 Jing Li
Jing Li Shuaiming Chen
Shuaiming Chen Huiwen Tan1
Huiwen Tan1 Yerong Yu
Yerong Yu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 18 March 2025
Sec. Pituitary Endocrinology
Volume 16 - 2025 | https://doi.org/10.3389/fendo.2025.1487120
Background: Posterior pituitary tumors (PPTs) are extremely rare, with fewer than 400 cases reported to date. In 2022, the World Health Organization (WHO) classified four types of tumors originating from the posterior pituitary: traditional pituicytoma, oncocytic pituicytoma, granular pituicytoma, and ependymal pituicytoma. To our knowledge, only one subject with coexistence of Cushing’s disease and oncocytic pituicytoma (spindle cell oncocytoma) has been reported, but the clinical features of this patient were not described in detail.
Case presentation: We presented a case of a patient with Cushing’s syndrome and a pituitary mass. Transsphenoidal surgery was performed, and pathologic examination revealed two distinct tumors: a corticotroph adenoma with a diameter of less than 2 mm and a larger oncocytic pituicytoma. Post-surgery serum cortisol was 51 nmol/L, indicating complete remission. Corticotroph adenoma or corticotroph hyperplasia was identified after surgery in less than half of the subjects with Cushing’s disease and PPT.
Conclusions: Our study indicates that Cushing’s disease in patients with PPT may be caused by the existence of collision lesions, with corticotroph adenoma or hyperplasia being difficult to detect due to their small dimensions.
In 2022, the World Health Organization (WHO) redefined four types of tumors derived from the posterior pituitary, namely, traditional pituicytoma, oncocytic pituicytoma, granular pituicytoma, and ependymal pituicytoma (1). This update builds upon the WHO’s 2017 classification, which originally categorized these tumors as pituicytoma, granular cell tumor (GCT), spindle cell oncocytoma (SCO), and sellar ependymoma (2). However, this earlier classification was criticized for being nonspecific and potentially confusing, as it was not based on histogenesis. Consequently, in 2022, the 5th Edition of the WHO Classification of Endocrine and Neuroendocrine Tumors proposed that the oncocytic form, the granular cell form, and the ependymal type be classified as subtypes of pituicytoma. This change reflects a deeper understanding of the biomarkers associated with the pituicyte lineage, recognizing these tumors as subtypes of classical pituicytoma (1).
Posterior pituitary tumors (PPTs) are extremely rare, with fewer than 400 cases reported to date (3). These tumors are all low-grade nonneuroendocrine neoplasms, and their clinical symptoms and signs are primarily related to mass effect, which is similar to nonfunctioning pituitary macroadenomas (2). Studies have shown a relative high incidence of coexistence between PPT and adenohypophyseal hyperfunction, such as Cushing’s syndrome, acromegaly, and hyperprolactinemia (4). Most PPTs that coexist with adenohypophyseal hyperfunction are traditional pituicytoma and granular pituicytoma. To our knowledge, only one subject with coexistence of Cushing’s disease and SCO, which should be renamed as oncocytic pituicytoma in the new WHO classification, has been reported by far (5). However, the clinical features of this patient were not described in detail.
We presented a rare case of Cushing’s syndrome associated with a mass in the pituitary gland. Transsphenoidal surgery was performed, and pathologic examination revealed two distinct tumors: a corticotroph adenoma with a diameter of less than 2 mm and a larger oncocytic pituicytoma.
Written informed consent was obtained from the patient for the publication of any potentially identifiable images or data included in this article. A 41-year-old woman presented to the endocrinology department of West China Hospital with a 4-month history of a round face and hirsutism. Physical examination revealed the presence of a moon face, excessive facial hair, a buffalo hump, thinning skin, and scattered acne on the back. She had regular menstruation, and no previous history of hypertension or diabetes. Cushing’s syndrome was suspected, and initial workup included the following results: adrenocorticotropin hormone (ACTH), 44.39 ng/L; morning serum cortisol, 519.00 nmol/L; late-night serum cortisol, 375.00 nmol/L; and elevated 24-h urinary free cortisol, 833.4 μg/24 h. Serum cortisol at 8:00 a.m. the next day after 1 mg overnight dexamethasone suppression test (DST) was 549.00 nmol/L. Desmopressin acetate stimulation test (DDAVP) demonstrated a sevenfold increase of ACTH levels. Serum cortisol at 8:00 a.m. after standard large-dose DST (2 mg every 6 h consecutively for 2 days) was 189.00 nmol/L, and the suppression rate of 24-h urinary free cortisol was 69.8%. Therefore, Cushing’s disease was highly suspected.
Contrast-enhanced magnetic resonance imaging (MRI) of the sellar region indicated right sphenoiditis and a slightly enhanced lesion with a diameter of 0.6 cm in the right lower lobe of the pituitary gland, and a microadenoma was suspected (Figure 1). Computed tomography (CT) scan of the nose confirmed the diagnosis of right sphenoiditis and also indicated maxillary sinusitis. Her sex hormones were within normal limits. Thyroid-stimulating hormone (TSH) was 1.56 mU/L (reference range: 0.27–4.2 mU/L), and free thyroxine (FT4) was 11.60 pmol/L (reference range: 12.0–22.0 pmol/L). Additional tests were also performed to screen for potential complications or comorbidities of Cushing’s disease. Oral glucose tolerance test showed impaired glucose tolerance and hyperinsulinemia, with a 2-h blood glucose level of 9.74 mmol/L and insulin at 123.0 μU/mL. Bone mass was normal, as shown by dual-energy x-ray absorptiometry (DXA), with Z-scores of −0.8 in the lumbar, −0.2 in the femoral neck, and −0.6 in the total hip. No spinal compression fractures were detected by x-rays of the spine. Additionally, she was diagnosed with hypertension, with a mean blood pressure of 133/93 mmHg, as measured by 24-h ambulatory blood pressure monitoring.
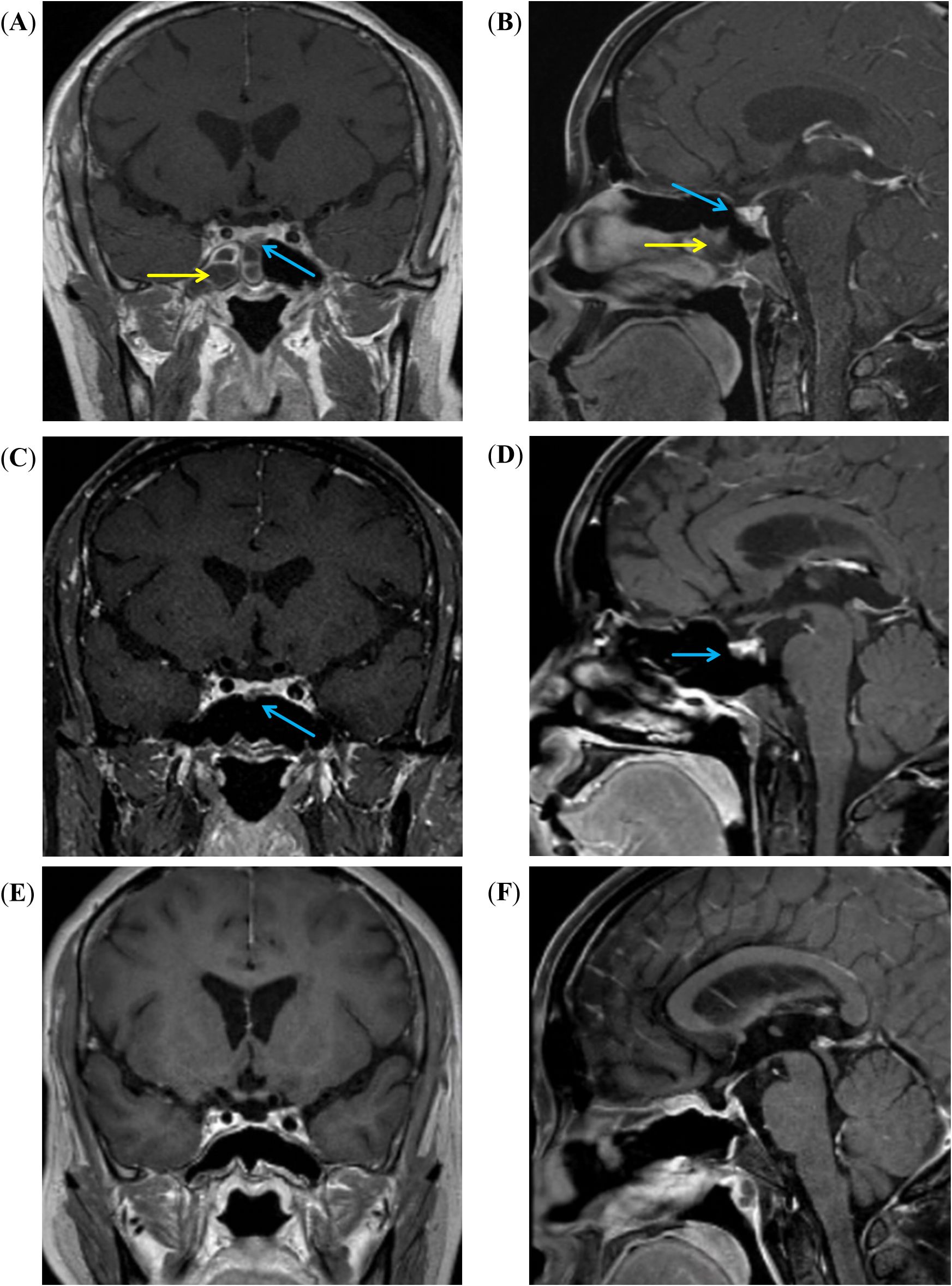
Figure 1. MRI images of the sellar region. (A, B) Coronal and sagittal T1-weighted enhanced MRI images show right sphenoid sinusitis (yellow arrow), and a low-signal intensity mass (blue arrow) is noted in the anterior-middle portion of the pituitary gland. (C, D) After 3 months of treatment with voriconazole, the right sphenoid sinusitis has resolved, but the mass (blue arrow) remains visible. (E, F) After the surgery, the pituitary tumor was resolved.
Endoscopic sinus surgery was performed to remove the mass in the right paranasal sinus, and Aspergillus was identified in the mass located in the right sphenoid sinus by immunohistochemistry. Because of concerns about a potential intracranial fungal infection following surgery, the patient was advised to undergo treatment with voriconazole 200 mg twice a day prior to undergoing surgery for Cushing’s disease. Three months later, she received another nasal endoscope, which showed clear maxillary and sphenoid sinuses. CT scan of the nose found no maxillary sinusitis and remarkable improved sphenoiditis. Consequently, the patient received a transsphenoidal surgery to remove the mass in the pituitary gland. During the surgery, a single, highly vascularized mass, measuring 5×6×4 mm, was identified in the sellar region. It compressed the normal pituitary tissue and invaded the dura in the floor of sellar turcica. The mass was completely removed. Hematoxylin–eosin (HE) staining of the excised tissue revealed the tumor, and immunohistochemical analysis confirmed the presence of two distinct types of tumors, which were adjacent but not in direct contact, with clear margins between them (Figure 2). One was a very small corticotroph adenoma, measuring less than 2 mm in diameter, which was positive for Syn, CK8/18, T-pit, and ACTH, but negative for TTF-1, GFAP, and other pituitary hormones. Ki-67 was less than 3%. The other tumor consisted of spindle-to-epithelioid tumor cells with variable eosinophilic cytoplasm, arising from poorly defined lobules and interlacing fascicles, as seen on HE staining. Immunohistochemistry showed positivity for Syn, S100, and TTF-1; scatteredly positive for GFAP; and negativity for all anterior pituitary hormones. Ki-67 was also less than 3%. Therefore, both corticotroph adenoma from anterior pituitary and oncocytic pituicytoma from posterior pituitary were simultaneously identified. The patient’s serum cortisol the morning after surgery was 51 nmol/L, indicating complete remission. No recurrence was observed based on clinical examination and MRI during the 32-month follow-up period.

Figure 2. Immunohistochemical analysis. The two types of tumors were adjacent but not in direct contact. (A [×100] and B [×400]) Basophilic cells with granular cytoplasm and round nuclei; maximum diameter of the tumor was less than 2 mm. (C [×400]) IHC staining for ACTH was positive. (D [×100] and E [×400]) Spindle cells with interlacing fascicles and poorly defined lobules; cytoplasm was eosinophilic, and nuclei were oval, spindle, and scatteredly elongated. (F [×200], G [×200], H [×200], and I [×200]) Positive IHC staining for S100, TTF-1 and Syn, and scatteredly positive for GFAP.
We conducted a search on PubMed from the inception until June 2024, using search terms “Cushing’s disease” or “hypercortisolism” in combination with “posterior pituitary tumors”, “pituicytoma”, “granular cell tumor”, “spindle cell oncocytoma”, or “sellar ependymoma”. No restrictions were placed on language or publication type. The authors added gray literature with their expertise. We also reviewed the reference lists of relevant narrative reviews related to the topics identified in our search.
Based on the search strategy described above, only 23 cases of PPT coexisting with hypercortisolism, including the present case, have been reported to date (Table 1). Among these, 19 (82.6%) were identified as pituicytomas, 2 were identified as granular pituicytomas (GCTs), and 2 were identified as oncocytic pituicytomas (SCOs). To date, there have been no reported cases of Cushing’s disease associated with ependymal pituicytomas (sellar ependymomas). It is important to note that not all of the original publications for the 23 patients listed in Table 1 provided a detailed account of the preoperative endocrine evaluation. For example, in the cases of patient #8, #17, and #18, the original reports mentioned a diagnosis of Cushing’s disease but did not include detailed descriptions of the preoperative endocrine workup, which is essential for confirming the diagnosis. Similarly, for patient #2, a definitive diagnosis of Cushing’s disease was not established before surgery. However, postoperative findings, including a significant reduction in serum cortisol levels and resolution of Cushing’s syndrome, strongly suggest a diagnosis of Cushing’s disease. Despite the incomplete documentation of endocrine assessments in some cases, all 23 patients were ultimately diagnosed with Cushing’s disease and underwent surgical treatment.
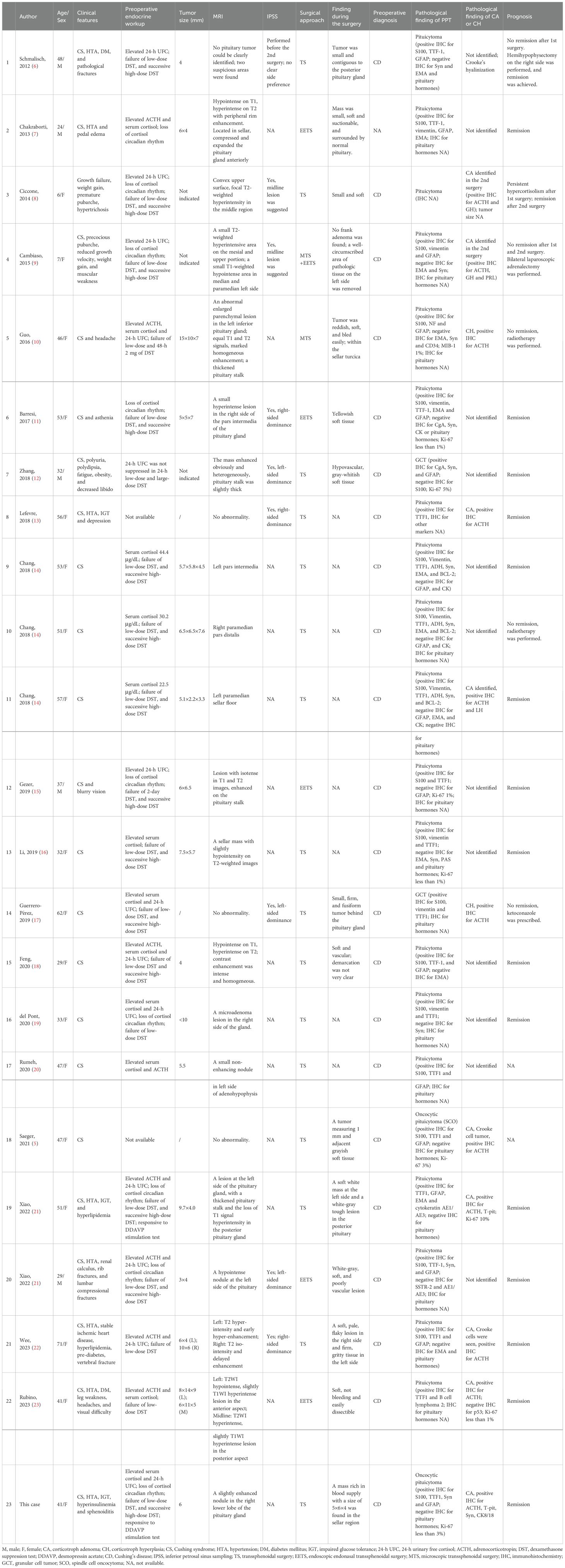
Table 1. Review of cases in the literature of coexistent pituicytoma and Cushing’s syndrome.
Pathological analysis revealed corticotroph adenomas in nine cases and corticotroph hyperplasia in two cases. Among the nine patients with corticotroph adenomas, two required a second surgery before tumor identification. In 12 cases, neither corticotroph adenomas nor hyperplasia was identified. Notably, nine patients achieved remission of Cushing’s syndrome after surgery, although prognosis data were unavailable for one patient. Pathological examination of all 23 PPTs showed no evidence of pituitary hormone secretion, consistent with previous reports that PPTs lack ACTH secretion capability.
Approximately 20 cases of PPTs have been reported following surgery for Cushing’s disease, as determined by pathological examination. The majority of these cases were diagnosed as pituicytomas, with only one case identified as an oncocytic pituicytoma (previously named SCO) (5). It is important to note that although most subjects in these studies achieved remission of Cushing’s syndrome after surgery, corticotroph adenoma or hyperplasia was identified in a fraction of cases by pathological examination. As a result, some researchers have hypothesized that PPT could produce some substances that might stimulate anterior pituitary hormone hypersecretion (18). In this study, we presented the second case of Cushing’s disease coexisting with oncocytic pituicytoma. Pathological examination demonstrated a corticotroph adenoma with a diameter of less than 2 mm, alongside a larger oncocytic pituicytoma.
PPTs are exceedingly rare tumors. While functional pituitary adenomas, such as corticotroph and GH adenomas, are relatively more common than PPTs, they remain uncommon overall. In addition to hyperprolactinemia caused by mass effect, patients with PPT have been found to exhibit a relatively high prevalence of adenohypophyseal hyperfunction. A systematic review that summarized all published clinical cases with PPT indicated a prevalence of 5.6% (15/226), including 10 cases with hypercortisolism and 5 cases with acromegaly (3). The German Pituitary Tumors Registry identified 69 PPTs, among which four subjects had coexisting functional pituitary adenomas—two with prolactin-producing tumors, one with GH tumor, and another one with Crooke cell tumor (5). To our knowledge, only three cases of concomitant PPTs and non-functional anterior pituitary adenomas have been reported (5, 24). In other words, because both PPTs and anterior pituitary adenomas are rare, and based on published literature, several cases of coexisting PPT and functional pituitary adenoma have been documented. Some researchers believe that the association between PPTs and adenohypophyseal hyperfunction is not merely coincidental (4). Possible mechanisms have been proposed to explain this coexistence. For example, cells from PPTs might induce stimulation signals and promote cell proliferation in adjacent adenohypophyseal neurosecretory cells or regulate hormones in the hypothalamus (18). However, it may also be due to the reason that adenohypophyseal hyperfunction makes individuals with PPTs more likely to receive surgery at an early stage. This could be supported by the evidence showing that the mean tumor size of sporadic PPT reported so far is much larger than that of PPTs with adenohypophyseal hyperfunction (4). The actual prevalence of PPTs may not be as low as it appears. Since almost all the subjects with PPTs were diagnosed incidentally after surgery due to mass effect, there may be additional undiagnosed cases of PPTs, which were not identified because of their benign and slow-growing nature.
In our literature review, only one subject (patient #18) has been reported where corticotroph adenoma coexisted with oncocytic pituicytoma (SCO). Specifically, the patient was considered to have Crooke cell adenoma, which is a very rare type of corticotroph adenoma characterized by Crooke hyaline changes in tumor cells. Clinical features of this case were not described in detail. Therefore, our study is the first to present detailed clinical data of a patient with corticotroph adenoma associated with oncocytic pituicytoma (SCO). Comprehensive endocrine evaluations confirmed the diagnosis of Cushing’s disease before surgery. MRI before surgery identified a mass in the sellar region, which was likely an image of the oncocytic pituicytoma based on the tumor’s size. The corticotroph adenoma was likely too small to be visualized by the MRI. We assume that the corticotroph adenoma was located near the oncocytic pituicytoma; thus, when the oncocytic pituicytoma was resected, the corticotroph adenoma was also removed simultaneously. Preoperative MRI scans identified two pituitary lesions in only two cases (patient #21 and #22). While the median tumor size of sporadic PPTs reported by far is 22.0 mm (3), the largest mass among the 23 cases was 15 mm in diameter. MRI did not detect any lesions in three patients (patient #3, #4, and #22), likely because hypercortisolism-related symptoms led to the early discovery of PPTs before the tumors grew large enough to cause mass effects.
Both patient #18 and our case of oncocytic pituicytoma (SCO) were positive for S100, TTF1, and GFAP, and negative for pituitary hormones by immunohistochemistry (IHC). Ki-67 was 3% in one patient and less than 3% in the other. Notably, the histomorphology of the PPTs among some of the 23 cases appears more typical for a oncocytic pituicytoma (SCO) than for a pituicytoma, according to both our research team and other investigators. Historically, the term pituicytoma has been used to describe various tumors with spindle cells in the sellar region (25). As a result, pituicytoma diagnosed in previous studies might not meet the traditional pituicytoma based on the 2022 WHO criteria. Additionally, the differential diagnosis between SCO and pituicytoma can sometimes be challenging, as they may present a wide range of overlapping histological features (26). Some researchers have even considered SCO to be a morphological variant of pituicytoma (26, 27). Therefore, in 2022, the 5th Edition of the WHO Classification of Endocrine and Neuroendocrine Tumors proposed that SCO, GCT, and sellar ependymoma should be classified as subtypes of pituicytoma (1). In other words, there might be more than two cases of Cushing’s syndrome associated with oncocytic pituicytoma.
The immunoprofile of oncocytic pituicytomas typically includes markers such as vimentin, S-100 protein, EMA, TTF-1, and somatostatin receptors (28). GFAP expression is less commonly observed and is typically limited to a small subset of cells. In the present case, the immunohistochemical findings were positive for Syn, S-100, and TTF-1, with scattered GFAP positivity and negativity for anterior pituitary hormones. These results are consistent with a diagnosis of oncocytic pituicytoma. However, distinguishing between PPTs based solely on histological and immunohistochemical features can be challenging due to overlapping characteristics. Recent genetic and epigenetic studies have identified subtle DNA methylation variations, mutation patterns, and differing clinical outcomes among PPTs. These findings suggest that molecular diagnostics could provide valuable insights for tumor subclassification (29). Nevertheless, the routine clinical application of these molecular techniques has not been fully explored and requires further research to determine their role in the classification and management of posterior pituitary neoplasms.
Our study highlights the challenges in diagnosing Cushing’s disease. Inferior petrosal sinus sampling (IPSS), considered the gold standard for confirming the source of ACTH secretion, remains underutilized due to its technical complexity, procedural risks, and the requirement for specialized expertise (30, 31). In our study, only 9 of 23 patients underwent IPSS, reflecting these limitations. According to the 2021 Pituitary Society guidelines, the diagnostic approach should prioritize non-invasive tests, such as late-night salivary cortisol, DSTs, and 24-h urinary free cortisol (32). These tests provide essential information, while pituitary MRI may be instrumental in distinguishing between ectopic and pituitary ACTH sources. IPSS is essential when biochemical and imaging results are inconclusive but may not be necessary if a pituitary tumor ≥10 mm or <6 mm is identified along with supportive biochemical evidence. In contrast, for tumors measuring between 6 and 9 mm, expert opinions vary; nevertheless, the majority recommend using IPSS for diagnosis in such cases (32). However, practical challenges persist, as not all centers have the resources or expertise to perform all recommended tests, particularly IPSS, leading to potential diagnostic delays. This underscores the importance of a multidisciplinary approach to ensure accurate diagnosis and optimal management of Cushing’s disease, taking into account the available resources.
In our case, the PPT was located in the lower anterior part of the pituitary, which could easily be mistaken for a pituitary adenoma (Figure 1). According to previous literature, pituicytomas (PPTs) are more commonly found in the suprasellar region (33–35). Salge-Arrieta et al. conducted a systematic analysis of imaging findings in 104 cases of pituicytoma and reported that PPTs were purely suprasellar in 22.1%, purely intrasellar in 41.3%, mixed sellar/suprasellar in 30.8%, and invaded the cavernous sinuses in 5.8% (33). Therefore, PPTs can originate anywhere along the hypophyseal–infundibulum–median eminence axis (33). In addition, the clinical presentation of PPTs is influenced primarily by tumor size and location. Among the reported cases, the most common presenting symptom was visual impairment (48.8%), followed by hypopituitarism (31.5%) and headache (25.6%) (35). Patients with suprasellar PPTs frequently experience isolated visual symptoms due to optic nerve compression, whereas intrasellar tumors more often result in headache and hypopituitarism due to compression of the pituitary gland or adjacent structures.
Recent studies highlight the role of frailty in outcomes for various conditions, including Cushing’s disease (36). Mild frailty, as assessed by the 11-factor modified frailty index, has been shown to predict surgical outcomes and may assist in preoperative risk stratification (36). In our study, we aimed to assess frailty in all the cases included in the review; however, variability in the reported data limited our ability to apply standardized scoring. Future research will incorporate frailty assessments to provide more evidence on its impact in Cushing’s disease.
In summary, our study highlights the rare coexistence of Cushing’s disease and oncocytic pituicytoma, a condition reported in only one other case. Hypercortisolism associated with PPT presents significant diagnostic challenges, as corticotroph adenomas or corticotroph hyperplasia are often undetected both preoperatively and, in some cases, postoperatively due to their small size. This emphasizes the need for improved diagnostic tools and surgical techniques to identify these lesions more reliably. Future research should focus on advanced imaging modalities with enhanced sensitivity and innovative intraoperative methods, such as real-time fluorescence-guided surgery and advanced pathological staining. Additionally, larger, multicenter studies are essential to uncover the mechanisms underlying the coexistence of these conditions and to inform the development of optimal diagnostic and therapeutic strategies.
JL: Data curation, Writing – review & editing, Visualization, Writing – original draft, Project administration. SC: Writing – review & editing, Writing – original draft, Data curation, Investigation, Visualization. HT: Conceptualization, Supervision, Writing – review & editing. YY: Project administration, Supervision, Writing – review & editing. YT: Data curation, Methodology, Resources, Writing – review & editing. BC: Data curation, Visualization, Writing – review & editing. JWL: Funding acquisition, Methodology, Project administration, Writing – review & editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by Grant 1.3.5 project for Disciplines of Excellence Clinical Research Incubation Project, West China Hospital of Sichuan University (grant number 2020HXFH034), and the Gangbao Project of the Health Commission of Sichuan Province (grant number Chuanganyan 2021-102).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Asa SL, Mete O, Perry A, Osamura RY. Overview of the 2022 who classification of pituitary tumors. Endocr Pathol. (2022) 33(1):6–26. doi: 10.1007/s12022-022-09703-7
2. Mete O, Lopes MB. Overview of the 2017 who classification of pituitary tumors. Endocr Pathol. (2017) 28:228–43. doi: 10.1007/s12022-017-9498-z
3. Guerrero-Pérez F, Marengo AP, Vidal N, Iglesias P, Villabona C. Primary tumors of the posterior pituitary: A systematic review. Rev Endocr Metab Disord. (2019) 20:219–38. doi: 10.1007/s11154-019-09484-1
4. Iglesias P, Guerrero-Pérez F, Villabona C, Díez JJ. Adenohypophyseal hyperfunction syndromes and posterior pituitary tumors: prevalence, clinical characteristics, and pathophysiological mechanisms. Endocrine. (2020) 70:15–23. doi: 10.1007/s12020-020-02399-x
5. Saeger W, von Schöning J, Flitsch J, Jautzke G, Bergmann M, Hagel C, et al. Co-occurrence of pituitary neuroendocrine tumors (Pitnets) and tumors of the neurohypophysis. Endocr Pathol. (2021) 32:473–9. doi: 10.1007/s12022-021-09677-y
6. Schmalisch K, Schittenhelm J, Ebner FH, Beuschlein F, Honegger J, Beschorner R. Pituicytoma in a patient with Cushing’s disease: case report and review of the literature. Pituitary. (2012) 15 Suppl 1:S10–S6. doi: 10.1007/s11102-010-0262-3
7. Chakraborti S, Mahadevan A, Govindan A, Sridhar K, Mohan NVS, Satish IR, et al. Pituicytoma: report of three cases with review of literature. Pathol Res Pract. (2013) 209:52–8. doi: 10.1016/j.prp.2012.10.006
8. Ciccone S, Cambiaso P, Longo D, Marini R, Pedicelli S, Deodati A, et al. Association of Pituicytoma and Cushing’s Disease: A Rare Pediatric Case. Dublin, Ireland: Horm Res Paediatr (2014). p. 284. doi: 10.1159/000365775
9. Cambiaso P, Amodio D, Procaccini E, Longo D, Galassi S, Camassei FD, et al. Pituicytoma and Cushing’s disease in a 7-year-old girl: A mere coincidence? Pediatrics. (2015) 136:e1632–e6. doi: 10.1542/peds.2015-0638
10. Guo X, Fu H, Kong X, Gao L, Wang W, Ma W, et al. Pituicytoma coexisting with corticotroph hyperplasia: literature review with one case report. Med (Baltimore). (2016) 95:e3062. doi: 10.1097/MD.0000000000003062
11. Barresi V, Lionti S, Messina E, Esposito F, Angileri FF, Cannavò S. A 53-year-old woman with Cushing’s disease and a pituitary tumor. Neuropathology. (2017) 37:86–90. doi: 10.1111/neup.12319
12. Zhang Y, Teng Y, Zhu H, Lu L, Deng K, Pan H, et al. Granular cell tumor of the neurohypophysis: 3 cases and a systematic literature review of 98 cases. World Neurosurg. (2018) 118:e621–e30. doi: 10.1016/j.wneu.2018.07.004
13. Lefevre E, Bouazza S, Bielle F, Boch A-L. Management of pituicytomas: A multicenter series of eight cases. Pituitary. (2018) 21:507–14. doi: 10.1007/s11102-018-0905-3
14. Chang T-W, Lee C-Y, Jung S-M, Lai H-Y, Chen C-T, Yeap M-C, et al. Correlations between clinical hormone change and pathological features of pituicytoma. Br J Neurosurg. (2018) 32:501–8. doi: 10.1080/02688697.2018.1472212
15. Gezer E, Selek A, Cetinarslan B, Canturk Z, Tarkun I, Ceylan S. The coexistence of infundibular pituicytoma and Cushing’s disease due to pituitary adenoma: A case report. Endocr Regul. (2019) 53:263–7. doi: 10.2478/enr-2019-0026
16. Li X, Liu Y, Miao Y, Wang J, Wang L, Wang E-H. A rare case of pituicytoma presenting with severe Cushing disease: A case report and review of literature. Med (Baltimore). (2019) 98:e17772. doi: 10.1097/MD.0000000000017772
17. Guerrero-Pérez F, Vidal N, Marengo AP, Pozo CD, Blanco C, Rivero-Celada D, et al. Posterior pituitary tumours: the spectrum of a unique entity. A clinical and histological study of a large case series. Endocrine. (2019) 63:36–43. doi: 10.1007/s12020-018-1774-2
18. Feng Z, Mao Z, Wang Z, Liao B, Zhu Y, Wang H. Non-adenomatous pituitary tumours mimicking functioning pituitary adenomas. Br J Neurosurg. (2020) 34:487–91. doi: 10.1080/02688697.2018.1464121
19. Marco Del Pont F, Villalonga JF, Ries-Centeno T, Arakaki N, Katz D, Cervio A. Pituicytoma associated with acromegaly and Cushing disease. World Neurosurg. (2020) 136:78–82. doi: 10.1016/j.wneu.2019.12.085
20. Rumeh ASAL, Bafaqeeh M, Khairan SJA, Al Shakweer W. Pituicytoma associated with Cushing’s disease: A case report and literature review. J Surg Case Rep. (2020) 2020:rjaa104. doi: 10.1093/jscr/rjaa104
21. Xiao T, Duan L, Chen S, Lu L, Yao Y, Mao X, et al. Pituicytoma associated with suspected Cushing’s disease: two case reports and a literature review. J Clin Med. (2022) 11(16):4805. doi: 10.3390/jcm11164805
22. Wee Z, Tang PY, Lai SH, Ang BT, Chandran SR. Co-existence of pituicytoma and corticotroph adenoma in a patient with Cushing’s disease. Pathology. (2023) 55:432–5. doi: 10.1016/j.pathol.2022.08.005
23. Rubino F, Eichberg DG, Saad AG, Komotar RJ, Ivan ME. Synchronous posterior and anterior pituitary tumors: A case report of a hypothetic paracrine relationship. Asian J Neurosurg. (2023) 18:377–82. doi: 10.1055/s-0043-1768601
24. Neidert MC, Leske H, Burkhardt J-K, Kollias SS, Capper D, Schrimpf D, et al. Synchronous pituitary adenoma and pituicytoma. Hum Pathol. (2016) 47:138–43. doi: 10.1016/j.humpath.2015.08.017
25. Mete O, Lopes MB, Asa SL. Spindle cell oncocytomas and granular cell tumors of the pituitary are variants of pituicytoma. Am J Surg Pathol. (2013) 37:1694–9. doi: 10.1097/PAS.0b013e31829723e7
26. Yoshimoto T, Takahashi-Fujigasaki J, Inoshita N, Fukuhara N, Nishioka H, Yamada S. Ttf-1-positive oncocytic sellar tumor with follicle formation/ependymal differentiation: non-adenomatous tumor capable of two different interpretations as a pituicytoma or a spindle cell oncocytoma. Brain Tumor Pathol. (2015) 32:221–7. doi: 10.1007/s10014-015-0219-3
27. Viaene AN, Lee EB, Rosenbaum JN, Nasrallah IM, Nasrallah MP. Histologic, immunohistochemical, and molecular features of pituicytomas and atypical pituicytomas. Acta Neuropathol Commun. (2019) 7:69. doi: 10.1186/s40478-019-0722-6
28. Roncaroli F, Giannini C. Posterior pituitary tumors and other rare entities involving the pituitary gland. Brain Pathol. (2025) 35:e13307. doi: 10.1111/bpa.13307
29. Schmid S, Solomon DA, Perez E, Thieme A, Kleinschmidt-DeMasters BK, Giannini C, et al. Genetic and epigenetic characterization of posterior pituitary tumors. Acta Neuropathol. (2021) 142:1025–43. doi: 10.1007/s00401-021-02377-1
30. Loriaux DL. Diagnosis and differential diagnosis of Cushing’s syndrome. N Engl J Med. (2017) 376:1451–9. doi: 10.1056/NEJMra1505550
31. Vassiliadi DA, Mourelatos P, Kratimenos T, Tsagarakis S. Inferior petrosal sinus sampling in Cushing’s syndrome: usefulness and pitfalls. Endocrine. (2021) 73:530–9. doi: 10.1007/s12020-021-02764-4
32. Fleseriu M, Auchus R, Bancos I, Ben-Shlomo A, Bertherat J, Biermasz NR, et al. Consensus on diagnosis and management of Cushing’s disease: A guideline update. Lancet Diabetes Endocrinol. (2021) 9:847–75. doi: 10.1016/S2213-8587(21)00235-7
33. Salge-Arrieta FJ, Carrasco-Moro R, Rodríguez-Berrocal V, Pian H, Martínez-San-Millán JS, Iglesias P, et al. Clinical features, diagnosis and therapy of pituicytoma: an update. J Endocrinol Invest. (2019) 42:371–84. doi: 10.1007/s40618-018-0923-z
34. Xie W, Li ZF, Bian L, He B, Zhao W, Zhang ZG, et al. Neuroimaging features of pituicytomas. Chin Med J (Engl). (2016) 129:1867–9. doi: 10.4103/0366-6999.186644
35. Chen B, Fan X, Zhang Z. Pituicytoma: report of three cases and a systematic literature review. Clin Neurol Neurosurg. (2021) 205:106650. doi: 10.1016/j.clineuro.2021.106650
36. Findlay MC, Rennert RC, Lucke-Wold B, Couldwell WT, Evans JJ, Collopy S, et al. Impact of frailty on surgical outcomes of patients with Cushing disease using the multicenter registry of adenomas of the pituitary and related disorders registry. Neurosurgery. 96(2):386–95. doi: 10.1227/neu.0000000000003090
Keywords: Cushing’s disease, oncocytic pituicytoma, spindle cell oncocytoma, pituitary adenoma, posterior pituitary tumors
Citation: Li J, Chen S, Tan H, Yu Y, Tang Y, Cai B and Li J (2025) Oncocytic pituicytoma in a patient with Cushing’s disease: a case report and narrative literature review. Front. Endocrinol. 16:1487120. doi: 10.3389/fendo.2025.1487120
Received: 27 August 2024; Accepted: 17 February 2025;
Published: 18 March 2025.
Edited by:
Xiang'En Shi, Capital Medical University, ChinaReviewed by:
Karol Piotr Sagan, Pomeranian Medical University, PolandCopyright © 2025 Li, Chen, Tan, Yu, Tang, Cai and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianwei Li, amVycnlsaTY3OEBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.