 Jonathan D. Douros
Jonathan D. Douros Jonathan N. Flak
Jonathan N. Flak Patrick J. Knerr
Patrick J. Knerr- 1Indiana Biosciences Research Institute, Indianapolis, IN, United States
- 2Department of Pharmacology and Toxicology, Indiana University School of Medicine, Indianapolis, IN, United States
1 Introduction
Incretin peptides are secreted from the intestine after nutrient ingestion and enhance glucose-stimulated insulin secretion (1). In healthy individuals glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic polypeptide (GIP) are the primary incretin factors (2, 3). However, GIP receptor (GIPR) activity is diminished in patients with type 2 diabetes (T2D), while GLP-1 receptor (GLP-1R) function remains intact (4). This finding, along with the obesogenic physiologic role of GIP (5, 6), motivated the development of GLP-1R agonists over GIPR agonists for the treatment of T2D (7–11). Long acting GLP-1 analogues were later approved for obesity based upon early observations that physiologic and pharmacologic GLP-1R agonism reduces food intake in preclinical models (12) and pioneering clinical trials of long acting GLP-1 analogues (13). Despite the substantial improvements in body weight and glucose control elicited by GLP-1R agonists, patient uptake and compliance are challenged by frequent gastrointestinal adverse events (GI AEs), including nausea and vomiting, that necessitate careful dose titration regimens to achieve efficacious exposure (14). Emerging preclinical and clinical evidence suggests that GIPR agonism can play a role in reducing the GI AEs of GLP-1R agonism and enable greater therapeutic potential.
2 Weight loss pharmacotherapies
2.1 GLP-1R and GIPR agonism reduce body weight in patients with obesity
Early GLP-1R agonists were approved to treat T2D (13, 15, 16), but the potential weight lowering efficacy for this class of molecules first seen in rodents was not confirmed in humans until clinical trials with the first long-acting GLP-1 analogue, liraglutide (12). Over 56 weeks, liraglutide (3 mg daily) induced 6.0% weight loss in patients with obesity and T2D (17) and 8.0% weight loss in patients with obesity alone (unless otherwise defined, weight loss is placebo controlled from baseline) (18). Further optimization of GLP-1 analogues to permit once-weekly time action led to the discovery of Fc protracted dulaglutide and the fatty diacid acylated semaglutide. Dulaglutide (1.5 mg weekly) produced middling weight reduction (3.1%) over 26 weeks in patients with obesity and T2D (13). However, semaglutide (2.4 mg weekly) is said to have “broken the sound barrier” (19), inducing 9.6% weight loss in patients with obesity and T2D (20) and 14.9% weight loss in patients with obesity alone (21) over 68 weeks of treatment. In fact, liraglutide (1.8 mg daily) and semaglutide (0.5 mg weekly) both significantly outperformed dulaglutide (1.5 mg weekly) for weight loss in head-to-head trials at doses that induce comparable HbA1c lowering (13, 22).
The mechanism underlying the impressive weight loss potential of GLP-1R agonists is primarily, if not exclusively, mediated by a reduction in energy intake. For instance, native GLP-1 infusion acutely reduced food intake in individuals without (23) or with T2D (24), and the short acting GLP-1R agonist exenatide exerted the same effect in patients with obesity (25). A similar effect was seen with chronic dosing of optimized, long acting GLP-1 analogues liraglutide and semaglutide in patients with obesity (26, 27) or T2D (28, 29). Clinical and preclinical data indicate that GLP-1R agonism does not impact energy expenditure or nutrient absorption/accretion over extended treatment (30). In fact, there is a well-documented counter-regulatory reduction in metabolic rate in both humans (31, 32) and rodents (30, 33) treated with GLP-1R agonists for extended periods due to the reduction food intake. Thus, all current evidence points to the suppression of energy intake being the underlying mechanism responsible for the beneficial effects of GLP-1R agonists on body weight in patients with obesity and/or T2D.
While GLP-1R agonism effectively reduces body weight in patients, there is a persistent need for greater efficacy of weight loss and comorbidity resolution. Unimolecular, multi-receptor agonists have emerged to meet this growing need. While numerous proof-of-concept candidate molecules have been disclosed (34–43), the only approved example of multi-receptor agonism for obesity and T2D is the dual GLP-1R/GIPR co-agonist tirzepatide (marketed as Mounjaro® and Zepbound®) (34, 44). Tirzepatide (15 mg weekly) drives 15.7% weight loss over 72 weeks in patients with obesity and T2D (45) and 22.5% weight loss in patients with obesity alone (46). Receptor occupancy analysis demonstrates that 5 mg weekly tirzepatide engages the GLP-1R to a similar degree as 1 mg weekly semaglutide (47); at this dose, tirzeaptide drives ~8.3% weight loss compared to 6.6% for semaglutide over 40 weeks in the head-to-head SURPASS-2 trial in patients with obesity and T2D. Thus, tirzepatide appears to outperform semaglutide at doses that comparably engage the GLP-1R. This phenomenon has been widely attributed to two hypotheses that are not mutually exclusive. The first is that tirzepatide is a partial, biased GLP-1R agonist, a profile that confers superior glucose and weight lowering in preclinical models (47–51). Second, tirzepatide is a full, potent GIPR agonist. GIPR monoagonism drives weight loss and food intake reduction on its own in both humans (52) and rodents (53–55). Tirzepatide stimulates insulin secretion through the GIPR in human islets from healthy donors, supporting a role for GIPR agonism in its pharmacology (56). In further support of this ex vivo finding, the GIPR monoagonist LY3537021 reduced body weight by ~4.1% over 8 weeks in patients with T2D (n = 18) (57). It should be noted that GIPR antagonism paradoxically produces additive weight loss when paired to GLP-1R agonism in preclinical studies (58–60) and that this mechanism appears relevant in patients with obesity (61). This phenomenon has been discussed in detail elsewhere (62). While dual receptor co-agonism offers enhanced weight loss and metabolic benefits, it also appears to offer upside in mitigating GI AEs.
2.2 GIPR agonism serves to reduce GLP-1R mediated GI AEs in the clinic
Treatment with GLP-1R agonists can drive nausea, diarrhea, vomiting, and constipation, which often results in discontinuation of treatment (6-10% of patients) or reduction in dose (~15% of patients) (63). The most pronounced effects occur acutely upon treatment initiation and wane over the course of the first 90 days of exposure. This phenomenon is clearly observed in the STEP 2 trial of semaglutide (2.4 mg weekly), where a cumulative 33.7% of patients with obesity and T2D reported nausea throughout the duration of the trial (20). An elegant data analysis of the STEP-2 protocol shows the temporal dynamic of this effect, where the incidence of nausea grew from ~5% at the beginning of the dose escalation period (week 1; 0.25 mg weekly) to ~15% by the end of the dose escalation (week 13; 2.4 mg weekly). Over the next 56 weeks, patients reporting nausea steadily decreased to ~8% by the end of the trial. We will treat nausea as a proxy for GI AEs because similar patterns were reported for diarrhea, vomiting, and constipation across trials.
It has been hypothesized that the deleterious, nauseating effects of GLP-1R agonists and the beneficial, food-intake reducing effects are inextricably linked. However, this hypothesis is losing traction in light of emerging clinical data. In the SURPASS-2 trial, patients with obesity and T2D who received tirzeapatide (5 mg weekly) achieved greater HbA1c and body weight reductions than those receiving semaglutide (1 mg weekly) during the 40 week study (64) despite comparable putative GLP-1R occupancy at these doses (47). Interestingly, patients receiving tirzepatide report reduced overall GI AEs (40%), nausea (17.4%), diarrhea (13.2%) and vomiting (5.7%) compared to patients on semaglutide (43%, 19.2%, 13.7%, and 8.1% respectively). This is suggestive but not demonstrative that GIPR agonism reduces the GI AE incidence associated with GLP-1R agonism. Seminal studies by Knop et al. demonstrate that the addition of a long-acting GIPR agonist (LY3537021) significantly reduced the total number of GI AEs induced with liraglutide treatment by 16% and numerically reduced nausea by 15% and vomiting by 3%, but not diarrhea (65). It should be noted that, while the tolerability and efficacy profile of the GLP-1R agonist/GIPR antagonizing antibody maridebart cafraglutide warrants enthusiasm, it has not yet been directly compared to GLP-1R agonism or dual GLP-1R/GIPR agonism in the clinical setting. Collectively, these data demonstrate GIPR agonism can improve the tolerability profile of GLP-1R agonists in humans.
With these points in mind, we undertook a broad assessment of published clinical data, curating the body weight loss (% from baseline), glycosylated hemoglobin levels (HbA1c), and nausea responses (% patients reporting) from the SUSTAIN 3, SUSTAIN 7, SCALE Diabetes, SURPASS 2, and STEP 2 trials (17, 22, 64, 66, 67) (Figures 1A–C). We assessed the GLP-1R agonists (exenatide, liraglutide, dulaglutide, semaglutide) and the dual GIPR/GLP-1R co-agonist tirzepatide as separate classes. We selected these trials due to their similarities in assessing patients with both obesity and T2D, treatment duration, clinical development phase (phase 3), and use of semaglutide as an active treatment arm. We also provide an assessment of body weight loss and nausea induced by semaglutide and tirzeapatide across patients with obesity and T2D in the STEP 2 and SURPASS 2 trials compared to those with obesity but not T2D in the STEP 1 and SURMOUNT 1 trials (Figure 2). All drugs followed a similar trend in which the calculated circulating concentration of the drug at stead state (Css) exhibits significant positive association with body weight loss (Figure 1D). This relationship was not present for Css and HbA1c, potentially due to a plateau in glycemic control for both drug classes (Figure 1E). The GLP-1R monoagonists displayed a significant positive correlation between Css and nausea, which is not observed for the GIPR/GLP-1R dual-agonist (Figure 1F). Additionally, it is clear that tirzeaptide induces less nausea per nmol in circulation compared to the GLP-1R monoagonists despite the improvement in weight loss. In contrast, co-treatment of semaglutide with the amylin receptor agonist cagrilintide resulted in 29% of patients with obesity and T2D reporting nausea compared to 13% for cagrilintide alone and 16% for semaglutide alone (68). This analysis supports the hypothesis that nausea suppression by GIPR agonism (57, 65, 69) contributes to the better tolerability profile of tirzepatide (15 mg) at doses 6.25x higher than semaglutide (2.4 mg).
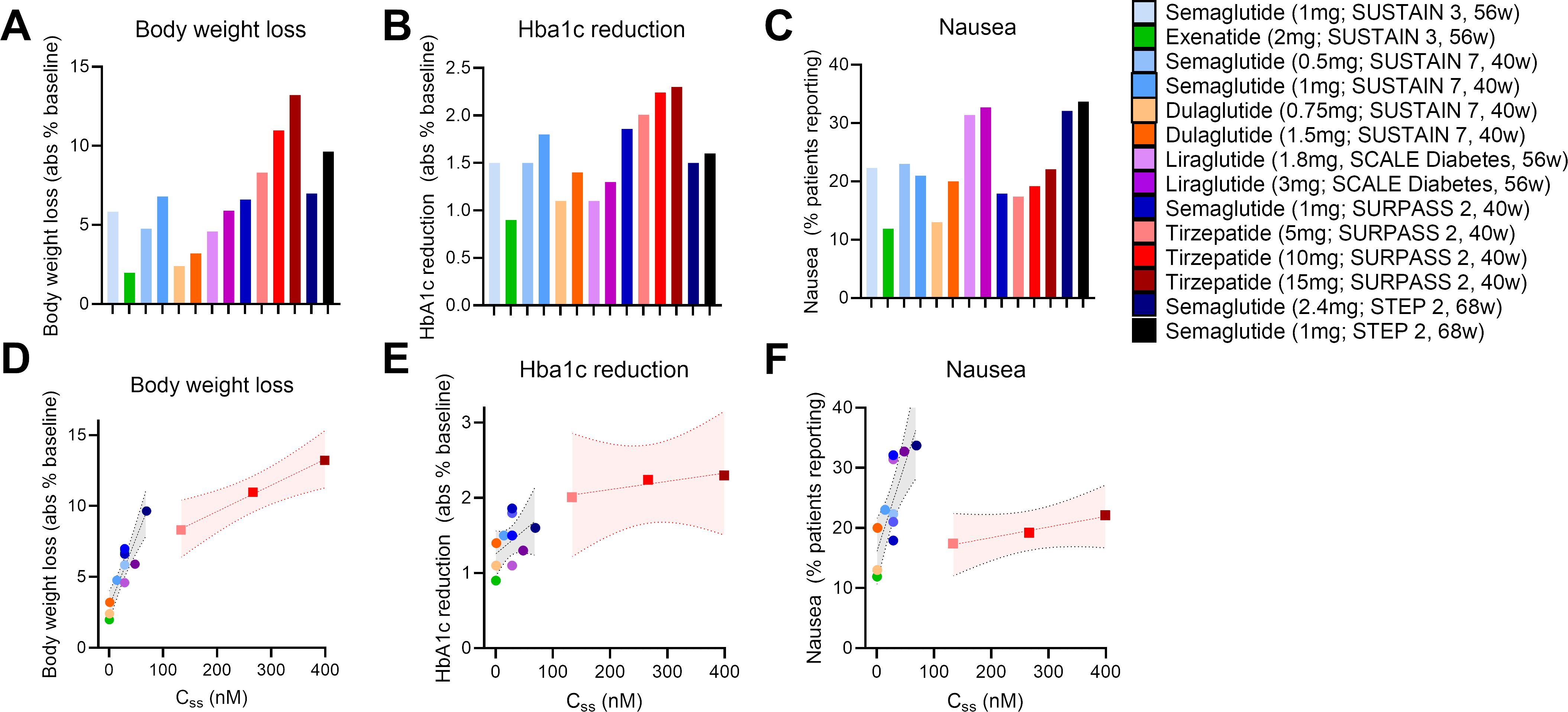
Figure 1. Tirzepatide exhits lower nausea compared to GLP-1R monoagonists across clinical trials. (A) Body weight loss, (B) HbA1c reduction, and (C) nausea reported across clinical trials as outlined. (D) Body weight loss, (E) HbA1c reduction, and (F) nausea as a function of the calculated circulating drug exposure (Concentration at steady state; Css).
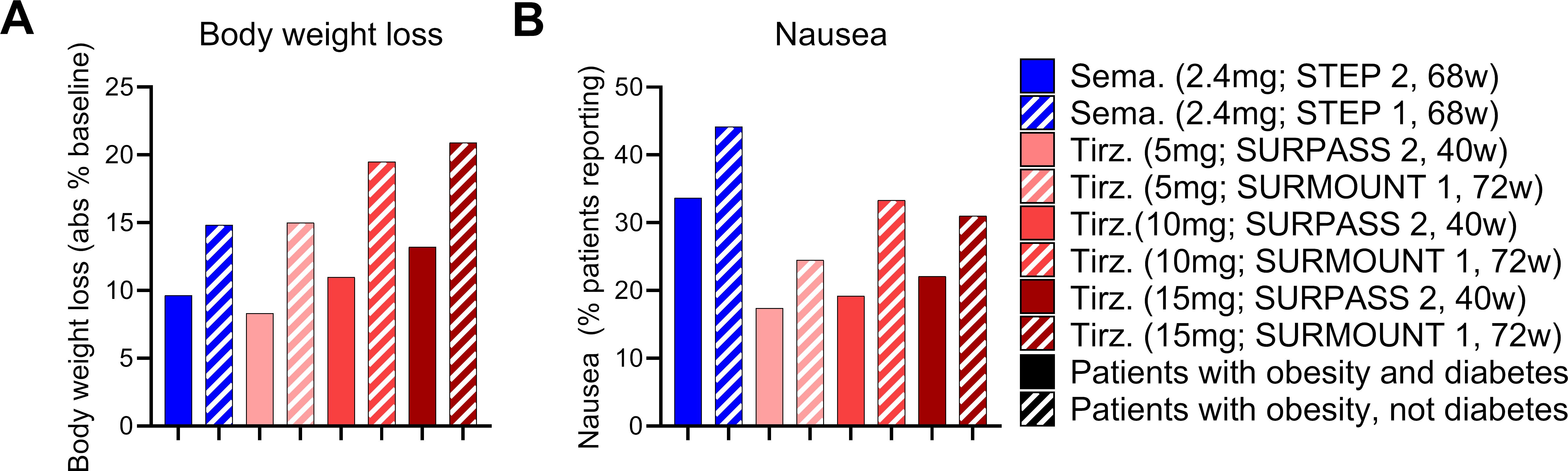
Figure 2. The effects of tirzeaptide and semaglutide are reduced in patients with obesity and diabetes compared to patients with obesity alone. (A) Body weight loss and (B) nausea reported across clinical trials as outlined. Groups in blue were treated with semaglutide while groups in red were treated with tirzepatide. Patients with obesity and T2D are shown in solid bars, while patients with obesity but not T2D are shown in the hatched bars.
3 Mechanisms of action for GLP-1R agonists to control food intake
GLP-1R agonists primarily act to reduce body weight by suppressing energy intake. Preclinical studies show that both short acting agonists (i.e. exenatide) (50) and long acting agonists (i.e. liraglutide, semaglutide) (35, 37) acutely reduce food intake in rodents. Depending on the molecular properties (33), these agonists exert their effects by activating GLP-1R+ glutamatergic neuronal populations (70, 71), but not GABAergic neurons (72). It has been hypothesized that the effects of GLP-1R agonists to reduce energy intake are at least partially driven by nausea and other GI AEs. Under this model, GI AEs are an essential feature, not a bug, of the pharmacology. However, key preclinical studies in mice indicate otherwise. GLP-1R agonists reduce food intake by triggering both aversive or emetic neural and satiety signals which are dissociable in rodents (73–76). This finding opens the door for the development of weight lowering pharmacotherapies that suppress food intake without triggering nausea by targeting specific subsets of GLP-1R+ neurons. Tailoring a molecule to this specific purpose requires a substantial understanding of how the current class of GLP-1R agonists access and interact with the satiating and aversive neuronal receptor populations.
Acute treatment of diet-induced obese mice with labelled semaglutide has been shown to allow the drug to access circumventricular organs that lack a substantial blood brain barrier, including the area postrema (AP), median eminence (ME), vascular organ of the lamina terminalis (OV), and subfornical organ (SFO) (33). In addition, labelled semaglutide can also cross the blood brain barrier to the caudal lateral septal nucleus (LSc), septofimbrial nucleus (SF), arcuate nucleus (ARC), median preoptic nucleus (MnPO), dorsal vagal complex (DVC). At steady state, labelled semaglutide also appeared in the choroid plexus (CHPL), dorsomedial hypothalamic nucleus (DMH), medial mammillary nucleus (MM), paraventricular hypothalamus (PVH), supraoptic nucleus (SO), and tuberal nucleus (TU) (Figure 3). In rodents, intracerebroventricular (ICV) administration of the GLP-1R antagonist exendin-9 suppressed the effects of peripheral GLP-1R agonist administration (liraglutide and exenatide) on food intake reduction. Exendin-9 administered IP was unable to block this effect, indicating a dominant role for CNS GLP-1R populations (77). Indeed, CNS glutamatergic neuron receptor populations, but not GABAergic neurons, are necessary for liraglutide-induced food intake and body weight reduction (71, 72). Furthermore, studies using a peripherally administered exenatide analogue conjugated to vitamin B12, which is sterically hindered from accessing regions protected by the blood brain barrier, improves glucose control in comparable fashion to unmodified exenatide, presumably at the level of the β-cell, but does not induce emesis in the house musk shrew (Suncus murinus) or rats (78, 79). It should be noted that physiologic GLP-1R agonism in the periphery mediates food intake reduction via vagal afferent signaling as rats with a surgical subdiaphragmatic vagal deafferentation (SDA) exhibit a more mild food intake suppression in response to acute administration of GLP-1 analogues than sham operated animals (80, 81). Collectively, this indicates a role for CNS GLP-1R populations to mediate food intake suppression by pharmacologic, and to a lesser degree physiologic, GLP-1R agonism.
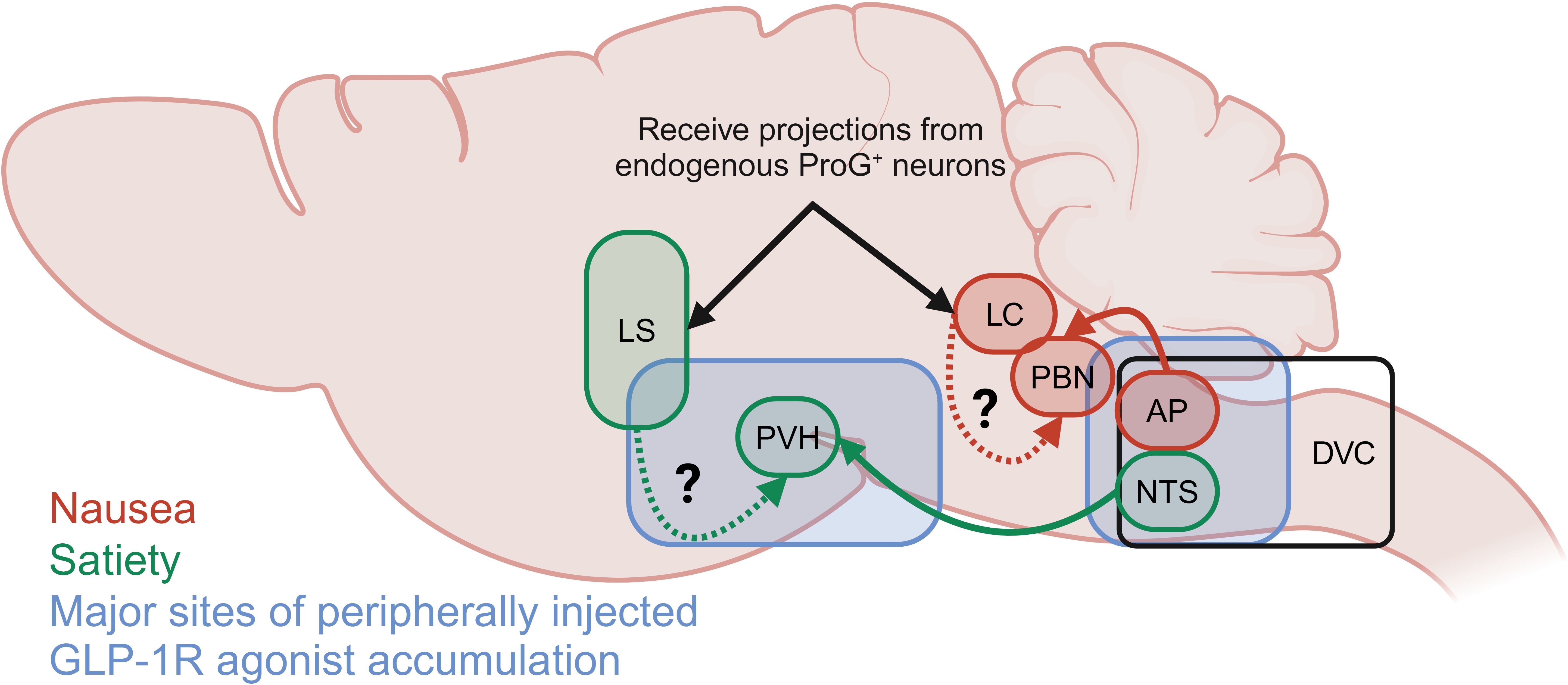
Figure 3. Summary of the primary brain depots for semaglutide as reported by Gabery et al. (33) and neuronal populations reported to mediate GLP-1R agonist-induced satiety (green) and food aversive, avoidant or nauseating behavior (red). Neuronal populations indicated by black arrows receive projections from pre-proglucagon positive (ProG+) neurons.
The specific effects of GLP-1R agonism on food avoidance, food aversion and emetic behaviors have been delineated more clearly in recent years. While mice and rats do not vomit, GLP-1R agonism induces emesis in the musk shrew and is associated with an increase in cFOS staining in the AP, nucleus of the solitary tract (NTS), DMH, VMH, lateral hypothalamus (LH), and PVH that is blocked by GLP-1R antagonism (77). Interestingly, daily peripheral exenatide administration induced a transient reduction in food intake, which regresses to baseline over time. This differs from the pattern of liraglutide, which induced an initial reduction in food intake with a transient increase in kaolin intake that both undergo partial tachyphylaxis over time (82). This comports with the human data showing that different GLP-1 analogues can have different food intake suppression and nausea profiles (82).
Location of the GLP-1R population plays an essential role in satiating versus aversive effects of GLP-1R agonists (Figure 3). Hypothalamic ARC or nodose ganglion (NG) GLP-1Rs are not necessary for the food intake reducing effects of GLP-1R agonists, while the hindbrain DVC is essential for this function (76). GLP-1R + neurons in the DVC, including the AP, are predominately (40-65%) responsive to aversive signals (LiCl, cinacalcet). Additionally, GLP-1R + neurons in the hindbrain locus coeruleus (LC) (83) partially mediate food avoidance behaviors of direct exenatide injection and peripherally injected semaglutide assessed by kaolin intake (84). These LC neurons receive projections from pre-proglucagon expressing neurons, indicating a possible role in the physiologic control of food intake by the endogenous GLP-1 system. It should be noted that the GLP-1R+ DVC/AP neurons project to the lateral parabrachial nucleus (PBN), which is in close proximity to the LC; thus, the PBN may serve an integrating function in the food aversive and avoidant effects of GLP-1R agonism (Figure 3).
On the other hand, GLP-1R+ neurons in the nucleus of the solitary tract (NTS) predominantly (~60-70%) respond to satiety signals and project to the PVH. Additionally, the forebrain lateral septum (LS) appears to mediate the non-avoidant food intake reducing effects of GLP-1Rs in that direct exenatide injection into the rat LS suppresses chow intake but does not enhance kaolin intake (85). Additionally, blockade of GLP-1Rs in the LS with exendin-9 alone enhanced sucrose and oil intake indicating a physiologic role for these neurons to suppress food intake. Interestingly, exenatide in the LS does not impair anxiety-like behaviors or operant responding, which are key functions for cells within this region that indicate a fine tuning of GLP-1R action in this region.
Finally, very few DVC neurons (~6-12%) are responsive to both aversive and satiating cells, indicating that there are indeed separate nausea and satiety effects by GLP-1R agonism. In further support of this notion, GLP-1R agonists can still reduce food intake when the aversive pathways of the AP are inhibited; ablation of the nausea-mediating AP, but not NTS, GLP-1R + neurons eliminates the conditioned taste avoidance response to semaglutide in rodents (70, 74). This finding is not unique to mice, as a similar response has been reported in musk shrews treated with direct injections of exenatide with and without exendin-9 into the AP and NTS (86). Thus, it appears possible to divorce nausea from satiation in mice treated with GLP-1R agonists by discovery of GLP-1R agonists that preferentially access and activate the NTS but not the AP.
In summary, long-acting GLP-1R agonists primarily reach the circumventricular organs of the hindbrain and food-intake controlling centers of the hypothalamus. In the hindbrain these agonists act to reduce food intake through both satiating and aversive actions. While it has been postulated that these two actions are inextricably linked more recent data shows they can indeed be divorced which opens the opportunity for tailored pharmacology that provide food intake reduction without the adverse events typically associated with GLP-1R agonism.
4 GI AEs associated with GIPR modulation
Because the food aversive and avoidant effects of GLP-1R agonism appear to be dissociable from satiety, it is theoretically possible to leverage this finding in a next generation, tailored pharmacology. Indeed, GIPR agonism can blunt the nauseating effects of GLP-1R agonists in humans (52). In preclinical models, GIPR agonism reduces food intake and body weight via CNS GABAergic neurons (53–55); this effect is additive with GLP-1R agonism and mediated by the same CNS GABAergic GIPR+ population (34, 54). In preclinical studies, GIPR/GLP-1R co-agonism produces less nauseating effects compared to GLP-1R agonism alone as measured by kaolin intake in mice and rats and emetic events in musk shrews (69). Critically, the reduction in nauseating or emetic responses induced by GIPR agonism does not limit the short term (72h) weight loss in any preclinical model (69). This supports the conclusions of Huang et al., that satiety (and by extension efficacy) and adverse events can be dissociated (76) and the clinical data outlined above. The precise mechanisms for this effect are unclear; however, intriguing hypotheses emerge from single cell RNA sequencing data in rodents. First, GIPR+ neurons in the rat hindbrain are located primarily in both inhibitory and excitatory neurons in addition to some expression in oligodendrocytes (69, 87, 88), while GLP-1R is primarily expressed in excitatory neurons. There is limited co-expression of GIPR and GLP-1R (88). It has also been demonstrated that inhibitory GABAergic CNS GIPR + neurons are necessary for the food intake reducing effects of GIPR agonists and dual incretin co-agonists (54, 55), while excitatory glutamatergic CNS GLP-1R + neurons are necessary for the food intake reducing effects of GLP-1R agonists (71). This suggests an inhibitory GABAergic signal from the GIPR agonists dampens GLP-1R-mediated aversive, avoidant, or emetic signals, which may be accomplished by unique projections of GIPR neurons to GLP-1R + neurons in the AP (Figure 4). Therefore, this circuit could curb nausea caused by GLP-1R agonism but not interfere with GLP-1 signaling in the NTS, which induces satiety. This model is also satisfying from an evolutionary biology perspective in which GLP-1 and GIP do not have redundant functions in the brain, but rather fit into complimentary niches. It is noteworthy that GIPR agonism can also attenuate PYY mediated nausea in preclinical models (89), suggesting optimism for either dual or triagonist analogues of PYY, GLP-1, and GIP.
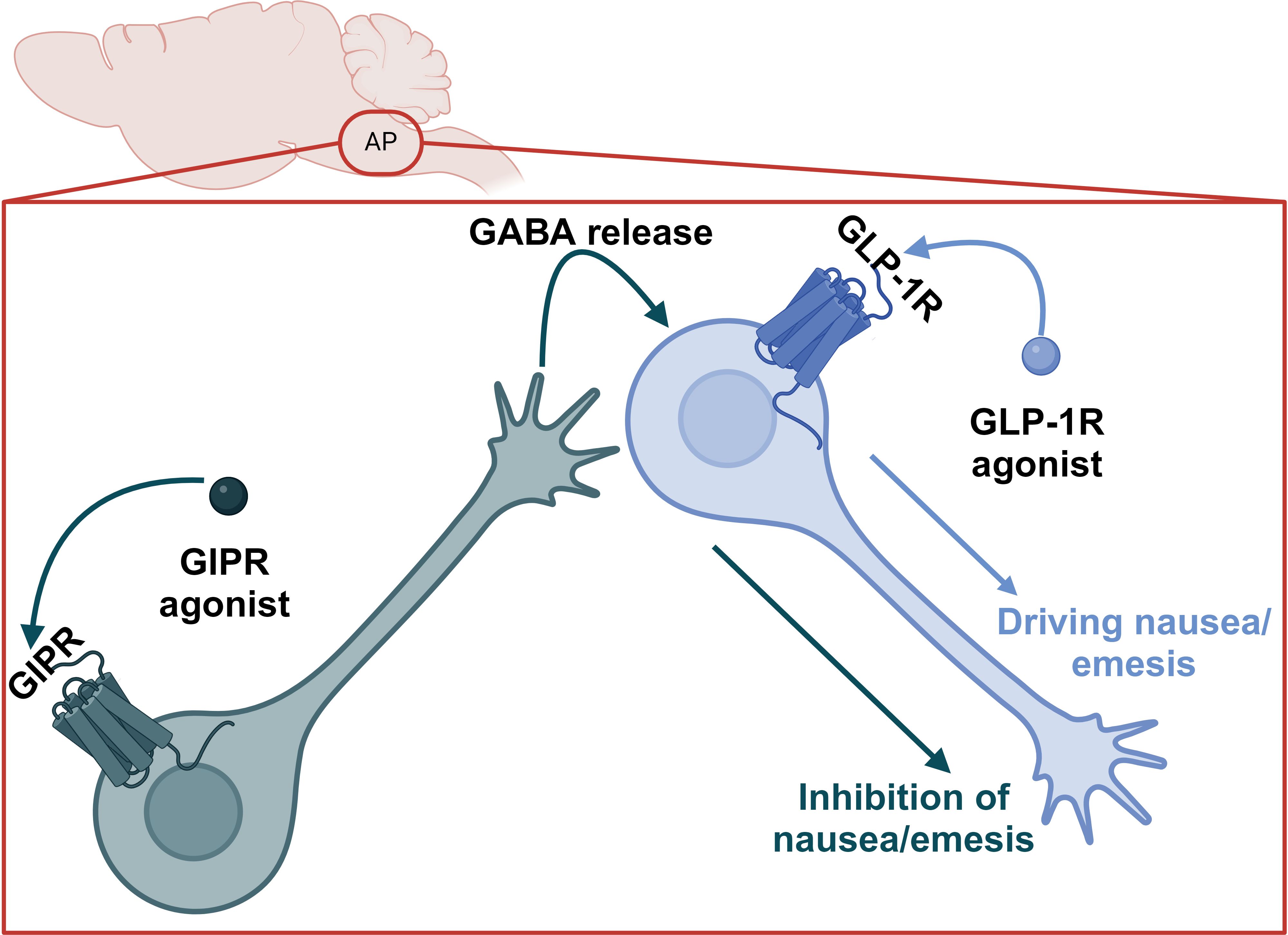
Figure 4. Proposed model for the suppression of GLP-1R agonist-induced nausea and emesis by GIPR agonism in the AP.
5 Conclusions
Incretin receptor agonists including GLP-1R agonists and dual GLP-1R/GIPR co-agonists reduce food intake and drive weight loss in patients with obesity. These therapeutic interventions also drive a variety of GI AEs. Emerging data in clinical and preclinical studies suggests the adverse and efficacious effects of these pharmacotherapies can be dissociated, and that GIPR agonism improves the GI AE profile of GLP-1R and dual incretin receptor agonists. The preclinical data suggests the hypothesis that inhibitory GIPR + neurons act specifically on GLP-1R+ neurons that drive nausea and food aversion or avoidance to dampen GLP-1R mediated nausea without reducing efficacy, potentially lifting the ceiling of GLP-1R agonist-induced reductions in body weight.
Author contributions
JD: Conceptualization, Data curation, Formal analysis, Funding acquisition, Writing – original draft, Writing – review & editing. JF: Conceptualization, Writing – original draft, Writing – review & editing. PK: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
We wish to thank Dr. Barent DuBois for shaping our discussions of exposure/response relationships.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Nauck MA, Homberger E, Siegel EG, Allen RC, Eaton RP, Ebert R, et al. Incretin effects of increasing glucose loads in man calculated from venous insulin and C-peptide responses. J Clin Endocrinol Metab. (1986) 63:492–8. doi: 10.1210/jcem-63-2-492
2. Gasbjerg LS, Helsted MM, Hartmann B, Jensen MH, Gabe MBN, Sparre-Ulrich AH, et al. Separate and combined glucometabolic effects of endogenous glucose-dependent insulinotropic polypeptide and glucagon-like peptide 1 in healthy individuals. Diabetes. (2019) 68:906–17. doi: 10.2337/db18-1123
3. Gasbjerg LS, Helsted MM, Hartmann B, Sparre-Ulrich AH, Veedfald S, Stensen S, et al. GIP and GLP-1 receptor antagonism during a meal in healthy individuals. J Clin Endocrinol Metab. (2020) 105. doi: 10.1210/clinem/dgz175
4. Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest. (1993) 91:301–7. doi: 10.1172/JCI116186
5. Miyawaki K, Yamada Y, Ban N, Ihara Y, Tsukiyama K, Zhou H, et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med. (2002) 8:738–42. doi: 10.1038/nm727
6. Gasbjerg LS, Christensen MB, Hartmann B, Lanng AR, Sparre-Ulrich AH, Gabe MBN, et al. GIP(3-30)NH(2) is an efficacious GIP receptor antagonist in humans: a randomised, double-blinded, placebo-controlled, crossover study. Diabetologia. (2018) 61:413–23. doi: 10.1007/s00125-017-4447-4
7. Goke R, Fehmann HC, Linn T, Schmidt H, Krause M, Eng J, et al. Exendin-4 is a high potency agonist and truncated exendin-(9-39)-amide an antagonist at the glucagon-like peptide 1-(7-36)-amide receptor of insulin-secreting beta-cells. J Biol Chem. (1993) 268:19650–5. doi: 10.1016/S0021-9258(19)36565-2
8. Lau J, Bloch P, Schaffer L, Pettersson I, Spetzler J, Kofoed J, et al. Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) analogue semaglutide. J Med Chem. (2015) 58:7370–80. doi: 10.1021/acs.jmedchem.5b00726
9. Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, et al. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem. (2000) 43:1664–9. doi: 10.1021/jm9909645
10. Glaesner W, Vick AM, Millican R, Ellis B, Tschang SH, Tian Y, et al. Engineering and characterization of the long-acting glucagon-like peptide-1 analogue LY2189265, an Fc fusion protein. Diabetes Metab Res Rev. (2010) 26:287–96. doi: 10.1002/dmrr.v26:4
11. Holst JJ. GLP-1 physiology in obesity and development of incretin-based drugs for chronic weight management. Nat Metab. (2024) 6:1866–85. doi: 10.1038/s42255-024-01113-9
12. Turton MD, O'Shea D, Gunn I, Beak SA, Edwards CM, Meeran K, et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature. (1996) 379:69–72. doi: 10.1038/379069a0
13. Dungan KM, Povedano ST, Forst T, Gonzalez JG, Atisso C, Sealls W, et al. Once-weekly dulaglutide versus once-daily liraglutide in metformin-treated patients with type 2 diabetes (AWARD-6): a randomised, open-label, phase 3, non-inferiority trial. Lancet. (2014) 384:1349–57. doi: 10.1016/S0140-6736(14)60976-4
14. Gorgojo-Martinez JJ, Mezquita-Raya P, Carretero-Gomez J, Castro A, Cebrian-Cuenca A, de Torres-Sanchez A, et al. Clinical recommendations to manage gastrointestinal adverse events in patients treated with glp-1 receptor agonists: A multidisciplinary expert consensus. J Clin Med. (2022) 12.
15. Buse JB, Rosenstock J, Sesti G, Schmidt WE, Montanya E, Brett JH, et al. Liraglutide once a day versus exenatide twice a day for type 2 diabetes: a 26-week randomised, parallel-group, multinational, open-label trial (LEAD-6). Lancet. (2009) 374:39–47. doi: 10.1016/S0140-6736(09)60659-0
16. Buse JB, Bergenstal RM, Glass LC, Heilmann CR, Lewis MS, Kwan AY, et al. Use of twice-daily exenatide in Basal insulin-treated patients with type 2 diabetes: a randomized, controlled trial. Ann Intern Med. (2011) 154:103–12. doi: 10.7326/0003-4819-154-2-201101180-00300
17. Davies MJ, Bergenstal R, Bode B, Kushner RF, Lewin A, Skjoth TV, et al. Efficacy of liraglutide for weight loss among patients with type 2 diabetes: the SCALE diabetes randomized clinical trial. JAMA. (2015) 314:687–99. doi: 10.1001/jama.2015.9676
18. Pi-Sunyer X, Astrup A, Fujioka K, Greenway F, Halpern A, Krempf M, et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. (2015) 373:11–22. doi: 10.1056/NEJMoa1411892
19. Kolata G, Nolen S. Research Behind Drugs For Obesity Wins Prize. New York, NY: The New York Times (2024). p. A19.
20. Davies M, Faerch L, Jeppesen OK, Pakseresht A, Pedersen SD, Perreault L, et al. Semaglutide 2.4 mg once a week in adults with overweight or obesity, and type 2 diabetes (STEP 2): a randomised, double-blind, double-dummy, placebo-controlled, phase 3 trial. Lancet. (2021) 397:971–84. doi: 10.1016/S0140-6736(21)00213-0
21. Wilding JPH, Batterham RL, Calanna S, Davies M, Van Gaal LF, Lingvay I, et al. Once-weekly semaglutide in adults with overweight or obesity. N Engl J Med. (2021) 384:989–1002. doi: 10.1056/NEJMoa2032183
22. Pratley RE, Aroda VR, Lingvay I, Ludemann J, Andreassen C, Navarria A, et al. Semaglutide versus dulaglutide once weekly in patients with type 2 diabetes (SUSTAIN 7): a randomised, open-label, phase 3b trial. Lancet Diabetes Endocrinol. (2018) 6:275–86. doi: 10.1016/S2213-8587(18)30024-X
23. Flint A, Raben A, Astrup A, Holst JJ. Glucagon-like peptide 1 promotes satiety and suppresses energy intake in humans. J Clin Invest. (1998) 101:515–20. doi: 10.1172/JCI990
24. Gutzwiller JP, Drewe J, Goke B, Schmidt H, Rohrer B, Lareida J, et al. Glucagon-like peptide-1 promotes satiety and reduces food intake in patients with diabetes mellitus type 2. Am J Physiol. (1999) 276:R1541–4. doi: 10.1152/ajpregu.1999.276.5.R1541
25. Schlogl H, Kabisch S, Horstmann A, Lohmann G, Muller K, Lepsien J, et al. Exenatide-induced reduction in energy intake is associated with increase in hypothalamic connectivity. Diabetes Care. (2013) 36:1933–40. doi: 10.2337/dc12-1925
26. Friedrichsen M, Breitschaft A, Tadayon S, Wizert A, Skovgaard D. The effect of semaglutide 2.4 mg once weekly on energy intake, appetite, control of eating, and gastric emptying in adults with obesity. Diabetes Obes Metab. (2021) 23:754–62. doi: 10.1111/dom.14280
27. van Can J, Sloth B, Jensen CB, Flint A, Blaak EE, Saris WH. Effects of the once-daily GLP-1 analog liraglutide on gastric emptying, glycemic parameters, appetite and energy metabolism in obese, non-diabetic adults. Int J Obes (Lond). (2014) 38:784–93. doi: 10.1038/ijo.2013.162
28. Gibbons C, Blundell J, Tetens Hoff S, Dahl K, Bauer R, Baekdal T. Effects of oral semaglutide on energy intake, food preference, appetite, control of eating and body weight in subjects with type 2 diabetes. Diabetes Obes Metab. (2021) 23:581–8. doi: 10.1111/dom.14255
29. Quast DR, Nauck MA, Schenker N, Menge BA, Kapitza C, Meier JJ. Macronutrient intake, appetite, food preferences and exocrine pancreas function after treatment with short- and long-acting glucagon-like peptide-1 receptor agonists in type 2 diabetes. Diabetes Obes Metab. (2021) 23:2344–53. doi: 10.1111/dom.v23.10
30. Jacobsen JM, Petersen N, Torz L, Gerstenberg MK, Pedersen K, Ostergaard S, et al. Housing mice near vs. below thermoneutrality affects drug-induced weight loss but does not improve prediction of efficacy in humans. Cell Rep. (2024) 43:114501. doi: 10.1016/j.celrep.2024.114501
31. Leibel RL, Rosenbaum M, Hirsch J. Changes in energy expenditure resulting from altered body weight. N Engl J Med. (1995) 332:621–8. doi: 10.1056/NEJM199503093321001
32. Rosenbaum M, Hirsch J, Gallagher DA, Leibel RL. Long-term persistence of adaptive thermogenesis in subjects who have maintained a reduced body weight. Am J Clin Nutr. (2008) 88:906–12. doi: 10.1093/ajcn/88.4.906
33. Gabery S, Salinas CG, Paulsen SJ, Ahnfelt-Ronne J, Alanentalo T, Baquero AF, et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.133429
34. Finan B, Ma T, Ottaway N, Muller TD, Habegger KM, Heppner KM, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med. (2013) 5:209ra151. doi: 10.1126/scitranslmed.3007218
35. Finan B, Yang B, Ottaway N, Smiley DL, Ma T, Clemmensen C, et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med. (2015) 21:27–36. doi: 10.1038/nm.3761
36. Jall S, Sachs S, Clemmensen C, Finan B, Neff F, DiMarchi RD, et al. Monomeric GLP-1/GIP/glucagon triagonism corrects obesity, hepatosteatosis, and dyslipidemia in female mice. Mol Metab. (2017) 6:440–6. doi: 10.1016/j.molmet.2017.02.002
37. Knerr PJ, Mowery SA, Douros JD, Premdjee B, Hjollund KR, He Y, et al. Next generation GLP-1/GIP/glucagon triple agonists normalize body weight in obese mice. Mol Metab. (2022) 63:101533. doi: 10.1016/j.molmet.2022.101533
38. Petersen J, Ludwig MQ, Juozaityte V, Ranea-Robles P, Svendsen C, Hwang E, et al. GLP-1-directed NMDA receptor antagonism for obesity treatment. Nature. (2024) 629:1133–41. doi: 10.1038/s41586-024-07419-8
39. Quarta C, Stemmer K, Novikoff A, Yang B, Klingelhuber F, Harger A, et al. GLP-1-mediated delivery of tesaglitazar improves obesity and glucose metabolism in male mice. Nat Metab. (2022) 4:1071–83. doi: 10.1038/s42255-022-00617-6
40. Sachs S, Bastidas-Ponce A, Tritschler S, Bakhti M, Bottcher A, Sanchez-Garrido MA, et al. Targeted pharmacological therapy restores beta-cell function for diabetes remission. Nat Metab. (2020) 2:192–209. doi: 10.1038/s42255-020-0171-3
41. Tschöp MH, Finan B, Clemmensen C, Gelfanov V, Perez-Tilve D, Müller TD, et al. Unimolecular polypharmacy for treatment of diabetes and obesity. Cell Metab. (2016) 24:51–62. doi: 10.1016/j.cmet.2016.06.021
42. Finan B, Clemmensen C, Zhu Z, Stemmer K, Gauthier K, Muller L, et al. Chemical hybridization of glucagon and thyroid hormone optimizes therapeutic impact for metabolic disease. Cell. (2016) 167:843–857.e14. doi: 10.1016/j.cell.2016.09.014
43. Finan B, Yang B, Ottaway N, Stemmer K, Muller TD, Yi CX, et al. Targeted estrogen delivery reverses the metabolic syndrome. Nat Med. (2012) 18:1847–56. doi: 10.1038/nm.3009
44. Coskun T, Sloop KW, Loghin C, Alsina-Fernandez J, Urva S, Bokvist KB, et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept. Mol Metab. (2018) 18:3–14. doi: 10.1016/j.molmet.2018.09.009
45. Garvey WT, Frias JP, Jastreboff AM, le Roux CW, Sattar N, Aizenberg D, et al. Tirzepatide once weekly for the treatment of obesity in people with type 2 diabetes (SURMOUNT-2): a double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. (2023) 402:613–26. doi: 10.1016/S0140-6736(23)01200-X
46. Jastreboff AM, Aronne LJ, Ahmad NN, Wharton S, Connery L, Alves B, et al. Tirzepatide once weekly for the treatment of obesity. N Engl J Med. (2022) 387:205–16. doi: 10.1056/NEJMoa2206038
47. Willard FS, Douros JD, Gabe MB, Showalter AD, Wainscott DB, Suter TM, et al. Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.140532
48. Hinds CE, Peace E, Chen S, Davies I, El Eid L, Tomas A, et al. Abolishing beta-arrestin recruitment is necessary for the full metabolic benefits of G protein-biased glucagon-like peptide-1 receptor agonists. Diabetes Obes Metab. (2023).
49. Jones B, Buenaventura T, Kanda N, Chabosseau P, Owen BM, Scott R, et al. Targeting GLP-1 receptor trafficking to improve agonist efficacy. Nat Commun. (2018) 9:1602. doi: 10.1038/s41467-018-03941-2
50. Lucey M, Pickford P, Bitsi S, Minnion J, Ungewiss J, Schoeneberg K, et al. Disconnect between signalling potency and in vivo efficacy of pharmacokinetically optimised biased glucagon-like peptide-1 receptor agonists. Mol Metab. (2020) 37:100991. doi: 10.1016/j.molmet.2020.100991
51. Douros JD, Mokrosinski J, Finan B. The GLP-1R as a model for understanding and exploiting biased agonism in next-generation medicines. J Endocrinol. (2024) 261. doi: 10.1530/JOE-23-0226
52. Knop FK, Urva S, Rettiganti M, Benson CT, Roell WC, Mather KJ, et al. (2023). A long-acting glucose dependent insulinotropic polypeptide receptor agonist shows weight loss without nausea or vomiting, in: American Diabetes Association – 83rd Annual Scientific Sessions, San Diego, CA, USA, 23 – 26 June 2023.
53. Mroz PA, Finan B, Gelfanov V, Yang B, Tschop MH, DiMarchi RD, et al. Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism. Mol Metab. (2019) 20:51–62. doi: 10.1016/j.molmet.2018.12.001
54. Liskiewicz A, Khalil A, Liskiewicz D, Novikoff A, Grandl G, Maity-Kumar G, et al. Glucose-dependent insulinotropic polypeptide regulates body weight and food intake via GABAergic neurons in mice. Nat Metab. (2023) 5:2075–85. doi: 10.1038/s42255-023-00931-7
55. Zhang Q, Delessa CT, Augustin R, Bakhti M, Collden G, Drucker DJ, et al. The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling. Cell Metab. (2021). doi: 10.2139/ssrn.3667144
56. El K, Douros JD, Willard FS, Novikoff A, Sargsyan A, Perez-Tilve D, et al. The incretin co-agonist tirzepatide requires GIPR for hormone secretion from human islets. Nat Metab. (2023) 5:945–54. doi: 10.1038/s42255-023-00811-0
57. Knop FK, Urva S, Rettiganti M, Benson C, Roell W, Mather KJ, et al. 56-OR: A long-acting glucose-dependent insulinotropic polypeptide receptor agonist shows weight loss without nausea or vomiting. Diabetes. (2023) 72. doi: 10.2337/db23-56-OR
58. Killion EA, Chen M, Falsey JR, Sivits G, Hager T, Atangan L, et al. Chronic glucose-dependent insulinotropic polypeptide receptor (GIPR) agonism desensitizes adipocyte GIPR activity mimicking functional GIPR antagonism. Nat Commun. (2020) 11:4981. doi: 10.1038/s41467-020-18751-8
59. Killion EA, Wang J, Yie J, Shi SD, Bates D, Min X, et al. Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Sci Transl Med. (2018) 10. doi: 10.1126/scitranslmed.aat3392
60. Lu SC, Chen M, Atangan L, Killion EA, Komorowski R, Cheng Y, et al. GIPR antagonist antibodies conjugated to GLP-1 peptide are bispecific molecules that decrease weight in obese mice and monkeys. Cell Rep Med. (2021) 2:100263. doi: 10.1016/j.xcrm.2021.100263
61. Veniant MM, Lu SC, Atangan L, Komorowski R, Stanislaus S, Cheng Y, et al. A GIPR antagonist conjugated to GLP-1 analogues promotes weight loss with improved metabolic parameters in preclinical and phase 1 settings. Nat Metab. (2024). doi: 10.1038/s42255-023-00966-w
62. Novikoff A, Muller TD. Antagonizing GIPR adds fire to the GLP-1R flame. Trends Endocrinol Metab. (2024) 35:566–8. doi: 10.1016/j.tem.2024.04.016
63. Hayes MR, Borner T, De Jonghe BC. The role of GIP in the regulation of GLP-1 satiety and nausea. Diabetes. (2021) 70:1956–61. doi: 10.2337/dbi21-0004
64. Frias JP, Davies MJ, Rosenstock J, Perez Manghi FC, Fernandez Lando L, Bergman BK, et al. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med. (2021). doi: 10.1056/NEJMoa2107519
65. Knop FK, Urva S, Rettiganti M, Benson CT, Roell WC, Mather KJ, et al. A long-acting glucose-dependent insulinotropic polypeptide receptor agonist improves the gastrointestinal tolerability of glucagon-like peptide-1 receptor agonist therapy. Diabetes Obes Metab. (2024).
66. Ahmann AJ, Capehorn M, Charpentier G, Dotta F, Henkel E, Lingvay I, et al. Efficacy and safety of once-weekly semaglutide versus exenatide ER in subjects with type 2 diabetes (SUSTAIN 3): A 56-week, open-label, randomized clinical trial. Diabetes Care. (2018) 41:258–66. doi: 10.2337/dc17-0417
67. Davies M, Færch L, Jeppesen OK, Pakseresht A, Pedersen SD, Perreault L, et al. Semaglutide 2·4 mg once a week in adults with overweight or obesity, and type 2 diabetes (STEP 2): a randomised, double-blind, double-dummy, placebo-controlled, phase 3 trial. Lancet. (2021) 397:971–84. doi: 10.1016/S0140-6736(21)00213-0
68. Frias JP, Deenadayalan S, Erichsen L, Knop FK, Lingvay I, Macura S, et al. Efficacy and safety of co-administered once-weekly cagrilintide 2.4 mg with once-weekly semaglutide 2.4 mg in type 2 diabetes: a multicentre, randomised, double-blind, active-controlled, phase 2 trial. Lancet. (2023) 402:720–30. doi: 10.1016/S0140-6736(23)01163-7
69. Borner T, Geisler CE, Fortin SM, Cosgrove R, Alsina-Fernandez J, Dogra M, et al. GIP receptor agonism attenuates GLP-1 receptor agonist-induced nausea and emesis in preclinical models. Diabetes. (2021) 70:2545–53. doi: 10.2337/db21-0459
70. Cheng W, Gordian D, Ludwig MQ, Pers TH, Seeley RJ, Myers MG Jr. Hindbrain circuits in the control of eating behaviour and energy balance. Nat Metab. (2022) 4:826–35. doi: 10.1038/s42255-022-00606-9
71. Sisley S, Gutierrez-Aguilar R, Scott M, D'Alessio DA, Sandoval DA, Seeley RJ. Neuronal GLP1R mediates liraglutide's anorectic but not glucose-lowering effect. J Clin Invest. (2014) 124:2456–63. doi: 10.1172/JCI72434
72. Adams JM, Pei H, Sandoval DA, Seeley RJ, Chang RB, Liberles SD, et al. Liraglutide modulates appetite and body weight through glucagon-like peptide 1 receptor-expressing glutamatergic neurons. Diabetes. (2018) 67:1538–48. doi: 10.2337/db17-1385
73. Campos CA, Bowen AJ, Han S, Wisse BE, Palmiter RD, Schwartz MW. Cancer-induced anorexia and malaise are mediated by CGRP neurons in the parabrachial nucleus. Nat Neurosci. (2017) 20:934–42. doi: 10.1038/nn.4574
74. Chen JY, Campos CA, Jarvie BC, Palmiter RD. Parabrachial CGRP neurons establish and sustain aversive taste memories. Neuron. (2018) 100:891–899.e5. doi: 10.1016/j.neuron.2018.09.032
75. Campos CA, Bowen AJ, Schwartz MW, Palmiter RD. Parabrachial CGRP neurons control meal termination. Cell Metab. (2016) 23:811–20. doi: 10.1016/j.cmet.2016.04.006
76. Huang KP, Acosta AA, Ghidewon MY, McKnight AD, Almeida MS, Nyema NT, et al. Dissociable hindbrain GLP1R circuits for satiety and aversion. Nature. (2024) 632:585–93. doi: 10.1038/s41586-024-07685-6
77. Lu Z, Chan SW, Tu L, Ngan MP, Rudd JA. GLP-1 receptors are involved in the GLP-1 (7-36) amide-induced modulation of glucose homoeostasis, emesis and feeding in Suncus murinus (house musk shrew). Eur J Pharmacol. (2020) 888:173528. doi: 10.1016/j.ejphar.2020.173528
78. Borner T, Workinger JL, Tinsley IC, Fortin SM, Stein LM, Chepurny OG, et al. Corrination of a GLP-1 receptor agonist for glycemic control without emesis. Cell Rep. (2020) 31:107768. doi: 10.1016/j.celrep.2020.107768
79. Mietlicki-Baase EG, Liberini CG, Workinger JL, Bonaccorso RL, Borner T, Reiner DJ, et al. A vitamin B12 conjugate of exendin-4 improves glucose tolerance without associated nausea or hypophagia in rodents. Diabetes Obes Metab. (2018) 20:1223–34.
80. Krieger JP, Arnold M, Pettersen KG, Lossel P, Langhans W, Lee SJ. Knockdown of GLP-1 receptors in vagal afferents affects normal food intake and glycemia. Diabetes. (2016) 65:34–43. doi: 10.2337/db15-0973
81. Kanoski SE, Fortin SM, Arnold M, Grill HJ, Hayes MR. Peripheral and central GLP-1 receptor populations mediate the anorectic effects of peripherally administered GLP-1 receptor agonists, liraglutide and exendin-4. Endocrinology. (2011) 152:3103–12. doi: 10.1210/en.2011-0174
82. Kanoski SE, Rupprecht LE, Fortin SM, De Jonghe BC, Hayes MR. The role of nausea in food intake and body weight suppression by peripheral GLP-1 receptor agonists, exendin-4 and liraglutide. Neuropharmacology. (2012) 62:1916–27. doi: 10.1016/j.neuropharm.2011.12.022
83. Llewellyn-Smith IJ, Reimann F, Gribble FM, Trapp S. Preproglucagon neurons project widely to autonomic control areas in the mouse brain. Neuroscience. (2011) 180:111–21. doi: 10.1016/j.neuroscience.2011.02.023
84. Fortin SM, Chen JC, Petticord MC, Ragozzino FJ, Peters JH, Hayes MR. The locus coeruleus contributes to the anorectic, nausea, and autonomic physiological effects of glucagon-like peptide-1. Sci Adv. (2023) 9:eadh0980. doi: 10.1126/sciadv.adh0980
85. Terrill SJ, Jackson CM, Greene HE, Lilly N, Maske CB, Vallejo S, et al. Role of lateral septum glucagon-like peptide 1 receptors in food intake. Am J Physiol Regul Integr Comp Physiol. (2016) 311:R124–32. doi: 10.1152/ajpregu.00460.2015
86. Chan SW, Lin G, Yew DT, Yeung CK, Rudd JA. Separation of emetic and anorexic responses of exendin-4, a GLP-1 receptor agonist in Suncus murinus (house musk shrew). Neuropharmacology. (2013) 70:141–7. doi: 10.1016/j.neuropharm.2013.01.013
87. Dowsett GKC, Lam BYH, Tadross JA, Cimino I, Rimmington D, Coll AP, et al. A survey of the mouse hindbrain in the fed and fasted states using single-nucleus RNA sequencing. Mol Metab. (2021) 53:101240. doi: 10.1016/j.molmet.2021.101240
88. Ludwig MQ, Cheng W, Gordian D, Lee J, Paulsen SJ, Hansen SN, et al. A genetic map of the mouse dorsal vagal complex and its role in obesity. Nat Metab. (2021) 3:530–45. doi: 10.1038/s42255-021-00363-1
Keywords: GLP-1 - glucagon-like peptide-1, GIP - glucose-dependent insulinotropic peptide, obesity, incretin, pharmacology
Citation: Douros JD, Flak JN and Knerr PJ (2025) The agony and the efficacy: central mechanisms of GLP-1 induced adverse events and their mitigation by GIP. Front. Endocrinol. 16:1530985. doi: 10.3389/fendo.2025.1530985
Received: 19 November 2024; Accepted: 02 January 2025;
Published: 03 February 2025.
Edited by:
Kirk M. Habegger, University of Alabama at Birmingham, United StatesReviewed by:
Thomas Alexander Lutz, University of Zurich, SwitzerlandJarrad Scarlett, University of Washington, United States
Copyright © 2025 Douros, Flak and Knerr. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan D. Douros, amRvdXJvc0BpbmRpYW5hYmlvc2NpZW5jZXMub3Jn