 Hui-Chen Wang1,2
Hui-Chen Wang1,2 Wen-Sen Lee1,3,4,5*
Wen-Sen Lee1,3,4,5*- 1Graduate Institute of Medical Sciences, College of Medicine, Taipei Medical University, Taipei, Taiwan
- 2Second Degree Bachelor of Science in Nursing, College of Medicine, National Taiwan University, Taipei, Taiwan
- 3Taipei Cancer Center, Taipei Medical University Hospital, Taipei, Taiwan
- 4Cell Physiology and Molecular Image Research Center, Wan Fang Hospital, Taipei Medical University, Taipei, Taiwan
- 5Department of Physiology, School of Medicine, Tzu Chi University, Hualien, Taiwan
Introduction: We previously demonstrated that progesterone (P4) can promote breast cancer cell proliferation and migration through activating the P4 receptor (PR)/cSrc-mediated signaling pathway. It has been suggested that high level of Src homology region 2 domain-containing phosphatase-2 (SHP2) might be involved in breast oncogenesis. This study aimed to investigate whether SHP2 is involved in the P4-mediated cSrc activation in breast cancer cells.
Methods: T47D, MCF-7 and BT-483 breast cancer cell lines were used in this study. Cell proliferation and migration were examined using MTT technique and wound healing assay, respectively. Immunoprecipitation assay and Western blot analysis were performed to evaluate protein-protein interaction and protein expression, respectively. Small interfering RNA (siRNA) technique was used to knock down protein expression.
Results: Knockdown of SHP2 expression abolished the P4-promoted cell proliferation and migration in T47D, MCF and BT-483 cell lines, suggesting that presence of SHP2 is essential for the P4-increased proliferation and migration of breast cancer cell lines. P4 (50 nM) treatment increased the complex formations of PR-cSrc-SHP2-caveolin-1, SHP2-p140Cap, and SHP2-Csk, and the level of p-cSrcY416 (activated form of cSrc). However, knockdown of SHP2 expression increased the complex formations of PR-cSrc-caveolin-1-Csk-p140Cap and the levels of p-caveolin-1, p-Csk and p-cSrcY527 (inactivated form of cSrc).
Discussion: Our data suggest that SHP2 can bind to cSrc-negative regulatory proteins (p140Cap and Csk), hence preventing the interaction between cSrc and cSrc-negative regulatory proteins, leading to decreased phosphorylation of cSrc Y527 and prolonged cSrc activation. These findings highlight the role of SHP2 in the P4-promoted breast cancer cell proliferation and migration.
1 Introduction
In the developed countries, breast cancer is the most common cancer diagnosed and the most invasive cancer in women. The survival rates of breast cancer have increased largely due to a better understanding of the disease, earlier detection and better treatment.
Female sex hormones have been indicated as the risk factors for breast cancer development (1). Exposure of P4 and progestin has been recognized as instrumental in the breast cancer risk associated with exposure to the female sex hormones (2). Traditionally, P4 has been thought to regulate biological effects through binding to its nuclear receptors, PR-A or PR-B, to alter gene transcription. Lately, a growing body of evidence suggests that P4 could bind to the PR located in the cytoplasm or membrane to initiate a quick acting non-genomic signaling cascade. We previously showed that P4 increased migration in arterial smooth muscle cells through a cSrc-mediated non-genomic pathway (3). The rapid action of P4-induced cSrc activation has been implicated in the regulation of cell cycle and migration (3, 4). Moreover, it has been indicated that physiological levels of P4 might increase migration (5) and proliferation (6–8) of breast cancer cells via an extra-nuclear signaling pathway. We previously also demonstrated that P4 promoted breast cancer cell proliferation (9) and migration (10) through activating the P4 receptor (PR)/cSrc-mediated signaling pathways. However, the molecular mechanism underlying P4-induced cSrc activation is still unclear.
SHP2, encoded by the PTPN11 gene, is a pro-migratory signal. SHP2 activates Src family kinases and downstream targets, hence enhancing cell invasion and migration (11). Experimental and clinical data have indicated that high levels of SHP2 mRNA correlate with poor progression-free survival and overall survival in lung adenocarcinomas (12) and SHP2 promotes tumor progression in many types of cancer including breast cancer (13). It has been reported that SHP2 can function as a positive regulator in cSrc signaling by interfering with the Csk-caveolin-1 complex formation in the H2O2-treated astrocytes (14). Knockdown of SHP2 in established breast cancers reduces their growth and metastasis (15). Moreover, reducing SHP2 by small interference RNA and adenovirus-mediated expression of a catalytically inactive mutant of SHP2 decreased cSrc activation (16). It has been demonstrated that transcription factors PR and CREB1 bind to the PTPN11 promoter to regulate the expression of SHP2 in response to decidual signals (17). However, whether SHP2 contributes to the P4-promoted breast cancer cell proliferation and migration is still unknown.
In the present study, we investigated the possible role of SHP2 in the P4-promoted breast cancer cell proliferation and migration. The experimental findings reported below highlight certain molecular mechanisms underlying P4-promoted breast cancer cell proliferation and migration.
2 Materials and methods
2.1 Cell culture
Three breast cancer cell lines (BT-483, MCF-7 and T47D) used in this study were purchased from the American Type Culture Collection/Bioresource Collection and Research Center (BCRC) (Taiwan), where the STR-PCR profiles were performed. The breast cancer cell lines were cultured as previously described (18). Briefly, breast cancer cell lines were grown in the medium containing Dulbecco’s Modified Eagle Medium (DMEM), 10% fetal bovine serum (FBS), penicillin (100 U/mL) and streptomycin (100 μg/mL), and incubated at 37°C in a humidified 5% CO2 incubator. At 24 h prior to the experiment, the culture medium was changed to DMEM supplemented with 10% charcoal-stripped FBS.
2.2 Reagents
Dulbecco’s Modified Eagle Medium (DMEM), FBS, penicillin, and streptomycin were purchased from GIBCO (Grand Island, NY). P4 was obtained from Sigma-Aldrich (St. Louis, MO); Anti-PR (sc-810) antibody was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-cSrc (#2109), anti-p-cSrcY416 (#2101), anti-p-cSrcY527 (#2105), anti-Csk (#4980), anti-caveolin-1 (#3267), anti-p-caveolin-1 (#3251), anti-p140Cap (#3757) and anti-SHP2 (#9793) antibodies and SHP-2 siRNA (#7917) were purchased from Cell Signaling Technology Inc. (Beverly, MA). Anti-p-Csk antibody (#PA5-40214) was from Invitrogen (Carlsbad, CA). Antibody against G3PDH (GTX10011) was from GeneTex (Hsinchu, Taiwan).
2.3 Western blot analysis
Western blot analyses were performed as described previously (19). After incubation, the cells were washed with PBS (pH 7.4), incubated with extraction buffer (Tris 50 mM, pH 7.5, NaCl 150 mM, PMSF 1 mM, NP-40 1%, 0.1% SDS, 10 μg/mL Aprotinin and EDTA 10 mM) on ice, and then centrifuged at 12,000 ×g for 30 min. The cell extract was then boiled in a ratio of 3:1 with sample buffer (Tris–HCl 250 mM, pH 6.8, glycerol 40%, β-mercaptoethanol 20%, SDS 8% and bromophenol blue 0.04%). After electrophoresis in a 12% SDS–polyacrylamide gel (50 μg of protein for each lane), the separated proteins were then transferred onto PVDF membranes. The membrane was cut according to molecular weight with the help of protein marker prior to incubation with antibodies, pre-incubated with BSA (1%) and NaNO3 (0.02%) to block the non-specific IgGs, incubated with specific primary antibody (0.2 μg/mL) for 1 h at room temperature followed by horseradish peroxidase-conjugated second antibody (Jackson ImmunoResearch Laboratories, West Grove, PA) (1:10,000) for additional 1 h, and then developed using the enhanced chemiluminescence (ECL) system (GE, Healthcare, NJ). The intensity of each band was quantify using Image Pro-Plus 4.5 Software.
2.4 Co-immunoprecipitation
Co-immunoprecipitation was conducted as described previously (20). Briefly, protein (200 μg) was immunoprecipitated using primary antibody (2 μg) and protein A agarose beads (20 μL). The precipitates were then washed 5 times with lysis buffer and 1 time with PBS. The pellets were re-suspended in sample buffer (50 mM Tris, pH 6.8, 100 mM bromophenol blue, and 10% glycerol), incubated for 10 min at 90°C before electrophoresis to release the proteins from the beads, and vortexed in the centrifuge for 10 min at 12,500 rpm to discard the beads. The supernatant was fractionated by 8.5% SDS-PAGE, and the protein–protein interaction was examined using Western blot analyses.
2.5 Cell transfection
For transient transfection of SHP2 siRNA or/and ER siRNA into breast cancer cell lines, jetPEI transfection reagent (Polyplus Transfection, Bioparc, France) was used and the transfection was performed according to the manufacture’s protocol. Briefly, the cells were grown in the culture medium and then switched to transfection medium prior to siNRA transfection. The cell culture was treated with a mixture containing 6 μL of SHP2 siRNA or/and ER siRNA, 12 μL of jetPEI transfection reagent and 500 μL of transfection medium for 4-5 h. The transfection medium was removed, switched to DMEM containing 10% FBS for additional 24 h, and then rendered quiescent for 24 h using DMEM containing 0.04% FBS. The culture medium was switched to DMEM containing 10% FBS prior to P4 treatment. The SHP2 siRNA and ER siRNA were purchased from Cell Signal Technology (# 7917) and Genepharma (A10001-2099), respectively.
2.6 MTT assay
MTT was performed as previously described (18). Briefly, the cells were seeded onto 24-well dishes and grown in culture medium supplemented with 10% charcoal-stripped FBS. After the cells were treated with P4 (50 nM) for 96 h, the MTT reagent [3-(4,5-Dimethyl-2- thiazolyl)-2,5-diphenyl-tetrazolium bromide] (USB/Amersham Life Science, Cleveland, OH) at a concentration of 2.5 mg/mL was added to each well. The optical density (570 nM) was measured by ELISA reader. Four samples were analyzed in each experiment.
2.7 Wound healing assay
Wound healing assay was conducted as described previously (21). Briefly, when the cells grown in 12-well plates reached 100% confluence, 10 μL micropipette were used to scalp a “wound” in a monolayer of cells. The images captured at the beginning of wound healing assay were compared to the images after 12 h incubation of the cells in a humidified 5% CO2 incubator at 37°C. The cells migrating to close the wound were examined and counted under microscope to quantify the migration rate of the cells.
2.8 Statistical analysis
All data were expressed as the mean value ± s.e.mean. Comparisons were subjected to one way analysis of variance (ANOVA) followed by Fisher’s least significant difference test. Significance was accepted at P < 0.05.
3 Results
3.1 Involvement of SHP2 in the P4-induced cSrc activation in breast cancer cell lines
We previously showed that P4 increased proliferation and migration of breast cancer cell lines through activating the cSrc-mediated signaling pathways (9, 10). In the present study, we further investigated the molecular mechanism underlying P4-regulated the cSrc-mediated signaling pathways. It has been suggested that high level of SHP2 in breast cancer cells might be involved in breast oncogenesis. Since we previously demonstrated that P4 (0-100 nM) concentration-dependently promoted the proliferation of breast cancer cell lines (T47D and MCF-7), and reached a plateau at 50 nM (9) that overlaps the physiologic range of plasma P4 concentrations in premenopausal women (22), we used P4 at 50 nM to investigate the role of SHP2 in the P4-induced cSrc activation. Using Western blot analysis and siRNA knockdown technique, we demonstrated that treatment with P4 (50 nM) for 5 min increased the levels of p-cSrcY416 (active form of cSrc) in T47D (Figure 1A), MCF-7 (Figure 1B) and BT-483 (Figure 1C) cell lines, and these effects were abolished by knockdown of SHP2. In contrast, knockdown of SHP2 increased the levels of p-cSrcY527 (inactive form of cSrc). Moreover, P4 increased the activities of cSrc negative regulatory proteins (p-Csk and p-caveolin-1) in these SHP2 knockdown breast cancer cell lines. These results suggest that SHP2 is required for the P4-induced cSrc activation in breast cancer cell lines.

Figure 1. SHP2 contributes to P4-induced cSrc activation in breast cancer cell lines. Treatment with P4 (50 nM) for 5 min increased the level of p-cSrcY416, but did not significantly affect the levels of p-Csk and p-caveolin-1 protein in T47D (A), MCF-7 (B), and BT-483 (C) cell lines. Knockdown of SHP2 increased the levels of p-Csk and p-caveolin-1, and induced cSrc inactivation in the P4-treated T47D cells. The protein levels were examined by Western blot analyses and values shown in parentheses represent the quantified results adjusted with their own total protein or G3PDH and expressed as fold of control. Data are representative of 2 independent experiments with similar results. The gels have been run in the same experimental conditions and the cropped blots, which were cut prior to hybridization with antibodies, were shown. Con, control; NT siRNA, non-target small interfering RNA; siRNA, small interfering RNA.
3.2 Involvement of SHP2 in the P4-promoted proliferation and migration in breast cancer cell lines
Using MTT assay and wound healing migration assay, we investigated the effects of P4 treatment on breast cancer cell proliferation and migration. Our results show that P4 (50 nM) time-dependently increased the proliferation of T47D (Figure 2A, top panel), MCF-7 (Figure 2B, top panel) and BT-483 (Figure 2C, top panel). Knockdown of SHP2 expression abolished the P4-increased the proliferation of T47D (Figure 2A, bottom panel), MCF-7 (Figure 2B, bottom panel) and BT-483 (Figure 2C, bottom panel). P4 also increased the migration of T47D (Figure 3A), MCF-7 (Figure 3B) and BT-483 (Figure 3C) cell lines, and these effects were abolished by knockdown of SHP2 expression. These results suggest that SHP2 plays an essential role in the P4-increased proliferation and migration in breast cancer cell lines. Since three breast cancer cell lines (T47D, MCF-7 and BT-483) used in this study are classified as Luminal A estrogen receptor+ (ER+), PR+, HER2-, invasive ductal carcinoma, we tested whether these data are specific to this type of tumor by using ER knockdown T47D. Our data showed that double knocked-down of ER and SHP2 and single knocked-down of SHP2 did not show significantly different effects on the P4-increased T47D proliferation (Supplementary Figure 1) and migration (Supplementary Figure 2), suggesting that the requirement of SHP2 in the P4-promoted breast cancer cell proliferation and migration was not specific to ER+, PR+ breast cancer cells. Moreover, knocked-down of ER did not significantly affect the SHP2 siRNA-increased phosphorylation of Csk, caveolin and SrcY527 and the SHP2 siRNA-abolished P4-increased phosphorylation of Y416 (Supplementary Figure 3). These data suggest that the essential role of SHP2 on the P4-promoted breast cancer cell proliferation and migration is not specific to ER+, PR+, HER2- breast cancer cell lines.
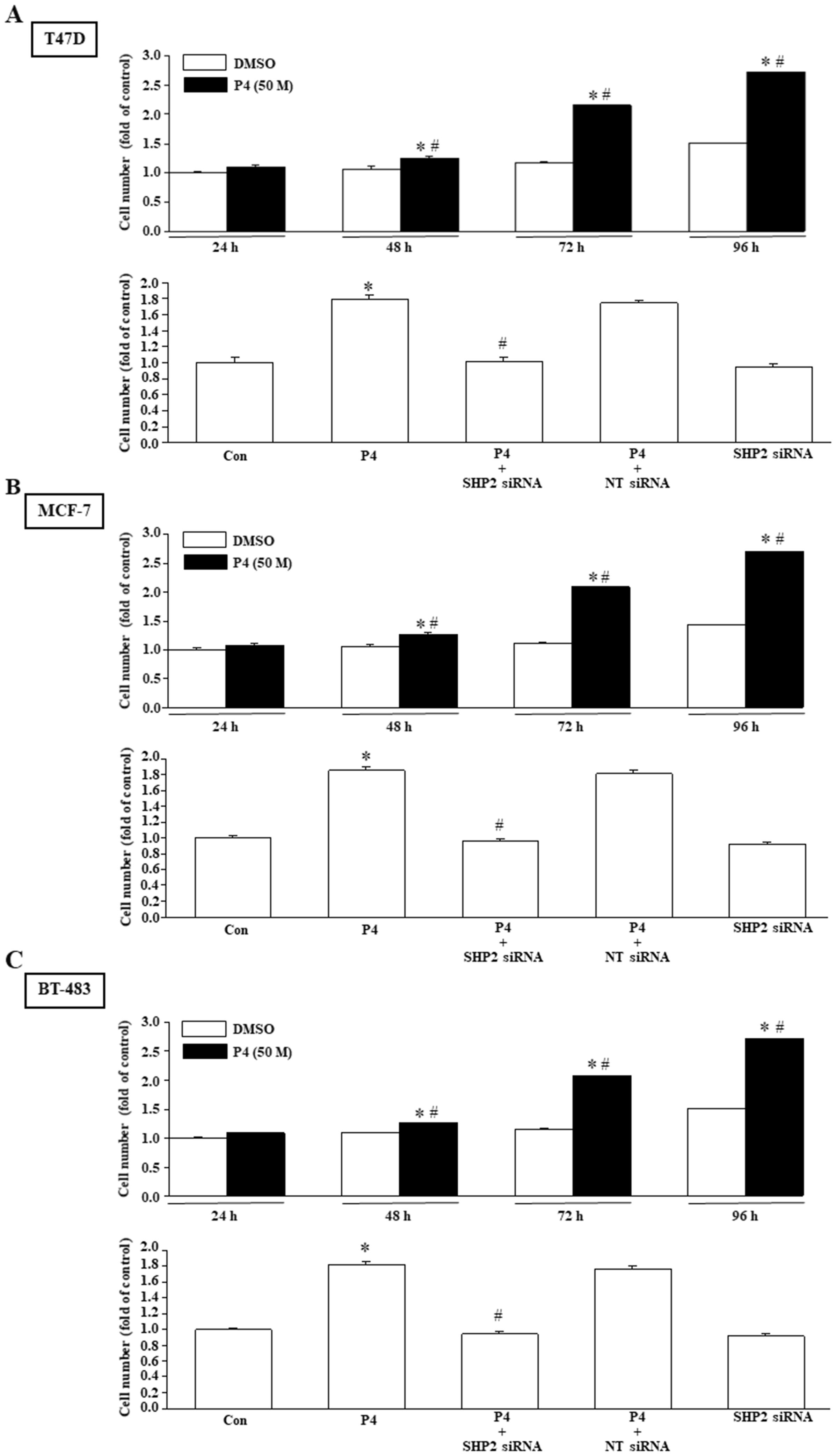
Figure 2. SHP2 is involved in the P4-promoted breast cancer cell proliferation. Treatment with P4 (50 nM) time-dependently promoted the cell proliferation in T47D (A, top panel), MCF-7 (B, top panel), and BT-483 (C, top panel). Knockdown of SHP2 using the siRNA technique prevented the P4-promoted proliferation in T47D (A, bottom panel), MCF-7 (B, bottom panel), and BT-483 (C, bottom panel). For proliferation assay, the cell number was measured using the MTT assay after daily treatment of the cell with P4 (50 nM) for three days. Values represent the means ± s.e.mean. (n = 4). *P < 0.05 different from control. #P < 0.05 different from the DMSO-treated group. NT siRNA, non-target small interfering RNA.
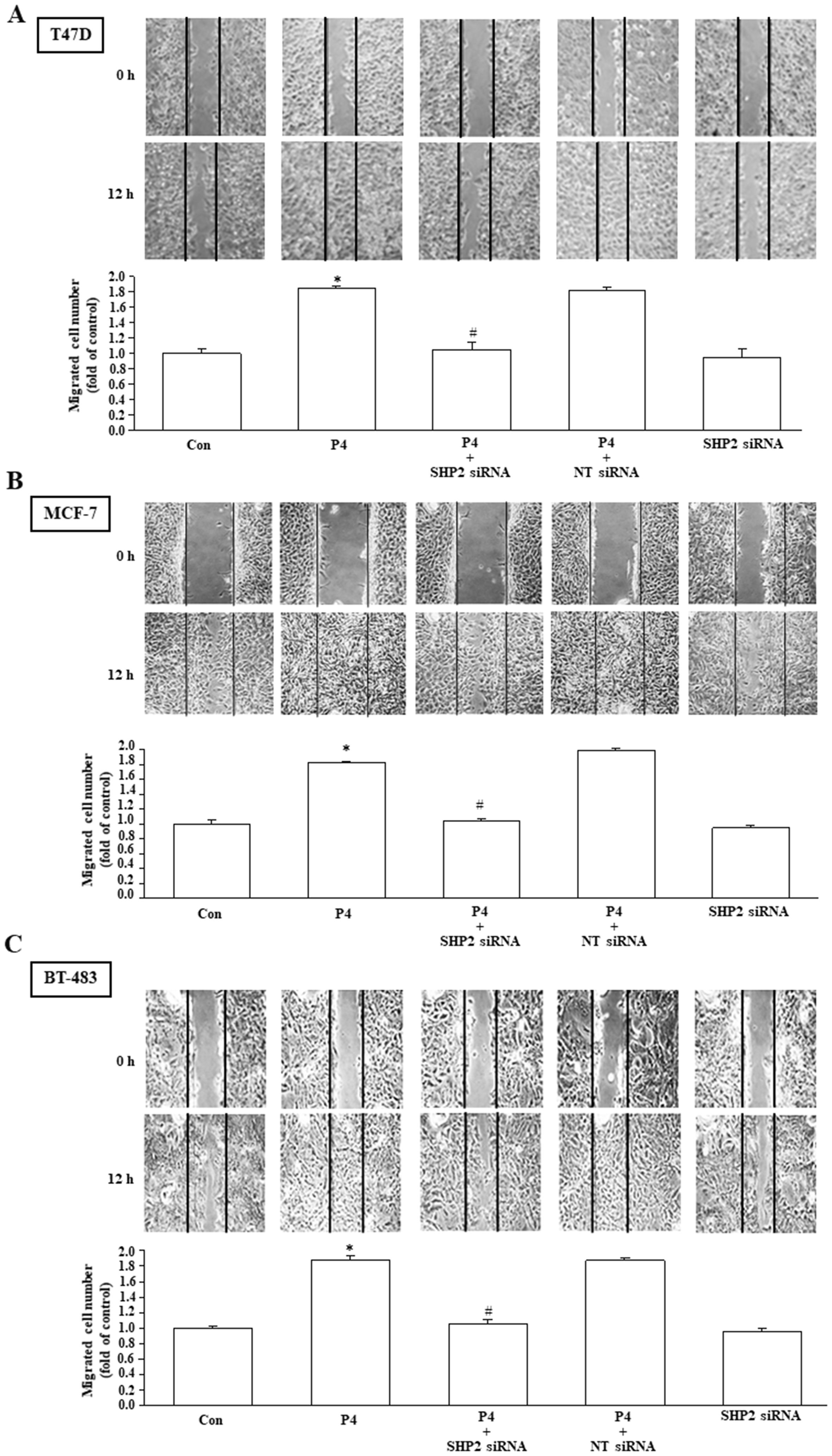
Figure 3. SHP2 is involved in the P4-promoted breast cancer cell migration. Knockdown of SHP2 using the siRNA technique prevented the P4-promoted migration in T47D (A), MCF-7 (B), and BT-483 (C). Top pane: representative photographs of wound healing migration assay. Bottom panel: quantified results expressed by fold of control. For migration assay, the cells migrating to close the wound were counted to quantify the migration rate of the cells. Values represent the means ± s.e.mean. (n = 3). *P < 0.05 different from control. #P < 0.05 different from the P4-treated group. NT siRNA, non-target small interfering RNA.
3.3 Effects of SHP2 knockdown on the interaction among cSrc and cSrc regulatory proteins in T47D cell line
We further examined the effects of SHP2 knockdown on the interaction among cSrc and its regulatory proteins in breast cancer cell lines (T47D, MCF-7 and BT-483). Co-immunoprecipitation assay demonstrated that treatment with P4 increased the complex formations of cSrc-SHP2, cSrc-PRA, cSrc-PRB, and cSrc-cavolin-1, PR-SHP2, and PR-caveolin-1, and the levels of p-PRA, p-PRB, and p-cSrcY416, but did not significantly affect the complex formations of cSrc and cSrc-negative proteins (Csk and p140Cap) in breast cancer cell lines, and the levels of p-Csk, p-caveolin-1, and p-cSrcY547 (inactive form of cSrc). In the SHP2 knockdown breast cancer cell lines, however, the P4-increased p-cSrcY416 level was abolished, and P4 increased the levels of p-caveolin-1, p-Csk and p-cSrcY527 (Figures 4A–D, 5A–D, 6A–D), PR-cSrc negative regulatory proteins (Figures 4B, 5B, 6B), Csk-PRA, Csk-PRB, Csk-p-PRA, Csk-p-PRB, and Csk-p140Cap (Figures 4C, 5C, 6C), caveolin-1-Csk, and caveolin-1-p140Cap (Figures 4D, 5D, 6D). The P4-increased complex formations of cSrc-caveolin-1, cSrc-PRA and cSrc-PRB were not affected by knockdown of SHP2. Although P4 increased the complex formation of cSrc-cavolin-1, the level of p-caveolin-1 was not affected. However, P4 increased the level of p-cavolin-1 in the SHP2 knockdown breast cancer cell lines.

Figure 4. Knockdown of SHP2 increases the interaction between cSrc and cSrc-negative regulatory proteins in T47D cells. (A) Knockdown of SHP2 increased the levels of p-caveolin-1, p-Csk and p-cSrcY527, and the complex formations of cSrc-p-caveolin-1, cSrc-p140Cap, cSrc-Csk and cSrc-p-Csk in the P4-treated T47D cells, but did not affect the P4-increased complex formations of cSrc-PR and cSrc-p-PR. (B) Knockdown of SHP2 increased the interaction between caveolin-1 and cSrc-negative regulatory proteins (p140Cap and Csk), but did not affect the P4-increased complex formations of caveolin-1-PR and caveolin-1-p-PR. (C) Knockdown of SHP2 increased the complex formations of Csk-caveolin-1, Csk-p-caveolin-1, Csk-PR, Csk-p-PR, Csk-p140Cap and Csk-cSrc. The protein-protein interaction was detected by co-immunoprecipitation technique. The protein levels were examined by Western blot analyses and values shown in parentheses represent the quantified results adjusted with their own total protein or G3PDH and expressed as fold of control. Data are representative of 2 independent experiments with similar results. The gels have been run in the same experimental conditions and the cropped blots, which were cut prior to hybridization with antibodies, were shown. Con, control; NT siRNA, non-target small interfering RNA; siRNA, small interfering RNA.

Figure 5. Knockdown of SHP2 increases the interaction between cSrc and cSrc-negative regulatory proteins in MCF-7 cells. (A) Knockdown of SHP2 increased the levels of p-caveolin-1, p-Csk and p-cSrcY527, and the complex formations of cSrc-p-caveolin-1, cSrc-p140Cap, cSrc-Csk and cSrc-p-Csk in the P4-treated MCF-7 cells, but did not affect the P4-increased complex formations of cSrc-PR and cSrc-p-PR. (B) Knockdown of SHP2 increased the interaction between caveolin-1 and cSrc-negative regulatory proteins (p140Cap and Csk), but did not affect the P4-increased complex formations of caveolin-1-PR and caveolin-1-p-PR. (C) Knockdown of SHP2 increased the complex formations of Csk-caveolin-1, Csk-p-caveolin-1, Csk-PR, Csk-p-PR, Csk-p140Cap and Csk-cSrc. The protein-protein interaction was detected by co-immunoprecipitation technique. The protein levels were examined by Western blot analyses and values shown in parentheses represent the quantified results adjusted with their own total protein or G3PDH and expressed as fold of control. Data are representative of 2 independent experiments with similar results. The gels have been run in the same experimental conditions and the cropped blots, which were cut prior to hybridization with antibodies, were shown. Con, control; NT siRNA, non-target small interfering RNA.

Figure 6. Knockdown of SHP2 increases the interaction between cSrc and cSrc-negative regulatory proteins in BT-483 cells. (A) Knockdown of SHP2 increased the levels of p-caveolin-1, p-Csk and p-cSrcY527, and the complex formations of cSrc-p-caveolin-1, cSrc-p140Cap, cSrc-Csk and cSrc-p-Csk in the P4-treated BT-483 cells, but did not affect the P4-increased complex formations of cSrc-PR and cSrc-p-PR. (B) Knockdown of SHP2 increased the interaction between caveolin-1 and cSrc-negative regulatory proteins (p140Cap and Csk), but did not affect the P4-increased complex formations of caveolin-1-PR and caveolin-1-p-PR. (C) Knockdown of SHP2 increased the complex formations of Csk-caveolin-1, Csk-p-caveolin-1, Csk-PR, Csk-p-PR, Csk-p140Cap and Csk-cSrc. The protein-protein interaction was detected by co-immunoprecipitation technique. Western blot analyses were conducted to examine the protein levels. Values shown in parentheses represent the quantified results adjusted with their own total protein or G3PDH and expressed as fold of control. Data are representative of 2 independent experiments with similar results. The gels were run in the same experimental conditions and the cropped blots, which were cut prior to incubation with antibodies, were shown. Con, control; NT siRNA, non-target small interfering RNA.
3.4 Role of PR in the P4-increased formation of cSrc-SHP2 complex
We also examined the effect of PR knockdown on the P4-induced formation of c-Src-SHP2 complex. Co-immunoprecipitation assay demonstrated that in the absence of P4, knockdown of PR abolished the formation of cSrc-SHP2 complex in T47D cell line. Moreover, the P4-increased complex formations of cSrc-SHP2 in T47D cell line was also abolished by knockdown of PR (Supplementary Figures 4A, B). These findings suggest that PR is essential for the formation of cSrc-SHP2 complex.
4 Discussion
We previously demonstrated that P4 increased the proliferation (9) and migration (10) in breast cancer cell lines through activating the cSrc-mediated signaling pathways. However, the molecular mechanism underlying P4-induced cSrc activation is still unclear. Since SHP2 is abundant in breast cancer cells and has been suggested to be involved in breast oncogenesis (23), we investigated the possible contribution of SHP2 in the P4-promoted breast cancer cell proliferation and migration. Our data showed that knockdown of SHP2 abolished the P4-induced cSrc activation and enhancement of proliferation and migration in breast cancer cell lines. To our knowledge, this was the first demonstration that the presence of SHP2 is essential for the P4-promoted breast cancer cell proliferation and migration.
Progestins have also been shown to induce a direct interaction between the polyproline motif of PR-B and the SH3 domain of cSrc, which results in cSrc activation in T47D cells (24). We previously demonstrated that P4 increased the complex formation of PR-cSrc and phosphorylation of cSrc at tyrosine 416 in breast cancer cell lines (14, 18). In the present study, we showed that treatment of T47D cells with P4 increased the levels of p-cSrcY416, but did not significantly affect the levels and activities of cSrc-negative regulatory proteins in T47D, MCF-7 and BT-483. However, the interaction between SHP2 and cSrc as well as cSrc-negative regulatory proteins were increased in the P4-treated T47D, MCF-7, and BT-483 breast cancer cell lines.
SHP2 contains two SH2 domains (N-SH2 and C-SH2), a PTP catalytic domain and a C-terminal tail (25, 26). In the basal condition, SHP2 is kept in an auto-inhibited conformation by intramolecular interactions between the N-SH2 domain and the catalytic cleft of the PTP domain (27). Upon stimulation, binding of growth factor receptor or adaptor proteins to the N-SH2 domain releases auto-inhibition, hence activating SHP2. SHP2 also contains proline-rich domain, which can bind to the SH3 domain of cSrc (28). It has been indicated that SHP2 can function as a positive regulator in cSrc signaling by interfering with the Csk-caveolin-1 complex formation in the H2O2-treated astrocytes (14). SHP2 overexpression is commonly observed in HER2 (+) ERα⁄PR (+) infiltrating ductal carcinoma, and the elevated level of SHP2 protein in breast cancer is positively correlated with lymph node metastasis and higher tumor grade (23). Knockdown of SHP2 in established breast cancers reduced their growth and metastasis (15). In the present study, we showed that treatment of T47D, MCF-7 and BT-483 cells with P4 increased the complex formations of cSrc-PR, cSrc-SHP2, and cSrc-caveolin-1. The interaction between SHP2 and cSrc-negative regulatory proteins (p140Cap, caveolin-1 and Csk) was also increased by P4 treatment. However, the levels of p-caveolin-1 and p-cSrc were not significantly increased by P4 treatment. These data suggest that SHP2 could prevent the interaction between cSrc and cSrc-negative regulatory proteins, hence causing the cSrc activation.
PR has been shown to directly bind to SH3 domain of c-Src through PR polyproline domain at the N-terminus and directly activate c-Src without dephosphorylation at tyrosine 527 (26). Treatment with P4 increased the formation of cSrc-PR complex and activated PR. These effects were not affected by knockdown of SHP2. However, P4 increased the formation of PR-Csk and PR-caveolin-1 in the SHP2 knockdown breast cancer cell lines. Our data suggest that the presence of SHP2 prevents the phosphorylation of cSrc at tyrosine 527, and P4 can directly activate cSrc without dephsophorylation at tyrosine 527. Knockdown of SHP2 increased the levels of p-caveolin-1 and p-Csk protein in T47D, MCF-7 and BT-483, and the complex formations of cSrc-cSrc negative regulatory proteins (p-caveolin-1 and p-Csk) in the P4-treated T47D cells. These data suggest that knockdown of SHP2 activated caveolin-1, subsequently enhancing the complex formation of cSrc-cSrc negative regulatory proteins, thereby increasing the phosphorylation of cSrcY527 and causing cSrc inactivation.
Caveolins, a family of integral membrane proteins, are the principal components of caveolae membrane and involved in regulation of a number of signaling pathways including those involved in cell proliferation, migration and transformation, and vesicular transport (29, 30). Caveolin-1 can act as a docking protein for both Csk and SHP2. It has been demonstrated that SHP2 can bind to caveolin-1 and contribute to the activation of cSrc in the H2O2-treated astrocytes through the competitive binding of Csk to caveolin-1 (14). In breast cancer cell lines, P4 can increase the complex formation of SHP2-caveolin-1, which prevented the P4-increased formations of Csk-p-caveolin-1 and Csk-cSrc) complex, hence increasing the phosphorylation of cSrcY416 and causing cSrc activation. Knockdown of SHP2 increased the phosphorylation of cSrc-negative regulatory proteins (caveolin-1 and Csk) and the complex formations of cSrc-p140Cap, cSrc-Csk, caveolin-1-p140Cap, and Csk-p140Cap, thereby increasing the phosphorylation of cSrcY527 and causing cSrc inactivation. We previously demonstrated that P4 promoted breast cancer cell proliferation (9) and migration (10) through activating the P4 receptor (PR)/cSrc-mediated signaling pathways. Taken together, knockdown of SHP2 prevented the P4-increased cSrcY416 and increased the level of cSrcY527 in the P4-treated breast cancer cell lines, suggesting that the presence of SHP2 played an important role in the P4-promoted breast cancer cell proliferation and migration.
p140Cap is an adaptor protein, and its carboxy-terminal domain contains a proline-rich sequence, which can bind to the cSrc SH3 domain, hence reducing the activity of cSrc (31, 32). Deletion of this proline-rich domain carboxy-terminal, the inhibition of cSrc activity induced by p140Cap mutants disappears (31). These results suggest that p140Cap can directly bind to cSrc, hence reducing the cSrc kinase activity. The p140Cap-caused inhibition of cSrc kinase activity is mediated by elevating the Csk phosphorylation. Csk, an endogenous regulator of the cSrc activity, can increase phosphorylation of the inhibitory Tyr527 in the cSrc carboxy-terminal domain, thereby allowing the binding of the cSrc SH2 domain and the acquirement of an inactive, closed conformation (33, 34). p140Cap was demonstrated to be able directly bind to Csk (33) and p140Cap overexpression can inhibit cell proliferation via reducing the cSrc kinase activity caused by Csk activation. In contrast, knockdown expression of p140Cap can enhance the activity of cSrc kinase (35). The complex formation of p140Cap-Csk contributes to the p140Cap-regulated cSrc inactivation. In the present study, we showed that knockdown of SHP2 increased the complex formations of cSrc-cSrc negative regulatory proteins (Csk and caveolin-1) and Csk-p140Cap, thereby increasing phosphorylation of cSrc at tyrosine 527 and cSrc inactivation.
In conclusion, the results from this study suggest that P4 increased the complex formation of PR-cSrc-SHP2, which increased the binding between SHP2 and caveolin-1, subsequently reducing complex formation of cSrc-Csk and cSrc-p140Cap, leading to decreased phosphorylation of cSrc527 and prolonged cSrc activation, and eventually promoted breast cancer cell proliferation and migration. Knockdown of SHP2 increased the complex formations of cSrc-Csk, cSrc-p140Cap and cSrc-p-caveolin-1, subsequently inducing phosphorylation of cSrc at tyrosine 527 and inactivation of cSrc, and eventually abolished the P4-promoted breast cancer proliferation and migration.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Ethics statement
Ethical approval was not required for the studies on humans in accordance with the local legislation and institutional requirements because only commercially available established cell lines were used.
Author contributions
HW: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Validation, Writing – original draft. WL: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the research grant from the Ministry of Science and Technology, R.O.C. (MOST 107-2320-B-038 -051 -MY3) to Dr. W-SL, and National Science Council Grant (NSC 112-2314-B-002-057-) to Dr. H-CW.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2025.1523589/full#supplementary-material
Abbreviations
Con, control; DMEM, Dulbecco’s Modified Eagle Medium; ER, estrogen receptor; FBS, fetal bovine serum; NT siRNA, non-target small interfering RNA; P4, progesterone; PR, progesterone receptor; SHP2, Src homology region 2 domain-containing phosphatase-2; siRNA, small interfering RNA.
References
1. MacMahon B, Cole P, Brown J. Etiology of human breast cancer: a review. J Natl Cancer institute. (1973) 50:21–42. doi: 10.1093/jnci.50.1.21
2. Khan SA. Progesterone exposure and breast cancer risk-addressing barriers. JAMA Network Open. (2020) 3:e203608. doi: 10.1001/jamanetworkopen.2020.3608
3. Hsu SP, Chen TH, Chou YP, Chen LC, Kuo CT, Lee TS, et al. Extra-nuclear activation of progesterone receptor in regulating arterial smooth muscle cell migration. Atherosclerosis. (2011) 217:83–9. doi: 10.1016/j.atherosclerosis.2011.02.051
4. Boonyaratanakornkit V, Bi Y, Rudd M, Edwards DP. The role and mechanism of progesterone receptor activation of extra-nuclear signaling pathways in regulation gene transcription and cell cycle progression. Steroids. (2008) 73:922–8. doi: 10.1016/j.steroids.2008.01.010
5. Fu XD, Giretti MS, Baldacci C, Garibaldi S, Flamini M, Sanchez AM, et al. Extra-nuclear signaling of progesterone receptor to breast cancer cell movement and invasion through the actin cytoskeleton. PloS One. (2008) 3:e2790. doi: 10.1371/journal.pone.0002790
6. Boonyaratanakornkit V, McGowan E, Sherman L, Mancini MA, Cheskis BJ, Edwards DP. The role of extranuclear signaling actions of progesterone receptor in mediating progesterone regulation of gene expression and the cell cycle. Mol Endocrinology. (2007) 21:359–75. doi: 10.1210/me.2006-0337
7. Skildum A, Faivre E, Lange CA. Progesterone receptors induce cell cycle progression via activation of mitogen-activated protein kinases. Mol Endocrinololgy. (2005) 19:327–39. doi: 10.1210/me.2004-0306
8. Saitoh M, Ohmichi M, Takahashi K, Kawagoe J, Ohta T, Doshida M, et al. Medroxyprogesterone acetate induces cell proliferation through up-regulation of cyclin D1 expression via phosphatidylinositol 3-kinase/Akt/nuclear factor-kappaB cascade in human breast cancer cells. Endocrinology. (2005) 146:4917–25. doi: 10.1210/en.2004-1535
9. Wang HC, Lee WS. Molecular mechanisms underlying progesterone-induced cytoplasmic retention of p27 in breast cancer cells. J Steroid Biochem Mol Biol. (2018) 183:202–9. doi: 10.1016/j.jsbmb.2018.06.015
10. Wang HC, Lee WS. Molecular mechanisms underlying progesterone-enhanced breast cancer cell migration. Sci Rep. (2016) 6:31509. doi: 10.1038/srep31509
11. Sausgruber N, Coissieux MM, Britschgi A, Wyckoff J, Aceto N, Leroy C, et al. SHP2 increases cell motility in triple-negative breast cancer through the activation of SRC-family kinases. Oncogene. (2015) 34:2272–8. doi: 10.1038/onc.2014.170
12. Karachaliou N, Cardona AF, Wilhelmina J, Bracht P, Aldeguer E, Drozdowskyj A, et al. Integrin-linked kinase (ILK) and src homology 2 domain-containing phosphatase 2 (SHP2): novel targets in EGFR-mutation positive non-small cell lung cancer (NSCLC). EBio Med. (2019) 39:207–14. doi: 10.1016/j.ebiom.2018.11.036
13. Zhang F, Niu R. Functions of shp2 in cancer. J Cell Mol Med. (2015) 19:2075–83. doi: 10.1111/jcmm.12618
14. Jo A, Park H, Lee SH, Ahn SH, Kim HJ, Park EM, et al. SHP-2 binds to caveolin-1 and regulates Src activity via competitive inhibition of CSK in response to H2O2 in astrocytes. PloS One. (2014) 9:e91582. doi: 10.1371/journal.pone.0091582
15. Aceto N, Sausgruber N, Brinkhaus H, Gaidatzis D, Martiny-Baron G, Mazzarol G, et al. Tyrosine phosphatase SHP2 promotes breast cancer progression and maintains tumor-initiating cells via activation of key transcription factors and a positive feedback signaling loop. Nat Med. (2012) 18:529–37. doi: 10.1038/nm.2645
16. Ha H, Bennett AM, Jin ZG. A novel role of vascular endothelial cadherin in modulating cSrc activation and downstream signaling of vascular endothelial growth factor. J Biol Chem. (2008) 283:7261–70. doi: 10.1074/jbc.M702881200
17. Zhou P, Ouyang L, Jiang T, Tian Y, Deng W, Wang H, et al. Progesterone and cAMP synergistically induce SHP2 expression via PGR and CREB1 during uterine stromal decidualization. FEBS J. (2024) 291:142–57. doi: 10.1111/febs.16966
18. Wang HC, Huo YN, Lee WS. Folic acid prevents the progesterone-promoted proliferation and migration in breast cancer cell lines. Eur J Nutr. (2020) 59:2333–44. doi: 10.1007/s00394-019-02077-3
19. Hsu SP, Lee WS. Progesterone receptor activation of extranuclear signaling pathways in regulating p53 expression in vascular endothelial cells. Mol Endocrinology. (2011) 25:421–432.99. doi: 10.1210/me.2010-0424
20. Wang HC, Lee WS. Progesterone induces RhoA inactivation in male rat aortic smooth muscle cells through up-regulation of p27kip1. Endocrinology. (2014) 155:4473–82. doi: 10.1210/en.2014-1344
21. Wen HC, Huo YN, Chou CM, Lee WS. PMA inhibits endothelial cell migration through activating the PKC-δ/Syk/NF-κB-mediated upregulation of Thy-1. Sci Rep. (2018) 8:16247. doi: 10.1038/s41598-018-34548-8
22. Shaaban MM, Klopper A. Plasma oestradiol and progesterone concentration in the normal menstrual cycle. J Obstetrics Gynoecology Br Commonwealth. (1973) 80:776–82. doi: 10.1111/j.1471-0528.1973.tb11218.x
23. Zhou X, Coad J, Ducatman B, Agazie YM. SHP2 is up-regulated in breast cancer cells and in infiltrating ductal carcinoma of the breast, implying its involvement in breast oncogenesis. Histopathology. (2008) 53:389–402. doi: 10.1111/j.1365-2559.2008.03103.x
24. Boonyaratanakornkit V, Scott MP, Ribon V, Sherman L, Anderson SM, Maller JL, et al. Progesterone receptor contains a proline-rich motif that directly interacts with SH3 domains and activates c-Src family tyrosine kinases. Mol Cell. (2001) 8:269–80. doi: 10.1016/s1097-2765(01)00304-5
25. Frankson R, Yu ZH, Bai Y, Li Q, Zhang RY, Zhang Z. Therapeutic targeting of oncogenic tyrosine phosphatases. Cancer Res. (2017) 77:5701–5. doi: 10.1158/0008-5472.CAN-17-1510
26. Bollu LR, Mazumdar A, Savage MI, Brown PH. Molecular pathways: targeting protein tyrosine phosphatases in cancer. Clin Cancer Res. (2017) 23:2136–42. doi: 10.1158/1078-0432.CCR-16-0934
27. Hof P, Pluskey S, Paganon SD, Eck MJ, Shoelson SE. Crystal structure of the tyrosine phosphatase SHP-2. Cell. (1998) 92:441–50. doi: 10.1016/s0092-8674(00)80938-1
28. Alastair WP, Matthew LJ. A SHPing tale: Perspectives on the regulation of SHP-1 and SHP-2 tyrosine phosphatases by the C-terminal tail. Cell Signal. (2005) 17:1323–32. doi: 10.1016/j.cellsig.2005.05.016
29. Bouras T, Lisanti MP, Pestell RG. Caveolin-1 in breast cancer. Cancer Biol Ther. (2004) 3:931–41. doi: 10.4161/cbt.3.10.1147
30. Williams TM, Lisanti MP. Caveolin-1 in oncogenic transformation, cancer, and metastasis. Am J Physiology-Cell Physiol. (2005) 288:C494–506. doi: 10.1152/ajpcell.00458.2004
31. Kay BK, Williamson MP, Sudol M. The importance of being proline: the interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J. (2000) 14:231–41. doi: 10.1096/fasebj.14.2.231
32. Chong YP, Ia KK, Mulhern TD, Cheng HC. Endogenous and synthetic inhibitors of the Src-family protein tyrosine kinases. Biochimican Biophys Acta. (2005) 1754:210–20. doi: 10.1016/j.bbapap.2005.07.027
33. Nada S, Okada M, MacAuley A, Cooper JA, Nakagawa H. Cloning of a complementary DNA for a protein-tyrosine kinase that specifically phosphorylates a negative regulatory site of p60c-src. Nature. (1991) 351:69–72. doi: 10.1038/351069a0
34. Okada M. Regulation of the SRC family kinases by Csk. Int J Biol Sci. (2012) 8:1385–97. doi: 10.7150/ijbs.5141
Keywords: breast cancer cell lines, Csk, cSrc, p-cSrcY416, p-cSrcY527, SHP2
Citation: Wang H-C and Lee W-S (2025) SHP2 is essential for the progesterone-promoted proliferation and migration in breast cancer cell lines. Front. Endocrinol. 16:1523589. doi: 10.3389/fendo.2025.1523589
Received: 06 November 2024; Accepted: 14 January 2025;
Published: 10 February 2025.
Edited by:
Eva Surmacz, Allysta Pharmaceuticals, Inc., United StatesReviewed by:
Loredana Mauro, University of Calabria, ItalyJian Gao, University of Texas MD Anderson Cancer Center, United States
Copyright © 2025 Wang and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen-Sen Lee, d3NsZWVAdG11LmVkdS50dw==