
155. Faivre E, Hölscher C. Neuroprotective effects of D-Ala2GIP on Alzheimer’s disease biomarkers in an APP/PS1 mouse model. Alzheimers Res Ther. (2013) 5:20. doi: 10.1186/alzrt174
156. Olofsson LE, Unger EK, Cheung CC, Xu AW. Modulation of AgRP-neuronal function by SOCS3 as an initiating event in diet-induced hypothalamic leptin resistance. Proc Natl Acad Sci. (2013) 110:E697–706. doi: 10.1073/pnas.1218284110
157. Faouzi M, Leshan R, Björnholm M, Hennessey T, Jones J, Münzberg H. Differential accessibility of circulating leptin to individual hypothalamic sites. Endocrinology. (2007) 148:5414–23. doi: 10.1210/en.2007-0655
158. Merino B, Díez-Fernández C, Ruiz-Gayo M, Somoza B. Choroid plexus epithelial cells co-express the long and short form of the leptin receptor. Neurosci Lett. (2006) 393:269–72. doi: 10.1016/j.neulet.2005.10.003
159. Langlet F, Levin BE, Luquet S, Mazzone M, Messina A, Dunn-Meynell AA, et al. Tanycytic VEGF-A boosts blood-hypothalamus barrier plasticity and access of metabolic signals to the arcuate nucleus in response to fasting. Cell Metab. (2013) 17:607–17. doi: 10.1016/j.cmet.2013.03.004
160. Maolood N, Meister B. Protein components of the blood–brain barrier (BBB) in the brainstem area postrema–nucleus tractus solitarius region. J Chem Neuroanat. (2009) 37:182–95. doi: 10.1016/j.jchemneu.2008.12.007
161. Imbernon M, Saponaro C, Helms HCC, Duquenne M, Fernandois D, Deligia E, et al. Tanycytes control hypothalamic liraglutide uptake and its anti-obesity actions. Cell Metab. (2022) 34:1054–1063.e7. doi: 10.1016/j.cmet.2022.06.002
162. Mullier A, Bouret SG, Prevot V, Dehouck B. Differential distribution of tight junction proteins suggests a role for tanycytes in blood-hypothalamus barrier regulation in the adult mouse brain. J Comp Neurol. (2010) 518:943–62. doi: 10.1002/cne.22273
163. Kohnke S, Buller S, Nuzzaci D, Ridley K, Lam B, Pivonkova H, et al. Nutritional regulation of oligodendrocyte differentiation regulates perineuronal net remodeling in the median eminence. Cell Rep. (2021) 36:109362. doi: 10.1016/j.celrep.2021.109362
164. Steuernagel L, Lam BYH, Klemm P, Dowsett GKC, Bauder CA, Tadross JA, et al. HypoMap—a unified single-cell gene expression atlas of the murine hypothalamus. Nat Metab. (2022) 4:1402–19. doi: 10.1038/s42255-022-00657-y
165. Hansford R, Buller S, Tsang A, Benoit S, Roberts A, Erskine E, et al. Glucose-dependent Insulinotropic Polypeptide Receptor Signalling in Oligodendrocytes Increases the Weight Loss Action of GLP-1R Agonism. [Preprint]. Available online at: https://papers.ssrn.com/sol3/papers.cfm?abstract_id=4704168.
Keywords: Glucose-dependent insulinotropic polypeptide (GIP)/GIP-receptor (GIPR), Glucagon-like peptide-1 receptor (GLP1R), food intake, body weight, central nervous system, obesity, diabetes
Citation: James-Okoro P-P, Lewis JE, Gribble FM and Reimann F (2025) The role of GIPR in food intake control. Front. Endocrinol. 16:1532076. doi: 10.3389/fendo.2025.1532076
Received: 21 November 2024; Accepted: 25 February 2025;
Published: 17 March 2025.
Edited by:
Tamer Coskun, Eli Lilly, United StatesReviewed by:
Simon M. Luckman, The University of Manchester, United KingdomLuiza Ghila, University of Bergen, Norway
Copyright © 2025 James-Okoro, Lewis, Gribble and Reimann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Frank Reimann, ZnIyMjJAY2FtLmFjLnVr; Fiona Mary Gribble, Zm1nMjNAY2FtLmFjLnVr