
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol., 04 March 2025
Sec. Neuroendocrine Science
Volume 16 - 2025 | https://doi.org/10.3389/fendo.2025.1503096
This article is part of the Research TopicOxytocin and Metabolic Dysregulation: From Pathophysiology to PharmacotherapyView all 6 articles
 Jared D. Slattery1
Jared D. Slattery1 June R. Rambousek1Edison Tsui1Mackenzie K. Honeycutt1
June R. Rambousek1Edison Tsui1Mackenzie K. Honeycutt1 Matvey Goldberg1
Matvey Goldberg1 James L. Graham2
James L. Graham2 Tomasz A. Wietecha3,4
Tomasz A. Wietecha3,4 Tami Wolden-Hanson1Amber L. Williams1Kevin D. O’Brien4,5
Tami Wolden-Hanson1Amber L. Williams1Kevin D. O’Brien4,5 Peter J. Havel2,6
Peter J. Havel2,6 James E. Blevins1,3*
James E. Blevins1,3*Previous studies have implicated hindbrain oxytocin (OT) receptors in the control of food intake and brown adipose tissue (BAT) thermogenesis. We recently demonstrated that hindbrain [fourth ventricle (4V)] administration of oxytocin (OT) could be used as an adjunct to drugs that directly target beta-3 adrenergic receptors (β3-AR) to elicit weight loss in diet-induced obese (DIO) rodents. What remains unclear is whether systemic OT can be used as an adjunct with the β3-AR agonist, CL 316243, to increase BAT thermogenesis and elicit weight loss in DIO rats. We hypothesized that systemic OT and β3-AR agonist (CL 316243) treatment would produce an additive effect to reduce body weight and adiposity in DIO rats by decreasing food intake and stimulating BAT thermogenesis. To test this hypothesis, we determined the effects of systemic (subcutaneous) infusions of OT (50 nmol/day) or vehicle (VEH) when combined with daily systemic (intraperitoneal) injections of CL 316243 (0.5 mg/kg) or VEH on body weight, adiposity, food intake and brown adipose tissue temperature (TIBAT). OT and CL 316243 monotherapy decreased body weight by 8.0 ± 0.9% (P<0.05) and 8.6 ± 0.6% (P<0.05), respectively, but OT in combination with CL 316243 produced more substantial weight loss (14.9 ± 1.0%; P<0.05) compared to either treatment alone. These effects were associated with decreased adiposity, energy intake and elevated TIBAT during the treatment period. The findings from the current study suggest that the effects of systemic OT and CL 316243 to elicit weight loss are additive and appear to be driven primarily by OT-elicited changes in food intake and CL 316243-elicited increases in BAT thermogenesis.
The obesity epidemic and its associated complications increase the risk for cardiovascular disease (CVD), hypertension, cancer, type 2 diabetes, and COVID-19 (1, 2). Many of the monotherapies to treat obesity are of limited effectiveness, associated with adverse and/or unwanted side effects (i.e. diarrhea, nausea, vomiting, sleep disturbance and depression) and/or are poorly tolerated. Improvements have been made in monotherapies to treat obesity, particularly within the family of drugs that target the glucagon-like peptide-1 receptor (GLP-1R). Although the FDA recently approved the use of the long-acting and highly effective GLP-1R agonist, semaglutide (3), it can also be associated with mild to moderate gastrointestinal (GI) side effects (3, 4), underscoring the need for continued optimization of existing treatments.
Recent studies suggest that combination therapy (co-administration of different compounds) and monomeric therapy (dual or triple agonists in single molecule) are more effective than monotherapy for prolonged weight loss (5, 6). Marked weight loss has been reported in long-term (20 weeks to ≥ 1 year) clinical studies in humans treated with the amylin analogue, cagrilintide, and semaglutide (≈ 15.6 to 17.1% of initial body weight (7, 8)) and the FDA-approved drug, Qsymia (topiramate + phentermine) (≈ 10.9% of initial body weight (9). Alternatively, the monomeric compound, tirzepatide (Zepbound™), targets both GLP-1R and glucose-dependent insulinotropic polypeptide receptor (GIPR), and was recently reported to elicit 20.9% and 25.3% weight loss in humans with obesity over 72- (10) and 88-week trials (11). Similarly, a recently described drug conjugate, GLP-1-MK-801, which serves as both a GLP-1R agonist and an NMDA receptor antagonist, resulted in 23.2% weight loss following a 14-day treatment regimen in DIO mice (12). In addition, retatrutide, a triple-agonist that targets GIPR, glucagon receptors (GCGR) and GLP-1R was reported to reduce body weight by 24.2% over a 48-week trial (13). Despite the considerable improvements that have been made with respect to weight loss, these treatments are still associated with adverse gastrointestinal side effects (10), leading, in some cases, to the discontinuation of the drug in up to 7.1% of participants (10).
While the hypothalamic neuropeptide, oxytocin (OT) is largely associated with reproductive behavior (14), recent studies implicate an important role for OT in the regulation of body weight (15–18). Studies to date indicate that OT elicits weight loss, in part, by reducing food intake and increasing lipolysis (19–21) and energy expenditure (19, 22–24). While OT is effective at evoking prolonged weight loss in DIO rodents (20, 23–29) and nonhuman primates (19), its overall effectiveness as a monotherapy to treat obesity is relatively modest following 4-8 week treatments in DIO mice (≈4.9%) (29), rats (≈8.7%) (29) and rhesus monkeys (≈3.3%) (19) thus making it more suited as a combination therapy with other drugs that work through other mechanisms. Head and colleagues recently reported that systemic OT and the opioid antagonist, naltrexone, resulted in an enhanced reduction of high-fat, high-sugar meal in rats (30). Recently, we found that hindbrain (fourth ventricle; 4V) OT treatment in combination with systemic treatment with CL 316243, a drug that directly targets beta-3 adrenergic receptors (β3-AR) to increase brown adipose tissue (BAT) thermogenesis (29, 31–34), resulted in greater weight loss (15.5 ± 1.2% weight loss) than either OT (7.8 ± 1.3% weight loss) or CL 316243 (9.1 ± 2.1% weight loss) alone (35).
The goal of the current study aimed to determine whether systemic OT treatment could be used as an adjunct with the β3-AR agonist, CL 316243, to increase BAT thermogenesis and elicit weight loss in DIO rats when using a more translational route of administration for OT delivery. We hypothesized that systemic OT and β3-AR agonist (CL 316243) treatment would produce an additive effect to reduce body weight and adiposity in DIO rats by decreasing food intake and stimulating BAT thermogenesis. To test this, we determined the effects of systemic (subcutaneous) infusions of OT (50 nmol/day) or vehicle (VEH) when combined with daily systemic (intraperitoneal (IP)) injections of CL 316243 (0.5 mg/kg) or VEH on body weight, adiposity, food intake, brown adipose tissue temperature (TIBAT) and thermogenic gene expression.
Adult male Long-Evans rats [~ 8-9 weeks old, 292-349 grams at start of high fat dietary intervention/~ 8-10 months old, 526-929 g body weight at study onset] were initially obtained from Envigo (Indianapolis, IN) and maintained for at least 4 months on a high fat diet (HFD) prior to study onset. All animals were housed individually in Plexiglas cages in a temperature-controlled room (22 ± 2°C) under a 12:12-h light-dark cycle. All rats were maintained on a 1 a.m./1 p.m. light cycle (lights on at 1 a.m./lights off at 1 p.m.). Rats had ad libitum access to water and a HFD providing 60% kcal from fat (approximately 6.8% kcal from sucrose and 8.9% of the diet from sucrose) (D12492; Research Diets, Inc., New Brunswick, NJ). The research protocols were approved both by the Institutional Animal Care and Use Committee of the Veterans Affairs Puget Sound Health Care System (VAPSHCS) and the University of Washington in accordance with NIH’s Guide for the Care and Use of Laboratory Animals (NAS, 2011) (36). We have used the ARRIVE Essential 10 checklist for reporting animal studies.
Fresh solutions of OT acetate salt (Bachem Americas, Inc., Torrance, CA) were solubilized in sterile water. Each minipump was placed into a test tube containing sterile 0.9% saline and then into a water bath at 37° C for approximately 40 hours prior to minipump implantation based on manufacturer’s recommended priming instructions for ALZET® model 2004 minipumps. CL 316243 (Tocris/Bio-Techne Corporation, Minneapolis, MN) was solubilized in sterile water each day of each experiment. CL 316243 was used in place of the FDA-approved β3-AR agonist, Mirabegron, due to the well-established effects of CL 316243 on lipolysis in isolated white adipocytes cells (37) and on BAT thermogenesis and energy expenditure in rodent models in our lab (35) and other labs (33, 34, 38, 39). In contrast to CL 316243, Mirabegron is not water soluble.
The approach for implanting minipumps has been described previously (35). Briefly, animals received a subcutaneous implantation of osmotic minipump (model 2004, DURECT Corporation Cupertino, CA) one week prior to CL 316243 treatment as previously described (35). Animals were treated once with the analgesic ketoprofen (2 mg/kg; Fort Dodge Animal Health) at the completion of the minipump implantations.
Animals were anesthetized with isoflurane and subsequently received implantations of a sterile PDT-4000 HR E-Mitter (26 mm long × 8 mm wide; Starr Life Sciences Company) or PDT-4000 E-Mitter (23 mm long × 8 mm wide) into the intraperitoneal cavity. The abdominal opening was closed with 4-0 Vicryl absorbable suture and the skin was closed with 4-0 monofilament nonabsorbable suture. Vetbond glue was used to seal the wound and bind any tissue together between the sutures. Animals were treated with the analgesic ketoprofen (2 mg/kg; Fort Dodge Animal Health; once/day for 3 consecutive days, including day of surgery) and the antibiotic enrofloxacin (5 mg/kg; Bayer Healthcare LLC., Animal Health Division Shawnee Mission, KS, United States; once per day for 4 consecutive days, including day of surgery) at the completion of the intra-abdominal implantations. Sutures were removed within two weeks after the PDT-4000 HR and PDT-4000 E-Mitter implantation. All PDT-4000 HR and PDT-4000 E-Mitters were confirmed to have remained within the abdominal cavity at the conclusion of the study.
The approach for implanting temperature transponders has been described previously (35). Rats were anesthetized with isoflurane prior to having the dorsal surface along the upper midline of the back shaved and scrubbed with 70% ethanol followed by betadine swabs as previously described (29). Following an incision (1 “) along the midline of the interscapular area a temperature transponder (14 mm long/2 mm wide) (HTEC IPTT-300; Bio Medic Data Systems, Inc., Seaford, DE) was implanted underneath the left IBAT pad as previously described (29, 40, 41). The transponder was subsequently secured in place by suturing it to the brown fat pad with sterile silk suture. The interscapular incision was closed with Nylon sutures (5-0), which were removed in awake animals approximately 10-14 days post-surgery. Animals were treated once with the analgesic ketoprofen (2 mg/kg; Fort Dodge Animal Health) at the completion of the temperature transponder implantations. HTEC IPTT-300 transponders were used in place of IPTT-300 transponders to enhance accuracy in our measurements as previously described (29).
CL 316243 (or saline vehicle; 0.1 ml/kg injection volume) was administered immediately prior to the start of the dark cycle following 4 hours of food deprivation. Animals remained without access to food for an additional 1 (Study 3) or 4 h (Studies 1-2) during the course of the TIBAT measurements. The purpose of the fast was to minimize the potential confound of diet-induced thermogenesis (42) on TIBAT, which was a key measurement in our studies. A handheld reader (DAS-8007-IUS Reader System; Bio Medic Data Systems, Inc.) was used to collect measurements of TIBAT. Measurements were taken under dimmed red light.
Determinations of lean body mass and fat mass were made on un-anesthetized rats by quantitative magnetic resonance using an EchoMRI 4-in-1-700™ instrument (Echo Medical Systems, Houston, TX) at the VAPSHCS Rodent Metabolic Phenotyping Core. Measurements were taken prior to subcutaneous minipump implantations as well as at the end of the infusion period.
Rats (N=10 at study onset) (~ 9 mo old; 517-823 g at start of study) were fed ad libitum and maintained on HFD for approximately 8 months prior to being implanted with temperature transponders (HTEC IPTT-300) underneath the left IBAT depot. Following a 2-week period, animals were subsequently implanted with a PDT-4000 HR E-Mitter telemetry device into the abdominal cavity. Following a 3-week recovery period, CL 316243 or vehicle was administered once per animal at approximately 1-week intervals so that each animal served as its own control. TIBAT was measured daily at baseline (-4 h; 9:00 a.m.), immediately prior to IP injections (0 h; 12:45-1:00 p.m.), and at 0.25, 0.5, 0.75, 1, 20 and 24-h post-injection. The doses of CL 316243 (0.01, 0.1, 0.5 and 1 mg/kg) were selected based on doses of CL 316243 found to be effective at increasing TIBAT when administered intraperitoneally (35) into DIO rats.
The protocol for measuring core temperature and gross motor activity has been described previously (43). Briefly, core temperature (surrogate marker of energy expenditure) and gross motor activity were recorded non-invasively in unanesthetized rats in the home cage every 30 sec throughout the study.
Rats (N=20 at study onset) (~ 10.5 mo old; 577-962 g at start of study) were fed ad libitum and maintained on HFD for approximately 8.5 months prior to prior to being implanted with a temperature transponder underneath the left IBAT depot. Following a 2-week period, animals were matched for both body weight and adiposity prior to being implanted with minipumps. Rats were subsequently maintained on a daily 4-h fast and received minipumps to infuse vehicle or OT (16 or 50 nmol/day) over 29 days. These doses were selected based on a dose of OT found to be effective at reducing body weight or body weight gain when administered subcutaneously (19, 20) or into the 4V (44) of HFD-fed rats. Daily food intake and body weight were collected across the 29-day infusion period.
Rats (N=43 at study onset) (~ 10 mo old; 526-929 g at start of study) were fed ad libitum and maintained on HFD for approximately 9 months prior to receiving implantations of temperature transponders underneath the left IBAT pad. Following a 1-week recovery period, a subset of animals (N=15) was implanted with a PDT-4000 E-Mitter telemetry device into the abdominal cavity or received sham implantations (N=10). Following up to a 1-month recovery period, animals were matched for both body weight and adiposity and were subsequently implanted with minipumps to infuse vehicle or OT (50 nmol/day) over 29 days, respectively. After having matched animals for OT-elicited reductions in body weight (infusion day 7), DIO rats subsequently received single daily IP injections of VEH or CL 316243 (0.5 mg/kg). We selected this dose of CL 316243 because it reduced energy intake and body weight gain and elevated both TIBAT and core temperature and (Study 1). Importantly, we found that this dose of CL 3162243 and CNS administration of OT were found to produce an additive effect on weight loss in DIO rats (35). In addition, we selected the dose of OT (50 nmol/day) because it produced comparable weight loss to that of OT alone in Study 1. TIBAT was measured daily at baseline (-4 h; 9:00 a.m.), immediately prior to IP injections (0 h; 12:45-1:00 p.m.), and at 0.25, 0.5, 0.75, 1, 20 and 24-h post-injection. In addition, daily food intake and body weight were collected across the 29-day infusion period. Data from animals that received the single dose of CL-316243 were analyzed over the 29-day infusion period.
Kaolin (K50001, Research Diets, Inc.) intake (g) was assessed over 29 days following implantation of minipumps containing vehicle (saline) or OT (16 or 50 nmol/day). Placement of kaolin and high fat diet was reversed every other day within each treatment condition.
Inguinal white adipose tissue (IWAT) and epididymal white adipose tissue (EWAT) depots were collected at the end of the infusion period in rats from Study 3. Rats from each group were euthanized following a 3-h fast. Rats were euthanized with intraperitoneal injections of ketamine cocktail [ketamine hydrochloride (214.3 mg/kg), xylazine (10.71 mg/kg) and acepromazine (3.3 mg/kg) in an injection volume up to 2 mL/rat] and transcardially exsanguinated with PBS followed by perfusion with 4% paraformaldehyde in 0.1 M PBS. Adipose tissue (IBAT, IWAT, and EWAT) was dissected and placed in 4% paraformaldehyde-PBS for 24 h and then placed in 70% ethanol (EtOH) prior to paraffin embedding. Sections (5 μm) sampled were obtained using a rotary microtome, slide-mounted using a floatation water bath (37°C), and baked for 30 min at 60°C to give approximately 15-16 slides/fat depot with two sections/slide.
Adipocyte size analysis was performed on deparaffinized, airdried, unstained and uncovered sections. Slightly underexposed photographs of dry sectioned produced clear, highly contrasted black and white images suitable for a built-in particle counting method of ImageJ software (National Institutes of Health, Bethesda, MD). Images were first converted to 16-bit files and then modified and analyzed with a cell shape factor 0.35-1 (a shape factor of 0 represents a straight line and a shape factor of 1 indicates a circle) (methods modified from (45)). Fixed (4% PFA), paraffin-embedded adipose tissue was sectioned and stained with aprimary rabbit anti-UCP-1 antibody (1:100; Abcam, Cambridge, MA (#ab10983/RRID: AB_2241462)] as has been previously described in lean C57BL/6J mice (46) and both lean and DIO C57BL/6 mice after having been screened in both IBAT and IWAT of Ucp1+/- and Ucp1-/- mice (47). Immunostaining specificity controls included omission of the primary antibody and replacement of the primary antibody with normal rabbit serum at the same dilution as the respective primary antibody. Area quantification for UCP-1 staining was performed on digital images of immunostained tissue sections using image analysis software (Image Pro Plus software, Media Cybernetics, Rockville, MD, USA). Slides were visualized using bright field on an Olympus BX51 microscope (Olympus Corporation of the Americas; Center Valley, PA) and photographed using a Canon EOS 5D SR DSLR (Canon U.S.A., Inc., Melville, NY) camera at 100X magnification. Values for each tissue within a treatment were averaged to obtain the mean of the treatment group.
Blood was collected from 4-h (Study 1) or 6-h fasted rats (Studies 2-3) within a 2-h window towards the end of the light cycle (10:00 a.m.-12:00 p.m.) as previously described in DIO CD® IGS rats and mice (25, 29). Animals from Study 3 were euthanized at 2-h post-CL 316243 or VEH treatment. Treatment groups were counterbalanced at time of euthanasia to avoid time of day bias. Blood samples [up to 3 mL] were collected immediately prior to transcardial perfusion by cardiac puncture in chilled K2 EDTA Microtainer Tubes (Becton-Dickinson, Franklin Lakes, NJ). Whole blood was centrifuged at 6,000 rpm for 1.5-min at 4°C; plasma was removed, aliquoted and stored at −80°C for subsequent analysis.
Plasma leptin and insulin were measured using electrochemiluminescence detection [Meso Scale Discovery (MSD®), Rockville, MD] using established procedures (29, 48). Intra-assay coefficient of variation (CV) for leptin was 2.7% and 3.2% for insulin. The range of detectability for the leptin assay is 0.137-100 ng/mL and 0.069-50 ng/mL for insulin. Plasma fibroblast growth factor-21 (FGF-21) (R&D Systems, Minneapolis, MN) and irisin (AdipoGen, San Diego, CA) levels were determined by ELISA. The intra-assay CV for FGF-21 and irisin were 4.5% and 8.4%, respectively; the ranges of detectability were 31.3-2000 pg/mL (FGF-21) and 0.078-5 μg/mL (irisin). Plasma adiponectin was also measured using electrochemiluminescence detection Meso Scale Discovery (MSD®), Rockville, MD] using established procedures (29, 48). Intra-assay CV for adiponectin was 1.1%. The range of detectability for the adiponectin assay is 2.8-178 ng/mL. The data were normalized to historical values using a pooled plasma quality control sample that was assayed in each plate.
Blood was collected for glucose measurements by tail vein nick following a 4 (Study 1) or 6-h fast (Studies 2-3) and measured with a glucometer using the AlphaTRAK 2 blood glucose monitoring system (Abbott Laboratories, Abbott Park, IL) (49). Tail vein glucose was measured at 2-h post-CL 316243 or VEH treatment (Study 3). Total cholesterol (TC) [Fisher Diagnostics (Middletown, VA)] and free fatty acids (FFAs) [Wako Chemicals USA, Inc., Richmond, VA)] were measured using an enzymatic-based kits. Intra-assay CVs for TC and FFAs were 1.4 and 2.3%, respectively. These assay procedures have been validated for rodents (50).
IBAT, EWAT and IWAT tissue was collected from 4 (Study 1) or 6-h fasted rats (Study 3). In addition, animals from Study 3 were euthanized at 2-h post-CL 316243 (0.5 mg/kg) or VEH administration. IBAT, EWAT and IWAT were collected within a 2-h window towards the end of the light cycle (10:00 a.m.-12:00 p.m.) as previously described in DIO CD® IGS/Long-Evans rats and C57BL/6J mice (25, 29, 44). Tissue was rapidly removed, wrapped in foil and frozen in liquid N2. Samples were stored frozen at -80°C until analysis.
RNA extracted from samples of IBAT, EWAT and IWAT (Study 3) were analyzed using the RNeasy Lipid Mini Kit (Qiagen Sciences Inc, Germantown, MD) followed by reverse transcription into cDNA using a high-capacity cDNA archive kit (Applied Biosystems, Foster City, CA). Quantitative analysis for relative levels of mRNA in the RNA extracts was measured in duplicate by qPCR on an Applied Biosystems 7500 Real-Time PCR system (Thermo Fisher Scientific, Waltham, MA). The TaqMan® probe for rat Nono (Rn01418995_g1), Ucp1 (catalog no. Rn00562126_m1), Adrb1 (catalog no. Rn00824536_s1), Adrb3 (catalog no. Rn01478698_g1), type 2 deiodinase (Dio2) (catalog no. Rn00581867_m1), G-protein coupled receptor 120 (Gpr120; catalog no. Rn01759772_m1), cell death-inducing DNA fragmentation factor α-like effector A (Cidea; catalog no. Rn04181355_m1), peroxisome proliferator-activated receptor gamma coactivator 1α (Ppargc1a; catalog no. Rn00580241_m1) and PR domain containing 16 (Prdm16; catalog no. Rn01516224_m1) were acquired from Thermo Fisher Scientific (Thermo Fisher Scientific Gene Expression Assay probes). Relative amounts of target mRNA were determined using the Comparative CT or 2-ΔΔCT method (51) following adjustment for the housekeeping gene, Nono. Specific mRNA levels of all genes of interest were normalized to the cycle threshold value of Nono mRNA in each sample and expressed as changes normalized to controls (vehicle/vehicle treatment).
All temperature transponders, PDT-4000 HR E-Mitters and PDT-4000 E-Mitters were confirmed to have remained underneath the IBAT depot and within the abdominal cavity, respectively, at the conclusion of the study.
All results are expressed as means ± SE. Comparisons between multiple groups involving between-subjects designs were made using one- or two-way ANOVA as appropriate, followed by a post-hoc Fisher’s least significant difference test. Comparisons involving within-subjects designs were made using a one-way repeated-measures ANOVA followed by a post-hoc Fisher’s least significant difference test. Analyses were performed using the statistical program SYSTAT (Systat Software, Point Richmond, CA). Differences were considered significant at P<0.05, 2-tailed.
The goal of this study was to identify a dose range of the beta-3 receptor agonist, CL 316243, that resulted in weight loss, reduced energy intake and an elevation in IBAT and core temperature in DIO rats. The effective dosing data from this study was used to select a dose range (0.01-1 mg/kg, IP) for use in the subsequent chronic dose escalation study (Study 3). By design, DIO rats were obese as determined by both body weight (782.1 ± 22.2 g) and adiposity (288.4 ± 12.4 g fat mass; 36.8 ± 0.8% adiposity) after maintenance on the HFD for approximately 8 months.
As in our previous study in DIO rats (35), there was an overall significant main effect of CL 316243 to reduce body weight gain at 20- [(F(4,36) = 4.347, P=0.006], 44- [(F(4,36) = 4.734, P=0.004], 68- [(F(4,36) = 7.316, P<0.01], 92- [(F(4,36) = 3.858, P=0.010], 116- [(F(4,36) = 8.203, P<0.01], 140- [(F(4,36) = 7.531, P<0.01], 164- [(F(4,36) = 6.100, P=0.001] and 188-h post-injection [(F(4,36) = 5.129, P=0.002].
Specifically, the highest dose (1 mg/kg) reduced body weight gain across all post-injection time intervals (P<0.05 vs VEH) (Figure 1A). The second highest dose of CL 316243 (0.5 mg/kg) also reduced weight gain at 20, 44, 68, 92, 1116, 140, and 164-h post-injection. A lower dose of CL 31243 (0.1 mg/kg) was also effective at reducing weight gain at 20-, 44-, 68-, 116, 140, 164 and 188-h post-0injection. The lowest dose (0.01 mg/kg) reduced body weight gain at 44-, 116-h post-injection and tended to reduce body weight gain at 68-h post-injection.
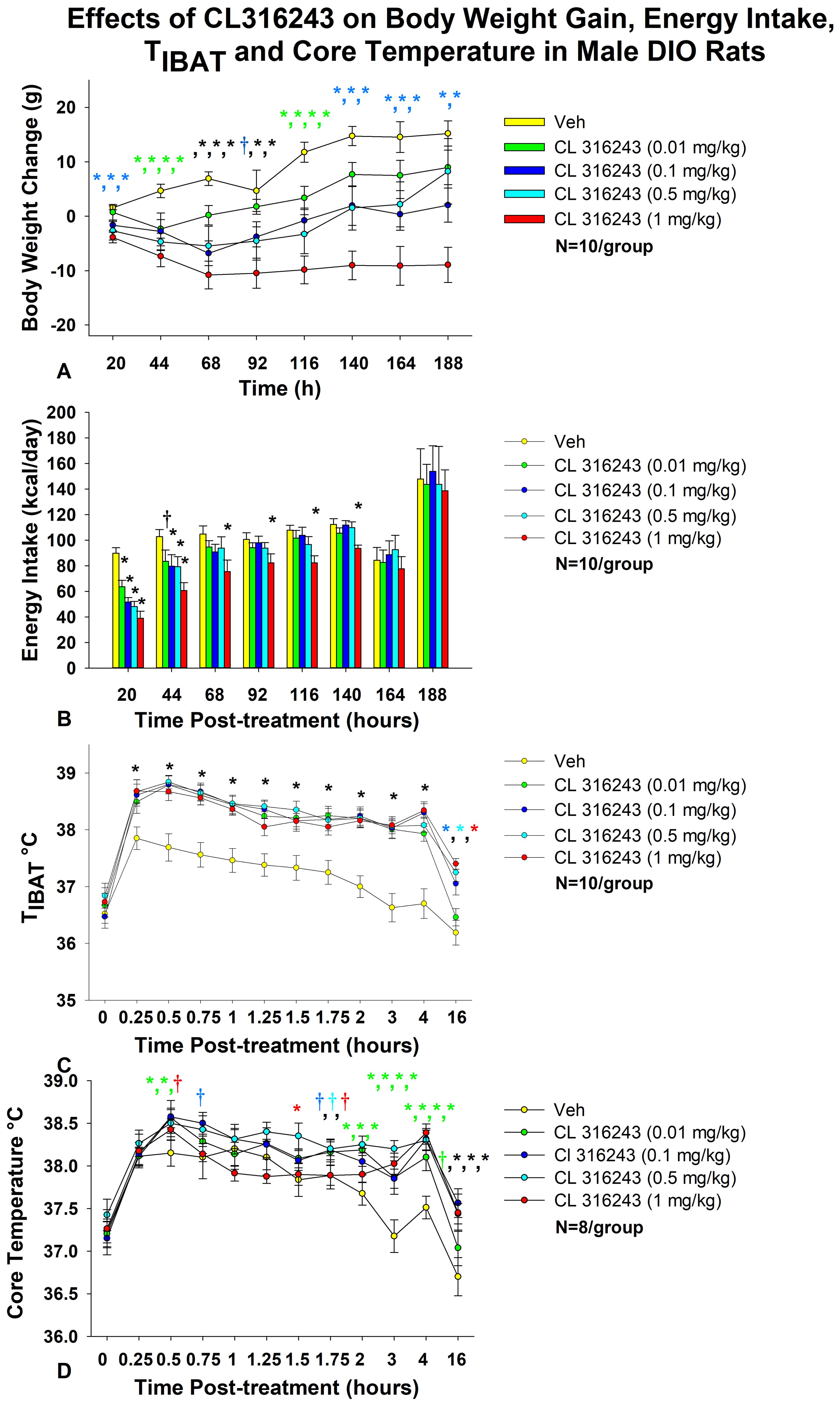
Figure 1. Dose-response effects of the beta-3 receptor agonist, CL 316243, on body weight, energy intake, TIBAT, core temperature and gross motor activity in male DIO rats. Ad libitum fed rats were maintained on HFD (60% kcal from fat; N=10-12/group) for approximately 8 months prior to receiving IP injections of CL 316243 (0.01-1 mg/kg) or vehicle (sterile water) where each animal received each treatment approximately once per week. (A) Effect of CL 316243 on body weight change in DIO rats, (B) Effect of CL 316243 on energy intake (kcal/day) in DIO rats, (C) Effect of CL 316243 on TIBAT in DIO rats; (D) Effect of CL 316243 on core temperature in DIO rats. Data are expressed as mean ± SEM. *P<0.05, †0.05<P<0.1 CL 316243 vs. vehicle. The comma symbols delineate multiple points of significance at the same time point. The different colors represent each dose that was significant at that particular time point.
As in our previous study in DIO rats (35), there was a significant main effect of CL 316243 to reduce energy intake at 20-h [(F(4,36) = 17.431, P<0.01], 44-h [(F(4,36) = 3.688, P=0.013], 116-h [(F(4,36) = 3.094, P=0.027] and 140-h post-injection [(F(4,36) = 4.416, P=0.007]. In addition, there was a near significant effect at 68-h post-injection [(F(4,36) = 2.131, P=0.097].
Specifically, CL 316243 reduced 20-h energy intake at 0.01, 0.1, 0.5 and 1 mg/kg (P<0.05) by 28.4 ± 5.5, 41.4 ± 5.4, 45.0 ± 6.0, and 56.5 ± 5.8% (Figure 1B). The higher doses (0.1, 0.5 and 1 mg/kg) also reduced energy intake by 18.0 ± 11.1, 17.8 ± 12.1, and 37.8 ± 7.7% (P<0.05) at 44-h post-injection (P<0.05). Furthermore, the highest dose (1 mg/kg) reduced energy intake by 25.7 ± 10.3, 13.0 ± 11.6, 22.8 ± 5.7 and 13.0 ± 11.6% at 68-, 92-, 116- and 140-h post-injection (P<0.05).
As in our previous study in DIO rats (35), there was a significant main effect of CL 316243 to elevate TIBAT at 0.25- [(F(4,36) = 22.639, P<0.01], 0.5- [(F(4,36) = 31.681, P<0.01], 0.75- [(F(4,36) = 19.777, P<0.01], 1- [(F(4,36) = 14.901, P<0.01], 1.25- [(F(4,36) = 19.606, P<0.01], 1.5- [(F(4,36) = 12.651, P<0.01], 1.75- [(F(4,36) = 18.074, P<0.01], 2- [(F(4,36) = 25.583, P<0.01], 3- [(F(4,36) = 23.501, P<0.01], 4- [(F(4,36) = 30.853, P<0.01] and 16-h post-injection [(F(4,36) = 13.296, P<0.01]. We also found a significant effect of time [(F(10,450) = 99.883, P<0.01] and a significant interactive effect between time and dose [(F(40,450) = 2.635, P<0.01] across 11 time points over the 16-h measurement period.
Specifically, systemic injections of CL 316243 (0.01-1 mg/kg) (Figure 1C) increased TIBAT at all time points between 0.25-h and 4-h post-injection. The higher doses (0.1-1 mg/kg) also increased TIBAT at 16-h post-injection (P<0.05).
There was a significant main effect of CL 316243 to elevate core temperature at 2- [(F(4,28) = 5.833, P<0.01], 3- [(F(4,28) = 7.394, P<0.01], 4- [(F(4,28) = 9.207, P<0.01], 5- [(F(4,28) = 3.534, P=0.019], 14- [(F(4,28) = 4.497, P=0.006], 16- [(F(4,28) = 7.136, P<0.01], 22- [(F(4,28) = 3.382, P=0.022], 24- [(F(4,28) = 14.967, P<0.01] and 40-h post-injection [(F(4,28) = 4.718, P=0.005].
CL 316243 produced a near significant main effect to elevate core temperature at 1.75- [(F(4,28) = 2.211, P=0.093] and 6-h post-injection [(F(4,28) = 2.419, P=0.072]. We also found a significant effect of time [(F(12,420) = 25.873, P<0.01] and a significant interactive effect between time and dose [(F(48,420) = 1.830, P=0.001] across 13 time points over the initial 16-h measurement period.
Specifically, systemic injections of CL 316243 increased core temperature at 0.5- (0.01 and 0.1 mg), 1.5- (0.5 mg/kg), 2- (0.01, 0.1 and 0.5 mg/kg), 3- (0.01, 0.1, 0.5 and 1 mg/kg), 4- (0.01, 0.1, 0.5 and 1 mg/kg), 5- (0.1 and 1 mg/kg), 6- (0.1, 0.5 and 1 mg/kg), 14- (0.01, 0.1, 0.5 and 1 mg/kg), 16- (0.1, 0.5 and 1 mg/kg), 22- (0.5 and 1 mg/kg), 24- (0.5 and 1 mg/kg), and 40-h post-injection (1 mg/kg) (Figure 1D).
There was also a near significant effect of CL 316243 to stimulate core temperature at 0.5-h (0.5 mg/kg), 0.75-h (0.1 mg/kg), 1.75-h (0.01, 0.1 and 0.5 mg/kg), 5-h (0.01 and 0.5 mg/kg), 6-h (0.01 mg/kg), 12-h (0.1 and 0.5 mg/kg), 16- (0.01 mg/kg), 20-h post-injection (0.5 mg/kg) and 24-h post-injection (0.01 mg/kg).
Two animals were removed from the core temperature and gross motor activity analysis due to defective telemetry devices.
There was a significant main effect of CL 316243 to reduce gross motor activity at 1-h post-injection [(F(4,28) = 5.603, P=0.002]. There was also a near significant main effect of CL 316243 to reduce gross motor activity at 0.75-h post-injection [(F(4,28) = 2.375, P=0.076].
Specifically, CL 316243 reduced gross motor activity at 0.75- (0.01, 0.1 and 0.5 mg/kg), and 1-h post-injection (0.01, 0.1, 0.5 and 1 mg/kg) (P<0.05; data not shown). There was also a near significant effect of CL 316243 (0.01 and 1 mg/kg) to reduce core temperature at 22-h post-injection (0.05<P<0.01). Otherwise, CL 316243 was ineffective at altering gross motor activity at any other time point (P=NS vs vehicle; data not shown).
By design, rats were DIO as determined by both body weight (771 ± 24.5 g) and adiposity (312.3 ± 16.7 g fat mass; 40.1 ± 1.1% adiposity) after maintenance on the HFD for approximately 8.5 months. Following temperature transponder implantations and prior to minipump implantations, groups were again matched for body weight and adiposity (vehicle: 760 ± 56.5 grams/36.8 ± 1.7% fat/283.3 ± 31.9 g fat mass); OT (16 nmol/day): 758.6 ± 39.9 grams/36.8 ± 2.0% fat/282.0 ± 28.1 g fat mass; OT (50 nmol/day): 763.6 ± 45.7 grams/36.6 ± 1.8% fat/284.5 ± 31.1 g fat mass. There was no difference in body weight [(F(2,17) = 0.027, P=NS)] or percent adiposity [(F(2,17) = 0.003, P=NS)] between groups prior to treatment onset. As expected, body weight of DIO rats remained stable over the month of vehicle treatment relative to pre-treatment [(F(1,5) = 2.865, P=0.151)] (Figure 2A). In contrast to vehicle treatment, systemic OT (16 nmol/day) resulted in a significant reduction of body weight relative to OT pre-treatment [(F(1,6) = 140.799, P<0.01)] (Figure 2A; P<0.05). Furthermore, SC OT, at a 3-fold higher dose (50 nmol/day), also resulted in a significant reduction of body weight relative to pre-treatment [(F(1,6) = 47.271, P<0.01)].
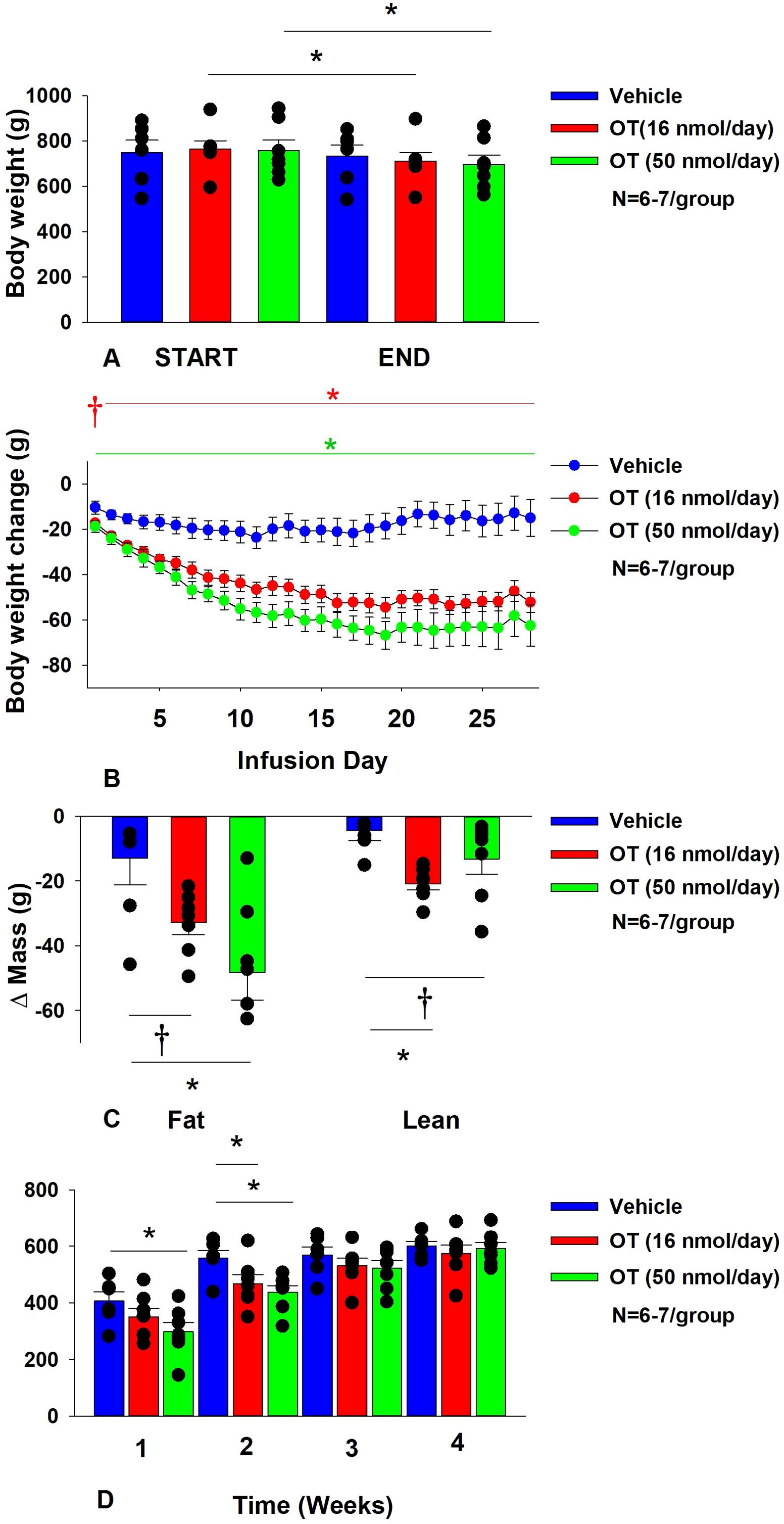
Figure 2. (A–D) Determine the dose-response effects of systemic infusions of OT (16 and 50 nmol/day) on body weight, adiposity and energy intake in DIO rats. (A) Rats were maintained on HFD (60% kcal from fat; N=6-7/group) for approximately 5.5 months prior to being implanted with temperature transponders and allowed to recover for 1-2 weeks prior to being implanted with subcutaneous minipumps. (A), Effect of chronic subcutaneous OT or vehicle on body weight in DIO rats; (B) Effect of chronic subcutaneous OT or vehicle on body weight change in DIO rats; (C) Effect of chronic subcutaneous OT or vehicle on change in fat mass and lean mass in DIO rats; (D) Effect of chronic subcutaneous OT or vehicle on change in weekly energy intake (kcal/week) in DIO rats. Data are expressed as mean ± SEM. *P<0.05, †0.05<P<0.1 OT vs. vehicle.
In addition, SC OT (16 and 50 nmol/day) was able to reduce weight gain (Figure 2B) relative to vehicle treatment throughout the 28-day infusion period. SC OT (50 nmol/day), at a dose that was at least 3-fold higher than the centrally effective dose (16 nmol/day), reduced weight gain throughout the entire 28-day infusion period. SC OT (16 nmol/day) treated rats had reduced weight gain between days 2-29 (P<0.05) while SC OT (50 nmol/day) reduced weight gain between days 1-29 (P<0.05). There was an overall effect of OT to reduce relative fat mass (pre- vs post-intervention) [(F(2,17) = 6.052, P=0.010)]. SC OT (50 nmol/day) reduced fat mass (P<0.05) and there was also a tendency for the lower dose (16 nmol/day) to reduce relative fat mass (P=0.066) (Figure 2C; P<0.05).
There was also an overall effect of OT to reduce relative lean mass (pre- vs post-intervention) [(F(2,17) = 5.572, P=0.014)]. Specifically, SC OT (16 nmol/day) reduced relative lean mass at the lower dose (16 nmol/day; P<0.01) while the higher dose (50 nmol/day) tended to reduce relative lean mass (P=0.090). Note that there was no significant reduction in total fat mass or lean mass (P=NS).
The changes in body weight and relative fat mass were not associated with any changes in plasma leptin, insulin, glucose or total cholesterol (Table 1). These effects that were mediated, at least in part, by a modest reduction of energy intake that was apparent during weeks 1 (50 nmol/day) and 2 (16 and 50 nmol/day) of OT treatment (Figure 2D; P<0.05).
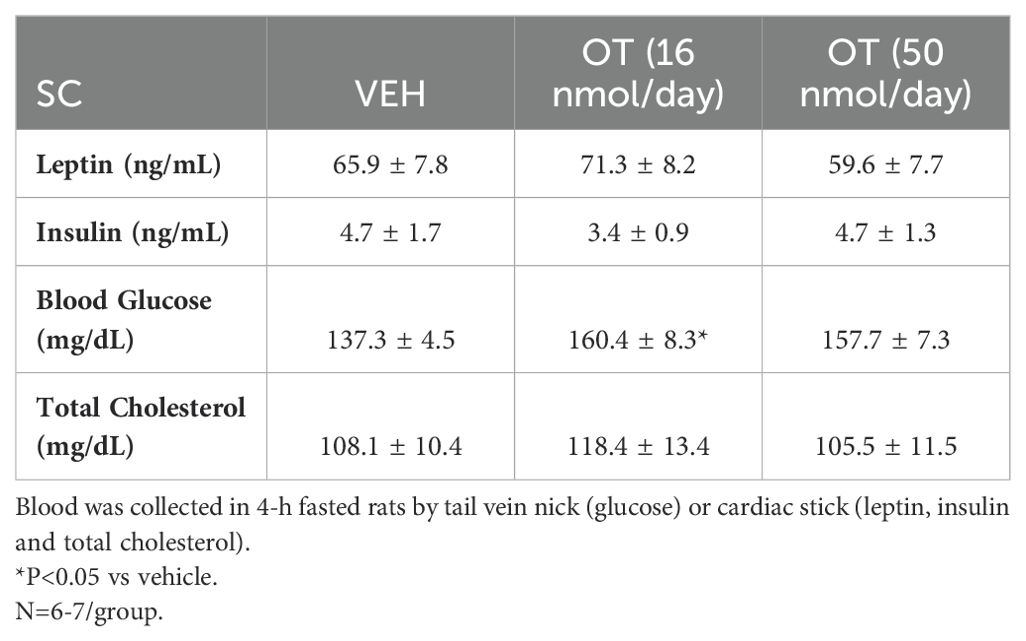
Table 1. Plasma measurements following SC OT (16 and 50 nmol/day).
The reduction of energy intake does not appear to be due to an aversive effect of systemic OT (16 or 50 nmol/day), since there was no effect on kaolin consumption relative to vehicle-treated DIO rats (P=NS; data not shown).
Energy intake data from a vehicle-treated animal during week 3 was deleted on account of an error that was made when recording the data. Kaolin data from a subset of animals was excluded on account of these animals shredding the kaolin diet (2 measurements during week 2, 1 measurement during week 3 and 1 measurement during week 4) and being significant outliers (Grubb’s test for outliers).
The goal of this study was to determine the effects of chronic OT treatment (single dose identified from Study 2) in combination with a single dose of the β3-AR agonist, CL 316243, on body weight and adiposity in DIO rats. By design, DIO rats were obese as determined by both body weight (804 ± 14 g) and adiposity (310 ± 11 g fat mass; 38.3 ± 4.7% adiposity) after maintenance on the HFD for approximately 7 months. Prior to the onset of CL 316243 treatment on infusion day 7, both OT treatment groups were matched for OT-elicited reductions of weight gain.
OT and CL 316243 alone reduced body weight by ≈ 7.8 ± 1.3% (P<0.05) and 9.1 ± 2.3% (P<0.05), respectively, but the combined treatment produced more pronounced weight loss (pre- vs post-intervention) (15.5 ± 1.2%; P<0.05) (Figure 3A) than either treatment alone (P<0.05). OT alone tended to reduce weight gain on days 16-28 (0.05<P<0.1) while CL 316243 alone tended to reduce or reduced weight gain on day 24 (0.05<P<0.1), day 25 (P=0.05), and days 26-28 (P<0.05) (Figure 3B). OT and CL 316243 together tended to reduce weight gain on day 9 (0.05<P<0.1) reduced weight gain on days 10-28 (P<0.05). The combination treatment appeared to produce a more pronounced reduction of weight gain relative to OT alone on day 25 (0.05<P<0.1) and this reached significance on days 26-28 (P<0.05).
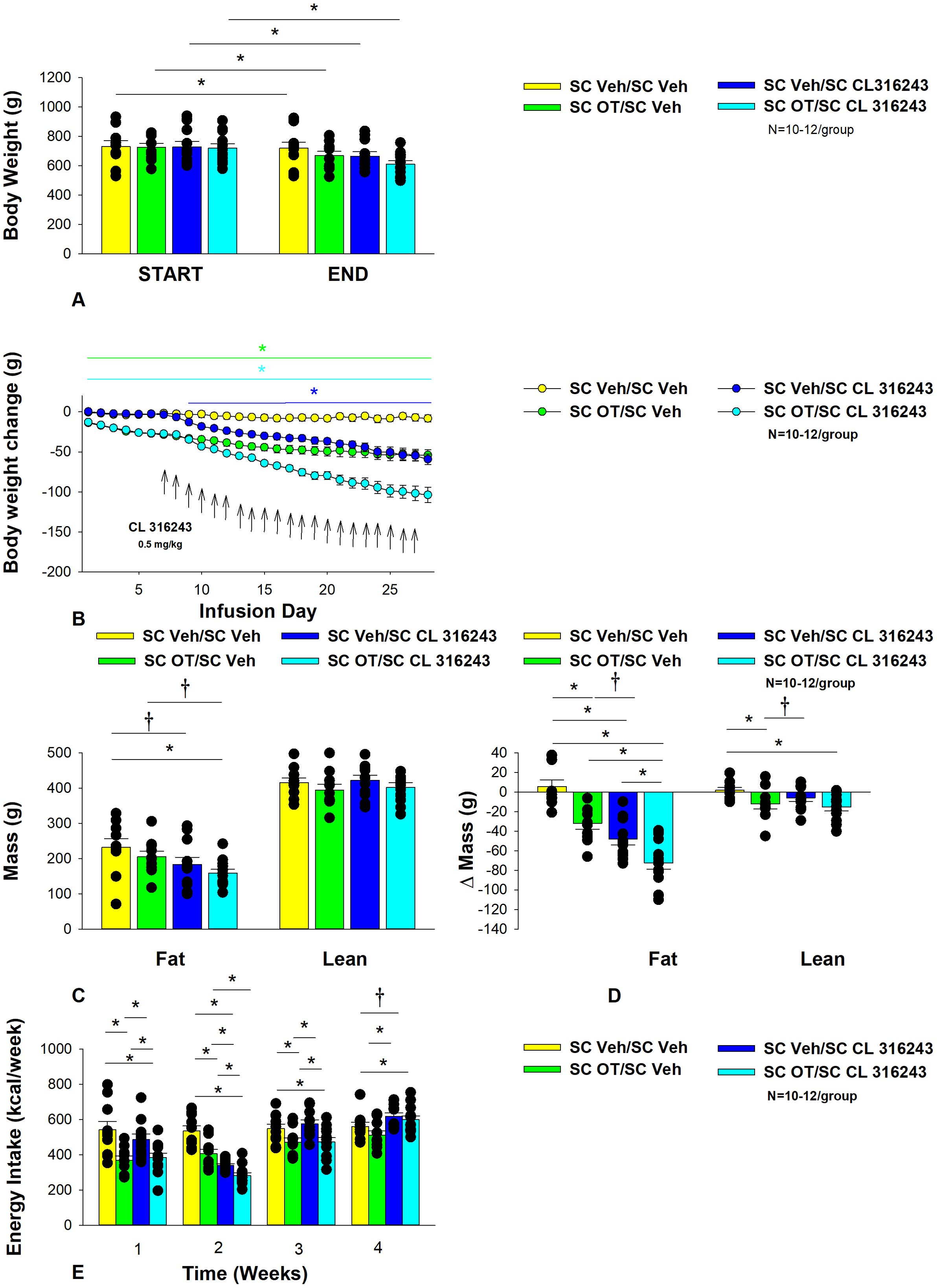
Figure 3. (A–E) Effect of chronic systemic OT infusions (50 nmol/day) and systemic beta-3 receptor agonist (CL 316243) administration (0.5 mg/kg) on body weight, body adiposity and energy intake in male DIO rats. Ad libitum fed rats were maintained on HFD (60% kcal from fat; N=8-10/group) for approximately 8 months prior to receiving continuous infusions of vehicle or OT (50 nmol/day) in combination with a single dose of CL 316243 (0.5 mg/kg). (A) Effect of chronic subcutaneous OT or vehicle in combination with systemic CL 316243 or vehicle on body weight in DIO rats; (B) Effect of chronic subcutaneous OT or vehicle in combination with systemic CL 316243 or vehicle on body weight change in HFD-fed DIO rats; (C) Effect of chronic subcutaneous OT or vehicle in combination with systemic CL 316243 or vehicle on fat mass and lean mass in DIO rats; (D) Effect of chronic subcutaneous OT or vehicle in combination with systemic CL 316243 or vehicle on change in fat mass and lean mass in DIO rats; (E) Effect of chronic subcutaneous OT or vehicle in combination with systemic CL 316243 or vehicle on change in weekly energy intake (kcal/week) in DIO rats. ↑ indicate 1x daily injections. Colored bars represent specific group comparisons vs vehicle. Data are expressed as mean ± SEM. *P<0.05, †0.05<P<0.1 vs. vehicle or baseline [pre-treatment; (A)].
In addition, the combination treatment tended to produce a greater reduction of weight gain relative to CL 316243 alone on days 21-22 and 26-28 (0.05<P<0.1). While OT alone did not significantly reduce fat mass (P=NS), there was a tendency for CL 316243 alone (0.05<P<0.1), and the combination of OT and CL 316243 (P<0.05) to reduce fat mass without impacting lean body mass (Figure 3C; P=NS). However, the combination treatment did not result in a significant reduction of fat mass relative to OT alone or CL 316243 alone (P=NS). OT and CL 316243 alone did produce a reduction in relative fat mass (pre- vs post-intervention; P<0.05). OT also produced a modest reduction in relative lean mass (Figure 3D; P<0.05). The combination treatment also produced a significant reduction of relative fat mass (P<0.05) which exceeded that of OT and CL 316243 alone (P<0.05).
Systemic OT treatment alone reduced energy intake during week 1 (P<0.05). OT, CL 316243 and the combined treatment were effective at reducing energy intake at week 2 (Figure 3E; P<0.05). OT and the combined treatment reduced energy intake during week 3 (P<0.05) but CL 316243 failed to reduce energy intake during this time (P=NS). All treatments were ineffective at reducing energy intake over weeks 3 and 4 (P=NS). The reduction of energy intake in response to OT alone, CL 316243 alone or the combined treatment does not appear to be due to an aversive effect, since there was no effect on kaolin consumption relative to vehicle-treated DIO rats (P=NS; data not shown).
Two-way ANOVA revealed an overall significant effect of OT [(F(1,39) = 63.434, P<0.01)], CL 316243 [(F(1,39) = 74.939, P<0.01)] but no significant interactive effect between OT and CL 316243 [(F(1,39) = 0.058, P=NS)] on weight loss. Consistent with this finding, two-way ANOVA revealed consistent overall effects of OT and CL-3162343 on reduction of body weight gain between days 10-29 but no significant overall effect. In addition, two-way ANOVA revealed no overall significant effect of OT [(F(1,39) = 2.003, P=0.165)], a significant effect of CL 316243 [(F(1,39) = 6.859, P=0.012)] and no significant interactive effect between OT and CL 316243 [(F(1,39) = 0.005, P=0.943)] on fat mass. There was no significant overall effect of OT [(F(1,39) = 2.069, P=0.158)] or CL 316243 [(F(1,39) = 0.217, P=0.644)] on lean mass and no interactive effect of OT and CL 316243 [(F(1,39) = 0.004, P=0.950)] on lean mass. Lastly, two-way ANOVA revealed an overall significant effect of OT [(F(1,39) = 21.464, P<0.01)], CL 316243 [(F(1,39) = 62.681, P<0.01)] and a near significant interactive effect between OT and CL 316243 [(F(1,39) = 3.190, P=0.082)] on energy intake (week 2).
Overall, these findings suggest an additive effect of OT and CL 316243 to produce sustained weight loss in DIO rats. The effects of the combination treatment on adiposity and energy intake appear to be driven largely by CL 316243 and OT, respectively.
CL 316243 elevated TIBAT on injection day 1 at 0.5, 0.75 and 1-h post-injection (P<0.05; Supplementary Figure 1A) and tended to elevate TIBAT at 0.25-h post-injection (0.05<P<0.1). Similarly, CL 316243, when given in combination with OT, also increased TIBAT at 0.5, 0.75 and 1-h post-injection and tended to elevate TIBAT at 0.25-h post-injection on injection day 1 (0.05<P<0.1). Both CL 316243 and CL 316243 + OT treatments elevated TIBAT relative to vehicle treated animals when the TIBAT data from injection day 1 were averaged over 1-h post-injection (P<0.05). There was no significant difference in TIBAT response to CL 316243 CL 316243 + OT treatments when the TIBAT data were averaged over 60 min (P=NS).
CL 316243 also elevated TIBAT on injection day 22 at 0.25, 0.5, 0.75 and 1-h post-injection (P<0.05; Supplementary Figure 1B). Similarly, CL 316243, when given in combination with OT, also increased TIBAT at 0.5 and 0.75-h post-injection and tended to elevate TIBAT at 0.25 and 1-h post-injection on injection day 22 (0.05<P<0.1). Both CL 316243 and CL 316243 + OT treatments elevated TIBAT relative to vehicle treated animals when the TIBAT data from injection day 22 were averaged over 1-h post-injection (P<0.05). There was no significant difference in the TIBAT response to CL 316243 and CL 316243 +OT treatments when the TIBAT data were averaged over 60 min (P=NS).
In addition, in a limited number of subjects (N=3-5/group), we found that CL 31243 in combination with OT elevated core temperature at 20-h post-injection relative to vehicle-treated animals (P<0.05; data not shown). CL 316243 alone also tended to elevate 20-h core temperature (P=0.1; data not shown). No changes in 20-h activity occurred in response to the treatments (data not shown).
H&E-stained sections from the four treatment conditions are shown in Figures 4A–D (EWAT) and Figures 4E–H (IWAT). CL 316243 alone (P<0.05) and in combination with OT (P<0.05) reduced EWAT adipocyte size in DIO rats relative to vehicle treatment (Supplementary Figure 2A; Figure 5A). There were no significant differences in the ability of the combined treatment to reduce EWAT adipocyte size relative to CL 316243 alone (P=0.156). Similarly, OT and CL 316243 given in combination reduced IWAT adipocyte size whereas there was no significant effect of OT or CL 316343 on adipocyte size when given alone (P=NS) (Supplementary Figure 2B; Figure 5B). A subset of EWAT (N=6) and IWAT (N=5) samples were excluded from the adipocyte size analysis due to poor tissue quality or an insufficient amount of tissue.

Figure 4. (A–D): Representative image of H&E-stained section from EWAT and IWAT following chronic systemic OT infusions (50 nmol/day) and systemic beta-3 receptor agonist (CL 316243) administration (0.5 mg/kg). Images taken from fixed (4% PFA) paraffin embedded sections (5 μm) containing EWAT (A–D) or IWAT (E–H) in HFD-fed rats treated with systemic OT (50 nmol/day) or vehicle in combination with IP CL 316243 (0.5 mg/kg) or IP vehicle. A/E, Veh/Veh. B/F, OT/Veh. C/G, Veh/CL 316243. D/H, OT-CL 316243; (A–H) all visualized at 100X magnification. Images were obtained using Image Pro Plus software.
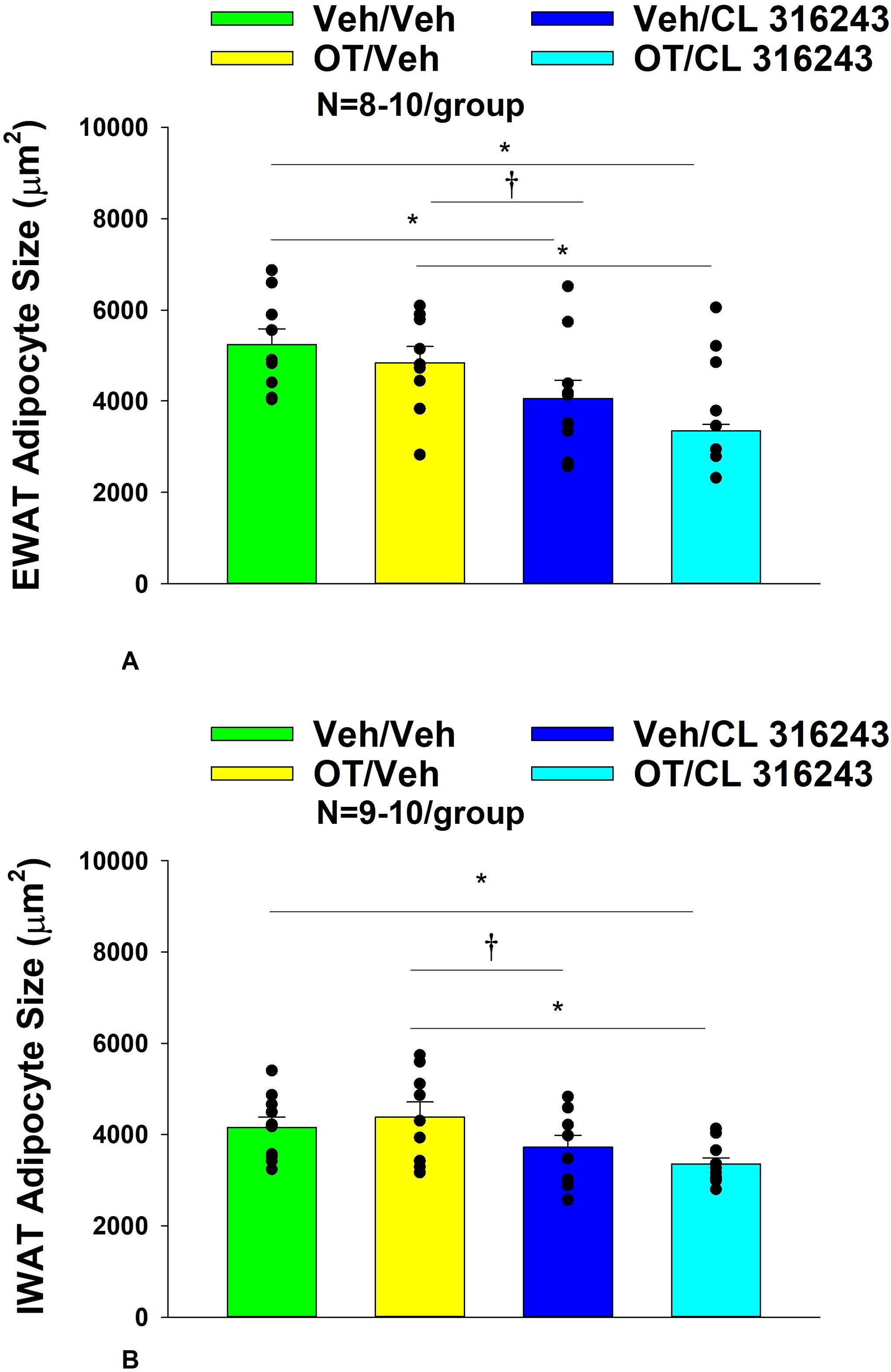
Figure 5. (A, B) Effect of chronic systemic OT infusions (50 nmol/day) and systemic beta-3 receptor agonist (CL 316243) administration (0.5 mg/kg) on adipocyte size in EWAT and IWAT in male DIO rats. (A) Adipocyte size (μm2) was measured in EWAT from rats that received chronic systemic infusion of OT (50 nmol/day) or vehicle in combination with daily CL 316243 (0.5 mg/kg) or vehicle treatment (N=9-10/group). (B) Adipocyte size (μm2) was measured in IWAT from rats that received chronic systemic infusion of OT (50 nmol/day) or vehicle in combination with daily CL 316243 (0.5 mg/kg) or vehicle treatment (N=9-10/group). Data are expressed as mean ± SEM. *P<0.05, †0.05<P<0.1.
CL 316243 alone and in combination with OT (P<0.05) increased UCP-1 expression in EWAT relative to VEH. In addition, CL 316243 alone also increased UCP-1 relative to OT (P<0.05). The combination of CL 316243 and OT also increased UCP-1 relative to OT treatment alone (Figures 6A–D; Figure 7A), but was not different from CL 316243 alone (P=0.167). Similarly, CL 316243 in combination with OT (P<0.05) increased UCP-1 expression in IWAT relative to VEH treatment and OT treatment (Figures 6E–H; Figure 7B). A subset of EWAT (N=10) and IWAT (N=3) samples were excluded in the UCP-1 expression analysis due to poor tissue quality or an insufficient amount of tissue.
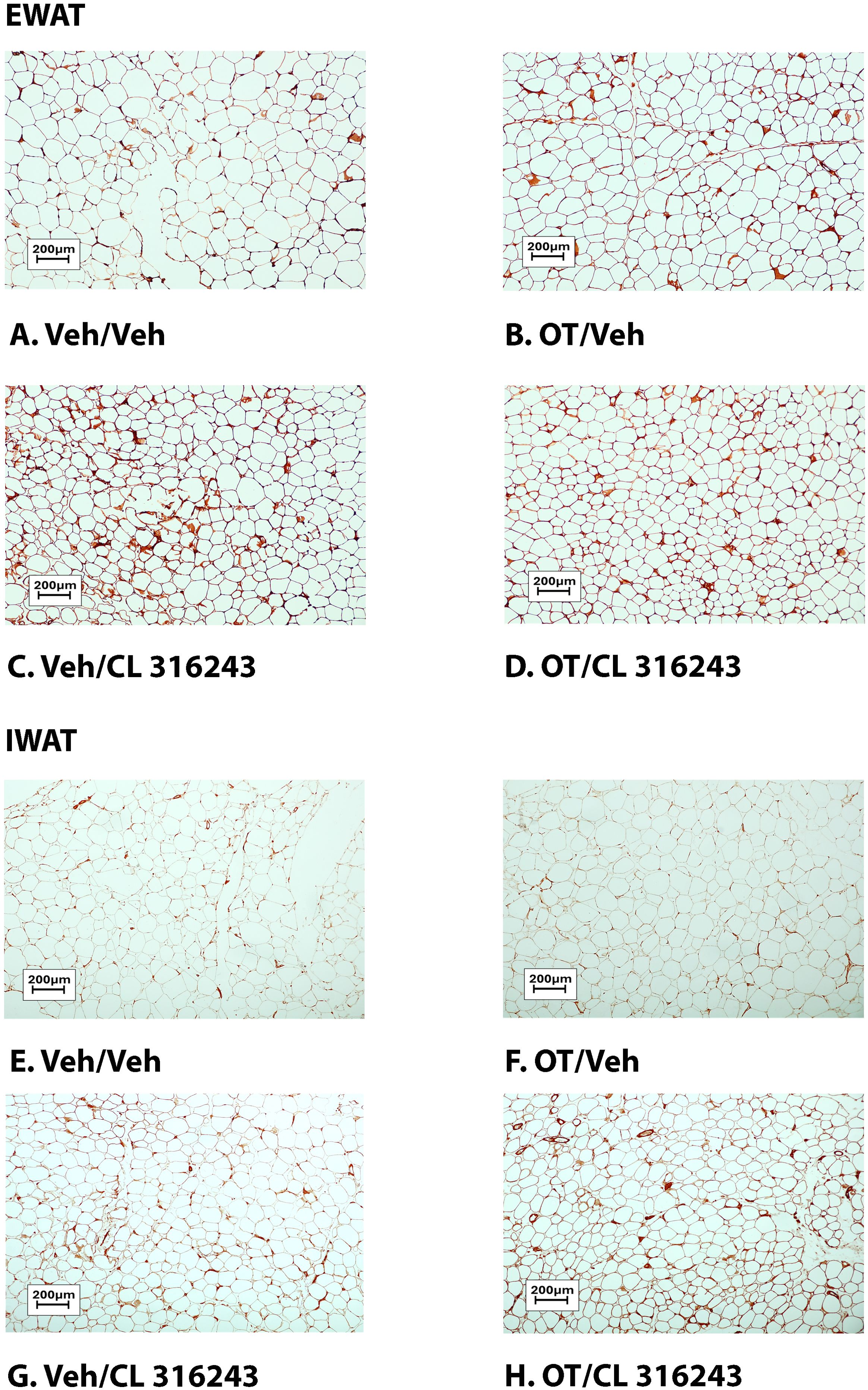
Figure 6. (A–D) Representative image to illustrate the effect of chronic systemic OT infusions (50 nmol/day) and systemic beta-3 receptor agonist (CL 316243) administration (0.5 mg/kg) on UCP-1 content in EWAT and IWAT in male DIO rats. UCP-1 was analyzed using Image Pro Plus software. Images were taken from fixed (4% PFA) paraffin embedded sections (5 μm) containing EWAT (A–D) in HFD-fed rats treated with SC OT (50 nmol/day) or SC vehicle in combination with IP CL 316243 (0.5 mg/kg) or IP vehicle. (A) Veh/Veh. (B) OT/Veh. (C) Veh/CL 316243 (D) OT/CL 316243; (A–H) all visualized at 100X magnification.

Figure 7. (A, B) Effect of chronic systemic OT infusions (50 nmol/day) and systemic beta-3 receptor agonist (CL 316243) administration (0.5 mg/kg) on UCP-1 content in EWAT and IWAT in male DIO rats. (A) UCP-1 expression was measured in EWAT from rats that received chronic systemic infusion of OT (50 nmol/day) or vehicle in combination with daily CL 316243 (0.5 mg/kg) or vehicle treatment (N=9-11/group). (B) UCP-1 expression was measured in IWAT from rats that received chronic systemic infusion of OT (50 nmol/day) or vehicle in combination with daily CL 316243 (0.5 mg/kg) or vehicle treatment (N=7-10/group). Data are expressed as mean ± SEM. *P<0.05, †0.05<P<0.1.
To characterize the endocrine and metabolic effects of systemic OT (50 nmol/day) and systemic beta-3 receptor agonist (CL 316243) in DIO rats in a chronic study using as single dose of CL 316243 (Study 3; Table 2), we measured blood glucose levels and plasma concentrations of leptin, insulin, FGF-21, irisin, adiponectin, TC, triglycerides, and FFAs. CL 316243 alone or in combination with OT resulted in a reduction of plasma leptin relative to OT (P<0.05) or vehicle alone (P<0.05). The combination treatment was also associated with a reduction of blood glucose and insulin relative to vehicle (P<0.05) and OT treatment (P<0.05) but it was not statistically different from CL 316243 (P=NS). We also found that FGF-21 was reduced in response to CL 316243 and CL 316243 in combination with OT relative to vehicle and OT treatment (P<0.05). In addition, the combination treatment was associated with an elevation of adiponectin relative to vehicle (P<0.05) and OT treatment (P<0.05) but it was not statistically different from CL 316243 (P=NS). A subset of data (N=2) was excluded from the plasma hormone analysis from Study 3 due to gross hemolysis or samples having been misplaced.
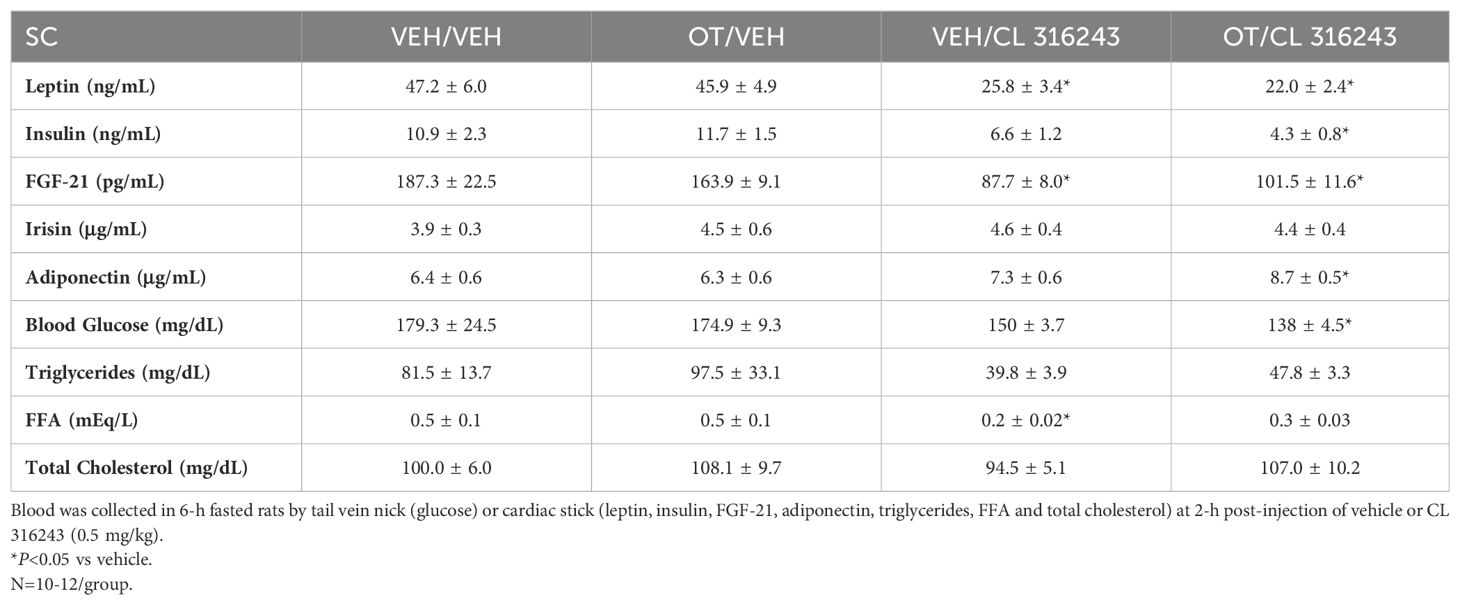
Table 2. Plasma measurements following SC OT +/- CL 316243 in DIO rats.
We next determined the extent to which CL 316243 (0.5 mg/kg), OT (50 nmol/day), or the combination treatment increased thermogenic gene expression in IBAT, EWAT and IWAT relative to vehicle at 2-hour post-CL 316243/vehicle injections.
IBAT: Consistent with published findings in mice and rats, chronic CL 316243 administration elevated relative levels of the thermogenic markers Ucp1 (33, 52, 53), Dio2 (35, 54), Ppargc1a (53) and Gpr120 (35, 53) (Table 3A; P<0.05). CL 316243 in combination with OT also elevated Ucp1, Dio2, Ppargc1a and Gpr120 (P<0.05). In addition, chronic CL 316243 alone and in combination with OT reduced Adrb3 (β3-AR) mRNA expression in IBAT (Table 3A; P<0.05).
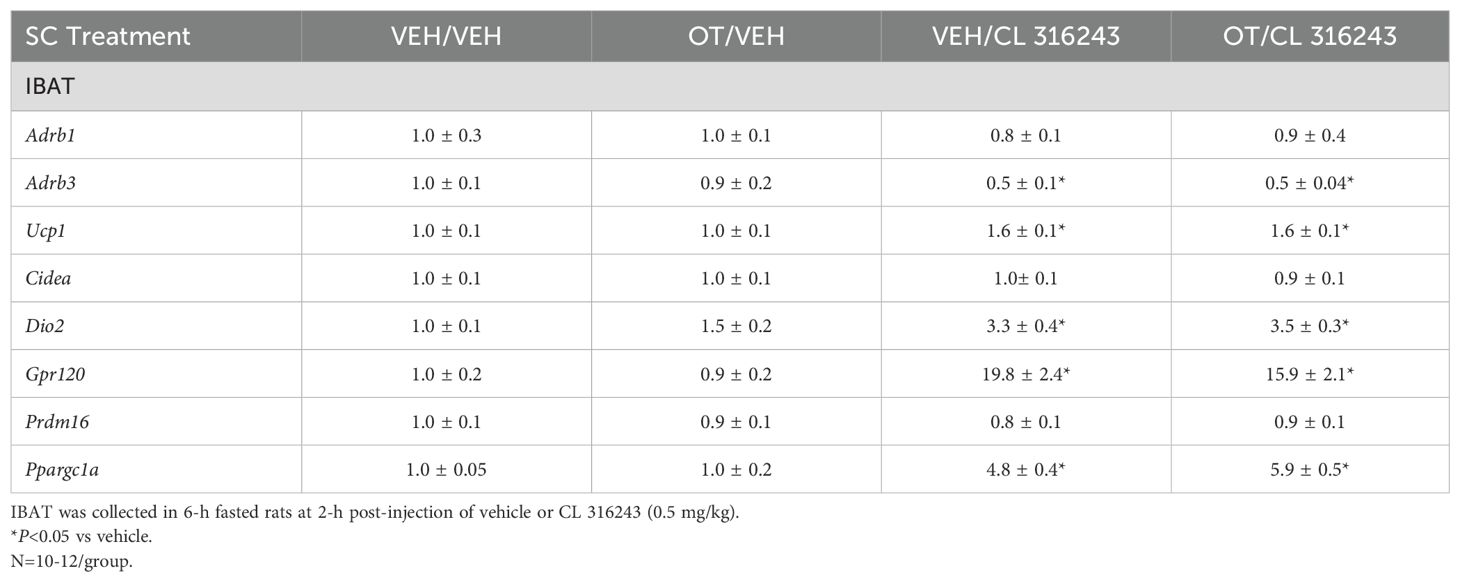
Table 3A. Changes in IBAT mRNA expression following SC OT and CL 316243 treatment in male DIO rats.
EWAT: CL 316243 elevated relative levels of the thermogenic marker Ucp1 relative to vehicle treatment (P<0.05; Table 3B). CL 316243 in combination with OT also elevated Ucp1, Dio2, Ppargc1a and Adrb3 relative to vehicle controls (P<0.05; Table 3B).
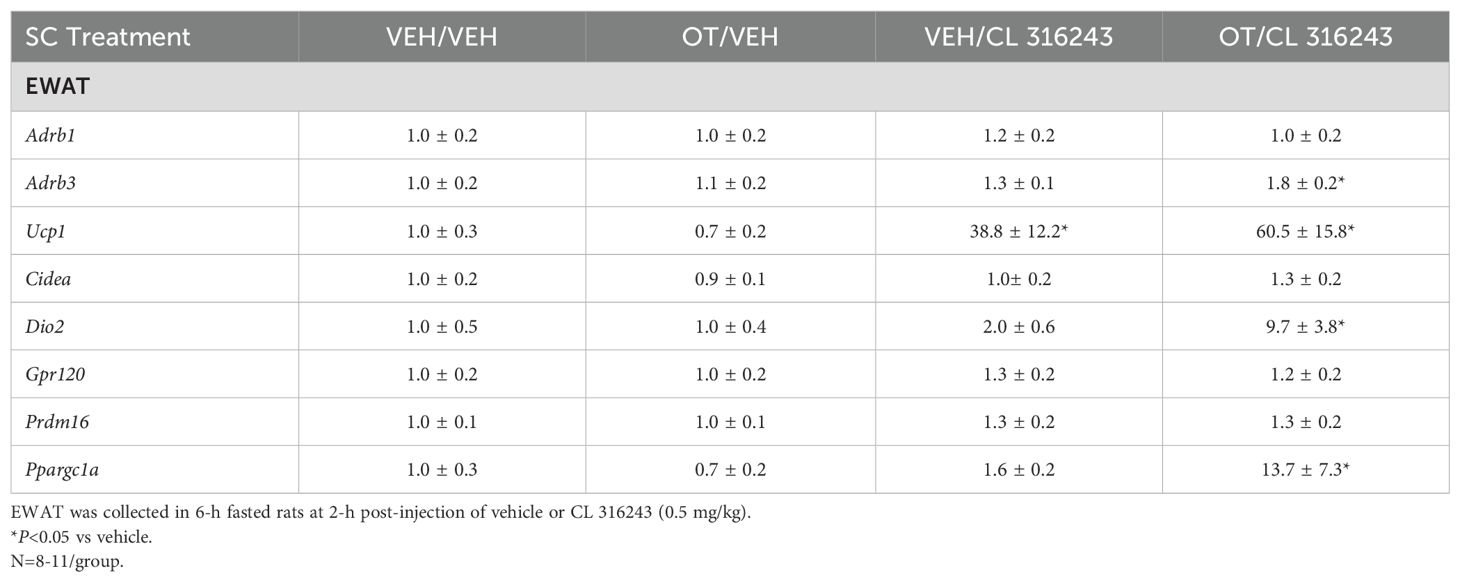
Table 3B. Changes in EWAT mRNA expression following SC OT and CL 316243 treatment in male DIO rats.
A subset of EWAT data [(N=5 (Ucp1); N=3 (Dio2); N=3 (Ppargc1a); N=1 (Adrb3)] was excluded from the EWAT gene expression analysis on account of missing samples, samples with undetectable values or statistical outliers (Grubbs’ test for outliers).
IWAT: There were no significant differences in the thermogenic markers Ucp1, Dio2, Ppargc1a or Adrb3 in response to chronic CL 316243 alone or in combination with OT (Table 3C; P=NS).
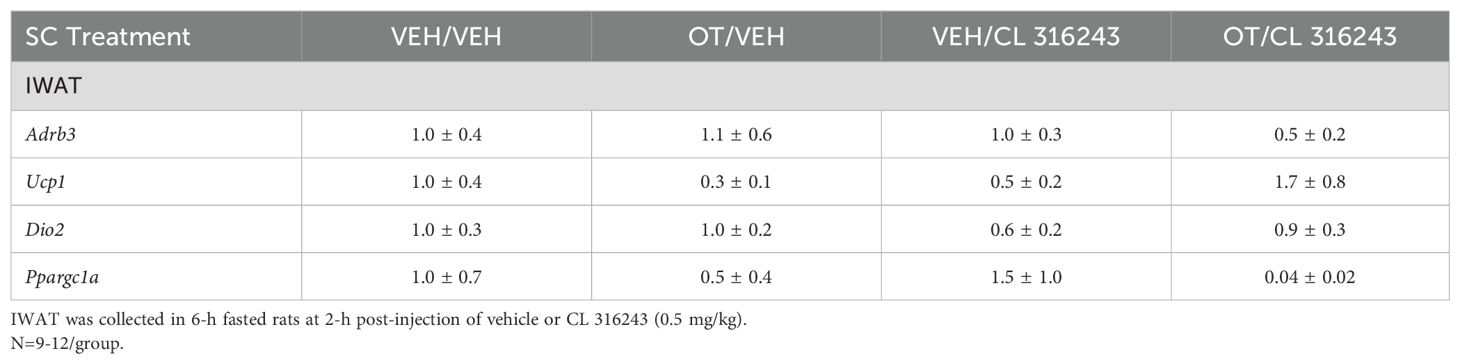
Table 3C. Changes in IWAT mRNA expression following SC OT and CL 316243 treatment in male DIO rats.
A subset of IWAT data [(N=3 (Ucp1); N=3 (Dio2); N=1 (Ppargc1a); N=2 (Adrb3)] was excluded from the IWAT gene expression analysis on account of missing samples, samples with undetectable values or statistical outliers (Grubbs’ test for outliers).
As a functional readout of BAT thermogenesis for the gene expression analyses, we measured TIBAT in response to CL 316243 alone or CL 316243 + OT during the time period that preceded tissue collection. CL 316243 alone resulted in an increase in TIBAT at 0.25, 0.5, 0.75 and 1-h post-injection (Supplementary Figure 3; P<0.05). Similarly, CL 316243 + OT resulted in an increase in TIBAT at 0.5-h post-injection (Supplementary Figure 3; P<0.05) and it also tended to increase TIBAT at 0.75-h post-injection (Supplementary Figure 3; 0.05<P<0.1).
The goal of the current study was to determine the extent to which systemic OT could be used as an adjunct with the β3-AR agonist, CL 316243, to increase BAT thermogenesis and elicit weight loss in DIO rats. We hypothesized that systemic OT and beta-3 agonist (CL 316243) treatment would produce an additive effect to reduce body weight and adiposity in DIO rats by decreasing food intake and stimulating BAT thermogenesis. To test this hypothesis, we determined the effects of systemic (subcutaneous) infusions of OT (50 nmol/day) or vehicle (VEH) when combined with daily systemic (intraperitoneal) injections of CL 316243 (0.5 mg/kg) or VEH on body weight, adiposity, food intake and TIBAT. OT and CL 316243 monotherapy decreased body weight by 8.0 ± 0.9% (P<0.05) and 8.6 ± 0.6% (P<0.05), respectively, but OT in combination with CL 316243 produced more substantial weight loss (14.9 ± 1.0%; P<0.05) compared to either treatment alone. These effects were associated with decreased adiposity, energy intake and elevated TIBAT during the treatment period. In addition, systemic OT and CL 316243 combination therapy increased IBAT thermogenic gene expression suggesting that increased BAT thermogenesis may also contribute to these effects. The findings from the current study suggest that the effects of systemic OT and CL 316243 to reduce body weight and adiposity are additive and appear to be driven primarily by OT-elicited changes in food intake and CL 316243-elicited increases in BAT thermogenesis.
Recent findings also indicate that other agents, namely the GLP-1R agonist, liraglutide, and the fat signal, oleoylethanolamide (OEA), act in an additive fashion with CL 316243 to reduce body weight or body weight gain. Oliveira recently reported that the combination of liraglutide and CL 316243 produce additive effects to reduce the change in body weight in a mouse model (55). These effects appeared to be attributed to additive effects on energy intake and increased oxygen consumption in IBAT and IWAT. In addition, the combined treatment increased expression of UCP-1 in IWAT (indicative of browning). Similarly, Suarez and colleagues demonstrated that the peroxisome proliferator-activating receptor-α (PPARα) agonist and fat signal, OEA, act in an additive fashion with CL 316243 produced to reduce food intake and weight gain in rats (33). These effects were associated with pronounced reductions in fat mass and increases in expression of thermogenic genes (PPARα and Ucp1) in EWAT (33). Of interest is the finding that systemic OT and central OT can increase OEA expression in EWAT (20). Furthermore, Deblon reported that the effectiveness of OT to decrease body weight was partially blocked in PPARα null mice (20), indicating that PPARα may partially mediate OT’s thermogenic effects in EWAT. OEA has also been found to stimulate 1) hypothalamic expression of OT mRNA (56), 2) PVN OT neurons (57), and 3) OT release within the PVN (57). In addition, OEA also decreases food intake, in part, through OT receptor signaling (56). Thus, it is possible that high fat diet-elicited stimulation of OEA (58) may reduce food intake, in part, through an OTR signaling and that the effects of OT to stimulate WAT thermogenesis might occur through PPARα. Additional studies that utilize adipose depot knockdown of PPARα will enable us to determine if PPARα in specific adipose depots may contribute to the ability of OT and CL 316243 to reduce body weight and adiposity.
Our findings and others raise the possibility that systemic OT could be reducing food intake and adiposity, in part, through a direct effect on peripheral OT receptors. Asker reported that OT-B12, a BBB-impermeable OT analogue, reduced food intake in rats, thus providing evidence that peripheral OTR signaling is important in the control of food intake (59). Consistent with these findings, Iwasaki found that the ability of peripheral administration of OT to reduce food intake was attenuated in vagotomized mice (60, 61). In addition, Brierley extended these findings and found that the effect of systemic administration of OT to suppress food intake required NTS preproglucagon neurons that receive direct synaptic input OTR-expressing vagal afferent neurons (62). We also found that systemic administration of a non-BBB penetrant OTR antagonist, L-371257, stimulated food intake and body weight gain in rats (63). Several studies have also found that subcutaneous infusion of OT reduced adipocyte size in 1) visceral fat in female Wistar rats that were peri- and postmenopausal (64) and 2) visceral fat in a dihydrotestosterone-elicited model of polycystic ovary syndrome in female Wistar rats (65), 3) subcutaneous fat in female ovariectomized Wistar rats (66), and 4) EWAT in male Zucker fatty rats (67). More recent studies have confirmed that peripheral administration of long-acting OT analogues (including ASK2131 and ASK1476) also reduced both food intake and body weight (68, 69). Together, these findings suggest that OT may also act in the periphery to decrease adipocyte size by a direct effect on OTRs found on adipose tissue (20, 21, 70). Of translational importance is the finding that subcutaneous (20, 25–28, 71) or intraperitoneal (28) administration of OT or long-acting OT analogues can recapitulate the effects of chronic CNS administration of OT on reductions of food intake and body weight.
The combination treatment and CL 316243 monotherapy reduced body weight and adiposity, in part, through increased BAT thermogenesis. Both CL 316243 alone and in combination with OT elevated TIBAT throughout the course of the injection study and increased IBAT thermogenic genes (Ucp1, Dio2 and Ppargc1a) and UCP-1 content in IBAT. These findings coincided with CL- 316243-elicited increases in TIBAT from the same animals during the time that preceded tissue collection. These findings are consistent with our previously published findings in rats (35) and other studies mice and rats that found chronic CL 316243 administration to increase the thermogenic markers Dio2 (54) and Gpr120 (33, 52, 53). Similar to our findings, others also reported that systemic CL 316243 increased Ucp1 mRNA expression in mice (72). We also found that the combination treatment and CL 316243 alone caused a downregulation of Adrb3 mRNA expression at 2-h post-injection. This finding is consistent with we have previously reported (35) and others who have reported that both cold exposure and norepinephrine reduced Adrb3 mRNA expression and mouse IBAT (73, 74) and mouse brown adipocytes (75). In summary, our results indicate that systemic administration of OT in combination with systemic CL 316243 treatment results in more profound reductions of body weight compared to either OT or CL 316243 alone. Moreover, the combined treatment of OT and CL 316243 stimulated BAT thermogenesis as determined by increased TIBAT and thermogenic gene expression in IBAT. Together, our data support the hypothesis that systemic OT and β3-AR agonist (CL 316243) treatment produce an additive effect to reduce body weight and adiposity in DIO rats. The effects of the combined treatment on body weight and adiposity appeared to be additive and driven predominantly by OT-elicited reductions of food intake and CL-316243-elicited increases in BAT thermogenesis.
Collectively, these findings suggest that systemic OT treatment could be a viable adjunct to other anti-obesity treatment strategies. While intranasal OT has been found to reduce body weight by approximately 9.3% in a small study with limited subjects (9-11/group) (76), it was not found, however, to have any effect on body weight in a larger scale well-controlled clinical study (N=30-31/group) in which subjects were matched well for body weight, adiposity and gender (77). Importantly, Plessow, Lawson and colleagues did find a significant effect of intranasal OT to reduce energy intake at 6-weeks post-treatment, which served as an important control. It is possible that a more extended length of treatment might have been required to take advantage of the reductions of energy intake that were not observed until the 6-week post-treatment time point. In addition, changes in dose, dosing frequency, or co-administration with Mg2+ (78, 79) might need to be taken into consideration in order to maximize the effects of intranasal OT on body weight in humans who are overweight or obese. Given that OT can be an effective delivery approach to reduce energy intake and elicit weight loss in several rodent models (see (16, 80, 81) for review) and obese nonhuman primates (19), it will also be important to determine if chronic systemic OT treatment can elicit weight loss when given in combination with CL 316243 at doses that are sub-threshold for producing adverse effects on heart rate or blood pressure (82). Recent findings indicate that OT can be effective at reducing food intake and/or body weight in female rats (83) and DIO male and female mice (26), respectively. Thus, it will be important to examine if this combination treatment produces an additive effect to reduce body weight and adiposity in female DIO rodents and nonhuman primates.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The animal study was approved by Institutional Animal Care and Use Committee of the Veterans Affairs Puget Sound Health Care System. The study was conducted in accordance with the local legislation and institutional requirements.
JS: Data curation, Investigation, Methodology, Supervision, Validation, Writing – review & editing. JR: Data curation, Investigation, Methodology, Supervision, Validation, Writing – review & editing. ET: Data curation, Investigation, Methodology, Supervision, Validation, Writing – review & editing. MH: Data curation, Investigation, Methodology, Supervision, Validation, Writing – review & editing. MG: Data curation, Investigation, Methodology, Supervision, Validation, Writing – review & editing. JG: Data curation, Formal analysis, Investigation, Methodology, Resources, Validation, Writing – review & editing. TW: Data curation, Formal analysis, Investigation, Methodology, Resources, Validation, Writing – review & editing. TW: Data curation, Formal analysis, Investigation, Methodology, Resources, Validation, Writing – review & editing. AW: Data curation, Investigation, Methodology, Validation, Writing – review & editing. KO’: Data curation, Investigation, Methodology, Resources, Supervision, Validation, Writing – review & editing. PH: Data curation, Investigation, Methodology, Resources, Supervision, Validation, Writing – review & editing. JB: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Resources, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This material was based upon work supported by the Office of Research and Development, Medical Research Service, Department of Veterans Affairs (VA) and the VA Puget Sound Health Care System Rodent Metabolic Phenotyping Core and the Cellular and Molecular Imaging Core of the Diabetes Research Center at the University of Washington and supported by National Institutes of Health (NIH) grant P30DK017047. This work was also supported by the VA Merit Review Award 5 I01BX004102, from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development Service and NIH 5R01DK115976 grant to James Blevins. PJH’s research program also received research support during the project period from NIH grants DK-095980, HL-091333, HL-107256 and a Multi-campus grant from the University of California Office of the President. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
The authors thank the technical support of Nishi Ivanov.
JB had a financial interest in OXT Therapeutics, Inc., a company that developed highly specific and stable analogs of oxytocin to treat obesity and metabolic disease but this is no longer the case.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2025.1503096/full#supplementary-material
Supplementary Figure 1 | (A, B) TIBAT measurements following acute systemic administration of the beta‐3 receptor agonist (CL 316243) or vehicle in male DIO rats. (A), injection day 1 (0.5 mg/kg) and (B), injection day 22 (0.5 mg/kg). Data are expressed as mean ± SEM. *P<0.05 vs VEH; †0.05<P<0.1 vs VEH.
Supplementary Figure 2 | (A–H): Representative image to illustrate the effect of chronic systemic OT infusions (50 nmol/day) and systemic beta-3 receptor agonist (CL 316243) administration (0.5 mg/kg) on adipocyte size in EWAT and IWAT in male DIO rats. Adipocyte size was analyzed using ImageJ. Images were taken from fixed (4% PFA) paraffin embedded sections (5 μm) containing EWAT (A–D) or IWAT (E–H) in HFD-fed rats treated with systemic OT (50 nmol/day) or vehicle in combination with IP CL 316243 (0.5 mg/kg) or IP vehicle. A/E, Veh/Veh. B/F, OT/Veh. C/G, Veh/CL 316243. D/H, OT-CL 316243; (A–H) all visualized at 100X magnification.
Supplementary Figure 3 | TIBAT measurements following acute systemic administration of the beta‐3 receptor agonist (CL 316243) or vehicle in male DIO rats. A, injection day 23 prior to euthanasia (0.5 mg/kg), euthanasia day. Data are expressed as mean ± SEM. *P<0.05 vs VEH; †0.05<P<0.1 vs VEH.
1. Smyth S, Heron A. Diabetes and obesity: the twin epidemics. Nat Med. (2006) 12:75–80. doi: 10.1038/nm0106-75
2. Targher G, Mantovani A, Wang XB, Yan HD, Sun QF, Pan KH, et al. Patients with diabetes are at higher risk for severe illness from COVID-19. Diabetes Metab. (2020) 46(4):335–7. doi: 10.1016/j.diabet.2020.05.001
3. Wilding JPH, Batterham RL, Calanna S, Davies M, Van Gaal LF, Lingvay I, et al. Once-weekly semaglutide in adults with overweight or obesity. New Engl J Med. (2021) 384:989. doi: 10.1056/NEJMoa2032183
4. Frias JP, Davies MJ, Rosenstock J, Perez Manghi FC, Fernandez Lando L, Bergman BK, et al. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. New Engl J Med. (2021) 385:503–15. doi: 10.1056/NEJMoa2107519
5. Chepurny OG, Bonaccorso RL, Leech CA, Wollert T, Langford GM, Schwede F, et al. Chimeric peptide EP45 as a dual agonist at GLP-1 and NPY2R receptors. Sci Rep. (2018) 8:3749. doi: 10.1038/s41598-018-22106-1
6. Frias JP, Bastyr EJ, Vignati L, Tschop MH, Schmitt C, Owen K, et al. The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090-2746, in patients with type 2 diabetes. Cell Metab. (2017) 26:343. doi: 10.1016/j.cmet.2017.07.011
7. Enebo LB, Berthelsen KK, Kankam M, Lund MT, Rubino DM, Satylganova A, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of concomitant administration of multiple doses of cagrilintide with semaglutide 2.4 mg for weight management: a randomised, controlled, phase 1b trial. Lancet. (2021) 397:1736–48. doi: 10.1016/S0140-6736(21)00845-X
8. Frias JP, Deenadayalan S, Erichsen L, Knop FK, Lingvay I, Macura S, et al. Efficacy and safety of co-administered once-weekly cagrilintide 2.4 mg with once-weekly semaglutide 2.4 mg in type 2 diabetes: a multicentre, randomised, double-blind, active-controlled, phase 2 trial. Lancet. (2023) 402:720–30. doi: 10.1016/S0140-6736(23)01163-7
9. Allison DB, Gadde KM, Garvey WT, Peterson CA, Schwiers ML, Najarian T, et al. Controlled-release phentermine/topiramate in severely obese adults: a randomized controlled trial (EQUIP). Obesity. (2012) 20:330–42. doi: 10.1038/oby.2011.330
10. Jastreboff AM, Aronne LJ, Ahmad NN, Wharton S, Connery L, Alves B, et al. Tirzepatide once weekly for the treatment of obesity. New Engl J Med. (2022) 387:205–16. doi: 10.1056/NEJMoa2206038
11. Aronne LJ, Sattar N, Horn DB, Bays HE, Wharton S, Lin WY, et al. Continued treatment with tirzepatide for maintenance of weight reduction in adults with obesity: the SURMOUNT-4 randomized clinical trial. JAMA. (2024) 331(1):38–48. doi: 10.1001/jama.2023.24945
12. Petersen J, Ludwig MQ, Juozaityte V, Ranea-Robles P, Svendsen C, Hwang E, et al. GLP-1-directed NMDA receptor antagonism for obesity treatment. Nature. (2024) 629:1133–41. doi: 10.1038/s41586-024-07419-8
13. Jastreboff AM, Kaplan LM, Hartman ML. Triple-hormone-receptor agonist retatrutide for obesity. Reply. New Engl J Med. (2023) 389:1629–30. doi: 10.1056/NEJMoa2301972
14. Gimpl G, Fahrenholz F. The oxytocin receptor system: structure, function, and regulation. Physiol Rev. (2001) 81:629–83. doi: 10.1152/physrev.2001.81.2.629
15. Blevins JE, Baskin DG. Translational and therapeutic potential of oxytocin as an anti-obesity strategy: Insights from rodents, nonhuman primates and humans. Physiol Behav. (2015) 152(Pt B):438–49. doi: 10.1016/j.physbeh.2015.05.023
16. Lawson EA. The effects of oxytocin on eating behaviour and metabolism in humans. Nat Rev Endocrinol. (2017) 13:700–9. doi: 10.1038/nrendo.2017.115
17. Lawson EA, Olszewski PK, Weller A, Blevins JE. The role of oxytocin in regulation of appetitive behaviour, body weight and glucose homeostasis. J Neuroendocrinol. (2020) 32:e12805. doi: 10.1111/jne.12805
18. McCormack SE, Blevins JE, Lawson EA. Metabolic effects of oxytocin. Endocrine Rev. (2020) 41(2):121–45. doi: 10.1210/endrev/bnz012
19. Blevins JE, Graham JL, Morton GJ, Bales KL, Schwartz MW, Baskin DG, et al. Chronic oxytocin administration inhibits food intake, increases energy expenditure, and produces weight loss in fructose-fed obese rhesus monkeys. Am J Physiol Regul Integr Comp Physiol. (2015) 308:R431–8. doi: 10.1152/ajpregu.00441.2014
20. Deblon N, Veyrat-Durebex C, Bourgoin L, Caillon A, Bussier AL, Petrosino S, et al. Mechanisms of the anti-obesity effects of oxytocin in diet-induced obese rats. PloS One. (2011) 6:e25565. doi: 10.1371/journal.pone.0025565
21. Yi KJ, So KH, Hata Y, Suzuki Y, Kato D, Watanabe K, et al. The regulation of oxytocin receptor gene expression during adipogenesis. J Neuroendocrinol. (2015) 27:335–42. doi: 10.1111/jne.2015.27.issue-5
22. Noble EE, Billington CJ, Kotz CM, Wang C. Oxytocin in the ventromedial hypothalamic nucleus reduces feeding and acutely increases energy expenditure. Am J Physiol Regul Integr Comp Physiol. (2014) 307:R737–45. doi: 10.1152/ajpregu.00118.2014
23. Zhang G, Bai H, Zhang H, Dean C, Wu Q, Li J, et al. Neuropeptide exocytosis involving synaptotagmin-4 and oxytocin in hypothalamic programming of body weight and energy balance. Neuron. (2011) 69:523–35. doi: 10.1016/j.neuron.2010.12.036
24. Zhang G, Cai D. Circadian intervention of obesity development via resting-stage feeding manipulation or oxytocin treatment. Am J Physiol Endocrinol Metab. (2011) 301:E1004–12. doi: 10.1152/ajpendo.00196.2011
25. Blevins JE, Thompson BW, Anekonda VT, Ho JM, Graham JL, Roberts ZS, et al. Chronic CNS oxytocin signaling preferentially induces fat loss in high fat diet-fed rats by enhancing satiety responses and increasing lipid utilization. Am J Physiol-Reg I. (2016) 310:R640–58. doi: 10.1152/ajpregu.00220.2015
26. Maejima Y, Aoyama M, Sakamoto K, Jojima T, Aso Y, Takasu K, et al. Impact of sex, fat distribution and initial body weight on oxytocin’s body weight regulation. Sci Rep. (2017) 7:8599. doi: 10.1038/s41598-017-09318-7
27. Maejima Y, Iwasaki Y, Yamahara Y, Kodaira M, Sedbazar U, Yada T. Peripheral oxytocin treatment ameliorates obesity by reducing food intake and visceral fat mass. Aging. (2011) 3:1169–77. doi: 10.18632/aging.100408
28. Morton GJ, Thatcher BS, Reidelberger RD, Ogimoto K, Wolden-Hanson T, Baskin DG, et al. Peripheral oxytocin suppresses food intake and causes weight loss in diet-induced obese rats. Am J Physiol-Endoc M. (2012) 302:E134–44. doi: 10.1152/ajpendo.00296.2011
29. Roberts ZS, Wolden-Hanson TH, Matsen ME, Ryu V, Vaughan CH, Graham JL, et al. Chronic hindbrain administration of oxytocin is sufficient to elicit weight loss in diet-induced obese rats. Am J Physiol Regul Integr Comp Physiol. (2017) 313: R357–71. doi: 10.1152/ajpregu.00169.2017
30. Head MA, Levine AS, Christian DG, Klockars A, Olszewski PK. Effect of combination of peripheral oxytocin and naltrexone at subthreshold doses on food intake, body weight and feeding-related brain gene expression in male rats. Physiol Behav. (2021) 238:113464. doi: 10.1016/j.physbeh.2021.113464
31. Enriori PJ, Sinnayah P, Simonds SE, Garcia Rudaz C, Cowley MA. Leptin action in the dorsomedial hypothalamus increases sympathetic tone to brown adipose tissue in spite of systemic leptin resistance. J Neurosci. (2011) 31:12189–97. doi: 10.1523/JNEUROSCI.2336-11.2011
32. Mirbolooki MR, Upadhyay SK, Constantinescu CC, Pan ML, Mukherjee J. Adrenergic pathway activation enhances brown adipose tissue metabolism: A [F-18]FDG PET/CT study in mice. Nucl Med Biol. (2014) 41:10–6. doi: 10.1016/j.nucmedbio.2013.08.009
33. Suarez J, Rivera P, Arrabal S, Crespillo A, Serrano A, Baixeras E, et al. Oleoylethanolamide enhances beta-adrenergic-mediated thermogenesis and white-to-brown adipocyte phenotype in epididymal white adipose tissue in rat. Dis Models Mech. (2014) 7:129–41. doi: 10.1242/dmm.013110
34. Xiao C, Goldgof M, Gavrilova O, Reitman ML. Anti-obesity and metabolic efficacy of the beta3-adrenergic agonist, CL316243, in mice at thermoneutrality compared to 22 degrees C. Obesity. (2015) 23:1450–9. doi: 10.1002/oby.21124
35. Edwards MM, Nguyen HK, Dodson AD, Herbertson AJ, Wietecha TA, Wolden-Hanson T, et al. Effects of combined oxytocin and beta-3 receptor agonist (CL 316243) treatment on body weight and adiposity in male diet-induced obese rats. Front Physiol. (2021) 12:725912. doi: 10.3389/fphys.2021.725912
36. Guide for the care and use of laboratory animals. Eighth Edition. Washington, D.C.: The National Academies Press (2011).
37. Susulic VS, Frederich RC, Lawitts J, Tozzo E, Kahn BB, Harper ME, et al. Targeted disruption of the beta 3-adrenergic receptor gene. J Biol Chem. (1995) 270:29483–92. doi: 10.1074/jbc.270.49.29483
38. Lateef DM, Abreu-Vieira G, Xiao C, Reitman ML. Regulation of body temperature and brown adipose tissue thermogenesis by bombesin receptor subtype-3. Am J Physiol Endocrinol Metab. (2014) 306:E681–7. doi: 10.1152/ajpendo.00615.2013
39. Grujic D, Susulic VS, Harper ME, HimmsHagen J, Cunningham BA, Corkey BE, et al. beta 3-adrenergic receptors on white and brown adipocytes mediate beta 3-selective agonist-induced effects on energy expenditure, insulin secretion, and food intake - A study using transgenic and gene knockout mice. J Biol Chem. (1997) 272:17686–93. doi: 10.1074/jbc.272.28.17686
40. Brito MN, Brito NA, Baro DJ, Song CK, Bartness TJ. Differential activation of the sympathetic innervation of adipose tissues by melanocortin receptor stimulation. Endocrinology. (2007) 148:5339–47. doi: 10.1210/en.2007-0621
41. Vaughan CH, Shrestha YB, Bartness TJ. Characterization of a novel melanocortin receptor-containing node in the SNS outflow circuitry to brown adipose tissue involved in thermogenesis. Brain Res. (2011) 1411:17–27. doi: 10.1016/j.brainres.2011.07.003
42. Fischer AW, Schlein C, Cannon B, Heeren J, Nedergaard J. Intact innervation is essential for diet-induced recruitment of brown adipose tissue. Am J Physiol Endocrinol Metab. (2019) 316:E487–503. doi: 10.1152/ajpendo.00443.2018
43. Edwards MM, Nguyen HK, Dodson AD, Herbertson AJ, Honeycutt MK, Slattery JD, et al. Sympathetic innervation of interscapular brown adipose tissue is not a predominant mediator of oxytocin-elicited reductions of body weight and adiposity in male diet-induced obese rats. Front Drug Delivery. (2024) 4:1497746. doi: 10.3389/fddev.2024.1497746
44. Edwards MM, Nguyen HK, Herbertson AJ, Dodson AD, Wietecha T, Wolden-Hanson T, et al. Chronic hindbrain administration of oxytocin elicits weight loss in male diet-induced obese mice. Am J Physiol Regul Integr Comp Physiol. (2021) 320: R471–87. doi: 10.1152/ajpregu.00294.2020
45. Parlee SD, Lentz SI, Mori H, MacDougald OA. Quantifying size and number of adipocytes in adipose tissue. Methods enzymology. (2014) 537:93–122. doi: 10.1016/B978-0-12-411619-1.00006-9
46. Cao Q, Jing J, Cui X, Shi H, Xue B. Sympathetic nerve innervation is required for beigeing in white fat. Physiol Rep. (2019) 7:e14031. doi: 10.14814/phy2.14031
47. Veniant MM, Sivits G, Helmering J, Komorowski R, Lee J, Fan W, et al. Pharmacologic effects of FGF21 are independent of the “Browning” of white adipose tissue. Cell Metab. (2015) 21:731–8. doi: 10.1016/j.cmet.2015.04.019
48. Bremer AA, Stanhope KL, Graham JL, Cummings BP, Wang W, Saville BR, et al. Fructose-fed rhesus monkeys: a nonhuman primate model of insulin resistance, metabolic syndrome, and type 2 diabetes. Clin Trans Sci. (2011) 4:243–52. doi: 10.1111/j.1752-8062.2011.00298.x
49. Blevins JE, Moralejo DH, Wolden-Hanson TH, Thatcher BS, Ho JM, Kaiyala KJ, et al. Alterations in activity and energy expenditure contribute to lean phenotype in Fischer 344 rats lacking the cholecystokinin-1 receptor gene. Am J Physiol Regul Integr Comp Physiol. (2012) 303:R1231–40. doi: 10.1152/ajpregu.00393.2012
50. Cummings BP, Digitale EK, Stanhope KL, Graham JL, Baskin DG, Reed BJ, et al. Development and characterization of a novel rat model of type 2 diabetes mellitus: the UC Davis type 2 diabetes mellitus UCD-T2DM rat. Am J Physiol Regul Integr Comp Physiol. (2008) 295:R1782–93. doi: 10.1152/ajpregu.90635.2008
51. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. (2001) 25:402–8. doi: 10.1006/meth.2001.1262
52. Gonzalez-Hurtado E, Lee J, Choi J, Wolfgang MJ. Fatty acid oxidation is required for active and quiescent brown adipose tissue maintenance and thermogenic programing. Mol Metab. (2018) 7:45–56. doi: 10.1016/j.molmet.2017.11.004
53. Rosell M, Kaforou M, Frontini A, Okolo A, Chan YW, Nikolopoulou E, et al. Brown and white adipose tissues: intrinsic differences in gene expression and response to cold exposure in mice. Am J Physiol Endocrinol Metab. (2014) 306:E945–64. doi: 10.1152/ajpendo.00473.2013
54. de Jong JMA, Wouters RTF, Boulet N, Cannon B, Nedergaard J, Petrovic N. The beta3-adrenergic receptor is dispensable for browning of adipose tissues. Am J Physiol Endocrinol Metab. (2017) 312:E508–18. doi: 10.1152/ajpendo.00437.2016
55. Oliveira FCB, Bauer EJ, Ribeiro CM, Pereira SA, Beserra BTS, Wajner SM, et al. Liraglutide activates type 2 deiodinase and enhances beta3-adrenergic-induced thermogenesis in mouse adipose tissue. Front Endocrinol. (2021) 12:803363. doi: 10.3389/fendo.2021.803363
56. Gaetani S, Fu J, Cassano T, Dipasquale P, Romano A, Righetti L, et al. The fat-induced satiety factor oleoylethanolamide suppresses feeding through central release of oxytocin. J Neurosci. (2010) 30:8096–101. doi: 10.1523/JNEUROSCI.0036-10.2010
57. Romano A, Cassano T, Tempesta B, Cianci S, Dipasquale P, Coccurello R, et al. The satiety signal oleoylethanolamide stimulates oxytocin neurosecretion from rat hypothalamic neurons. Peptides. (2013) 49:21–6. doi: 10.1016/j.peptides.2013.08.006
58. Sospedra I, Moral R, Escrich R, Solanas M, Vela E, Escrich E. Effect of high fat diets on body mass, oleylethanolamide plasma levels and oxytocin expression in growing rats. J Food Sci. (2015) 80(6):H1425–31. doi: 10.1111/jfds.2015.80.issue-6
59. Asker M, Krieger JP, Liles A, Tinsley IC, Borner T, Maric I, et al. Peripherally restricted oxytocin is sufficient to reduce food intake and motivation, while CNS entry is required for locomotor and taste avoidance effects. Diabetes Obes Metab. (2023) 25:856–77. doi: 10.1111/dom.14937
60. Iwasaki Y, Kumari P, Wang L, Hidema S, Nishimori K, Yada T. Relay of peripheral oxytocin to central oxytocin neurons via vagal afferents for regulating feeding. Biochem Biophys Res Commun. (2019) 519:553–8. doi: 10.1016/j.bbrc.2019.09.039
61. Iwasaki Y, Maejima Y, Suyama S, Yoshida M, Arai T, Katsurada K, et al. Peripheral oxytocin activates vagal afferent neurons to suppress feeding in normal and leptin-resistant mice: a route for ameliorating hyperphagia and obesity. Am J Physiol Regul Integr Comp Physiol. (2015) 308:R360–9. doi: 10.1152/ajpregu.00344.2014
62. Brierley DI, Holt MK, Singh A, de Araujo A, McDougle M, Vergara M, et al. Central and peripheral GLP-1 systems independently suppress eating. Nat Metab. (2021) 3:258–73. doi: 10.1038/s42255-021-00344-4
63. Ho JM, Anekonda VT, Thompson BW, Zhu M, Curry RW, Hwang BH, et al. Hindbrain oxytocin receptors contribute to the effects of circulating oxytocin on food intake in male rats. Endocrinology. (2014) 155:2845–57. doi: 10.1210/en.2014-1148
64. Erdenebayar O, Kato T, Kawakita T, Kasai K, Kadota Y, Yoshida K, et al. Effects of peripheral oxytocin administration on body weight, food intake, adipocytes, and biochemical parameters in peri- and postmenopausal female rats. Endocrine J. (2020) 68(1):7–16. doi: 10.1507/endocrj.EJ19-0586
65. Iwasa T, Matsuzaki T, Mayila Y, Kawakita T, Yanagihara R, Irahara M. The effects of chronic oxytocin administration on body weight and food intake in DHT-induced PCOS model rats. Gynecological endocrinology: Off J Int Soc Gynecological Endocrinol. (2020) 36:55–60. doi: 10.1080/09513590.2019.1631276
66. Iwasa T, Matsuzaki T, Mayila Y, Yanagihara R, Yamamoto Y, Kawakita T, et al. Oxytocin treatment reduced food intake and body fat and ameliorated obesity in ovariectomized female rats. Neuropeptides. (2019) 75:49–57. doi: 10.1016/j.npep.2019.03.002
67. Balazova L, Krskova K, Suski M, Sisovsky V, Hlavacova N, Olszanecki R, et al. Metabolic effects of subchronic peripheral oxytocin administration in lean and obese zucker rats. J Physiol pharmacology: an Off J Polish Physiol Soc. (2016) 67:531–41. doi: 10.1016/j.npep.2019.03.002
68. Elfers CT, Blevins JE, Salameh TS, Lawson EA, Silva D, Kiselyov A, et al. Novel long-acting oxytocin analog with increased efficacy in reducing food intake and body weight. Int J Mol Sci. (2022) 23:11249. doi: 10.3390/ijms231911249
69. Elfers CT, Blevins JE, Lawson EA, Pittner R, Silva D, Kiselyov A, et al. Robust reductions of body weight and food intake by an oxytocin analog in rats. Front Physiol. (2021) 12:726411. doi: 10.3389/fphys.2021.726411
70. Schaffler A, Binart N, Scholmerich J, Buchler C. Hypothesis paper Brain talks with fat–evidence for a hypothalamic-pituitary-adipose axis? Neuropeptides. (2005) 39:363–7. doi: 10.1016/j.npep.2005.06.003
71. Altirriba J, Poher AL, Caillon A, Arsenijevic D, Veyrat-Durebex C, Lyautey J, et al. Divergent effects of oxytocin treatment of obese diabetic mice on adiposity and diabetes. Endocrinology. (2014) 155:4189–201. doi: 10.1210/en.2014-1466
72. Yoshitomi H, Yamazaki K, Abe S, Tanaka I. Differential regulation of mouse uncoupling proteins among brown adipose tissue, white adipose tissue, and skeletal muscle in chronic beta 3 adrenergic receptor agonist treatment. Biochem Biophys Res Commun. (1998) 253:85–91. doi: 10.1006/bbrc.1998.9746
73. Kasahara Y, Sato K, Takayanagi Y, Mizukami H, Ozawa K, Hidema S, et al. Oxytocin receptor in the hypothalamus is sufficient to rescue normal thermoregulatory function in male oxytocin receptor knockout mice. Endocrinology. (2013) 154:4305–15. doi: 10.1210/en.2012-2206
74. Kasahara Y, Tateishi Y, Hiraoka Y, Otsuka A, Mizukami H, Ozawa K, et al. Role of the oxytocin receptor expressed in the rostral medullary raphe in thermoregulation during cold conditions. Front Endocrinol. (2015) 6:180. doi: 10.3389/fendo.2015.00180
75. Bengtsson T, Cannon B, Nedergaard J. Differential adrenergic regulation of the gene expression of the beta-adrenoceptor subtypes beta1, beta2 and beta3 in brown adipocytes. Biochem J. (2000) 347 Pt 3:643–51.
76. Zhang H, Wu C, Chen Q, Chen X, Xu Z, Wu J, et al. Treatment of obesity and diabetes using oxytocin or analogs in patients and mouse models. PloS One. (2013) 8:e61477. doi: 10.1371/journal.pone.0061477
77. Plessow F, Kerem L, Wronski ML, Asanza E, O’Donoghue ML, Stanford FC, et al. Intranasal oxytocin for obesity. NEJM Evid. (2024) 3:EVIDoa2300349. doi: 10.1056/EVIDoa2300349
78. Bharadwaj VN, Meyerowitz J, Zou B, Klukinov M, Yan N, Sharma K, et al. Impact of magnesium on oxytocin receptor function. Pharmaceutics. (2022) 14(5):1105. doi: 10.3390/pharmaceutics14051105
79. Antoni FA, Chadio SE. Essential role of magnesium in oxytocin-receptor affinity and ligand specificity. Biochem J. (1989) 257:611–4. doi: 10.1042/bj2570611
80. Niu J, Tong J, Blevins JE. Oxytocin as an anti-obesity treatment. Front Neurosci. (2021) 15:743546. doi: 10.3389/fnins.2021.743546
81. Olszewski PK, Noble EE, Paiva L, Ueta Y, Blevins JE. Oxytocin as a potential pharmacological tool to combat obesity. J Neuroendocrinol. (2022) 34(9):e13106. doi: 10.1111/jne.13106
82. Cypess AM, Weiner LS, Roberts-Toler C, Franquet Elia E, Kessler SH, Kahn PA, et al. Activation of human brown adipose tissue by a beta3-adrenergic receptor agonist. Cell Metab. (2015) 21:33–8. doi: 10.1016/j.cmet.2014.12.009
Keywords: obesity, brown adipose tissue (BAT), white adipose tissue (WAT), food intake, thermogenesis, oxytocin
Citation: Slattery JD, Rambousek JR, Tsui E, Honeycutt MK, Goldberg M, Graham JL, Wietecha TA, Wolden-Hanson T, Williams AL, O’Brien KD, Havel PJ and Blevins JE (2025) Effects of systemic oxytocin and beta‐3 receptor agonist (CL 316243) treatment on body weight and adiposity in male diet‐induced obese rats. Front. Endocrinol. 16:1503096. doi: 10.3389/fendo.2025.1503096
Received: 28 September 2024; Accepted: 31 January 2025;
Published: 04 March 2025.
Edited by:
Zhi Yi Ong, University of New South Wales, AustraliaReviewed by:
Dana Hutchinson, Monash University, AustraliaCopyright © 2025 Slattery, Rambousek, Tsui, Honeycutt, Goldberg, Graham, Wietecha, Wolden-Hanson, Williams, O’Brien, Havel and Blevins. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: James E. Blevins, amVibGV2aW5AdXcuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.