 Huifang Peng
Huifang Peng Wenyuan Peng
Wenyuan Peng Jiali Chen
Jiali Chen Keyan Hu
Keyan Hu Yingyu Zhang
Yingyu Zhang Yujin Ma
Yujin Ma Hongwei Jiang
Hongwei Jiang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 14 February 2025
Sec. Pediatric Endocrinology
Volume 16 - 2025 | https://doi.org/10.3389/fendo.2025.1507749
This article is part of the Research Topic Disorders of Sex Development In Children: Advancing Multidisciplinary Approaches For Complex Diagnosis And Management View all 4 articles
The RNA helicase DHX37 gene is involved in ribosomal biological processes, and linked to human genetic diseases associated with 46,XY disorders of sex development (46,XY DSD) or neurodevelopment. Recently, relevant reports have primarily focused on 46,XY DSD. However, there is still a lack of overall understanding of the genetic characteristics, phenotype, etc. of the DHX37 gene in human genetic diseases, and its molecular mechanism is not fully understood. We searched literature databases and summarized and analyzed all the literature related to DHX37 to date, including case reports, cohort studies, and molecular mechanism studies, to comprehensively demonstrate the role of DHX37 in human genetic diseases. Sixty patients were reported to have DHX37-related 46,XY DSD, with p.R308Q, p.R674W variants being the two most common mutation hotspots, accounting for 36.67% and 11.67% of cases respectively. In DSD cohorts, DHX37 gene mutations have different detection frequencies (0.77%–45.45%), whereas in testicular regression syndrome and 46,XY gonadal dysgenesis cohorts, they have a high detection rate. The gonadal development and fertility of female (46,XX) carriers with DHX37 gene mutations are not affected; however, incomplete penetrance may be observed in males (46,XY). The treatments are primarily surgical intervention and hormone replacement therapy administered at appropriate times; however, the long-term prognosis remains unknown. Although the molecular mechanism of DHX37 mutation related 46,XY DSD is unclear, ribosome synthesis, cell cycle regulation, and the NF-κB and Wnt pathways may be affected. This review summarizes the profile of DHX37 defects in human genetic diseases.
DHX37 (NM_032656.4) belongs to the DexD/H-box RNA helicase family, a conserved protein group with an Asp-Glu-Ala-Asp motif (DEAD) (1). DHX37 is associated with 46,XY disorders of sex development (46,XY DSD; 46,XY sex reversal 11 in OMIM #273250) as autosomal dominant inheritance, and developmental delay (“neurodevelopmental disorder with brain anomalies, with or without vertebral or cardiac anomalies” in OMIM #618731) as autosomal recessive inheritance (2, 3). We searched PubMed, Embase, and other literature databases and referred to the ClinVar gene variation database to summarize and display the DHX37 gene variations and related clinical phenotype diagrams in Figure 1. There were 11 cohort studies and 6 case reports on DHX37-related 46,XY DSD, along with a little articles on neurodevelopmental disorders. We observed that the reported pathogenic or likely pathogenic mutations were mainly missense mutations, with only one frameshift mutation mentioned (Figure 1A). This characteristic is consistent across the phenotypes of 46,XY DSD or neurological system related phenotypes. Furthermore, another substantial feature of these variants is that mutations associated with both 46,XY DSD and the neurological system are highly concentrated in the two primary functional domains of DHX37, RecA1 (262-429 amino acid) and RecA2 (459-716 amino acid. The data is sourced from the InterPro and UniProt databases, website: https://www.ebi.ac.uk/interpro/protein/UniProt/Q8IY37/ and https://www.uniprot.org/uniprotkb/Q8IY37/entry). Although a greater number of variants have been reported to be associated with 46, XY DSD, there is no significant difference in the distribution of variants related to neurodevelopmental disorder in the domain structure.

Figure 1. DHX37 gene variants associated with 46,XY differential sex development (46,XY DSD) and the nervous system, along with their clinical manifestations. (A) Correspondence diagram showing DHX37 mutation sites, protein location, and clinical phenotypes. The left column describes other relevant clinical manifestations in patients with these variants, such as developmental delay, intellectual disability, hypotonia, and spinal abnormalities. The relevant specific genetic variants are listed in detail in the middle column. The specific locations of these variants and their functional domains are shown on the DHX37 gene diagram in middle. The right column outlines the variant sites associated with DSD and their clinical manifestations, including abnormalities of the external and internal genitalia, such as micropenis, ambiguous genitalia, and absence of the uterus. It also indicates the clinical significance of the variant (LP or P). (B) The frequency of DHX37 gene mutation sites. AIC, Aicardi syndrome, the related case involved was clinically diagnosed with AIC subsequently tested for DHX37 gene mutation, and we have retained this information. DD, developmental delay. ID, intellectual disability. LP, likely pathogenic. P, pathogenic. The DHX37 domain information data is sourced from the InterPro (https://www.ebi.ac.uk/interpro/protein/UniProt/Q8IY37/) and UniProt (https://www.uniprot.org/uniprotkb/Q8IY37/entry) databases.
This study mainly focuses on the situation of DHX37 in 46,XY DSD related fields. Of the 25 mutation sites associated with 46,XY DSD, 16 were located within these two domains. The RecA2 region has a higher frequency of mutations. Regarding the DHX37-related 46,XY DSD external genital phenotypes, micropenis was present in the majority of variant phenotypes, commonly accompanied by cryptorchidism. Both symptoms are widely present in mutations located within the RecA1 and RecA2 regions. Mutations in the RecA1 region display a broader range of external genitalia phenotypes, including clitoromegaly, absent vaginal opening, and hypospadias, which encompass more “female” phenotypes. Mutations in RecA2 predominantly manifest as micropenis and cryptorchidism. This characteristic combination is commonly observed in mutations affecting both the RecA1 and RecA2 regions of the DHX37 gene. The co-occurrence of these two phenotypic features suggests a possible shared underlying mechanism in the developmental pathways influenced by mutations in these regions, potentially affecting penile growth and testicular descent. Regarding the internal genitalia, most mutations clustered in these two regions showed the absence of Müllerian structures, whereas Wolffian structures were present in some cases.
Sixty patients were reported to carry DHX37 variants related to 46,XY DSD, with a total of 25 variant sites. Among them, the p.R308Q variant had the highest frequency, accounting for 36.67% (22/60); the p.R674W variant accounted for 11.67% (7/60); p.T304W variant;accounted for 5% (3/60); p.R334W, p.S408L, p.T477M, p.S595F, p.R674Q and p.V999M variants accounted for 3.33% (2/60) each; and the other 16 mutations, p.I254V, p.R334L, p.G386S, p.R390H, p.L467V, p.G478R, p.R487H, p.A492P, p.D506N, p.S626L, p.L627F, p.T727fs, p.A737T, p.N900D, p.Q921R and p.G1030E, accounted for 1.67% (1/60) each (Figure 1B). The two most common variants, p.R308Q and p.R674W, classified by the American College of Medical Genetics and Genomics classification as pathogenic (P) or likely pathogenic (LP), were located in the RecA1 and RecA2 regions, respectively. This aligns with our previous assertion that mutations within the RecA1 region tend to manifest more severe and diverse phenotypes. This distribution pattern highlights the potential functional importance of these specific amino acid positions within the DHX37 protein and their impact on sexual development. The clinical manifestations of the p.R308Q mutation locus include micropenis, cryptorchidism, hypoplasia of testicular tissue, testicular fibrosis, blurring of genitalia, and other clinical manifestations, such as clitoral hypertrophy and labial fusion in female patients without other systemic changes.
The first confirmation of an association between DHX37 gene defect and 46,XY DSD was found via genetic screening of cohorts in 2019. In the cohort of 87 patients diagnosed with 46,XY DSD, including 55 patients with 46,XY GD and 32 patients with 46,XY DSD, after ruling out LHCGR, AR, CYP17A1, HSD17B3, HSD3B2, and SRD5A2 gene defects, there were 17 patients with the “P” or “LP” mutation of DHX37 gene, with a mutation frequency of 19.54% (17/87). Among the 14 patients with embryonic regression testicular syndrome (ETRS) in this cohort, seven had DHX37 mutations, suggesting that DHX37 may be the main molecular cause of ETRS (50%, 7/14) (2). In another 46,XY DSD cohort of 145 patients, 13 (13/145, 8.97%) had a DHX37 mutation. Of the 81 patients with 46,XY GD, nine carried a DHX37 mutation (9/81, 11%), whereas in 16 patients with 46,XY testicular regression syndrome (TRS), four carried a DHX37 mutation (4/16, 25%) (4). The above two cohort studies established that DHX37 mutation is one reason of 46,XY DSD, and is more common in TRS. In a small cohort of patients with TRS and partial gonadal dysplasia (PGD) in Japan (n=11), five patients (5/11, 45.45%) had DHX37 mutations based on NGS-panel detection (5). In five ETRS patients from China, two of them (40%, 2/5) were detected p.R308Q of DHX37 at two months old and one year and 5 months old (6). In a 46,XY PGD cohort (n=25) from the United Kingdom and Chile, the DHX37 mutation accounted for 16% (4/25), ranking second only to NR5A1 gene mutation frequency (20%) (7). Using whole-exome sequencing (WES) in 140 patients with 46,XY DSD, DHX37 mutations were detected in seven (7/140, 5%), with clinical phenotypes involving TRS (five patients), complete gonadal dysplasia (CGD) (one patient), and 46,XY DSD (one patient) (8). This study expanded the phenotype of 46,XY DSD caused by DHX37 gene mutation by adding CGD. In a subsequent cohort of 46,XY CGD, a 31-year-old “female” with primary amenorrhea from the 46,XY “female” cohort (n=25) presented with ambiguous genitalia, Tanner stage as B1P3A2, Müllerian absence, ovaries absence, and a follicle stimulating hormone (FSH) level of 86.56 mIU/mL (normal range 2.5–10 mIU/mL). This patient had a 46,XY chromosome karyotype and DHX37 c.1877C>T, p.S626L mutation (9). In a Brazilian cohort of 209 cases of 46,XY DSD, four patients carried DHX37 “P” or “LP” mutations, with a frequency of 1.91% (4/209), identified using Sanger sequencing or massively parallel sequencing. Three additional patients were found to carry VUSs in DHX37, including two with mutations in other genes, which cannot be confirmed as a monogenic genetic cause. The detection frequency of DHX37 mutation ranked sixth in this cohort (10). In a study of 54 Chinese 46,XY DSD cohorts, four patients carried the DHX37 pathogenic variants detected using WES, with the fourth highest detection frequency of 7.41% (4/54). The top three detected genes were AR, SRD5A2, and NR5A1 (11). There have also been reports of DHX37 mutations in a posterior hypospadias cohort (2.38%, 1/42) (12). In a DSD cohort from Ukraine, WES was performed on 79 cases of 46,XY DSD, and the DHX37 p.R674Q variant was detected in a three-year-old female patient (1/79, 1.27%) with an inguinal hernia and bilateral vesicoureteral reflux (13). In some patients, excised gonadal tissues were not pathologically examined before molecular diagnostic confirmation, and the extent of gonadal tissue differentiation and development is unknown. Notably, patients with the p.R308Q variant in some cohorts are almost exclusively females, who often present with primary amenorrhea, no uterine or ovarian tissue, enlarged clitoris, underdeveloped labia, and, in some cases, a vagina. These cohorts included female patients with 46,XY DSD of unknown etiology (4) and excluded patients with 17ß-hydroxysteroid dehydrogenase 3 deficiency, 5α-reductase type 2 deficiency, androgen insensitivity syndrome (7). In a cohort with 521 primary spermatogenic failure (SPGF) patient, 64 (12.28%, 64/521) men were molecular diagnosed finding in 39 genes, using clinical exomes sequencing. There were 4 patients carried DHX37 mutations as p.G386S, p.T727Dfs*60, p.N900D, p.Q921R (14). Overall, 12 studies of the 46,XY DSD cohort that mentioned different DHX37 variations are shown in Table 1. The overall detection rate of 46,XY DSD was 12.280%–81.82%, whereas the detection rate of DHX37 mutations was 0.77–45.45% in the differential cohort, which had various inclusion and exclusion criteria and patient numbers. Patients whose testes were not present in the scrotum or inguinal region on imaging or laparoscopic exploration, whose abdominal exploration showed testicular atrophy or gonadal dysgenesis, and whose anti-Müllerian hormone levels were below normal were included. This suggests that, in the presence of the above conditions, along with laboratory and imaging tests, genetic testing is necessary to determine whether a patient has gonadal dysgenesis. Variants in the DHX37 gene were found in TRS and 46,XY GD cohorts, with a high rate of detection. For 46,XY GD, various studies have confirmed that variants in NR5A1, SRY, and MAP3K1 are the most common causes of non-syndromic GD, with DHX37 mutations also showing a relatively high detection frequency. For TRS or ETRS, DHX37 mutation is the main genetic molecular etiology, accounting for 25%–50%; however, the sample sizes of the relevant TRS/ETRS cohorts were relatively small, necessitating large-scale screening.
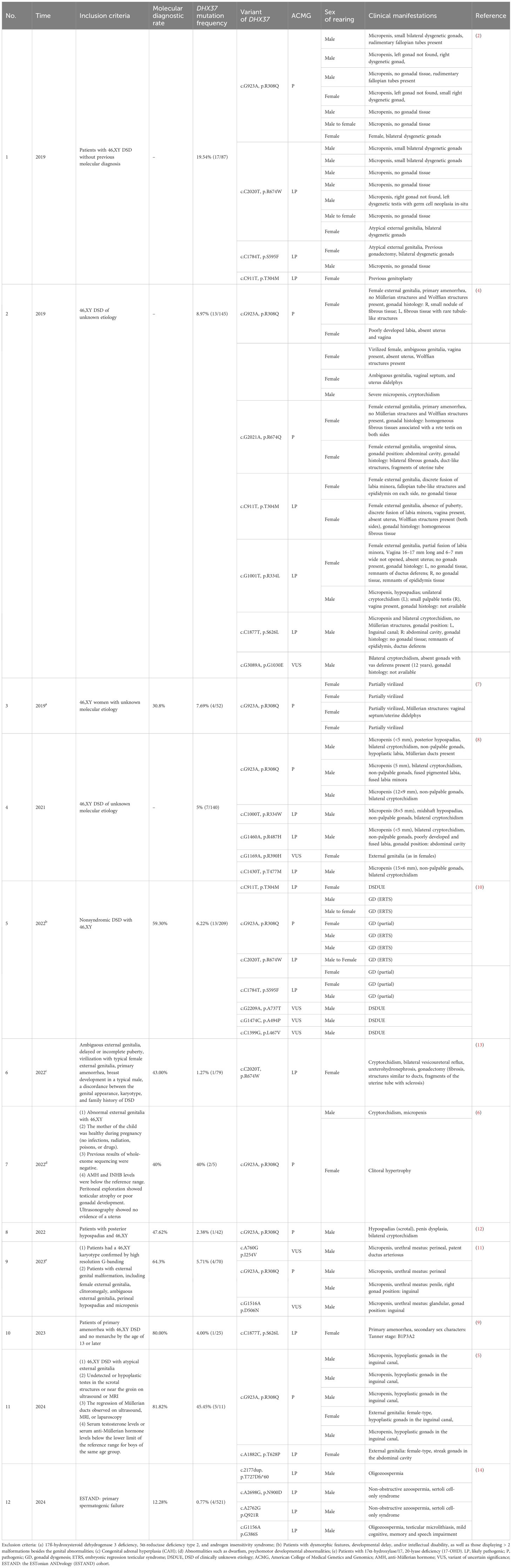
Table 1. The DHX37 gene in the 46,XY DSD cohorts.
Table 2 summarizes the profiles of female carriers. None had any clinical manifestations of DSD, and they were capable of normal reproduction. Thirteen families had 15 female individuals carrying the “P” or “LP” variant of the DHX37 gene who were not affected and could have a normal pregnancy. However, male offspring who inherited the DHX37 gene variant exhibited the DSD phenotype, And the mutation sites include p.R308Q, p.L467V, p.R674W, p.R671T, p.G478R, p.R627F, and p.S595F (2, 8, 15–18). This suggests that the DHX37 mutation does not affect gonadal development in females with 46,XX. Therefore, this gene mutation should be mentioned during genetic counseling to avoid passing the mutation to offspring, which will lead to gonadal dysgenesis, burdening the family and child.
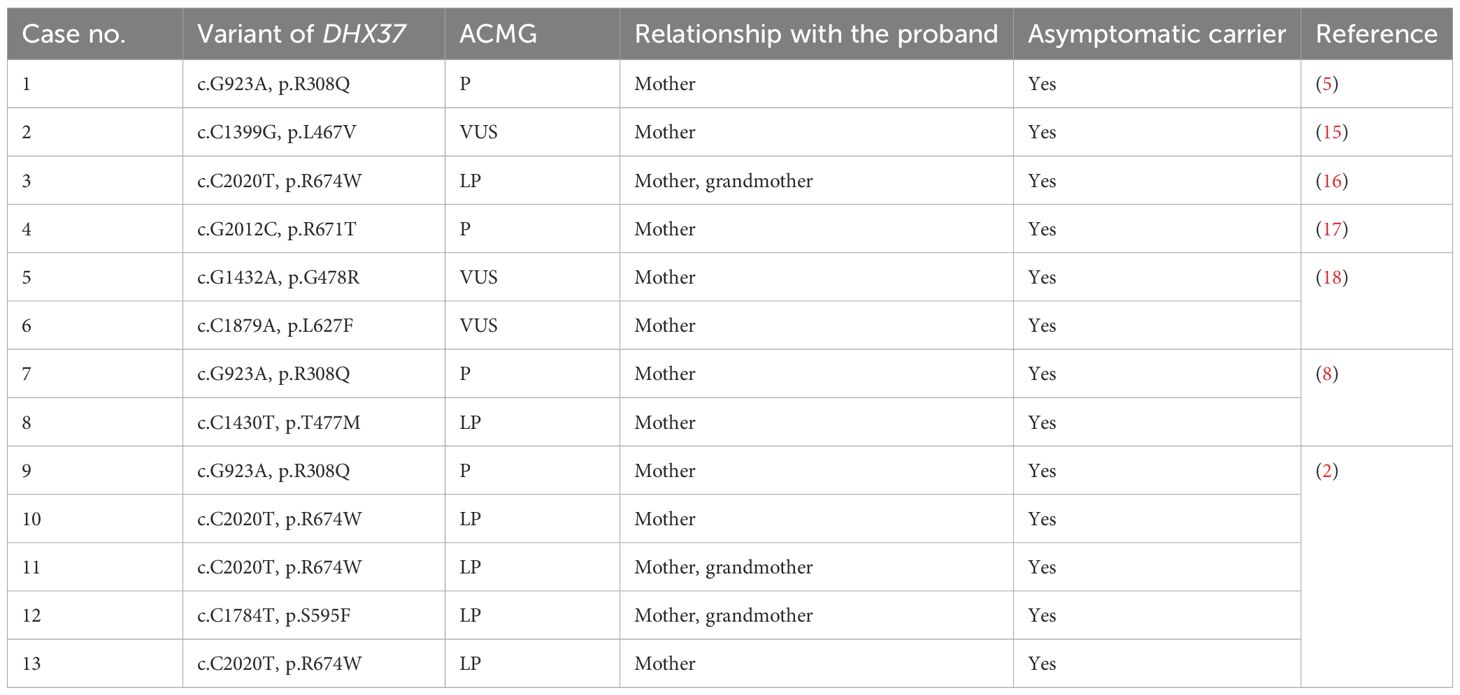
Table 2. Status of female carriers of DHX37 gene mutations.
There was no significant difference in the expression of DHX37 between XX and XY individuals in the sex-determining embryonic gonads of mice (4). In human fetal testicular tissue, DHX37 is detected in Sertoli cells and some spermatogonia, but not in germ cells (2). The DHX37 gene may only function after sex differentiation, following the SRY gene, and simultaneously with SOX8 or SOX9 (19), which may be the main reason why it only affects gonadal development in 46,XY individuals but exerts no effect on 46,XX individuals.
Several cases of male carriers (the father of the proband) with no evident phenotype or with normal fertility have been reported. In a Brazilian family, there were two male siblings with 46,XY DSD (ETRS, micropenis, and non-palpable) caused by the p.R308Q mutation in DHX37. Their father also carried the p.R308Q mutation but exhibited no related phenotype and had three healthy children (2). A French patient with 46,XY DSD (TRS) had a p.T477M homozygous mutation in DHX37, inherited from both parents, the fertile father of this patient was noted to have unilateral testicular agenesis (8). In an Algerian patient with 46,XY DSD, the main clinical manifestations were micropenis, bilateral cryptorchidism, non-palpable gonads, and poorly developed, fused labia caused by the p.R487H mutation in DHX37, inherited from a phenotypically an asymptomatic father (8). DHX37 mutations may also result in incomplete penetrance in males with 46,XY.
The majority of members of the RNA helicase family have been reported to be associated with neurological diseases (20, 21), and DHX37 is currently the only helicase gene that is associated with both neurological disorders and DSD (2, 22), its molecular mechanisms have been reported very little. In pseudomales (female-to-male sex reversals) of the Chinese tongue sole (Cynoglossus semilaevis), the expression of DHX37 and other Z chromosome-specific genes, which are important for spermatogenesis maintenance, is lower than that in normal males (23). In zebrafish, DHX37 can interact with GlyR1,3,4a transcripts, and DHX37 gene defects (dhx37nig1 mutation) can cause splicing defects in the transcription process of GlyR1,3,4a subunits. The defects can also reduce mRNA levels and regulate glycinergic synaptic transmission, leading to an abnormal motor response (24). UTP14A activates the ATPase activity of DHX37 by binding to its carboxyl-terminal domain via conserved regions, thereby enhancing the binding of DHX37 to RNA and promoting ribosome synthesis (25). Activated DHX37 can displace box C/D snoRNA U3 from pre-ribosomal particles to ensure correct and orderly folding of ribosomal subunits (26, 27). In hepatocellular carcinoma cells, DHX37 is highly expressed and promotes proliferation and cancer progression by interacting with PLRG1 and activating the expression of CCND1 (28). In human CD8 T cells, DHX37 could be a regulator affecting NF-κB signaling, T cell activation, and cytotoxicity (29). Nuclear stress, transient activation of the Wnt pathway, and elevated P53 have also been reported in individuals with DHX37 defects (30). Overall, the pathogenic molecular mechanisms of DHX37 and 46,XY DSD are not fully understood. These mechanisms may be involved in ribosome synthesis, cell cycle regulation, and the NF-κB and Wnt pathways. It is currently unclear how this gene is integrated into the genetic network of differences in sexual development; therefore, further validation experiments are required.
The treatment of 46,XY DSD related to DHX37 deficiency is very difficult; there are currently no specific treatment methods available, with very limited reference cases (5, 31). We reviewed all current publications and only six cases with specific treatments have been reported, as summarized in Table 3. Surgical procedures, such as testicular fixation or orchidectomy, are one of the main coping strategies to avoid malignant changes in gonadal tissue; however, there has been no significant improvement in testicular function after surgery. Androgen therapy is also necessary and can help increase the penis size of patients to some extent, maintain normal adrenal function, and support pubertal development; however, it cannot improve fertility. Patients who present as female usually need to undergo external genital plastic surgery. Detailed prognostic and follow-up information is lacking, possibly because DHX37 has only been associated with 46,XY DSD in the last few years, and documenting the dynamic development of infants, children, and adolescents is time-consuming and labor-intensive. Molecular mutations can lead to diseases, and only treatment at the genetic level can truly solve this problem. With advancements in science and technology, and the deepening of knowledge on genetic diseases, we look forward to a cure for genetic diseases in the future.
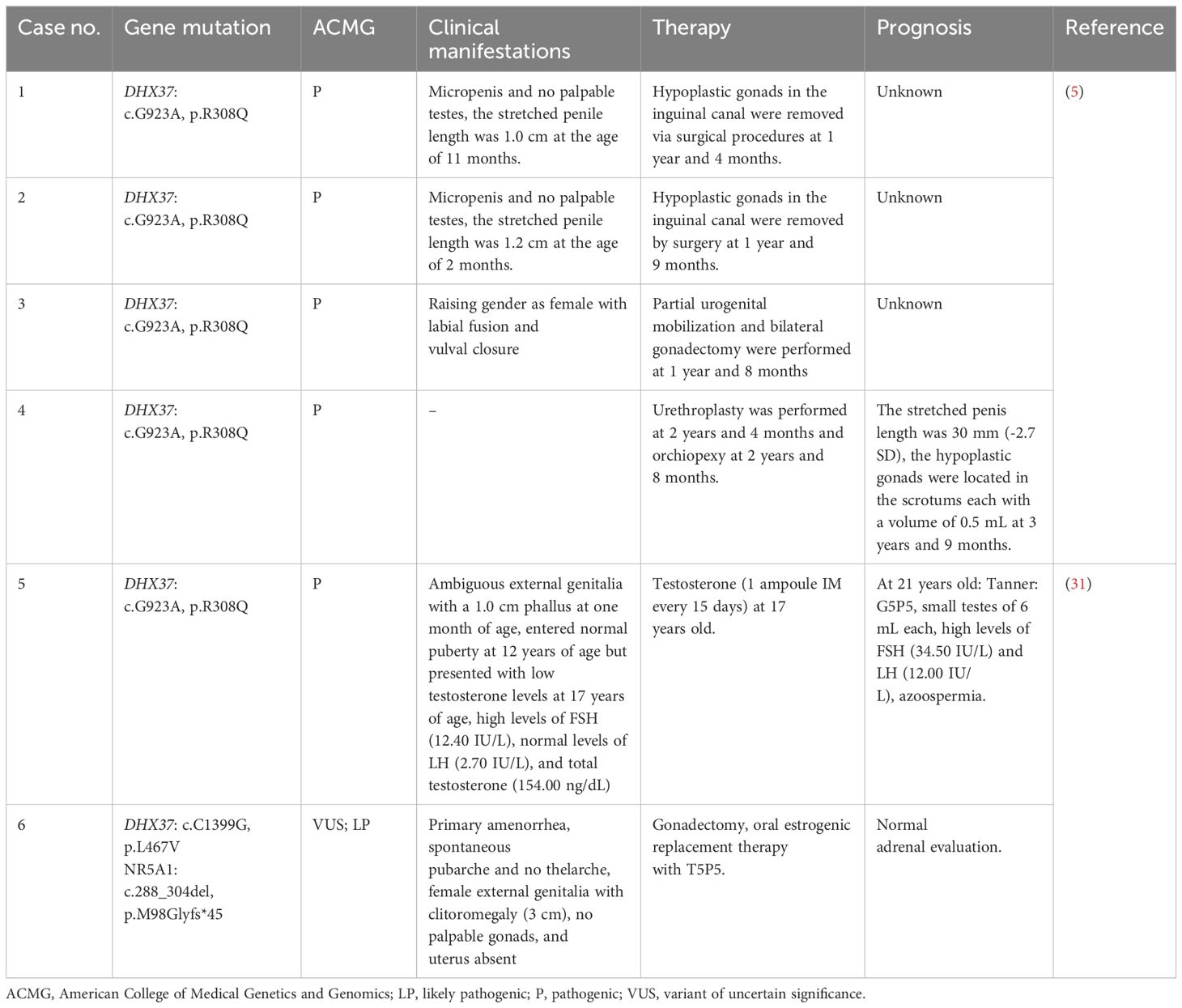
Table 3. Treatment and prognosis of patients with 46,XY associated with DHX37 gene mutations.
In summary, sixty patients had DHX37-related 46,XY DSD, with a total of 25 variant sites. The p.R308Q and p.R674W variants were the two most common mutation hotspots, accounting for 36.67% and 11.67% of cases, respectively. The gonadal development and fertility of female (46,XX) carriers of DHX37 mutations are not affected; however, incomplete penetrance may be observed in males (46,XY). In DSD cohorts, DHX37 gene mutations have different detection frequencies (0.77%–45.45%), whereas in TRS and 46,XY GD cohorts, they have a high rate of detection. The molecular mechanism of DHX37 pathogenesis, the specific pathways of action, and target molecules remain uncertain; however, ribosome synthesis, cell cycle regulation, and the NF-κB and Wnt pathways are suspected to be involved. Surgical intervention and appropriate timing of hormone replacement therapy are commonly used for DHX37-related 46,XY DSDs, although there are limited reported cases. The maintenance of male function and fertility after treatment remains unknown. Gene therapy for genetic diseases may provide new opportunities for the treatment of these diseases in the future.
HP: Data curation, Writing – original draft, Writing – review & editing. WP: Data curation, Writing – original draft. JC: Data curation, Writing – original draft. KH: Methodology, Writing – review & editing. YZ: Visualization, Writing – review & editing. YM: Methodology, Writing – review & editing. HJ: Resources, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Application Research Project of the Science and Technology Research and Development Plan Joint Fund of Henan Province in China (No. 232103810049), and Key Project of Science and Technology Research and Development Joint Fund of Henan Province in China (No. 225200810054).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Paine I, Posey JE, Grochowski CM, Jhangiani SN, Rosenheck S, Kleyner R, et al. Paralog studies augment gene discovery: DDX and DHX genes. Am J Hum Genet. (2019) 105:302–16. doi: 10.1016/j.ajhg.2019.06.001
2. da Silva TE, Gomes NL, Lerário AM, Keegan CE, Nishi MY, Carvalho FM, et al. Genetic evidence of the association of DEAH-box helicase 37 defects with 46,XY gonadal dysgenesis spectrum. J Clin Endocrinol Metab. (2019) 104:5923–34. doi: 10.1210/jc.2019-00984
3. Karaca E, Harel T, Pehlivan D, Jhangiani SN, Gambin T, Coban Akdemir Z, et al. Genes that affect brain structure and function identified by rare variant analyses of mendelian neurologic disease. Neuron. (2015) 88:499–513. doi: 10.1016/j.neuron.2015.09.048
4. McElreavey K, Jorgensen A, Eozenou C, Merel T, Bignon-Topalovic J, Tan DS, et al. Pathogenic variants in the DEAH-box RNA helicase DHX37 are a frequent cause of 46,XY gonadal dysgenesis and 46,XY testicular regression syndrome. Genet Med. (2020) 22:150–9. doi: 10.1038/s41436-019-0606-y
5. Shimura K, Ichihashi Y, Nakano S, Sato T, Hamajima T, Numasawa K, et al. DHX37 variant is one of common genetic causes in Japanese patients with testicular regression syndrome / partial gonadal dysgenesis without müllerian derivatives. Horm Res Paediatr. (2024). doi: 10.1159/000537761
6. Pan L, Su Z, Jiao Y, Sun J, Yin J, Wang H, et al. DHX37 gene heterozygous variant—a frequent cause of embryonic testicular regression syndrome. Chin J Endocrinol Metab. (2022) 37:1413–5. doi: 10.3760/cma.j.cn101070-20210806-00929
7. Buonocore F, Clifford-Mobley O, King TFJ, Striglioni N, Man E, Suntharalingham JP, et al. Next-generation sequencing reveals novel genetic variants (SRY, DMRT1, NR5A1, DHH, DHX37) in adults with 46,XY DSD. J Endocr Soc. (2019) 3:2341–60. doi: 10.1210/js.2019-00306
8. Zidoune H, Martinerie L, Tan DS, Askari M, Rezgoune D, Ladjouze A, et al. Expanding DSD phenotypes associated with variants in the DEAH-box RNA helicase DHX37. Sex Dev. (2021) 15:244–52. doi: 10.1159/000515924
9. Kulkarni V, Chellasamy SK, Dhangar S, Ghatanatti J, Vundinti BR. Comprehensive molecular analysis identifies eight novel variants in XY females with disorders of sex development. Mol Hum Reprod. (2023) 29:gaad001. doi: 10.1093/molehr/gaad001
10. Gomes NL, Batista RL, Nishi MY, Lerário AM, Silva TE, de Moraes Narcizo A, et al. Contribution of clinical and genetic approaches for diagnosing 209 index cases with 46,XY differences of sex development. J Clin Endocrinol Metab. (2022) 107:e1797–806. doi: 10.1210/clinem/dgac064
11. Zhang W, Mao J, Wang X, Zhao Z, Zhang X, Sun B, et al. The genetic spectrum of a Chinese series of patients with 46, XY disorders of the sex development. Andrology. (2024) 12:98–108. doi: 10.1111/andr.13446
12. Shaomei W, Yongbin P, Daiyue Y, Zhaorong H, Huirong Y, Nan L, et al. Whole exome sequencing applied to 42 Han Chinese patients with posterior hypospadias. Steroids. (2022) 184:109041. doi: 10.1016/j.steroids.2022.109041
13. Globa E, Zelinska N, Shcherbak Y, Bignon-Topalovic J, Bashamboo A, MсElreavey K. Disorders of sex development in a large ukrainian cohort: clinical diversity and genetic findings. Front Endocrinol (Lausanne). (2022) 13:810782. doi: 10.3389/fendo.2022.810782
14. Lillepea K, Juchnewitsch AG, Kasak L, Valkna A, Dutta A, Pomm K, et al. Toward clinical exomes in diagnostics and management of male infertility. Am J Hum Genet. (2024) 111:877–95. doi: 10.1016/j.ajhg.2024.03.013
15. Wan Y, Yu R, Luo J, Huang P, Zheng X, Sun L, et al. A novel DEAH-box helicase 37 mutation associated with differences of sex development. Front Endocrinol (Lausanne). (2023) 14:1059159. doi: 10.3389/fendo.2023.1059159
16. Yang Y, Huang H, Wu T, Yang L, Xie L, Shuai X, et al. A case of 46, XY disorders of sexual development caused by the heterozygous mutation of DHX37 gene. Chin J Pract Pediatrics. (2022) 37:1413–5. doi: 10.3760/cma.j.cn101070-20210806-00929
17. Jiang W, Yu J, Mao Y, Tang Y, Cao L, Du Q, et al. Identification and functional analysis of a rare variant of gene DHX37 in a patient with 46,XY disorders of sex development. Mol Genet Genomic Med. (2024) 12:e2453. doi: 10.1002/mgg3.2453
18. Yang H, Ma X, Tian H, Yuan J, Wu D, Dong G, et al. Two novel heterozygous variants in recA2 domain of DHX37 cause 46,XY gonadal dysgenesis and testicular regression syndrome. Sex Dev. (2023) 17:198–202. doi: 10.1159/000534086
19. de Oliveira FR, Guaragna MS, Maciel-Guerra AT, Barros BA, de Mello MP, Guerra-Junior G, et al. DHX37 and the implications in Disorders of Sex Development: an update review. Horm Res Paediatr. (2023) 97:433–44. doi: 10.1159/000535969
20. Lederbauer J, Das S, Piton A, Lessel D, Kreienkamp HJ. The role of DEAD- and DExH-box RNA helicases in neurodevelopmental disorders. Front Mol Neurosci. (2024) 17:1414949. doi: 10.3389/fnmol.2024.1414949
21. Mannucci I, Dang NDP, Huber H, Murry JB, Abramson J, Althoff T, et al. Genotype-phenotype correlations and novel molecular insights into the DHX30-associated neurodevelopmental disorders. Genome Med. (2021) 13:90. doi: 10.1186/s13073-021-00900-3
22. Hiz Kurul S, Oktay Y, Töpf A, Szabó NZ, Güngör S, Yaramis A, et al. High diagnostic rate of trio exome sequencing in consanguineous families with neurogenetic diseases. Brain. (2022) 145:1507–18. doi: 10.1093/brain/awab395
23. Wang HY, Liu X, Chen JY, Huang Y, Lu Y, Tan F, et al. Single-cell-resolution transcriptome map revealed novel genes involved in testicular germ cell progression and somatic cells specification in Chinese tongue sole with sex reversal. Sci China Life Sci. (2023) 66:1151–69. doi: 10.1007/s11427-021-2236-4
24. Hirata H, Ogino K, Yamada K, Leacock S, Harvey RJ. Defective escape behavior in DEAH-box RNA helicase mutants improved by restoring glycine receptor expression. J Neurosci. (2013) 33:14638–44. doi: 10.1523/JNEUROSCI.1157-13.2013
25. Boneberg FM, Brandmann T, Kobel L, van den Heuvel J, Bargsten K, Bammert L, et al. Molecular mechanism of the RNA helicase DHX37 and its activation by UTP14A in ribosome biogenesis. RNA. (2019) 25:685–701. doi: 10.1261/rna.069609.118
26. Roychowdhury A, Joret C, Bourgeois G, Heurgué-Hamard V, Lafontaine DLJ, Graille M. The DEAH-box RNA helicase Dhr1 contains a remarkable carboxyl terminal domain essential for small ribosomal subunit biogenesis. Nucleic Acids Res. (2019) 47:7548–63. doi: 10.1093/nar/gkz529
27. Choudhury P, Hackert P, Memet I, Sloan KE, Bohnsack MT. The human RNA helicase DHX37 is required for release of the U3 snoRNP from pre-ribosomal particles. RNA Biol. (2019) 16:54–68. doi: 10.1080/15476286.2018.1556149
28. Liu Z, Ye Y, Liu Y, Liu Y, Chen H, Shen M, et al. RNA helicase DHX37 facilitates liver cancer progression by cooperating with PLRG1 to drive superenhancer-mediated transcription of cyclin D1. Cancer Res. (2022) 82:1937–52. doi: 10.1158/0008-5472.CAN-21-3038
29. Dong MB, Wang G, Chow RD, Ye L, Zhu L, Dai X, et al. Systematic immunotherapy target discovery using genome-scale in vivo CRISPR screens in CD8 T cells. Cell. (2019) 178:1189–1204.e23. doi: 10.1016/j.cell.2019.07.044
30. McElreavey K, Pailhoux E, Bashamboo A. DHX37 and 46,XY DSD: A new ribosomopathy? Sex Dev. (2022) 16:194–206. doi: 10.1159/000522004
Keywords: DHX37, 46,XY DSD, RNA helicase, human genetic diseases, genetic defects
Citation: Peng H, Peng W, Chen J, Hu K, Zhang Y, Ma Y and Jiang H (2025) Profile of DHX37 gene defects in human genetic diseases: 46,XY disorders of sex development. Front. Endocrinol. 16:1507749. doi: 10.3389/fendo.2025.1507749
Received: 08 October 2024; Accepted: 22 January 2025;
Published: 14 February 2025.
Edited by:
Christopher Joseph Romero, Mount Sinai Kravis Children’s Hospital, United StatesReviewed by:
Anatoly Tiulpakov, Research Centre for Medical Genetics, RussiaCopyright © 2025 Peng, Peng, Chen, Hu, Zhang, Ma and Jiang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongwei Jiang, amlhbmdod0BoYXVzdC5lZHUuY24=; Huifang Peng, cGVuZ2h1aWZhbmdfc2t5QDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.