 Africa Manero-Azua1†
Africa Manero-Azua1† Yerai Vado1†
Yerai Vado1† Judith Gonzàlez Morlà2Eduard Mogas3
Judith Gonzàlez Morlà2Eduard Mogas3 Arrate Pereda1
Arrate Pereda1 Guiomar Perez de Nanclares1*
Guiomar Perez de Nanclares1*- 1Rare Disease Research Group, Molecular (Epi) Genetics Laboratory, Bioaraba Health Research Institute, Araba University Hospital, Vitoria-Gasteiz, Spain
- 2Pediatric Endocrinology Section, Hospital Comarcal de Palamós, Girona, Spain
- 3Pediatric Endocrinology Section, Hospital Universitari Vall d’Hebron, Barcelona, Spain
Objective: To identify the genetic cause underlying the methylation defect in a patient with clinical suspicion of PHP1B/iPPSD3.
Design: Imprinting is an epigenetic mechanism that allows the regulation of gene expression. The GNAS locus is one of the loci within the genome that is imprinted. When the methylation pattern is affected, it causes pseudohypoparathyroidism type 1B (PHP1B) or inactivating PTH/PTHrP signaling disorder 3 (iPPSD3). Paternal uniparental isodisomy (iUPDpat) of the chromosomal region comprising the GNAS locus has been described as one of the possible underlying genetic causes of the methylation alteration.
Methods: We present the case of a patient clinically diagnosed with iPPSD3. We performed a commercial methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA), single-nucleotide polymorphism (SNP) array, and microsatellite study. In addition, we designed a custom MS-MLPA to analyze GNAS and nearby differentially methylated regions (DMRs).
Results: A methylation defect at the four GNAS-DMRs was detected, confirming the clinical diagnosis. Complementary techniques revealed the presence of a mixed isodisomy and heterodisomy of chromosome 20. Surprisingly, the GNAS locus was located on the heterodisomic zone.
Conclusions: Paternal uniparental heterodisomy (hUPD) at the GNAS locus is also a genetic defect associated with iPPSD3. In the absence of parental samples, our custom MS-MLPA allows for the detection of a methylation defect at the GNAS locus and flanking DMRs, suggestive of uniparental disomy (UPD). We also suggest updating the actual guidelines to include hUPD at the GNAS locus as a cause of iPPSD3.
1 Introduction
The GNAS locus is an imprinted locus located in the long arm of chromosome 20 (20q13.2-20q13.3) (1). Due to its complex imprinted pattern, the expression of the multiple transcripts derived from the locus is dependent on the parental origin (2).
There are four first alternative exons, which are spliced onto exons 2-13, that are common for all the transcripts. These alternative first exons are regulated by four differentially methylated regions (DMRs), which are cis-acting elements that control the parent-of-origin expression: GNAS-NESP:TSS-DMR, GNAS-AS1:TSS-DMR, GNAS-XL:Ex1-DMR and GNAS A/B:TSS-DMR. The first one is methylated in the paternal allele and in consequence, is expressed from the maternal allele. In contrast, the other three are imprinted in the maternal allele and expressed from the paternal one (3–7).
When the methylation of the complex locus is abnormal in the DMRs, it gives rise to the disease known as pseudohypoparathyroidism type 1B (PHP1B, OMIM#603233) or according to the recent recommendations, inactivating PTH/PTHrP signaling disorder 3 (iPPSD3) (8). The methylation defect can affect one or multiple DMRs and it can be partial or complete (9), but all patients affected with iPPSD3 show loss of methylation (LOM) at the GNAS A/B:TSS-DMR (10). Clinically, iPPSD3 patients have been characterized by parathyroid hormone (PTH) resistance, leading to hypocalcemia and hyperphosphatemia. Sometimes, thyroid stimulating hormone (TSH) resistance is also present. In these patients, Albright’s Hereditary Osteodystrophy (AHO) phenotype is commonly absent, and if present, it is usually milder (10, 11). Some additional signs include excessive intrauterine growth (12, 13).
Of the iPPSD3 patients, 15%-20% of the cases are familial, with LOM at the GNAS A/B:TSS-DMR secondary to deletion of 3.3 kb in the STX16 gene. This genetic defect is inherited from the maternal lineage in an autosomal dominant manner (10), and is accompanied by a GNAS-AS2:TSS-DMR partial LOM (14). However, there are some rare familial cases with different deletions and duplications affecting only the GNAS A/B:TSS-DMR or all the DMRs (10). Apart from the familial cases, there are also sporadic iPPSD3 patients (15). In the majority of the sporadic cases, a methylation alteration usually affects the four GNAS-DMRs and the genetic cause of the methylation defect remains unknown. Nevertheless, in approximately 8%-20% of them, paternal uniparental isodisomy (iUPD) including 20q13 has been detected (15–17).
Uniparental disomy (UPD) is an abnormal genetic condition in which the individual carries a pair of chromosomes (or segments of chromosomes) from the same progenitor without a contribution from the other parent. Two different types of UPD can be distinguished: uniparental heterodisomy (hUPD), when both homologous chromosomes are inherited from one parent; and, uniparental isodisomy (iUPD), when a single chromosome is inherited from the parent and it duplicates (18, 19). Although it is very rare, translocations giving rise to paternal uniparental disomy of chromosome 20, upd(20)pat, have also been described (15).
For the detection of paternal iUPD in 20q13 [iUPD(20q)pat], current international guidelines for the diagnosis of PHP and related disorders recommend the analysis of microsatellites or short tandem repeats (STR) in the trio (if the samples are available) and/or single nucleotide polymorphism (SNP) array (15). However, if the samples of the parents are not available, STR analysis can be non-informative (20).
2 Materials and methods
2.1 Case presentation
We present the clinical case of a 3-year-old boy who was admitted to the hospital emergency department due to a second seizure episode in the context of a viral illness caused by influenza A. The initial blood tests in the emergency room revealed hypocalcemia with a calcium level of 5.48 mg/dL (NV 9.4-10.8 mg/dl). Further examinations were conducted, showing a low ionized calcium level of 4.15 mg/dL (4.8-5.3 mg/dl), phosphorus of 6.1 mg/dL (3.5-6 mg/dL), PTH of 859 pg/mL (10-65 pg/ml), alkaline phosphatase of 181 UI/L (150-600 UI/L), and vitamin D (25 OH cholecalciferol) of 17 ng/mL (25-85 ng/ml). The conclusion from these results was a possible diagnosis of pseudohypoparathyroidism.
2.1.1 Family and perinatal history
There was no consanguinity or relevant family history reported. Dichorionic diamniotic opposite-sex twin pregnancy was ended by Cesarean section due to the malposition of his twin. The full-term neonates were born at 37 + 5 weeks of gestation. The patient was born small for his gestational age with a birth weight of 2.250 g (-2.0 SD), birth length of 45 cm (-2.4 SD), and a head circumference of 31.5 cm (-1.46 SD). The birth weight of the female twin was normal at 2.700 g (-0.6 SD) with a length of 46 cm (SD –1.1 SD) and her head circumference was 32 cm (-0.9 SD). Both twins had a normal perinatal evolution.
2.1.2 Past medical history
The patient had a first episode of seizure at 2 years and 10 months. In that context, he was febrile, concluding a diagnosis of atypical febrile seizure due to the seizure’s long duration, his slow recovery, and residual Todd´s paralysis on the left side of his body, which evolved successfully. Brain computed tomography (CT) and magnetic resonance imaging (MRI) were performed with normal results. However, electroencephalography showed epileptiform paroxysms in the occipital regions with right-sided predominance, and treatment with levetiracetam was started. The patient also had a history of mild congenital heart disease due to a bicuspid aortic valve and mild aortic stenosis, which is currently followed by the pediatric cardiology department with no current treatment.
2.1.3 Physical examination
No apparent dysmorphia was observed. His current weight is 20 kg (-0.86 SD), with a height of 109 cm (0.99 SD). Tanner Stage 1 was assessed for both testes in the scrotum.
2.1.4 Additional examinations
Blood test: glucose = 152 mg/dL, urea = 24.79 mg/dL, creatinine = 0.73 mg/dL, sodium = 135 mEq/L, potassium = 3.6 mEq/L, chloride = 103 mEq/L, total calcium = 5.48 mg/dL, ionized calcium = 4.15 mg/dL, phosphorus = 6.1 mg/dL, magnesium = 0.91 mg/dL, albumin = 4.02 g/dL, PTH = 859 pg/mL, alkaline phosphatase = 181 U/L, vitamin D (25 OH) = 17 ng/mL, AST = 38 U/L, and ALT = 12 U/L.
Urine test: creatinine = 34.79 mg/dL, calcium = 5.07 mg/dL, Ca/Cre ratio= 0.14 (normal values < 0.2), and phosphorus = 19.40 mg/dL. Complete blood count: 23,750 leukocytes/mm³ (32% L, 58% N, 0% C). Normal thyroid function. Nasopharyngeal swab: positive PCR for influenza A.
Normal chest X-ray.
2.1.5 Evolution
Upon arrival at the emergency department, the patient presented with a generalized seizure and a temperature of 39°C. Antipyretics and intravenous midazolam (0.1 mg/kg) were administered, resolving the seizure.
Hypocalcemia was corrected with intravenous calcium infusion, followed by oral supplementation, with progressive correction of calcium levels, while maintaining phosphorus and magnesium within normal ranges.
Given the clinical suspicion of pseudohypoparathyroidism, treatment with calcitriol [1,25(OH)2 VitD]) 0.25 mcg/24 h, calcium carbonate 500 mg/12 h, and calcidiol (25OH VitD) 4000 IU/24 h was prescribed.
Follow-up of the patient was satisfactory, with normalization of the phosphate-calcium metabolism parameters after 2 months of the treatment (calcium 9.3 mg/dl, ionized calcium 5.07 mg/dl, phosphate 5.38 mg/dl, PTH 96 pg/ml, alkaline phosphatase 151 U/L, and 25OH vitamin D 42 ng/mL). Neurologically, while on the treatment with levetiracetam, the patient has not presented with new seizures, but a current electroencephalogram (EEG) (at the age of 4 years) continues to show the presence of epileptiform discharges.
2.2 Nucleic acid extraction
The genomic DNA of the patient and parents was extracted from peripheral blood using the QIAamp DNA Mini Kit (QIAGEN, Hilden, Germany), following the manufacturer’s instructions.
2.3 Molecular studies
2.3.1 Methylation-specific multiplex ligation-dependent probe amplification
Dosage and methylation analyses for chromosome 20 were carried out using two different methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) experiments, following the manufacturer’s recommendations (MRC-Holland, Amsterdam, The Netherlands). For the study of the GNAS locus, the ME031-C1 kit was used. A custom MS-MLPA was designed to detect upd(20q)pat, including probes in the MCTS2P:TSS-DMR (20q11.21), NNAT: TSS-DMR (20q11.2-q12), L3MBTL1:alt-TSS-DMR (20q13.12), and GNAS A/B:TSS-DMR (20q13.32) (Table 1). These custom-designed probes were combined with the SALSA MLPA Probemix P200 Reference-1 kit, which contains reference probes for unique human DNA sequences and specific quality control fragments.
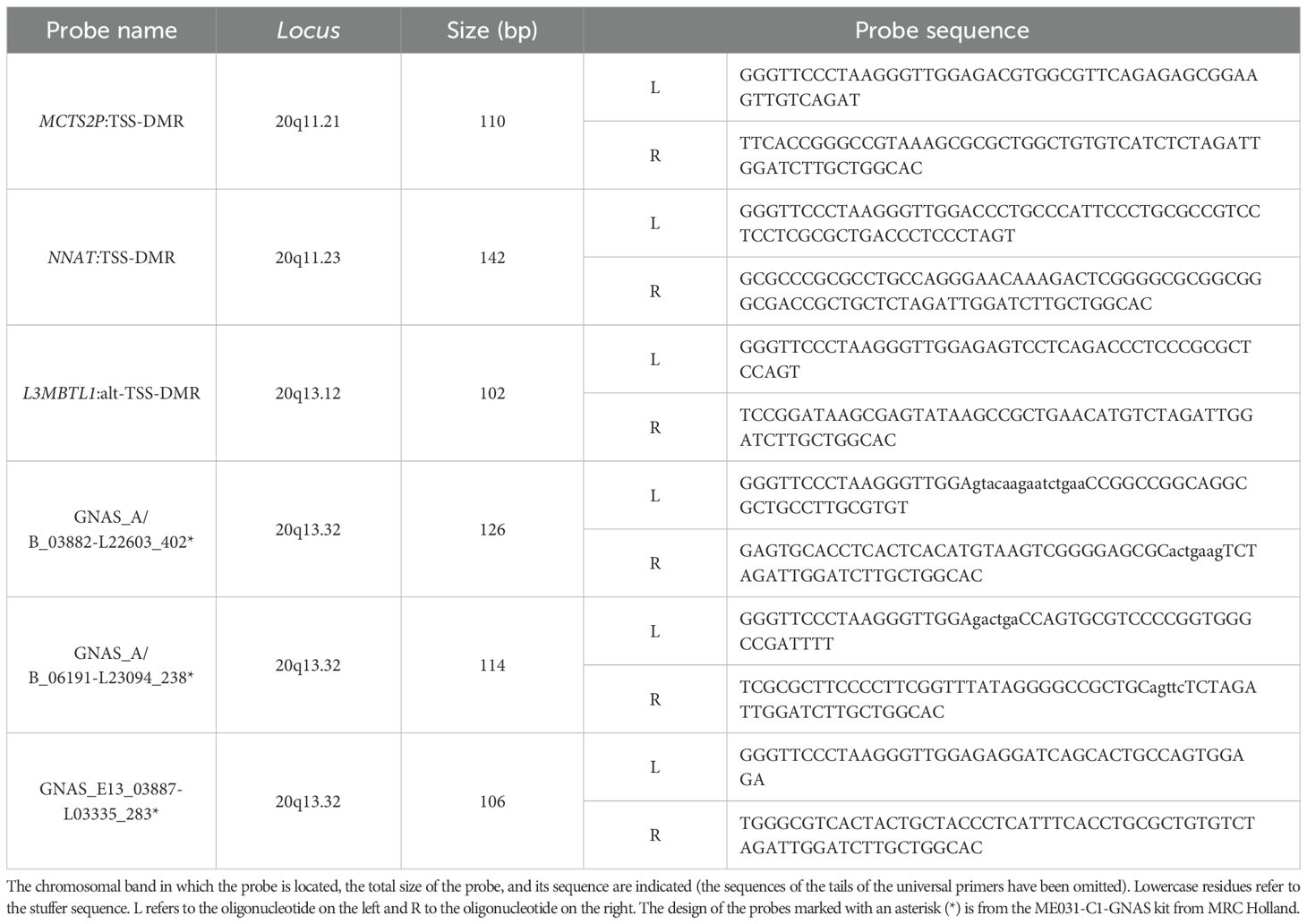
Table 1. Probe set of the custom-designed MS-MLPA kit (MS-MLPA UPD20q).
The size of the fragments was evaluated with an ABI 3500 Genetic analyzer (Applied Biosystems, Foster City, California, USA), according to the manufacturer’s protocols. Data were analyzed using Coffalyser.net version 9.4 software (MRC-Holland).
2.3.2 SNP array
The SNP array technique was used to confirm and determine the possible extent of the obtained findings by MS-MLPA. For this purpose, the CytoScan High-Density assay (ThermoFisher Scientific) was performed to genotype 906,600 SNPs, and the subsequent analysis was carried out using the recommended Chromosome Analysis Suite (ChAS) software version 4.5 (ThermoFisher Scientific).
2.3.3 Microsatellite study
The STR study was performed for the trio by fluorescent polymerase chain reaction (PCR), using different markers along the long arm (D20S496, D20S459, D20S443, D20S171, D20S173, D20S1044, D20S468, D20S746, D20S195, D20S174, D20S866, and D20S891) and short arm (D20S864, D20S103, D20S105, D20S894, D20S114 and D20S180) of chromosome 20 (Figure 1).
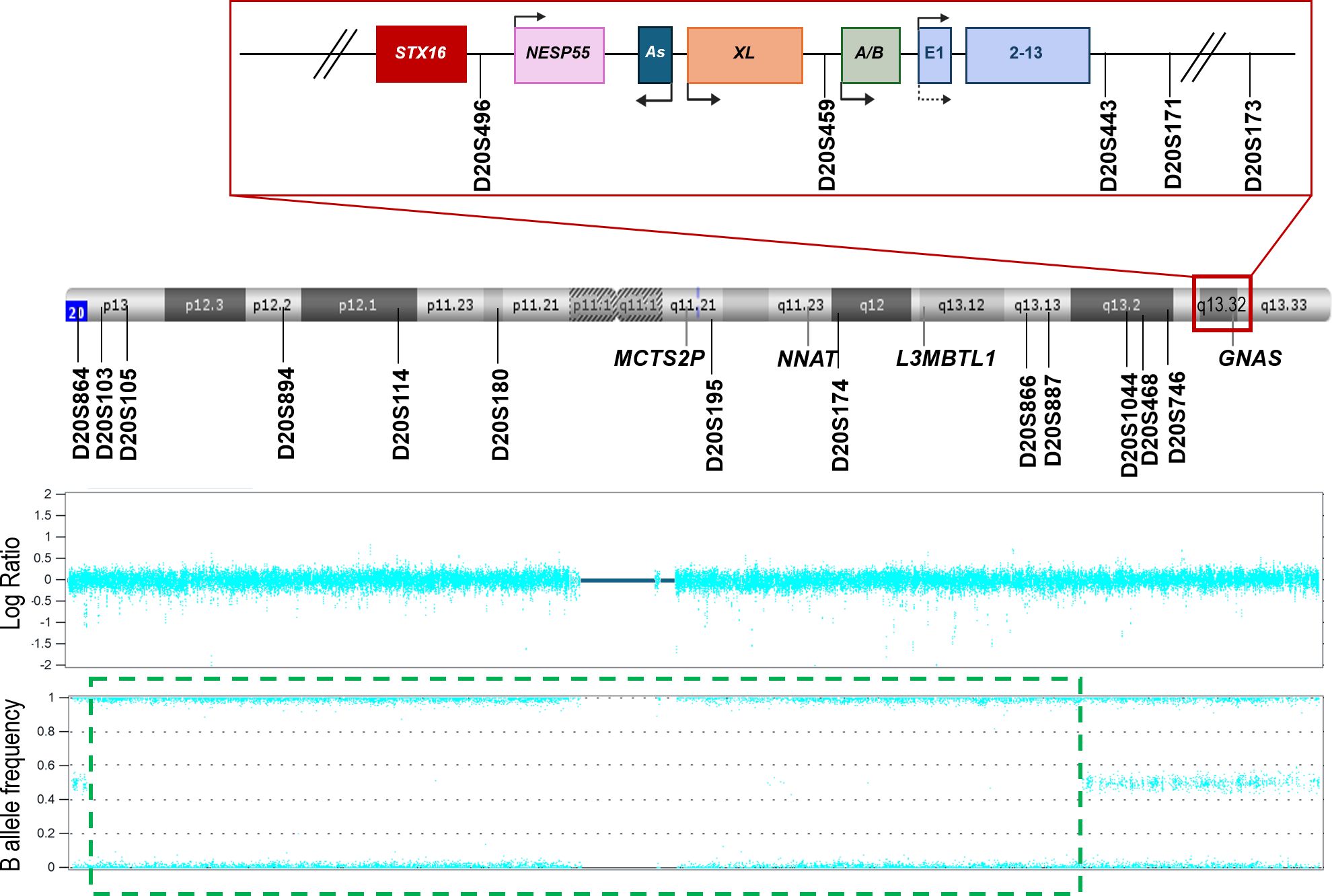
Figure 1. Schematic representation of chromosome 20, the distribution of the microsatellites studied, and the results obtained from the SNP array. The GNAS locus (red square) has been magnified to view it in more detail. The different DMRs of the locus are denoted by pink (NESP55), dark blue (AS), orange (XL), and green (A/B) squares. The regulator gene STX16 (upstream GNAS locus) is represented by a red square. The GNAS gene is drawn in light blue. Arrows indicate the transcription sense. MCTS2P, NNAT, and L3MBTL1 are other imprinted loci in the chromosome. The position of each STR analyzed throughout the chromosome and in the GNAS locus is indicated with a straight line. In the lower part of the image, the SNP array results are presented. In the Log Ratio panel, no copy number variations are shown. In the lower panel, the green dashed square delimits a zone with no heterozygosity.
The thermocycling program was: 95°C (5 min); 35x{95°C (30 s); 55°C (30 s); 72°C (30 s)}; 72°C (7 min); 60°C (1 h); 4°C (∞). After thermocycling, the product was visualized in an agarose gel (1.5% w/v) electrophoresis with 1x TAE (Thermo Fisher Scientific, Waltham, Massachusetts, USA) and subsequent dilutions were performed.
The fragments’ size was determined with an ABI 3500 Genetic analyzer. The results were analyzed with GeneMapper Software 5 (Applied Biosystems).
3 Results
3.1 GNAS MS-MLPA
The methylation status of the GNAS locus was evaluated and complete methylation alteration was found in all the DMRs (Data not shown). Specifically, gain of methylation (GOM) was present in the GNAS-NESP: TSS-DMR and LOM in the GNAS-AS1:TSS-DMR, GNAS-XL:Ex1-DMR and GNAS A/B:TSS-DMR. We also detected that no copy number variations were present. As the methylation was completely altered, a paternal UPD needed to be checked.
3.2 SNP array
To confirm/discard UPD, a SNP array in the proband case was performed, revealing loss of heterozygosity from 20p13 to 20q13.13 (Figure 1), suggesting either the possibility of homozygosity due to a high level of consanguinity or an isodisomy in that zone (not including GNAS).
No other regions with loss of heterozygosity were detected.
3.3 Custom upd(20q)pat MS-MLPA
The custom MS-MLPA for detecting UPD in chromosome 20q gave the results represented in Figure 2. Briefly, it can be observed that the GNAS locus and the DMRs upstream of GNAS present a complete LOM in the patient, suggesting that the observed loss of heterozygosity could be the consequence of an upd(20q)pat. The digestion control probes and the reference probes showed the expected methylation and genomic levels, indicating that the experiment worked well. A control individual with no disease was included, and, as expected, no methylation alterations or copy number variants (CNVs) were detected.
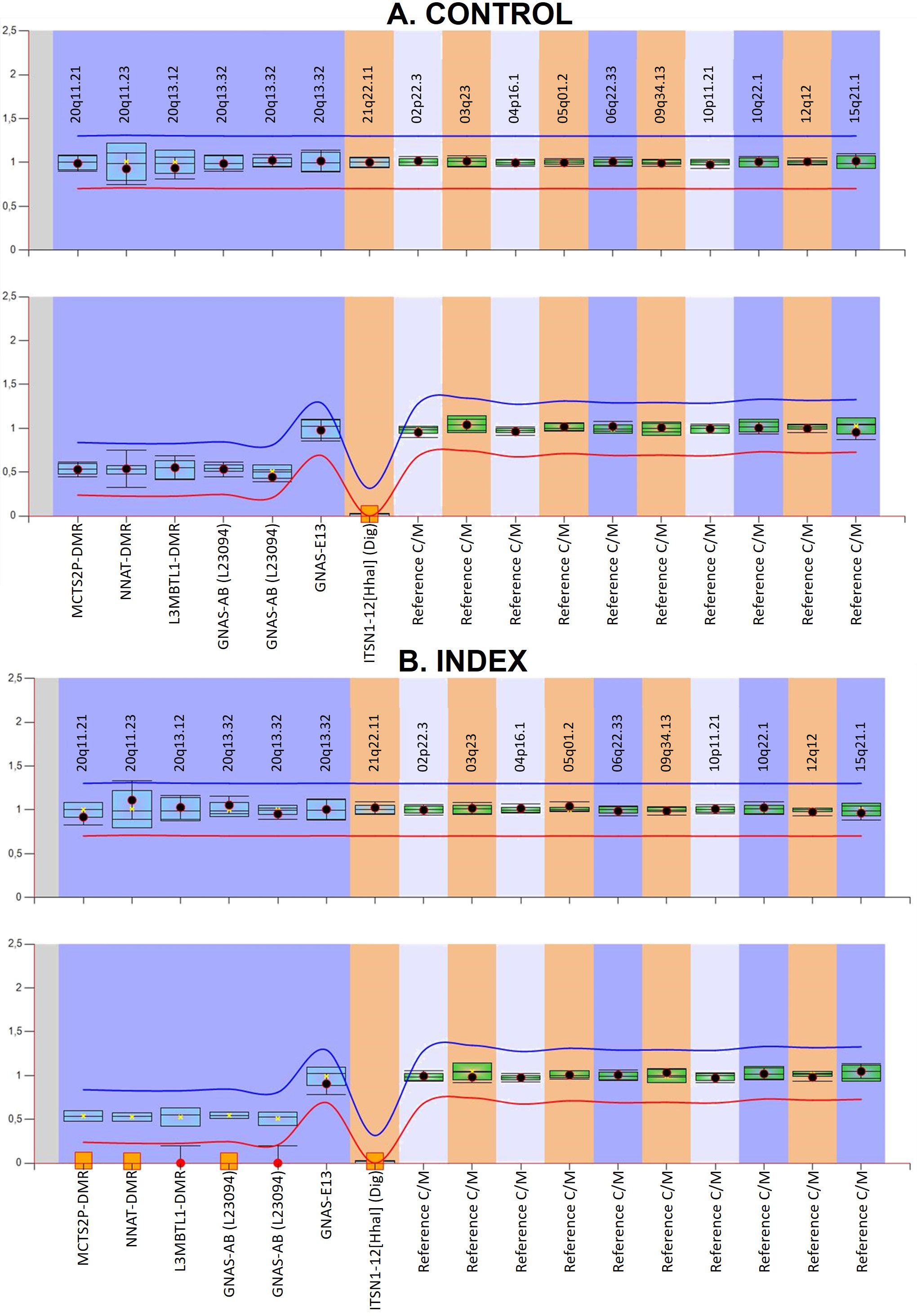
Figure 2. Graphical representation of the custom MS-MLPA results viewed with Coffalyser analysis software. (A) shows the results of a control and (B) corresponds to the index case presenting with iPPSD3. In the upper panel for each individual, the genomic dosage is shown. In the lower panels, the methylation status is represented. In the methylation graphic, the imprinted DMRs should be 0.5, while in biallelically expressed ones, the result should be 1 if they do not have the HhaI digestion site. In contrast, digestion control probes must show a 0. The obtained from the reference samples for each probe is represented by the blue (if on chromosome 20) or green (if on other genomic regions) bars and the 95% confidence interval for the sample value under study is represented by the point error bars. The mean values of five control subjects were used for the assessment of relative copy number and methylation percentage. The red and blue lines represent the arbitrary limits that are within ±0.3 of the mean value of each probe in the reference samples. Loss-of-methylation is represented by red circles and orange squares.
3.4 Microsatellite study
Later on, access to parental samples was possible, so, to confirm the results obtained from the SNP array and custom upd(20q) MS-MLPA, different STRs throughout the GNAS locus, 20p, and 20q regions were studied. The distribution of the microsatellites along the chromosome is depicted in Figure 1 and the results obtained are summarized in Table 2.
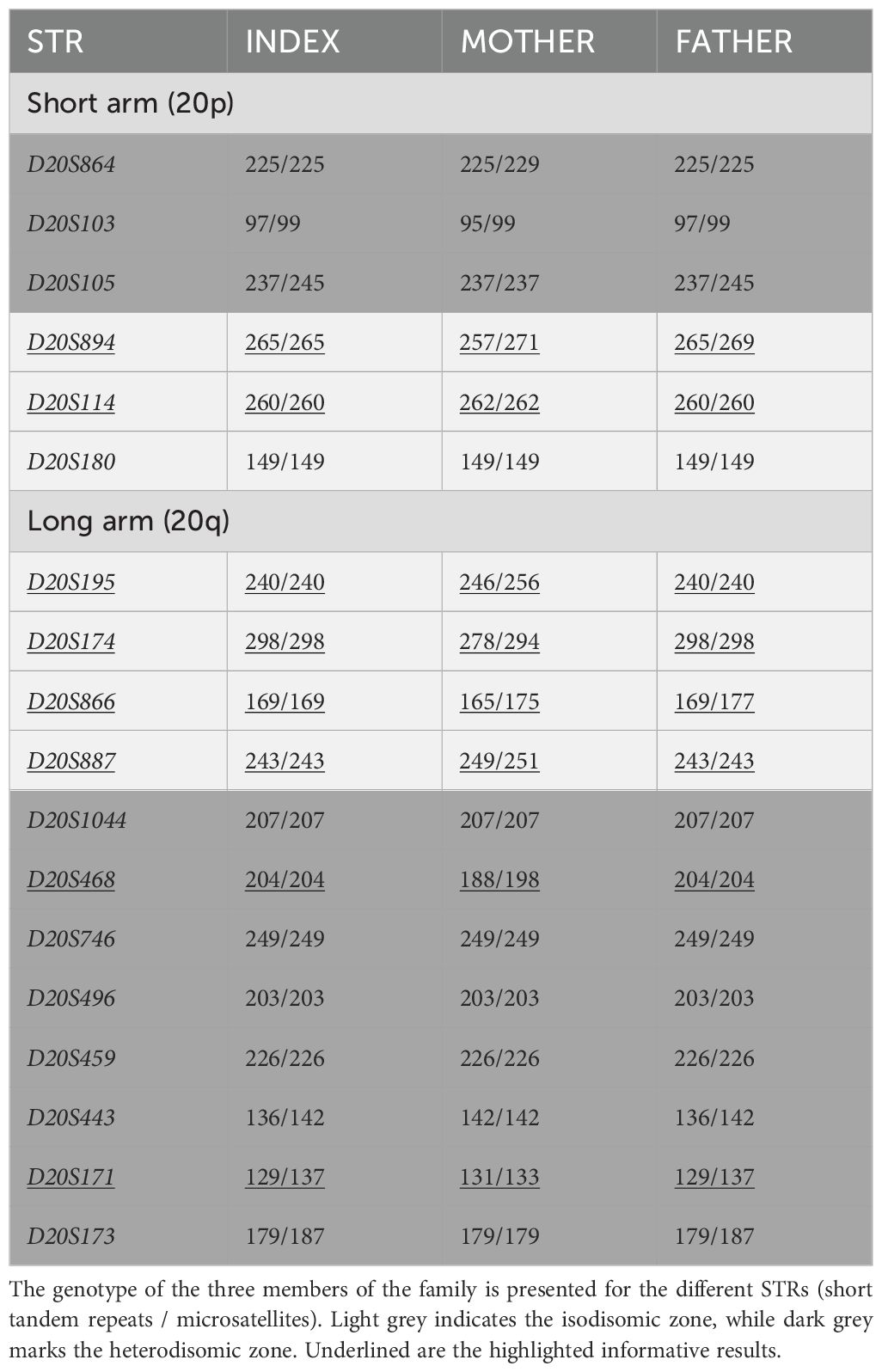
Table 2. Genotypes obtained for the microsatellites studied in chromosome 20.
As it can be seen in Table 2, the results confirmed that the patient presents a mixed upd(20q)pat, with an isodisomic region that does not include GNAS and a heterodisomic zone in both telomeric regions.
4 Discussion
GNAS is a complex locus characterized by a specific imprinting pattern. When the methylation is altered, at least when hypomethylation in the GNAS A/B:TSS-DMR happens, patients develop iPPSD3 (10). The patient described in this work presented with a complete methylation defect affecting all four DMRs detected by the MS-MLPA specific for the GNAS locus, so he was classified as a sporadic case. In patients with complete alteration, paternal UPD should be tested, specifically, isodisomy of chromosome 20 [iUPD (20)] affecting the chromosomic region that includes the GNAS locus (10, 21–23).
Following the current guidelines (10), the genetic material of the proband was analyzed with an SNP array. An SNP array is a good technique to detect iUPD since in cases where a single homolog chromosome is transmitted in duplicate from a parent, whole (or a large part of) chromosomal homozygosity can be seen (23). As is shown in Figure 1, there is a central zone with no heterozygosity in the patient. There are two possible reasons for this. First, when blocks of homozygosity are present in different chromosomes, parental consanguinity is suggested (24). However, at the genetic counseling consultation, the parents denied any relatedness and the SNP array results only showed loss-of-heterozygosity (LOH) in chromosome 20. Second, if large blocks of homozygosity are restricted to a single chromosome, it could represent UPD (25). Still, it was surprising that in the SNP array, there was a region with no LOH in which the GNAS locus was included. This could be a consequence of a chromosome with both isodisomy and heterodisomy. To confirm this, a custom MS-MLPA was designed and performed, confirming the loss of methylation at the four maternal DMRs located at 20q [MCTS2P:TSS-DMR (20q11.21), NNAT:TSS-DMR (20q11.2-q12), L3MBTL1:alt-TSS-DMR (20q13.12), and GNAS A/B:TSS-DMR (20q13.32)], which supported the possibility of UPD and was suggestive of a mixed disomy throughout chromosome 20.
Fortunately, to confirm this, samples from the parents were finally available and STR analysis was performed, with the STRs located around chromosome 20. Thus, as shown in Figure 1; Table 2, the patient under study shows a combination of hetero- and isodisomy of chromosome 20. We would like to draw attention to this study of STRs. If, instead of the SNP array, we had initially analyzed the markers flanking GNAS (D20S496, D20S459, D20S443, D20S171, D20S173) in the absence of parental sample (as was in this situation), we would have ruled out iUPD as the patient carried a heterozygous genotype for some of them. And, since hUPD had been demonstrated once by STRs and the SNP array for the GNAS locus (17), this patient would have been characterized as spor-PHP1B/iPPSD3 with no underlying genetic cause. The correct (epi)genetic diagnosis of this disease allows for appropriate genetic counseling, as it is possible to establish the risk of recurrence and transmission of the disease in the presence of an underlying genetic alteration, whereas in the case of epimutation it would, for the time being, be unknown. Furthermore, if the patient carries iUPD, additional clinical manifestations secondary to an unmasked recessive disease may arise.
Moreover, from a research perspective, the correct subclassification of the different types of iPPSD3 cohorts [according to their (epi)genotype] could help to define clinical-epigenetic relationships and improve prognostic information. Thus, in the absence of parental samples, complete methylation defects at GNAS cannot be determined if it is due to an hUPD (20, 26), and, even for iUPD detection, a SNP array can be more informative than STRs (23), both have their limitations for hUPD identification. As an alternative, we propose the use of an MS-MLPA to examine additional DMRs at this chromosomal arm, as in our case.
It has already been well-demonstrated in the literature that this combination, called mixed UPD, is a molecular disease-causing mechanism described for several chromosomes (25, 27, 28). Regarding iPPSD3, researchers have also detected this genetic defect in chromosome 20, with an interstitial isodisomy in 20q affecting GNAS (29). Mixed UPD occurs when a chromosome contains both isodisomic and heterodisomic parts as a consequence of meiotic nondisjunction events. If the error occurs during meiosis I, a zone of homozygosity will be seen in the terminal region of the chromosomes. However, if the error is in meiosis II, the region of homozygosity is present in the centromeric region (23).
UPDs occur as a consequence of at least two independent errors during meiosis or after fertilization, so they do not follow a Mendelian inheritance (29) and are not necessarily disease-causing because no genetic material is lost or gained. However, some specific events lead to disease, as is the case in imprinting. The chromosomes that contain imprinted loci, as is the case for chromosome 20 and the GNAS locus, are sensitive to UPD. This happens because, due to UPD, the balance of methylation that leads to parental-dependent transcription is altered (23, 25).
In our study, we describe a patient with iPPSD3 with an heterodisomy in the GNAS locus. This is novel since even though iUPD(20q)pat is widely described, it is rare to find an heterodisomy as the cause of iPPSD3. In fact, only one case is described in the literature, and, as in our case, it was caused by a mixed UPD, and GNAS was found in the heterodisomic zone (17). In the reported patient, there were no informative STRs nor SNPs in the GNAS locus. There was one informative STR in the STX16 gene. The mutation rate of STR must be taken into account, which is quite a common event (30, 31) and could lead to a difficulty in heterodisomy confirmation. To solve this problem, we propose the inclusion of a novel custom-designed MS-MLPA to detect upd(20q). This custom-designed MS-MLPA includes probes that hybridize in different DMRs along the long arm of chromosome 20: MCTS2P:TSS-DMR, NNAT: TSS-DMR and L3MBTL1:alt-TSS-DMR. Thanks to the imprinted nature of these DMRs (32–34), it has been possible to detect an alteration in the methylation status (Figure 2), showing complete hypomethylation. The combined information from the STRs/SNP array and this novel assay allowed us to confirm the mixed upd (20).
Although it is extremely rare, maternal UPD in chromosome 20 (upd(20)mat) has been detected in some patients. This genetic condition causes a poorly characterized syndrome known as Mulchandani-Bhoj-Conlin (MBCS, OMIM#617352). The main characteristics of this syndrome are growth failure (intrauterine/postnatal growth retardation) and feeding difficulties (35, 36). As the cause of MBCS is an UPD in chromosome 20, the MS-MLPA developed for the diagnosis of iPPSD3 can also be used for the detection of upd(20)mat and proceed to its diagnosis. At the moment, the specific (epi)genetic cause of MBCS still remains unknown, thus, a limitation of this custom MS-MLPA for the diagnosis of MBCS is that only 20q has been studied.
In conclusion, if the study of STRs is only done in the index case and it shows an heterozygote genotype, it is not enough to discard an UPD, as we present an extra confirmation that hUPD is possible at this locus. In this work, we have also presented a custom MS-MLPA for DMRs in the long arm of chromosome 20. Its efficiency in detecting upd(20q)pat as the underlying genetic cause in iPPSD3 cases has been demonstrated.
Data availability statement
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Ethics committee for clinical research of Euskadi- Basque Country (CEIC-E). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
AM-A: Formal analysis, Investigation, Methodology, Writing – review & editing. YV: Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. JGM: Investigation, Writing – review & editing. EM: Investigation, Writing – review & editing. AP: Formal analysis, Investigation, Methodology, Validation, Writing – review & editing. GPdN: Conceptualization, Formal analysis, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Instituto de Salud Carlos III (ISCIIII) of the Ministry of Economy and Competitiveness (Spain), which was co-financed by the European Regional Development Fund (grant number PI20/00950) and the Department of Health of the Basque Government (grant number GV2021/111056).
Acknowledgments
The authors would like to thank the family for their consent to carry out these studies and to publish this work.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Levine MA, Modi WS, O’Brien SJ. Mapping of the gene encoding the α subunit of the stimulatory G protein of adenylyl cyclase (GNAS1) to 20q13.2 → q13.3 in human by in situ hybridization. Genomics. (1991) 11:478–9. doi: 10.1016/0888-7543(91)90164-A
2. Reik W, Walter J. Genomic imprinting: parental influence on the genome. Nat Rev Genet. (2001) 2:21–32. doi: 10.1038/35047554
3. Monk D, Morales J, den Dunnen JT, Russo S, Court F, Prawitt D, et al. Recommendations for a nomenclature system for reporting methylation aberrations in imprinted domains. Epigenetics. (2018) 13:117–21. doi: 10.1080/15592294.2016.1264561
4. Hayward BE, Moran V, Strain L, Bonthron DT. Bidirectional imprinting of a single gene: GNAS1 encodes maternally, paternally, and biallelically derived proteins. Proc Natl Acad Sci U.S.A. (1998) 95:15475–80. doi: 10.1073/pnas.95.26.15475
5. Turan S, Bastepe M. GNAS spectrum of disorders. Curr Osteoporos Rep. (2015) 13:146–58. doi: 10.1007/s11914-015-0268-x
6. Bastepe M. The GNAS locus: quintessential complex gene encoding gsalpha, XLalphas, and other imprinted transcripts. Curr Genomics. (2007) 8:398–414. doi: 10.2174/138920207783406488
7. Monk D, Mackay DJG, Eggermann T, Maher ER, Riccio A. Genomic imprinting disorders: lessons on how genome, epigenome and environment interact. Nat Rev Genet. (2019) 20:235–48. doi: 10.1038/s41576-018-0092-0
8. Thiele S, Mantovani G, Barlier A, Boldrin V, Bordogna P, De Sanctis L, et al. From pseudohypoparathyroidism to inactivating PTH/PTHrP signalling disorder (iPPSD), a novel classification proposed by the EuroPHP network. Eur J Endocrinol. (2016) 175:P1–17. doi: 10.1530/EJE-16-0107
9. Garin I, Mantovani G, Aguirre U, Barlier A, Brix B, Elli FM, et al. European guidance for the molecular diagnosis of pseudohypoparathyroidism not caused by point genetic variants at GNAS: an EQA study. Eur J Hum Genet. (2015) 23:438–44. doi: 10.1038/ejhg.2014.127
10. Mantovani G, Bastepe M, Monk D, De Sanctis L, Thiele S, Usardi A, et al. Diagnosis and management of pseudohypoparathyroidism and related disorders: First international Consensus Statement. Nat Rev Endocrinol. (2018) 14:476–500. doi: 10.1038/s41574-018-0042-0
11. Perez de Nanclares G, Fernández-Rebollo E, Santin I, García-Cuartero B, Gaztambide S, Menéndez E, et al. Epigenetic defects of GNAS in patients with pseudohypoparathyroidism and mild features of Albright’s hereditary osteodystrophy. J Clin Endocrinol Metab. (2007) 92:2370–3. doi: 10.1210/jc.2006-2287
12. de Lange IM, Verrijn Stuart AA, van der Luijt RB, Ploos van Amstel HK, van Haelst MM. Macrosomia, obesity, and macrocephaly as first clinical presentation of PHP1b caused by STX16 deletion. Am J Med Genet A. (2016) 170:2431–5. doi: 10.1002/ajmg.a.v170.9
13. De Nanclares GP, Fernández-Rebollo E, Santin I, García-Cuartero B, Gaztambide S, Menéndez E, et al. Epigenetic defects of GNAS in patients with pseudohypoparathyroidism and mild features of Albright’s hereditary osteodystrophy. J Clin Endocrinol Metab. (2007) 92:2370–3. https://academic.oup.com/jcem/article/92/6/2370/2598178.
14. Rochtus A, Martin-Trujillo A, Izzi B, Elli F, Garin I, Linglart A, et al. Genome-wide DNA methylation analysis of pseudohypoparathyroidism patients with GNAS imprinting defects. Clin Epigenet. (2016) 8:10. doi: 10.1186/s13148-016-0175-8
15. Mantovani G, Bastepe M, Monk D, de Sanctis L, Thiele S, Usardi A, et al. Diagnosis and management of pseudohypoparathyroidism and related disorders: first international Consensus Statement. Nat Rev Endocrinol. (2018) 14:476–500. https://www.nature.com/articles/s41574-018-0042-0.
16. Elli FM, Linglart A, Garin I, De Sanctis L, Bordogna P, Grybek V, et al. The prevalence of GNAS deficiency-related diseases in a large cohort of patients characterized by the EuroPHP network. J Clin Endocrinol Metab. (2016) 101:3657–68. doi: 10.1210/jc.2015-4310
17. Colson C, Decamp M, Gruchy N, Coudray N, Ballandonne C, Bracquemart C, et al. High frequency of paternal iso or heterodisomy at chromosome 20 associated with sporadic pseudohypoparathyroidism 1B. Bone. (2019) 123:145–52. doi: 10.1016/j.bone.2019.03.023
18. Benn P. Uniparental disomy: Origin, frequency, and clinical significance. Prenat Diagn. (2021) 41:564–72. doi: 10.1002/pd.v41.5
19. Shaffer LG, Agan N, Goldberg JD, Ledbetter DH, Longshore JW, Cassidy SB. American College of Medical Genetics statement of diagnostic testing for uniparental disomy. Genet Med. (2001) 3:206–11. doi: 10.1097/00125817-200105000-00011
20. del Gaudio D, Shinawi M, Astbury C, Tayeh MK, Deak KL, Raca G. Diagnostic testing for uniparental disomy: a points to consider statement from the American College of Medical Genetics and Genomics (ACMG). Genet Med. (2020) 22:1133–41. doi: 10.1038/s41436-020-0782-9
21. Bastepe M, Lane AH, Jüppner H. Paternal uniparental isodisomy of chromosome 20q–and the resulting changes in GNAS1 methylation–as a plausible cause of pseudohypoparathyroidism. Am J Hum Genet. (2001) 68:1283–9. doi: 10.1086/320117
22. Fernández-Rebollo E, Lecumberri B, Garin I, Arroyo J, Bernal-Chico A, Goñi F, et al. New mechanisms involved in paternal 20q disomy associated with pseudohypoparathyroidism. Eur J Endocrinol. (2010) 163:953–62. doi: 10.1530/EJE-10-0435
23. Yauy K, de Leeuw N, Yntema HG, Pfundt R, Gilissen C. Accurate detection of clinically relevant uniparental disomy from exome sequencing data. Genet Med. (2020) 22:803–8. doi: 10.1038/s41436-019-0704-x
24. Li L-H, Ho S-F, Chen C-H, Wei C-Y, Wong W-C, Li L-Y, et al. Long contiguous stretches of homozygosity in the human genome. Hum Mutat. (2006) 27:1115–21. doi: 10.1002/humu.v27:11
25. Tucker T, Schlade-Bartusiak K, Eydoux P, Nelson TN, Brown L. Uniparental disomy: Can SNP array data be used for diagnosis? Genet Med. (2012) 14:753–6. doi: 10.1038/gim.2012.35
26. Eggermann T. Prenatal detection of uniparental disomies (Upd): Intended and incidental finding in the era of next generation genomics. Genes (Basel). (2020) 11:1–12. doi: 10.3390/genes11121454
27. Liehr T. Uniparental disomy is a chromosomic disorder in the first place. Mol Cytogenet. (2022) 15:5. doi: 10.1186/s13039-022-00585-2
28. Zhou L, Zheng Z, Xu Y, Lv X, Xu C, Xu X. Prenatal diagnosis of 7 cases with uniparental disomy by utilization of single nucleotide polymorphism array. Mol Cytogenet. (2021) 14:19. doi: 10.1186/s13039-021-00537-2
29. Fernández-Rebollo E, Lecumberri B, Garin I, Arroyo J, Bernal-Chico A, Goñi F, et al. New mechanisms. Eur J Endocrinol. (2010) 163:953–62. doi: 10.1530/EJE-10-0435
30. Fungtammasan A, Ananda G, Hile SE, Su MS-W, Sun C, Harris R, et al. Accurate typing of short tandem repeats from genome-wide sequencing data and its applications. Genome Res. (2015) 25:736–49. doi: 10.1101/gr.185892.114
31. Fan H, Chu J-Y. A brief review of short tandem repeat mutation. Genomics Proteomics Bioinf. (2007) 5:7–14. doi: 10.1016/S1672-0229(07)60009-6
32. Teichroeb JH, Betts DH, Vaziri H. Suppression of the imprinted gene NNAT and X-chromosome gene activation in isogenic human iPS cells. PloS One. (2011) 6:e23436. doi: 10.1371/journal.pone.0023436
33. Cheong CY, Chng K, Ng S, Chew SB, Chan L, Ferguson-Smith AC. Germline and somatic imprinting in the nonhuman primate highlights species differences in oocyte methylation. Genome Res. (2015) 25:611–23. doi: 10.1101/gr.183301.114
34. Monk D, Arnaud P, Frost JM, Wood AJ, Cowley M, Martin-Trujillo A, et al. Human imprinted retrogenes exhibit non-canonical imprint chromatin signatures and reside in non-imprinted host genes. Nucleic Acids Res. (2011) 39:4577–86. doi: 10.1093/nar/gkq1230
35. Mulchandani S, Bhoj EJ, Luo M, Powell-Hamilton N, Jenny K, Gripp KW, et al. Maternal uniparental disomy of chromosome 20: A novel imprinting disorder of growth failure. Genet Med. (2016) 18:309–15. doi: 10.1038/gim.2015.103
Keywords: PHP1B, iPPSD3, GNAS, heterodisomy, upd(20)pat, MS-MLPA
Citation: Manero-Azua A, Vado Y, Gonzàlez Morlà J, Mogas E, Pereda A and Perez de Nanclares G (2024) Heterodisomy in the GNAS locus is also a cause of pseudohypoparathyroidism type 1B (iPPSD3). Front. Endocrinol. 15:1505244. doi: 10.3389/fendo.2024.1505244
Received: 02 October 2024; Accepted: 18 November 2024;
Published: 16 December 2024.
Edited by:
Tsutomu Ogata, Hamamatsu University School of Medicine, JapanReviewed by:
Masayo Kagami, National Center for Child Health and Development (NCCHD), JapanDaniela Regazzo, University Hospital of Padua, Italy
Copyright © 2024 Manero-Azua, Vado, Gonzàlez Morlà, Mogas, Pereda and Perez de Nanclares. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guiomar Perez de Nanclares, Z25hbmNsYXJlc0Bvc2FraWRldHphLmV1cw==
†These authors have contributed equally to this work