 Xiaopeng Jia
Xiaopeng Jia Gongzi Zhang2*
Gongzi Zhang2*- 1Trauma Orthopedics, The First Affiliated Hospital of Jinzhou Medical University, Jinzhou, China
- 2Department of Rehabilitation Medicine, Chinese People’s Liberation Army General Hospital, Beijing, China
As the population ages, the occurrence of osteoporosis is becoming more common. Diabetes mellitus is one of the factors in the development of osteoporosis. Compared with the general population, the incidence of osteoporosis is significantly higher in diabetic patients. Diabetic osteoporosis (DOP) is a metabolic bone disease characterized by abnormal bone tissue structure due to hyperglycemia and insulin resistance, reduced bone strength and increased risk of fractures. This is a complex mechanism that occurs at the cellular level due to factors such as blood vessels, inflammation, and hyperglycemia and insulin resistance. Although the application of some drugs in clinical practice can reduce the occurrence of DOP, the incidence of fractures caused by DOP is still very high. Extracellular vesicles (EVs) are a new communication mode between cells, which can transfer miRNAs and proteins from mother cells to target cells through membrane fusion, thereby regulating the function of target cells. In recent years, the role of EVs in the pathogenesis of DOP has been widely demonstrated. In this article, we first describe the changes in the bone microenvironment of osteoporosis. Second, we describe the pathogenesis of DOP. Finally, we summarize the research progress and challenges of EVs in DOP.
1 Introduction
Osteoporosis is a systemic bone disorder characterized by low bone mass and deterioration of the microstructure of bone tissue, often resulting in bone fragility and an increased risk of fractures (1). Osteoporosis leads to fragility fractures, with a significantly increased risk of fractures of the proximal femur and distal radius, with vertebral compression being the most common (2). Globally, osteoporosis causes more than 8.9 million fractures each year (3). Osteoporosis occurs primarily in older patients, affecting more than 14 million people in the United States and more than 200 million people worldwide (4, 5). According to the International Osteoporosis Foundation, osteoporosis is the leading cause of fracture in older adults in patients over the age of 50 (6). Osteoporotic fractures are more common in women than in men, with 1 in 3 women and 1 in 5 men at the risk of osteoporotic fractures. This number continues to rise as the population ages (7). It is estimated that the annual economic consumption of osteoporosis in the United States alone will climb to $25.3 billion by 2025 (8). Osteoporosis leads to a reduced quality of life for patients, and the financial burden of medical expenses for its treatment is significant. Clearly, osteoporosis has become a global public problem.
Factors such as genetics, hormone deficiencies, and aging can all contribute to disruption of bone homeostasis maintained by osteoblasts and osteoclasts, leading to osteopenia and deterioration of the microstructure of bone tissue, ultimately leading to osteoporosis (9). Diabetes is one of the main causes of osteoporosis. Studies have shown that the incidence of osteoporosis in diabetic patients is 60%, which is significantly higher than that of non-diabetic patients (10). Diabetes mellitus leads to abnormal blood glucose metabolism, abnormally elevated levels of extracellular metabolites, and disruption of bone remodeling (11). Diabetic osteoporosis (DOP) is a systemic chronic metabolic bone disease characterized by increased cortical porosity, imbalance of bone metabolism, and distortion of bone microstructure (12). DOP is often associated with osteoporotic fractures due to abnormal glucose metabolism, accumulation of advanced glycation end products (AGEs), and damage to the osteovascular system (13). Although the specific mechanism of DOP has not yet been clarified, many reports have confirmed the relationship between the two (14). At present, the clinical treatment of osteoporosis is mainly based on bisphosphonate, alendronate sodium inhibition of bone resorption capacity of osteoclasts, osteotriol and calcium and other bone drugs, and drugs that promote osteoblast function (15–17). Despite the success of these treatments, the incidence of fractures due to DOP remains high.
At present, extracellular vesicles (EVs) have attracted attention as a special way of signaling between cells (18). EVs have a double-layer lipid membrane structure, which has the advantages of low immunogenicity and good cell compatibility in vivo (19). EVs are vesicles produced by paracrine in living cells, and EVs can deliver miRNAs and proteins in cells to target cells through membrane fusion, and regulate the biological activity of target cells (20). The role of EVs in DOP has been well established (Figure 1). In this article, we first describe the changes in the osteoporotic bone microenvironment. Second, we describe the pathogenesis of DOP. Finally, we summarize the research progress and challenges of EVs in DOP.
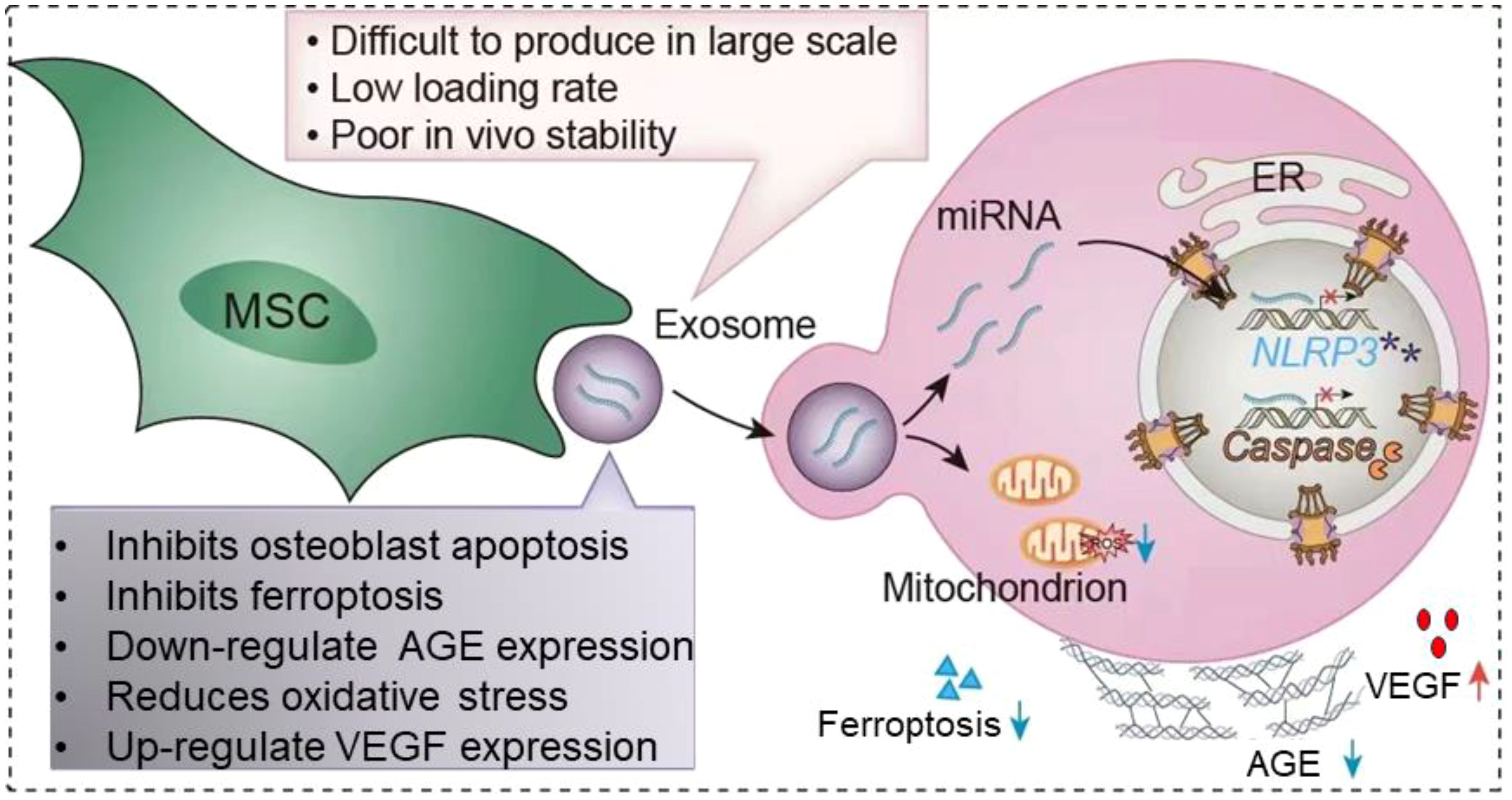
Figure 1. Application of extracellular vesicles in diabetic osteoporosis. MSC-EVs is able to be captured by target cells via endocytosis. EVs are able to carry the miRNAs and proteins they carry into target cells. It leads to decreased apoptosis of osteoblasts, inhibits ferroptosis, downregulates AGE, reduces oxidative stress, promotes VEGF secretion and thus promotes angiogenesis. However, the clinical application of EVs is faced with factors such as how to mass produce in large quantities, low loading rates and poor stability in vitro. The red cross means represents inhibition, and purple asterisk means NLRP3 post-catabolic products.
2 Osteoporotic microenvironment
Bone provides a physical scaffold for the daily activities of vertebrates. The cells in the bone mainly include osteoblasts, osteoclasts, and bone marrow mesenchymal stem cells (21). Osteoblasts are the main cells responsible for bone formation, and these cells secrete cell matrices such as type I collagen, osteopontin, osteocalcin, and alkaline phosphatase (22). Osteoclasts play a major role in the body for bone resorption (23). Bone has a unique ability to regenerate and repair, and the regulation of the balance of osteoblasts and osteoclasts is the basis for this unique repair ability (24). The stem cells in bone are mainly bone marrow mesenchymal stem cells and bone stem cells (25). Bone marrow mesenchymal stem cells (BMSCs) form other cells in the bone microenvironment (ECM), including osteoblasts, chondroblasts, adipocytes, and other stromal cells (26). The ECM regulates the biological behavior of cells, providing biochemical signals to cells and physical and mechanical signals to cells (27). The ECM is mainly composed of type I collagen fibers (85-90%), and hydroxyapatite, as a component that enhances the mechanical properties of collagen fibers, can increase the tensile modulus of collagen matrix by 4 times (28). In addition to collagen fibers, ECM also contains a large amount of non-collagen components such as osteocalcin, osteopondin, osteopontin, adhesion proteins, and proteoglycans (29). Non-collagen components are not only involved in the composition of the ECM, but also play an integral role in the bone remodeling process (30). Bone remodeling is the dynamic equilibrium process of bone ECM, which occurs during a person’s lifetime activities. Bone remodeling can be separated into five phases: activation; resorption; reversal; formation; and termination (31). Abnormal bone remodeling, which leads to a decrease in bone mass, density, mass, and strength, is the most important factor in osteoporosis (32).
Osteoblasts are derived from BMSCs in vivo and are thought to be the only cells in vertebrates that can promote bone regeneration (33). BMSCs are pluripotent cells in the bone marrow that can differentiate into osteoblasts, adipocytes, and chondrocytes (34). Differentiation of BMSCs into osteoblasts is first required to induce Runx family transcription factor 2 (RUNX2) (35). 60% to 80% of osteoblasts differentiated by BMSCs undergo apoptosis, and the remaining osteoblasts become mature osteoblasts (36). Osteoblasts secrete extracellular matrix proteins, type I collagen, and other non-collagen proteins (37).ECM is first secreted in an unmineralized osteoid form and subsequently mineralized into bone by the accumulation of calcium phosphate in the form of hydroxyapatite (38). In addition, osteoblasts promote osteoclast maturation and proliferation. Osteoblasts secrete macrophage colony-stimulating factor (M-CSF) that binds to the colony-stimulating factor-1 receptor (c-FMS) in osteoclasts, thereby activating phosphoinositide 3-kinase (PI3K) and growth factor receptor-binding protein 2 (Grb2) and further promoting osteoclast differentiation and maturation (39). Osteoclasts arise from the bloodline monocyte-macrophage system and are the primary functional cells for bone resorption (40). Factors such as age, gender, trauma, and medications can all lead to disruption of bone remodeling balance, dysfunction of bone cell activity and abnormal cytokine secretion, which ultimately leads to osteopenia and bone imbalance (41). The decrease in collagen fibers and bone mineral salt content, the reduction, fracture or disappearance of trabeculae leads to the relaxation of bone tissue structure, which in turn affects the mechanical nature of the skeletal system and leads to fractures (42). The changes in the osteoporosis microenvironment are mainly caused by the destruction of bone remodeling balance caused by the decrease of osteoblast activity and the enhancement of osteoclast activity, which in turn leads to the decrease in the number of osteocytes and the occurrence of osteoporosis (Figure 2).
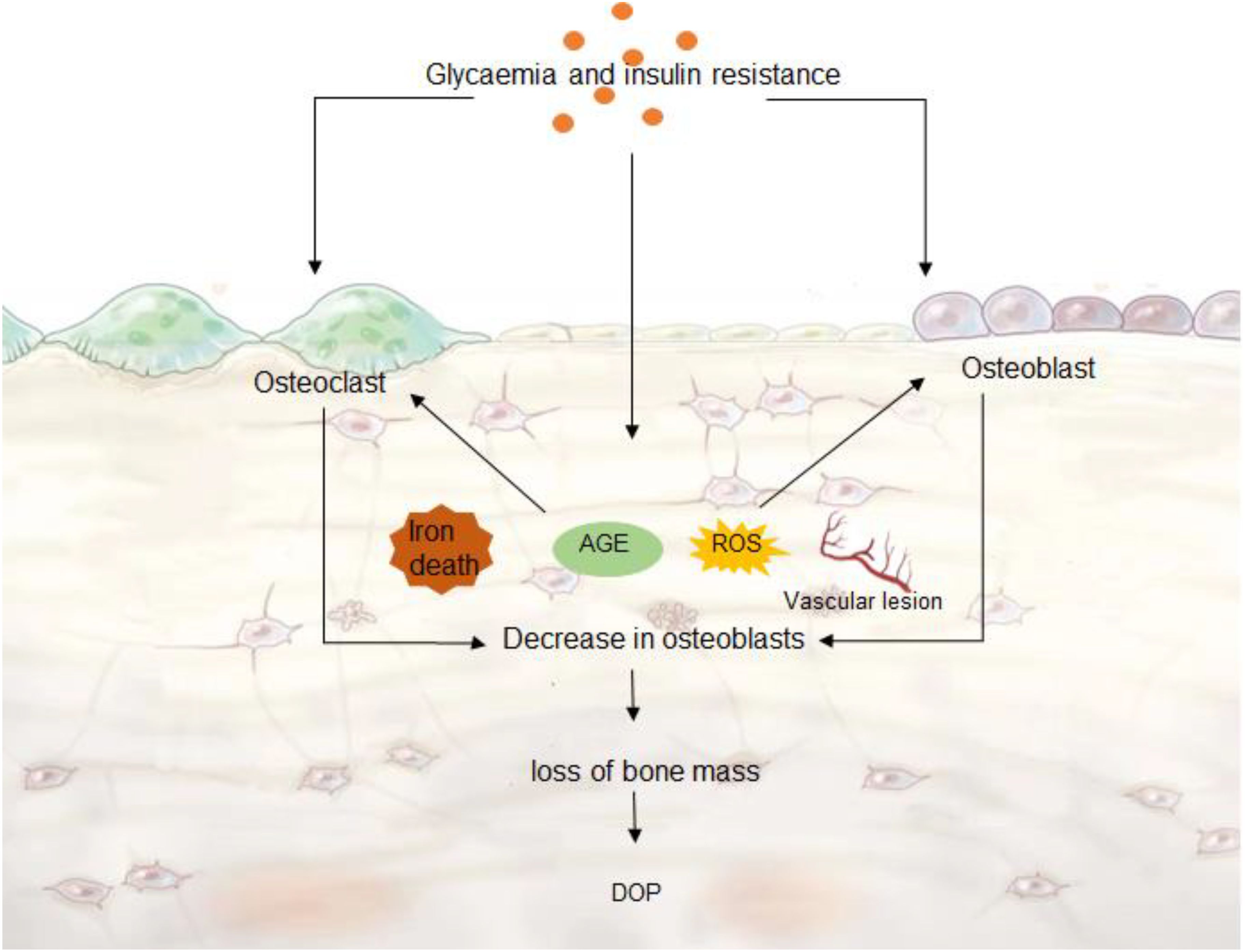
Figure 2. DOP microenvironment. Hyperglycemia and insulin resistance can cause ferroptosis, ROS and AGE production, vascular lesions, which further accelerate the decrease of osteoblast activity, and the enhancement of osteoclast activity leads to abnormal bone remodeling. Decreased bone cell content leads to DOP.
3 Pathogenesis of DOP
3.1 Hyperglycemia and insulin resistance
The main factors of diabetes mellitus are insulin deficiency due to decreased function of β cells in the pancreas to secrete insulin, or insulin resistance due to decreased physiological effects of insulin in cells in the body due to insulin insensitivity leading to insulin-related physiological effects (43). Insulin resistance and chronic inflammatory processes go hand in hand (44). Adipose tissue releases IL-1β, IL-6, and TNFα by activating M1 macrophages, creating a pro-inflammatory environment that blocks the action of insulin (45). The release of inflammatory factors exacerbates aberrant activation of nucleotide-binding oligomeric domain-like receptor protein 3 (NLRP3) inflammasomes, and autoinflammatory mechanisms may exacerbate pancreatic injury (46). Insulin resistance may affect bone health by impairing the insulin-like growth factor-1 (IGF-1) signaling pathway (47). Insulin inhibits the expression of IGF-binding protein-1 (IGFBP-1) in osteoblasts, and insulin deficiency increases IGFBP-1 levels, thereby reducing the anabolic effects of unbound IGF-1 in bone (48). In addition, studies have shown that reduced nitric oxide (NO) bioavailability is one of the mechanisms of diabetes-induced osteoporosis (49). Endoxela Nocintas (Enos) de Rivedno can reduce osteoclast activity, thereby reducing bone destruction and reducing the incidence of osteoporosis (50). Insulin resistance can also eventually lead to an increase in glucose in the body.
Glucose is the main source of bone development and growth and plays a vital role in the skeletal system (51). Glycolysis plays an important role in osteoclast differentiation. Blocking the glycolytic pathway has been shown to inhibit osteoclast production (52). Mature osteoclasts have been shown to have higher rates of glycolysis (40). Diabetes disrupts the balance of bone metabolism and can lead to osteoporosis (53). Glucose stimulates the non-canonical Wnt/protein kinase C pathway and upregulates the peroxisome proliferator-activated receptor γ (PPARγ), resulting in increased BMSC differentiation to adipose and bone loss (54). While some adipogenic factors such as adiponectin, endolipids, and omentin-1 have negative effects on bones (55). Not only that, higher blood glucose levels in type 2 diabetes are inversely correlated with serum osteocalcin levels (56). Osteocalcin is considered a biochemical marker of bone remodeling, and a hallmark of type 2 diabetic bone disease is decreased bone formation and decreased serum osteocalcin (57). In addition, hyperglycemia can lead to osmotic diuresis, impaired reabsorption of calcium ions in the renal tubules and collecting ducts, which in turn leads to a decrease in blood calcium levels (58). Decreased serum calcium stimulates parathyroid hormone secretion, leading to increased osteoclast activity, which in turn exacerbates bone destruction (59).
Insulin resistance and hyperglycemia have been widely demonstrated in the pathogenesis of TOP. Control of blood glucose levels reduces the incidence of DOP. Therefore, insulin resistance and hyperglycemia have a non-negligible role in the occurrence of DOP.
3.2 Oxidative stress
Reactive oxygen species (ROS) are normal metabolites of oxygen, and when they are produced in excess, they can lead to a disruption of the oxidant-antioxidant balance (60). Diabetic patients have an increased release of oxidative factors in the body due to disorders in blood glucose metabolism (61). Studies have shown that biomarkers of oxidative stress are positively correlated with glucose concentration, and antioxidant enzymes are negatively correlated with glucose concentration (62). Hyperglycemia can lead to excessive production of ROS, which is not only a major factor in aging, but also one of the leading causes of diabetes complications, insulin resistance, and mitochondrial dysfunction (63). The massive accumulation of ROS can lead to protein inactivation and lipid membrane peroxidation, which can lead to DNA damage (64). Not only that, the accumulation of a large amount of ROS can also lead to the inactivation of various enzyme systems in the body, which in turn will disrupt the normal level of substance metabolism in the body (65). In addition, excess ROS can alter the microenvironment of osteoblasts, destroy bone microstructure, reduce bone formation, and prolong fracture healing time (66). Wang et al. showed that hyperglycemia and oxidative stress can exacerbate the accumulation of advanced glycation and glycooxidation end products (AGEs/AGOEs) in bones (67). The increase in AGEs/AGOEs in the bone microenvironment leads to increased bone fragility and thus osteoporosis (68). Zhou et al. showed that the expression level of oxidative stress-related genes in osteoporosis patients was significantly higher than that of normal people, such as Supero Hidededis, Mutassé (Saud), Ineris Rosites, Catalas (Kate), Totar, Antio, Hitant, Status (Tass), Andesidro Prosides (Sea) (69). Therefore, oxidative stress plays an important role in the development of osteoporosis.
In bone metabolism, ROS mainly relies on mitochondria, xanthine oxidase and NADPH oxidase to promote osteoblast apoptosis and inhibit osteoblast differentiation (70). In addition, ROS promotes osteoclast differentiation mainly by inducing multinuclear fusion of BMSCs and increasing the expression of NFATc1, CTSK and MMP9 through MAPK and NF-κB signaling pathways (71). Therefore, the accumulation of ROS can cause bone healing disorders and promote bone aging (72). Jiang et al. confirmed that inhibition of autophagy and oxidative stress in vivo can reduce the incidence of DOP (73). ROS not only affects osteogenic differentiation but also stimulates osteoblast apoptosis (74). Reducing inflammatory oxidative stress can reduce the occurrence of DOP.
Hypoxia-inducible factor (HIF-1) is the main regulator of hypoxia adaptation response, and as a transcription factor, HIFs are involved in maintaining cellular oxygen homeostasis (75). Hyperglycemia promotes ubiquitination of the carboxy-terminus of HIF-1α ligase and subsequent proteasomal degradation, thereby destabilizing HIF-1α (76). Wang et al. showed that SENP3 overexpression inhibited osteoclast differentiation in type II DOP by increasing the stability and expression of HIF-1α (77). Metformin (Met) is a potent anti-diabetic and redox modulator (78). Met has been shown to improve insulin resistance and oxidative stress levels in type 2 diabetes through the Keap1/Nrf2/Ho-1 pathway (79). Chen et al. confirmed that Met can inhibit hyperglyce-induced oxidative stress by activating the Nrf2/HO-1 signaling pathway in type 2 DOP, reducing bone loss and bone microstructure destruction (72). In addition, Jiang et al. also confirmed that inhibition of oxidative stress can enhance collagen fibers, changes in trabecular bone density, and improve changes in bone microenvironment (73).
Oxidative stress has been shown to accelerate the progression of degenerative diseases. Preclinical studies have reported a large number of studies in which reducing oxidative stress and delaying the progression of DOP have been reported. Moreover, oxidative stress is one of the initiating factors affecting the DOP microenvironment. Reducing oxidative stress can not only maintain the cellular activity of osteoblasts, osteoclasts and BNSCs, but also reduce bone loss and damage to bone microstructure, and delay the progression of DOP. Therefore, reducing oxidative stress plays an important role in the occurrence of DOP.
3.3 Ferroptosis
Iron participates in various biochemical processes such as enzymatic reactions and oxygen transport in the body, and is one of the indispensable trace elements in human life (80). Iron enters cells by binding to transferrin receptor 1 (TFR1) on the cell membrane to reduce ferric iron to iron (81). Subsequently, divalent metal transporter 1 (DMT1) releases ferric iron into the labile iron pool (LIP) in the cytoplasm. Lysosomes recycle ferritin and iron from mitochondria (81, 82). Ferroptosis is a form of apoptosis that relies on intracellular iron, and iron overload caused by abnormal iron metabolism is one of the main features of ferroptosis (83). Iron overload decreases cell viability, glutathione, and superoxide dismutase levels, increasing ROS production and lipid peroxidation (84). Iron can reach the mitochondrial matrix through the outer mitochondrial membrane and inner mitochondrial membrane, where it subsequently regulates the physiological functions of important organelles in the mitochondria (85). Diane et al. confirmed that mitochondrial transferrin 1/2 (Mfrn1/2) on the inner mitochondrial membrane is destroyed, resulting in abnormal mitochondrial iron metabolism (86). Although the specific mechanism of ferroptosis-induced osteoporosis is elusive, the occurrence of ferroptosis and osteoporosis has been widely confirmed (87).
Ferroptosis relies primarily on iron overload and lipid peroxidation (88). Biological features of ferroptosis include decreased cystine uptake, reduced glutacystine depletion, inhibition of cystine/glutamate reverse transport system (Xc system) activity, and aberrant accumulation of iron ions and ROS (12). Ferroptosis can also lead to the breakdown of the barrier between bone resorption and bone formation, which can lead to osteoporosis (89). Iron overload-mediated oxidative stress increases ROS levels, impairs mitochondrial dysfunction, and decreases the mineralization and osteogenic differentiation capacity of BMSCs (90). Hyperglycemia can cause overexpression of divalent metal transporter 1 (DMT1) in osteoblasts, which can induce ferroptosis (91). Ma et al. demonstrated that melatonin inhibits hyperglyce-induced ferroptosis by activating the Nrf2/HO-1 signaling pathway in type 2 DOP, thereby delaying the onset of osteoporosis (89). Nuclear factor erythroid-2 related factor 2 (NRF2) is an antioxidant stress gene in vivo that regulates oxidative stress by controlling the expression of heme oxygenase-1 (HO-1) and a variety of other proteins with antioxidant and anti-inflammatory effects (92). Zhang et al. confirmed that activation of NRF2 can inhibit hyperglucose-induced ferroptosis in osteoblasts, thereby reducing the occurrence of osteoporosis (93). Lin et al. demonstrated that hyperglycemia induces ferroptosis (upregulation of ferroptosis-related genes SLC7A11 and GPX4 expression in osteoporoblasts by activating the METTL3/ASK1-p38 signaling pathway, resulting in osteoporosis (94). Ferroptosis has been widely demonstrated in the occurrence of DP (95–97).
3.4 Build-up of advanced glycation products
In the hyperglycemic state, non-enzymatic glycosylation of proteins, phospholipids, and nucleic acids (whose classical pathway is also known as the Maillard reaction) leads to the formation of advanced glycation end products (AGEs) (98). Because of its higher surface-to-volume ratio, AGE accumulates significantly higher in trabecular bone than in cortical bone (67). AGE leads to decreased bone formation by decreasing osteoblast differentiation and proliferation, downregulating matrix production, and increasing osteoblast apoptosis (99). Studies have shown that AGEs not only induce osteoclast production by upregulating RANKL mRNA, but also affect osteoblasts by inhibiting cell growth, promoting apoptosis, and downregulating differentiation (100). Elevated blood sugar in diabetic patients can induce excessive accumulation of AGEs in bones, which in turn leads to osteoporosis (101). Moreover, studies have found that pathological activation of AGE and AGE receptor (RAGE) signaling inhibits the osteogenic differentiation of BMSCs by modulating DNA methylation and Wnt signaling (102). Inhibition of AGE-RAGE activation can reduce the occurrence of DP (103). In addition, activation of RAGE enhances apoptosis, inflammation, and oxidative stress that directly damages cells and tissues (104). Notsu et al. showed that AGE-RAGE increased TGF-β expression and secretion and inhibited stromal cell mineralization (105). Ding et al. demonstrated through in vivo experiments in mice that the number of osteoclasts decreased, bone resorption decreased, bone mass increased, biomechanical strength was improved, and the occurrence of cancellous femoral bone was reduced in mice that were knocked out or inhibited (106). Inhibition of RAGE signaling is to improve the ability of BMSCs to differentiate into osteoblasts and osteocytes under diabetic conditions (100).
The study of AGE in DOP has gradually been recognized. AGE affects trabecular bone distribution in the bone microenvironment, alters bone mineral density, and leads to DOP. Reducing the expression of AGE can inhibit osteoclast acquisition and delay the progression of AD. Reducing the expression of AGE plays an important role in the occurrence of DOP.
3.5 Osteovascular
Hyperglycemia can lead to microvascular and macrovascular lesions. Angiogenesis in bone and its role in bone metabolism fundamentally affect the pathogenesis of osteoporosis (107). Neovascularization is closely related to osteogenic activity, which not only directly provides oxygen, nutrients and growth factors to the osteogenic active area, but also promotes osteogenic activity by secreting vascular secretion signals to create a microenvironment suitable for the pre-survival of mesenchymal stem cells and osteoblasts (108). There is a capillary subtype in the bone that is involved in bone formation and stimulates the proliferation of bone progenitor cells and differentiation factors within the bone marrow to actively guide bone formation, and this blood vessel is known as H-shaped capillaries (109). Reduction of H-type capillaries is associated with bone loss in a mouse model of aging and postmenopausal osteoporosis (110). Hu et al. found that damage to H-type blood vessels by NADPH oxidase 2 (NOX2)-mediated endothelial oxidative stress may be a target for type 1 diabetes-induced bone disease (111). Activation of Notch and Hif-1α signaling pathways in bone tissue and exogenous PDGF-BB treatment increased the number of H-type blood vessels while restoring bone mass (112). Type H capillary injury is associated with a variety of factors. Lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1) is a major receptor for the recognition of oxidized LDL and is involved in lipid metabolism and the development of vascular inflammation (113). LOX-1 can promote osteoclast activity and enhance bone destruction (114). Qiu et al. confirmed that knockdown of LOX-1 gene can enhance bone mineral structure and improve the expression of trabecular microstructure and osteogenesis-related genes such as bone morphin 2 (BMP2) and osteopontin (OPN) (115). βII-Spectrin (SPTBN1) is an actin cross-linked molecular scaffolding protein involved in the arrangement of transmembrane proteins and organelle organization in vivo (116). Xu et al. found that SPTBN1 could not regulate osteoblast differentiation through TGF-β/Smad3 and STAT1/Cxcl9 signaling pathways, and could also promote the expression of vascular endothelial growth factor (VEGF) to promote the formation of skeletal blood vessels (117).
Blood vessels maintain the transition between nutrients and metabolic wastes in bone tissue, providing growth essentials for the bone microenvironment. Bone tissue is inseparable from the support of blood vessels. In the DOP setting, the generation of new blood vessels is a key factor in reversing disease progression.
4 Application of extracellular vesicles in DOP
4.1 EVs
EVs are a heterogeneous population of membrane vesicles secreted by living cells and play an important role in maintaining cellular function and tissue homeostasis in multicellular organisms (118). According to their subcellular origin and size, they are divided into three main groups: apoptotic bodies, membrane vesicles, and EVs. EVs are membrane-vesicle-like structures with a diameter of about 30-150 nm, which contain proteins, lipids, and RNA in cells (19, 119). EVs originate from nanoscale vesicles formed by inward budding from endosomal membranes and are secreted into the extracellular environment when membrane vesicles fuse with the plasma membrane (120). As a special way of information transmission between cells, Evs can deliver the goods carried by itself to target cells through endocytosis and cytocytosis, thereby regulating the life process of target cells (119). Because EVs are produced by living cells and have a double-layer lipid membrane structure, EVs have the advantages of low immunogenicity and good cell compatibility in vivo (19). In addition, EVs can also transfer the carnage into the EVs through exogenous loading pathways, such as co-incubation, thermal shock, sonication, and electroporation (121). The advantage of EVs is that they can transport the contents to target cells without being degraded (122). In addition, chemical modifications give EVs unique targeting capabilities (123). EVs from different cell sources have different targeting abilities in vivo (124). This type of cell homing is species-independent. Chemical modification and genetic engineering are the main improvement methods for EV membrane modification (125). For example, Alvarez-Erviti et al. targeted the central nervous system by genetically engineering dendritic cells to express Lamp2b and RVG peptides (126).
The ability of stem cells to self-proliferate and differentiate in a directed manner plays an important role in tissue engineering and regenerative medicine (127). However, dead cells produced by stem cells after local metabolism can trigger local inflammation and tumorigenesis (128). Exogenous stem cells are not conducive to survival and proliferation in the ischemic and inflammatory microenvironment, and may even lead to the death of many cells in the body (118). Not only that, but the retention and undersurvival of mesenchymal stem cells at the site of administration can hinder their therapeutic efficacy (129). EVs can avoid the toxic side effects of cell therapy. EVs are paracrine production that not only maintain part of the function of the cell from which they originate, but can fuse with the cell membrane of the target cell after it has functioned (130). The role of EVs has been proven in many areas, such as tumors, obligatory spondylitis, diseases of the cardiovascular system, osteoarthritis, etc (131–134).
4.2 Application of EVs in DOP
The use of EVS in DOP has been demonstrated in preclinical studies and is considered a meaningful therapeutic trend for DOP. Adipose tissue-derived MSCs have shown strong potential in reducing inflammation in the body and improving the paracrine effects of chondrocytes (10). Tofino et al. demonstrated that adipose tissue-derived MSCs (AD-MSCs)-derived EVs significantly reduce inflammatory mediators and inhibit nitric oxide synthase activity in osteoarthritis chondrocytes (135). Decreased NO bioavailability is one of the mechanisms of diabetes-induced osteoporosis (49). Therefore, can EVs derived from AD-MSCs also play a role in DOP? To verify this effect, Zhang et al. isolated AD-MSCs-derived EVs and confirmed that they can alleviate diabetic osteoporosis in mice by inhibiting NLRP3 inflammasome activation in osteoclasts in diabetic mice (136). AD-MSCs-derived EVs inhibit the production of pro-inflammatory cytokines IL-1β and IL-18 and inhibit the production of apoptotic factor caspase-1 for 72 hours in vivo. Moreover, AD-MSCs-derived EVs reduced the loss of bone mineral content and the loss of bone mineral density at day 28 in mice. Zhang et al. transduced miR-146a into AD-MSCs-derived EVs in vitro to enhance its anti-inflammatory effect and delay the progression of DOP (137). miR-146a can modulate inflammation and osteogenesis (138). AD-MSCs-derived EVs overexpressing miR-146a could significantly inhibit NLRP3 inflammasome activation and alleviate the pathogenesis of DOP. Therefore, EVs can reduce inflammation in DOP and promote osteoblast activity, delaying the progression of LOP.
Abnormal macrophage polarization and pro-inflammatory M1 macrophage hypersensitivity can be detected in DOP. Appropriate interventions to promote macrophage polarization to the M2 subtype will help promote diabetic fracture healing (139). Wang et al. cultured M2 macrophages to produce EVs that induce the transformation of M1 macrophages into M2 macrophages by stimulating the PI3K/AKT pathway (140). M2 macrophages increase bone morphogenetic protein 2 (BMP-2) expression, leading to cartilage and bone formation. Bone marrow mesenchymal stem cells (BMSCs) have the potential for multidirectional differentiation and self-renewal, playing a central role in bone homeostasis and regeneration (141). Studies have shown that during the process of cellular senescence, BMSCs tend to differentiate into adipocytes, resulting in low bone mass and bone marrow fat accumulation (142). MSC-derived exosome metastases miR-146a, miR-126, miR-128-3p, and miR-196a, among others, to alleviate DOP and reduce the incidence of fractures (143). It was found that the elevated expression of miR-382-3p and miR-550a-5p in the serum of patients with type 2 diabetes affected the osteogenic differentiation of BMSCs (144). miR-221, as one of the highly expressed miRNAs in diabetic BMSC exosomes, has the ability to inhibit osteogenesis and promote adipogenesis (145). Han et al. showed that inhibition of miR-221 expression in BMSC-EVs increased bone mass, reduced bone marrow fat accumulation, and promoted bone regeneration in diabetic mice (143).
EVS acts as a carrier to transfer the cargo to the osteoporosis microenvironment, which delays the progression of osteoporosis under the premise of protecting the carrier from destruction. Although studies of Evs in osteoporosis have been widely reported, there are few preclinical studies of EVs in DOP. The therapeutic role of EVs in DOP has been confirmed in all published preclinical studies.
4.3 Challenges in the application of EVs in DOP
The most important factors for EVS are how to stabilize production on a large scale and have low load efficiency (118). Despite the multiple advantages of EVs, there are still no standardized recommendations on how to produce, purify, and store them on a large scale (146). In a laboratory setting, less than 1 ug of exosomal protein should only be harvested from 1 ml of medium (147). Although there are many biochemical strategies, physical strategies (hypoxia, heat stress, and starvation), mechanical strategies (shear stress and 3D culture), and instrumental strategies, among others (148). However, due to the differences in size, surface marking, content, and origin of EVs, separating them is also a major challenge (149). Grangier et al. reported a 3D incubation platform that can minimize operation time, incubation cycle, and cost to enable large-scale production of EVs (150). However, mass-produced Evs also need to undergo ultracentrifugation for purification. Although ultracentrifugation is the gold standard for the separation and purification of EVs (151). However, the purification of EVs is limited by the time-consuming, costly, inefficient, and lipoprotein co-separation of large-scale EVS (149). In recent years, immunoaffinity chromatography, antigen/protein conjugation, ultrafiltration, and size exclusion chromatography have all been used for mass production of EVs (146). While no single technique is perfect, combining the above techniques with others may be the solution to meet multiple requirements for exosome batch production and purification simultaneously (151).
In addition, the low loading efficiency of EVs and how they are chemically modified to be targeted are another factor limiting their application (152). The modification of exosomes is divided into internal strategies (drug loading) and external strategies (surface modifications). The primary purpose of surface modification is to selectively deliver exosomes to target cells for precise processing (153). Although co-incubation, sonication, thermal shock, and electroporation can improve the loading rate, their effect is limited (154). For example, the loading efficiency of co-incubation is low, and ultrasonic electroporation disrupts the membrane structure of EVs (155). In addition, the long-term storage of EVs is also a limiting factor for their application. At present, the methods used for long-term storage of EVs mainly include cryopreservation, lyophilization and spray drying (156). -80°C is considered the optimal temperature with minimal impact on the morphology and content of exosomes (156).
How long manufactured EVs can be stored in vitro and in vivo stability after storage remain unknown. At present, there are no consistent reports on related studies. Even though there are some limitations to the clinical application of EVs, there are still many clinical trials of EVs (146). At present, the ability of EVs to reverse bone imbalance, stimulate osteoblast activity, inhibit osteoclast activity, and deliver siRNA to regulate the osteoporosis microenvironment in osteoporosis has been widely reported, but the reports in Evs in DOP are still insufficient. We believe that EVs will make a breakthrough in the field of DOP in the near future.
5 Prospect
EVS has demonstrated its potential in tissue engineering and regenerative medicine in preclinical studies in recent years. EVs have low immunogenicity, biological histocompatibility, and can avoid the side effects of cell therapy. EVS is capable of being chemically modified to be targeted. At the same time, EVs can also act as carriers to release their contents into target cells. Preclinical studies have demonstrated that EVs can reduce the incidence of DOP and reduce the complications of fractures in DOP. Although there are few reports of preclinical studies on the use of EVs in DOP, there have been more clinical trials of EVs in other fields. At present, the problems faced by EVS have been recognized by researchers, and many improvement solutions have been designed. We believe that as people’s awareness of EVs continues to rise, there will definitely be a breakthrough in the application of EVs in DOP.
Author contributions
XJ: Conceptualization, Writing – original draft. GZ: Formal analysis, Writing – review & editing. DY: Formal analysis, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This article was supported by the National Key Research and Development Program of China (2022YFB4703500).
Acknowledgments
We would like to express our appreciation to everyone involved in drafting and preparing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ensrud KE, Crandall CJ. Osteoporosis. Ann Intern Med. (2024) 177:Itc1–itc16. doi: 10.7326/aitc202401160
2. Sheik Ali A. Osteoporosis: A narrative review. Cureus. (2023) 15:e43031. doi: 10.7759/cureus.43031
3. Johnell O, Kanis JA. An estimate of the worldwide prevalence and disability associated with osteoporotic fractures. Osteoporos Int. (2006) 17:1726–33. doi: 10.1007/s00198-006-0172-4
4. Sözen T, Özışık L, Başaran N. An overview and management of osteoporosis. Eur J Rheumatol. (2017) 4:46–56. doi: 10.5152/eurjrheum.2016.048
5. Yu B, Gao Q, Sheng S, Zhou F, Geng Z, Wei Y, et al. Smart osteoclasts targeted nanomedicine based on amorphous CaCO(3) for effective osteoporosis reversal. J Nanobiotechnology. (2024) 22:153. doi: 10.1186/s12951-024-02412-9
6. Shen Y, Huang X, Wu J, Lin X, Zhou X, Zhu Z, et al. The global burden of osteoporosis, low bone mass, and its related fracture in 204 countries and territories, 1990-2019. Front Endocrinol (Lausanne). (2022) 13:882241. doi: 10.3389/fendo.2022.882241
7. Li H, Xiao Z, Quarles DL, Li W. Osteoporosis: mechanism, molecular target and current status on drug development. Curr Medicinal Chem. (2021) 28:1489–507. doi: 10.2174/0929867327666200330142432
8. Adejuyigbe B, Kallini J, Chiou D, Kallini JR. Osteoporosis: molecular pathology, diagnostics, and therapeutics. Int J Mol Sci. (2023) 24(19):14583. doi: 10.3390/ijms241914583
9. Wang H, Luo Y, Wang H, Li F, Yu F, Ye L. Mechanistic advances in osteoporosis and anti-osteoporosis therapies. MedComm (2020). (2023) 4:e244. doi: 10.1002/mco2.244
10. Hamann C, Kirschner S, Günther KP, Hofbauer LC. Bone, sweet bone–osteoporotic fractures in diabetes mellitus. Nat Rev Endocrinol. (2012) 8:297–305. doi: 10.1038/nrendo.2011.233
11. Cortet B, Lucas S, Legroux-Gerot I, Penel G, Chauveau C, Paccou J. Bone disorders associated with diabetes mellitus and its treatments. Joint Bone Spine. (2019) 86:315–20. doi: 10.1016/j.jbspin.2018.08.002
12. Chen Y, Zhao W, Hu A, Lin S, Chen P, Yang B, et al. Type 2 diabetic mellitus related osteoporosis: focusing on ferroptosis. J Trans Med. (2024) 22:409. doi: 10.1186/s12967-024-05191-x
13. Pan Q, Chen H, Fei S, Zhao P, Deng M, Xiao F, et al. Medications and medical expenditures for diabetic patients with osteoporosis in Beijing, China: A retrospective study. Diabetes Res Clin Pract. (2023) 206:110980. doi: 10.1016/j.diabres.2023.110980
14. Liu P, Zhang Z, Cai Y, Li Z, Zhou Q, Chen Q. Ferroptosis: Mechanisms and role in diabetes mellitus and its complications. Ageing Res Rev. (2024) 94:102201. doi: 10.1016/j.arr.2024.102201
15. Moore AE, Dulnoan D, Voong K, Ayis S, Mangelis A, Gorska R, et al. The additive effect of vitamin K supplementation and bisphosphonate on fracture risk in post-menopausal osteoporosis: a randomised placebo controlled trial. Arch Osteoporos. (2023) 18:83. doi: 10.1007/s11657-023-01288-w
16. Gether L, Storgaard H, Kezic S, Jakasa I, Hartmann B, Skov-Jeppesen K, et al. Effects of topical corticosteroid versus tacrolimus on insulin sensitivity and bone homeostasis in adults with atopic dermatitis-A randomized controlled study. Allergy. (2023) 78:1964–79. doi: 10.1111/all.15690
17. Reid IR, Horne AM, Mihov B, Bava U, Stewart A, Gamble GD. Duration of fracture prevention after zoledronate treatment in women with osteopenia: observational follow-up of a 6-year randomised controlled trial to 10 years. Lancet Diabetes Endocrinol. (2024) 12:247–56. doi: 10.1016/s2213-8587(24)00003-2
18. Yao J, Chen Y, Lin Z. Exosomes: Mediators in microenvironment of colorectal cancer. Int J Cancer. (2023) 153:904–17. doi: 10.1002/ijc.34471
19. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Sci (New York N.Y.). (2020) 367:eaau6977. doi: 10.1126/science.aau6977
20. Jeppesen DK, Zhang Q, Franklin JL, Coffey RJ. Extracellular vesicles and nanoparticles: emerging complexities. Trends Cell Biol. (2023) 33:667–81. doi: 10.1016/j.tcb.2023.01.002
21. Ambrosi TH, Longaker MT, Chan CKF. A revised perspective of skeletal stem cell biology. Front Cell Dev Biol. (2019) 7:189. doi: 10.3389/fcell.2019.00189
22. Long F. Building strong bones: molecular regulation of the osteoblast lineage. Nat Rev Mol Cell Biol. (2011) 13:27–38. doi: 10.1038/nrm3254
23. Salhotra A, Shah HN, Levi B, Longaker MT. Mechanisms of bone development and repair. Nat Rev Mol Cell Biol. (2020) 21:696–711. doi: 10.1038/s41580-020-00279-w
24. Murphy MP, Irizarry D, Lopez M, Moore AL, Ransom RC, Longaker MT, et al. The role of skeletal stem cells in the reconstruction of bone defects. J Craniofac Surg. (2017) 28:1136–41. doi: 10.1097/scs.0000000000003893
25. Dallas SL, Xie Y, Shiflett LA, Ueki Y. Mouse cre models for the study of bone diseases. Curr osteoporosis Rep. (2018) 16:466–77. doi: 10.1007/s11914-018-0455-7
26. Gori JL, Butler JM, Chan YY, Chandrasekaran D, Poulos MG, Ginsberg M, et al. Vascular niche promotes hematopoietic multipotent progenitor formation from pluripotent stem cells. J Clin Invest. (2015) 125:1243–54. doi: 10.1172/jci79328
27. Lopes D, Martins-Cruz C, Oliveira MB, Mano JF. Bone physiology as inspiration for tissue regenerative therapies. Biomaterials. (2018) 185:240–75. doi: 10.1016/j.biomaterials.2018.09.028
28. Nair AK, Gautieri A, Chang SW, Buehler MJ. Molecular mechanics of mineralized collagen fibrils in bone. Nat Commun. (2013) 4:1724. doi: 10.1038/ncomms2720
29. Hidalgo-Bastida LA, Cartmell SH. Mesenchymal stem cells, osteoblasts and extracellular matrix proteins: enhancing cell adhesion and differentiation for bone tissue engineering. Tissue Eng Part B Rev. (2010) 16:405–12. doi: 10.1089/ten.TEB.2009.0714
30. Zhu G, Zhang T, Chen M, Yao K, Huang X, Zhang B, et al. Bone physiological microenvironment and healing mechanism: Basis for future bone-tissue engineering scaffolds. Bioactive materials. (2021) 6:4110–40. doi: 10.1016/j.bioactmat.2021.03.043
31. Siddiqui JA, Partridge NC. Physiological bone remodeling: systemic regulation and growth factor involvement. Physiology. (2016) 31:233–45. doi: 10.1152/physiol.00061.2014
32. Ledger GA, Burritt MF, Kao PC, O'Fallon WM, Riggs BL, Khosla S. Role of parathyroid hormone in mediating nocturnal and age-related increases in bone resorption. J Clin Endocrinol Metab. (1995) 80:3304–10. doi: 10.1210/jcem.80.11.7593443
33. Pacifici R. T cells and post menopausal osteoporosis in murine models. Arthritis Res Ther. (2007) 9:102. doi: 10.1186/ar2126
34. Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. (1997) 89:755–64. doi: 10.1016/s0092-8674(00)80258-5
35. Kitase Y, Prideaux M. Targeting osteocytes vs osteoblasts. Bone. (2023) 170:116724. doi: 10.1016/j.bone.2023.116724
36. Manolagas SC, Parfitt AM. What old means to bone. Trends Endocrinol Metab. (2010) 21:369–74. doi: 10.1016/j.tem.2010.01.010
37. Nefussi JR, Sautier JM, Nicolas V, Forest N. How osteoblasts become osteocytes: a decreasing matrix forming process. J Biol Buccale. (1991) 19:75–82.
38. Blair HC, Larrouture QC, Li Y, Lin H, Beer-Stoltz D, Liu L, et al. Osteoblast differentiation and bone matrix formation in vivo and in vitro. Tissue Eng Part B: Rev. (2016) 23:268–80. doi: 10.1089/ten.teb.2016.0454
39. Xu J, Yu L, Liu F, Wan L, Deng Z. The effect of cytokines on osteoblasts and osteoclasts in bone remodeling in osteoporosis: a review. Front Immunol. (2023) 14:1222129. doi: 10.3389/fimmu.2023.1222129
40. Park-Min KH. Metabolic reprogramming in osteoclasts. Semin Immunopathol. (2019) 41:565–72. doi: 10.1007/s00281-019-00757-0
41. Liu W, Wang T, Yang C, Darvell BW, Wu J, Lin K, et al. Alkaline biodegradable implants for osteoporotic bone defects–importance of microenvironment pH. Osteoporos Int. (2016) 27:93–104. doi: 10.1007/s00198-015-3217-8
42. Luo ZH, Ma JX, Zhang W, Tian AX, Gong SW, Li Y, et al. Alterations in the microenvironment and the effects produced of TRPV5 in osteoporosis. J Transl Med. (2023) 21:327. doi: 10.1186/s12967-023-04182-8
43. Zhang J, Li Y, Lai D, Lu D, Lan Z, Kang J, et al. Vitamin D status is negatively related to insulin resistance and bone turnover in chinese non-osteoporosis patients with type 2 diabetes: A retrospective cross-section research. Front Public Health. (2021) 9:727132. doi: 10.3389/fpubh.2021.727132
44. Ren N, Kim E, Li B, Pan H, Tong T, Yang CS, et al. Flavonoids alleviating insulin resistance through inhibition of inflammatory signaling. J Agric Food Chem. (2019) 67:5361–73. doi: 10.1021/acs.jafc.8b05348
45. Harford KA, Reynolds CM, McGillicuddy FC, Roche HM. Fats, inflammation and insulin resistance: insights to the role of macrophage and T-cell accumulation in adipose tissue. Proc Nutr Soc. (2011) 70:408–17. doi: 10.1017/S0029665111000565
46. Wu M, Cai YL, Yang Y, Hu HM, Yao Y, Yang J, et al. Vitamin D ameliorates insulin resistance-induced osteopenia by inactivating the nucleotide-binding oligomerization domain-like receptor protein 3 inflammasome. Heliyon. (2023) 9:e13215. doi: 10.1016/j.heliyon.2023.e13215
47. Guo H, Wang C, Jiang B, Ge S, Cai J, Zhou Y, et al. Association of insulin resistance and β-cell function with bone turnover biomarkers in dysglycemia patients. Front Endocrinol (Lausanne). (2021) 12:554604. doi: 10.3389/fendo.2021.554604
48. Moyer-Mileur LJ, Slater H, Jordan KC, Murray MA. IGF-1 and IGF-binding proteins and bone mass, geometry, and strength: relation to metabolic control in adolescent girls with type 1 diabetes*. J Bone Mineral Res. (2008) 23:1884–91. doi: 10.1359/jbmr.080713
49. Yousefzadeh N, Jeddi S, Kashfi K, Ghasemi A. Diabetoporosis: Role of nitric oxide. Excli J. (2021) 20:764–80. doi: 10.17179/excli2021-3541
50. Tai Y-T, Cherng Y-G, Chang C-C, Hwang Y-P, Chen J-T, Chen R-M. Pretreatment with low nitric oxide protects osteoblasts from high nitric oxide-induced apoptotic insults through regulation of c-Jun N-terminal kinase/c-Jun-mediated Bcl-2 gene expression and protein translocation. J Orthopaedic Res. (2007) 25:625–35. doi: 10.1002/jor.20365
51. Karner CM, Long F. Glucose metabolism in bone. Bone. (2018) 115:2–7. doi: 10.1016/j.bone.2017.08.008
52. Indo Y, Takeshita S, Ishii KA, Hoshii T, Aburatani H, Hirao A, et al. Metabolic regulation of osteoclast differentiation and function. J Bone Miner Res. (2013) 28:2392–9. doi: 10.1002/jbmr.1976
53. An Y, Zhang H, Wang C, Jiao F, Xu H, Wang X, et al. Activation of ROS/MAPKs/NF-κB/NLRP3 and inhibition of efferocytosis in osteoclast-mediated diabetic osteoporosis. FASEB J. (2019) 33:12515–27. doi: 10.1096/fj.201802805RR
54. Fazeli PK, Horowitz MC, MacDougald OA, Scheller EL, Rodeheffer MS, Rosen CJ, et al. Marrow fat and bone—New perspectives. J Clin Endocrinol Metab. (2013) 98:935–45. doi: 10.1210/jc.2012-3634
55. Carnevale V, Romagnoli E, Del Fiacco R, Pepe J, Cipriani C, Piemonte S, et al. Relationship between bone metabolism and adipogenesis. J Endocrinol Invest. (2010) 33:4–8.
56. Liang Y, Tan A, Liang D, Yang X, Liao M, Gao Y, et al. Low osteocalcin level is a risk factor for impaired glucose metabolism in a Chinese male population. J Diabetes Investig. (2016) 7:522–8. doi: 10.1111/jdi.12439
57. de Araújo IM, Salmon CE, Nahas AK, Nogueira-Barbosa MH, Elias J Jr., de Paula FJ. Marrow adipose tissue spectrum in obesity and type 2 diabetes mellitus. Eur J Endocrinol. (2017) 176:21–30. doi: 10.1530/eje-16-0448
58. Puglisi S, Rossini A, Poli R, Dughera F, Pia A, Terzolo M, et al. Effects of SGLT2 inhibitors and GLP-1 receptor agonists on renin-angiotensin-aldosterone system. Front Endocrinol (Lausanne). (2021) 12:738848. doi: 10.3389/fendo.2021.738848
59. Wang Y, Hu Y, Lan S, Chen Z, Zhang Y, Guo X, et al. A recombinant parathyroid hormone-related peptide locally applied in osteoporotic bone defect. Advanced Sci (Weinheim Baden-Wurttemberg Germany). (2023) 10:e2300516. doi: 10.1002/advs.202300516
60. Toker H, Ozdemir H, Balcı H, Ozer H. N-acetylcysteine decreases alveolar bone loss on experimental periodontitis in streptozotocin-induced diabetic rats. J Periodontal Res. (2012) 47:793–9. doi: 10.1111/j.1600-0765.2012.01497.x
61. Chen X-F, Li X-L, Yang M, Song Y, Zhang Y. Osteoprotective effects of salidroside in ovariectomized mice and diabetic mice. Eur J Pharmacol. (2018) 819:281–8. doi: 10.1016/j.ejphar.2017.12.025
62. Rizwan H, Pal S, Sabnam S, Pal A. High glucose augments ROS generation regulates mitochondrial dysfunction and apoptosis via stress signalling cascades in keratinocytes. Life Sci. (2020) 241:117148. doi: 10.1016/j.lfs.2019.117148
63. Rains JL, Jain SK. Oxidative stress, insulin signaling, and diabetes. Free Radical Biol Med. (2011) 50:567–75. doi: 10.1016/j.freeradbiomed.2010.12.006
64. Nassar H, Kantarci A, van Dyke TE. Diabetic periodontitis: a model for activated innate immunity and impaired resolution of inflammation. Periodontol 2000. (2007) 43:233–44. doi: 10.1111/j.1600-0757.2006.00168.x
65. Yang K, Pei L, Zhou S, Tao L, Zhu Y. Metformin attenuates H2O2−induced osteoblast apoptosis by regulating SIRT3 via the PI3K/AKT pathway. Exp Ther Med. (2021) 22:1316. doi: 10.3892/etm.2021.10751
66. Waddington RJ, Alraies A, Colombo JS, Sloan AJ, Okazaki J, Moseley R. Characterization of oxidative stress status during diabetic bone healing. Cells Tissues Organs. (2011) 194:307–12. doi: 10.1159/000324251
67. Wang B, Vashishth D. Advanced glycation and glycoxidation end products in bone. Bone. (2023) 176:116880. doi: 10.1016/j.bone.2023.116880
68. Vashishth D, Gibson GJ, Khoury JI, Schaffler MB, Kimura J, Fyhrie DP. Influence of nonenzymatic glycation on biomechanical properties of cortical bone. Bone. (2001) 28:195–201. doi: 10.1016/S8756-3282(00)00434-8
69. Zhou Q, Zhu L, Zhang D, Li N, Li Q, Dai P, et al. Oxidative stress-related biomarkers in postmenopausal osteoporosis: A systematic review and meta-analyses. Dis Markers. (2016) 2016:7067984. doi: 10.1155/2016/7067984
70. Zhang C, Li H, Li J, Hu J, Yang K, Tao L. Oxidative stress: A common pathological state in a high-risk population for osteoporosis. Biomedicine Pharmacotherapy. (2023) 163:114834. doi: 10.1016/j.biopha.2023.114834
71. Liu H-D, Ren M-X, Li Y, Zhang R-T, Ma N-F, Li T-L, et al. Melatonin alleviates hydrogen peroxide induced oxidative damage in MC3T3-E1 cells and promotes osteogenesis by activating SIRT1. Free Radical Res. (2022) 56:63–76. doi: 10.1080/10715762.2022.2037580
72. Chen B, He Q, Yang J, Pan Z, Xiao J, Chen W, et al. Metformin suppresses Oxidative Stress induced by High Glucose via Activation of the Nrf2/HO-1 Signaling Pathway in Type 2 Diabetic Osteoporosis. Life Sci. (2023) 312:121092. doi: 10.1016/j.lfs.2022.121092
73. Jiang B, Mou YJ, Zhang XM, Lu K, Xie P, Rao YL, et al. Ziyin Bushen Fang improves Diabetic Osteoporosis by Inhibiting Autophagy and Oxidative Stress In vitro and In vivo. Comb Chem High Throughput Screen. (2024) 27:786–96. doi: 10.2174/0113862073261310231113062630
74. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. (2005) 120:483–95. doi: 10.1016/j.cell.2005.02.001
75. Semenza GL. Pharmacologic targeting of hypoxia-inducible factors. Annu Rev Pharmacol Toxicol. (2019) 59:379–403. doi: 10.1146/annurev-pharmtox-010818-021637
76. Catrina SB, Zheng X. Hypoxia and hypoxia-inducible factors in diabetes and its complications. Diabetologia. (2021) 64:709–16. doi: 10.1007/s00125-021-05380-z
77. Wang C, Zhu X, Chen R, Zhang X, Lian N. Overexpression of SENP3 promotes PPAR-γ transcription through the increase of HIF-1α stability via SUMO2/3 and participates in molecular mechanisms of osteoporosis. Mol Cell Endocrinol. (2023) 577:112014. doi: 10.1016/j.mce.2023.112014
78. Yan Y, Jun C, Lu Y, Jiangmei S. Combination of metformin and luteolin synergistically protects carbon tetrachloride-induced hepatotoxicity: Mechanism involves antioxidant, anti-inflammatory, antiapoptotic, and Nrf2/HO-1 signaling pathway. BioFactors. (2019) 45:598–606. doi: 10.1002/biof.1521
79. Abdelgawad LM, Mohy Abd El-hamed M, Sabry D, Abdelgwad M. Efficacy of photobiomodulation and metformin on diabetic cell line of human periodontal ligament stem cells through keap1/nrf2/ho-1 pathway. rbmb. (2021) 10:30–40. doi: 10.52547/rbmb.10.1.30
80. Nakamura T, Naguro I, Ichijo H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim Biophys Acta (BBA) - Gen Subj. (2019) 1863:1398–409. doi: 10.1016/j.bbagen.2019.06.010
81. Liu P, Wang W, Li Z, Li Y, Yu X, Tu J, et al. Ferroptosis: A new regulatory mechanism in osteoporosis. Oxid Med Cell Longev. (2022) 2022:2634431. doi: 10.1155/2022/2634431
82. Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J. (2016) 473:769–77. doi: 10.1042/BJ20150658
83. Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell. (2003) 3:285–96. doi: 10.1016/S1535-6108(03)00050-3
84. Jiang Z, Wang H, Qi G, Jiang C, Chen K, Yan Z. Iron overload-induced ferroptosis of osteoblasts inhibits osteogenesis and promotes osteoporosis: An in vitro and in vivo study. IUBMB Life. (2022) 74:1052–69. doi: 10.1002/iub.2656
85. Paul BT, Manz DH, Torti FM, Torti SV. Mitochondria and Iron: current questions. Expert Rev Hematol. (2017) 10:65–79. doi: 10.1080/17474086.2016.1268047
86. Ward DM, Cloonan SM. Mitochondrial iron in human health and disease. Annu Rev Physiol. (2019) 81:453–82. doi: 10.1146/annurev-physiol-020518-114742
87. Gao Z, Chen Z, Xiong Z, Liu X. Ferroptosis - A new target of osteoporosis. Exp Gerontol. (2022) 165:111836. doi: 10.1016/j.exger.2022.111836
88. Latunde-Dada GO. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta (BBA) - Gen Subj. (2017) 1861:1893–900. doi: 10.1016/j.bbagen.2017.05.019
89. Ma H, Wang X, Zhang W, Li H, Zhao W, Sun J, et al. Melatonin suppresses ferroptosis induced by high glucose via activation of the nrf2/HO-1 signaling pathway in type 2 diabetic osteoporosis. Oxid Med Cell Longev. (2020) 2020:9067610. doi: 10.1155/2020/9067610
90. Li X, Zeng J, Liu Y, Liang M, Liu Q, Li Z, et al. Inhibitory effect and mechanism of action of quercetin and quercetin diels-alder anti-dimer on erastin-induced ferroptosis in bone marrow-derived mesenchymal stem cells. Antioxidants (Basel). (2020) 9(3):205. doi: 10.3390/antiox9030205
91. Zhang W-L, Meng H-Z, Yang M-W. Regulation of DMT1 on bone microstructure in type 2 diabetes. Int J Med Sci. (2015) 12(16):441–9. doi: 10.7150/ijms.11986
92. Zhang Y, Yu W, Liu Y, Chang W, Wang M, Zhang L. Regulation of nuclear factor erythroid-2-related factor 2 as a potential therapeutic target in intracerebral hemorrhage. Front Mol Neurosci. (2022) 15:995518. doi: 10.3389/fnmol.2022.995518
93. Zhang Z, Ji C, Wang YN, Liu S, Wang M, Xu X, et al. Maresin1 suppresses high-glucose-induced ferroptosis in osteoblasts via NRF2 activation in type 2 diabetic osteoporosis. Cells. (2022) 11(16):2560. doi: 10.3390/cells11162560
94. Lin Y, Shen X, Ke Y, Lan C, Chen X, Liang B, et al. Activation of osteoblast ferroptosis via the METTL3/ASK1-p38 signaling pathway in high glucose and high fat (HGHF)-induced diabetic bone loss. FASEB J. (2022) 36:e22147. doi: 10.1096/fj.202101610R
95. Zhao Y, Du Y, Gao Y, Xu Z, Zhao D, Yang M. ATF3 regulates osteogenic function by mediating osteoblast ferroptosis in type 2 diabetic osteoporosis. Dis Markers. (2022) 2022:9872243. doi: 10.1155/2022/9872243
96. Du YX, Zhao YT, Sun YX, Xu AH. Acid sphingomyelinase mediates ferroptosis induced by high glucose via autophagic degradation of GPX4 in type 2 diabetic osteoporosis. Mol Med. (2023) 29:125. doi: 10.1186/s10020-023-00724-4
97. Xu CY, Xu C, Xu YN, Du SQ, Dai ZH, Jin SQ, et al. Poliumoside protects against type 2 diabetes-related osteoporosis by suppressing ferroptosis via activation of the Nrf2/GPX4 pathway. Phytomedicine. (2024) 125:155342. doi: 10.1016/j.phymed.2024.155342
98. Tang SY, Allen MR, Phipps R, Burr DB, Vashishth D. Changes in non-enzymatic glycation and its association with altered mechanical properties following 1-year treatment with risedronate or alendronate. Osteoporos Int. (2009) 20:887–94. doi: 10.1007/s00198-008-0754-4
99. Meng H-Z, Zhang W-L, Liu F, Yang M-W. Advanced glycation end products affect osteoblast proliferation and function by modulating autophagy via the receptor of advanced glycation end products/raf protein/mitogen-activated protein kinase/extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase (RAGE/raf/MEK/ERK) pathway*. J Biol Chem. (2015) 290:28189–99. doi: 10.1074/jbc.M115.669499
100. Asadipooya K, Uy EM. Advanced glycation end products (AGEs), receptor for AGEs, diabetes, and bone: review of the literature. J Endocr Soc. (2019) 3:1799–818. doi: 10.1210/js.2019-00160
101. Hunt HB, Torres AM, Palomino PM, Marty E, Saiyed R, Cohn M, et al. Altered tissue composition, microarchitecture, and mechanical performance in cancellous bone from men with type 2 diabetes mellitus. J Bone Mineral Res. (2019) 34:1191–206. doi: 10.1002/jbmr.3711
102. Zhang M, Li Y, Rao P, Huang K, Luo D, Cai X, et al. Blockade of receptors of advanced glycation end products ameliorates diabetic osteogenesis of adipose-derived stem cells through DNA methylation and Wnt signalling pathway. Cell proliferation. (2018) 51:e12471. doi: 10.1111/cpr.12471
103. Sun Y, Zhu Y, Liu X, Chai Y, Xu J. Morroniside attenuates high glucose-induced BMSC dysfunction by regulating the Glo1/AGE/RAGE axis. Cell proliferation. (2020) 53:e12866. doi: 10.1111/cpr.12866
104. Rabbani N, Xue M, Thornalley PJ. Methylglyoxal-induced dicarbonyl stress in aging and disease: first steps towards glyoxalase 1-based treatments. Clin Sci (Lond). (2016) 130:1677–96. doi: 10.1042/cs20160025
105. Notsu M, Yamaguchi T, Okazaki K, Tanaka K, Ogawa N, Kanazawa I, et al. Advanced glycation end product 3 (AGE3) suppresses the mineralization of mouse stromal ST2 cells and human mesenchymal stem cells by increasing TGF-β expression and secretion. Endocrinology. (2014) 155:2402–10. doi: 10.1210/en.2013-1818
106. Ding KH, Wang ZZ, Hamrick MW, Deng ZB, Zhou L, Kang B, et al. Disordered osteoclast formation in RAGE-deficient mouse establishes an essential role for RAGE in diabetes related bone loss. Biochem Biophys Res Commun. (2006) 340:1091–7. doi: 10.1016/j.bbrc.2005.12.107
107. Ramasamy SK, Kusumbe AP, Itkin T, Gur-Cohen S, Lapidot T, Adams RH. Regulation of hematopoiesis and osteogenesis by blood vessel–derived signals. Annu Rev Cell Dev Biol. (2016) 32:649–75. doi: 10.1146/annurev-cellbio-111315-124936
108. Liu X, Zhang P, Gu Y, Guo Q, Liu Y. Type H vessels: functions in bone development and diseases. Front Cell Dev Biol. (2023) 11:1236545. doi: 10.3389/fcell.2023.1236545
109. Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature. (2014) 507:323–8. doi: 10.1038/nature13145
110. Huang J, Yin H, Rao SS, Xie PL, Cao X, Rao T, et al. Harmine enhances type H vessel formation and prevents bone loss in ovariectomized mice. Theranostics. (2018) 8:2435–46. doi: 10.7150/thno.22144
111. Hu XF, Xiang G, Wang TJ, Ma YB, Zhang Y, Yan YB, et al. Impairment of type H vessels by NOX2-mediated endothelial oxidative stress: critical mechanisms and therapeutic targets for bone fragility in streptozotocin-induced type 1 diabetic mice. Theranostics. (2021) 11:3796–812. doi: 10.7150/thno.50907
112. Ding W, Xu C, Zhang Y, Chen H. Advances in the understanding of the role of type-H vessels in the pathogenesis of osteoporosis. Arch Osteoporos. (2020) 15:5. doi: 10.1007/s11657-019-0677-z
113. Civelek S, Kutnu M, Uzun H, Erdenen F, Altunoglu E, Andican G, et al. Soluble lectin-like oxidized LDL receptor 1 as a possible mediator of endothelial dysfunction in patients with metabolic syndrome. J Clin Lab Anal. (2015) 29:184–90. doi: 10.1002/jcla.21748
114. Li X, Wang X, Hu Z, Chen Z, Li H, Liu X, et al. Possible involvement of the oxLDL/LOX-1 system in the pathogenesis and progression of human intervertebral disc degeneration or herniation. Sci Rep. (2017) 7(1):7403. doi: 10.1038/s41598-017-07780-x
115. Qiu J, Liu J, Tian L, Yu J, Duan Q, Liu Y, et al. Knockdown of LOX-1 ameliorates bone quality and generation of type H blood vessels in diabetic mice. Arch Biochem Biophysics. (2024) 752:109870. doi: 10.1016/j.abb.2023.109870
116. Susuki K, Zollinger DR, Chang K-J, Zhang C, Huang CY-M, Tsai C-R, et al. Glial βII spectrin contributes to paranode formation and maintenance. J Neurosci. (2018) 38:6063–75. doi: 10.1523/jneurosci.3647-17.2018
117. Xu X, Yang J, Ye Y, Chen G, Zhang Y, Wu H, et al. SPTBN1 prevents primary osteoporosis by modulating osteoblasts proliferation and differentiation and blood vessels formation in bone. Front Cell Dev Biol. (2021) 9:653724. doi: 10.3389/fcell.2021.653724
118. Xia Y, Yang R, Hou Y, Wang H, Li Y, Zhu J, et al. Application of mesenchymal stem cell-derived exosomes from different sources in intervertebral disc degeneration. Front bioengineering Biotechnol. (2022) 10:1019437. doi: 10.3389/fbioe.2022.1019437
119. Liang Y, Duan L, Lu J, Xia J. Engineering exosomes for targeted drug delivery. Theranostics. (2021) 11:3183–95. doi: 10.7150/thno.52570
120. Urabe F, Kosaka N, Ito K, Kimura T, Egawa S, Ochiya T. Extracellular vesicles as biomarkers and therapeutic targets for cancer. Am J Physiol Cell Physiol. (2020) 318:C29–c39. doi: 10.1152/ajpcell.00280.2019
121. Armstrong JPK, Holme MN, Stevens MM. Re-engineering extracellular vesicles as smart nanoscale therapeutics. ACS Nano. (2017) 11:69–83. doi: 10.1021/acsnano.6b07607
122. Rankin-Turner S, Vader P, O'Driscoll L, Giebel B, Heaney LM, Davies OG. A call for the standardised reporting of factors affecting the exogenous loading of extracellular vesicles with therapeutic cargos. Advanced Drug Delivery Rev. (2021) 173:479–91. doi: 10.1016/j.addr.2021.04.012
123. Kwok ZH, Wang C, Jin Y. Extracellular vesicle transportation and uptake by recipient cells: A critical process to regulate human diseases. Processes. (2021) 9(2):273. doi: 10.3390/pr9020273
124. Yong T, Wang D, Li X, Yan Y, Hu J, Gan L, et al. Extracellular vesicles for tumor targeting delivery based on five features principle. J Control Release. (2020) 322:555–65. doi: 10.1016/j.jconrel.2020.03.039
125. Zhang X, Zhang H, Gu J, Zhang J, Shi H, Qian H, et al. Engineered extracellular vesicles for cancer therapy. Advanced Materials. (2021) 33:2005709. doi: 10.1002/adma.202005709
126. Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJA. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol. (2011) 29:341–5. doi: 10.1038/nbt.1807
127. Scala P, Rehak L, Giudice V, Ciaglia E, Puca AA, Selleri C, et al. Stem cell and macrophage roles in skeletal muscle regenerative medicine. Int J Mol Sci. (2021) 22(19):10867. doi: 10.3390/ijms221910867
128. Krylova SV, Feng D. The machinery of exosomes: biogenesis, release, and uptake. Int J Mol Sci. (2023) 24(2):1337. doi: 10.3390/ijms24021337
129. Arshi A, Petrigliano FA, Williams RJ, Jones KJ. Stem cell treatment for knee articular cartilage defects and osteoarthritis. Curr Rev Musculoskelet Med. (2020) 13:20–7. doi: 10.1007/s12178-020-09598-z
130. Li Z, Huang Z, Bai L. Cell interplay in osteoarthritis. Front Cell Dev Biol. (2021) 9:720477. doi: 10.3389/fcell.2021.720477
131. Tavasolian F, Lively S, Pastrello C, Tang M, Lim M, Pacheco A, et al. Proteomic and genomic profiling of plasma exosomes from patients with ankylosing spondylitis. Ann Rheum Dis. (2023) 82:1429–43. doi: 10.1136/ard-2022-223791
132. Ye L, Li Y, Zhang S, Wang J, Lei B. Exosomes-regulated lipid metabolism in tumorigenesis and cancer progression. Cytokine Growth Factor Rev. (2023) 73:27–39. doi: 10.1016/j.cytogfr.2023.05.002
133. Li H, Zhang J, Tan M, Yin Y, Song Y, Zhao Y, et al. Exosomes based strategies for cardiovascular diseases: Opportunities and challenges. Biomaterials. (2024) 308:122544. doi: 10.1016/j.biomaterials.2024.122544
134. Lu J, Zhang Y, Yang X, Zhao H. Harnessing exosomes as cutting-edge drug delivery systems for revolutionary osteoarthritis therapy. BioMed Pharmacother. (2023) 165:115135. doi: 10.1016/j.biopha.2023.115135
135. Tofiño-Vian M, Guillén MI, Pérez del Caz MD, Silvestre A, Alcaraz MJ. Microvesicles from human adipose tissue-derived mesenchymal stem cells as a new protective strategy in osteoarthritic chondrocytes. Cell Physiol Biochem. (2018) 47:11–25. doi: 10.1159/000489739
136. Zhang L, Wang Q, Su H, Cheng J. Exosomes from adipose derived mesenchymal stem cells alleviate diabetic osteoporosis in rats through suppressing NLRP3 inflammasome activation in osteoclasts. J Biosci Bioeng. (2021) 131:671–8. doi: 10.1016/j.jbiosc.2021.02.007
137. Zhang L, Wang Q, Su H, Cheng J. Exosomes from adipose tissues derived mesenchymal stem cells overexpressing microRNA-146a alleviate diabetic osteoporosis in rats. Cell Mol Bioeng. (2022) 15:87–97. doi: 10.1007/s12195-021-00699-4
138. Jiang C, Lin Y, Shan H, Xia W, Pan C, Wang N, et al. miR-146a Protects against Staphylococcus aureus-Induced Osteomyelitis by Regulating Inflammation and Osteogenesis. ACS Infect Dis. (2022) 8:918–27. doi: 10.1021/acsinfecdis.1c00459
139. Meng F, Xue X, Yin Z, Gao F, Wang X, Geng Z. Research progress of exosomes in bone diseases: mechanism, diagnosis and therapy. Front bioengineering Biotechnol. (2022) 10:866627. doi: 10.3389/fbioe.2022.866627
140. Wang Y, Lin Q, Zhang H, Wang S, Cui J, Hu Y, et al. M2 macrophage-derived exosomes promote diabetic fracture healing by acting as an immunomodulator. Bioact Mater. (2023) 28:273–83. doi: 10.1016/j.bioactmat.2023.05.018
141. Xia Y, Zhu J, Yang R, Wang H, Li Y, Fu C. Mesenchymal stem cells in the treatment of spinal cord injury: Mechanisms, current advances and future challenges. Front Immunol. (2023) 14:1141601. doi: 10.3389/fimmu.2023.1141601
142. Li CJ, Xiao Y, Sun YC, He WZ, Liu L, Huang M, et al. Senescent immune cells release grancalcin to promote skeletal aging. Cell Metab. (2022) 34:184–5. doi: 10.1016/j.cmet.2021.12.003
143. Han F, Wang C, Cheng P, Liu T, Wang WS. Bone marrow mesenchymal stem cells derived exosomal miRNAs can modulate diabetic bone-fat imbalance. Front Endocrinol (Lausanne). (2023) 14:1149168. doi: 10.3389/fendo.2023.1149168
144. Heilmeier U, Hackl M, Skalicky S, Weilner S, Schroeder F, Vierlinger K, et al. Serum miRNA signatures are indicative of skeletal fractures in postmenopausal women with and without type 2 diabetes and influence osteogenic and adipogenic differentiation of adipose tissue–derived mesenchymal stem cells in vitro. J Bone Mineral Res. (2016) 31:2173–92. doi: 10.1002/jbmr.2897
145. Guo M, Liu N, Guo Z. MiR-221-5p/Smad3 axis in osteoclastogenesis and its function: Potential therapeutic target for osteoporosis. Steroids. (2022) 185:109063. doi: 10.1016/j.steroids.2022.109063
146. Tan F, Li X, Wang Z, Li J, Shahzad K, Zheng J. Clinical applications of stem cell-derived exosomes. Signal Transduct Target Ther. (2024) 9:17. doi: 10.1038/s41392-023-01704-0
147. Gurunathan S, Kang M-H, Jeyaraj M, Qasim M, Kim J-H. Review of the isolation, characterization, biological function, and multifarious therapeutic approaches of exosomes. Cells. (2019) 8(4):307. doi: 10.3390/cells8040307
148. Bei HP, Hung PM, Yeung HL, Wang S, Zhao X. Bone-a-petite: engineering exosomes towards bone, osteochondral, and cartilage repair. Small. (2021) 17:2101741. doi: 10.1002/smll.202101741
149. Yang X-X, Sun C, Wang L, Guo X-L. New insight into isolation, identification techniques and medical applications of exosomes. J Controlled Release. (2019) 308:119–29. doi: 10.1016/j.jconrel.2019.07.021
150. Grangier A, Branchu J, Volatron J, Piffoux M, Gazeau F, Wilhelm C, et al. Technological advances towards extracellular vesicles mass production. Adv Drug Delivery Rev. (2021) 176:113843. doi: 10.1016/j.addr.2021.113843
151. Doyle LM, Wang MZ. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells. (2019) 8(7):727. doi: 10.3390/cells8070727
152. Araldi RP, Delvalle DA, da Costa VR, Alievi AL, Teixeira MR, Dias Pinto JR, et al. Exosomes as a nano-carrier for chemotherapeutics: A new era of oncology. Cells. (2023) 12(17):2144. doi: 10.3390/cells12172144
153. Zhu Q, Ling X, Yang Y, Zhang J, Li Q, Niu X, et al. Embryonic stem cells-derived exosomes endowed with targeting properties as chemotherapeutics delivery vehicles for glioblastoma therapy. Advanced Sci. (2019) 6:1801899. doi: 10.1002/advs.201801899
154. Olumesi KR, Goldberg DJ. A review of exosomes and their application in cutaneous medical aesthetics. J Cosmet Dermatol. (2023) 22:2628–34. doi: 10.1111/jocd.15930
155. Luan X, Sansanaphongpricha K, Myers I, Chen H, Yuan H, Sun D. Engineering exosomes as refined biological nanoplatforms for drug delivery. Acta Pharmacol Sin. (2017) 38:754–63. doi: 10.1038/aps.2017.12
Keywords: diabetic osteoporosis, extracellular vesicles, osteoblasts, osteoclasts, oxidative stress
Citation: Jia X, Zhang G and Yu D (2024) Application of extracellular vesicles in diabetic osteoporosis. Front. Endocrinol. 15:1466775. doi: 10.3389/fendo.2024.1466775
Received: 18 July 2024; Accepted: 15 November 2024;
Published: 10 December 2024.
Edited by:
Åke Sjöholm, Gävle Hospital, SwedenReviewed by:
Zhen Geng, Shanghai University, ChinaSanjiv Neupane, Stony Brook University, United States
Copyright © 2024 Jia, Zhang and Yu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gongzi Zhang, emd6d29vZHZAMTYzLmNvbQ==; Deshui Yu, ZGVzaHVpeXVAMTI2LmNvbQ==