
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 03 February 2025
Sec. Bone Research
Volume 16 - 2025 | https://doi.org/10.3389/fendo.2025.1488489
 Haoling Zhang1,2,3
Haoling Zhang1,2,3 Rui Zhao4Xuemei Wang5Yaqian Qi4
Rui Zhao4Xuemei Wang5Yaqian Qi4 Doblin Sandai2*Wei Wang6*
Doblin Sandai2*Wei Wang6* Zhijing Song4,7*Qiudong Liang1*
Zhijing Song4,7*Qiudong Liang1*Mitochondria maintain bacterial traits because of their endosymbiotic origins, yet the host cell recognizes them as non-threatening since the organelles are compartmentalized. Nevertheless, the controlled release of mitochondrial components into the cytoplasm can initiate cell death, activate innate immunity, and provoke inflammation. This selective interruption of endosymbiosis as early as 2 billion years ago allowed mitochondria to become intracellular signaling hubs. Recent studies have found that the interruption of mitochondrial symbiosis may be closely related to the occurrence of various diseases, especially osteoporosis (OP). OP is a systemic bone disease characterized by reduced bone mass, impaired bone microstructure, elevated bone fragility, and susceptibility to fracture. The interruption of intra-mitochondrial symbiosis affects the energy metabolism of bone cells, leads to the imbalance of bone formation and bone absorption, and promotes the occurrence of osteoporosis. In this paper, we reviewed the mechanism of mitochondrial intersymbiosis interruption in OP, discussed the relationship between mitochondrial intersymbiosis interruption and bone marrow mesenchymal stem cells, osteoblasts and osteoclasts, as well as the inheritance and adaptation in the evolutionary process, and prospected the future research direction to provide new ideas for clinical treatment.
Mitochondria serve a crucial function as the cellular powerhouses, with their optimal performance being indispensable for sustaining cellular health and ensuring the overall homeostasis of organisms. In recent years, research on mitochondria has progressively broadened to encompass their symbiotic interactions with other cellular entities, particularly their relationships with host cells (1). For example, Nitric oxide interrupts mitochondrial apoptosis signaling through S-nitrosation of caspase-8, thereby preventing tumor necrosis factor-α-induced apoptosis of rat hepatocytes (2). Bay 11-7082 blocks the NF-κB pathway and promotes UCN-01-mediated mitochondrial interruption and apoptosis in human multiple myeloma cells (3). Mitochondrial symbiosis refers to the interaction between mitochondria within the cell or between mitochondria and other organelles, and can describe cooperative exchange such as mitochondrial fusion, fission, or the exchange of metabolites between mitochondria and other organelles such as the endoplasmic reticulum (4, 5). Mitochondrial symbiosis will be limited to intracellular processes and interactions within the cells of individual organisms, rather than interactions between different organisms. Endosymbiosis involves one organism living inside the cells of another, such as how mitochondria are thought to have evolved. Mitochondrial symbiosis can describe the interactions between mitochondria or their relationships with other organelles within the cell. Unlike symbiosis, which involves two different organisms, or endosymbiosis, in which one organism lives in another, mitochondrial symbiosis would be limited to intracellular processes and interactions within the cells of a single organism, rather than interactions between different organisms (6, 7).
The disruption of mitochondrial endosymbiosis refers to the breakdown of the evolutionary mutual dependence between mitochondria and the host cell, which originally allowed for the integration of free-living bacteria into a symbiotic organelle. This disruption can lead to the loss of key mitochondrial functions or extensive degradation of the mitochondrial genome, ultimately causing a significant decline in mitochondrial efficiency within the host cell (8). Mitochondria, which evolved from primitive bacteria via endosymbiosis, have transferred many of their genetic functions to the host cell’s nucleus over time. However, when this process is interrupted—whether by environmental stressors, failures in gene transfer, or reduced cellular reliance on mitochondria—it results in severe impairment of mitochondrial activity.
The interruption of mitochondrial symbiosis, particularly in chronic pathological conditions, underscores its profound effect on cellular survival and overall physiological homeostasis. Such interruption leads to a substantial reduction in intracellular energy availability, directly weakening the cell’s capacity for proliferation and differentiation, especially in energy-demanding cell types like osteoblasts and myocytes (9). Furthermore, the interruption of mitochondrial symbiosis signifies an adaptive response to fluctuations in both internal and external environments affecting biological energy metabolism. In the face of environmental stressors, cells frequently depend on the adaptive modulation of mitochondria to preserve internal stability. However, when this adaptive mechanism degradation, cells may enter a state of “energy deficiency,” further exacerbating pathological damage (10).
The interruption of mitochondrial symbiosis primarily arises from a multitude of mechanisms, including oxidative stress, mutations in mitochondrial DNA, and the depletion of mitochondrial membrane potential (11–13). The heightened production of ROS (reactive oxygen species) induced by oxidative stress can cause damage on the mitochondrial membrane, impairing its functionality and consequently diminishing ATP synthesis (14). Concurrently, mitochondrial mutations may result in the dysfunction of the electron transport chain, further exacerbating metabolic disorders within cells (15). Additionally, a reduced mitochondrial membrane potential disrupts the internal storage and release of calcium, thereby impairing the normal operation of cellular signaling pathways (16). Collectively, these factors lead to in insufficient cellular energy, heightened apoptotic signaling, and ultimately, tissue damage.
Oxidative stress and inflammation caused by aging, combined with cellular damage caused by mitochondrial dysfunction, create a vicious cycle that severely impairs bone health. As the global population ages, the prevention and management of OP (osteoporosis) have become increasingly crucial. Osteoporosis is a metabolic bone disorder characterized by a reduction in bone mass and the deterioration of the microarchitecture of bone tissue (17). Its pathogenesis is intricately linked to mitochondrial dysfunction (18). Research indicates that the decline in mitochondrial efficiency directly inhibits the activity of both osteoblasts and osteoclasts. The energy deficiency experienced by osteoblasts is closely associated with dysfunctional mitochondrial function, leading to diminished proliferation, differentiation, and a reduce in bone matrix synthesis. Moreover, ROS generated by mitochondria play a dual role in bone metabolism; while moderate levels of ROS can promote osteoblast differentiation, excessive ROS production initiates osteoclasts, thereby increasing bone resorption (19). Recent studies have provided compelling evidence that the interruption of mitochondrial symbiosis is directly connected with the onset of osteoporosis. For instance, mitochondrial function is typically impaired in individuals with osteoporosis, and mitochondrial DNA content shows a significant positive correlation with bone density (20, 21). In animal models, enhancing mitochondrial function or inhibiting ROS production has been shown to effectively improve bone mineral density, suggesting that restoring mitochondrial function may represent a viable strategy for the prevention and treatment of osteoporosis (22).
In recent years, mounting evidence has indicated that the interruption of mitochondrial symbiosis and its interactions with host cells may significantly influence the regulatory mechanisms governing bone metabolism. Particularly during the aging process, the decline in mitochondrial function is closely linked to the pathogenesis of osteoporosis. This insight underscores the importance of conducting an in-depth examination of how mitochondrial symbiosis interruption inhibits bone metabolism, not only to elucidate the pathophysiological mechanisms underlying osteoporosis but also to generate innovative strategies for its prevention and treatment. In summary, this study aims to meticulously elucidate the association between the interruption of mitochondrial symbiosis and the progression of osteoporosis, with the intent of providing new perspectives on the mechanisms of related diseases and clinical interventions. By uncovering the intricate relationship between mitochondrial function and bone metabolism, we aspire to lay the groundwork for the development of future therapeutic strategies for osteoporosis.
This paragraph primarily explores the mechanisms of intracellular signal transduction resulting from the interruption of mitochondrial symbiosis, which encompasses seven key mechanisms (Figure 1). These mechanisms include: (a) MOMP (Mitochondrial outer membrane permeability), which enhances the permeability of the mitochondrial outer membrane, thereby facilitating the release of various essential proteins, such as Cytc (cytochrome c); (b) The opening of the MPTP (mitochondrial permeability transition pore), which promotes the efflux of Ca2+ ions and activates cell signaling pathways; (c) The release of mtDNA (mitochondrial DNA) into the cytoplasm, which triggers the expression of numerous pro-inflammatory genes; (d) CL (Cardiolipin), which plays a crucial role in regulating the curvature of the mitochondrial membrane; (e) Mitochondrial metabolites, including citric acid and succinic acid, that modulate the activity of specific protein targets; (f, g) Complexes I and III, which facilitate the directed transfer of protons into the mitochondrial intermembrane space.
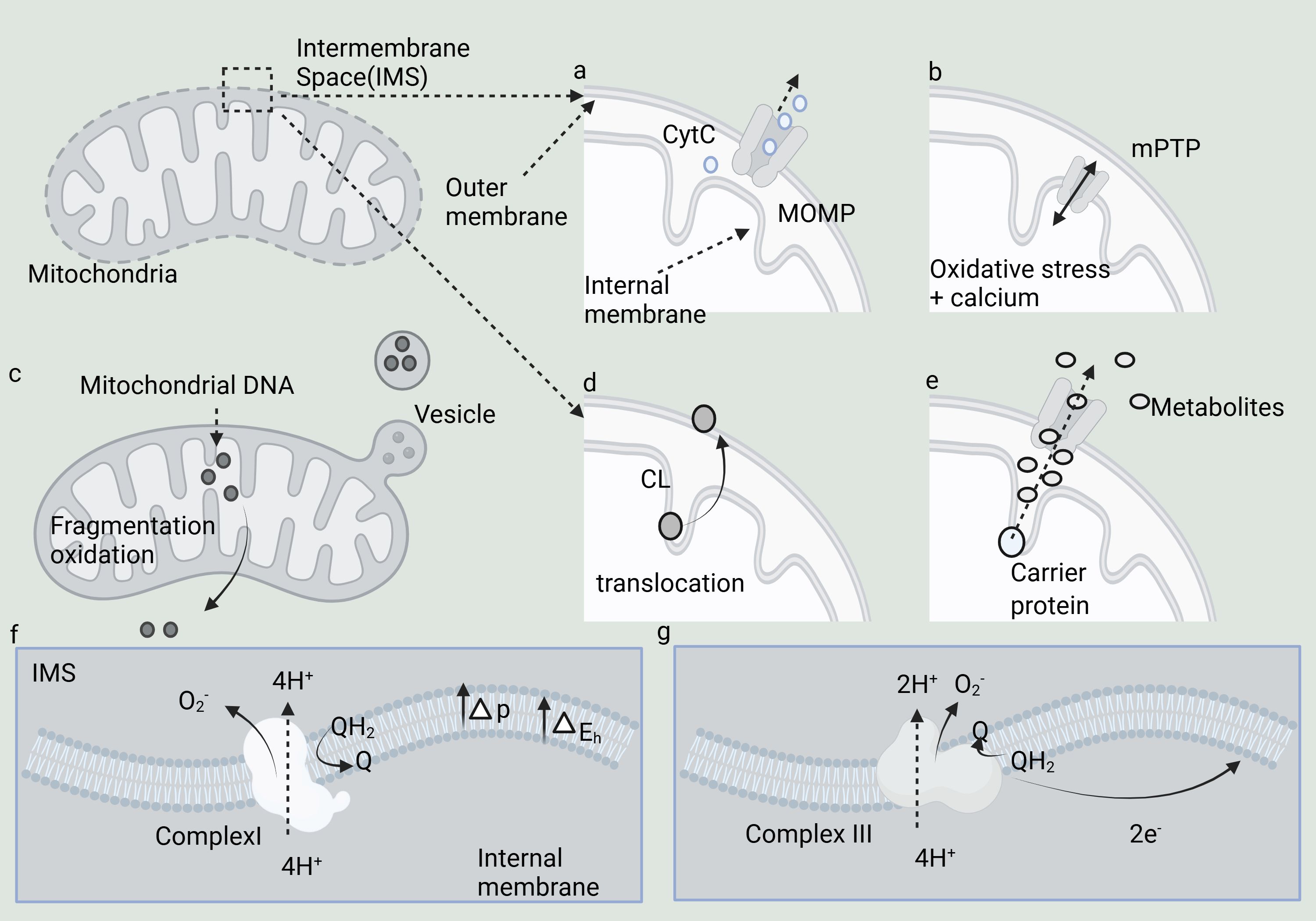
Figure 1. Mechanisms underlying the evolution of disrupted mitochondrial symbiosis and its signaling to the cytoplasm. (A–G) Seven key mechanisms.
The interruption of mitochondrial symbiosis implies that the delicate relationship between mitochondria and host cells is disrupted, triggering the activation of multiple cellular signaling mechanisms. Mitochondria are pivotal in immune signaling and programmed cell death, with their functional state directly influencing the intensity of immune responses and determining cell survival outcomes. Through various mechanisms, mitochondria can release molecules into the cytoplasm, thereby activating numerous immune signaling pathways or programmed cell death processes, highlighting the profound impact of mitochondrial “heterogeneity” on the rest of the cell (23, 24). MOMP is a critical event in apoptosis and programmed cell death (25, 26). MOMP leads to enhanced permeability of the mitochondrial outer membrane, allowing the release of various crucial proteins such as Cyt c, AIF, Smac/DIABLO, cytochrome b5, and mtDNA. These proteins play crucial roles in cell survival, apoptosis, and stress responses (27–29). The process of MOMP is controlled by the Bcl-2 protein family, which includes both pro-apoptotic proteins (like Bax and Bak) and anti-apoptotic proteins (such as Bcl-2 and Bcl-xL) (30). MOMP can be induced by both endogenous stimuli (such as DNA damage, oxidative stress) and exogenous signals (such as cytokines, drugs) (31).
The MPTP is a calcium-dependent, non-selective ion channel in the membrane that serves various functions. Despite over 50 years of research, its molecular structure remains elusive. MPTP spans both the inner and outer mitochondrial membranes, functioning as a non-specific channel for signal transduction and material transfer between the mitochondrial matrix and cytoplasm. It plays a key role in maintaining Ca2+ homeostasis, regulating oxidative stress signals, and mediating certain stimulation-induced protein translocations (32). The short-term (reversible) opening of MPTP protects cells from oxidative damage by promoting Ca2+ ion efflux from the mitochondrial matrix, facilitating cell signaling. mtDNA, which lacks CpG methylation, closely resembles bacterial DNA. When mtDNA is released into the cytoplasm, it is recognized by bacterial DNA sensors such as TLR9 and cGAS, triggering the expression of multiple pro-inflammatory genes (33–35).
The release of mtRNA (mitochondrial RNA) triggers viral sensing pathways via RIG-I-like receptors, which interact with MAVS (Mitochondrial Antiviral Signaling Protein) on the surface of the outer mitochondrial membrane. The N-formylation of mitochondrial translation polypeptides results in N-terminal N-formylated methionine residues, a hallmark of bacterial proteins that underscores the connection between mitochondria and their bacterial origin (8, 36, 37). CL plays a crucial role in regulating membrane curvature in both bacterial and mitochondrial membranes, and is vital for the function of various membrane proteins, which explains why mitochondria have retained CL. CL is found in the mitochondria of all eukaryotes and possesses a unique dimer structure, comprising two phosphatidic acid residues linked by glycerol bridges, which imparts distinct physicochemical properties (38). In the mitochondrial respiratory chain, complexes I and III are key sources of superoxide production and may play a role in mitochondrial redox signaling. Complex I (nicotinamide adenine dinucleotide, NADH) and Complex III (Ubiquinol: cytochrome c oxidoreductase; Cytochrome BC-1 complex) are thought to generate superoxide anions, which are released into the mitochondrial matrix and intermembrane space, respectively. The main role of these two complexes is to capture energy from redox reactions to facilitate the directional transfer of protons into the mitochondrial intermembrane space (39).
Mitochondrial metabolites, such as citric acid and succinic acid, function as signals by modulating the activity of protein targets or influencing DNA and histone methylation, thereby serving as potential epigenetic signals. Mitochondria orchestrate various metabolic pathways to generate metabolites essential for cell survival and proliferation. As a distinct metabolic hub, the TCA (tricarboxylic acid) cycle not only supports energy metabolism and the production of NADH to facilitate the respiratory chain but also provides metabolites with specialized signal transduction roles, such as acetyl-CoA, citrate, cis-aconitate, isocitrate, 2-oxoglutarate, succinate, and fumarate (40). The Warburg effect signifies the metabolic shift of cells from oxidative phosphorylation to glycolysis, without indicating a complete loss of mitochondrial function, as active mitochondria are still necessary in most tumor and inflammatory states.In this metabolic profile, referred to as the Warburg effect, most of the pyruvate generated from glycolysis is redirected toward lactic fermentation, circumventing mitochondrial oxidative metabolism (41).This section explores the complexities of intracellular signal transduction mechanisms precipitated by the interruption of mitochondrial symbiosis, encompassing seven pivotal processes. Each of these processes plays a crucial role in cellular responses, ranging from apoptosis to immune signaling and metabolic regulation.
In this section, we will investigate the mechanisms behind the breakdown of mitochondrial symbiosis and its connection to osteoporosis. Specifically, we will examine how the interruption of mitochondrial symbiosis affects osteoblasts, osteoclasts, and bone marrow mesenchymal stem cells, leading to a decline in bone mineral density, increased bone resorption, and slowed regeneration of bone marrow mesenchymal stem cells. These factors collectively contribute to the worsening of bone loss. This exploration underscores the significant role of mitochondrial dysfunction in influencing bone health and advancing the progression of osteoporosis.
In recent years, the focus on mitochondrial endosymbiosis has expanded beyond its traditional role in bioenergetics, revealing its profound implications in various physiological contexts, particularly in bone biology. Osteoblasts, the bone-forming cells, rely heavily on mitochondrial function for their differentiation and activity. Interruption of mitochondrial endosymbiosis can lead to significant alterations in osteoblast function, potentially contributing to pathological conditions such as osteoporosis.
AOPPs (Advanced oxidized protein products) promote the generation of reactive oxygen ROS from NOX (nicotinamide adenine dinucleotide phosphate oxidase), resulting in the depolarization of the mitochondrial membrane potential (ΔΨm). This activates the mitochondria-dependent intrinsic apoptotic pathway, leading to osteoblast apoptosis, which contributes to osteopenia and deterioration of bone microstructure. PINK1/Parkin-mediated mitophagy reduces plasma AOPP levels, inhibits AOPP-induced osteoblast apoptosis, and mitigates bone loss, bone microstructure degradation, and the decrease in BMD (bone mineral density) associated with AOPP accumulation (42). Nitric oxide, released by sodium nitroprusside, induces osteoblast apoptosis through a mitochondria-dependent cascade, resulting in mitochondrial dysfunction, increased intracellular ROS levels, and the release of Cytc from mitochondria into the cytoplasm, thereby activating caspase-3 (43). AGEs (Advanced glycation end products) promote osteoblast apoptosis and are critical in the pathogenesis of diabetic osteoporosis. Sufibin directly downregulates RAGE (receptor of advanced glycation endproducts) expression, modulates the RAGE-mediated mitochondrial pathway, and prevents AGE-induced osteoblast apoptosis (44). These findings highlight that the protective effects of mitophagy contrast sharply with the destructive impact of AOPPs on mitochondrial integrity.
MOMP enhances the permeability of the mitochondrial outer membrane, facilitating the release of Cyt c, AIF, Smac/DIABLO, cytochrome b5, and mtDNA. Hypoxia triggers apoptosis through caspase activation, which is accompanied by the release of mitochondrial cytochrome C in MC3T3-E1 osteoblasts (45). miR-181a-5p targets Runx1, a transcription factor that negatively regulates AIF-1 expression. The downregulation of Runx1 counteracts the osteogenic differentiation promoted by miR-181a-5p inhibitors, while downregulation of AIF-1 reverses the inhibition of osteogenic differentiation induced by miR-181a-5p mimics (46). The apoptosis of osteoblasts and osteocytes associated with androgen action in bone is linked to an increased Bax/Bcl-2 ratio (47).
P53-dependent MPTP opening is essential for Dex (dexamethasone)-induced osteoblast death. In MC3T3-E1 osteoblasts, Dex-induced MPTP opening is characterized by a decrease in MMP, the formation of mitochondrial complexes between CyPD and ANT-1, and the release of Cytc. The MPTP inhibitor SfA significantly inhibits Dex-induced MMP loss, Cyt c release, and subsequent MC3T3-E1 cell death (48). In osteoblasts exposed to angiotensin II, protein and RNA levels of mitochondrial catalase and MnSOD (manganese superoxide dismutase) were reduced. Angiotensin II suppresses SIRT1 protein levels, leading to hyperacetylation of FoxO3a and inhibition of catalase and MnSOD expression. This uncovers a new SIRT1-FoxO3a-MnSOD pathway involved in mitochondrial oxidative stress and mtDNA damage caused by angiotensin II in osteoblasts (49). Nicotine significantly diminishes MnSOD activity, while Sirt3 enhances it through the deacetylation of MnSOD. Mn (III) tetrakis (4-benzoic acid) porphyrin (MnTBAP, an MnSOD analog) markedly mitigates the detrimental effects of nicotine on osteoblasts. The Sirt3-MnSOD pathway has been recognized as a key regulator of mitochondrial oxidative stress and mtDNA damage caused by nicotine, with MnTBAP showing promise as a potential treatment for osteoporosis (50).
Antimycin A treatment reduces CL peroxidation, allowing actin to play a significant protective role in osteoblasts by inhibiting CL (51). TiO2 (Titanium dioxide) nanoparticles, widely utilized in various engineering and bioengineering applications, enhance superoxide anion production and disrupt antioxidant systems in human osteoblasts (52). Ras induces the expression of ERK-dependent angiogenic transcription factors HIF-1α and VEGF-A in osteoblasts exposed to superoxide activation shock waves (53). Glycolysis supplies 80% of the energy required by osteoblasts under aerobic conditions, with lactic acid being the primary glucose metabolite in bone. During osteoblast maturation, mitochondrial respiration is diminished, and Me2 directs glucose carbon into the malate-aspartate shuttle to sustain glycolysis (54). Citrate, the predominant component of vertebrate bones, plays a crucial role in normal bone development and maintenance by stabilizing precursors and inhibiting their conversion to hydroxyapatite, which results in mineral deposition with smaller size and lower crystallinity (55). WNT3A enhances aerobic glycolysis by elevating the levels of essential glycolytic enzymes, a process referred to as the Warburg effect. This metabolic regulation relies on LRP5 rather than β-catenin and is facilitated by mTORC2-AKT signaling downstream of RAC1. Blocking WNT3A-induced metabolic enzymes hinders osteoblast differentiation in vitro (56). Interruption of intra-mitochondrial symbiosis affects osteoblast function through various mechanisms, including energy metabolism dysregulation, impaired cell signaling, and increased oxidative stress, potentially exacerbating bone-related diseases such as OP. Understanding mitochondrial regulatory mechanisms in osteoblasts could illuminate the pathogenesis of bone diseases and identify potential targets for novel therapeutic strategies.
Osteoclasts are multinucleated cells primarily responsible for bone resorption, breaking down the bone matrix to release minerals, thereby maintaining the balance of bone remodeling. Impairment of mitochondrial function can disrupt the differentiation process of osteoclasts, potentially inhibiting their development and, consequently, affecting the normal process of bone remodeling. This section primarily explores the connection between the intracellular signaling pathways resulting from the interruption of mitochondrial symbiosis and their implications for osteoclasts.
NFATc1 is a crucial transcription factor involved in osteoclast differentiation and is essential for the regulation of osteoclast-specific genes. The ITAM collaborates with FcRγ and DAP12-associated immunoglobulin-like receptors to facilitate NF-κB receptor activator (RANKL) co-stimulatory signaling, subsequently activating calcium signaling via PLCγ (57). Mitochondrial calcium (Ca²+) cycling facilitates the activation of the transcription factor NFAT (58). In osteoclasts, mitochondrial ROS contribute to apoptosis (59). Dysfunction in Cyt c oxidase enhances macrophage phagocytosis and promotes osteoclast formation (60). Phosphorylation of BCL2 is involved in the autophagy and differentiation of osteoclasts from OCPs induced by RANKL. RANKL initiates autophagy in OCPs through phosphorylation of BCL2 at the S70 site, which enhances osteoclastogenesis. This suggests that targeting the inactivation of BCL2 at the S70 site in OCPs may offer a therapeutic strategy for addressing pathological bone loss (61). Curcumin blocks RANK signaling and the subsequent JNK-BCL2-Beclin1 pathway, effectively reducing RANKL-induced autophagy in OCPs (62). In Tfam cKO mice, levels of Tfam, mtDNA copy number, and ATP in osteoclasts are significantly decreased. While Tfam cKO mice are smaller than the control group, their trabecular bone volume does not change despite the lack of Tfam. Histological examination of the proximal tibia and lumbar vertebrae shows a notable decrease in the number of osteoclasts in Tfam cKO mice. Despite a pro-apoptotic tendency in Tfam cKO osteoclasts, their bone resorption activity is increased (63).
Recent studies have increasingly highlighted the pivotal role of mitochondrial dynamics in regulating cellular functions, particularly in osteoclasts. Mitochondrial fusion and the dynamic regulation of the mitochondrial network, mediated by fusion proteins such as MFN1 and MFN2, are critical for energy production, cell survival, and numerous intracellular signaling pathways, including calcium homeostasis. Knockout of MFN1 and MFN2 in osteoclast precursors of mice has been shown to increase bone mass in young female mice and prevent the differentiation of osteoclast precursors into mature osteoclasts in vitro. Notably, female mice deficient in MFN2 exhibited increased bone mass after one week and showed resistance to osteolysis induced by the receptor activator of NF-κB ligand (RANKL) at 8 weeks. Restoration of osteoclast differentiation by reintroducing intact MFN2 or defective mitochondrial autophagy variants was associated with enhanced calcium entry and normalization of NFATc1 expression, suggesting that MFN2 plays a critical role in controlling the mitochondria-endoplasmic reticulum crosstalk within osteoclasts. This underscores the essential role of MFN2 in maintaining osteoclast function and bone homeostasis (64).
Mitochondrial complex I activity reduces inflammation and promotes bone resorption by influencing the polarization of macrophages into osteoclasts (65). Estrogen deficiency speeds up bone resorption and plays a role in the onset of OP. RANKL triggers complex I activity in OCPs, enhancing OXPHOS and mitochondrial ATP production within three hours of exposure. The effects of mitochondrial bioenergetics are associated with an elevated ability to oxidize substrates from the TCA cycle, along with fatty acids and amino acids (66). Osteoclasts, which are bone-resorptive multinucleated cells derived from monocyte/macrophage lineages, play a crucial role in both normal and pathological bone turnover. Alpha-tocopherol succinate (αTP-suc) has demonstrated potent anticancer activity in vitro and in vivo (67). The Warburg phenomenon remains essential for providing a clearer understanding of metabolism and bone remodeling. Inducing the Warburg effect to promote collective glucose metabolism could enhance cell proliferation. While leveraging the energy advantages of the Warburg effect presents challenges, it appears that targeting osteocalcin induces phenotypic changes by linking WNT, Notch, AKT, and insulin signaling pathways, thereby enhancing its efficacy in osteoblast phenotypes. Osteocalcin modulates ATP utilization through the SOST (sclerostin protein) gene within the bone microenvironment. Specifically activating ATP production during the maturation of osteoblasts is an important approach to combat OP (68). In cell biology, the mitochondrial endosymbiosis theory posits that mitochondria originated from an endosymbiotic relationship between primitive eukaryotic cells and archaea. However, interruption of this endosymbiosis can severely impair cellular function. In osteoclasts, the loss of mitochondrial function diminishes their bone resorption capacity. As the cells responsible for degrading bone matrix during bone remodeling, osteoclasts rely on mitochondrial energy to sustain their activity. When mitochondrial function is compromised, osteoclast activity declines, potentially leading to bone metabolic diseases such as osteoporosis. Therefore, maintaining proper mitochondrial function is vital for bone health.
Interruption of mitochondrial endosymbiosis profoundly impacts the function and differentiation potential of BM-MSCs (bone marrow mesenchymal stem cells). BM-MSCs possess multidirectional differentiation potential, allowing them to differentiate into osteoblasts, adipocytes, and chondrocytes. Mitochondria serve not only as the core of energy metabolism in BM-MSCs but also regulate critical biological processes, including cell proliferation, differentiation, apoptosis, and autophagy. BM-MSCs mitigate hypoxia-induced mitochondrial damage and apoptosis, possibly through upregulation of Mfn2 expression in mouse trophoblast cells, which alters mitochondrial structure (69). Pinocytosis, a process vital for the removal of dead and dying cells, is essential for maintaining tissue homeostasis. However, pinocytosis impairs osteoblast differentiation, as transcriptional analysis and functional assays reveal that mitochondrial function in MSCs is downregulated following pinocytosis. Experimentally, pinocytosis induced mitochondrial fission in BM-MSCs, and pharmacological inhibition of this fission reduced cell activity and preserved osteoblast differentiation, indicating that pinocytosis-mediated mitochondrial remodeling is crucial for BM-MSC differentiation (70). Bcl-Xl is expressed at comparable levels in both undifferentiated and differentiated hMSCs, while Bcl-2 expression is limited to differentiated cells. The downregulation of Bcl-Xl in undifferentiated hMSCs leads to increased sensitivity to apoptosis, whereas the overexpression of Bcl-2 triggers apoptosis (71). BMSC (Bone marrow stromal cells)-BMP2 induces GSC apoptosis, inhibits proliferation, reduces the formation of GSC neurospheres and their diameters, and downregulates the expression of GSC markers Nanog and OCT4, indicating stemness inhibition (72). Activation of AT1R signaling stimulates BM-MSC apoptosis, linked to increased mtROS production and reduced mtDNA content (73). Mitochondrial dysfunction is a defining feature of numerous tissue injuries and the aging of stem cells. Although the tissue-repairing properties of MSC-EVs (mesenchymal stem cell-derived extracellular vesicles) are well established, the TFAM-OE (overexpression of TFAM) amplifies the protective benefits of MSC-EVs against mitochondrial damage and inflammation. Studies indicate that MSC-EVs have potential for addressing diseases associated with mitochondrial dysfunction, with TFAM signaling playing a vital role in sustaining their regenerative abilities (74). MSCs (Human mesenchymal stem cells) have the ability to transfer mitochondria to cells with significant impairments. However, this transfer has not been seen in cells with pathogenic mtDNA mutations, like A3243G mutations or 4,977 bp deletions, and is restricted to situations where mitochondrial function is nearly absent. Gaining insight into the mechanisms behind mitochondrial transfer may facilitate the development of cell therapy for mitochondrial repair or gene therapy aimed at treating diseases linked to mitochondrial dysfunction (75). MSC transplantation alleviates metabolic defects in recipient cells through intercellular mitochondrial transport (IMT). However, due to reduced mitochondrial CL content, MSC-derived osteoblasts (MSC-OBs) fail to sequester damaged mitochondria into LC3-dependent autophagy, the presumed mitochondrial autophagy receptor in MSCs. Consequently, the potential of MSC-OBs to rescue metabolic defects and prevent cell death in stress-induced epithelial cells is diminished. Small molecule screening identified PQQ (pyrroloquinoline quinone) as a regulator of mitochondrial autophagy and IMT. Long-term culture of MSC-OBs with PQQ (MSC-ObPQQ type) restored CL content, facilitated mitochondrial sequestration into autophagosomes, and activated mitochondrial autophagy (76). NAD/NADH reoxidation modifies the in vitro metabolic reconfiguration, rejuvenating aging MSCs. Niacinamide treatment increases intracellular NAD+ levels, rebalances the NAD+/NADH ratio, enhances Sirt-1 activity in high-passage hMSCs, partially restores mitochondrial function, and rejuvenates aging hMSCs. In contrast, human fibroblasts exhibit limited aging due to a relatively stable NAD+/NADH balance during expansion (77, 78). When intermitochondrial symbiosis is disrupted, the energy metabolism of BM-MSCs is compromised, leading to a significant decline in cell proliferation and differentiation. This dysfunction can impair bone tissue repair and regeneration, potentially resulting in conditions such as osteoporosis or delayed fracture healing. Therefore, maintaining mitochondrial function stability is crucial for the proper functioning of BM-MSCs and is a key factor in bone tissue engineering and regenerative medicine.
The relationship between the interruption of mitochondrial symbiosis and OP involves two primary factors: heredity and adaptation, both of which have garnered significant attention in the biomedical field. Mitochondrial dysfunction is maternally transmitted through genetic pathways, with mtDNA mutations or damage leading to disorders in energy metabolism during OXPHOS, which is closely linked to bone metabolism (79–82). Abnormal mitochondrial function directly impairs osteoblast activity and the bone mineralization process, thereby increasing the risk of OP (83). For example, the AKT-GSK3β signaling pathway regulates OPA1 cleavage associated with mitochondrial dysfunction, which may contribute to OS-induced osteoblast apoptosis (84). Calcium and phosphate supplements promote bone cell mineralization (85).The interruption of mitochondrial function triggers intracellular compensatory responses, leading to oxidative stress and prompting cells to activate protective mechanisms such as the antioxidant response and autophagy (86, 87). While these reactions may support short-term cell survival, they can result in chronic inflammation and ultimately disrupt the balance of bone metabolism over time. Mitochondrial dysfunction reduces overall energy levels, impacts the metabolic processes essential for maintaining BMD, and exacerbates the progression of OP (88, 89). The interruption of mitochondrial symbiosis is not only a direct consequence of genetic factors but also an adaptive response to environmental changes and insufficient energy supply, manifesting in two interrelated scenarios.
The interruption of mitochondrial symbiosis is intricately linked to inflammatory responses in OP, highlighting the complex interplay between bone health and the immune system. Mitochondrial dysfunction results in the excessive production of ROS within cells, leading to oxidative stress (90). Accumulation of ROS directly damages bone cells, activates inflammatory signaling pathways, and fosters a chronic inflammatory response (91). This overproduction of inflammatory mediators, such as TNF-α (tumor necrosis factor α), IL-6 (interleukin-6), and NF-κB (nuclear factor κB), stimulates osteoclast activation and proliferation, thereby accelerating bone resorption (92, 93). The breakdown of mitochondrial symbiosis influences bone cell survival by modulating autophagy and apoptosis, which in turn exacerbates the inflammatory response. interruption of intra-mitochondrial symbiosis causes mitochondria to release Cyt c, initiating apoptosis and ultimately leading to cell death. Whether there is a distinct Cyt c pool within mitochondria specifically involved in apoptosis activation, or if chemically modified Cyt c drives apoptosis, remains unresolved. The production of H2O2 (hydrogen peroxide) increases before the release of Cyt c, and both events occur prior to apoptosis. The addition of mitochondrial uncoupling agents prevents the formation of H2O2 and subsequent apoptosis, underscoring the critical role of mitochondria-derived H2O2. Capturing hydroxyl radicals derived from H2O2 reduces cell apoptosis. Cytoplasmic Cyt c originates from a single mitochondrial compartment, supporting a unified pool involved in both respiration and apoptosis, and is chemically identical to its natural state (94). Mitochondria generate hydrogen peroxide (H2O2), which leads to oxidative damage and initiates anti-parasitic and antibacterial responses, ultimately causing cell death. In cultures of primary human bone marrow cells, H2O2 stimulated the concentration-dependent activation of TRAP-positive multinucleated giant cells and expanded the pit area in osteoclast activity assays. Additionally, H2O2 promotes the expression of M-CSF (macrophage colony-stimulating factor) and RANKL, while increasing the RANKL/OPG ratio, reinforcing the idea that oxidative stress is linked to enhanced bone resorption and decreased bone mass in healthy women (95). In conclusion, the intricate relationship between mitochondrial symbiosis interruption and inflammatory responses in osteoporosis underscores the critical connection between bone health and immune function.
The interruption of mitochondrial symbiosis triggers the stimulation of RANKL and M-CSF, which may play a crucial role in human osteoclast formation. The identification of RANKL as a central regulator of osteoclast activity has opened new avenues for therapeutic intervention. Denosumab, a highly specific anti-RANKL antibody, has been shown to rapidly and significantly reduce bone resorption. Its pharmacokinetics allow for subcutaneous administration every six months, making RANKL inhibition a therapeutic approach for OP and related conditions (96). CSF1R (Colony-stimulating factor 1 receptor), also referred to as c-FMS, is a receptor tyrosine kinase that interacts with M-CSF and IL-34 as its ligands (97). M-CSF attaches to the cell surface receptor c-Fms, triggering a signaling cascade that promotes osteoclast differentiation. When TNF-responsive stromal cells are present, TNF-α boosts M-CSF gene expression. Subsequently, M-CSF activates the RANK in OCPs. Reflecting the proliferative and survival-promoting effects of M-CSF, TNF-α increases the number of OCPs when stromal cells expressing TNF receptors are present. Targeting the M-CSF signaling pathway offers a therapeutic strategy against TNF-α, as demonstrated by the complete prevention of osteolysis in mice treated with anti-c-Fms after TNF injection, with minimal impact on macrophage numbers. Therefore, M-CSF and its receptor c-Fms emerge as promising therapeutic targets in the context of inflammatory bone erosion (98).
The interruption of intra-mitochondrial symbiosis leads to the release of metabolites such as succinate and fumarate, which heighten inflammation through the SUCRN1 pathway. Succinate levels can increase 24-fold during the TCA cycle, acting as an extracellular ligand that promotes osteoclast formation by binding to specific receptors on osteoclast lineage cells. Targeting receptor activation has proven effective in inhibiting osteoclastogenesis (99). Dimethyl fumarate mitigates ROS signaling by boosting antioxidant responses, thereby suppressing osteoclast activity (100). Additionally, the interruption of mitochondrial symbiosis results in the release of mtRNA and mtDNA, which activate the MDA-5/RIG-I and cGAS-STING signaling pathways, leading to cytokine and IFN (interferon) production and triggering inflammation. The release of phosphocreatine activates ATP and NLRP3, inducing pyroptosis and the production of IL-1β and IL-18, further exacerbating inflammation. Sting deficiency has been shown to promote osteoclast to genesis and inhibit osteoblast genesis, with accompanying increases in p-38 phosphorylation in osteoclasts and reductions in β-Catenin phosphorylation in BMSC. While cGAS gene deletion does not significantly affect BMD, Sting gene deficiency has been linked to reduced bone formation in mice, presenting a novel target for OP treatment (101) (Figure 2). The breakdown of mitochondrial symbiosis is closely associated with the inflammatory response in OP. Mitochondria, being the central hubs of cellular energy metabolism, when dysfunctional, lead to elevated oxidative stress, which in turn triggers the release of inflammatory factors. In the context of OP, chronic inflammation is recognized as a key factor contributing to bone loss. The interruption of mitochondrial symbiosis results in the excessive release of inflammatory mediators, intensifying osteoclast activity, increasing bone resorption, and ultimately precipitating or exacerbating osteoporosis. Therefore, maintaining mitochondrial function and inhibiting the inflammatory response could emerge as promising strategies for the prevention and treatment of osteoporosis.
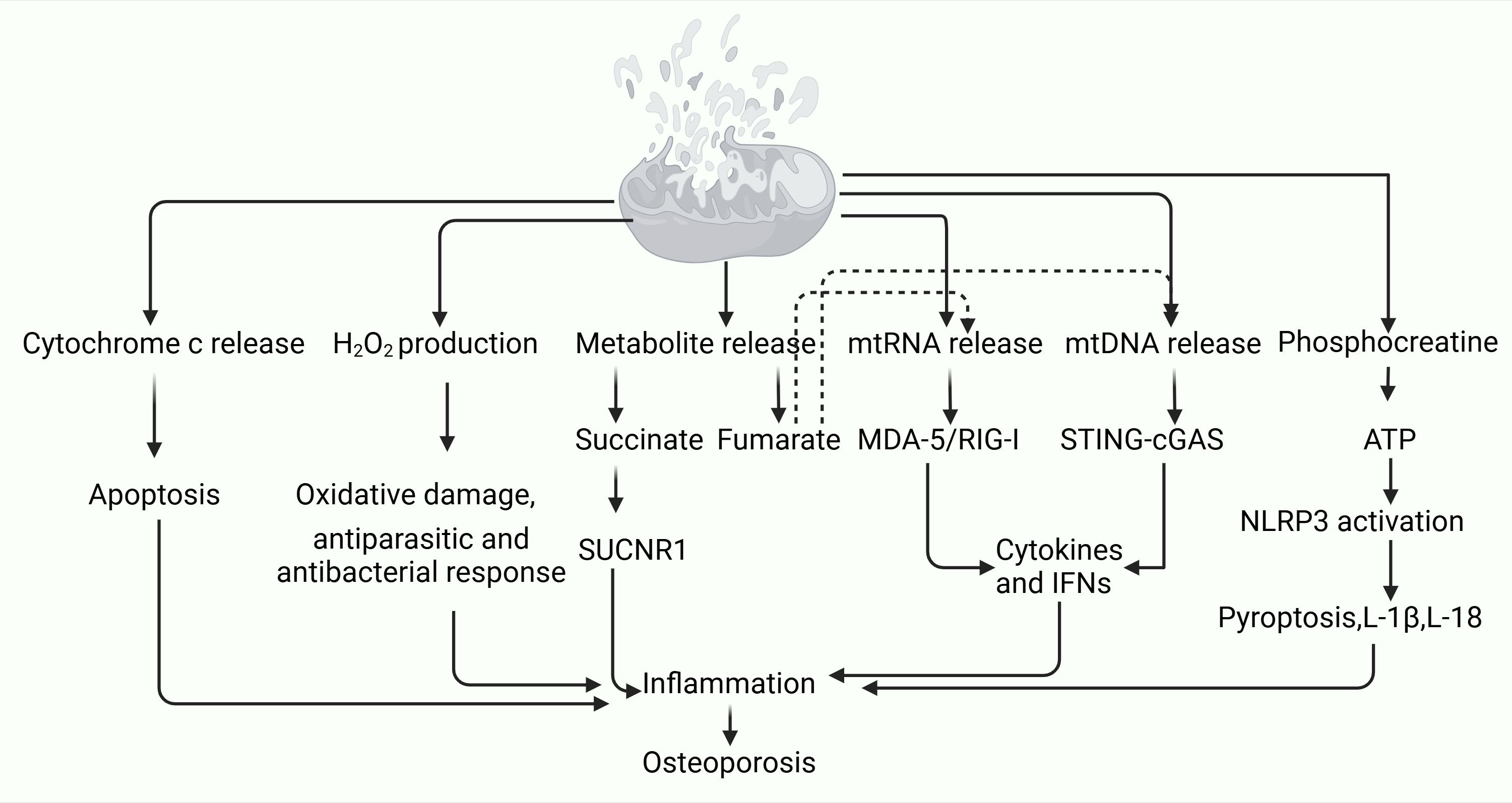
Figure 2. Breakdown of endosymbiosis mediates the mechanism by which inflammation leads to osteoporosis.
Targeting the interruption of mitochondrial endosymbiosis as a therapeutic approach for osteoporosis underscores the crucial roles of cellular energy metabolism, oxidative stress regulation, and apoptotic pathways in maintaining bone health. Osteoporosis, a metabolic bone disease marked by reduced BMD and heightened fracture risk, is closely linked to decreased bone formation, increased bone resorption, and dysfunction of bone cells. The interruption of mitochondrial symbiosis is a central factor in these pathological processes. A comprehensive understanding of the related molecular targets and the development of targeted therapies are of paramount importance for the effective treatment of OP.
Osteocyte apoptosis, triggered by various factors, plays a pivotal role in the pathogenesis of OP. Modulating bone metabolism disorders by inhibiting the apoptosis of osteoblasts and osteocytes, or by inducing apoptosis in osteoclasts, is fundamental to OP therapy. Therapeutic agents targeting mitochondrial symbiosis and disrupting apoptosis pathways include Gastrodin, Sulforaphane, Crocin, Chlorogenic acid, Luteolin, Geniposide, Curcumin, Tabersonine, Icariin, Grape seed proanthocyanidins, Naringin, and Vanillin, among others. Additionally, Table 1 provides a list of potential therapeutic drugs aimed at targeting the interruption of intramitochondrial symbiosis in OP.
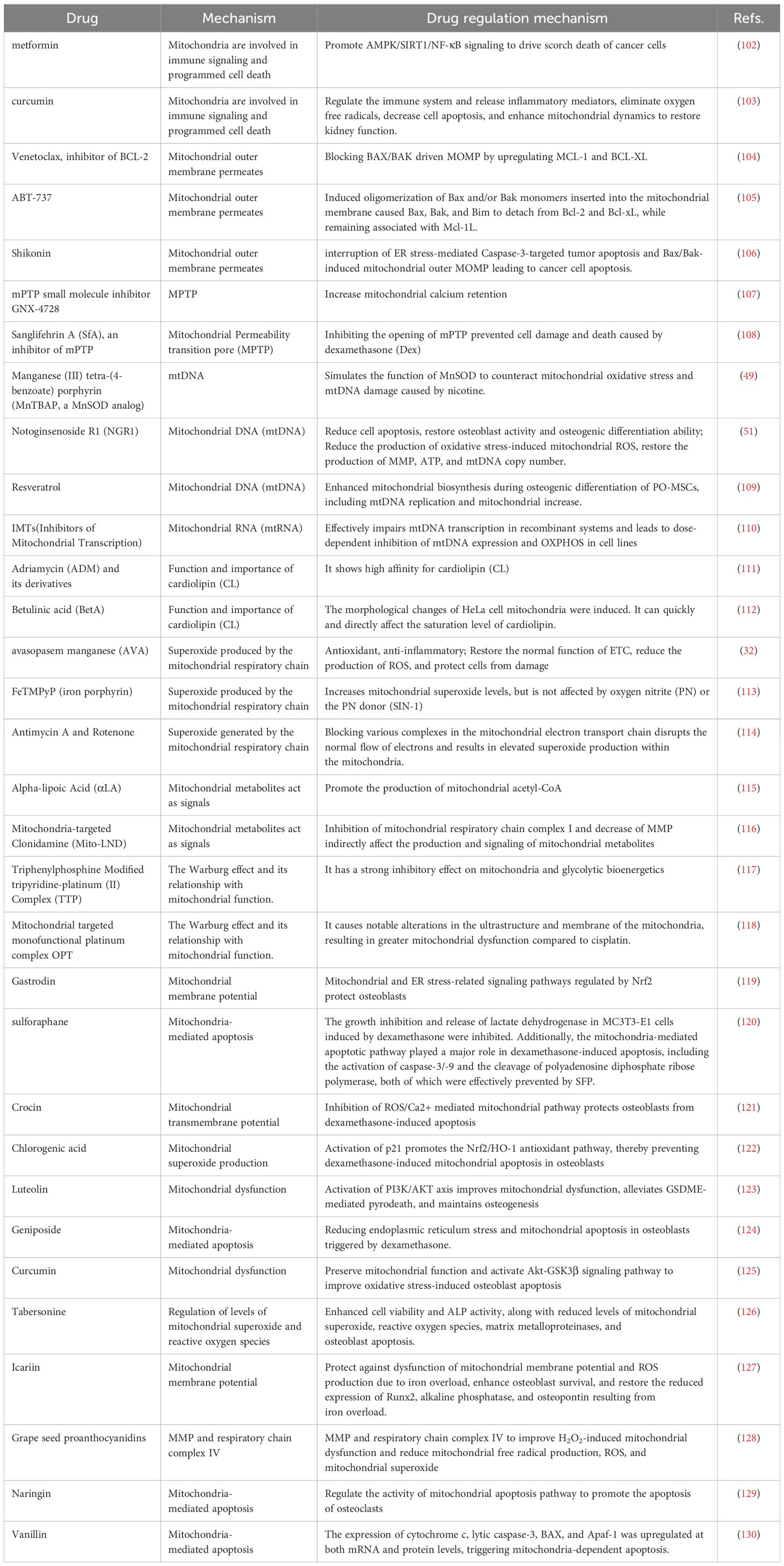
Table 1. Interruption of mitochondrial symbiosis as a potential therapeutic target and drug for osteoporosis.
The mitochondrial permeability transition, triggered by the opening of MPTP, results in a collapse of bioenergy and subsequent cell death. Edaravone has been shown to inhibit Dex-induced MPTP opening and reduce MMP in osteoblasts. However, the protective effect of Edaravone on Dex-induced osteoblast damage was mitigated when MPTP was inhibited by Cyclosporin A or cyclophiline D siRNA (131). NGR1 was found to decrease the expression of JNK and P-JNK, while simultaneously increasing MMP, mtROS, ATP levels, and mtDNA. It also promoted the levels and activities of ALP, OCN, COLI, and Runx2. NGR1 alleviates mitochondrial dysfunction induced by oxidative stress and restores osteoblast function by inhibiting the JNK signaling pathway. Resveratrol, a natural polyphenol with antioxidant, anti-aging, and anticancer properties, has also shown significant effects in enhancing the osteogenic capacity of BM-MSCs and upregulating mitochondrial biogenesis. Treatment with resveratrol (5μM) increased ALP activity and calcium deposition, indicating enhanced osteogenesis. During the osteogenic differentiation of PO-MSCs, resveratrol treatment (5μM) improved mitochondrial mass and mtDNA copy number, further promoting mitochondrial biogenesis (110).
Although the interruption of mitochondrial intersymbiosis holds potential as a therapeutic target for OP, drug development in this area remains limited. The intricate nature of mitochondrial functions presents significant challenges for the design of mitochondria-targeted therapies. These therapeutic approaches remain largely experimental, with significant challenges persisting, particularly in the development of mitochondria-targeted interventions. Among these, compounds such as metformin, resveratrol, and crocin have been investigated in mitochondrial therapy through randomized controlled trials (RCTs) and prospective studies. While the clinical relevance of metformin is well-established, including its widespread applicability, short-term exposure to clinical doses has been shown to increase skeletal muscle mitochondrial H2O2 emission and production in healthy elderly individuals (132). In contrast, the therapeutic promise of resveratrol, although supported by earlier in vitro studies in mitochondrial myopathy, has not translated into meaningful in vivo outcomes. A recent study confirmed the lack of clinical efficacy, and a cross-over RCT demonstrated no significant benefit of resveratrol in patients with mitochondrial myopathy (133).
The fully human monoclonal antibody denosumab, approved in 2010 for the treatment of osteoporosis, has demonstrated robust anti-resorptive effects, leading to significant increases in BMD and reductions in fracture risk at critical skeletal sites. However, it is now evident that transitioning to an alternative anti-osteoporosis therapy following denosumab discontinuation is crucial to mitigate the transient rebound in bone turnover. This rebound effect can cause rapid BMD loss and significantly elevate the risk of multiple vertebral fractures (MVF) (134). Denosumab-related side effects, including hypocalcemia, fracture risk, and potential drug resistance, are particularly relevant in bone diseases where RANKL signaling is a predominant driver. Emerging approaches, such as mitochondrial-targeted therapies, hold promise in this context by modulating osteoclast metabolic activity and viability through the regulation of mitochondrial dynamics, including fusion, fission, and ROS production. Although preclinical studies suggest that mitochondrial-targeted interventions can effectively restore the balance between osteoblast and osteoclast activity within the bone microenvironment, leading to significant improvements in bone density in experimental models, these strategies remain in the early stages of development. Critical gaps persist in understanding their long-term safety and efficacy, and technical hurdles in the design of mitochondria-specific drugs further complicate their clinical translation. While the experimental results are encouraging, extensive research is necessary to establish the feasibility and therapeutic value of mitochondrial-targeted therapies in clinical osteoporosis management.
This study highlights the critical association between mitochondrial dysfunction and the pathogenesis of osteoporosis. Disruption of mitochondrial homeostasis undermines the delicate balance of bone metabolism, characterized by impaired osteoblast activity and heightened osteoclast function. This imbalance results in decreased bone density, structural deterioration, and ultimately, the development of osteoporosis. Moreover, the findings underscore the pivotal roles of mitochondrial integrity, energy metabolism, and oxidative stress in the pathological mechanisms underlying osteoporosis. These insights suggest that preserving mitochondrial function could serve as a promising therapeutic strategy for preventing and managing osteoporosis. This research provides a strong theoretical foundation for the development of innovative, mitochondria-targeted therapeutic interventions aimed at maintaining bone health. Mitochondria, often referred to as the “powerhouses” of the cell, are central not only to energy metabolism but also to a wide range of cellular processes. Targeting mitochondrial dysfunction holds significant promise as a therapeutic strategy, yet numerous challenges and limitations remain. A key hurdle lies in the heterogeneity of mitochondrial dysfunction, driven by factors such as genetic variation and environmental influences, which complicates the development of universal treatment approaches. Equally concerning are the limitations inherent in current animal models and the scarcity of robust clinical trial data, which hinder the translation of laboratory findings into viable clinical applications. Recent discoveries have revealed that mitochondria exist in distinct subtypes within cells, each specializing in specific functions. Some are primarily involved in energy production under nutrient-starved conditions, while others are dedicated to biosynthetic processes. This functional specialization underscores the critical need to develop therapies capable of selectively targeting specific mitochondrial populations to maximize efficacy and minimize off-target effects. Addressing these challenges will require innovative approaches to drug design and a deeper understanding of mitochondrial biology to ensure precision in therapeutic interventions. Future studies should continue to focus on the molecular mechanism of mitochondrial intersymbiosis interruption, explore more therapeutic strategies targeting mitochondrial function, and combine the results of multi-disciplinary research to develop more accurate and efficient treatment programs. Additionally, personalized treatment plans for different patient groups also need to be further studied in order to achieve better clinical treatment results. A limitation of this study is that, while mitochondrial dysfunction is linked to the development of osteoporosis, establishing a causal relationship between the two remains challenging. It is still unclear whether mitochondrial destruction directly results in osteoporosis or if osteoporosis itself contributes to the decline in mitochondrial function, necessitating further investigation.
HZ: Writing – original draft. RZ: Writing – original draft. XW: Writing – original draft. YQ: Writing – original draft. DS: Writing – review & editing. WW: Writing – review & editing. ZS: Writing – review & editing. QL: Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study is supported by National Natural Science Foundation of China, No. 81960877; University Innovation Fund of Gansu Province, No. 2021A-076; Gansu Province Science and Technology Plan (Innovation Base and Talent Plan), No. 21JR7RA561; Natural Science Foundation of Gansu Province and No. 21JR1RA267; Education Technology Innovation Project of Gansu Province, No. 2022A-067; and Open Project of Key Laboratory of Dunhuang Medicine and Transformation of Ministry of Education, No. DHYX21-07, No. DHYX22-05, and No. DHYX21-01. This is also supported by Gansu Province Joint Fund General Program, No. 24JRRA878; Gansu Provincial Science and Technology Program Project, No. 24JRRA1020; Gansu Province Key Talent Program, No. 2025RCXM006; Teaching Research and Reform Program for Postgraduate Education at Gansu University of Traditional Chinese Medicine (GUSTCM), No. YBXM-202406; and Special Fund for Mentors of “Qihuang Talents” in the First-Level Discipline of Chinese Medicine, No. ZYXKBD-202415 and ZYXKSD-202415.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Jiang M, Wang L, Sheng H. Mitochondria in depression: The dysfunction of mitochondrial energy metabolism and quality control systems. CNS Neurosci Ther. (2024) 30:e14576. doi: 10.1111/cns.14576
2. Kim YM, Kim TH, Chung HT, Talanian RV, Yin XM, Timothy R, et al. Nitric oxide prevents tumor necrosis factor α–induced rat hepatocyte apoptosis by the interruption of mitochondrial apoptotic signaling through S-nitrosylation of caspase-8. Hepatology. (2000) 32:770–8. doi: 10.1053/jhep.2000.18291
3. Dai Y, Pei XY, Rahmani M, Conrad DH, Dent P, Gran S, et al. Interruption of the NF-κB pathway by Bay 11-7082 promotes UCN-01-mediated mitochondrial dysfunction and apoptosis in human multiple myeloma cells. Blood. (2004) 103:2761–70. doi: 10.1182/blood-2003-09-3037
4. Meyer JN, Leuthner TC, Luz AL. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology. (2017) 391:42–53. doi: 10.1016/j.tox.2017.07.019
5. Adebayo M, Singh S, Singh AP, Dasgupta S. Mitochondrial fusion and fission: The fine-tune balance for cellular homeostasis. FASEB journal: Off Publ Fed Am Societies Exp Biol. (2021) 35:e21620. doi: 10.1096/fj.202100067R
6. Zachar I, Boza G. Endosymbiosis before eukaryotes: mitochondrial establishment in protoeukaryotes. Cell Mol Life Sci. (2020) 77:3503–23. doi: 10.1007/s00018-020-03462-6
7. Song Q, Zhao F, Hou L, Miao M. Cellular interactions and evolutionary origins of endosymbiotic relationships with ciliates. ISME J. (2024) 18:1–17. doi: 10.1093/ismejo/wrae117
8. Murphy MP, O’Neill LA. A break in mitochondrial endosymbiosis as a basis for inflammatory diseases. Nature. (2024) 626:271–9. doi: 10.1038/s41586-023-06866-z
9. Iovine B, Iannella ML, Nocella F, Pricolo MR, Bevilacqua MA. Carnosine inhibits KRAS-mediated HCT116 proliferation by affecting ATP and ROS production. Cancer Lett. (2012) 315:122–8. doi: 10.1016/j.canlet.2011.07.021
10. Da W, Tao L, Zhu Y. The role of osteoclast energy metabolism in the occurrence and development of osteoporosis. Front Endocrinol. (2021) 12:675385. doi: 10.3389/fendo.2021.675385
11. Chen GH, Song CC, Pantopoulos K, Wei XL, Zheng H, Luo Z, et al. Mitochondrial oxidative stress mediated Fe-induced ferroptosis via the NRF2-ARE pathway. Free Radical Biol Med. (2022) 180:95–107. doi: 10.1016/j.freeradbiomed.2022.01.012
12. Kim M, Mahmood M, Reznik E, Gammage PA. Mitochondrial DNA is a major source of driver mutations in cancer. Trends Cancer. (2022) 8:1046–59. doi: 10.1016/j.trecan.2022.08.001
13. Hong J, Guan X, Chen Y, Tan X, Zhang S, Feng G, et al. Mitochondrial membrane potential independent near-infrared mitochondrial viscosity probes for real-time tracking mitophagy. Analytical Chem. (2023) 95:5687–94. doi: 10.1021/acs.analchem.2c05568
14. Yuan Y, Ming Z, Gong-Hua H, Lan G, Lu D, Peng L, et al. Cr (VI) induces the decrease of ATP level and the increase of apoptosis rate mediated by ROS or VDAC1 in L-02 hepatocytes. Environ Toxicol Pharmacol. (2012) 34:579–87. doi: 10.1016/j.etap.2012.06.016
15. Stepien KM, Heaton R, Rankin S, Murphy A, Bentley J, Sexton D, et al. Evidence of oxidative stress and secondary mitochondrial dysfunction in metabolic and non-metabolic disorders. J Clin Med. (2017) 6:71. doi: 10.3390/jcm6070071
16. Matuz-Mares D, González-Andrade M, Araiza-Villanueva MG, Vázquez-Meza Vilchis-LanderosHéctor MM. Mitochondrial calcium: effects of its imbalance in disease. Antioxidants. (2022) 11:801. doi: 10.3390/antiox11050801
17. Wang W, Zhang H, Sandai D, Zhao R, Bai J, Wang Y, et al. ATP-induced cell death: a novel hypothesis for osteoporosis. Front Cell Dev Biol. (2023) 11:1324213. doi: 10.3389/fcell.2023.1324213
18. Yan C, Shi Y, Yuan L, Lv D, Sun B, Wang J, et al. Mitochondrial quality control and its role in osteoporosis[. Front Endocrinol. (2023) 14:1077058. doi: 10.3389/fendo.2023.1077058
19. Jia P, Xu YJ, Zhang ZL, Li K, Li B, Zhang W, et al. Ferric ion could facilitate osteoclast differentiation and bone resorption through the production of reactive oxygen species. J Orthopaedic Res. (2012) 30:1843–52. doi: 10.1002/jor.v30.11
20. He D, Liu H, Zhao Y, Wei W, Cai Q, Shi S, et al. Assessing the interaction effects of mitochondrial DNA polymorphisms and lifestyle on heel bone mineral density. J Clin Endocrinol Metab. (2024) 110(2):dgae195. doi: 10.1210/clinem/dgae195
21. Geng X, Zhang Y, Yan J, Chu C, Gao F, Jiang Z, et al. Mitochondrial DNA mutation m. 3243A> G is associated with altered mitochondrial function in peripheral blood mononuclear cells, with heteroplasmy levels and with clinical phenotypes. Diabetic Med. (2019) 36:776–83. doi: 10.1111/dme.2019.36.issue-6
22. Ma Q, Liang M, Tang X, Luo F, Dou C. Vitamin B5 inhibit RANKL induced osteoclastogenesis and ovariectomy induced osteoporosis by scavenging ROS generation. Am J Trans Res. (2019) 11:5008.
23. Nagata S, Tanaka M. Programmed cell death and the immune system. Nat Rev Immunol. (2017) 17:333–40. doi: 10.1038/nri.2016.153
24. Zhu M, Barbas AS, Lin L, Scheuermann U, Bishawi M, Brennan TV. Mitochondria released by apoptotic cell death initiate innate immune responses. Immunohorizons. (2018) 2:384–97. doi: 10.4049/immunohorizons.1800063
25. Suhaili SH, Karimian H, Stellato M, Lee T-H, Aguilar M-I. Mitochondrial outer membrane permeabilization: a focus on the role of mitochondrial membrane structural organization. Biophys Rev. (2017) 9:443–57. doi: 10.1007/s12551-017-0308-0
26. Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. (2010) 11:621–32. doi: 10.1038/nrm2952
27. James D, Parone PA, Terradillos O, Lucken-Ardjomande S, Montessuit S, Martinou JC. Mechanisms of mitochondrial outer membrane permeabilization. Paper presented at Mitochondrial Biology: New Perspectives: Novartis Foundation Symposium. (2007) 287:170–6. doi: 10.1002/9780470725207.Ch12
28. Riley JS, Quarato G, Cloix C, Lopez J, O'Prey J, Pearson M, et al. Mitochondrial inner membrane permeabilisation enables mt DNA release during apoptosis. EMBO J. (2018) 37:e99238. doi: 10.15252/embj.201899238
29. Ripple MO, Abajian M, Springett R. Cytochrome c is rapidly reduced in the cytosol after mitochondrial outer membrane permeabilization. Apoptosis. (2010) 15:563–73. doi: 10.1007/s10495-010-0455-2
30. Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. (2008) 18:157–64.
31. Ichim G, Lopez J, Ahmed SU, Muthalagu N, Giampazolias E, Delgado ME, et al. Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol Cell. (2015) 57:860–72. doi: 10.1016/j.molcel.2015.01.018
32. Yang Y, He P-Y, Zhang Y, Li N. Natural products targeting the mitochondria in cancers. Molecules. (2020) 26:92. doi: 10.3390/molecules26010092
33. Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. (2010) 141:280–9. doi: 10.1016/j.cell.2010.02.026
34. Kim J, Kim H-S, Chung JH. Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp Mol Med. (2023) 55:510–9. doi: 10.1038/s12276-023-00965-7
35. Wu G, Zhu Q, Zeng J, Gu X, Miao Y, Xu W, et al. Extracellular mitochondrial DNA promote NLRP3 inflammasome activation and induce acute lung injury through TLR9 and NF-κB. J Thorac Dis. (2019) 11:4816. doi: 10.21037/jtd.2019.10.26
36. Castanier C, Garcin D, Vazquez A, Arnoult D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. (2010) 11:133–8. doi: 10.1038/embor.2009.258
37. Li S, Kuang M, Chen L, Li Y, Liu S, Du H, et al. The mitochondrial protein ERAL1 suppresses RNA virus infection by facilitating RIG-I-like receptor signaling. Cell Rep. (2021) 34:1–15. doi: 10.1016/j.celrep.2020.108631
38. Ren M, Phoon CK, Schlame M. Metabolism and function of mitochondrial cardiolipin. Prog Lipid Res. (2014) 55:1–16. doi: 10.1016/j.plipres.2014.04.001
39. Dröse S, Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Mitochondrial Oxid phosphorylation: nuclear-encoded genes Enzyme regulation pathophysiology. (2012) 748:145–69.
40. Frezza C. Mitochondrial metabolites: undercover signalling molecules. Interface Focus. (2016) 7:20160100. doi: 10.1098/rsfs.2016.0100
41. Lu J, Tan M, Cai Q. The Warburg effect in tumor progression: mitochondrial oxidative metabolism as an anti-metastasis mechanism. Cancer Lett. (2015) 356:156–64. doi: 10.1016/j.canlet.2014.04.001
42. Li W, Jiang W-S, Su Y-R, Tu K-W, Zou L, Liao C-R, et al. PINK1/Parkin-mediated mitophagy inhibits osteoblast apoptosis induced by advanced oxidation protein products. Cell Death Dis. (2023) 14:88. doi: 10.1038/s41419-023-05595-5
43. Ho WP, Chen TL, Chiu WT, Tai YT, Chen RM. Nitric oxide induces osteoblast apoptosis through a mitochondria-dependent pathway. Ann New York Acad Sci. (2005) 1042:460–70. doi: 10.1196/annals.1338.039
44. Mao Y, Cai W, Sun X, Dai P, Li X, Wang Q, et al. RAGE-dependent mitochondria pathway: a novel target of silibinin against apoptosis of osteoblastic cells induced by advanced glycation end products. Cell Death Dis. (2018) 9:674. doi: 10.1038/s41419-018-0718-3
45. Chae H-J, Kim S-C, Han K-S, Chae S-W, An N-H, Kim H-M, et al. Hypoxia induces apoptosis by caspase activation accompanying cytochrome C release from mitochondria in MC3T3E1 osteoblasts. p38 MAPK is related in hypoxia-induced apoptosis. Immunopharmacol immunotoxicology. (2001) 23:133–52. doi: 10.1081/IPH-100103855
46. Liu J, Chang X, Dong D. MicroRNA-181a-5p curbs osteogenic differentiation and bone formation partially through impairing Runx1-dependent inhibition of AIF-1 transcription. Endocrinol Metab. (2023) 38:156–73. doi: 10.3803/EnM.2022.1516
47. Wiren KM, Toombs AR, Semirale AA, Zhang X. Osteoblast and osteocyte apoptosis associated with androgen action in bone: requirement of increased Bax/Bcl-2 ratio. Bone. (2006) 38:637–51. doi: 10.1016/j.bone.2005.10.029
48. Zhen YF, Wang GD, Zhu LQ, Tan SP, Zhang FY, Zhou XZ, et al. P53 dependent mitochondrial permeability transition pore opening is required for dexamethasone-induced death of osteoblasts. J Cell Physiol. (2014) 229:1475–83. doi: 10.1002/jcp.v229.10
49. Li Y, Shen G, Yu C, Li G, Shen J, Gong J, et al. Angiotensin II induces mitochondrial oxidative stress and mtDNA damage in osteoblasts by inhibiting SIRT1–FoxO3a–MnSOD pathway. Biochem Biophys Res Commun. (2014) 455:113–8. doi: 10.1016/j.bbrc.2014.10.123
50. Li Y, Yu C, Shen G, Li G, Shen J, Xu Y, et al. Sirt3–MnSOD axis represses nicotine-induced mitochondrial oxidative stress and mtDNA damage in osteoblasts. Acta Biochim Biophys Sin. (2015) 47:306–12. doi: 10.1093/abbs/gmv013
51. Lee YS, Choi EM. Actein isolated from black cohosh promotes the function of osteoblastic MC3T3-E1 cells. J medicinal Food. (2014) 17:414–23. doi: 10.1089/jmf.2013.2841
52. Niska K, Pyszka K, Tukaj C, Wozniak M, Radomski MW, Inkielewicz-Stepniak I. Titanium dioxide nanoparticles enhance production of superoxide anion and alter the antioxidant system in human osteoblast cells. Int J nanomedicine. (2015) 10:1095–107.
53. Wang F-S, Wang C-J, Chen Y-J, Chang P-R, Huang Y-T, Sun Y-C, et al. Ras induction of superoxide activates ERK-dependent angiogenic transcription factor HIF-1α and VEGF-A expression in shock wave-stimulated osteoblasts. J Biol Chem. (2004) 279:10331–7. doi: 10.1074/jbc.M308013200
54. Lee W-C, Ji X, Nissim I, Long F. Malic enzyme couples mitochondria with aerobic glycolysis in osteoblasts. Cell Rep. (2020) 32:1–10. doi: 10.1016/j.celrep.2020.108108
55. Wu X, Dai H, Yu S, Zhao Y, Long Y, Li W, et al. Citrate regulates extracellular matrix mineralization during osteoblast differentiation in vitro. J Inorganic Biochem. (2021) 214:111269. doi: 10.1016/j.jinorgbio.2020.111269
56. Esen E, Chen J, Karner CM, Okunade AL, Patterson BW, Long F. WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab. (2013) 17:745–55. doi: 10.1016/j.cmet.2013.03.017
57. Shinohara M, Takayanagi H. Novel osteoclast signaling mechanisms. Curr osteoporosis Rep. (2007) 5:67–72. doi: 10.1007/s11914-007-0005-1
58. Kim M-S, Usachev YM. Mitochondrial Ca2+ cycling facilitates activation of the transcription factor NFAT in sensory neurons. J Neurosci. (2009) 29:12101–14. doi: 10.1523/JNEUROSCI.3384-09.2009
59. Arnett TR, Orriss IR. Metabolic properties of the osteoclast. Bone. (2018) 115:25–30. doi: 10.1016/j.bone.2017.12.021
60. Miyazaki T, Neff L, Tanaka S, Horne WC, Baron R. Regulation of cytochrome c oxidase activity by c-Src in osteoclasts. J Cell Biol. (2003) 160:709. doi: 10.1083/jcb.200209098
61. Ke D, Yu Y, Li C, Han J, Xu J. Phosphorylation of BCL2 at the Ser70 site mediates RANKL-induced osteoclast precursor autophagy and osteoclastogenesis. Mol Med. (2022) 28:22. doi: 10.1186/s10020-022-00449-w
62. Ke D, Xu H, Han J, Dai H, Wang X, Luo J, et al. Curcumin suppresses RANKL-induced osteoclast precursor autophagy in osteoclastogenesis by inhibiting RANK signaling and downstream JNK-BCL2-Beclin1 pathway. Biomed J. (2024) 47:100605. doi: 10.1016/j.bj.2023.100605
63. Miyazaki T, Mori S, Shigemoto K, Larsson N-G, Nakamura T, Kato S, et al. Maintenance of mitochondrial DNA copy number is essential for osteoclast survival. Arthritis Res Ther. (2012) 14:1–54. doi: 10.1186/ar3650
64. Ballard A, Zeng R, Zarei A, Shao C, Cox L, Yan H, et al. The tethering function of mitofusin2 controls osteoclast differentiation by modulating the Ca2+–NFATc1 axis. J Biol Chem. (2020) 295:6629–40. doi: 10.1074/jbc.RA119.012023
65. Jin Z, Wei W, Yang M, Du Y, Wan Y. Mitochondrial complex I activity suppresses inflammation and enhances bone resorption by shifting macrophage-osteoclast polarization. Cell Metab. (2014) 20:483–98. doi: 10.1016/j.cmet.2014.07.011
66. Marques-Carvalho A, Silva B, Pereira FB, Kim HN, Almeida M, Sardão VA. Oestradiol and osteoclast differentiation: Effects on p53 and mitochondrial metabolism. Eur J Clin Invest. (2024) 54(6):e14195. doi: 10.1111/eci.14195
67. Kim H-N, Lee J-H, Jin WJ, Lee ZH. [amp]]alpha;-Tocopheryl succinate inhibits osteoclast formation by suppressing receptor activator of nuclear factor-kappaB ligand (RANKL) expression and bone resorption. J Bone Metab. (2012) 19:111–20. doi: 10.11005/jbm.2012.19.2.111
68. Vadivalagan C, Krishnan A, Chen S-J, Hseu Y-C, Muthu S, Dhar R, et al. The Warburg effect in osteoporosis: Cellular signaling and epigenetic regulation of energy metabolic events to targeting the osteocalcin for phenotypic alteration. Cell Signalling. (2022) 100:110488. doi: 10.1016/j.cellsig.2022.110488
69. Wang L, Xu X, Kang L, Xiang W. Bone marrow mesenchymal stem cells attenuate mitochondria damage induced by hypoxia in mouse trophoblasts. PloS One. (2016) 11:e0153729. doi: 10.1371/journal.pone.0153729
70. Quarato ER, Salama NA, Li AJ, Smith CO, Zhang J, Kawano Y, et al. Efferocytosis by bone marrow mesenchymal stromal cells disrupts osteoblastic differentiation via mitochondrial remodeling. Cell Death Dis. (2023) 14:428. doi: 10.1038/s41419-023-05931-9
71. Oliver L, Hue E, Rossignol J, Bougras G, Hulin P, Naveilhan P, et al. Distinct roles of Bcl-2 and Bcl-Xl in the apoptosis of human bone marrow mesenchymal stem cells during differentiation. PloS One. (2011) 6:e19820. doi: 10.1371/journal.pone.0019820
72. Feng J, Yao Z, Yang H, Ma J, Zhong X, Qin Y, et al. Bone marrow-derived mesenchymal stem cells expressing BMP2 suppress glioma stem cell growth and stemness through Bcl-2/Bax signaling. J Cancer Res Ther. (2022) 18:2033–40. doi: 10.4103/jcrt.jcrt_1983_21
73. Zhang F, Dong Z, Gao S, Chen G, Liu D. AT1R-Mediated Apoptosis of Bone Marrow Mesenchymal Stem Cells Is Associated with mtROS Production and mtDNA Reduction. Oxid Med Cell Longevity. (2019) 2019:4608165. doi: 10.1155/2019/4608165
74. Zhao M, Liu S, Wang C, Wang Y, Wan M, Liu F, et al. Mesenchymal stem cell-derived extracellular vesicles attenuate mitochondrial damage and inflammation by stabilizing mitochondrial DNA. ACS nano. (2020) 15:1519–38. doi: 10.1021/acsnano.0c08947
75. Cho YM, Kim JH, Kim M, Park SJ, Koh SH, Ahn HS, et al. Mesenchymal stem cells transfer mitochondria to the cells with virtually no mitochondrial function but not with pathogenic mtDNA mutations. PloS One. (2012) 7:e32778. doi: 10.1371/journal.pone.0032778
76. Sagar S, Faizan MI, Chaudhary N, Gheware A, Sharma K, Azmi I, et al. Obesity impairs therapeutic efficacy of mesenchymal stem cells by inhibiting cardiolipin-dependent mitophagy and intercellular mitochondrial transfer in mouse models of allergic airway inflammation. BioRxiv. (2021). doi: 10.1101/2021.11.27.470183
77. Ohkouchi S, Block GJ, Katsha AM, Kanehira M, Ebina M, Kikuchi T, et al. Mesenchymal stromal cells protect cancer cells from ROS-induced apoptosis and enhance the Warburg effect by secreting STC1. Mol Ther. (2012) 20:417–23. doi: 10.1038/mt.2011.259
78. Yuan X, Liu Y, Bijonowski BM, Tsai A-C, Fu Q, Logan TM, et al. NAD+/NADH redox alterations reconfigure metabolism and rejuvenate senescent human mesenchymal stem cells in vitro. Commun Biol. (2020) 3:774. doi: 10.1038/s42003-020-01514-y
79. Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E. Mitochondria, oxidative stress and neurodegeneration. J neurological Sci. (2012) 322:254–62. doi: 10.1016/j.jns.2012.05.030
80. Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, et al. Mitochondrial diseases. Nat Rev Dis Primers. (2016) 2:1–22. doi: 10.1038/nrdp.2016.80
81. Jiang Z, Shen H. Mitochondria: emerging therapeutic strategies for oocyte rescue. Reprod Sci. (2022) 29:711–22. doi: 10.1007/s43032-021-00523-4
82. Liu Z, Tian J, Peng F, Wang J. Hypermethylation of mitochondrial DNA facilitates bone metastasis of renal cell carcinoma. J Cancer. (2022) 13:304. doi: 10.7150/jca.62278
83. Liu J, Gao Z, Liu X. Mitochondrial dysfunction and therapeutic perspectives in osteoporosis. Front Endocrinol. (2024) 15:1325317. doi: 10.3389/fendo.2024.1325317
84. Cai WJ, Chen Y, Shi LX, Cheng HR, Banda I, Ji YH, et al. AKT-GSK3β signaling pathway regulates mitochondrial dysfunction-associated OPA1 cleavage contributing to osteoblast apoptosis: Preventative effects of hydroxytyrosol. Oxid Med Cell Longevity. (2019) 2019:4101738. doi: 10.1155/2019/4101738
85. Chang YL, Stanford CM, Keller JC. Calcium and phosphate supplementation promotes bone cell mineralization: Implications for hydroxyapatite (HA)-enhanced bone formation. J Biomed Materials Research: Off J Soc Biomaterials Japanese Soc Biomaterials Aust Soc Biomaterials Korean Soc Biomaterials. (2000) 52:270–8. doi: 10.1002/1097-4636(200011)52:2<270::AID-JBM5>3.0.CO;2-1
86. Jurcau A. Insights into the pathogenesis of neurodegenerative diseases: Focus on mitochondrial dysfunction and oxidative stress. Int J Mol Sci. (2021) 22:11847. doi: 10.3390/ijms222111847
87. Wohlgemuth SE, Calvani R, Marzetti E. The interplay between autophagy and mitochondrial dysfunction in oxidative stress-induced cardiac aging and pathology. J Mol Cell Cardiol. (2014) 71:62–70. doi: 10.1016/j.yjmcc.2014.03.007
88. Dobson PF. The effects of mitochondrial dysfunction on bone density, osteoblasts and osteoclasts. (PhD Thesis). Newcastle University (2018).
89. Ling W, Krager K, Richardson KK, Warren AD, Ponte F, Aykin-Burns N, et al. Mitochondrial Sirt3 contributes to the bone loss caused by aging or estrogen deficiency. JCI Insight. (2021) 6:1–18. doi: 10.1172/jci.insight.146728
90. Murphy MP. Mitochondrial dysfunction indirectly elevates ROS production by the endoplasmic reticulum. Cell Metab. (2013) 18:145–6. doi: 10.1016/j.cmet.2013.07.006
91. Zhang Y, Dong F, Wang Z, Xu B, Zhang T, Wang Q, et al. Fluoride exposure provokes mitochondria-mediated apoptosis and increases mitophagy in osteocytes via increasing ROS production. Biol Trace Element Res. (2023) 201:3994–4007. doi: 10.1007/s12011-022-03450-w
92. Wu Y, Yang Y, Wang L, Chen Y, Han X, Sun L, et al. Effect of Bifidobacterium on osteoclasts: TNF-α/NF-κB inflammatory signal pathway-mediated mechanism. Front Endocrinol. (2023) 14:1109296. doi: 10.3389/fendo.2023.1109296
93. Yao Z, Getting SJ, Locke IC. Regulation of TNF-induced osteoclast differentiation. Cells. (2021) 11:132. doi: 10.3390/cells11010132
94. Oursler MJ, Bradley EW, Elfering SL, Giulivi C. Native, not nitrated, cytochrome c and mitochondria-derived hydrogen peroxide drive osteoclast apoptosis. Am J Physiology-Cell Physiol. (2005) 288:C156–68. doi: 10.1152/ajpcell.00092.2004
95. Baek KH, Oh KW, Lee WY, Lee SS, Kim MK, Kwon HS, et al. Association of oxidative stress with postmenopausal osteoporosis and the effects of hydrogen peroxide on osteoclast formation in human bone marrow cell cultures. Calcified Tissue Int. (2010) 87:226–35. doi: 10.1007/s00223-010-9393-9
96. McClung M. Role of RANKL inhibition in osteoporosis. Arthritis Res Ther. (2007) 9:1–6. doi: 10.1186/ar2167
97. Mun SH, Park PSU, Park-Min K-H. The M-CSF receptor in osteoclasts and beyond. Exp Mol Med. (2020) 52:1239–54. doi: 10.1038/s12276-020-0484-z
98. Ross FP. M-CSF, c-Fms, and signaling in osteoclasts and their precursors. Ann New York Acad Sci. (2006) 1068:110–6. doi: 10.1196/annals.1346.014
99. Guo Y, Xie C, Li X, Yang J, Yu T, Zhang R, et al. Succinate and its G-protein-coupled receptor stimulates osteoclastogenesis. Nat Commun. (2017) 8:15621. doi: 10.1038/ncomms15621
100. Yamaguchi Y, Kanzaki H, Katsumata Y, Itohiya K, Fukaya S, Miyamoto Y, et al. Dimethyl fumarate inhibits osteoclasts via attenuation of reactive oxygen species signalling by augmented antioxidation. J Cell Mol Med. (2018) 22:1138–47. doi: 10.1111/jcmm.2018.22.issue-2
101. Liu L, Tong X, Huang M, Zhu C, Chen X, Yuan Y, et al. The effects of cGAS-Sting pathway on bone mineral density. (2023). doi: 10.21203/rs.3.rs-3404007/v1
102. Zheng Z, Bian Y, Zhang Y, Ren G, Li G. Metformin activates AMPK/SIRT1/NF-κB pathway and induces mitochondrial dysfunction to drive caspase3/GSDME-mediated cancer cell pyroptosis. Cell Cycle. (2020) 19:1089–104. doi: 10.1080/15384101.2020.1743911
103. Cai Y, Huang C, Zhou M, Xu S, Xie Y, Gao S, et al. Role of curcumin in the treatment of acute kidney injury: research challenges and opportunities. Phytomedicine. (2022) 104:154306. doi: 10.1016/j.phymed.2022.154306
104. Zhou D, Ran Y, Yu R, Liu G, Ran D, Liu Z. SIRT1 regulates osteoblast senescence through SOD2 acetylation and mitochondrial dysfunction in the progression of Osteoporosis caused by Cadmium exposure. Chemico-Biological Interact. (2023) 382:110632. doi: 10.1016/j.cbi.2023.110632
105. Milnerowicz S, Maszewska J, Skowera P, Stelmach M, Lejman M. AML under the scope: current strategies and treatment involving FLT3 inhibitors and venetoclax-based regimens. Int J Mol Sci. (2023) 24:15849. doi: 10.3390/ijms242115849
106. Buron N, Porceddu M, Brabant M, Desgué D, Racoeur C, Lassalle M, et al. Use of human cancer cell lines mitochondria to explore the mechanisms of BH3 peptides and ABT-737-induced mitochondrial membrane permeabilization. PloS One. (2010) 5:e9924. doi: 10.1371/journal.pone.0009924
107. Ma X, Yu M, Hao C, Yang W. Shikonin induces tumor apoptosis in glioma cells via endoplasmic reticulum stress, and Bax/Bak mediated mitochondrial outer membrane permeability. J Ethnopharmacology. (2020) 263:113059. doi: 10.1016/j.jep.2020.113059
108. Fang J, Chavez-Valdez R, Flock DL, Avaritt O, Saraswati M, Robertson C, et al. An inhibitor of the mitochondrial permeability transition pore lacks therapeutic efficacy following neonatal hypoxia ischemia in mice. Neuroscience. (2019) 406:202–11. doi: 10.1016/j.neuroscience.2019.02.030
109. Li X, Lin H, Zhang X, Jaspers RT, Yu Q, Ji Y, et al. Notoginsenoside R1 attenuates oxidative stress-induced osteoblast dysfunction through JNK signalling pathway. J Cell Mol Med. (2021) 25:11278–89. doi: 10.1111/jcmm.v25.24
110. Moon DK, Kim BG, Lee AR, In Choe Y, Khan I, Moon KM, et al. Resveratrol can enhance osteogenic differentiation and mitochondrial biogenesis from human periosteum-derived mesenchymal stem cells. J Orthopaedic Surg Res. (2020) 15:1–10. doi: 10.1186/s13018-020-01684-9
111. Bonekamp NA, Peter B, Hillen HS, Felser A, Bergbrede T, Choidas A, et al. Small-molecule inhibitors of human mitochondrial DNA transcription. Nature. (2020) 588:712–6. doi: 10.1038/s41586-020-03048-z
112. Praet M, Defrise-Quertain F, Ruysschaert JM. Comparison of adriamycin and derivatives uptake into large unilamellar lipid vesicles in response to a membrane potential. Biochim Biophys Acta (BBA)-Biomembranes. (1993) 1148:342–50. doi: 10.1016/0005-2736(93)90148-S
113. Mapuskar KA, Pulliam CF, Tomanek-Chalkley A, Rastogi P, Wen H, Dayal S, et al. The antioxidant and anti-inflammatory activities of avasopasem manganese in age-associated, cisplatin-induced renal injury. Redox Biol. (2024) 70:103022. doi: 10.1016/j.redox.2023.103022
114. Albuck AL, Sakamuri SS, Sperling JA, Evans WR, Kolli L, Sure VN, et al. Peroxynitrite decomposition catalyst enhances respiratory function in isolated brain mitochondria. Am J Physiology-Heart Circulatory Physiol. (2021) 320:H630–41. doi: 10.1152/ajpheart.00389.2020
115. Kim HY, Chung JM, Chung K. Increased production of mitochondrial superoxide in the spinal cord induces pain behaviors in mice: the effect of mitochondrial electron transport complex inhibitors. Neurosci Lett. (2008) 447:87–91. doi: 10.1016/j.neulet.2008.09.041
116. Stoner MW, Thapa D, Zhang M, Gibson GA, Calderon MJ, St. Croix CM, et al. [amp]]alpha;-Lipoic acid promotes α-tubulin hyperacetylation and blocks the turnover of mitochondria through mitophagy. Biochem J. (2016) 473:1821–30. doi: 10.1042/BCJ20160281
117. Guo T, Wu C, Zhou L, Zhang J, Wang W, Shen Y, et al. Preclinical evaluation of Mito-LND, a targeting mitochondrial metabolism inhibitor, for glioblastoma treatment. J Trans Med. (2023) 21:532. doi: 10.1186/s12967-023-04332-y
118. Wang K, Zhu C, He Y, Zhang Z, Zhou W, Muhammad N, et al. Restraining cancer cells by dual metabolic inhibition with a mitochondrion-targeted platinum (II) complex. Angewandte Chemie Int Edition. (2019) 58:4638–43. doi: 10.1002/anie.201900387
119. Liu S, Zhou L, Yang L, Mu S, Fang T, Fu Q. Gastrodin alleviates glucocorticoid induced osteoporosis in rats via activating the Nrf2 signaling pathways. Oncotarget. (2018) 9:11528. doi: 10.18632/oncotarget.23936
120. Lin H, Wei B, Li G, Zheng J, Sun J, Chu J, et al. Sulforaphane reverses glucocorticoid-induced apoptosis in osteoblastic cells through regulation of the Nrf2 pathway. Drug design Dev Ther. (2014) 8:973–82. doi: 10.2147/DDDT.S65410
121. Nie Z, Deng S, Zhang L, Chen S, Lu Q, Peng H. Crocin protects against dexamethasone−induced osteoblast apoptosis by inhibiting the ROS/Ca2+−mediated mitochondrial pathway. Mol Med Rep. (2019) 20:401–8. doi: 10.3892/mmr.2019.10267
122. Han D, Gu X, Gao J, Wang Z, Liu G, Barkema HW, et al. Chlorogenic acid promotes the Nrf2/HO-1 anti-oxidative pathway by activating p21Waf1/Cip1 to resist dexamethasone-induced apoptosis in osteoblastic cells. Free Radical Biol Med. (2019) 137:1–12. doi: 10.1016/j.freeradbiomed.2019.04.014
123. Chai S, Yang Y, Wei L, Cao Y, Ma J, Zheng X, et al. Luteolin rescues postmenopausal osteoporosis elicited by OVX through alleviating osteoblast pyroptosis via activating PI3K-AKT signaling. Phytomedicine. (2024) 128:155516. doi: 10.1016/j.phymed.2024.155516
124. Xiao Y, Ren Q, Zheng Y, Zhang S, Ouyang J, Jiao L, et al. Geniposide ameliorated dexamethasone-induced endoplasmic reticulum stress and mitochondrial apoptosis in osteoblasts. J Ethnopharmacology. (2022) 291:115154. doi: 10.1016/j.jep.2022.115154
125. Dai P, Mao Y, Sun X, Li X, Muhammad I, Gu W, et al. Attenuation of oxidative stress-induced osteoblast apoptosis by curcumin is associated with preservation of mitochondrial functions and increased Akt-GSK3β signaling. Cell Physiol Biochem. (2017) 41:661–77. doi: 10.1159/000457945
126. Sun X, Gan L, Li N, Sun S, Li N. Tabersonine ameliorates osteoblast apoptosis in rats with dexamethasone-induced osteoporosis by regulating the Nrf2/ROS/Bax signalling pathway. AMB Express. (2020) 10:165. doi: 10.1186/s13568-020-01098-0
127. Jing X, Du T, Chen K, Guo J, Xiang W, Yao X, et al. Icariin protects against iron overload-induced bone loss via suppressing oxidative stress. J Cell Physiol. (2019) 234:10123–37. doi: 10.1002/jcp.v234.7
128. Zhang Z, Zheng L, Zhao Z, Shi J, Wang X, Huang J. Grape seed proanthocyanidins inhibit H2O2-induced osteoblastic MC3T3-E1 cell apoptosis via ameliorating H2O2-induced mitochondrial dysfunction. J toxicological Sci. (2014) 39:803–13. doi: 10.2131/jts.39.803
129. Li F, Sun X, Ma J, Ma X, Zhao B, Zhang Y, et al. Naringin prevents ovariectomy-induced osteoporosis and promotes osteoclasts apoptosis through the mitochondria-mediated apoptosis pathway. Biochem Biophys Res Commun. (2014) 452:629–35. doi: 10.1016/j.bbrc.2014.08.117
130. Chen Y, Dou C, Yi J, Tang R, Yu T, Zhou L, et al. Inhibitory effect of vanillin on RANKL-induced osteoclast formation and function through activating mitochondrial-dependent apoptosis signaling pathway. Life Sci. (2018) 208:305–14. doi: 10.1016/j.lfs.2018.07.048
131. Sun W-X, Zheng H-Y, Lan J. Edaravone protects osteoblastic cells from dexamethasone through inhibiting oxidative stress and mPTP opening. Mol Cell Biochem. (2015) 409:51–8. doi: 10.1007/s11010-015-2511-2
132. McKenzie AI, Mahmassani ZS, Petrocelli JJ, de Hart N, Fix DK, Ferrara PJ, et al. Short-term exposure to a clinical dose of metformin increases skeletal muscle mitochondrial H2O2 emission and production in healthy, older adults: A randomized controlled trial. Exp gerontology. (2022) 163:111804. doi: 10.1016/j.exger.2022.111804
133. Løkken N, Khawajazada T, Storgaard JH, Raaschou Pedersen D, Christensen ME, Hornsyld TM, et al. No effect of resveratrol in patients with mitochondrial myopathy: A cross-over randomized controlled trial. J Inherited Metab Dis. (2021) 44:1186–98. doi: 10.1002/jimd.12393
Keywords: interruption of mitochondrial endosymbiosis, osteoporosis, cytoplasmic signal, bone tissue cells, heredity, ADAPT, targeted therapy
Citation: Zhang H, Zhao R, Wang X, Qi Y, Sandai D, Wang W, Song Z and Liang Q (2025) Interruption of mitochondrial symbiosis is associated with the development of osteoporosis. Front. Endocrinol. 16:1488489. doi: 10.3389/fendo.2025.1488489
Received: 30 August 2024; Accepted: 14 January 2025;
Published: 03 February 2025.
Edited by:
Yonggang Ma, Yangzhou University, ChinaReviewed by:
Antonietta Franco, Humanitas University, ItalyCopyright © 2025 Zhang, Zhao, Wang, Qi, Sandai, Wang, Song and Liang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Doblin Sandai, ZG9ibGluQHVzbS5teQ==; Wei Wang, d2FuZ3dlaUBnc3p5LmVkdS5jbg==; Zhijing Song, c29uZ3poaWppbmcyMDIwQDE2My5jb20=; Qiudong Liang, MTM4MzczODU1MjhAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.