 Aneta Kodytková1*†
Aneta Kodytková1*† Petra Dušátková1†
Petra Dušátková1† Shenali Anne Amaratunga1†
Shenali Anne Amaratunga1† Stanislava Koloušková1†Barbora Obermannová1†Renata Pomahačová2†Štěpánka Průhová1†Marta Šnajderová1†
Stanislava Koloušková1†Barbora Obermannová1†Renata Pomahačová2†Štěpánka Průhová1†Marta Šnajderová1† Zdeněk Šumník1†Jiřina Zapletalová3†Valerij Semjonov4†
Zdeněk Šumník1†Jiřina Zapletalová3†Valerij Semjonov4† Jan Lebl1†
Jan Lebl1†- 1Department of Pediatrics, 2nd Faculty of Medicine, Charles University and Motol University Hospital, Prague, Czechia
- 2Department of Pediatrics, Faculty of Medicine, Charles University and University Hospital Pilsen, Pilsen, Czechia
- 3Department of Pediatrics, Faculty of Medicine, Palacky University and Olomouc University Hospital, Olomouc, Czechia
- 4Department of Statistics, Motol University Hospital, Prague, Czechia
Introduction: Prader-Willi syndrome (PWS) is primarily caused by a paternal microdeletion of the 15q11-q13 region, maternal uniparental disomy (mUPD) or unbalanced translocations. The MKRN3 gene, located within 15q11-q13, is a master regulator of pubertal initiation. We aimed to compare variant pubertal onset and progression with recent normative data and to correlate it with abnormal MKRN3 gene status.
Methods: Age at pubarche, gonadarche, subsequent pubertal progression and bone age (BA) at gonadarche were investigated in 37 PWS patients (18 females) who already entered pubarche and/or gonadarche with median age 11.1 (95% CI: 6.4 – 18.8) years. All patients were re-tested to confirm genetic subtypes of PWS. The MKRN3 gene was analyzed using single gene sequencing.
Results: Out of 37 subjects, 22 had microdeletion and 15 mUPD. Regardless of genetic subtypes and MKRN3 gene status, no correlation between genotypes and the pubertal pattern was found. They initiated pubarche early – girls at 7.4 (95%CI:6.4–8.4), and boys at 9.2 (8.2–10.2) years. The subsequent progression from PH2 to PH4 (pubic hair development) was prolonged to 3.7 years in girls (1.5–5.9;p<0.05), and 2.9 in boys (2.2–3.6;p<0.001). The age at gonadarche was adequate – 10.0 years in girls (8.8–11.2), and 11.0 in boys (9.8–12.1). Progression rate of breast development from B2 to B4 was 3.9 (0.2–7.5) years in girls and of testicular volume from 4 ml to 15ml was 3.8 (0.0–8.1) years in boys. The BA at gonadarche is advanced by 0.6 ± 1.1 years (p<0.001).
Conclusions: Children with PWS, regardless of the genetic subtype and/or MKRN3 status, had an early pubarche and normally timed gonadarche. Pubarche progression was slower. Advanced BA was significantly correlated with gonadarche.
Introduction
The genetic background for Prader-Willi syndrome (PWS) is abnormal genomic imprinting during gametogenesis, which causes a loss of paternal gene expression on chromosome 15 at the molecular level, in the 15q11-q13 region. About 65 – 75% of patients with PWS have a de novo paternal deletion within this critical region. Based on three breakpoints (BP) in this area, three classes of deletions may be distinguished – common type 1 deletion (BP1-3), type 2 deletion (BP2-3) and a rare atypical microdeletion 15q11.2 (BP1-2) (1, 2). An additional 20 – 30% of PWS patients exhibit maternal uniparental disomy (mUPD) of chromosome 15, and the remaining 1 – 3% of cases have microdeletion or epimutation in the imprinting center (1, 3, 4). Very rarely, an unbalanced Robertsonian chromosomal translocation may occur (5).
PWS, with an estimated incidence of 1 in 10,000 to 20,000 newborns, is the most common cause of syndromic life-threatening obesity (6, 7). Eating disorder in PWS can be divided into 4 phases: 1. hypotonia, feeding difficulties, and failure to thrive (from birth to 15 months of age); 2. weight gain without significant change in appetite or caloric intake or with only a slight increase in interest in food (from 15 months to 5 years); 3. development of hyperphagia, typically accompanied by food-seeking and lack of satiety (from 5 to 13 years); some adults progress to phase 4, which is the loss of an insatiable appetite and the achievement of a feeling of satiety (8). Several reports have demonstrated oxytocin abnormalities in PWS patients, which may be causal for several symptoms - poor suckling response at birth, hyperphagia with food addiction, poor social skills and emotional dysregulation. The results of animal studies indicate that oxytocin therapy is one of the important therapeutic strategies for children with PWS (9, 10). Excessive eating behavior in PWS appears to result from hypothalamic dysfunction, as do other key clinical features such as short stature, testicular retention due to hypogonadotropic hypogonadism, and, in some patients, central hypothyroidism and/or central hypocorticism (4). Furthermore, due to hypothalamic syndrome, pubertal development may be incomplete, delayed or completely absent and this can also lead to complications in the reproductive sphere. On the contrary, signs of central precocious puberty (CPP) have been reported in a minority (approximately 3.5%) of children with PWS (11–13).
The candidate gene for abnormal pubertal development in PWS is the maternally imprinted MKRN3 that is located in 15q11.2, within the PWS critical region (14). MKRN3 contains a characteristic ring zinc finger motif and encodes a specific protein (macorin ring finger protein 3) (15), which is abundantly expressed in the hypothalamus and is involved in hormonal regulation of puberty by the inhibition of gonadotropin-releasing hormone (GnRH) secretion (16, 17). In murine models, increased expression of MKRN3 in the hypothalamus was reported in the neonatal and juvenile period with a subsequent decrease before pubertal onset, which is suggestive of its role to inhibit activation of hypothalamo-pituitary-gonadal axis (18). The inhibitory role of the MKRN3 protein is further supported by a decrease in its serum level in both girls and boys before the onset of puberty (19, 20), whereas girls with idiopathic CPP have lower serum MKRN3 when compared to prepubertal peers of similar age (21). The loss-of-function pathogenic variants in the MKRN3 gene are the most common genetic cause of CPP (22–24).
Real-life clinical experience shows aberrant pubertal development in Prader-Willi syndrome, but clear data on the timing of the pubertal milestones have not been clearly elucidated yet. Even more, identification of the MKRN3 gene within the Prader-Willi syndrome critical region as the master regulator of pubertal development led to the need of testing the potential link between the MKRN3 gene expression and/or sequence and individual pubertal development in PWS patients. In this study, we aimed to estimate pubertal milestones in 37 PWS patients (18 females) with median age 11.1 (95% CI: 6.4 – 18.8) years on the background of recent normative data, to correlate bone age (BA) and gonadarche onset and evaluate the link between variant pubarche and gonadarche and the MKRN3 gene sequence or expression level in individual genetic subtypes of PWS.
Materials and methods
Patients
This is a cross-sectional study including all available relevant retrospective data such as the onset of pubarche and/or gonadarche and their further progression, bone age at the time of gonadarche. All patients with genetically proven PWS who were currently treated by growth hormone (GH) between 2020 and 2021 in the three largest centers for GH treatment in Czechia and who already have entered pubarche (PH2) and/or gonadarche (glandular breast B2 or testicular volume 4 ml) were included in the study. Finally, a cohort of 37 children, adolescents, and young adults with median age 11.1 (95% CI: 6.4 – 18.8) years were evaluated with genetically confirmed PWS. Detailed information about the patients at the time of examination can be found in Table 1. All children started growth hormone (GH) treatment early in life at median age of 0.9 years (95% CI: 0.3 – 4.0 years). Their long-term clinical data have been collected according to a standard protocol every 6 months within the country-wide registry of GH recipients - database REPAR (25). Pubarche (PH2) and gonadarche (glandular breast B2 or testicular volume 4 ml) was evaluated according to Tanner (26, 27). One girl from our cohort (1/3) reached menarche and adult sexual maturity at an adequate age, but menarche was hormonally induced.
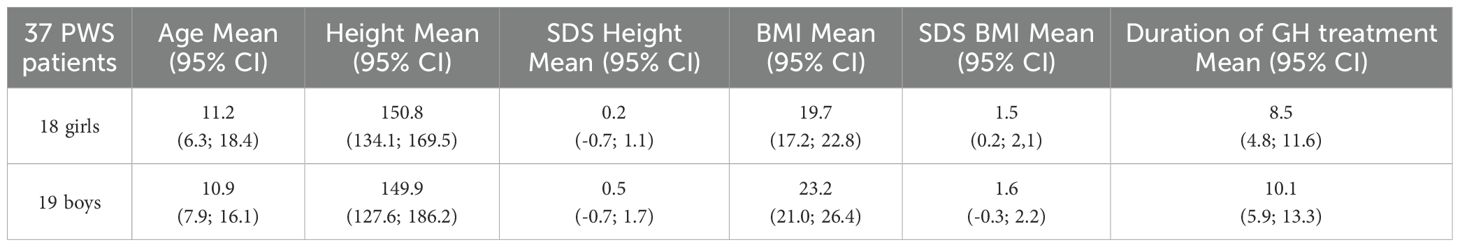
Table 1. Demographic data of PWS cohort at current age.
BA at gonadarche, based on a radiograph of the left hand and wrist, was assessed in 16 patients using the TW3 method (28).
All tested individuals and their parents gave written informed consent for enrollment into the study including genetic testing and all data used were anonymized. This study was approved by the Ethics Committee at the 2nd Faculty of Medicine, Charles University in Prague (date of approval: 27.10.2021; EK-1240.4/21). The research was conducted ethically in accordance with the World Medical Association Declaration of Helsinki.
Normative data
Calculations of height and BMI SDS were based on the 6th Czech National-wide Anthropological Survey of Children and Adolescents (29, 30). Normative data for pubarche and gonadarche were obtained from recent Danish studies (31, 32). According to these population-based studies, mean age of girls at entry into stage B2 is 9.9 ± 1.3 years (95% CI: 9.7 – 10.0) and into stage PH2 is 11.1 (95% CI: 11.0 – 11.2) years (31). In boys, puberty starts at age 11.7 (95% CI: 11.5 – 11.8) years (testicular size more than 3 ml) and stage PH2 is achieved at 12.4 (95% CI: 12.2 – 12.6) years (32, 33).
Genetic re-testing
All patients had their diagnosis of PWS already confirmed from previous genetic testing in different labs. For the purpose of this study, we re-tested all study participants. After obtaining informed consent, the patient’s blood was sampled, and DNA was extracted. Larger structural changes (deletions, duplications) causing PWS, as well as a different level of methylation in the MKRN3 gene from the reference, were detected by methylation-specific MLPA (Multiplex Ligation Probe-Dependent Amplification; MRC Holland, Amsterdam, Netherlands; ME028 according to manufacturer’s instructions). Patients were divided into 3 groups according to the discovered structural change: type 1 deletion (BP1-3), type 2 deletion (BP2-3) and mUPD. The Sanger direct sequencing method and subsequent analysis using Mutation Surveyor software (SoftGenetics, State College, USA) were used to detect DNA sequence changes (substitutions, deletions, insertions) in the MKRN3 gene.
Statistical evaluation of pubertal milestones
All ages at pubarche and gonadarche were converted to SDS (31, 32). Deviations of pubarche/gonadarche below or over ± 2 SDS were evaluated as precocious or delayed, respectively. A sample t-test was used to compare age at pubarche and age at gonadarche (B2/testes 4ml) with the healthy population. One sample t-test was also used to evaluate the duration of progression of breast development, testicular volume progression and pubarche development compared to normative data. We performed a paired t-test, separately for girls and boys, to examine whether the onset of puberty (B2/testes 4ml) occurs later than the PH2 pubic hair stage in our patients. A Pearson correlation test was done to evaluate the linear relationship between bone age and calendar age at gonadarche and one tailed paired t-test was used to test the hypothesis that bone age is greater than calendar age at gonadarche.
Results
Genetic subgroups
Based on the results of MS-MLPA, out of 37 patients tested, in 22 (59%) PWS was caused by 15q11-q13 microdeletion – type 1 deletion in 9, and type 2 deletion in 13 patients, respectively. The remaining 15 (41%) patients had mUPD. The Sanger sequencing did not detect any (likely) pathogenic point variant within the MKRN3 gene.
Regardless the genetic subtype, dose, or sequence of MKRN3 gene no correlation was found between individual genotypes and the abnormal pattern of pubarche or gonadarche in PWS.
Pubarche and gonadarche
Mean age at pubarche differs significantly between patients with PWS and healthy children (Table 2). In comparison with normative data, pubarche occurred 3.7 years earlier in girls, and 3.2 years earlier in boys (p<0.001; Table 2). On the contrary, gonadarche started at adequate age in both boys and girls (Table 3) compared to normative values.
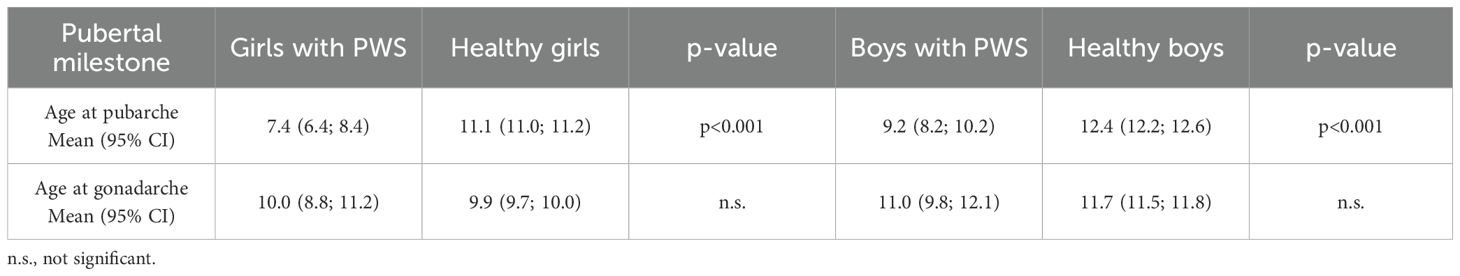
Table 2. Age of pubertal milestones – the comparison of patients with PWS and healthy population (28, 29).
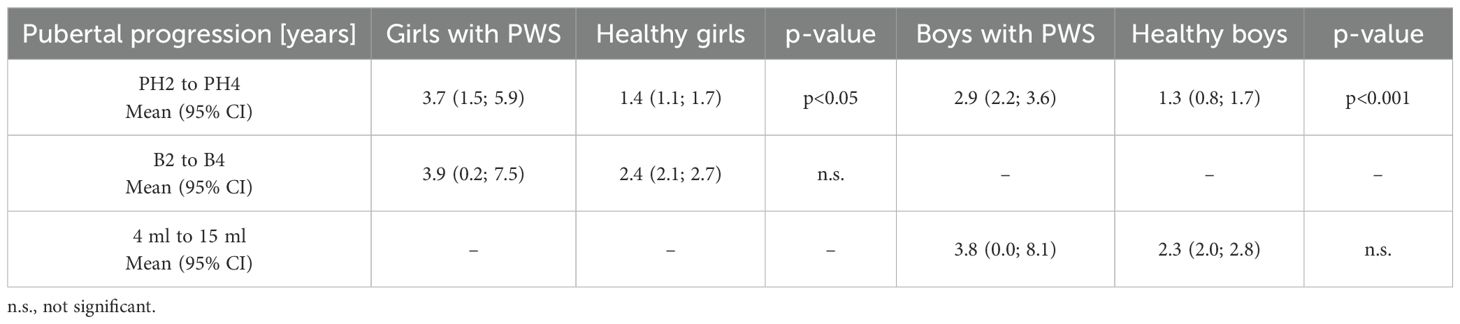
Table 3. Duration of pubarche progression, breast development and testicular volume progression – the comparison of patients with PWS patients and healthy population (28, 29).
The BA at gonadarche tended to be advanced by 0.6 ± 1.1 years (p<0.001), there was significant correlation between skeletal advancement and age of gonadarche (Pearson correlation coefficient 0.8; p<0.001).
In patients with PWS, the pubic hair growth (stage PH2) started significantly earlier than the gonadarche (B2/testes 4ml) in both girls (p<0.001) and boys (p<0.01) – Figure 1A. The estimated mean difference between ages at pubarche and gonadarche was -2.5 in girls (95% CI: -3.6 to -1.4) and -1.2 in boys (95% CI: -2.0 to -0.4).
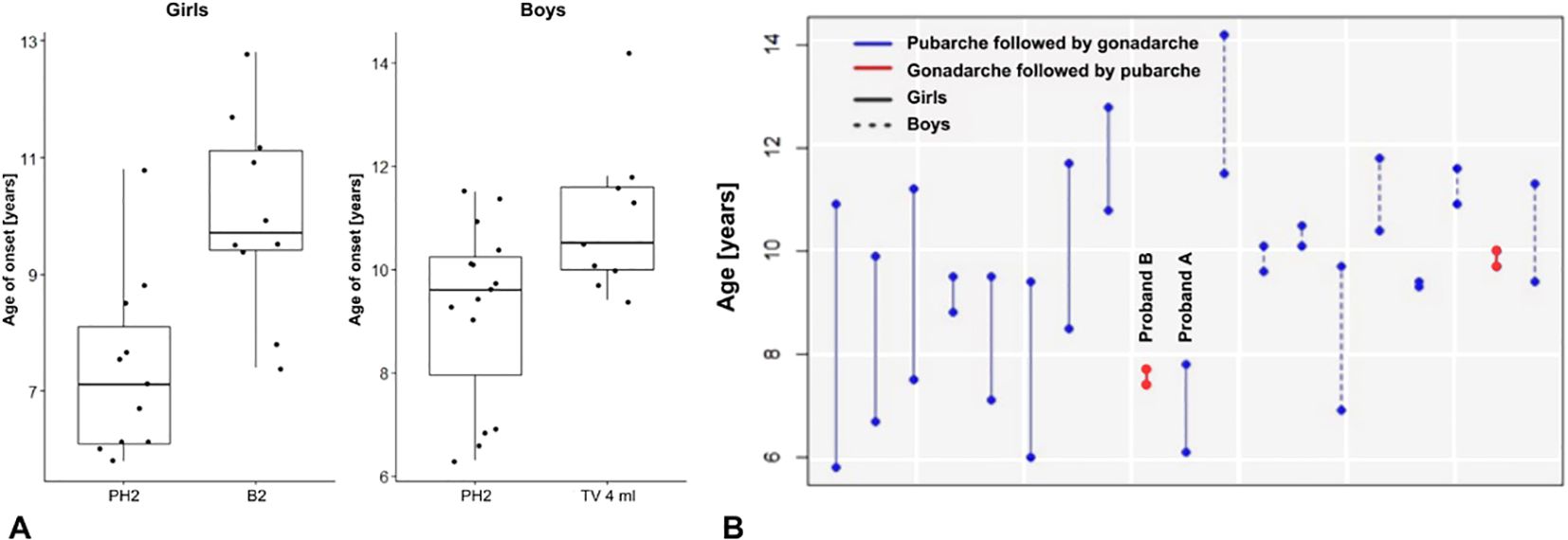
Figure 1. (A) Onset of pubarche and gonadarche in patients with PWS. (B) Differences in the age at onset of pubarche and puberty in 19 individuals (girls and boys) who already developed both - pubarche (PH2) and gonadarche (B2, testicular volume 4 ml). Only 2 girls had gonadarche followed by pubarche.
Pubertal progression
In patients with PWS, the average time of progression from PH2 to PH4 was significantly prolonged (Table 3), compared to the average time of pubarche progression in healthy girls and boys (Figures 2A, B), respectively.
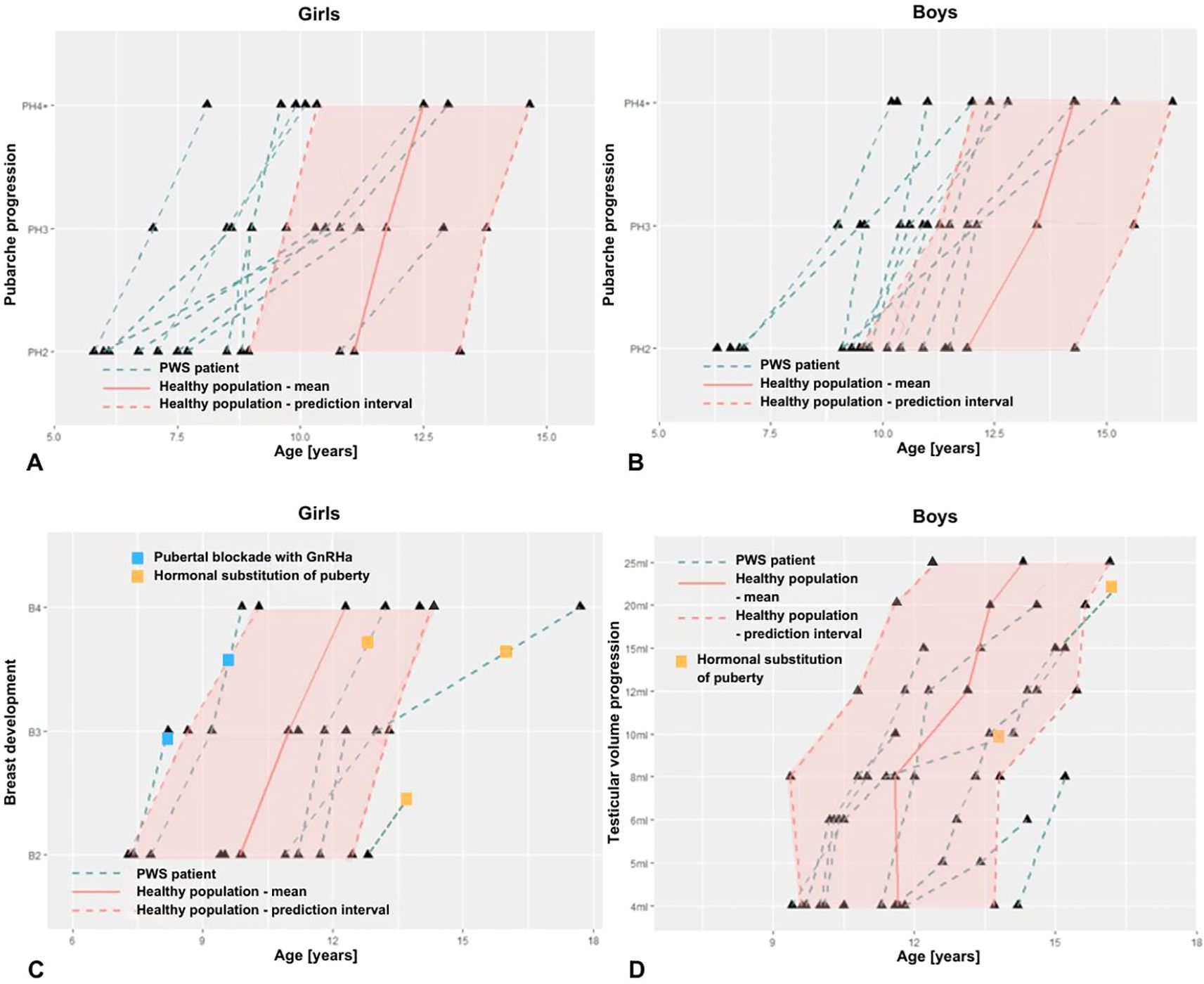
Figure 2. Onset and progression of pubic hair (A, B), of breast maturation in girls (C) and of testicular volume in boys (D) with PWS compared to normative values (healthy population).
Due to the limited number of patients within the late pubertal age, we failed to prove prolonged progression of breast development (stage B2 to B4), as well as, progression of testicular volume from 4 ml to 15 ml. In patients with PWS, there was no significant difference in duration of this period compared to the healthy population (Table 3, Figures 2C, D).
Hormonal replacement to accelerate non-advancing puberty was initiated in 4/5 adolescent girls at an average age of 13.7 years and in 2/3 adolescent boys at age 15.9 years (Figures 2 C, D). These children had spontaneous gonadarche at a physiological age, but further pubertal progression was insufficient, therefore hormone replacement was initiated at a later stage of pubertal development.
Two girls with mUPD developed rapidly progressing CPP with first clinical signs (stage B2) at ages 7.4 and 7.8 years. Both started receiving analogues of gonadotropin releasing hormone (GnRHa) by mean age 9.0 years. In one girl with CPP (proband A), puberty started with breast development followed by pubic hair consistently with the healthy population (Figure 1B). In the second girl with CPP (proband B) the pubertal pattern was typical for PWS – pubarche preceding gonadarche.
Discussion
In this cross-sectional study combined with relevant retrospective data, we investigated 37 patients with PWS with spontaneous pubarche and/or gonadarche. We originally hypothesised that pathogenic variants in the MKRN3 gene might be the cause of this abnormal pubertal pattern. MKRN3 is included in the cluster of genes causative for PWS and is known to act as a repressor of puberty initiation (24, 34). If a loss-of-function variant in the MKRN3 gene causes CPP, all patients with PWS caused by Type 1 and 2 deletions including MKRN3, would have to develop a CPP phenotype. However, we were unable to confirm this hypothesis, the percentage of those PWS adolescents with CPP is similar to the healthy population (5.4% in our cohort vs. 3.5% in healthy population) (13, 35). Interestingly, both PWS girls with CPP in our cohort had mUPD, but not all girls with mUPD have precocious puberty, suggesting that mUPD is not a predictor of CPP in patients with PWS.
We aimed to employ the newest normative data (“pubertograms”), thanks to which the age at onset of pubarche, gonadarche and further progression of puberty in our patients could be objectively evaluated. Most previous studies have concluded that puberty is delayed or incomplete in patients with PWS (36). However, our results have shown that the original concept of delayed puberty onset was modified by our results showing that in PWS patients the gonadarche occurs at a similar age to the healthy population consistently with others studies (36–38), occurring at a typical age of 10.0 years in girls and 11.7 in boys. On the other hand, pubarche occurs in both sexes substantially earlier than in the healthy population. Age at pubarche is correlated neither with age at GH treatment initiation, nor with BMI (39). However, BA in PWS patients was advanced by 0.7 years, and is significantly correlated with pubertal onset (gonadarche). In a previous study, accelerated BA was associated with GH treatment, which is known to accelerate skeletal maturation (40). This may also be causative in our patients. Alternatively, accelerated BA can be caused by the early onset of androgen production, premature pubarche (adrenarche) and obesity as observed by other observational association studies (41, 42), however the natural course of pubarche cannot be evaluated. Another factor influencing the early or late onset of puberty and reproductive physiology in PWS patients may be hyperleptinemia, which is derived from body composition and the number of adipocytes (43, 44).
We have confirmed that pubarche progression in adolescents with PWS is prolonged by over 1 year compared to the healthy population in both sexes. However, we failed to prove that there was no difference in breast development and testicular volume progression compared to the healthy population. The recent study (37) together with our paper, consistently show that these patients do not reach the adult stage of pubertal development without gonadal hormone replacement. Thus, hormone replacement is not needed to initiate puberty but is essential for its progression to later stages and for reaching the adult stage of maturation. Therefore, the early onset of pubarche and abnormal pubertal pattern is suggestive of a dissociation pattern from adrenal maturation leading up to adrenarche, and the functionality of the hypothalamic-pituitary-gonadal axis in these patients.
The relationship between genotype and phenotype in PWS patients is complex and is influenced by specific genetic mechanisms leading to the disorder (4). Understanding these genotype-phenotype correlations can help in managing and providing tailored care for individuals with PWS. However, we have shown that this is not applicable to pubarche, gonadarche, BA and pubertal progression. The onset and progression of puberty in PWS patients cannot be predicted by their genotype.
Limitations
The clinical assessment of puberty was one of the most significant challenges. Therefore, we conducted a search for recent studies of pubertal development in populations of similar ethnicity and socioeconomic standard to allow comparison for our cohort with patients with PWS. For comparison, we used reference standards from Denmark (28–30). Despite this, due to the fact that it is not the same population, this limitation remains. If we eliminated this limitation, we would not be able to evaluate pubertal development in a standard way.
Another limitation was that only pubarche or only incipient signs of gonadarche were present in some of our patients. Therefore, it was not possible to evaluate the further progression of these signs in the whole cohort and part of these results were influenced by the size of the group. We will have more accurate information about the progression of puberty in these patients in a few years.
Conclusion
Regardless of the genetic subtype and the MKRN3 gene status, children with PWS had early and slowly progressing pubarche. However, the age at gonadarche was normal and further progression of puberty was insufficient. The BA at the onset of gonadarche was advanced and was significantly associated with gonadarche. Genetic background was not suitable predictor for the timing of pubertal onset.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving humans were approved by Ethics Committee at the 2nd Faculty of Medicine, Charles University in Prague (date of approval: 27.10.2021; EK-1240.4/21). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
AK: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. PD: Methodology, Supervision, Writing – review & editing. SA: Data curation, Methodology, Writing – review & editing. SK: Data curation, Supervision, Writing – review & editing. BO: Data curation, Writing – review & editing. RP: Data curation, Investigation, Writing – review & editing. ŠP: Investigation, Supervision, Writing – review & editing. MŠ: Investigation, Supervision, Writing – review & editing. ZŠ: Data curation, Investigation, Supervision, Writing – review & editing. JZ: Data curation, Investigation, Supervision, Writing – review & editing. VS: Formal Analysis, Methodology, Validation, Visualization, Writing – review & editing. JL: Conceptualization, Methodology, Project administration, Supervision, Writing – original draft.
Funding
The author(s) declare that financial support was received for the research and/or publication of this article. The genetic testing of study participants was funded by an institutional grant of University Hospital Motol No. 64203/6001.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Kim S, Miller JL, Kuipers PJ, German JR, Beaudet AL, Sahoo T, et al. Unique and atypical deletions in Prader–Willi syndrome reveal distinct phenotypes. Eur J Hum Genet. (2012) 20:283–90. doi: 10.1038/ejhg.2011.187
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
2. Manzardo AM, Weisensel N, Ayala S, Hossain W, Butler MG. Prader-Willi syndrome genetic subtypes and clinical neuropsychiatric diagnoses in residential care adults. Clin Genet. (2018) 93:622–31. doi: 10.1111/cge.13142
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
3. Cassidy SB, Schwartz S, Miller JL, Driscoll DJ. Prader-willi syndrome. Genet Med. (2012) 14:10–26. doi: 10.1038/gim.0b013e31822bead0
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
4. Angulo MA, Butler MG, Cataletto ME. Prader-Willi syndrome: A review of clinical, genetic, and endocrine findings. J Endocrinol Invest. (2015) 38:1249–63. doi: 10.1007/s40618-015-0312-9
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
5. Nicholls R, Knoll J, Butler M, Karam S, Lalande M. Genetic imprinting suggested by maternal heterodisomy in nondeletion Prader-Willi syndrome. Nature. (1989) 342:281–5. doi: 10.1038/342281a0
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
6. Butler MG. Prader-Willi syndrome: Obesity due to Genomic Imprinting. Curr Genomics. (2011) 12:204–15. doi: 10.2174/138920211795677877
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
7. Lionti T, Reid SM, White SM, Rowell MM. A population-based profile of 160 Australians with Prader-Willi syndrome: Trends in diagnosis, birth prevalence and birth characteristics. Am J Med Genet Part A. (2015) 167A:371–8. doi: 10.1002/ajmg.a.36845
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
8. Miller JL, Lynn CH, Driscoll DC, Goldstone AP, Gold JA, Kimonis V, et al. Nutritional phases in Prader-Willi syndrome. Am J Med Genet Part A. (2011) 155:1040–9. doi: 10.1002/ajmg.a.33951
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
9. Miller JL, Tamura R, Butler MG, Kimonis V, Sulsona C, Gold JA, et al. Oxytocin treatment in children with Prader–Willi syndrome: A double-blind, placebo-controlled, crossover study. Am J Med Genet Part A. (2017) 173:1243–50. doi: 10.1002/ajmg.a.v173.5
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
10. Tauber M, Diene G. Prader–Willi syndrome: Hormone therapies. Handb Clin Neurol. (2021) 181:351–67. doi: 10.1016/B978-0-12-820683-6.00026-9
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
11. Lee HS, Hwang JS. Central precocious puberty in a girl with Prader-Willi syndrome. J Pediatr Endocrinol Metab. (2013) 30:1201–4. doi: 10.1515/jpem-2013-0040
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
12. Ludwig NG, Radaeli RF, Silva MM, Romero CM, Al E. A boy with Prader-Willi syndrome: unmasking precocious puberty during growth hormone replacement therapy. Arch Endocrinol Metab. (2016) 60:596–600. doi: 10.1590/2359-3997000000196
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
13. Crinò A, Di Giorgio G, Schiaffini R, Fierabracci A, Spera S, Maggioni A, et al. Central precocious puberty and growth hormone deficiency in a boy with Prader-Willi syndrome. Eur J Pediatr. (2008) 167:1455–8. doi: 10.1007/s00431-008-0679-0
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
14. Meader BN, Albano A, Hilal S, Delaney A. Heterozygous deletions in MKRN3 cause central precocious puberty without Prader-Willi syndrome. J Clin Endocrinol Metab. (2020) 105:2732–9. doi: 10.1210/clinem/dgaa331
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
15. Jong MTC, Carey AH, Caldwell KA, Lau MH, Handel MA, Driscoll DJ, et al. Imprinting of a RING zinc-finger encoding gene in the mouse chromosome region homologous to the Prader – Willi syndrome genetic region. Hum Mol Genet. (1999) 8:795–803. doi: 10.1093/hmg/8.5.795
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
16. Liu H, Kong X, Chen F. Mkrn3 functions as a novel ubiquitin E3 ligase to inhibit Nptx1 during puberty initiation. Oncotarget. (2017) 8:85102–9. doi: 10.18632/oncotarget.19347
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
17. Roberts SA, Naulé L, Chouman S, Johnson T, Johnson M, Carroll RS, et al. Hypothalamic overexpression of makorin ring finger protein 3 results in delayed puberty in female mice. Endocrinol (United States). (2022) 163:1–10. doi: 10.1210/endocr/bqac132
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
18. Abreu AP, Dauber A, Macedo DB, Sekoni D, Brito VN, Gill JC, et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N Engl J Med. (2013) 368:2467–75. doi: 10.1056/NEJMoa1302160
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
19. Busch AS, Hagen CP, Almstrup K, Juul A. Circulating MKRN3 levels decline during puberty in healthy boys. J Clin Endocrinol Metab. (2016) 101:2588–93. doi: 10.1210/jc.2016-1488
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
20. Grandone A, Cirillo G, Sasso M, Capristo C, Tornese G, Marzuillo P, et al. MKRN3 levels in girls with central precocious puberty and correlation with sexual hormone levels: a pilot study. Endocrine. (2018) 59:203–8. doi: 10.1007/s12020-017-1281-x
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
21. Jeong HR, Lee HJ, Shim YS, Kang MJ, Yang S. Serum Makorin ring finger protein 3 values for predicting central precocious puberty in girls. Gynecol Endocrinol. (2019) 35:732–6. doi: 10.1080/09513590.2019.1576615
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
22. Garcia JP, Guerriero KA, Keen KL, Kenealy BP, Seminara SB, Terasawa E. Kisspeptin and neurokinin b signaling network underlies the pubertal increase in gnrh release in female rhesus monkeys. Endocrinology. (2017) 158:3269–80. doi: 10.1210/en.2017-00500
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
23. Dauber A, Marina C-S, Macedo BD, Vinicius BN, Abreu AP. … Paternally inherited DLK1 deletion associated with familial central precocious puberty. J Clin Endocrinol Metab. (2017) 102:1557–67. doi: 10.1210/jc.2016-3677
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
24. Valadares LP, Meireles CG, Toledo IP De, Abreu AP, Carroll RS, Latronico AC, et al. MKRN3 mutations in central precocious puberty: A systematic review and meta-analysis. J Endocr Soc. (2019) 3:979–95. doi: 10.1210/js.2019-00041
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
25. Kodytková A, Al Lababidi E, Čermáková I, Černá J, Čížek J, Kalvachová B, et al. Analýza dat z celostátního registru pacientů léčených růstovým hormonem REPAR. Česko-Slovenská Pediatr. (2020) 75:205–12.
26. Marshall W, Tanner J. Variations in pattern of pubertal changes in girls. Arch Dis Child. (1969) 44:291–303. doi: 10.1136/adc.44.235.291
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
27. Marshall W, Tanner J. Variations in the pattern of pubertal changes in boys. Arch Dis Child. (1970) 45:13–23. doi: 10.1136/adc.45.239.13
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
28. Tanner J, Healy M, Goldstein H, Al. E. Assessment of skeletal maturity and prediction of adult height (TW3 method). 3rd ed. London, UK: WB Saunders (2001).
29. Bláha P, Vignerová J, Riedlová J, Kobzová J, Krejčovský L. VI. celostátní antropologický výzkum dětí a mládeže 2001. Ces-Slov Pediat. (2003) 58:766–70.
30. Bláha P, Hrušková M, Krejčovský L, Al. E. Growth and development of Czech children aged from birth to six years: Anthropological research 2001 - 2003. Prague, Czech Republic: Charles University in Prague (2010).
31. Aksglaede L, Sørensen K, Petersen J, Skakkebæk N, Juul A. Recent decline in age at breast development: The Copenhagen puberty study. Pediatrics. (2009) 123:932–9. doi: 10.1542/peds.2008-2491
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
32. Sørensen K, Aksglaede L, Petersen JH, Juul A. Recent changes in pubertal timing in healthy Danish boys: Associations with body mass index. J Clin Endocrinol Metab. (2010) 95:263–70. doi: 10.1210/jc.2009-1478
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
33. Goede J, Hack W, Sijstermans K, van der Voort-Doedens L, van der Ploeg T, Vries AM, et al. Normative values for testicular volume measured by ultrasonography in a normal population from infancy to adolescence. Horm Res Paediatr. (2011) 76:56–64. doi: 10.1159/000326057
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
34. Palumbo S, Cirillo G, Aiello F, Papparella A, Miraglia E, Grandone A. MKRN3 role in regulating pubertal onset: the state of art of functional studies. Front Endocrinol (Lausanne). (2022) 13:1–11. doi: 10.3389/fendo.2022.991322
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
36. Crinò A, Schiaffini R, Ciampalini P, Spera S, Beccaria L, Benzi F, et al. Hypogonadism and pubertal development in Prader-Willi syndrome. Eur J Pediatr. (2003) 162:327–33. doi: 10.1007/s00431-002-1132-4
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
37. Passone GBC, Aragão FFL, Franco RR, Leite SEJ, Gonzalez BAM, De Albuquerque Schil SP, et al. Puberty in girls with Prader-Willi syndrome: cohort evaluation and clinical recommendations in a Latin American tertiary center. Front Endocrinol (Lausanne). (2024) 15:1–8. doi: 10.3389/fendo.2024.1403470
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
38. Hirsch HJ, Eldar-Geva T, Bennaroch F, Pollak Y, Gross-Tsur V. Sexual dichotomy of gonadal function in Prader-Willi syndrome from early infancy through the fourth decade. Hum Reprod. (2015) 30:2587–96. doi: 10.1093/humrep/dev213
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
39. Griffing E, Halpin K, Lee BR, Paprocki E. Premature pubarche in Prader - Willi syndrome: Risk factors and consequences. Clin Endocrinol (Oxf). (2024) 101:162–9. doi: 10.1111/cen.v101.2
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
40. Eiholzer U, Obwegeser C, Witassek F, Meinhardt U. Bone age maturation in prader-willi syndrome on GH treatment is accelerated in pre-pubertal age without affecting final height. ESPE Abstr. (2015) 84:P–3-968.
41. DeSalvo DJ, Mehra R, Vaidyanathan P, Kaplowitz PB. In children with premature adrenarche, bone age advancement by 2 or more years is common and generally benign. J Pediatr Endocrinol Metab. (2013) 26:215–21. doi: 10.1515/jpem-2012-0283
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
42. Sopher A, Jean A, Zwany S, Winston D, Pomeranz C, Bell J, et al. Bone age advancement in prepubertal children with obesity and premature adrenarche: possible potentiating factors. Obes (Silver Spring). (2011) 19:1259–64. doi: 10.1038/oby.2010.305
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
43. Elias CF, Purohit D. Leptin signaling and circuits in puberty and fertility. Cell Mol Life Sci. (2013) 70:841–62. doi: 10.1007/s00018-012-1095-1
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
44. Yura S, Ogawa Y, Sagawa N, Masuzaki H, Itoh H, Ebihara K, et al. Accelerated puberty and late-onset hypothalamic hypogonadism in female transgenic skinny mice overexpressing leptin. J Clin Invest. (2000) 105:749–55. doi: 10.1172/JCI8353
PubMed Abstract | PubMed Abstract | Crossref Full Text | Google Scholar
Keywords: Prader-Willi syndrome puberty, pubarche, gonadarche, MKRN3 gene, puberty, bone age
Citation: Kodytková A, Dušátková P, Amaratunga SA, Koloušková S, Obermannová B, Pomahačová R, Průhová Š, Šnajderová M, Šumník Z, Zapletalová J, Semjonov V and Lebl J (2025) Variant pubertal development in Prader-Willi syndrome: early and slow progression of pubarche with normal age at gonadarche. Front. Endocrinol. 16:1527140. doi: 10.3389/fendo.2025.1527140
Received: 12 November 2024; Accepted: 25 March 2025;
Published: 15 April 2025.
Edited by:
Xi Li, Affiliated Kangning Hospital of Wenzhou Medical University, ChinaReviewed by:
Claudia Camerino, University of Bari Medical School, ItalyJhancy Malay, RAK Medical and Health Sciences University, United Arab Emirates
Copyright © 2025 Kodytková, Dušátková, Amaratunga, Koloušková, Obermannová, Pomahačová, Průhová, Šnajderová, Šumník, Zapletalová, Semjonov and Lebl. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aneta Kodytková, YW5ldGEua29keXRrb3ZhQGZubW90b2wuY3o=
†ORCID: Aneta Kodytková, orcid.org/0009-0007-9281-4376
Petra Dušátková, orcid.org/0000-0002-8647-9088
Shenali Anne Amaratunga, orcid.org/0000-0002-3388-5824
Stanislava Koloušková, orcid.org/0009-0002-1352-3313
Barbora Obermannová, orcid.org/0009-0002-5126-7349
Renata Pomahačová, orcid.org/0000-0002-7952-1364
Štěpánka Průhová, orcid.org/0000-0001-8019-8026
Marta Šnajderová, orcid.org/0009-0005-4111-1270
Zdeněk Šumník, orcid.org/0000-0002-6462-6462
Jiřina Zapletalová, orcid.org/0000-0003-2333-5586
Valerij Semjonov, orcid.org/0009-0008-3966-7116
Jan Lebl, orcid.org/0000-0002-3365-5375