
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Endocrinol. , 04 February 2025
Sec. Neuroendocrine Science
Volume 16 - 2025 | https://doi.org/10.3389/fendo.2025.1483305
 Mercedes Aguilar-Soto1
Mercedes Aguilar-Soto1 Julia M. Zuarth-Vázquez1
Julia M. Zuarth-Vázquez1 Laura Leyva-Figueroa1
Laura Leyva-Figueroa1 Karla Zarco-Ávila2
Karla Zarco-Ávila2 Armando Gamboa-Domínguez2Aldo Eguiluz-Melendez3
Armando Gamboa-Domínguez2Aldo Eguiluz-Melendez3 Laura C. Hernández-Ramírez4*
Laura C. Hernández-Ramírez4*Introduction: Patients with neurofibromatosis type 1 (NF1) are at risk for developing various neoplasms. Since the early twentieth century, multiple cases of pituitary neuroendocrine tumors (PitNETs) occurring in this context have been published. Yet, the role of NF1 (17q11.2) loss-of-function (LOF) variants in pituitary tumorigenesis remains unclear.
Aim: We report the clinical and molecular characterization of a case of PitNET diagnosed in a patient with NF1. We also review the available data for and against a causal association between NF1 defects and pituitary tumors.
Methods: Our patient was recruited via an ongoing prospective study of individuals with neuroendocrine neoplasms. Genetic testing was carried out by means of targeted next generation sequencing (NGS) and Sanger sequencing in blood and tumor DNA, respectively. NF1 expression was analyzed via quantitative polymerase chain reaction (qPCR) in blood and tumor cDNA. Similar cases were searched in the literature.
Results: A 54-year-old-man was incidentally diagnosed with a clinically non-functioning PitNET via brain imaging. He had a personal and family history of NF1 and carried the germline pathogenic variant NF1 (NM_001042492.3): c.147C>A, p.Y49*. Via transsphenoidal surgery, a 16 mm lesion was resected, showing strong granular cytoplasmic immunoreactivity with patchy distribution for NF1 and preserved heterozygosity for the NF1 defect. Additional NGS ruled out germline defects in PitNET-associated genes. By qPCR, NF1 was significantly overexpressed in the tumor when compared with another NF-PitNET, but not when compared with a corticotropinoma. We reviewed twenty-three case reports of PitNETs occurring in patients with either clinical NF1 without genetic study, individuals with NF1 germline variants with or without clinical NF1 or associated with somatic NF1 defects. Predominance of GH-secreting and large PitNETs, with young-onset in around half of the cases, were noticed. Two individuals developed multiple endocrine neoplasia-like phenotypes but tested negative for other relevant genetic defects.
Conclusions: Although the association of NF1 and PitNETs could be coincidental, the clinical characteristics of the reviewed cases differ from those of typical incidentalomas. NF1 could drive pituitary tumorigenesis via haploinsufficiency, but this hypothesis requires further research. Additional clinical and molecular data from large cohorts of affected individuals should help clarify this question.
Neurofibromatosis type (NF1) or Von Recklinghausen’s disease (MIM 162200) is an autosomal dominant syndrome predisposing patients to the development of benign and malignant tumors (1). Although the disease mainly affects the nervous system, several other organs such as the skin, cardiovascular, skeletal, and endocrine systems can be affected. It is one of the most common inherited disorders, with an estimated prevalence ranging from 1/3000-1/4000, which might be underestimated in countries without genetic testing protocols (2–4). NF1 patients have a reduction of 10-15 years in life expectancy compared with the general population (4). Germline loss-of-function (LOF) variants of NF1 (17q11.2) underlie this phenotype in most cases, occurring de novo in 42% (1). Cutaneous manifestations such as café-au-lait spots and neurofibromas, including plexiform neurofibromas, are hallmarks of NF1 (5). Optic gliomas, Lisch nodules, choroidal abnormalities, and skeletal dysplasia are also common features (6, 7). Fifteen to twenty percent of patients develop glial low-grade tumors, predominantly in the optic pathways, the brainstem, and the cerebellum. In adults, the risk of high-grade gliomas, including glioblastomas, is increased by 10-50-fold compared with the general population (7).
Multiple endocrine manifestations have been associated with NF1 (8). Pheochromocytomas and paragangliomas (PPGLs) occur in up to 6.6% of NF1 patients, while germline and somatic NF1 variants are detected in 3% and one-fourth of sporadic cases of PPGLs, respectively (9–13). Gastrointestinal neuroendocrine neoplasms of the periampullary region, usually somatostatinomas, are diagnosed in 1% of NF1 patients (14). Gastrointestinal stromal cell tumors can rarely occur. The association of NF1 LOF with other endocrine neoplasms is uncertain.
NF1 has been largely associated with central precocious puberty, with a reported frequency of 2.4-5.6% (15–18). Most, but not all of these cases are associated with optic pathway tumors. Growth hormone (GH) excess occurs in 6-11% of children with NF1 and optic pathway tumors, either with or without precocious puberty (19–21). The mechanism causing GH excess is unclear, but the most accepted hypothesis involves loss of somatostatinergic inhibition from the optic pathway tumors (22). A recent study, however, described a heterogeneous spectrum of structural defects among individuals with NF1 and GH excess (23). Out of ten patients reported, six had optic pathway tumors, one of which had also a pituitary neuroendocrine tumor (PitNET), which was negative for GH immunostaining. Another patient was diagnosed with possible pituitary hyperplasia and therefore did not undergo surgery. PitNETs were also documented in two patients without optic pathway tumors. These findings suggest that PitNETs or hyperplasia might have a role in GH excess in patients with NF1.
Indeed, multiple cases of PitNETs have been reported in patients with NF1 since the early 1900s. Initially thought as a rare and possibly coincidental association, the growing number of publications in the recent years, as well as the finding of somatic NF1 variants in sporadic PitNETs, suggest that a linking mechanism might exist between both diseases (24). Here, we present a patient with NF1 who developed a clinically non-functioning PitNET (NF-PitNET). Detailed molecular studies in this case and a thorough compilation of similar cases reported in the literature are presented.
A fifty-four-year-old man from San Juan del Río, Querétaro, a small city in central Mexico, was admitted to our Institute in 2023. His paternal grandmother died of an unspecified cancer and his mother died at age 48 years due to an unknown cause. He mentioned the presence of café-au-lait spots in his father (who died at age 85 years), eight of his ten siblings (three died before age 65 years), and his 29-year-old daughter (alive). One brother had primary biliary cirrhosis, and a nephew died at age 28 years from complications of epilepsy (Figure 1A). Consanguinity was mentioned. His past medical history was remarkable for the presence of NF1 stigmata (café-au-lait spots and cutaneous neurofibromas) since childhood, right corneal transplantation, and amaurosis of the same eye due to retinal detachment twenty years prior, well-controlled hypertension, prediabetes, dyslipidemia, and pernicious anemia.
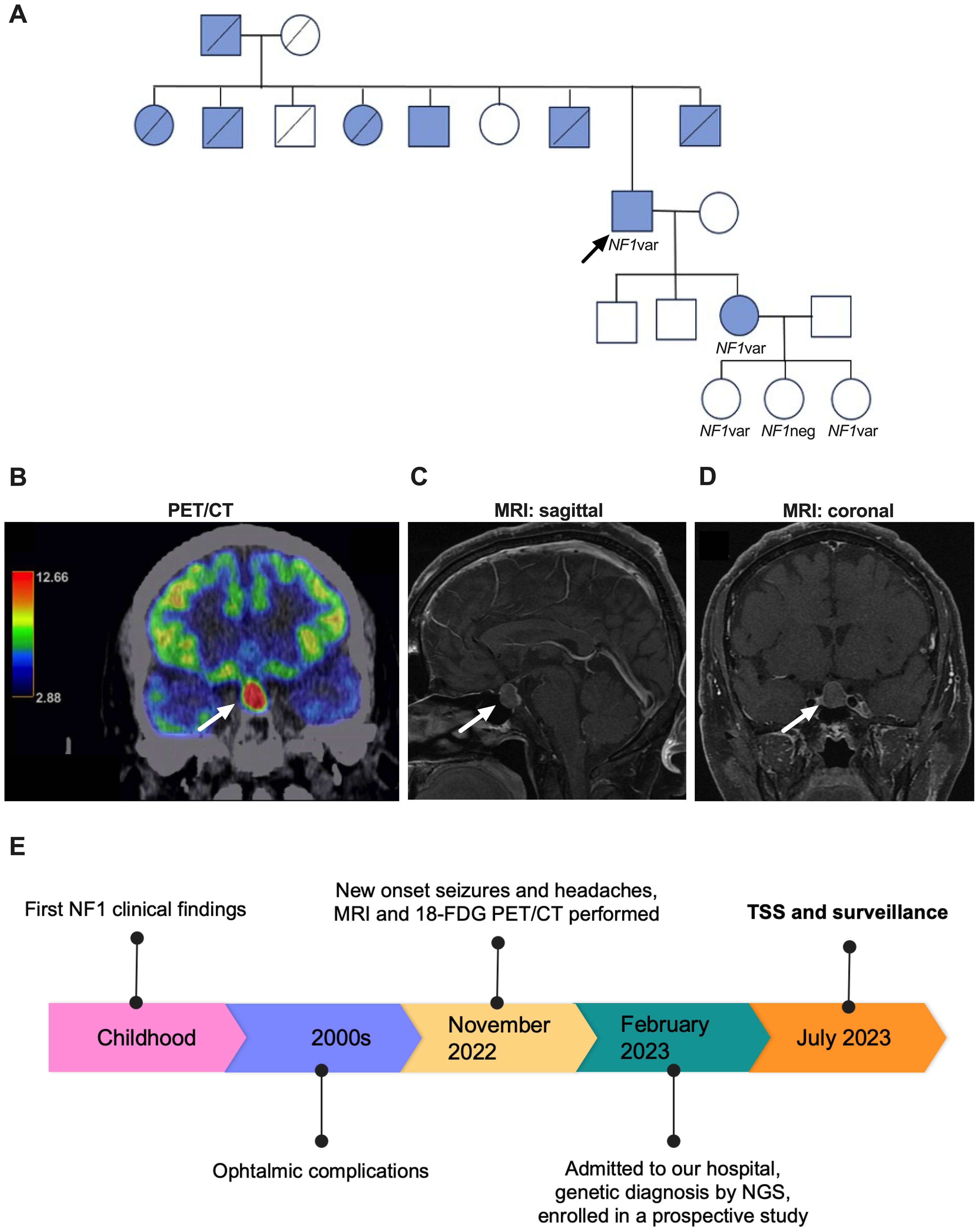
Figure 1. (A) Family tree with blue circles representing individuals affected with NF1. NF1var: NF1 variant carrier. NF1neg: NF1 variant screening negative. Arrow: proband. Crossed figures: deceased. (B) 18F-FDG PET/CT showing a pituitary lesion of with homogenous uptake of the contrast with a maximum standardized uptake value of 20.8. (C, D) sagittal and coronal gadolinium-enhanced MRI (T1), showing a 16x15x11 mm pituitary lesion on the left parasagittal aspect, contacting the wall of the cavernous sinus, with homogeneous enhancement and displacement of the pituitary stalk to the left and of the optic chiasm anteriorly. Arrowheads point to the lesion. (E) Timeline of patient evolution. TSS, transsphenoidal surgery; NGS, next generation sequencing.
Four months prior to admission, the patient developed short-term memory impairment, holocranial headaches, and new-onset seizures. These symptoms prompted the indication for brain magnetic resonance imaging (MRI) and 18F-fluorodeoxyglucose positron emission tomography/computed tomography (18F-FDG PET/CT), both revealing an incidental pituitary lesion; a pituitary MRI confirmed the diagnosis (Figures 1B–D). Laboratory tests revealed hyposomatotropinemia, but other pituitary hormones were normal (Table 1). On physical examination, hemianopsia of the left eye (which was the one with preserved vision) was evident by campimetry. He had multiple neurofibromas on the neck (0.5 cm), thorax, and limbs; the largest one (3x5 cm) was found in his lumbar region. The presence of multiple café-au-lait spots with positive Crowe sign was noted. He had a BMI of 30.2 kg/m² and his vital signs were normal.
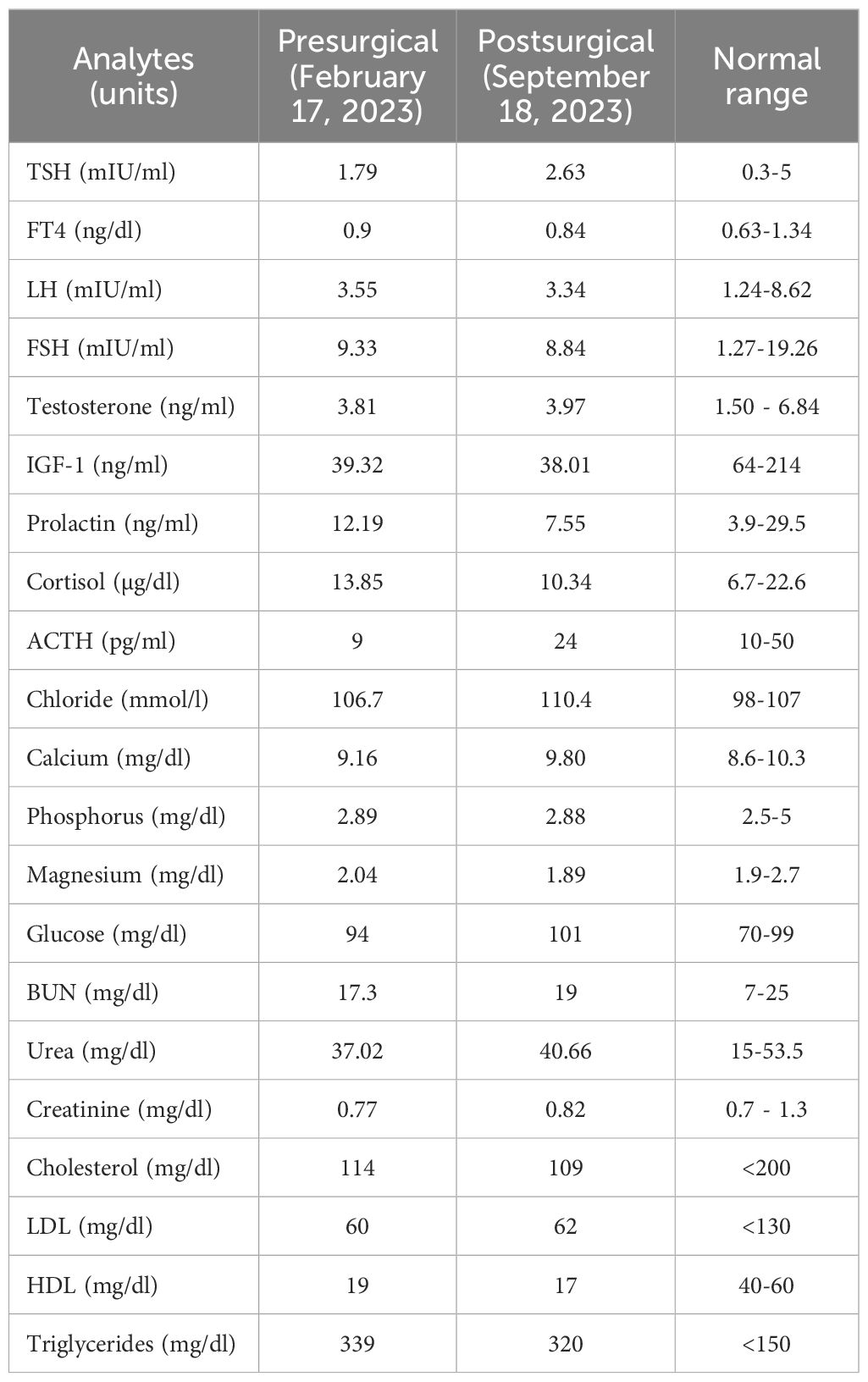
Table 1. Laboratory results.
Because of important visual impairment, an endonasal endoscopic transsphenoidal surgery was performed without complications (Figure 1E). Surgical pathology reported a PitNET with 0 mitoses per 2 mm2, weak, hypogranular periodic acid-Schiff staining and reticulin demonstrating focal loss of the usual acinar pattern. Immunostaining was positive for chromogranin and negative for ACTH, GH, and prolactin, and Ki-67 was <1%. Immunostaining for other pituitary hormones and transcription factors was unavailable (Figure 2A). Significant visual improvement was documented after surgery, although with a tumor remnant; hormonal levels remained unchanged. A diagnosis of idiopathic epilepsy was established, and the patient is currently under treatment with levetiracetam and valproic acid.
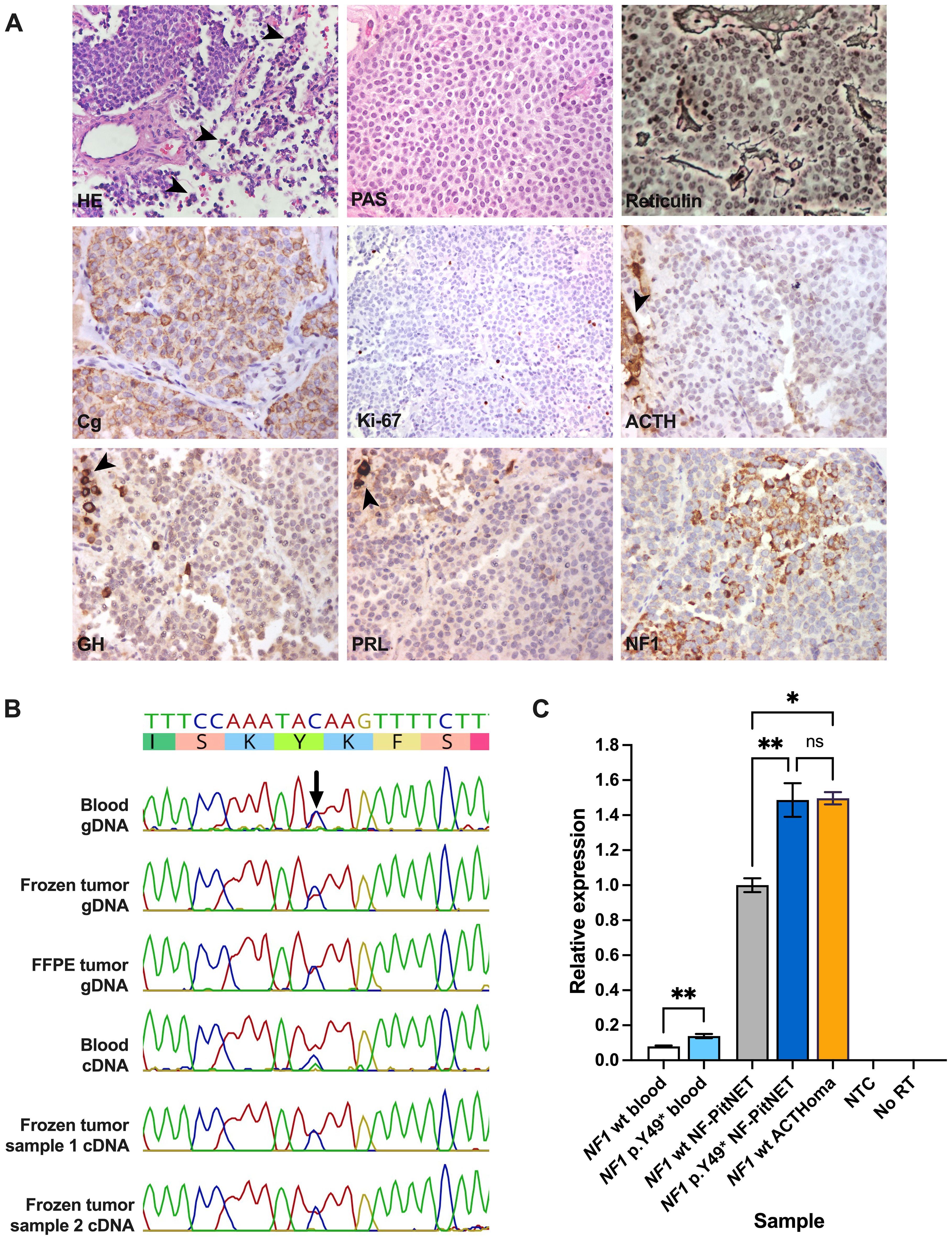
Figure 2. (A) Histopathological images of the resected PitNET. Groups of normal anterior pituitary cells surrounding the tumor (arrowheads) are shown in images for HE and negative ACTH, GH, and PRL immunostaining. Routine preparations were retrieved from the pathology archive, except for NF1 immunostaining. Magnification: 40x. (B) Sanger sequencing results of multiple blood and PitNET genomic and coding DNA samples from the patient. (C) Quantitative polymerase chain reaction depicting different blood and PitNET samples. ACTH, adrenocorticotropic hormone; ACTHoma, corticotropinoma; cDNA, coding DNA; Cg, chromogranin; FFPE, formalin-fixed paraffin-embedded; gDNA, genomic DNA; GH, growth hormone; HE, hematoxylin-eosin; NTC, no template control; NF1 wt, NF1 wild type; No RT, no reverse transcriptase control; PRL, prolactin. PAS, periodic acid-Schiff staining. ns, not significant; *, P≤0.05; **, P≤0.01.
A blood sample of the patient was first studied in a routine diagnostic laboratory via a next-generation sequencing (NGS) panel (Invitae Common Hereditary Cancers Panel). This test detected a pathogenic germline variant in NF1 (NM_001042492.3): c.147C>A, p.Y49* (dbSNP: rs1597626026, ClinVar accession: VCV002114679.2). This change, which is absent from Genome Aggregation Database v.4.1.0 (gnomAD) and the Mexico City Prospective Study (MCPS) databases, creates a premature stop codon in the exon 2, predictably leading to LOF (22, 24–26). The same variant has been reported in the literature as a germline change in two individuals with clinical NF1 (27, 28) and at the somatic level in one case of ovarian cancer (29). This defect was also reported as a somatic change in a patient with synchronous endometrial and ovarian cancer, although the germline sequence was not reported (30, 31). Two other germline variants affecting the same position and causing the same effect on the protein (c.147C>G and c.147del) have also been found in the context of NF1 (2, 5, 32, 33). The patient received genetic counseling and cascade screening for his relatives. One daughter, aged 31 years, underwent clinical evaluation and genetic confirmation. Two of the patient’s three granddaughters (aged eight and three years) have also been genetically confirmed, while the other two sons have not yet been tested.
Since PitNETs are not among the neoplasms typically associated with NF1, this patient was enrolled in an ongoing prospective study aimed to characterize the genetic bases of neuroendocrine neoplasms in a cohort of Mexican patients. The protocol has been approved by the internal review boards of the participating institutions (ClinicalTrials.gov NCT06523582). Under informed consent, blood samples for DNA and RNA, as well as fresh-frozen and formalin-fixed and paraffin-embedded PitNET samples were obtained. Samples from additional participants from the same study were used for comparison. Sanger sequencing was performed at the National Autonomous University of Mexico’s Research Support Network Molecular Biology unit. The rest of the experimental procedures were carried out in our lab.
DNA was extracted from blood using the DNA Isolation Kit for Mammalian Blood (Roche 11667327001) plus RNase (Roche 11119915001) treatment. For DNA extraction from tumors, samples were first mechanically disrupted with magnetic beads (fresh-frozen samples) or deparaffinized with HistoChoice (Merck H2779) and then processed with the Maga Zorb DNA Mini-Prep kit (Promega MB1004). Samples for RNA extraction were lysed with magnetic beads or red cell lysis buffer (0.1mM EDTA, 10 mM potassium bicarbonate, and 168 mM ammonium chloride), as appropriate, and further processed with the RNeasy Plus Mini Kit (QIAGEN 74134) plus DNase (QIAGEN 79254) treatment. Reverse transcription of RNA samples was done with the SuperScript III First-Strand Synthesis SuperMix for qRT-PCR kit (Invitrogen 11752050).
For NGS we used a custom-designed panel (details available on request). Library preparation was done with mechanical fragmentation and the Twist Biosciences target enrichment protocol. Sequencing was carried out in a MiSeq (Illumina) instrument, obtaining paired-end 150 bp reads with an average depth of 257x for the regions of interest. Sequencing quality was verified using FastQC, sequences were aligned to the GRCh38/hg38 human genome using Burrows-Wheeler aligner-maximum exact matches, PCR duplicates were marked with MarkDuplicates and FreeBayes was used for variant calling (34–37). Copy number variants were searched for with ExomeDepth (38).
Using the Franklin (Genoox) online platform, medium or high-quality nonsynonymous variants in exons and exon-intron junctions were identified (39). Variants with frequency <0.1% in both gnomAD and MCPS were then selected (25, 26). Variants of interest were searched for in ClinVar and their reported classification (according to the criteria of the American College of Medical Genetics and Genomics and Association for Molecular Pathology) was noted (40, 41). For variants with conflicting classification or not listed in ClinVar, functional effects were predicted using the tools linked to the Varsome online platform and data reported in the literature, when available (42). Aside from the previously known NF1 defect, no variants of interest were identified.
To analyze NF1 expression by immunostaining, a mouse polyclonal anti-NF1 antibody (Novus Biologicals NBP2-37914, 1:200) was used, following a previously reported protocol (23). For Sanger sequencing in blood and tumor DNA and cDNA samples, a 105 bp region in exon 2 was amplified using GoTaq Green Master Mix (Promega M7122) and the primers 5’-ACAGGACAGCAGAACACACA-3’ and 5’-AGTGAGGCCGCTTATAACCA-3’. Amplicons were column-purified (Wizard SV Gel and PCR Clean-Up System, Promega A9281) and subjected to unidirectional sequencing (BigDye Terminator 3.1 Cycle Sequencing Kit, Applied Biosystems 4337456) in a 3500xL Genetic Analyzer (Applied Biosystems). Sequences were analyzed using the Geneious Prime v.2024.0.5 (Biomatters, Ltd.) software. For quantitative polymerase chain reaction (qPCR) in blood and tumor samples, 10 µl reactions were prepared using 5 ng cDNA, 1X TaqMan Fast Advanced Master Mix (Applied Biosystems 4444557), and 1X TaqMan Hs01035108_m1 FAM (Applied Biosystems 4453320) and ACTB VIC (Applied Biosystems 4325788) assays. Reactions were prepared in triplicates for all samples and analyzed in a StepOnePlus Real-Time PCR Systems (Applied Biosystems). Relative expression (comparative Ct method) was analyzed with unpaired t test or one-way ANOVA, as appropriate.
First, we ruled out germline variants in other genes with confirmed or suggested association with PitNETs (AIP, ATRX, BRAF, CABLES1, CDKN1B, DICER1, CDKN1A, GNAS, GPR101, MAX, MEN1, MLH1, MSH2, MSH6, PRKAR1A, PIK3CA, PMS2, PTEN, RASD1, RET, SDHA, SDHAF2, SDHB, SDHC, SDHD, TMEM127, TP53, TSC1, TSC2, USP8, USP48, VHL). Using Sanger sequencing, the NF1 variant was observed in heterozygosis in genomic and coding DNA from blood and fresh-frozen and formalin-fixed paraffin-embedded PitNET tissue (Figure 2B). By immunostaining, the tumor displayed areas of moderate or strong granular cytoplasmic immunoreactivity for NF1 with a patchy distribution, similar to what was observed in two PitNETs from a previous publication (23) (Figure 2A).
Using qPCR, we observed significantly increased relative NF1 expression in the patient’s blood (one sample) and fresh frozen PitNET (average of two samples) compared with samples from NF1 wild type controls (P=0.0099 and 0.0040, respectively). NF1 expression, however, was not increased in the patient’s tumor when compared with a sporadic corticotropinoma (P>0.9999) that was negative for USP8, USP48, and BRAF hotspot variants (Figure 2C).
We report the case of a 54-year-old man with clinical manifestations and genetic confirmation of NF1, who was diagnosed with a clinically NF-PitNET. The tumor had preserved heterozygosity at the variant locus, and the abnormal mRNA did not undergo nonsense-mediated RNA decay. By qPCR, significantly elevated NF1 expression levels were observed in both the patient’s blood and PitNET, compared with NF1 wild type controls, but not when compared with a sporadic corticotropinoma. The increased NF1 expression in the tumor may be attributed to compensation for haploinsufficiency, but could also indicate a generic tumor response, since NF1 mRNA is indeed expressed in all PitNET subtypes (43).
Aside from this case, multiple instances of PitNETs have been documented in patients with NF1. Our literature search identified 24 cases (including this one) of PitNETs in individuals with established clinical and/or genetic diagnoses of NF1 or with somatic NF1 variants (Table 2). Only reports with a PitNET demonstrated by imaging studies and/or histopathological analysis were included. The earliest documented cases date back to 1912, and among those with comprehensive data available, there are records of eight females aged 7-70 years at diagnosis, and fourteen males aged 5-65 years. The reports include thirteen patients with clinical features of NF1 without genetic confirmation, eight with germline NF1 variants (one without NF1 manifestations) and three PitNETs harboring somatic NF1 variants. Only one patient with GH excess was reported to have coexistent optic pathway gliomas (Case 17).
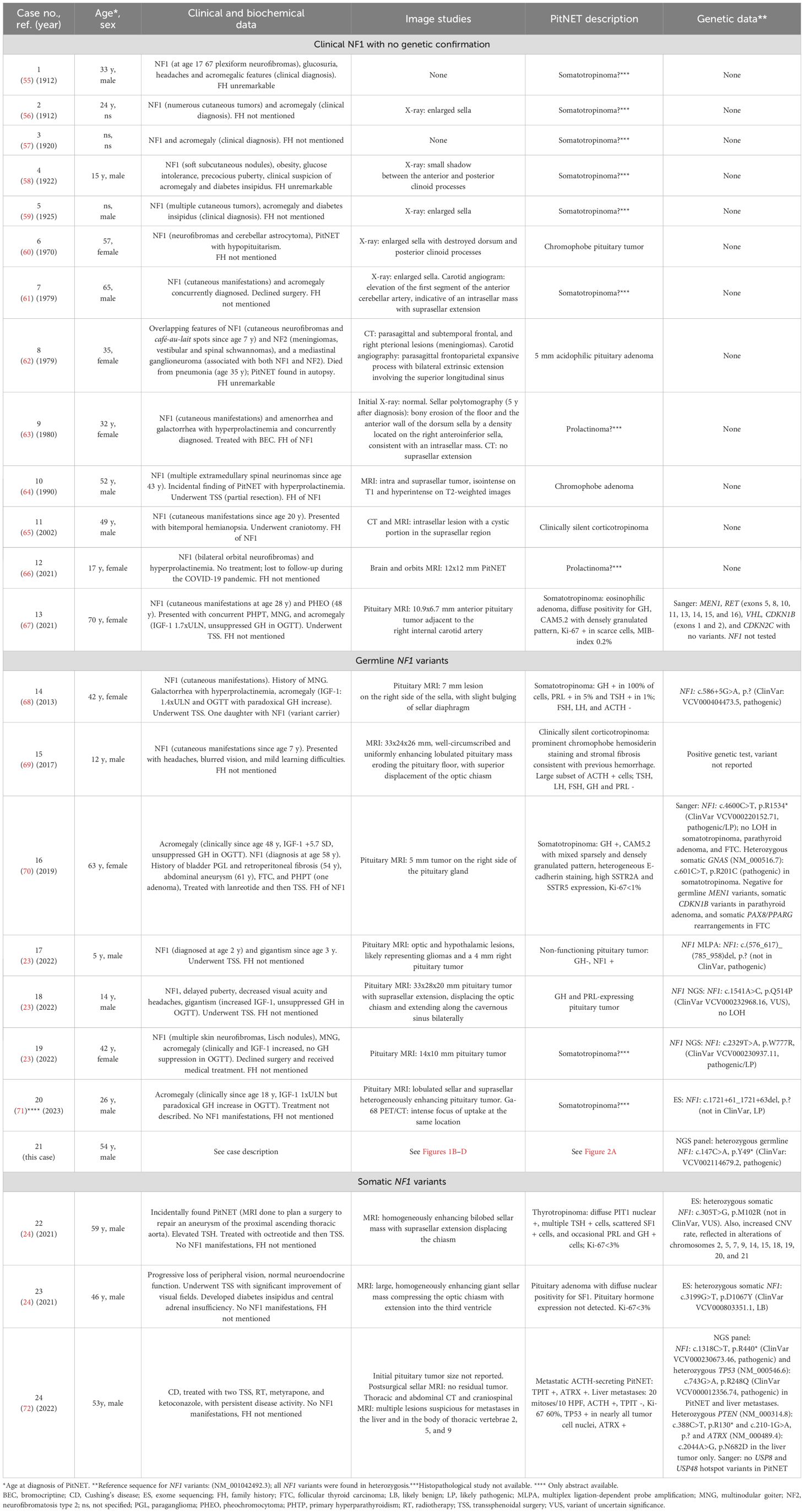
Table 2. Previously reported cases of the association of PitNETs with clinical NF1 and/or NF1 variants.
Not all tumors had a definitive clinical diagnosis due to historical difficulties in hormone assessment. Clinically, 13 (54%) of the patients exhibited acromegalic features, while 5 (21%) presented with compressive symptoms due to large tumor size. Four previously reported patients had tumors clearly categorized as clinically NF-PitNETs: two were a silent corticotropinomas, one was SF1 positive, and one had negative hormonal staining. One patient had a functioning thyrotropinoma, while another presented with hypercortisolism from a metastatic ACTH-secreting PitNET. Imaging was similarly challenging for the earliest cases due to limited techniques available at the time. In cases of PitNETs evaluated with MRI, the dimensions ranged from 5-7 mm for microadenomas and 10-33 mm for macroadenomas, with the latter often resulting in displacement of the optic chiasm. Only one patient with a germline NF1 defect had also genetic testing of the tumor, but no LOH was found.
Germline NF1 variants contribute to tumorigenesis by disrupting the negative regulation of the RAS/MAPK/ERK pathway, leading to enhanced mitotic activity and cellular proliferation (44). Since NF1 is a tumor suppressor, the lack of LOH at the NF1 defect locus in PitNETs from our patient and one previously studied individual (Case 18) may argue against a driver role for NF1 in PitNETs. However, LOH is not a universal finding among NF1-associated tumors. A study of 91 classical non-endocrine NF1 tumors identified LOH in one-fifth of cases, with heterogeneous somatic hits among different tumors, particularly in cases where multiple lesions from the same individual were examined (45). In contrast, LOH is common in PPGLs, occurring both in cases with only somatic variants and in those with coexisting germline defects (10, 11, 46).
Alternative mechanisms could explain a possible causal association of NF1 LOF and PitNETs. Firstly, additional defects could affect different regions of the NF1 gene (47). This possibility cannot be ruled out in cases where only a short sequence was investigated in the tumor (Case 18 and ours). Secondly, as seen in other NF1 manifestations, NF1 haploinsufficiency could be sufficient for the development of PitNETs (48). In line with this, recent studies in mouse models showed that Nf1+/- microenvironment accelerates the development of benign tumors while inhibiting their progression to malignancy (49). Furthermore, for haploinsufficient tumor suppressors, both underexpression (due to loss of one allele) and overexpression (as a compensatory response) can lead to imbalances in tightly regulated signaling pathways (50). An increased NF1 expression could therefore create a paradoxical situation, resulting in toxicity, rather than physiological compensation.
Thirdly, somatic NF1 variants could represent second hits in tumors with a different initial genetic insult. For instance, in a mouse strain with preexistent genomic instability and a high rate of breast cancer development, monoallelic or biallelic Nf1 deletions occur in almost all tumors (51). Similarly, somatic NF1 variants are frequent findings in multiple types of human cancers, often correlating with therapeutic resistance and increased aggressiveness (52). Unsurprisingly, at least one case of a PitNET harboring a somatic pathogenic NF1 variant has been documented (Case 24). In this setting, it remains unclear whether the NF1 sequence change represents a passenger or a driver defect.
Despite these data, the association of NF1 and PitNETs could be coincidental, given the frequent discovery of pituitary incidentalomas in the general population. Individuals with NF1 undergo brain imaging as part of their clinical surveillance, which could increase the risk for incidental pituitary lesions. Nevertheless, patients reported in Table 2 exhibit a high incidence of GH-secreting and large PitNETs, whereas pituitary incidentalomas are most often small NF-PitNETs. Remarkably, 10 (45%) patients were <40 years at PitNET diagnosis. These characteristics (predominance of somatotropinomas and young onset) resemble those of PitNETs caused by proven causative germline defects (53).
A key limitation of this study is the absence of immunostaining for LH, FSH, and relevant transcription factors. The latter immunostainings are part of the current recommendations for PitNET classification (54), but are not available in most centers worldwide, including ours. Despite this, the most likely diagnosis remains a gonadotropinoma, consistent with the majority of NF-PitNET cases. While our review of NF1-associated PitNETs is informative, a larger cohort would provide a more robust understanding of the potential relationship between NF1 and PitNETs. The available data are insufficient to definitively confirm or refute causality, and it remains unclear whether NF1 LOF plays an active role in pituitary tumorigenesis. Further investigations are required to elucidate the role of NF1 variants in the pathogenesis of PitNETs.
The association between NF1 and PitNETs represents a complex interplay that challenges our understanding of both conditions. While NF1 is traditionally associated with optic pathway gliomas and other neoplasms, the emergence of PitNETs in these patients adds a layer of clinical and genetic complexity. The case presented here highlights the need for further investigation into the genetic mechanisms underlying this association. Genetic testing revealed a pathogenic germline variant in NF1, suggesting a potential role in tumorigenesis. However, the absence of LOH in the PitNET points toward a nuanced genetic landscape that requires broader exploration, including deep clinical and genetic characterization of large cohorts of individuals with NF1. We were not able to determine a causal relationship between NF1 and the presence of a PitNET in this patient. Future studies will be essential for unraveling the mechanisms potentially linking NF1 and PitNETs, thereby guiding clinical management and genetic counseling strategies for affected individuals.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/biosample/43250447, SAMN43250447.
The studies involving humans were approved by Comité de Ética en Investigación del Instituto Nacional de Ciencias Médicas y Nutrición. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
MA: Conceptualization, Data curation, Formal Analysis, Investigation, Validation, Visualization, Writing – original draft, Writing – review & editing. JZ: Conceptualization, Data curation, Formal Analysis, Investigation, Validation, Visualization, Writing – original draft, Writing – review & editing. LL: Conceptualization, Data curation, Formal Analysis, Investigation, Validation, Visualization, Writing – original draft, Writing – review & editing. KZ: Formal Analysis, Investigation, Validation, Visualization, Writing – original draft, Writing – review & editing. AG: Formal Analysis, Investigation, Methodology, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AE: Project administration, Resources, Writing – review & editing. LH: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Our research is supported by departmental funding from the Coordination of Scientific Research from the National Autonomous University of Mexico (UNAM), research grants from UNAM’s Support Program for Research Projects and Technological Innovation (UNAM-PAPIIT projects TA200322 and TA200524) and the Mexican Society of Nutrition and Endocrinology, and an equipment grant from the United Kingdom’s Society for Endocrinology.
We are thankful for the kind help of Dr. Rosa G. Rebollar-Vega with NGS, of Dr. Andrea Gutierrez Maria with article retrieval, and of Dr. Donato Iacovazzo with the translation of histopathological data.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ly KI, Blakeley JO. The diagnosis and management of neurofibromatosis type 1. Med Clin North Am. (2019) 103:1035–54. doi: 10.1016/j.mcna.2019.07.004
2. Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM, et al. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A. (2010) 152A:327–32. doi: 10.1002/ajmg.a.33139
3. Kallionpää RA, Uusitalo E, Leppävirta J, Pöyhönen M, Peltonen S, Peltonen J. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet Med. (2018) 20:1082–6. doi: 10.1038/gim.2017.215
4. Bergqvist C, Servy A, Valeyrie-Allanore L, Ferkal S, Combemale P, Wolkenstein P. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J Rare Dis. (2020) 15:37. doi: 10.1186/s13023-020-1310-3
5. Saleh M, Dib A, Beaini S, Saad C, Faraj S, El Joueid Y, et al. Neurofibromatosis type 1 system-based manifestations and treatments: a review. Neurol Sci. (2023) 44:1931–47. doi: 10.1007/s10072-023-06680-5
6. Patel NB, Stacy GS. Musculoskeletal manifestations of neurofibromatosis type 1. AJR Am J Roentgenol. (2012) 199:W99–106. doi: 10.2214/ajr.11.7811
7. Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nat Rev Dis Primers. (2017) 3:17004. doi: 10.1038/nrdp.2017.4
8. Chevalier B, Dupuis H, Jannin A, Lemaitre M, Do Cao C, Cardot-Bauters C, et al. Phakomatoses and endocrine gland tumors: noteworthy and (not so) rare associations. Front Endocrinol (Lausanne). (2021) 12:678869. doi: 10.3389/fendo.2021.678869
9. Bausch B, Borozdin W, Neumann HP. Clinical and genetic characteristics of patients with neurofibromatosis type 1 and pheochromocytoma. N Engl J Med. (2006) 354:2729–31. doi: 10.1056/NEJMc066006
10. Burnichon N, Buffet A, Parfait B, Letouze E, Laurendeau I, Loriot C, et al. Somatic NF1 inactivation is a frequent event in sporadic pheochromocytoma. Hum Mol Genet. (2012) 21:5397–405. doi: 10.1093/hmg/dds374
11. Welander J, Larsson C, Backdahl M, Hareni N, Sivler T, Brauckhoff M, et al. Integrative genomics reveals frequent somatic NF1 mutations in sporadic pheochromocytomas. Hum Mol Genet. (2012) 21:5406–16. doi: 10.1093/hmg/dds402
12. Gruber LM, Erickson D, Babovic-Vuksanovic D, Thompson GB, Young WF Jr., Bancos I. Pheochromocytoma and paraganglioma in patients with neurofibromatosis type 1. Clin Endocrinol (Oxf). (2017) 86:141–9. doi: 10.1111/cen.13163
13. Tabebi M, Frikha F, Volpe M, Gimm O, Soderkvist P. Domain landscapes of somatic NF1 mutations in pheochromocytoma and paraganglioma. Gene. (2023) 872:147432. doi: 10.1016/j.gene.2023.147432
14. Patil S, Chamberlain RS. Neoplasms associated with germline and somatic NF1 gene mutations. Oncologist. (2012) 17:101–16. doi: 10.1634/theoncologist.2010-0181
15. Habiby R, Silverman B, Listernick R, Charrow J. Precocious puberty in children with neurofibromatosis type 1. J Pediatr. (1995) 126:364–7. doi: 10.1016/s0022-3476(95)70449-3
16. Cnossen MH, Stam EN, Cooiman LC, Simonsz HJ, Stroink H, Oranje AP, et al. Endocrinologic disorders and optic pathway gliomas in children with neurofibromatosis type 1. Pediatrics. (1997) 100:667–70. doi: 10.1542/peds.100.4.667
17. Carmi D, Shohat M, Metzker A, Dickerman Z. Growth, puberty, and endocrine functions in patients with sporadic or familial neurofibromatosis type 1: a longitudinal study. Pediatrics. (1999) 103:1257–62. doi: 10.1542/peds.103.6.1257
18. Virdis R, Sigorini M, Laiolo A, Lorenzetti E, Street ME, Villani AR, et al. Neurofibromatosis type 1 and precocious puberty. J Pediatr Endocrinol Metab. (2000) 13 Suppl 1:841–4. doi: 10.1515/jpem.2000.13.s1.841
19. Bizzarri C, Bottaro G. Endocrine implications of neurofibromatosis 1 in childhood. Horm Res Paediatr. (2015) 83:232–41. doi: 10.1159/000369802
20. Cambiaso P, Galassi S, Palmiero M, Mastronuzzi A, Del Bufalo F, Capolino R, et al. Growth hormone excess in children with neurofibromatosis type-1 and optic glioma. Am J Med Genet A. (2017) 173:2353–8. doi: 10.1002/ajmg.a.38308
21. Sani I, Albanese A. Endocrine long-term follow-up of children with neurofibromatosis type 1 and optic pathway glioma. Horm Res Paediatr. (2017) 87:179–88. doi: 10.1159/000458525
22. Hannah-Shmouni F, Stratakis CA. Growth hormone excess in neurofibromatosis 1. Genet Med. (2019) 21:1254–5. doi: 10.1038/s41436-018-0312-1
23. Hannah-Shmouni F, Trivellin G, Beckers P, Karaviti LP, Lodish M, Tatsi C, et al. Neurofibromatosis type 1 has a wide spectrum of growth hormone excess. J Clin Med. (2022) 11:2168. doi: 10.3390/jcm11082168
24. Hong CS, Kundishora AJ, Elsamadicy AA, Koo AB, McGuone D, Inzucchi SE, et al. Somatic NF1 mutations in pituitary adenomas: Report of two cases. Cancer Genet. (2021) 256-257:26–30. doi: 10.1016/j.cancergen.2021.03.004
25. Ziyatdinov A, Torres J, Alegre-Diaz J, Backman J, Mbatchou J, Turner M, et al. Genotyping, sequencing and analysis of 140,000 adults from Mexico City. Nature. (2023) 622:784–93. doi: 10.1038/s41586-023-06595-3
26. Chen S, Francioli LC, Goodrich JK, Collins RL, Kanai M, Wang Q, et al. A genomic mutational constraint map using variation in 76,156 human genomes. Nature. (2024) 625:92–100. doi: 10.1038/s41586-023-06045-0
27. Valero MC, Martin Y, Hernandez-Imaz E, Marina Hernandez A, Melean G, Valero AM, et al. A highly sensitive genetic protocol to detect NF1 mutations. J Mol Diagn. (2011) 13:113–22. doi: 10.1016/j.jmoldx.2010.09.002
28. Lyskjaer I, Lindsay D, Tirabosco R, Steele CD, Lombard P, Strobl AC, et al. H3K27me3 expression and methylation status in histological variants of Malignant peripheral nerve sheath tumours. J Pathol. (2020) 252:151–64. doi: 10.1002/path.5507
29. de Witte CJ, Kutzera J, van Hoeck A, Nguyen L, Boere IA, Jalving M, et al. Distinct genomic profiles are associated with treatment response and survival in ovarian cancer. Cancers (Basel). (2022) 14:1511. doi: 10.3390/cancers14061511
30. Ge LL, Xing MY, Zhang HB, Wang ZC. Neurofibroma development in neurofibromatosis type 1: insights from cellular origin and Schwann cell lineage development. Cancers (Basel). (2022) 14:4513. doi: 10.3390/cancers14184513
31. Sakamoto I, Hirotsu Y, Amemiya K, Nozaki T, Mochizuki H, Omata M. Elucidation of genomic origin of synchronous endometrial and ovarian cancer (SEO) by genomic and microsatellite analysis. J Gynecol Oncol. (2023) 34:e6. doi: 10.3802/jgo.2023.34.e6
32. Fahsold R, Hoffmeyer S, Mischung C, Gille C, Ehlers C, Kucukceylan N, et al. Minor lesion mutational spectrum of the entire NF1 gene does not explain its high mutability but points to a functional domain upstream of the GAP-related domain. Am J Hum Genet. (2000) 66:790–818. doi: 10.1086/302809
33. Sabbagh A, Pasmant E, Imbard A, Luscan A, Soares M, Blanche H, et al. NF1 molecular characterization and neurofibromatosis type I genotype-phenotype correlation: the French experience. Hum Mutat. (2013) 34:1510–8. doi: 10.1002/humu.22392
34. FastQC: a quality control tool for high throughput sequence data(2010). Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (Accessed September 3, 2023).
35. Garrison E, Marth G. Haplotype-based variant detection from short-read sequencing. (2012). doi: 10.48550/arXiv.1207.3907
36. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM(2013). Available online at: http://arxiv.org/abs/1303.3997 (Accessed September 3, 2023).
37. Picard. Broad Institute, GitHub repository. Available online at: https://broadinstitute.github.io/picard/ (Accessed September 3, 2023).
38. Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. (2012) 28:2747–54. doi: 10.1093/bioinformatics/bts526
39. Franklin by Genoox. Available online at: https://franklin.genoox.com/clinical-db/home (Accessed September 7, 2023).
40. Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. (2014) 42:D980–5. doi: 10.1093/nar/gkt1113
41. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
42. Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. VarSome: the human genomic variant search engine. Bioinformatics. (2019) 35:1978–80. doi: 10.1093/bioinformatics/bty897
43. Neou M, Villa C, Armignacco R, Jouinot A, Raffin-Sanson ML, Septier A, et al. Pangenomic classification of pituitary neuroendocrine tumors. Cancer Cell. (2020) 37:123–34.e5. doi: 10.1016/j.ccell.2019.11.002
44. Gottfried ON, Viskochil DH, Couldwell WT. Neurofibromatosis Type 1 and tumorigenesis: molecular mechanisms and therapeutic implications. Neurosurgical Focus FOC. (2010) 28:E8. doi: 10.3171/2009.11.FOCUS09221
45. Upadhyaya M, Han S, Consoli C, Majounie E, Horan M, Thomas NS, et al. Characterization of the somatic mutational spectrum of the neurofibromatosis type 1 (NF1) gene in neurofibromatosis patients with benign and Malignant tumors. Hum Mutat. (2004) 23:134–46. doi: 10.1002/humu.10305
46. Bausch B, Borozdin W, Mautner VF, Hoffmann MM, Boehm D, Robledo M, et al. Germline NF1 mutational spectra and loss-of-heterozygosity analyses in patients with pheochromocytoma and neurofibromatosis type 1. J Clin Endocrinol Metab. (2007) 92:2784–92. doi: 10.1210/jc.2006-2833
47. Pemov A, Li H, Patidar R, Hansen NF, Sindiri S, Hartley SW, et al. The primacy of NF1 loss as the driver of tumorigenesis in neurofibromatosis type 1-associated plexiform neurofibromas. Oncogene. (2017) 36:3168–77. doi: 10.1038/onc.2016.464
48. Frost M, Serra E, Viskochil D, Korf BR, Mattson-Hoss MK, Croston GE, et al. Rationale for haploinsufficiency correction therapy in neurofibromatosis type 1. J Trans Genet Genomics. (2022) 6:403–28. doi: 10.20517/jtgg.2022.14
49. Brosseau JP, Liao CP, Wang Y, Ramani V, Vandergriff T, Lee M, et al. NF1 heterozygosity fosters de novo tumorigenesis but impairs Malignant transformation. Nat Commun. (2018) 9:5014. doi: 10.1038/s41467-018-07452-y
50. Morrill SA, Amon A. Why haploinsufficiency persists. Proc Natl Acad Sci U.S.A. (2019) 116:11866–71. doi: 10.1073/pnas.1900437116
51. Wallace MD, Pfefferle AD, Shen L, McNairn AJ, Cerami EG, Fallon BL, et al. Comparative oncogenomics implicates the neurofibromin 1 gene (NF1) as a breast cancer driver. Genetics. (2012) 192:385–96. doi: 10.1534/genetics.112.142802
52. Philpott C, Tovell H, Frayling IM, Cooper DN, Upadhyaya M. The NF1 somatic mutational landscape in sporadic human cancers. Hum Genomics. (2017) 11:13. doi: 10.1186/s40246-017-0109-3
53. Korbonits M, Blair JC, Boguslawska A, Ayuk J, Davies JH, Druce MR, et al. Consensus guideline for the diagnosis and management of pituitary adenomas in childhood and adolescence: Part 1, general recommendations. Nat Rev Endocrinol. (2024) 20:278–89. doi: 10.1038/s41574-023-00948-8
54. Asa SL, Mete O, Perry A, Osamura RY. Overview of the 2022 WHO classification of pituitary tumors. Endocr Pathol. (2022) 33:6–26. doi: 10.1007/s12022-022-09703-7
55. De Castro A. Sur la coexistence de la maladie de Recklinghausen avec l’acromégalie. In: Nouvelle iconographie de la Salpétrière. Paris: Lecrosnier et Babè. vol. 25. France (1912). p. 41–4.
56. Wolfsohn GM. E. Neurofibromatosis und akromegalie. Berliner Klinische Wochenschrift. (1912) 49:1088–90.
57. Ormond AW. Case of von recklinghausen’s disease with acromegaly. Proc R Soc Med. (1920) 13:124–6.
58. Barber HW, Shaw M. Recklinghausen’s disease with pituitary tumour. Proc R Soc Med. (1922) 15:30–1.
59. Darquier LC. Syndrome de Recklinghausen et Acromegalie. Bull la Societe Francaise Dermatologie Syphiligraphie. (1925) 54–8.
60. Boudin G, Pepin B, Vernant CL. Multiple tumours of the nervous system in Recklinghausen’s disease. An anatomo-clinical case with chromophobe adenoma of the pituitary gland. Presse Med (1893). (1970) 78:1427–30.
61. Adeloye A. Coexistence of acromegaly and neurofibromatosis in a Nigerian. East Afr Med J. (1979) 56:38–9.
62. Barberis M, Gambacorta M, Versari P, Filizzolo F. About a case of Recklinghausen’s disease associated with pituitary adenoma (author’s transl). Pathologica. (1979) 71:265–72.
63. Pinnamaneni K, Birge SJ, Avioli LV. Prolactin-secreting pituitary tumor associated with von Recklinghausen’s disease. Arch Intern Med. (1980) 140:397–9. doi: 10.1001/archinte.1980.00330150111026
64. Nakajima M, Nakasu Y, Nakasu S, Matsuda M, Handa J. Pituitary adenoma associated with neurofibromatosis: case report. Nihon Geka Hokan. (1990) 59:278–82.
65. Kurozumi K, Tabuchi A, Ono Y, Tamiya T, Ohmoto T, Furuta T, et al. Pituitary adenoma associated with neurofibromatosis type 1: case report. No Shinkei Geka. (2002) 30:741–5.
66. Mishra Hk R, Kumar A, Kumar A, Suman S. Incidentally detected pituitary macroadenoma in a case of neurofibromatosis type 1: A case report and review of literature. Indian J Case Rep. (2021) 7:244–6. doi: 10.32677/IJCR.2021.v07.i06.008
67. Yasuda S, Inoue I, Shimada A. Neurofibromatosis type 1 with concurrent multiple endocrine disorders: adenomatous goiter, primary hyperparathyroidism, and acromegaly. Intern Med. (2021) 60:2451–9. doi: 10.2169/internalmedicine.4981-20
68. Checa Garrido A, del Pozo Pico C. Acromegaly and type 1 neurofibromatosis. Is association of both conditions due to chance? Endocrinol Nutr. (2013) 60:144–5. doi: 10.1016/j.endonu.2012.01.020
69. Smith N, Santoreneos S. Non-functioning pituitary macroadenoma in a child with neurofibromatosis type 1. ANZ J Surg. (2017) 87:E220–E1. doi: 10.1111/ans.13108
70. Hozumi K, Fukuoka H, Odake Y, Takeuchi T, Uehara T, Sato T, et al. Acromegaly caused by a somatotroph adenoma in patient with neurofibromatosis type 1. Endocrine J. (2019) 66:853–7. doi: 10.1507/endocrj.EJ19-0035
71. Alsagheir O, Alobaid LA, Alswailem M, Al-Hindi H, Alzahrani AS. A novel NF1 mutation as the underlying cause of dysmorphic features and acromegaly in an atypical case of neurofibromatosis type 1. J Endocrine Soc. (2023) 7:SAT613. doi: 10.1210/jendso/bvad114.1346
Keywords: endocrine neoplasia, genetic diagnosis, neurofibromatosis type 1, NF1, pituitary neuroendocrine tumors, tumor suppressor
Citation: Aguilar-Soto M, Zuarth-Vázquez JM, Leyva-Figueroa L, Zarco-Ávila K, Gamboa-Domínguez A, Eguiluz-Melendez A and Hernández-Ramírez LC (2025) Association of pituitary neuroendocrine tumors and neurofibromatosis type 1: assessing causation versus coincidence. Case report. Front. Endocrinol. 16:1483305. doi: 10.3389/fendo.2025.1483305
Received: 19 August 2024; Accepted: 16 January 2025;
Published: 04 February 2025.
Edited by:
Nils Lambrecht, United States Department of Veterans Affairs, United StatesReviewed by:
Heraldo Garmes, State University of Campinas, BrazilCopyright © 2025 Aguilar-Soto, Zuarth-Vázquez, Leyva-Figueroa, Zarco-Ávila, Gamboa-Domínguez, Eguiluz-Melendez and Hernández-Ramírez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Laura C. Hernández-Ramírez, bGF1cmEuaGVybmFuZGV6QGNpYy51bmFtLm14
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.