 Jingna Wang
Jingna Wang Rongmin Li1
Rongmin Li1- 1Baoding Hospital, Beijing Children’s Hospital Affiliated with Capital Medical University, Baoding, China
- 2Key Discipline of Pediatrics, Endocrinology Genetics and Metabolism Center, National Center for Children’s Health (Beijing), Beijing Children’s Hospital Affiliated with Capital Medical University, Beijing, China
Objective: The objective of this study is to investigate the clinical presentation and underlying genetic etiology of a Chinese child diagnosed with idiopathic central precocious puberty (ICPP).
Methods: Clinical data from a pediatric patient with ICPP, including medical history, physical examination findings, laboratory results, and imaging studies, were collected and analyzed. Whole exome sequencing (WES) was performed to identify potential pathogenic genetic variants underlying the patient’s ICPP.
Results: A 4 ¾-year-old female patient presented with precocious puberty, characterized by accelerated growth, Tanner stage II breast development, and Tanner stage I pubic hair. A café-au-lait macule was observed on the patient’s right flank. WES revealed a novel makorin RING finger protein 3 (MKRN3) gene heterozygous frameshift pathogenic variant c.1219delA (p.R407Gfs*75), which was inherited from the patient’s asymptomatic father, and leading to a truncated protein 73 amino acids downstream from the mutation site.
Conclusion: This case underscores the genetic heterogeneity of ICPP and further implicates MKRN3 gene mutations in its pathogenesis. The identification of this novel pathogenic variant expands the known mutational spectrum associated with ICPP, particularly within the Chinese pediatric population. Comprehensive genetic testing should be considered in pediatric patients presenting with early-onset ICPP to facilitate accurate diagnosis, inform genetic counseling, and guide personalized management strategies.
Introduction
Central precocious puberty (CPP) is a common pediatric endocrine disorder characterized by the premature activation of the hypothalamic-pituitary-gonadal (HPG) axis, resulting in the development of secondary sexual characteristics before the age of 8 years in girls and 9 years in boys. This pediatric endocrine disorder exhibits a notable gender disparity in its prevalence, with a significantly higher incidence in girls. Epidemiological data suggest that the prevalence ranges from 0.217 to 26.28 per 10,000 in girls, compared to a much lower range of 0.02 to 0.9 per 10,000 in boys (1). This gender disparity suggests that genetic factors may play a critical role in the pathogenesis of CPP. Although the precise molecular mechanisms remain under investigation, genetic contributions are emerging as a key area of research in understanding this condition. Previous studies have identified the makorin ring-finger protein 3 (MKRN3) gene as a crucial factor in the onset of puberty. MKRN3 negatively regulates the HPG axis, and loss-of-function (LOF) mutations in this gene can lead to increased gonadotropin-releasing hormone (GnRH) secretion, premature activation of the HPG axis, and subsequent development of CPP (2–4).
The MKRN3 gene, which encodes a RING (really interesting new gene) zinc finger protein, was first identified in 1999 by Jong MT et al. during their investigation into the molecular mechanisms of Prader-Willi syndrome (5). Located on chromosome 15q11.2, a critical region associated with Prader-Willi syndrome, MKRN3 is a single exon, paternally expressed imprinted gene that encodes an E3 ubiquitin ligase. MKRN3 protein contains multiple domains, including two C3H motifs at the N-terminus, followed by a unique makorin-type Cys-His (CH)-rich domain, a C3H4 RING zinc finger, and a terminal C3H motif. The RING zinc finger domain is responsible for its E3 ubiquitin ligase activity, while the C3H zinc finger motifs are involved in RNA binding. MKRN3 plays a role in protein degradation and is involved in various cellular processes, including signal transduction, cell cycle regulation, differentiation, and morphogenesis (6).
While numerous studies have explored the genetic underpinnings of CPP, data from the Chinese population remains limited. This report presents a case of CPP in a Chinese child attributed to a novel MKRN3 gene mutation, aiming to enhance clinical understanding of this condition and its genetic determinants.
Clinical presentation and genetic analysis
General information
A 4-year-9-month-old female patient presented to our hospital on March 17, 2021, with a chief complaint of “breast development for 2 months”. At birth, a congenital melanocytic nevus measuring approximately 1.5 cm × 2 cm with irregular borders was noted on her right waist. At 4 years and 7 months of age, the patient experienced thelarche, which spontaneously regressed without medical intervention. However, two weeks prior to this admission, the patient again noticed recurrent breast development.
Personal history
The patient is the firstborn from an uncomplicated, full-term cesarean section delivery, with no history of perinatal complications. Birth weight was 3.4 kg, and birth length was 50 cm. Developmental milestones, including gross motor skills and social interaction, were appropriate for age. The maternal prenatal course was unremarkable. There is no history of consanguinity between the parents. The father, with a height of 167 cm, experienced his pubertal growth spurt at approximately 12 years of age, with growth cessation at 14 years. The mother, with a height of 162 cm, experienced menarche at 13 years of age.
Physical examination
The patient’s height was 116 cm, which is slightly above the average for her age (109 ± 4.1 cm), with a weight of 20 kg and a BMI of 14.9, all in the normal range (17.8 ± 2.4 kg and 15.3 ± 1.6). No dysmorphic facial features were observed. A 3 cm × 4 cm irregular café-au-lait macule was present on the right waist. There was no evidence of acne or dorsocervical fat pad. The thyroid gland was non-palpable. Breast development was consistent with Tanner stage II, with bilateral breast buds measuring approximately 1.5 cm × 1.5 cm, and no areolar hyperpigmentation. The external genitalia were age-appropriate, with the labia majora fully covering the labia minora. Pubic hair development was consistent with Tanner stage I, and no axillary hair was present. The spine and limbs showed no deformities, and the neurological examination was unremarkable.
Laboratory investigations
Laboratory tests revealed a normal complete blood count, comprehensive metabolic panel, and urinalysis. Tumor markers, including alpha-fetoprotein (AFP), carcinoembryonic antigen (CEA), and carbohydrate antigen 19-9 (CA 19-9), were within normal limits. Thyroid function tests, including thyroid-stimulating hormone (TSH) at 1.75uIU/mL (reference range:0.4-6uIU/mL), free thyroxine (free T4) at 17.24pmol/L (reference range:8.37-29.6pmol/L), free triiodothyronine (free T3) at 6.96pmol/L (reference range:2.5-9 pmol/L) were all within normal ranges. Insulin-like growth factor 1 (IGF-1) at 262 ng/mL (reference range: 49-283ng/mL) was normal. Adrenocorticotropic hormone (ACTH) was measured at 13.92 pg/mL (reference range: 0 - 46 pg/mL), and cortisol was within the normal range at 273.30 nmol/L (reference range: 138.2 - 691 nmol/L).
Sex hormone analysis showed the following levels: luteinizing hormone (LH) at 0.3 IU/L (reference range: 0 - 0.33 IU/L), follicle-stimulating hormone (FSH) at 2.14 IU/L (reference range: 0.42 - 5.45 IU/L), prolactin at 7.64 ng/mL (reference range: 4.2 - 23.04 ng/mL), estradiol at 15.08 pg/mL (reference range: 0 - 10 pg/mL), total testosterone at 12.64 ng/dL (reference range: 1.1 - 62 ng/dL), and progesterone at 0.21 ng/mL (reference range: 0 - 0.35 ng/mL), as shown in Table 1.
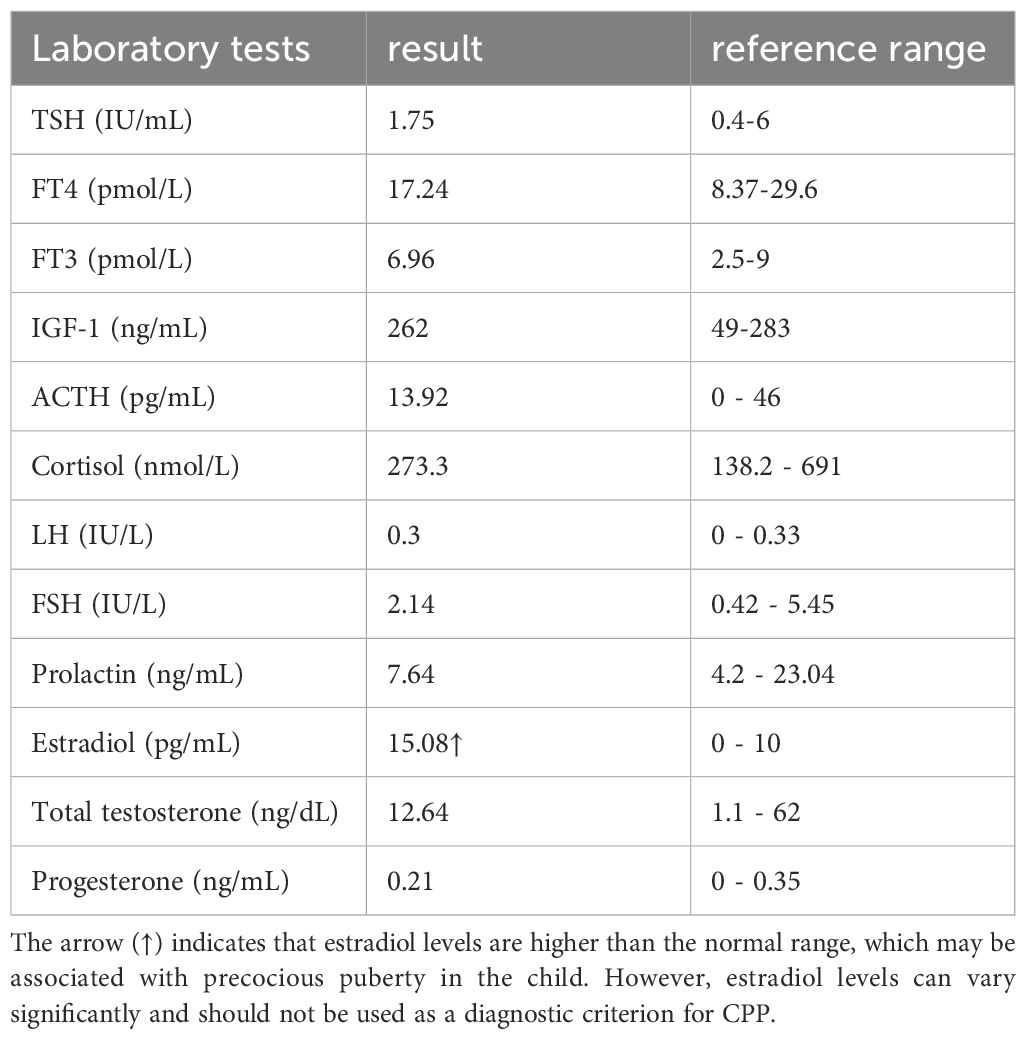
Table 1. The main laboratory data of the patient.
Imaging studies
Breast ultrasound revealed bilateral breast buds with the left breast measuring 2.9 cm × 0.7 cm × 3.4 cm and the right breast measuring 2.9 cm × 0.6 cm × 2.9 cm. The margins were ill-defined, with no significant internal abnormal echogenicity or increased vascularity detected.
Pelvic ultrasound showed a uterus measuring 3.0 cm × 1.5 cm × 0.9 cm with a thin endometrial stripe. The left ovary measured 2.4 cm × 0.7 cm × 1.0 cm, containing two follicles each measuring 0.3 cm, while the right ovary measured 2.9 cm × 1.0 cm × 1.2 cm, containing six follicles each measuring 0.3 cm.
Abdominal ultrasound showed no evidence of adrenal masses or enlargement. Brain MRI demonstrated a normal-appearing pituitary gland.
Genetic analysis
All clinical investigations and therapeutic interventions were performed with informed consent from the patient’s legal guardians and were approved by the institutional ethics committee.
After obtaining informed consents, 3 mL of EDTA-anticoagulated peripheral blood was collected from the patient and her parents. High-throughput whole exome sequencing (WES) was performed by Beijing Maikeno Technology Co., Ltd.
Briefly, genomic DNA was extracted from the blood samples and subjected to DNA library preparation. The library was then hybridized with biotinylated probes targeting the whole exome (P039-Exom, Maikeno) under optimized conditions. Streptavidin-coated magnetic beads were utilized to capture the biotinylated probe-target DNA complexes. The captured DNA fragments were then eluted, purified, and enriched. The enriched target DNA was sequenced using the Illumina NextSeq 500 platform. Sequencing reads were aligned to the human reference genome hg19 using the Burrows-Wheeler Aligner (BWA) software. The resulting alignment files were sorted, filtered, and locally realigned to reduce false-positive variant calls. Variants of potential clinical significance were confirmed by Sanger sequencing.
Genetic sequencing results
A heterozygous frameshift mutation (c.1219delA) was identified in exon 1 of the MKRN3 gene (the reference transcript: NM_005664; exon1) in the proband (Figure 1). This single nucleotide deletion causes a frameshift that leads to the substitution of arginine (R) at codon 407 with glycine (G), ultimately resulting in premature termination of protein translation 73 amino acids downstream (p.R407Gfs*75). Familial segregation analysis confirmed that the proband’s father is a heterozygous carrier of the mutation, while the mother carries the wild-type (WT) allele at this locus (Figures 2, 3).
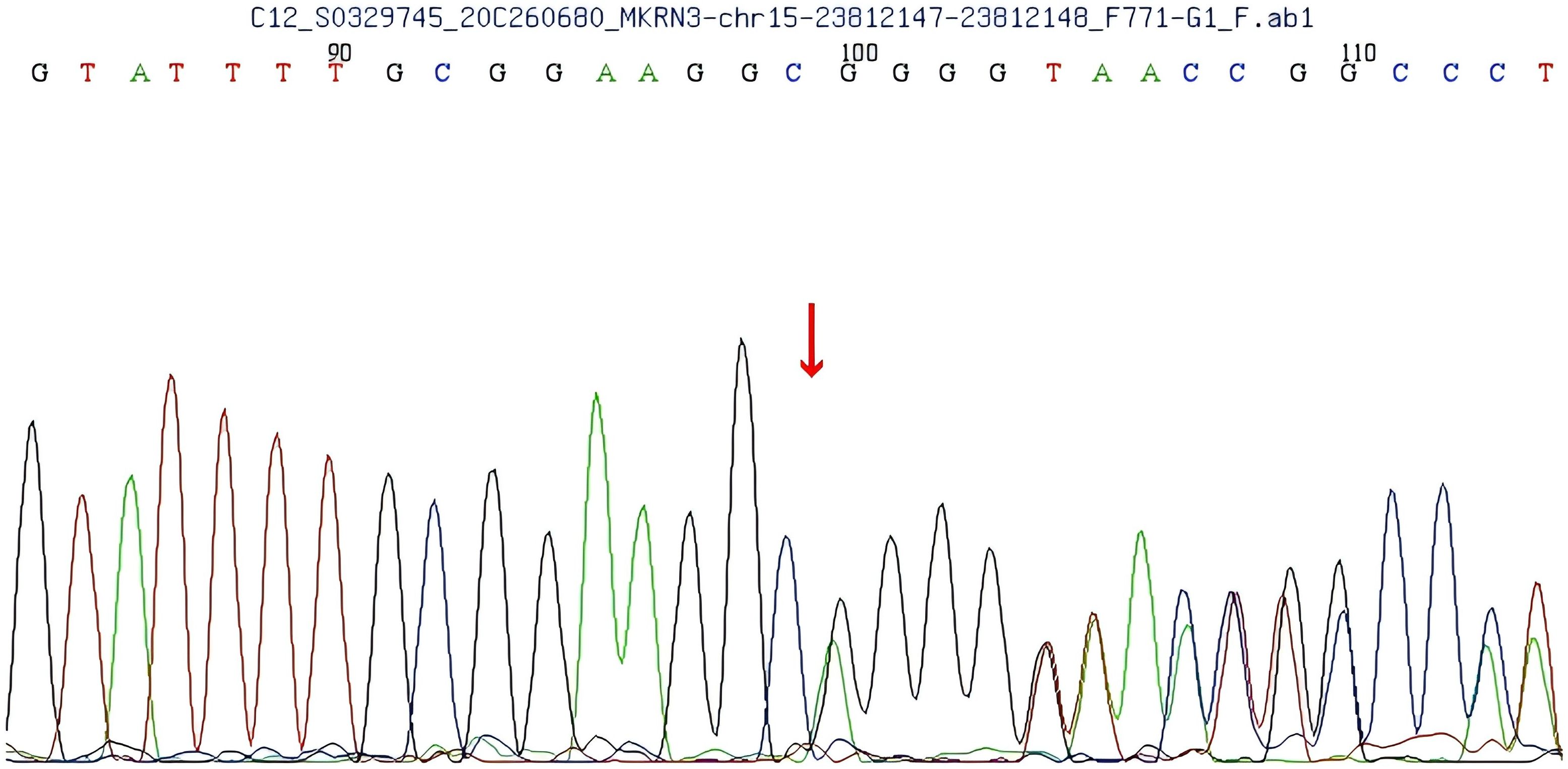
Figure 1. Sequencing results of the patient show a c.1219delA mutation in exon 1 of the makorin RING finger protein 3 (MKRN3) gene, leading to an Argine 407 to glycine (R407G) mutation.
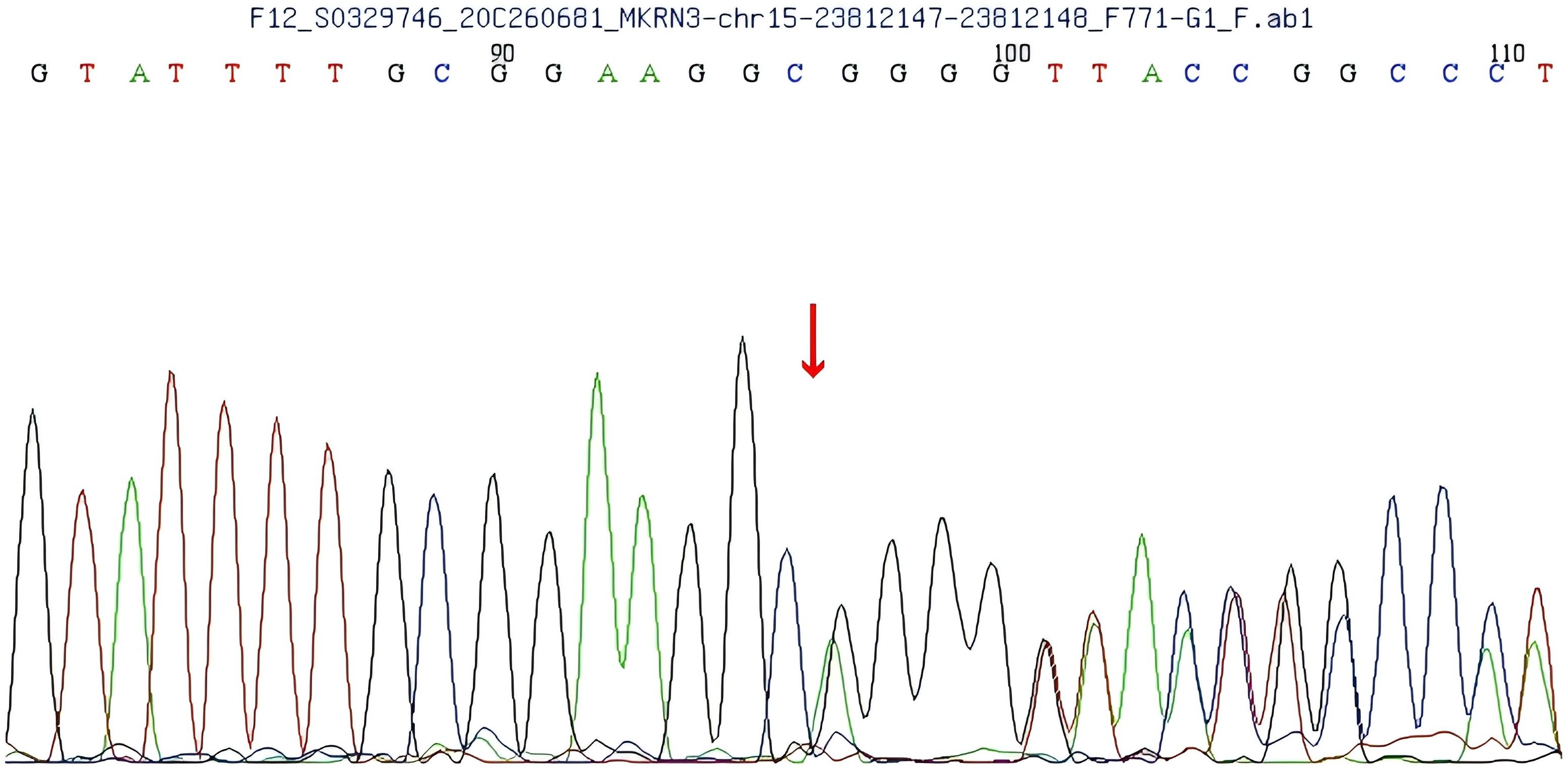
Figure 2. Gene sequencing of the patient’s father shows a single nucleotide deletion (1219delA) in exon 1 of the makorin RING finger protein 3 (MKRN3) gene, resulting in an Argine 407 to glycine (R407G) mutation.
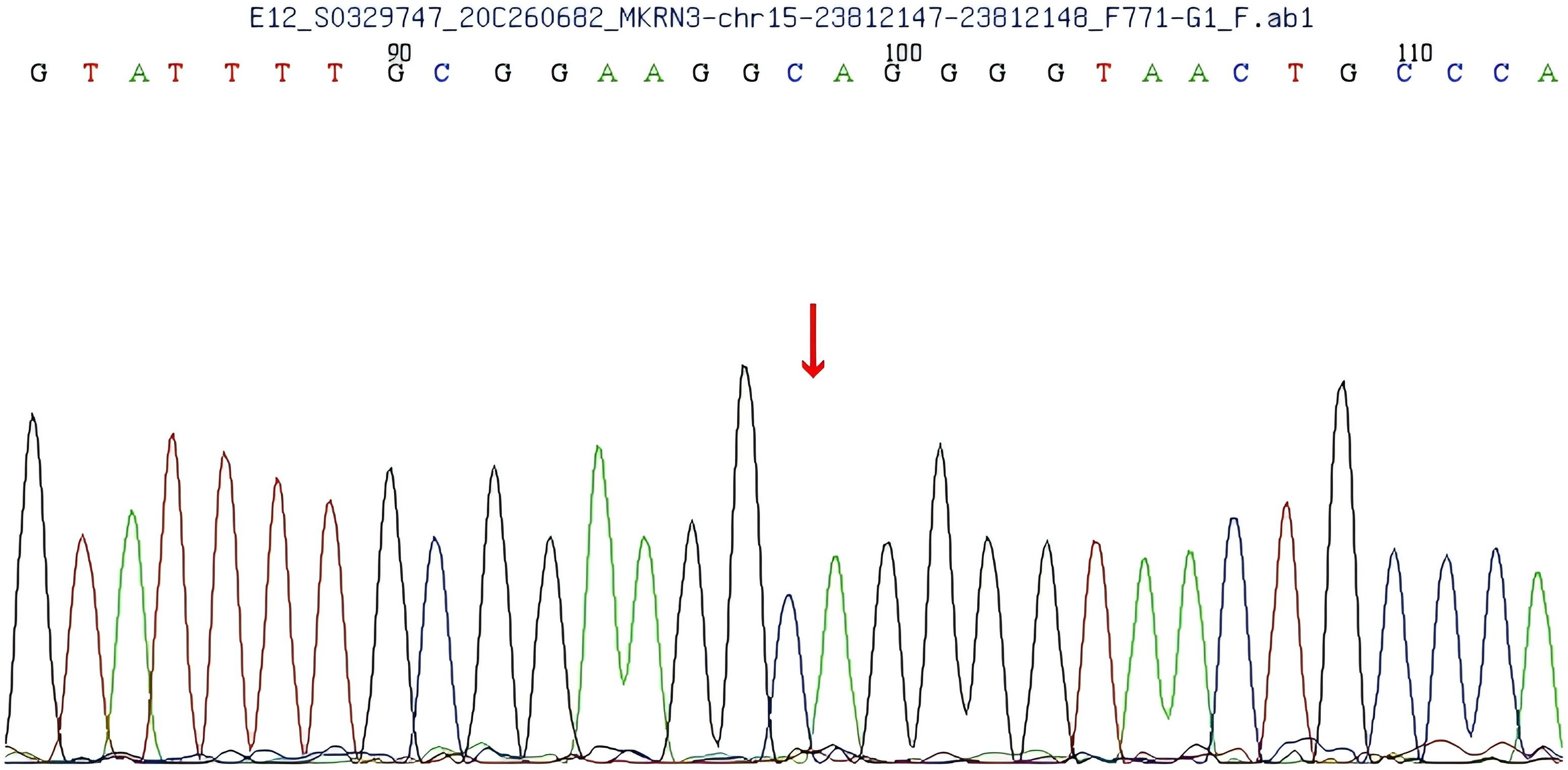
Figure 3. Gene sequencing of the patient’s mother shows no mutation in exon 1 of the makorin RING finger protein 3 (MKRN3) gene.
This variant is absent from population databases and has not been previously reported in the Human Gene Mutation Database (HGMD). Additionally, ClinVar does not provide pathogenicity assessments for this variant. Based on the American College of Medical Genetics and Genomics (ACMG) guidelines, the variant is classified as likely pathogenic.
Discussion
Uniqueness of the case and discovery of a novel MKRN3 mutation
In this study, we report a case of a 4-year-9-month-old Chinese girl diagnosed with ICPP caused by a novel heterozygous frameshift mutation in the MKRN3 gene (c.1219delA). This mutation leads to premature termination of the protein, resulting in a loss-of-function protein. The discovery of this mutation expands the known pathogenic mutation spectrum of the MKRN3 gene, particularly in the Chinese population where data on MKRN3 mutations are relatively scarce. This case provides a foundation for further exploration of the genetic characteristics and pathogenic mechanisms of MKRN3 in the Chinese population.
Further insights into the pathogenesis of CPP
Previous studies have shown that the MKRN3 gene plays an inhibitory role in suppressing GnRH secretion before puberty (7). In 2013, Abreu et al. pioneered the identification of pathogenic mutations in the MKRN3 gene as a causative gene for CPP through WES analysis of 40 affected individuals from 15 families (8). They specifically identified four distinct MKRN3 variants in 12 CPP patients across 5 families, including one missense and three frameshift mutations (8). Subsequent research across diverse populations has further expanded the repertoire of pathogenic MKRN3 variants to a total of 54, with these mutations primarily classified as frameshift, missense, or nonsense (9–12) (Table 2). The novel mutation identified in this study is not located in the last base pairs, the mutated mRNA probable undergoes a nonsense mediated decay (NMD), and we consider that this novel discovered mutation would be a loss-of-function mutation. Although no experimental structural data is available for MKRN3 protein, the three-dimensional (3D) structure predicted by AlphaFold suggests that the R407G mutation occurs in the c-terminal C3H domain (Figure 4). This variant may retain E3 ubiquitin ligase activity but likely lacks RNA-binding capability due to the disruption of the C3H motif.
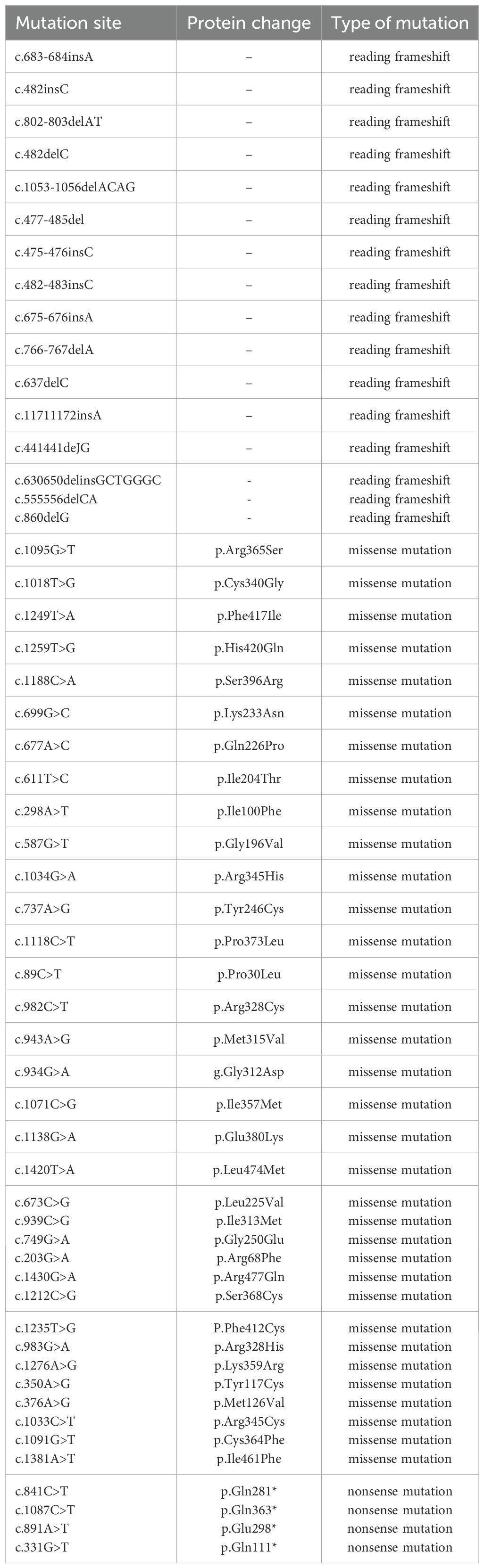
Table 2. Confirmed makorin RING finger protein 3 (MKRN3) Mutations.
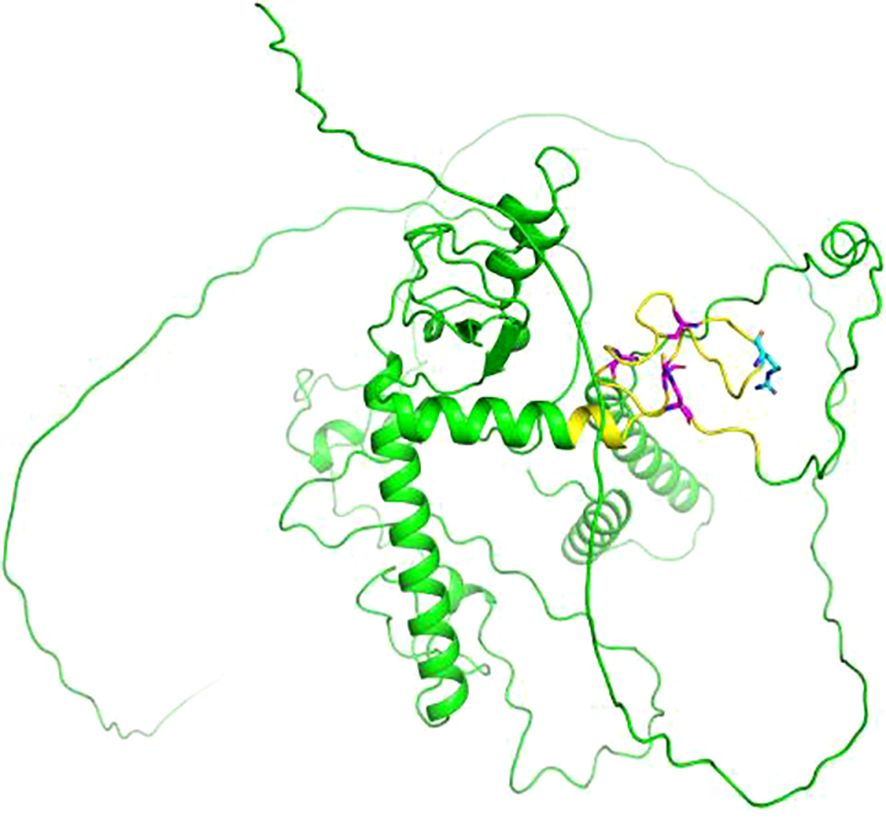
Figure 4. Cartoon representation of the three-dimensional (3D) structure of makorin RING finger protein 3 (MKRN3) predicted by AlphaFold (AF-Q13064-F1). The point mutation at position 407 is highlighted in cyan, and the C3H domain is highlighted in yellow. The cysteine (C) and histidine (H) residues of the C3H domain are highlighted in magenta. The image was generated using PyMOL (Version 2.5.5).
The exact role of MKRN3 in regulating the central neuroendocrine system and how its mutations lead to CPP is still unclear. Studies in animals show that MKRN3 can inhibit the secretion of gonadotropin-releasing hormone 1 (GnRH1). In mice, MKRN3 mRNA levels peak around postnatal days 10-12 and then drop sharply by day 15, aligning with the start of puberty. Under normal conditions, MKRN3 ubiquitinates methyl-CpG binding domain protein 3 (MBD3), disrupting its binding to the GnRH1 promoter. This prevents the recruitment of TET2, leading to increased promoter methylation and suppression of GnRH1 transcription. In humans, serum MKRN3 levels were found to exhibit substantial inter-individual variability but consistently decline prior to the onset of puberty, showing a negative correlation with gonadotropin secretion. This observation suggests that MKRN3 may exert an inhibitory effect on hypothalamic GnRH secretion during childhood (13). Decreased MKRN3 levels during puberty could lead to the release from suppression of the HPG axis, thereby triggering pubertal development (14). Moreover, some children with CPP exhibit markedly reduced or undetectable serum MKRN3 levels, potentially linked to MKRN3 gene mutations (14). It is thus hypothesized that LOF mutations in MKRN3 could compromise its inhibitory function over the HPG axis, leading to pulsatile GnRH release, premature activation of the axis, and consequently, precocious puberty. Further investigations into the relationship between MKRN3 and sex hormones have revealed an inverse correlation between MKRN3 levels and peripheral blood LH, FSH, and estradiol (E2) levels, but not with anti-Müllerian hormone (AMH) levels (15). These findings reinforce the concept that MKRN3 acts as a negative regulator of gonadotropins and E2, suppressing hypothalamic GnRH secretion during childhood and declining prior to pubertal onset. Elucidating the expression patterns and functional roles of MKRN3 protein is crucial for understanding the regulatory mechanisms underlying human pubertal initiation.
Clinical management implications of this case
The mutation in this patient was inherited from her asymptomatic father, consistent with the imprinting inheritance pattern of the MKRN3 gene. Since MKRN3 is a paternally expressed imprinted gene, mutations are typically passed from asymptomatic fathers to their offspring. Understanding the family’s genetic background, especially the developmental history of the father, is crucial for accurate diagnosis and family genetic counseling. This study highlights the importance of comprehensive genetic analysis in early-onset ICPP cases to accurately identify pathogenic mutations.
Identifying the genetic cause of CPP early is crucial for personalized treatment. The mutation in this patient has not been reported in public databases, indicating that it is a novel pathogenic variant. For cases like this, clinicians should consider early genetic testing to formulate personalized management strategies. Moreover, as CPP caused by MKRN3 mutations tends to have an earlier onset, close monitoring and follow-up are necessary, especially for asymptomatic individuals from the same family, particularly paternal family members, to ensure timely intervention.
Therapeutic and prognostic outlook
Currently, GnRH analogs are the standard treatment for CPP. These analogs exert their therapeutic effect primarily at the anterior pituitary, where they competitively bind to GnRH receptors on gonadotrophs. While an initial transient increase in LH, FSH, and sex hormones may be observed, continuous administration leads to pituitary desensitization, reducing gonadotropin secretion and subsequently suppressing sex hormone levels, thus effectively interrupting the prematurely activated HPG axis.
Importantly, extended GnRH analog therapy has been recommended for individuals with CPP caused by MKRN3 gene mutations (16). While the therapeutic response to GnRH analogs in MKRN3-induced CPP is generally comparable to that observed in idiopathic CPP, the earlier average age of onset in MKRN3-related cases necessitates vigilant monitoring of even asymptomatic children from affected families. This proactive approach ensures the timely identification of CPP onset and allows for prompt intervention, optimizing treatment outcomes.
Although GnRH analogs remain the primary treatment for CPP, long-term efficacy and therapeutic strategies for CPP caused by MKRN3 mutations may need further optimization. Since early intervention is critical in preventing premature epiphyseal closure and limiting adult height, this study supports the importance of early genetic screening and individualized treatment for such cases.
Conclusions
By linking this case to the specific findings, this study not only adds to the body of evidence on the genetic basis of CPP but also provides new insights and directions for future clinical management and research. This study underscores the importance of MKRN3 mutations in the Chinese population, but further functional studies are needed to elucidate the precise pathophysiological mechanisms. These studies will help improve our understanding of the genetic mechanisms underlying CPP and lead to more precise therapeutic approaches.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics statement
The studies involving humans were approved by Beijing Children’s Hospital Affiliated to Medical University Baoding Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the minor(s)’ legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Author contributions
JNW: Visualization, Writing – original draft, Conceptualization, Investigation, Supervision, Writing – review & editing. RL: Investigation, Writing – original draft. JYW: Supervision, Writing – review & editing. DW: Investigation, Writing – original draft. SL: Supervision, Writing – review & editing. YS: Writing – review & editing, Supervision. JC: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Baoding Self-financing Project of Science and Technology Plan(1951ZF076).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Brito VN, Canton AP, Seraphim CE, Abreu AP, Macedo DB, Mendonca BB, et al. The congenital and acquired mechanisms implicated in the etidlogy of central precocious puberty. Endocr Rev. (2023) 44:193–221. doi: 10.1210/endrev/bnac020
2. Liu J, Li T, Peng M, Luo M, Gui Z, Long S, et al. The key roles of MKRN3 ring finger protein 3(MKRN3) during the development of pubertal initiation and central precocious puberty(CPP). Curr Mol Med. (2023) 23:668–77. doi: 10.2174/1566524022666220624105430
3. Chen Z, You Q, Wang J, Dong Z, Wang W, Yang Y, et al. The functional study of a novel MKRN3 missense mutation associated with familial central precocious puberty. Am J Med Genet A. (2024) 194:e63460. doi: 10.1002/ajmg.a.v194.4
4. Eren SE, Simsek E. Comparison of makorin ring finger protein 3 levels between obese and normal weight patients with central precocious puberty. J Clin Res Pediatr Endocrinol. (2023) 15:182–9. doi: 10.4274/jcrpe.galenos.2023.2022-6-6
5. Jong MT, Carey AH, Caldwell KA, Lau MH, Handel MA, Driscoll DJ, et al. Imprinting of a RING zinc-finger encoding gene in the mouse chromosome region homologous to the Prader-Willi syndrome genetic region. Hum Mol Genet. (1999) 8:795–803. doi: 10.1093/hmg/8.5.795
6. Gray TA, Hernandez L, Carey AH, Schaldach MA,Smithwick MJ, Rus K MA, Smithwick MJ, Rus K, et al. The ancint source of a distinct gene family encoding proteins featuring RING and CH3 zinc-finger motifs with abundant expression in developing brain and nervous systerm. Genomics. (2000) 66:76–86. doi: 10.1006/geno.2000.6199
7. Zhang YY, Luo FH. Recent advances in etiology of central precocious puberty caused by genetic defects. Zhongguo Dang Dai Er Ke Za Zhi. (2024) 26:302–7. doi: 10.7499/j.issn.1008-8830.2309098
8. Abreu AP, Dauber A, Macedo DB, Noel SD, Brito VN, Gill JC, et al. Central precocious puberty caused by mutations in the imprinted gene MKRN3. N Engl Med. (2013) 368:2467–75. doi: 10.1056/NEJMoa1302160
9. Gernay C, Brachet C, Boros E, Tenoutasse S, Libioulle C, Heinrichs C, et al. Six novel variants in the MKRN3 gene causing central precocious puberty. Endocrine Soc. (2023) 7:1–8. doi: 10.1210/jendso/bvac168
10. Liu M, Fan L, Gong C. A novel heterozygous nonsense mutation in MKRN3 causing central precocious puberty: A case report. Chin Clin Case Database. (2022) 4:e06604. doi: 10.3760/cma.j.cmcr.2022.e06604
11. Maione L, Naule L, Kaiser UB. Makorin RING finger protein 3 and central precocious puberty. Curr Opin Endorc Metab Res. (2020) 14:152–9. doi: 10.1016/j.coemr.2020.08.003
12. Magnotto JC, Mancini A, Bird K, Montenegro L, Tutunculer F, Pereira SA, et al. Novel MKRN3 missense mutations associated with central precocious puberty reveal distinct effects on. Clin Endocrinology& Metab. (2023) 108:1646–56. doi: 10.1210/clinem/dgad151
13. Magnotto JC, Mancini A, Bird K, et al. Novel MKRN3 missense mutations associated with central precocious puberty reveal distinct effects on ubiquitination. J Clin Endocrinol Metab. (2023) 108:1646–56. doi: 10.1210/clinem/dgad151
14. Hagen CP, Sorensen K, Mieritz MG, Johannsen TH, Almstrup K, Juul A, et al. Circulating MKRN3 levels decline prior to pubertal onset and through puberty: a longitudinal study of healthy girls. J Clin Endocrinol Metab. (2015) 100:1902–6. doi: 10.1210/jc.2014-4462
15. Grandone A, Cirillo G, Sasso M, Capristo C, Tornese G, Marzuillo P, et al. MKRN3 levels in girls with central precocious puberty and correlation with sexual hormone levels: a pilot study. Endocrine. (2017) 59:203–8. doi: 10.1007/s12020-017-1281-x
Keywords: idiopathic central precocious puberty, MKRN3, Chinese child, c.1219delA, p.R407Gfs*75
Citation: Wang J, Li R, Wang J, Wu D, Lei S, Sang Y and Chang J (2024) Novel MKRN3 gene mutation associated with central precocious puberty in a Chinese child: a case report. Front. Endocrinol. 15:1491664. doi: 10.3389/fendo.2024.1491664
Received: 05 September 2024; Accepted: 25 November 2024;
Published: 20 December 2024.
Edited by:
Silvano Bertelloni, University of Pisa, ItalyReviewed by:
Manuel Pombo, University of Santiago de Compostela, SpainGiorgia Pepe, University of Messina, Italy
Copyright © 2024 Wang, Li, Wang, Wu, Lei, Sang and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanmei Sang, c2FuZ3lhbm1laUB5YWhvby5jb20=; Jie Chang, NzQ3MzI3NzI2QHFxLmNvbQ==