 Jianyu Wang
Jianyu Wang Chunhua Wang2
Chunhua Wang2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Endocrinol. , 14 February 2025
Sec. Clinical Diabetes
Volume 16 - 2025 | https://doi.org/10.3389/fendo.2025.1502783
Monogenic diabetes, which encompasses neonatal diabetes (NDM), maturity onset diabetes of the young (MODY), and several diabetes-associated syndromes, primarily arises from impaired function or abnormal development of the islets of Langerhans, particularly pancreatic β-cells responsible for insulin secretion. This condition is typically associated with a single pathogenic genetic mutation. Charcot-Marie-Tooth disease type 1A (CMT1A) is a hereditary demyelinating neuropathy that is caused by a duplication of the PMP22 gene located on chromosome 17. Herein, we report a case of a young Chinese patient with MODY6 harboring a novel mutation (c. 317C>T, p. Ala106Val) in the NEUROD1 gene. Additionally, this patient concurrently presents with CMT1A, which is characterized by a large segmental duplication within the exon of the PMP22 gene and its adjacent regions. Considering the patient’s compromised islet function, we treat him with insulin and oral hypoglycemic agents (metformin, acarbose). This represents the first reported instance of a patient with NEUROD1-MODY coexisting with CMT1A.
MODY6, a rare subtype of MODY, results from either a heterozygous or homozygous mutation in the NEUROD1 gene on chromosome 2q32 (1, 2). NEUROD1 functions as a transcription factor that binds to and activates the insulin promoter, playing a crucial role in maintaining normal glucose homeostasis (3). MODY should be strongly suspected in individuals under 35 years of age (with those under 25 being particularly indicative) who present with mild hyperglycemia at onset and lack the typical features of either type 1 or type 2 diabetes mellitus (2, 4). The clinical manifestations of MODY6 encompass a broad spectrum, varying from patients displaying typical MODY characteristics to those who resemble individuals with common type 2 diabetes mellitus (5). The two primary sequencing technologies used for genetic diagnosis of MODY are targeted sequencing of genes associated with monogenic diabetes and whole exome sequencing (6).
Hereditary motor and sensory neuropathies, commonly referred to as Charcot–Marie–Tooth disease (CMT), are marked by a length-dependent loss of axonal integrity in the peripheral nervous system (PNS), leading to progressive muscle weakness and sensory deficits. The genetic diagnosis of CMT has been facilitated by targeted next-generation sequencing and whole-exome sequencing methods. CMT1A, the most common form of Charcot-Marie-Tooth disease (CMT), is caused by an approximately 1.4 Mb duplication of the PMP22 gene on chromosome 17p11.2-12. This genetic alteration leads to disrupted myelin formation and impaired nerve function. Most patients with CMT1A exhibit a ‘typical’ phenotype, which includes onset in childhood, sensory loss, distal weakness, absent reflexes and foot deformities (7, 8).
Herein, we presented a MODY6 patient coexistence with Charcot-Marie-Toothe 1A syndrome. The whole-exome sequencing (WES) revealed a novel mutation in NEUROD1 (c. 317C>T, p. Ala106Val), along with an approximately 1.38Mb duplication (copy number: 3) in the patient’s 17p12 region, which encompasses the PMP22 gene. As the first reported case of a patient with NEUROD1-MODY coexisting with CMT1A, it exerts positive effects on improving clinicians’ understanding of MODY6 coexisting with CMT1A.
A 29-year-old male was admitted to QiLu hospital of Shangdong university Dezhou hospital (Dezhou People’s Hospital), presenting with a one-year history of polydipsia, polyuria, and blurred vision. Six months prior, the patient’s fasting blood glucose level was measured at 14+ mmol/L at a local community outpatient clinic. However, he did not accept any treatment and did not monitor blood glucose. Ten days ago, the patient reported experiencing numbness in the hands and feet and had a fasting blood glucose level measured at over 16 mmol/L at a local community outpatient clinic. Treatment with “metformin 1g twice daily and glibenclamide 5mg twice daily” was initiated to manage blood glucose levels, yet hyperglycemia persisted. Anamnestic history revealed progressive atrophy and weakness of the distal extremities, resulting in unsteady gait since childhood. Physical examination revealed a high arch, as shown in Figure 1, accompanied by numbness in the limbs and decreased sensation to pinprick and temperature in the lower extremities. Electromyography: The motor nerve of left and right common peroneal nerves and tibial nerve are not elicited, the sensory nerve of left and right superficial peroneal nerves are injured, the sensory nerve amplitude of left and right sural nerves reduced. The family history: The patient’s mother was diagnosed with diabetes at the age of 50, with a BMI of 24kg/m2. She was treated with metformin 500mg once daily and maintained normal blood glucose levels. Additionally, the patient’s father also exhibited high arch and decreased running ability, while his son developed a high arch at the age of 2. The clinical information was collected and recorded by the QiLu hospital of Shangdong university Dezhou hospital (Dezhou People’s Hospital). The patient is described in Table 1 and the timeline of the patient’s care is showed in Figure 2.
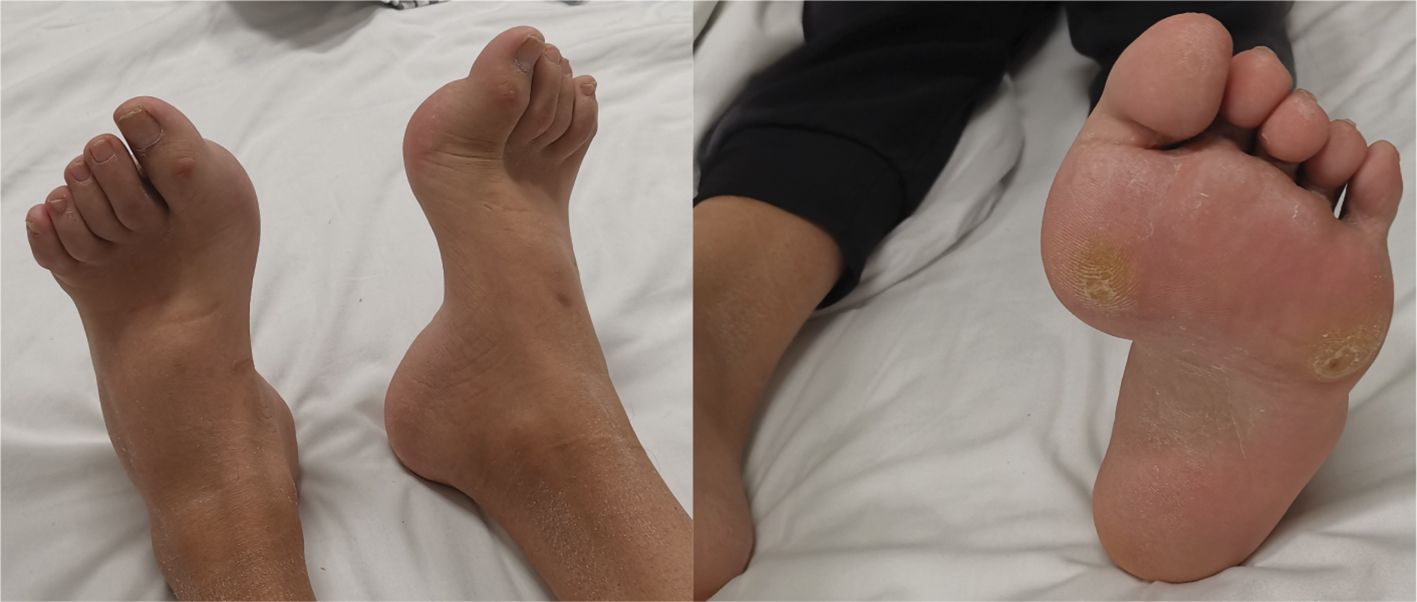
Figure 1. The images of patient’s high arch.
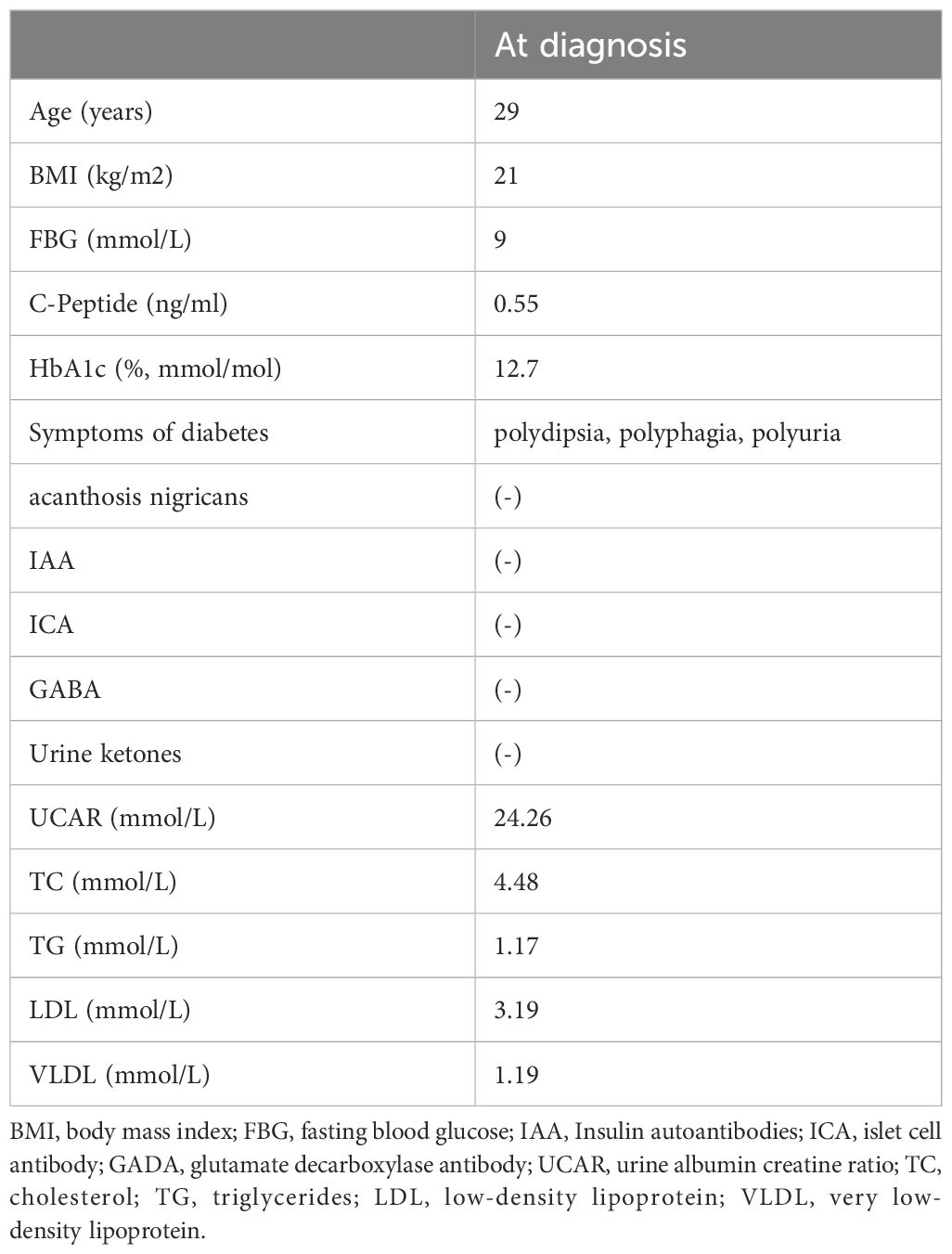
Table 1. Clinical characterization about diabetes of the patient.
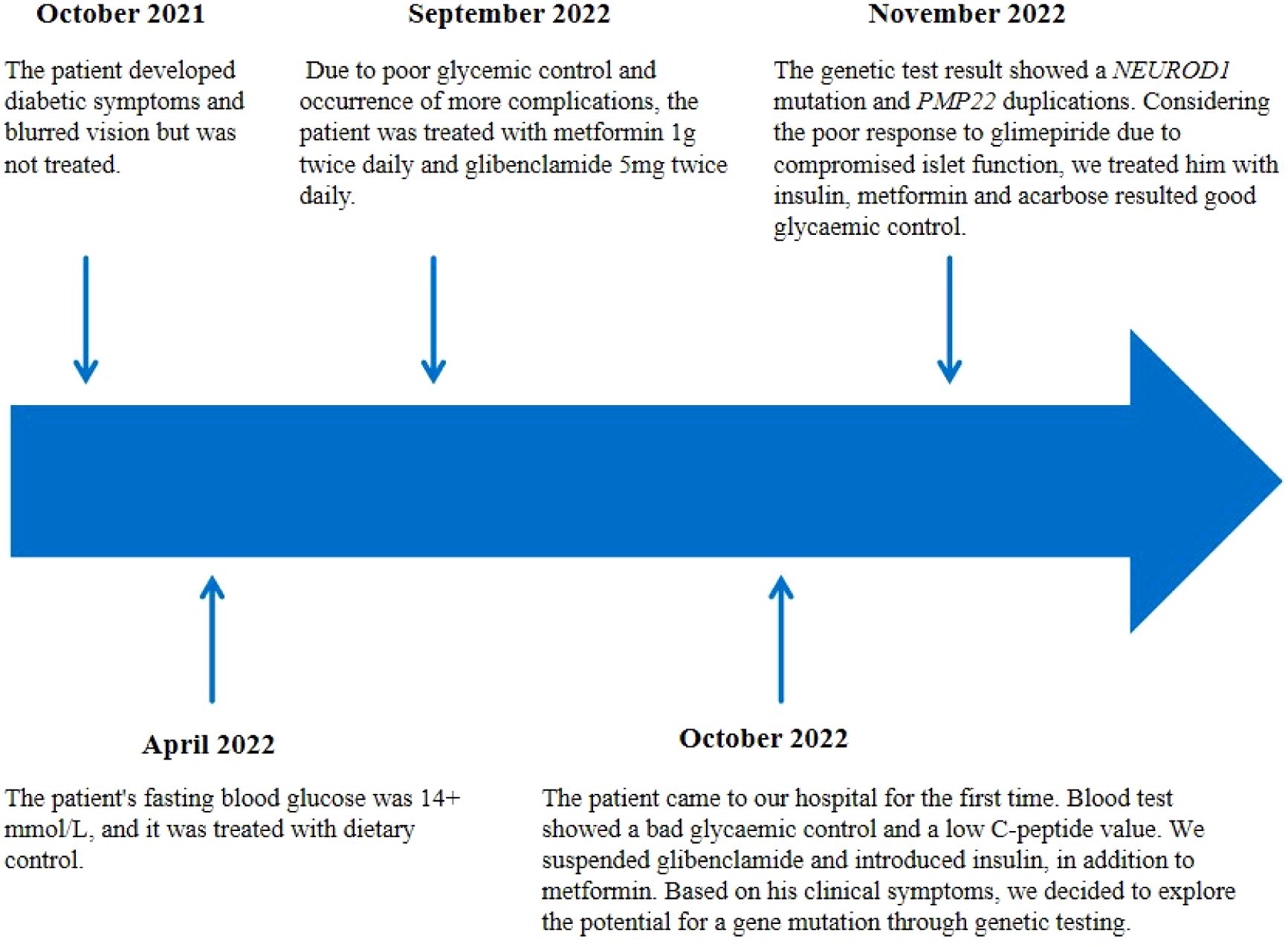
Figure 2. The timeline of the patient’s care.
Based on the patient’s clinical characterizations, which are inconsistent with those of type 1 diabetes, such as the absence of pancreatic antibodies, particularly when measured at diagnosis (9), the persistence of islet function, and the lack of ketoacidosis, as well as the inconsistencies with type 2 diabetes, including the onset of diabetes before the age of 45 years with a normal BMI, the absence of acanthosis nigricans, and normal triglyceride levels, we classify this case as a specific type of diabetes. Moreover, due to the low level of C-peptide and family history, we suspected him to have genetic defects in β cell function such like MODY. With the consent of the patient and his family, we collected peripheral blood samples from the patient, his parents and his son for gene testing.
The whole exome sequencing report revealed that a novel heterogeneous missense mutation (NM_002500. 5:c.317C>T, p.Ala106Val) in the exon2 of the NEUROD1 gene since it has not been reported in the 1000G, gnomAD, or ExAC databases, situated on chromosome chr2:182543271(rs:-) which is PM2 supported, PM3 moderate as shown in Figures 3A, C, as well as an approximately 1.38 Mb duplication(number of copies:3) on chromosome 17p11.2 that contains the peripheral myelin protein 22 (PMP22) gene which is pathogenic that ClinGen CNV scoring≥ 0.99 causing Charcot-Marie-Tooth disease type 1A (CMT1A) by CNV-seq, which are shown in Figures 3B, D. Moreover, the Ala106 residue in the DNA-binding domain of NEUROD1 is evolutionarily conserved in mammals as shown in Supplementary Figures 1, 2, both SIFT and Polyphen algorithms have shown that the Ala106Val mutant is damaging: SIFT score=0 (deleterious), Polyphen score=0.999 (probably damaging) and the PSIPRED analysis indicated that the analyzed substitution appeared to result in helix breakage as shown in Supplementary Figures 3, 4. Treatment protocols: mixed rotamine zinc recombinant human insulin lispro Injection (25R): 18 units (before breakfast), 16 units (before dinner); acarbose tablets: 50mg thrice daily; metformin: 500mg thrice daily; epalrestat: 50mg thrice daily; mecobalamin: 0.5mg thrice daily. After seven days of treatment, the patient’s fasting plasma glucose decreased to 7mmol/L and 2-hour plasma glucose was 6-12 mmol/L.
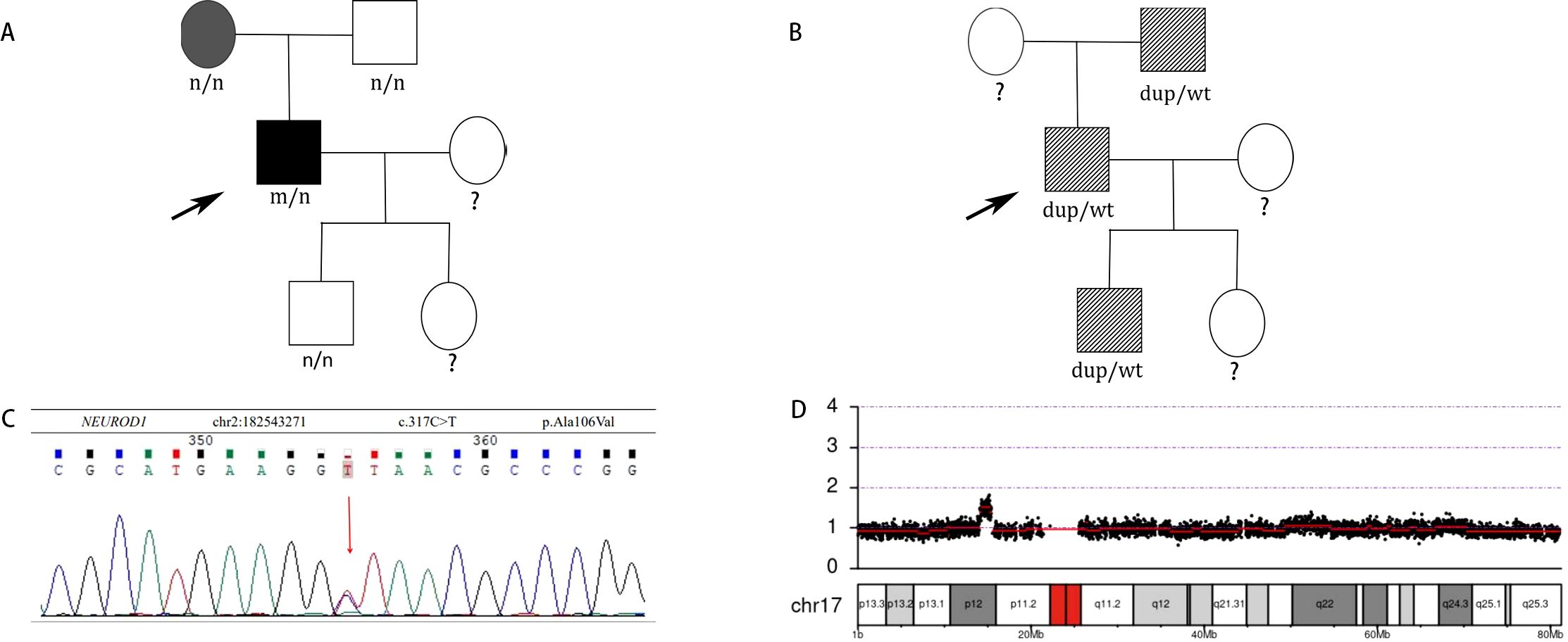
Figure 3. (A) Identification of a novel missense NEUROD1 mutation A106V associated with diabetes in patients. Subjects carrying A106V mutation with diabetes are shown in black. Subjects without carrying A106V mutation with diabetes are shown in gray. Black arrow indicates the probands. n, normal allele; m, mutant allele. (B) Identification of PMP22 duplication associated with CTM1A in patients. Subjects carrying PMP22 duplication with CMT1A phenotype are shown with slash. Black arrow indicates the probands. wt: wild type, dup: PMP22 duplication. (C) DNA sequences of the NEUROD1 mutation c.317 C>T found in the patient. (D) CNV-seq of the PMP22 gene duplication on chromosome 17p11.2.
In this study, we described a case of MODY6 associated with a novel mutation in NEUROD1 within the context of Charcot-Marie-Tooth 1A. The helix-loop-helix (HLH) protein NEUROD1, also known as BETA2, functions as a regulatory switch in the development of the endocrine pancreas (1). Additionally, NEUROD1 binds to the bHLH consensus E-box-binding site within the insulin promoter to activate transcription (3), as well as to the promoters of the sulfonylurea receptor 1 (SUR1) (9), glucokinase (GCK) (10), the glucose-6-phosphatase catalytic subunit-related protein (11), and PAX6 (12), all of these factors contribute to the maintenance of glucose homeostasis. MODY6 represents an extremely rare form of MODY, arising from heterozygous or homozygous mutations in the NEUROD1 gene (13).
The overall phenotype of MODY6 includes a wide clinical spectrum, ranging from patients displaying typical MODY features to those who exhibit characteristics akin to common type 2 diabetes mellitus (5). The diagnosis of MODY should consider the following atypical features: age under 35 (with age under 25 being more suggestive), negative autoantibodies, the presence of neonatal hypoglycemia, and/or multiple family members with diabetes that does not align with the characteristics of type 1 or type 2 diabetes (14–16). Features atypical for type 1 diabetes mellitus include the following: 1. No presence of pancreatic islet autoantibodies. 2. Demonstrated endogenous insulin production beyond the honeymoon phase. 3. Detectable C-peptide levels during hyperglycemia (C-peptide ≥0.60 ng/mL or 0.2 nmol/L). 4. Low insulin requirements for management (i.e., less than 0.5 U/kg/day). 5. Absence of ketoacidosis when insulin treatment is discontinued. Features atypical of type 2 diabetes mellitus include the following: 1. Onset of diabetes before the age of 45. 2. Absence of significant obesity. 3. Lack of acanthosis nigricans. 4. Normal triglyceride levels and/or normal of elevated high-density lipoprotein cholesterol (HDL-C) (17). Since type 2 diabetes has strong genetic component, shared risk alleles and shared environment that can cause multiple family members to develop type 2 diabetes. In some families, it can be difficult to distinguish from autosomal dominant inheritance. Furthermore, genetic background across different races can significantly influence the pathogenesis of even monogenic forms of diabetes (18). Whole-exome sequencing (WES) has emerged as a powerful tool for discovering novel genes associated with genetic disorders (19). The therapeutic approach for different forms of MODY varies based on their clinical features and underlying molecular causes. In some cases, insulin therapy is required for optimal glucose control, while other patients with MODY can effectively manage their condition with oral hypoglycemic agents (e.g., sulfonylureas) without the need for insulin (20). First-line treatment for MODY6 includes dietary management and oral hypoglycemic agents, such as sulfonylureas, while insulin is considered as second-line therapy (4). The goals of therapy are to enhance quality of life and prevent diabetes-related complications.
The patient’s mother without the mutation in the gene NEROUD1 has typical characteristics of type 2 diabetes such like: overweight (BMI≥24kg/m2) or obese (BMI≥28kg/m2), onset after age 45, insulin treatment not to be required, abdominal obesity, ketoacidosis seldom occurring spontaneously. We considered the patient’s mother to be a T2DM. However, the patient has clinical characteristics of MODY different from his mother, we considered the novel NEUROD1 heterozygous missense mutation contributed to the clinical features of MODY6. The Ala106Val mutation located in the DNA-binding domain of the NEUROD1 protein impacts the structure of the NEUROD1 to interfere with transcriptional activation. The most probable mechanism is that the side chain of valine is larger than that of alanine and exhibits greater hydrophobicity. Three diabetes pedigrees with different NEUROD1 mutations (Arg111Leu, Glu110Lys and Arg103Pro) at nearby locations have been reported, but their clinical features are different, ranging from MODY to type 2 diabetes, indicating the heterogeneity of the NEUROD1 mutations (1, 21, 22). The whole exome sequencing of family members carrying or not carrying the NEUROD1 mutation, along with functional analyses, could provide insight into how various genetic and/or environmental risk factors collaborate with the NEUROD1 mutation to drive the onset and advancement of diabetes.
Charcot–Marie–Tooth (CMT) disease is the most prevalent inherited neuromuscular disorder (23). The most common genetic form of CMT is Charcot-Marie-Tooth disease type 1A (CMT1A), caused by the duplication of the peripheral myelin protein 22 (PMP22) gene located on chromosome 17 (24, 25), Patients typically exhibit a ‘classical CMT phenotype’ characterized by progressive muscle weakness, atrophy, reduced sensory function, hyporeflexia, and skeletal deformities. The approach to diagnosing CMT is shifting from a purely clinical method in the past to a combined clinical and genetic approach in the present (26). The genetic diagnosis of CMT is performed using targeted next-generation sequencing and whole-exome sequencing techniques. Strategies to downregulate PMP22 gene expression have emerged as a logical extension of treatment research. Some therapies, including ascorbic acid (27), progesterone receptor antagonists (28), small interfering RNA (29) have shown promise in preclinical studies. Unfortunately, the successful translation of these therapies to human trials has been disappointing thus far (30).
PMP22 is expressed in the myelinating Schwann cells of the peripheral nervous system, where it plays an essential role in the formation and maintenance of compact myelin (31). Duplication of the PMP22 gene leads to a decrease in the amount of functional myelin, causing demyelination and eventually resulting in secondary axonal degeneration and loss during the process of remyelination (32). NEUROD1 is specifically expressed in developing neurons and is crucial for neuronal maturation and neurite outgrowth (33). Interestingly, overexpression of NEUROD1 in adult spinal neurons significantly accelerates axonal regeneration following sciatic nerve injury (34). Based on these findings, we propose that the combination of NEUROD1 and PMP22 mutations may exert an additive effect on the nervous system through axonal apoptosis, loss, and regeneration.
In a literature review, we found several reports of patients with CMT1A and diabetes mellitus (DM) (35–39). Reports have documented three families with both CMT1A and type 2 diabetes mellitus (T2DM), suggesting a potential chance association between the two conditions. A retrospective analysis of clinic patients indicated that neuropathy is more severe in diabetic patients with CMT1A compared to those without diabetes (40). A previously reported case of diabetes coexisting with CMT1A, presenting as a recurrent foot ulcer misdiagnosed as a diabetic foot, underscores the importance of differentiating between CMT1A and diabetic peripheral neuropathy (41). The severity of peripheral neuropathy is more significant than other complications such as nephropathy or retinopathy in CMT1A compared to diabetic peripheral neuropathy. However, there are no case reports about CMT patients with the gene mutations of NEUROD1 which are associated with MODY6. Because DM exacerbates motor and sensory impairment in CMT1A, the management of blood glucose is necessary to relieve the peripheral neuropathy.
In conclusion, according to our analysis about clinical features, gene and family history, we considered that the novel mutation of NEUROD1 is responsible for the diabetes phenotype in the patient. Interestingly and rarely, the patient, his father and his son also are diagnosed with CMT1A. But the patient’s father and son are not diagnosed with DM which are inconsistent with the possibility of a chance association between CMT1A and type 2 DM. However, both of diabetes and CMT1A are likely to have a cumulative negative effective effect on peripheral nerves (38), it is important to differential diagnosis between CMT1A and diabetic peripheral neuropathy and maintain normal blood glucose to relieve peripheral neuropathy. Despite the phenotypic normality observed in his 7-year-old son, it is prudent to focus attention on the peripheral neuromuscular system. Considering the patient’s compromised islet function, our therapeutic approach involved administering insulin to safeguard islet function, metformin to enhance insulin sensitivity, and acarbose to inhibit glucose absorption. The glycaemic control was remarkably improved. There are no FDA-approved treatments for CMT1A. However, further studies will yield insights into the mechanism between the heterozygous mutation of NEUROD1 gene and diabetes by biology experiments.
The data sets generated and analyzed during the current study are available from the corresponding author upon reasonable request. The data presented in this study are deposited in the GenBank repository, with accession number PRJNA1221855. The information is accessible at the following link: https://www.ncbi.nlm.nih.gov/sra/PRJNA1221855.
The studies involving humans were approved by Qilu Hospital of Shandong University Dezhou Hospital Ethics Committee. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
JW: Writing – original draft, Writing – review & editing. CW: Data curation, Writing – review & editing. YC: Formal Analysis, Writing – review & editing. SQ: Writing – review & editing. MW: Supervision, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We thank the proband and his families for their participating actively that enabled this work. We thank professor Ming Liu and AR Yumeng Huang at Tianjin medical university for providing professional guidance to us.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2025.1502783/full#supplementary-material
1. Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, Orban T, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet. (1999) 23:323–8. doi: 10.1038/15500
2. Bonnefond A, Unnikrishnan R, Doria A, Vaxillaire M, Kulkarni RN, Mohan V, et al. Monogenic diabetes. Nat Rev Dis Primers. (2023) 9:12. doi: 10.1038/s41572-023-00421-w
3. Naya FJ, Stellrecht CM, Tsai MJ. Tissue-specific regulation of the insulin gene by a novel basic helix-loop-helix transcription factor. Genes Dev. (1995) 9:1009–19. doi: 10.1101/gad.9.8.1009
4. Broome DT, Pantalone KM, Kashyap SR, Philipson LH. Approach to the patient with MODY-monogenic diabetes. J Clin Endocrinol Metab. (2021) 106:237–50. doi: 10.1210/clinem/dgaa710
5. Horikawa Y, Enya M. Genetic dissection and clinical features of MODY6 (NEUROD1-MODY). Curr Diabetes Rep. (2019) 19:12. doi: 10.1007/s11892-019-1130-9
6. Dillon OJ, Lunke S, Stark Z, Yeung A, Thorne N, Melbourne Genomics Health A, et al. Exome sequencing has higher diagnostic yield compared to simulated disease-specific panels in children with suspected monogenic disorders. Eur J Hum Genet. (2018) 26:644–51. doi: 10.1038/s41431-018-0099-1
7. Thomas PK, Marques W Jr., Davis MB, Sweeney MG, King RH, Bradley JL, et al. The phenotypic manifestations of chromosome 17p11.2 duplication. Brain. (1997) 120:465–78. doi: 10.1093/brain/120.3.465
8. Krajewski KM, Lewis RA, Fuerst DR, Turansky C, Hinderer SR, Garbern J, et al. Neurological dysfunction and axonal degeneration in Charcot-Marie-Tooth disease type 1A. Brain. (2000) 123:1516–27. doi: 10.1093/brain/123.7.1516
9. Kim JW, Seghers V, Cho JH, Kang Y, Kim S, Ryu Y, et al. Transactivation of the mouse sulfonylurea receptor I gene by BETA2/NeuroD. Mol Endocrinol. (2002) 16:1097–107. doi: 10.1210/mend.16.5.0934
10. Moates JM, Nanda S, Cissell MA, Tsai MJ, Stein R. BETA2 activates transcription from the upstream glucokinase gene promoter in islet beta-cells and gut endocrine cells. Diabetes. (2003) 52:403–8. doi: 10.2337/diabetes.52.2.403
11. Martin CC, Svitek CA, Oeser JK, Henderson E, Stein R, O’Brien RM. Upstream stimulatory factor (USF) and neurogenic differentiation/beta-cell E box transactivator 2 (NeuroD/BETA2) contribute to islet-specific glucose-6-phosphatase catalytic-subunit-related protein (IGRP) gene expression. Biochem J. (2003) 371:675–86. doi: 10.1042/BJ20021585
12. Marsich E, Vetere A, Di Piazza M, Tell G, Paoletti S. The PAX6 gene is activated by the basic helix-loop-helix transcription factor NeuroD/BETA2. Biochem J. (2003) 376:707–15. doi: 10.1042/BJ20031021
13. Bouillet B, Crevisy E, Baillot-Rudoni S, Gallegarine D, Jouan T, Duffourd Y, et al. Whole-exome sequencing identifies the first French MODY 6 family with a new mutation in the NEUROD1 gene. Diabetes Metab. (2020) 46:400–2. doi: 10.1016/j.diabet.2020.03.001
14. Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S. Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia. (2010) 53:2504–8. doi: 10.1007/s00125-010-1799-4
15. Pihoker C, Gilliam LK, Ellard S, Dabelea D, Davis C, Dolan LM, et al. Prevalence, characteristics and clinical diagnosis of maturity onset diabetes of the young due to mutations in HNF1A, HNF4A, and glucokinase: results from the SEARCH for Diabetes in Youth. J Clin Endocrinol Metab. (2013) 98:4055–62. doi: 10.1210/jc.2013-1279
16. Shepherd M, Shields B, Hammersley S, Hudson M, McDonald TJ, Colclough K, et al. Systematic population screening, using biomarkers and genetic testing, identifies 2.5% of the U.K. Pediatric diabetes population with monogenic diabetes. Diabetes Care. (2016) 39:1879–88. doi: 10.2337/dc16-0645
17. Naylor R, Knight Johnson A, del Gaudio D. Maturity-Onset Diabetes of the Young Overview. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews((R)). Seattle, WA: GeneReviews (1993).
18. Horikawa Y. Maturity-onset diabetes of the young as a model for elucidating the multifactorial origin of type 2 diabetes mellitus. J Diabetes Investig. (2018) 9:704–12. doi: 10.1111/jdi.12812
19. Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. (2011) 12:745–55. doi: 10.1038/nrg3031
20. Romer AI, Singer RA, Sui L, Egli D, Sussel L. Murine perinatal beta-cell proliferation and the differentiation of human stem cell-derived insulin-expressing cells require NEUROD1. Diabetes. (2019) 68:2259–71. doi: 10.2337/db19-0117
21. Kristinsson SY, Thorolfsdottir ET, Talseth B, Steingrimsson E, Thorsson AV, Helgason T, et al. MODY in Iceland is associated with mutations in HNF-1alpha and a novel mutation in NeuroD1. Diabetologia. (2001) 44:2098–103. doi: 10.1007/s001250100016
22. Szopa M, Ludwig-Galezowska AH, Radkowski P, Skupien J, Machlowska J, Klupa T, et al. A family with the Arg103Pro mutation in the NEUROD1 gene detected by next-generation sequencing - Clinical characteristics of mutation carriers. Eur J Med Genet. (2016) 59:75–9. doi: 10.1016/j.ejmg.2016.01.002
23. Braathen GJ, Sand JC, Lobato A, Hoyer H, Russell MB. Genetic epidemiology of Charcot-Marie-Tooth in the general population. Eur J Neurol. (2011) 18:39–48. doi: 10.1111/j.1468-1331.2010.03037.x
24. Gutmann L, Shy M. Update on charcot-marie-tooth disease. Curr Opin Neurol. (2015) 28:462–7. doi: 10.1097/WCO.0000000000000237
25. Nelis E, Van Broeckhoven C, De Jonghe P, Lofgren A, Vandenberghe A, Latour P, et al. Estimation of the mutation frequencies in Charcot-Marie-Tooth disease type 1 and hereditary neuropathy with liability to pressure palsies: a European collaborative study. Eur J Hum Genet. (1996) 4:25–33. doi: 10.1159/000472166
26. Reilly MM, Murphy SM, Laura M. Charcot-marie-tooth disease. J Peripher Nerv Syst. (2011) 16:1–14. doi: 10.1111/j.1529-8027.2011.00324.x
27. Passage E, Norreel JC, Noack-Fraissignes P, Sanguedolce V, Pizant J, Thirion X, et al. Ascorbic acid treatment corrects the phenotype of a mouse model of Charcot-Marie-Tooth disease. Nat Med. (2004) 10:396–401. doi: 10.1038/nm1023
28. Meyer zu Horste G, Prukop T, Liebetanz D, Mobius W, Nave KA, Sereda MW. Antiprogesterone therapy uncouples axonal loss from demyelination in a transgenic rat model of CMT1A neuropathy. Ann Neurol. (2007) 61:61–72. doi: 10.1002/ana.21026
29. Lee JS, Chang EH, Koo OJ, Jwa DH, Mo WM, Kwak G, et al. Pmp22 mutant allele-specific siRNA alleviates demyelinating neuropathic phenotype in vivo. Neurobiol Dis. (2017) 100:99–107. doi: 10.1016/j.nbd.2017.01.006
30. Morena J, Gupta A, Hoyle JC. Charcot-marie-tooth: from molecules to therapy. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20143419
31. Notterpek L, Roux KJ, Amici SA, Yazdanpour A, Rahner C, Fletcher BS. Peripheral myelin protein 22 is a constituent of intercellular junctions in epithelia. Proc Natl Acad Sci U.S.A. (2001) 98:14404–9. doi: 10.1073/pnas.251548398
32. Watila MM, Balarabe SA. Molecular and clinical features of inherited neuropathies due to PMP22 duplication. J Neurol Sci. (2015) 355:18–24. doi: 10.1016/j.jns.2015.05.037
33. Cho JH, Tsai MJ. The role of BETA2/NeuroD1 in the development of the nervous system. Mol Neurobiol. (2004) 30:35–47. doi: 10.1385/MN:30:1:035
34. Lai M, Pan M, Ge L, Liu J, Deng J, Wang X, et al. NeuroD1 overexpression in spinal neurons accelerates axonal regeneration after sciatic nerve injury. Exp Neurol. (2020) 327:113215. doi: 10.1016/j.expneurol.2020.113215
35. Celik M, Forta H, Parman Y, Bissar-Tadmouri N, Demirkirkan K, Battaloglu E. Charcot-Marie-Tooth disease associated with Type 2 diabetes mellitus. Diabetes Med. (2001) 18:685–6.
36. Ota T, Osawa K. Marked improvement of glycaemic control with pioglitazone in a Type 2 diabetic patient associated with Charcot-Marie-Tooth disease. Diabetes Med. (2003) 20:420–1. doi: 10.1046/j.1464-5491.2003.009173.x
37. Koc F, Sarica Y, Yerdelen D, Baris I, Battaloglu E, Sert M. A large family with Charcot-Marie-Tooth Type 1a and Type 2 diabetes mellitus. Int J Neurosci. (2006) 116:103–14. doi: 10.1080/00207450500341431
38. Sun AP, Tang L, Liao Q, Zhang H, Zhang YS, Zhang J. Coexistent Charcot-Marie-Tooth type 1A and type 2 diabetes mellitus neuropathies in a Chinese family. Neural Regener Res. (2015) 10:1696–9. doi: 10.4103/1673-5374.167771
39. Carvalho MA, Secchin JB, Lourenco CM, Leal RC, Bueno KC, Barreira AA, et al. Coexistence of CMT1A and diabetes mellitus in a young woman with a severe and progressive neuropathy and respiratory insufficiency. Muscle Nerve. (2013) 47:141–2. doi: 10.1002/mus.23459
40. Sheth S, Francies K, Siskind CE, Feely SM, Lewis RA, Shy ME. Diabetes mellitus exacerbates motor and sensory impairment in CMT1A. J Peripher Nerv Syst. (2008) 13:299–304. doi: 10.1111/j.1529-8027.2008.00196.x
Keywords: diabetes, NEUROD1 mutation, MODY6, PMP22, CMT1A
Citation: Wang J, Wang C, Chen Y, Qi S and Wang M (2025) A case report of a MODY6 patient coexistence with Charcot-Marie-Toothe 1A syndrome. Front. Endocrinol. 16:1502783. doi: 10.3389/fendo.2025.1502783
Received: 27 September 2024; Accepted: 24 January 2025;
Published: 14 February 2025.
Edited by:
Habib Ur Rehman, University of Veterinary and Animal Sciences, PakistanReviewed by:
Janine C Quijano, City of Hope National Medical Center, United StatesCopyright © 2025 Wang, Wang, Chen, Qi and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Wang, d21ybXl5QDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.