 Maria Paola Paronetto
Maria Paola Paronetto Clara Crescioli
Clara Crescioli- 1Department of Movement, Human and Health Sciences, University of Rome Foro Italico, Rome, Italy
- 2Laboratory of Molecular and Cellular Neurobiology, Fondazione Santa Lucia, IRCCS, Rome, Italy
The phosphodiesterases type 5 (PDE5) are catalytic enzymes converting the second messenger cyclic guanosine monophosphate (cGMP) to 5’ GMP. While intracellular cGMP reduction is associated with several detrimental effects, cGMP stabilization associates with numerous benefits. The PDE5 specific inhibitors, PDE5i, found their explosive fortune as first-line treatment for erectile dysfunction (ED), due to their powerful vasoactive properties. The favorable effect for ED emerged as side-effect when PDE5i were originally proposed for coronary artery disease (CAD). From that point on, the use of PDE5i captured the attention of researchers, clinicians, and companies. Indeed, PDE5-induced intracellular cGMP stabilization offers a range of therapeutic opportunities associated not only with vasoactive effects, but also with immune regulatory/anti-inflammatory actions. Chronic inflammation is acknowledged as the common link underlying most non-communicable diseases, including metabolic and cardiac diseases, autoimmune and neurodegenerative disorders, cancer. In this scenario, the clinical exploitation of PDE5i is undeniably beyond ED, representing a potential therapeutic tool in several human diseases. This review aims to overview the biological actions exerted by PDE5i, focusing on their ability as modulators of inflammation-related human diseases, with particular attention to inflammatory-related disorders, like cardiac diseases and cancer.
1 Introduction
The enzyme family of phosphodiesterases (PDEs) catalyzes the hydrolytic breakdown of cyclic adenosine monophosphate (cAMP) and cyclic guanosine monophosphate (cGMP) into the biologically inactive derivates 5′-AMP and 5′-GMP, respectively. In Eukaryotes, through their activity instrumental to regulate cAMP/cGMP intracellular levels, PDE5 can control the homeostasis of a variety of intracellular signaling and, therefore, multiple cellular functions.
Mammalian PDEs comprise 11 subfamilies, encoding distinct PDE isoforms through alternative pre-mRNA splicing, multiple promoter usage and alternative transcription start sites in human, rat, and mouse (1).
In human tissues, PDEs are almost ubiquitous and share the main catalytic function to hydrolyze the 3′ cyclic phosphate bond of cAMP, cGMP, or both, but with different substrate specificities and intracellular localization (2, 3).
PDE enzymes can be grouped into three broad categories based on their sequence homology, substrate specificity and selectivity: PDE4, PDE7 and PDE8 specific for cAMP hydrolysis; PDE5, PDE6 and PDE9 specific for cGMP hydrolysis; PDE1, PDE2, PDE3, PDE10 and PDE11 exhibiting dual specificity, for cAMP and cGMP, although with different affinities, depending on the isoform.
PDEs are structurally modular proteins sharing a conserved catalytic core of approximately 270 amino acids. The determinants responsible for subcellular localization and specific interactions of the isoforms are in the N-terminal portions; these regulatory regions can be post-translationally modified and modulate the enzymatic activity/localization in response to external and internal cues. In addition, the N-terminal domain can include dimerization domains and autoinhibitory modules, as in PDEs 1, 4, and 5.
PDEs function is not limited to cyclic nucleotide content regulation within cells, it rather allows the single cell to respond to and interact with intra- and extracellular signals, through the creation of subcellular compartmentalization (like individual pockets, nanodomains) of cyclic nucleotide signaling (1). Indeed, PDE location might change depending on several factors, i.e., tissue type, health/disease status or aging; this characteristic is of paramount importance when considering PDEs as potential therapeutic targets (1, 4–10). In this view, many companies exploited the ability of PDEs to control specific paths to develop selective drugs against specific PDE types and isoforms (11). The efficacy of the prototypical “first-in class” PDE inhibitors (PDEi) emerged in the early ‘70s, the first-generation of PDE inhibitors was produced between the 80s and 90s, followed by the development of highly selective isoenzyme-specific drugs (11). Indeed, since PDE dysfunction associates with several pathophysiological conditions affecting, i.e., cardiovascular system, fertility, nervous system, metabolism immunity and cancer, PDEs represent potential therapeutic targets in several human diseases (1).
Among the different inhibitors, PDE5i undeniably represent the first and most successfully studied molecules (1). Originally proposed for the treatment of CVD, these drugs found their explosive fortune for erectile dysfunction (ED) treatment, due to their vasoactive activity. Nowadays, PDE5i are licensed for ED and pulmonary artery hypertension (PAH). Importantly, PDE5i can efficaciously counteract inflammatory processes and can regulate cell growth/division/death (12). Based on these properties, the clinical exploitation of PDE5i might be undeniably beyond ED, with important implications in several human diseases. In fact, chronic inflammation, or meta-inflammation, is widely recognized as the common link in most non-communicable diseases (NCD).
This review aims to overview and highlight the biological actions exerted by PDE5i, focusing on their ability as modulators of inflammation-related human diseases, with particular attention dedicated to inflammatory-related disorders, cardiac diseases and cancer.
2 PDE5 and PDE5 inhibitors
PDE5 belongs to the metallo-hydrolase superfamily and controls different physiological function through cGMP to 5’ GMP catalytic conversion (13).
PDE5 dimeric structure includes the regulatory unit that expresses GAF-A and GAF-B domains, acting in concert to control the catalytic unit and PDE5 dimerization (14). The N-terminal GAF-A domain exhibits cGMP binding sites, which account for PDE5 affinity to cGMP (15). Many human tissues express PDE5, i.e., visceral and vascular smooth muscle, including corpora cavernosa, skeletal muscle, platelets, lung, brain, spinal cord, kidney, gastrointestinal tissue, prostate, bladder, urethra (14, 16–22).
The PDE5A gene, located on the human chromosome 4q26, contains 23 exons spanning approximately 100 kilobases and encoding three alternatively spliced coding variants (PDE5A1,2,3), which display different isoform-specific first exons, guided by specific promoters responsive to cGMP or cAMP stimulation (16, 23).
In mice, the splice variants of the PDE5A, mPde5a1, mPde5a2, and mPde5a3 show differential tissue distribution; the adult heart express different levels mPde5a1 and mPde5a2, while mPde5a3 seems undetectable (24).
In humans, PDE5A3 protein expression is restricted to smooth and cardiac muscle, while PDE5A1 and PDE5A2 are ubiquitously expressed (16).
The three PDE5A isoforms share the same cGMP-catalytic activities and differ only in the N-terminal region. All isoforms are similarly inhibited by sildenafil, a specific PDE5i, marketed as a treatment for ED in 1998. In fact, due to its selective inhibition of cGMP degradation in vascular smooth muscle cells in corpora cavernosa, sildenafil promotes vascular dilatation through nitric oxide (NO) increased availability (25). Thereafter, between 2003 and 2012, the other specific PDE5i tadalafil, vardenafil and avanafil were licensed for ED treatment (26). Currently, several PDE5i are FDA approved in and outside United Stated and marketed for ED (1).
Since cGMP is a second messenger involved in many physiological processes, beyond vasodilation, it is not surprising that cGMP signaling modulation might represent a useful tool to control other pathophysiological conditions.
Indeed, cGMP stabilization, beside benefits on endothelial, associates with several downstream favorable effects, as addressed later in this paper. Furthermore, the observation that cGMP accumulation can counteract inflammatory processes might be clinically relevant, considering that systemic chronic inflammation is the leading cause in several diseases, including neurological disorders, PAH, hypertension, cardiomyopathy, cancer, and lower urinary tract syndrome, in addition to ED (27–32) as summarized in Figure 1.
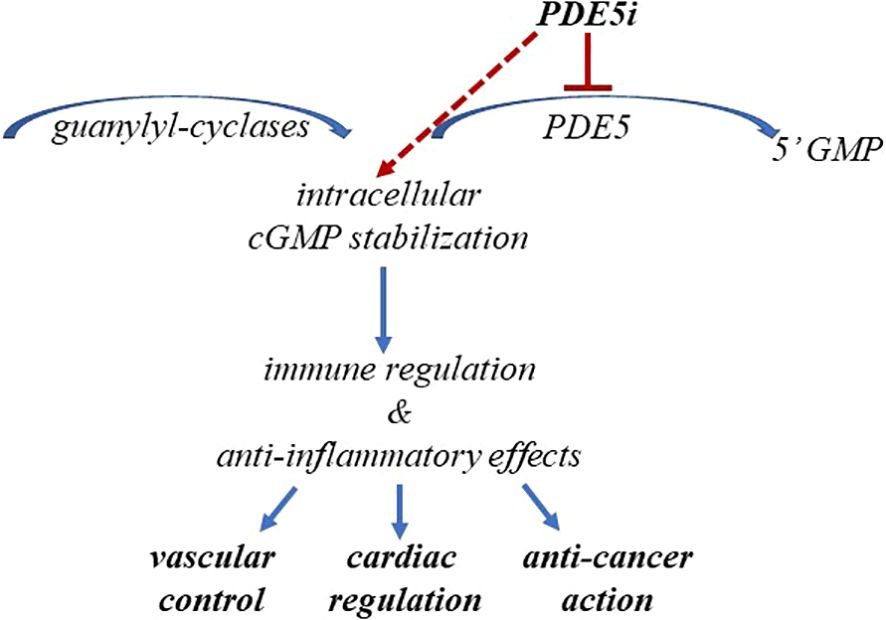
Figure 1. PDE5i-induced cGMP stabilization impacts human health. The figure schematically summarizes the benefits from cGMP stabilization in cardiovascular diseases and cancer.
3 The role of PDE5i in inflammation: a roundtrip from cardiac disease and erectile dysfunction
Since quite long ago, anti-inflammatory drugs have been recognized as useful tools to control and counteract the risk of cardiovascular diseases (33). The homeostasis between cardiovascular health and disease largely depends on the immune/inflammatory balance associated with the specific biomolecular mediators, such as cytokines and chemokines. Inflammation triggers the early phases of the atherosclerotic processes and increases the level of inflammatory mediators, that, in turn, associates with a higher risk of developing cardiovascular diseases. Several contributions sustain the proof of concept that inflammation and especially inflammatory cytokines represent a significant risk factor of cardiovascular event development in different conditions, including primary heart diseases, myocarditis secondary to infections, metabolic diseases or rheumatic diseases (34–36). The description of the role of inflammatory cascades involved in distinct conditions in heart diseases goes beyond the aim of this review; we remand therefore to exhaustive reviews focusing on this topic (37–41). In this context, the need to target inflammation emerges as a therapeutic strategy to manage cardiovascular diseases.
Even if PDE5i represent a milestone in ED treatment, this class of drugs was originally developed in the late ‘80s for the relief of angina pectoris (42, 43). Sildenafil was indicated for the treatment of coronary artery disease (CAD), in 1989. While the beneficial effects were quite disappointing for CAD, interesting vascular effects on male penile erection emerged (44).
Furthermore, the observation that PDE5 inhibition can also ameliorate pulmonary vascular resistance (45) led to a secondary approval of sildenafil and tadalafil for PAH treatment (1, 46).
Albeit the vascular targeting was the main indication for therapeutic purposes in ED and PAH, PDE5i-induced regulatory properties led to “rethinking” of their application for cardiac purposes. Thus, albeit initially the concerns on the use PDE5i for CAD treatment - due to the marginal beneficial effects - shifted their application from CVD to ED, thereafter, sildenafil was “reallocated” for the treatment of cardiac disorders, due to both endothelial and anti-inflammatory/pro-remodeling effects in cardiac cells (43).
The clinical relevance of PDE5i has been proven in ischemia-reperfusion injury, pressure overload-induced hypertrophy, heart failure, myocardial ischemia (47, 48). Undeniably, PDE5i-induced benefits on cardiac health can be explained by endothelial function improvements exerted by these drugs throughout the body. Vascular endothelial cells and vascular smooth muscle cells are the canonical targets of NO–cGMP signaling, which, when potentiated by PDE5i, results in cGMP stabilization, and, consequently, in vasodilatory, antioxidative and anti‐proliferative effects (49).
The vasodilatory properties of the NO–cGMP pathway can reduce peripheral vascular resistance and improve cardiac preload to ameliorate myocardial perfusion, resulting in clinically relevant advantages, like in ischemic heart disease. PDE5 expression is found in cardiac vessels, i.e., coronary vascular smooth muscle cells (50, 51). However, the cardiac benefits from PDEi-induced cGMP signaling potentiation are reported in cardiomyocytes as well (52–54).
While PDE5 is almost undetectable in heathy cardiomyocytes, localized at Z-disks, enzyme expression is significantly upregulated and diffusely distributed in pathologic conditions, such as congestive heart failure, right ventricular hypertrophy, overload hypertrophy, ischemic cardiomyopathy (55–58). As from previous studies, isolated human cardiomyocytes express PDE5 similarly to human endothelial cells and human peripheral blood mononuclear cells (59).
Thus far, PDE5 is likely upregulated during cardiac remodeling processes leading to diseases. Cardiac dysfunctions, albeit distinct, associate with “aberrant” cardiac remodeling, that comprehends many pathophysiological changes at cell level, including, i.e., cell death, energy metabolism, oxidative stress, inflammation, collagen and contractile protein content, calcium transport (60). Growing evidence shows that inflammation and oxidative stress play a pivotal role in these processes. The immune/inflammatory response represents per se a physiological adaptation aimed to restore homeostasis after short-term stress, but, under persistent stressful stimuli - i.e., due to metabolic morbidities, pressure/volume overload, injury, or overtraining – the consequent cellular and molecular modification result in cardiac loss function, at first asymptomatic, ending in heart failure (61–64). Of interest, cGMP/cGMP-dependent protein kinases G (PKG) signaling, which is enhanced by NO-soluble guanylate cyclase (GC) and natriuretic peptide NP-particulate GC (pGC), regulates cardiac remodeling and limits inflammation (42, 65). I.e., heart failure either with reduced ejection fraction or preserved ejection fraction (HFrEF and HFpEF, respectively) is characterized by impaired and dysfunctional cGMP-PKG signaling (65).
Following PDE5i, cGMP concentration rises and activates PKG, which, in turn, deactivates various cellular signaling cascades engaged in aberrant remodeling and hypertrophy development, through suppression of effects mediated by calcineurin, protein kinase C (PKC), Ras homolog gene family member A (RhoA) and Rho-associated kinase (ROCK), and extracellular signal regulated kinase (ERK)-mitogen-activated protein kinase (MAPK) (65).
Remarkably, sildenafil either combined or not with natriuretic peptide (NP), that stimulates per se cGMP production, phosphorylates sarcomeric proteins, such as titin, troponin-I, cardiac myosin-binding protein C, reducing myocyte/myofiber stiffness and improving systolic and diastolic function (66–69). Hence, PDE5 inhibition emerges to play a pivotal role against cellular processes underlying disease initiation and development, by counteracting biomolecular cascade and mediators engaged in shift from health to disease. This effect, in part, relies on the reduction in T helper (Th)1-type mediators dependent on cGMP stabilization (70), as addressed in the following paragraph.
3.1 cGMP stabilization, immune/inflammation and impact on health
Since cGMP stabilization inhibits inflammatory processes, it represents a potential tool to limit initiation/progression of several diseases driven by inflammation, including metabolic and cardiovascular diseases, autoimmune and neurodegenerative disorders, and cancer (27–30). Indeed, in various cell types, cGMP accumulation dependent on PDE5 inhibition associates with several therapeutic benefits, as previously addressed (13). Thus, the use of PDE5i has been pharmacologically exploited beyond ED and PHA.
We briefly recall that cGMP synthesis occurs upon NO stimulation and, through GC, activates downstream cGMP-PKG and ion channels with a variety of effects (71).
The reduction of intracellular cGMP is well known to be involved in detrimental events in impaired vascular responses, vascular smooth muscle proliferation, in enhanced platelet aggregation and in immune response (72–75). In immunity, cGMP signaling pathway acts through the modulation of nuclear factor (NF)-κB and plays a role in critical processes, i.e., stress, inflammation, immune cell differentiation and regulation tolerance (76, 77). The experimental increase of cellular cGMP (through a NO donor) results in the reduction of inflammatory cytokines, like TNFα or IL-6, which, conversely, are increased when cGMP is reduced (by a soluble GC inhibitor) (78). Albeit some roles and mechanisms are still to be fully elucidated, it is documented that the reduction in cGMP formation suppresses cGMP-induced anti-inflammatory effects in experimental conditions mimicking inflammation (79).
Furthermore, sildenafil-induced cGMP increase can impact the regulation of T cells and the production of proinflammatory cytokines (80). Animal studies report that sildenafil decreases serum IL-6 and impacts T cell subpopulations - i.e., CD4+/CD8+ T cell ratio, T regulatory (Treg), natural killer (NK) T cells, with some sex-dependent effects (81). The anti-inflammatory effect was confirmed by murine studies on neuroinflammation investigating TNF-α, IFN-γ, IL-2, and IL-1β in mice after sildenafil (82).
As from studies in human males and females, sildenafil influences the expression of Treg transcription factor Foxp3, involved in T effector cell deactivation, attenuates NK activity and TNFα level (83, 84), Albeit some controversies on reported data, there is evidence on sildenafil-induced reduction of local inflammatory mediators and receptors, either local or circulating, such as IL-1β, TNFα, monocyte chemoattractant protein 1(MCP-1) and its receptor CCR2, documented in different animal model of diseases (82).
It is worthwhile to mention that overproduction of reactive oxygen species (ROS), characterized by an imbalance in the synthesis and removal of ROS, commonly detected in cardiovascular disease, can be accompanied by reduced endothelium-derived NO responses (85), and impairment of soluble GC function, which might contribute to the increased tone seen in pathophysiological conditions associated with chronic overproduction of ROS. A direct link between enhanced ROS and decreased sGC expression was established by Gerassimou and colleagues (86), showing that exposure of smooth muscle cells to exogenously applied ROS-generating agents caused a decrease in α1 and β1 sGC subunit levels, thus attenuating cGMP formation. Since cGMP is engaged in the regulation of smooth muscle cell proliferation as well as vessel tone, the inhibition of sGC expression by ROS in pathophysiological states would be expected to modulate vascular remodeling and tone. Reduction in the availability and function of NO contributes to vasoconstriction, inflammation and vascular remodeling. Remarkably, whilst playing a key role in the pathogenesis of cardiovascular disease, ROS dysregulation can also limit the clinical efficacy of current therapies (87).
3.2 PDE5i and inflammatory mediators: an example from CXC chemokines for cardiac purposes
The effect of PDE5i onto immune status largely accounts for the indication “back” for cardiac purposes, as previously stated in this review.
Indeed, the role of fine-tuned immune/inflammatory processes, especially driven by Th1 type dominance, is undeniably recognized among the mechanism associated with or preceding cardiac disorders. We have reported on the importance of early changes in circulating and local concentrations of some biomediators, such as CXC-motif chemokines, early involved in the shift towards cardiac diseases, before clinical sign manifestations. Hence, it is of pivotal importance to control as soon as possible their biological effects to control/limit disease initiation.
This is the case of the chemokines CXCL10 and CXCL8, classified as ERL- and ERL+, based on absence or presence of Glut-Leu-Arg motif (88), both molecules with multifaceted functions in disease development, widely engaged in cardiac disease initiation and progression (89).
Interestingly, CXCL10 and CXCL8 significantly increased in sera of diabetic subjects at the onset of cardiomyopathy and are targeted by sildenafil (59, 90). Besides the decrease in systemic level of these CXC molecules, sildenafil can inhibit their release from human endothelial cells and cardiomyocytes, suggesting an anti-inflammatory action at local level (90). This effect is, indeed, quite remarkable since the local cellular components are dynamic units able to interact with and potentiate the signaling inflammatory network, when challenged by Th 1-driven microenvironment, i.e., due to sugar or lipid excess (91, 92). Cardiomyocytes or endothelial cells can contribute to the immune/inflammatory dialogue between local and systemic counterparts producing, among other proinflammatory mediators, CXCL10 and CXCL8, both virtually absent in non-inflammatory conditions; this effect recalls and amplifies local infiltration by immunocytes, triggering an aberrant cell remodeling and establishing a vicious loop (91). Interestingly, sildenafil-induced inhibition of IL-8 from human endothelial cells associates with cGMP increased concentration (90). Higher CXCL10 and CXCL8 levels detected in patients with different cardiac diseases vs. healthy subjects likely reflect early inflammatory processes, differently from classical biomarkers, which reveal already established damages (91). Interestingly, three-month treatment with sildenafil (100 mg/day) decreased CXCL10 and CXCL8 in subjects with diabetes at onset of cardiomyopathy, ameliorated metabolic indices like post-prandial glycemia, hemoglobin A1c, lipidemic profile, whereas failed to modify cardiac standard markers, such as mass and volume index, ejection fraction, blood pressure (93, 94). The latter result is likely due to the well-preserved left ventricular function of the subjects enrolled at the onset of cardiomyopathy, with no sign ischemia; at the same time, this observation suggests the importance of PDE5 inhibition at the onset of cardiomyopathy, to decline early inflammatory biomediators. Decreasing chemokines like CXCL10, known trigger the first auto- or alloimmune response, and CXCL8, present since early stages of inflammation and remaining active for long time (95), before clinical manifestations - i.e., at stages A or B, according to American Heart Association/American College of Cardiology (AHA/ACC) classification (https://www.heart.org/en/health-topics/heart-failure/what-is-heart-failure/classes-of-heart-failure) - might be of clinically relevant impact, as it could attenuate downstream inflammatory cascade and progression to HF. In line with this observation, higher level of inflammatory cytokines associates with increased risk of adverse cardiovascular outcome (96, 97), pretransplant higher CXCL10 correlates with higher risk of heart rejection and worse outcome (98), and trials designed to target inflammatory cytokines to improve outcome are reported (99) there are ongoing trials. Recently, integrated multi-omics data show decrease in CXCL10 is a promising drug target, since it accounts for about 70% of the effects mediated by pharmacological IL-6R inhibition in a clinical trial (100).
Indeed, albeit HF pathophysiology exhibits remarkable heterogeneity, anti-cytokine therapy emerges as one of the most promising approaches. Quite intriguingly, the hypothesis of a “proinflammatory cytokine storm” proposed by Seta et al. in 1996 has been strengthened as research on cardiac diseases has progressed, highlighting the importance of inflammation as one of the main therapeutic targets (101).
A similar process involving a chemokine storm is likely to trigger a significant overresponse in autoimmune diseases, such as Systemic Lupus Erythematosus (SLE) (102, 103). Remarkably, the chemokine CXCL10 seems to act as a trigger/enhancer of the immune storm. This effect is not surprising, since CXCL10 is engaged in early processes both in allo- and auto-immune response, as previously addressed (104).
In Systemic Sclerosis (SSc), an autoimmune disorder characterized by important vascular impairments and multiorgan severe failure (105), CXCL10 and CXCL11 can discriminate those subjects developing SSc from an initial “lighter” condition of very early diagnosis of SSc (VEDOSS) (106). Of note, PDE5i, which are recommended as first-line agents for the treatment of secondary PAH, and for the management of refractory Raynaud’s phenomenon (46, 107, 108), could result useful to maintain cardiac and skeletal muscle function, as previously reported (109).
4 Role of PDE5 in cancer
Cyclic nucleotides act as second messengers promoting protein kinase activation, which in turn can promote apoptotic signaling. The cGMP and cAMP signaling pathway is aberrantly regulated in several human cancers, displaying different characteristics. Both cAMP and cGMP were shown to display antiproliferative and pro-apoptotic properties (110). In line with these observations, altered expression of PDE isozymes was reported in different cancer specimens and in hematological malignancies (111, 112).
In this paragraph, we will provide a detailed overview of clinical data on PDE5 expression and function in the pathogenesis of human cancer. In addition, we will discuss whether PDE5 inhibitors could be exploited for therapeutic purpose in cancer, evaluating ongoing clinical trials, summarized in Table 1.
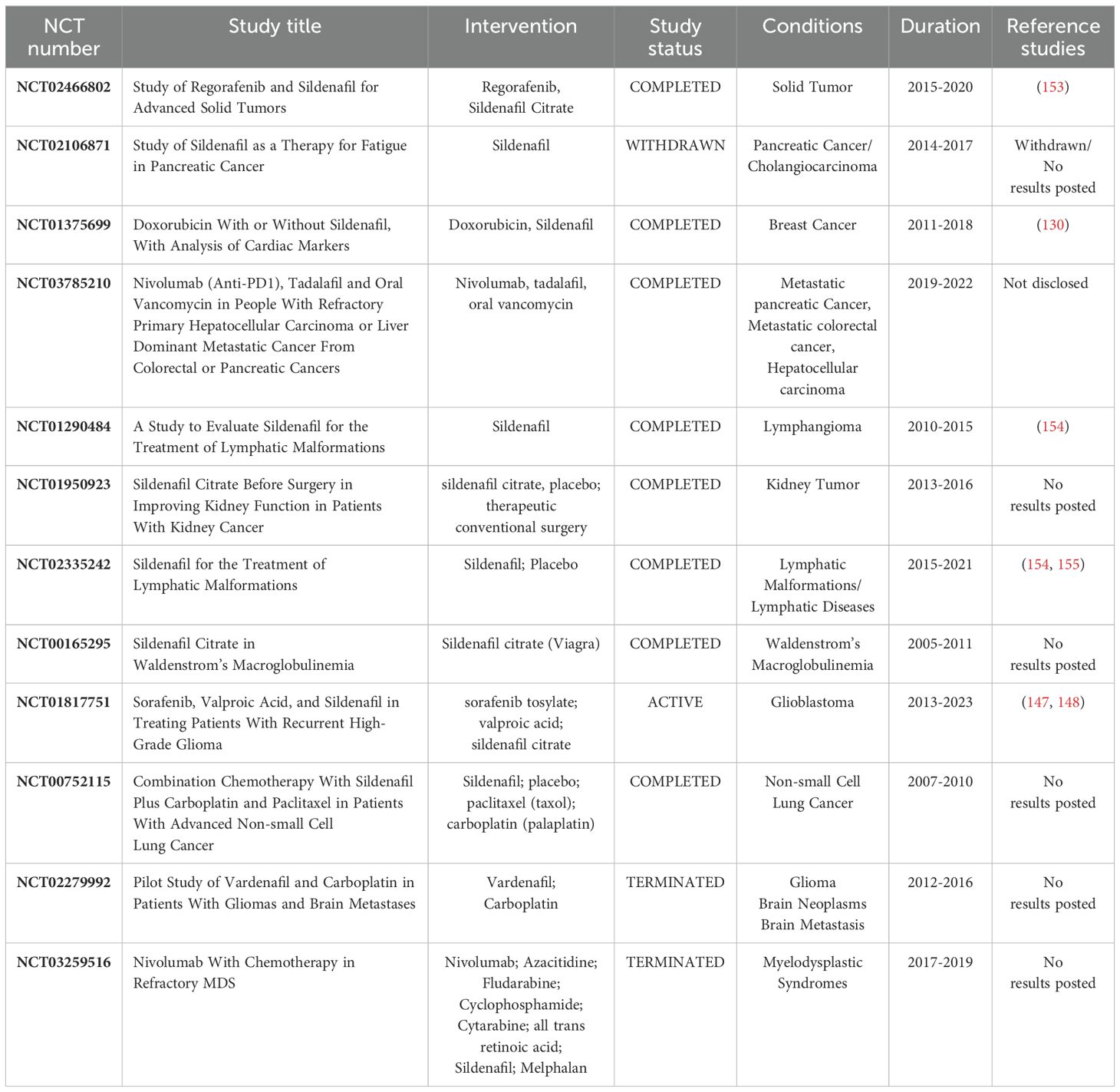
Table 1. Clinical trials for human cancer treatment using PDE5i.
4.1 Prostate cancer
Several prostate cancer (PCa) cell lines display increased level of PDE5 expression (113), and PDE5 inhibitors can sensitize cancer cells to chemotherapeutic agents, either by reducing extrusion of the chemotherapeutics agents by ABCB1, ABCC1 and ABCG2 transporters (114) or by promoting apoptosis (115). The pathway relies on the upregulation of caspase 3 and 9, downregulation of Bcl-XL and production of reactive oxygen species (ROS) (116). In xenograft mouse models of PCa, sildenafil significantly inhibited tumor growth by inducing apoptosis and increasing the expression of activated caspase 3, while protecting mice against reperfusion injury/ischemia, which is a side effect induced by doxorubicin treatment (116). In addition, in PCa cells, hypoxic conditions can inhibit the NO/cGMP pathway, leading to chemoresistance. Thus, treatment with PDE5 inhibitors sensitizes hypoxic cells to chemotherapeutic agents by restoring the NO/cGMP pathway (117). PDE5 inhibitors can also counteract PCa progression by increasing apoptosis, as shown in combined therapy with cisplatin or doxorubicin (118). However, VCaP cells, which display the TMPRSS2-ERG fusion, display increased risk of PCa progression after treatment with PDE5 inhibitors (119), demonstrating that PCa cells display different properties and behavior in response to PDE5 inhibition.
PCa can evolve into a castration resistance and more aggressive stage, in which androgen deprivation therapy is not efficacious anymore (castration-resistant prostate cancer -CRPC). During this stage, current treatment is represented by vincristine, which is an anti-mitotic agent, which induces cell cycle arrest and caspase-mediated cell death, but as side-effect can favor the occurrence of neuropathies. Combination of vincristine with sildenafil can reduce deleterious side effects of vincristine, by lowering effective concentration of the chemotherapeutic agent (120). Moreover, Sildenafil was shown amplify vincristine effect both in vitro (PC-3 and DU-145 cell lines) and in vivo (120).
Sildenafil has been extensively used in clinical trials for PCa patients to recover erectile dysfunction following hormone therapy, radiation therapy or prostatectomy. Thus, these trials have not been addressed in the present manuscript.
4.2 Breast cancer
Increased PDE5 expression was shown in clinical samples of breast cancer patients, at both RNA and protein level, and was correlated with tumor grade, stage of the disease and lymph node involvement (121, 122). PDE5 was also found differentially expressed in breast cancer subtypes, with higher expression levels in the more aggressive HER2-enriched and triple-negative subtypes and lower levels in the estrogen receptor (ER)-positive Luminal B- and the Luminal A subtypes (123). Remarkably, high PDE5 expression was correlated with worse prognosis in a cohort of 1,988 patients (123).
PDE5 inhibition with sulindac sulfide (SS) was shown to promote cGMP accumulation, activation of PKG which, in turn, caused inhibition of cell growth and apoptosis in breast cancer cell lines (124), representing a viable strategy for breast cancer treatment. Similarly, sildenafil and tadalafil treatment of breast cancer cell lines diminished cell growth and induced apoptosis, at least in part by modulating β-catenin expression transcription (125). Sildenafil treatment was also shown to affect HSP90 expression, thus promoting PKD2 degradation and impairing cancer cell proliferation and viability (126).
PDE5 activity supports stromal fibroblasts, which produce and secrete chemokines like CXCL16, promoting cancer progression. Sildenafil and vardenafil treatment of breast cancer associated fibroblasts reduced CXCL16 chemokine expression, thus inhibiting cancer progression and increasing the efficiency of both chemotherapy and radiotherapy (127). Importantly, sildenafil can act as an aromatase inhibitor, impairing the conversion of androgens into estrogens, thereby inhibiting breast cancer growth (128). By improving blood supply to the tumor vasculature, PDE5 inhibitors can also ameliorate antineoplastic drug administration in the tumor region, thus reducing tumor size in mouse xenograft models (129).
Despite these relevant observations, only one study clinically evaluated the safety and activity of PDE5 inhibitors in breast cancer. Particularly, it was shown that administration of exisulind in combination with capecitabine was well tolerated in a small number of patients with metastatic breast cancer (n = 35), but the synergism between these two drugs at the doses tested was modest. The clinical trial NCT01375699 evaluated the use of sildenafil as a cardioprotective factor after doxorubicin chemotherapy, in breast cancer patients. Although the results are still being processed, this study demonstrated that adding sildenafil to chemotherapy was safe (130) 5these reported findings suggest that PDE5 inhibitors could be used as adjuvants in breast cancer treatment, decreasing cancer aggressiveness and avoiding cardiovascular side effects caused by conventional chemotherapies.
4.3 Colorectal cancer
Inflammation can represent a driver of oncogenesis; in fact, patients with inflammatory bowel disease (IBD) display increased risk to develop colorectal cancer (CRC), thus providing rationale for using anti-inflammatory drugs in CRC prevention. Notably, intestinal cGMP signaling regulates epithelial homeostasis and was implicated in the suppression of colitis and colon cancer. In this context, inhibition of PDE5 could be instrumental to counteract CRC. Treatment of mice with sildenafil was sufficient to activate cGMP signaling in the colon mucosa and to protect against azoxymethane/dextran sulfate sodium (AOM/DSS) inflammation-driven colorectal cancer. Particularly, oral administration of sildenafil in mice after treatment with AOM/DSS, reduced polyp formation and inflammation, also counteracting myeloid-cell infiltration (131). These results highlight the potential therapeutic value of PDE5 inhibitors for the prevention of colitis-driven colon cancer. In line with these results, clinical studies confirmed the therapeutic value of PDE inhibitors, such as exisulind, in the prevention of colorectal polyp formation in patients with familial adenomatous polyposis (FAP) (132, 133).
Furthermore, PDE5 inhibitors were shown to inhibit the oncogenic activity of β-catenin either by repressing its transcription or promoting its proteosomal degradation in several colon cancer cells (134–136). Treatment of human CRC cells with sildenafil resulted in the inhibition of cell proliferation, cell cycle arrest and apoptosis; moreover, it showed reduction of tumor growth in CRC xenograft models in nude mice (137).
Combined therapy of tadalafil with nivolumab and vancomycin was evaluated in the clinical trial NCT03785210, demonstrating decreased liver metastasis from primary colorectal and pancreatic cancer (Table 1). This result suggests that sildenafil could be used in combination with other drugs in the treatment of CRC.
4.4 Lung cancer
Several lung cancer cell lines were shown to overexpress PDE enzymes, resulting in the decrease of cGMP. Hence, treatment with PDE inhibitors should result in increased intracellular cGMP and inhibition of tumor growth (138). This hypothesis was tested in several studies exploring the potential of combined therapies of PDEi with classical cytotoxic agents.
Combined use of platinum-based agents with PDEi displayed higher antiproliferative effect on lung cancer cells in comparison with the platinum monotherapy, which is currently in use as the standard care (139). Similar effects were also observed by combining sildenafil with radiotherapy in the treatment of Lewis lung carcinoma (LLC), where inhibition of PDE activity was instrumental to abolish the irradiation-derived immunosuppression, improving the T cells response. These results suggest that sildenafil could be exploited to delay tumor recurrence after radiotherapy (140).
The clinical trial NCT00752115 is currently evaluating the combined treatment of sildenafil and carboplatin in patients with advanced non-small-cell lung cancer (Table 1).
4.5 Brain cancer
PDE5 is highly expressed in several brain tumor cell lines, including glioma and glioblastoma. Moreover, it was found upregulated during murine neuroblastoma cell differentiation (141).
Remarkably, modulation of cGMP signaling via PDEi was shown to improve the permeability across the brain blood barrier, thus improving delivery of therapeutic agents. Oral administration of sildenafil and vardenafil was shown to enhance tumor capillary permeability in rat models of gliosarcoma, thus improving the efficacy of the chemotherapeutic treatment (142). Moreover, combined administration of adriamycin and vardenafil in rat model of brain tumors showed longer overall survival than adriamycin alone (142). Similarly, combined therapy of sildenafil and OSU-03012 improved the efficacy of the therapy in glioblastoma cells by decreasing the expression of plasma membrane receptors and drug efflux pumps (143). In hard-to-treat brain metastases from different primary tumors, PDE5 inhibitors effectively modulated the brain blood barrier permeability, enhancing delivery and therapeutic efficacy of monoclonal antibodies in mouse models (144, 145).
However, in glioblastoma multiforme, which is the most aggressive and lethal brain tumor, high levels of the PDE5 protein were associated with decreased aggressiveness, invasive potential, and resistance to radiation (146).
The phase II clinical trial NCT01817751 is evaluating the activity of sildenafil and other drugs (sorafenib and valproic acid) in patients with recurrent high-grade glioma (147, 148), whereas the clinical trial NCT02279992 is evaluating the use of vardenafil to increase systemic concentration of carboplatin in patients with recurrent malignant gliomas or metastatic brain cancer. The NCT01817751 study confirmed the safety and survival benefit of the 3-drug combination sildenafil-sorafenib-valproic acid for the treatment of high-grade glioma (148). The combination of sorafenib and valproic acid was predicated on the basis that sorafenib activity is enhanced by HDAC inhibition (149, 150), whereas the addition of sildenafil was based on its ability to increase peroxynitrate levels and to block ABCB1 and ABCG2 drug-efflux pumps, thus increasing drug delivery to the tumor (114, 151).
PDE5i are being evaluated also in other tumor subtypes, not discussed in this manuscript. These trials were summarized in Table 1.
5 Concluding remarks
So far, even though the fortune of PDE5i started and continues as drugs for ED treatment, it is undeniable that their potential administration in inflammatory-related diseases amplifies their range of possible applications. Particularly, PDE5i, as anti-inflammatory drugs, potentially have the power to revert destabilized cell homeostasis, that is the turning-point where several diseases start from considering the many favorable effects deriving from PDE5i-dependent cGMP stabilization, the clinical exploitation of PDE5i can go undeniably beyond ED, as we addressed here, particularly for cardiac diseases and cancer. Efforts from the scientific community are to recognize and close the gaps present in knowledge, to address, expand and diversify the potential clinical use of PDE5i.
Indeed, albeit there are promising data on PDE5i administration to reduce adverse cardiac outcome, particularly in comorbidity with diabetes, consistent translation to other cardiac diseases, such as infarction, CHF, arrhythmia, is still lacking. Some reasons for the limitation in PDE5i translatability from bench to bedside may include, i.e., drug dosages or the use of animal models, not always immediately translatable to humans. Despite previous disappointments in clinical translation, targeting inflammation is the most promising and attractive direction in cardiovascular therapeutics. Dissection of the paths and mechanisms underlying aberrant responses as well as a better profiling of patient subpopulations will improve the benefit from PDE5i-targeted inflammation.
Moreover, albeit large-scale studies and trials support the anti-cancer potential of PDE5i, the association between PDE5 inhibition and tumor microenvironment, universally acknowledged to be critical in driving tumor growth/invasion/metastasis, and response to therapies, is still lacking. Furthermore, since PDE5i are not free of side effects, i.e., headaches, flushing, dyspepsia, altered vision and optic neuropathy, back pain and myalgia, priapism, melanoma, blood pressure, rhinitis, prostate cancer, hearing loss (152). Thus, well-performed studies on selected populations are needed to identify the populations with a “safe” profile to undergo PDE5i treatment. In this scenario, further studies aimed to validate PDE5i clinical applications are mandatory to expand our understanding, in a view of precision and interdisciplinary approach.
Author contributions
MP: Conceptualization, Writing – original draft, Writing – review & editing. CC: Conceptualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Baillie GS, Tejeda GS, Kelly MP. Therapeutic targeting of 3’,5’-cyclic nucleotide phosphodiesterases: inhibition and beyond. Nat Rev Drug Discovery. (2019) 18:770–96. doi: 10.1038/s41573-019-0033-4
2. Levy I, Horvath A, Azevedo M, de Alexandre RB, Stratakis CA. Phosphodiesterase function and endocrine cells: links to human disease and roles in tumor development and treatment. Curr Opin Pharmacol. (2011) 11:689–97. doi: 10.1016/j.coph.2011.10.003
3. Delhaye S, Bardoni B. Role of phosphodiesterases in the pathophysiology of neurodevelopmental disorders. Mol Psychiatry. (2021) 26:4570–82. doi: 10.1038/s41380-020-00997-9
4. Perera RK, Sprenger JU, Steinbrecher JH, Hübscher D, Lehnart SE, Abesser M, et al. Microdomain switch of cGMP-regulated phosphodiesterases leads to ANP-induced augmentation of β-adrenoceptor-stimulated contractility in early cardiac hypertrophy. Circ Res. (2015) 116:1304–11. doi: 10.1161/CIRCRESAHA.116.306082
5. Penmatsa H, Zhang W, Yarlagadda S, Li C, Conoley VG, Yue J, et al. Compartmentalized cyclic adenosine 3’,5’-monophosphate at the plasma membrane clusters PDE3A and cystic fibrosis transmembrane conductance regulator into microdomains. Mol Biol Cell. (2010) 21:1097–110. doi: 10.1091/mbc.e09-08-0655
6. Ahmad F, Lindh R, Tang Y, Ruishalme I, Ost A, Sahachartsiri B, et al. Differential regulation of adipocyte PDE3B in distinct membrane compartments by insulin and the beta3-adrenergic receptor agonist CL316243: effects of caveolin-1 knockdown on formation/maintenance of macromolecular signalling complexes. Biochem J. (2009) 424:399–410. doi: 10.1042/BJ20090842
7. Bonkale WL, Winblad B, Ravid R, Cowburn RF. Reduced nitric oxide responsive soluble guanylyl cyclase activity in the superior temporal cortex of patients with Alzheimer’s disease. Neurosci Lett. (1995) 187:5–8. doi: 10.1016/0304-3940(95)11323-O
8. Yarla NS, Gali H, Pathuri G, Smriti S, Farooqui M, Panneerselvam J, et al. Targeting the paracrine hormone-dependent guanylate cyclase/cGMP/phosphodiesterases signaling pathway for colorectal cancer prevention. Semin Cancer Biol. (2019) 56:168–74. doi: 10.1016/j.semcancer.2018.08.011
9. Li N, Chen X, Zhu B, Ramírez-Alcántara V, Canzoneri JC, Lee K, et al. Suppression of β-catenin/TCF transcriptional activity and colon tumor cell growth by dual inhibition of PDE5 and 10. Oncotarget. (2015) 6:27403–15. doi: 10.18632/oncotarget.v6i29
10. Lee DI, Zhu G, Sasaki T, Cho GS, Hamdani N, Holewinski R, et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature. (2015) 519:472–6. doi: 10.1038/nature14332
11. Schudt C, Hatzelmann A, Beume R, Tenor H. Phosphodiesterase inhibitors: history of pharmacology. Handb Exp Pharmacol. (2011) 204:1–46. doi: 10.1007/978-3-642-17969-3_1
12. Saikia Q. (2022). A Review on the Pharmacological Importance of PDE5 and Its Inhibition to Manage Biomedical Conditions. Journal of Pharmacology and Pharmacotherapeutics, 13:246–57. doi: 10.1177/0976500X221129008
13. Nandi S, Kumar P, Amin SA, Jha T, Gayen S. First molecular modelling report on tri-substituted pyrazolines as phosphodiesterase 5 (PDE5) inhibitors through classical and machine learning based multi-QSAR analysis. SAR QSAR Environ Res. (2021) 32:917–39. doi: 10.1080/1062936X.2021.1989721
14. Ahmed WS, Geethakumari AM, Biswas KH. Phosphodiesterase 5 (PDE5): Structure-function regulation and therapeutic applications of inhibitors. BioMed Pharmacother. (2021) 134:111128. doi: 10.1016/j.biopha.2020.111128
15. Cruz-Burgos M, Losada-Garcia A, Cruz-Hernández CD, Cortés-Ramírez SA, Camacho-Arroyo I, Gonzalez-Covarrubias V, et al. New approaches in oncology for repositioning drugs: the case of PDE5 inhibitor sildenafil. Front Oncol. (2021) 11:627229. doi: 10.3389/fonc.2021.627229
16. Lin CS, Lin G, Xin ZC, Lue TF. Expression, distribution and regulation of phosphodiesterase 5. Curr Pharm Des. (2006) 12:3439–57. doi: 10.2174/138161206778343064
17. Dolci S, Belmonte A, Santone R, Giorgi M, Pellegrini M, Carosa E, et al. Subcellular localization and regulation of type-1C and type-5 phosphodiesterases. Biochem Biophys Res Commun. (2006) 341:837–46. doi: 10.1016/j.bbrc.2006.01.035
18. Carosa E, Castri A, Forcella C, Sebastiani G, Di Sante S, Gravina GL, et al. Platelet-derived growth factor regulation of type-5 phosphodiesterase in human and rat penile smooth muscle cells. J Sex Med. (2014) 11:1675–84. doi: 10.1111/jsm.12568
19. Menniti FS, Faraci WS, Schmidt CJ. Phosphodiesterases in the CNS: targets for drug development. Nat Rev Drug Discovery. (2006) 5:660–70. doi: 10.1038/nrd2058
20. Giuliano F, Ückert S, Maggi M, Birder L, Kissel J, Viktrup L. The mechanism of action of phosphodiesterase type 5 inhibitors in the treatment of lower urinary tract symptoms related to benign prostatic hyperplasia. Eur Urol. (2013) 63:506–16. doi: 10.1016/j.eururo.2012.09.006
21. Gresele P, Momi S, Falcinelli E. Anti-platelet therapy: phosphodiesterase inhibitors. Br J Clin Pharmacol. (2011) 72:634–46. doi: 10.1111/j.1365-2125.2011.04034.x
22. Foresta C, Caretta N, Zuccarello D, Poletti A, Biagioli A, Caretti L, et al. Expression of the PDE5 enzyme on human retinal tissue: new aspects of PDE5 inhibitors ocular side effects. Eye (Lond). (2008) 22:144–9. doi: 10.1038/sj.eye.6702908
23. Yanaka N, Kotera J, Ohtsuka A, Akatsuka H, Imai Y, Michibata H, et al. Expression, structure and chromosomal localization of the human cGMP-binding cGMP-specific phosphodiesterase PDE5A gene. Eur J Biochem. (1998) 255:391–9. doi: 10.1046/j.1432-1327.1998.2550391.x
24. Campolo F, Zevini A, Cardarelli S, Monaco L, Barbagallo F, Pellegrini M, et al. Identification of murine phosphodiesterase 5A isoforms and their functional characterization in HL-1 cardiac cell line. J Cell Physiol. (2018) 233:325–37. doi: 10.1002/jcp.25880
25. Andersson KE. PDE5 inhibitors - pharmacology and clinical applications 20 years after sildenafil discovery. Br J Pharmacol. (2018) 175:2554–65. doi: 10.1111/bph.14205
26. Goldstein I, Jones LA, Belkoff LH, Karlin GS, Bowden CH, Peterson CA, et al. Avanafil for the treatment of erectile dysfunction: a multicenter, randomized, double-blind study in men with diabetes mellitus. Mayo Clin Proc. (2012) 87:843–52. doi: 10.1016/j.mayocp.2012.06.016
27. Peixoto CA, Gomes FO. The role of phosphodiesterase-5 inhibitors in prostatic inflammation: a review. J Inflammation (Lond). (2015) 12:54. doi: 10.1186/s12950-015-0099-7
28. Benjamins JA, Nedelkoska L. Cyclic GMP-dependent pathways protect differentiated oligodendrocytes from multiple types of injury. Neurochem Res. (2007) 32:321–9. doi: 10.1007/s11064-006-9187-7
29. Zhao S, Zhang L, Lian G, Wang X, Zhang H, Yao X, et al. Sildenafil attenuates LPS-induced pro-inflammatory responses through down-regulation of intracellular ROS-related MAPK/NF-κB signaling pathways in N9 microglia. Int Immunopharmacol. (2011) 11:468–74. doi: 10.1016/j.intimp.2010.12.017
30. Khoshakhlagh P, Bahrololoumi-Shapourabadi M, Mohammadirad A, Ashtaral-Nakhai L, Minaie B, Abdollahi M. Beneficial effect of phosphodiesterase-5 inhibitor in experimental inflammatory bowel disease; molecular evidence for involvement of oxidative stress. Toxicol Mech Methods. (2007) 17:281–8. doi: 10.1080/15376510601003769
31. Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. (2019) 25:1822–32. doi: 10.1038/s41591-019-0675-0
32. Schlossmann J, Schinner E. cGMP becomes a drug target. Naunyn Schmiedebergs Arch Pharmacol. (2012) 385:243–52. doi: 10.1007/s00210-012-0730-6
33. Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. (1997) 336:973–9. doi: 10.1056/NEJM199704033361401
34. Sorriento D, Iaccarino G. Inflammation and cardiovascular diseases: the most recent findings. Int J Mol Sci. (2019) 20(16):3879. doi: 10.3390/ijms20163879
35. Henein MY, Vancheri S, Longo G, Vancheri F. The role of inflammation in cardiovascular disease. Int J Mol Sci. (2022) 23(21):12906. doi: 10.3390/ijms232112906
36. Alfaddagh A, Martin SS, Leucker TM, Michos ED, Blaha MJ, Lowenstein CJ, et al. Inflammation and cardiovascular disease: From mechanisms to therapeutics. Am J Prev Cardiol. (2020) 4:100130. doi: 10.1016/j.ajpc.2020.100130
37. Tschöpe C, Ammirati E, Bozkurt B, Caforio ALP, Cooper LT, Felix SB, et al. Myocarditis and inflammatory cardiomyopathy: current evidence and future directions. Nat Rev Cardiol. (2021) 18:169–93. doi: 10.1038/s41569-020-00435-x
38. Trachtenberg BH, Hare JM. Inflammatory cardiomyopathic syndromes. Circ Res. (2017) 121:803–18. doi: 10.1161/CIRCRESAHA.117.310221
39. Weber BN, Giles JT, Liao KP. Shared inflammatory pathways of rheumatoid arthritis and atherosclerotic cardiovascular disease. Nat Rev Rheumatol. (2023) 19:417–28. doi: 10.1038/s41584-023-00969-7
40. Adamo L, Rocha-Resende C, Prabhu SD, Mann DL. Reappraising the role of inflammation in heart failure. Nat Rev Cardiol. (2020) 17:269–85. doi: 10.1038/s41569-019-0315-x
41. Houston BA, Brittain EL, Tedford RJ. Right ventricular failure. N Engl J Med. (2023) 388:1111–25. doi: 10.1056/NEJMra2207410
42. Ghofrani HA, Osterloh IH, Grimminger F. Sildenafil: from angina to erectile dysfunction to pulmonary hypertension and beyond. Nat Rev Drug Discovery. (2006) 5:689–702. doi: 10.1038/nrd2030
43. Kass DA, Champion HC, Beavo JA. Phosphodiesterase type 5: expanding roles in cardiovascular regulation. Circ Res. (2007) 101:1084–95. doi: 10.1161/CIRCRESAHA.107.162511
44. Chrysant SG, Chrysant GS. The pleiotropic effects of phosphodiesterase 5 inhibitors on function and safety in patients with cardiovascular disease and hypertension. J Clin Hypertens (Greenwich). (2012) 14:644–9. doi: 10.1111/j.1751-7176.2012.00669.x
45. Soulaidopoulos S, Terentes-Printzios D, Ioakeimidis N, Tsioufis KP, Vlachopoulos C. Long-term effects of phosphodiesterase-5 inhibitors on cardiovascular outcomes and death: A systematic review and meta-analysis. Eur Heart J Cardiovasc Pharmacother. (2024) 10(5):403–12. doi: 10.1093/ehjcvp/pvae029
46. Galiè N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med. (2005) 353:2148–57. doi: 10.1056/NEJMoa050010
47. Degen CV, Bishu K, Zakeri R, Ogut O, Redfield MM, Brozovich FV. The emperor’s new clothes: PDE5 and the heart. PloS One. (2015) 10:e0118664. doi: 10.1371/journal.pone.0118664
48. Korkmaz-Icöz S, Li K, Loganathan S, Ding Q, Ruppert M, Radovits T, et al. Brain-dead donor heart conservation with a preservation solution supplemented by a conditioned medium from mesenchymal stem cells improves graft contractility after transplantation. Am J Transplant. (2020) 20:2847–56. doi: 10.1111/ajt.15843
49. Tzoumas N, Farrah TE, Dhaun N, Webb DJ. Established and emerging therapeutic uses of PDE type 5 inhibitors in cardiovascular disease. Br J Pharmacol. (2020) 177:5467–88. doi: 10.1111/bph.14920
50. Kamel R, Leroy J, Vandecasteele G, Fischmeister R. Cyclic nucleotide phosphodiesterases as therapeutic targets in cardiac hypertrophy and heart failure. Nat Rev Cardiol. (2023) 20:90–108. doi: 10.1038/s41569-022-00756-z
51. Shan X, Margulies KB. Differential regulation of PDE5 expression in left and right ventricles of feline hypertrophy models. PloS One. (2011) 6:e19922. doi: 10.1371/journal.pone.0019922
52. Blanton RM, Takimoto E, Lane AM, Aronovitz M, Piotrowski R, Karas RH, et al. Protein kinase g iα inhibits pressure overload-induced cardiac remodeling and is required for the cardioprotective effect of sildenafil in vivo. J Am Heart Assoc. (2012) 1:e003731. doi: 10.1161/JAHA.112.003731
53. Tsai EJ, Liu Y, Koitabashi N, Bedja D, Danner T, Jasmin JF, et al. Pressure-overload-induced subcellular relocalization/oxidation of soluble guanylyl cyclase in the heart modulates enzyme stimulation. Circ Res. (2012) 110:295–303. doi: 10.1161/CIRCRESAHA.111.259242
54. Takimoto E. Cyclic GMP-dependent signaling in cardiac myocytes. Circ J. (2012) 76:1819–25. doi: 10.1253/circj.CJ-12-0664
55. Nagendran J, Archer SL, Soliman D, Gurtu V, Moudgil R, Haromy A, et al. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation. (2007) 116:238–48. doi: 10.1161/CIRCULATIONAHA.106.655266
56. Shan X, Quaile MP, Monk JK, French B, Cappola TP, Margulies KB. Differential expression of PDE5 in failing and nonfailing human myocardium. Circ Heart Fail. (2012) 5:79–86. doi: 10.1161/CIRCHEARTFAILURE.111.961706
57. Pokreisz P, Vandenwijngaert S, Bito V, Van den Bergh A, Lenaerts I, Busch C, et al. Ventricular phosphodiesterase-5 expression is increased in patients with advanced heart failure and contributes to adverse ventricular remodeling after myocardial infarction in mice. Circulation. (2009) 119:408–16. doi: 10.1161/CIRCULATIONAHA.108.822072
58. Schwartz BG, Levine LA, Comstock G, Stecher VJ, Kloner RA. Cardiac uses of phosphodiesterase-5 inhibitors. J Am Coll Cardiol. (2012) 59:9–15. doi: 10.1016/j.jacc.2011.07.051
59. Di Luigi L, Corinaldesi C, Colletti M, Scolletta S, Antinozzi C, Vannelli GB, et al. Phosphodiesterase type 5 inhibitor sildenafil decreases the proinflammatory chemokine CXCL10 in human cardiomyocytes and in subjects with diabetic cardiomyopathy. Inflammation. (2016) 39:1238–52. doi: 10.1007/s10753-016-0359-6
60. Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an International Forum on Cardiac Remodeling. J Am Coll Cardiol. (2000) 35:569–82. doi: 10.1016/S0735-1097(99)00630-0
61. Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. (2016) 119:91–112. doi: 10.1161/CIRCRESAHA.116.303577
62. Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res. (2015) 116:1254–68. doi: 10.1161/CIRCRESAHA.116.302317
63. Frieler RA, Mortensen RM. Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation. (2015) 131:1019–30. doi: 10.1161/CIRCULATIONAHA.114.008788
64. Izzicupo P, D’Amico MA, Bascelli A, Di Fonso A, D’Angelo E, Di Blasio A, et al. Walking training affects dehydroepiandrosterone sulfate and inflammation independent of changes in spontaneous physical activity. Menopause. (2013) 20:455–63. doi: 10.1097/gme.0b013e31827425c9
65. Numata G, Takimoto E, Cyclic GMP. and PKG signaling in heart failure. Front Pharmacol. (2022) 13:792798. doi: 10.3389/fphar.2022.792798
66. Bishu K, Hamdani N, Mohammed SF, Kruger M, Ohtani T, Ogut O, et al. Sildenafil and B-type natriuretic peptide acutely phosphorylate titin and improve diastolic distensibility in vivo. Circulation. (2011) 124:2882–91. doi: 10.1161/CIRCULATIONAHA.111.048520
67. Layland J, Solaro RJ, Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc Res. (2005) 66:12–21. doi: 10.1016/j.cardiores.2004.12.022
68. Wijnker PJ, Murphy AM, Stienen GJ, van der Velden J. Troponin I phosphorylation in human myocardium in health and disease. Neth Heart J. (2014) 22:463–9. doi: 10.1007/s12471-014-0590-4
69. Thoonen R, Giovanni S, Govindan S, Lee DI, Wang GR, Calamaras TD, et al. Molecular screen identifies cardiac myosin-binding protein-C as a protein kinase G-Iα Substrate. Circ Heart Fail. (2015) 8:1115–22. doi: 10.1161/CIRCHEARTFAILURE.115.002308
70. Shenoy P, Agarwal V. Phosphodiesterase inhibitors in the management of autoimmune disease. Autoimmun Rev. (2010) 9:511–5. doi: 10.1016/j.autrev.2010.02.012
71. McDonald TS, Kumar V, Fung JN, Woodruff TM, Lee JD. Glucose clearance and uptake is increased in the SOD1. FASEB J. (2021) 35:e21707. doi: 10.1096/fj.202002450R
72. Kim HH, Liao JK. Translational therapeutics of dipyridamole. Arterioscler Thromb Vasc Biol. (2008) 28:s39–42. doi: 10.1161/ATVBAHA.107.160226
73. Liao JK, Bettmann MA, Sandor T, Tucker JI, Coleman SM, Creager MA. Differential impairment of vasodilator responsiveness of peripheral resistance and conduit vessels in humans with atherosclerosis. Circ Res. (1991) 68:1027–34. doi: 10.1161/01.RES.68.4.1027
74. Radomski MW, Rees DD, Dutra A, Moncada S. S-nitroso-glutathione inhibits platelet activation in vitro and in vivo. Br J Pharmacol. (1992) 107:745–9. doi: 10.1111/j.1476-5381.1992.tb14517.x
75. Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest. (1989) 83:1774–7. doi: 10.1172/JCI114081
76. Davies KJA. Cardiovascular adaptive homeostasis in exercise. Front Physiol. (2018) 9:369. doi: 10.3389/fphys.2018.00369
77. Kanoh H, Iwashita S, Kuraishi T, Goto A, Fuse N, Ueno H, et al. cGMP signaling pathway that modulates NF-κB activation in innate immune responses. iScience. (2021) 24:103473. doi: 10.1016/j.isci.2021.103473
78. Nguyen TH, Axell A, Turek I, Wright B, Meehan-Andrews T, Irving HR. Modulation of inflammatory cytokine production in human monocytes by cGMP and IRAK3. Int J Mol Sci. (2022) 23(5):2552. doi: 10.3390/ijms23052552
79. Siednienko J, Nowak J, Moynagh PN, Gorczyca WA. Nitric oxide affects IL-6 expression in human peripheral blood mononuclear cells involving cGMP-dependent modulation of NF-κB activity. Cytokine. (2011) 54:282–8. doi: 10.1016/j.cyto.2011.02.015
80. Kniotek M, Boguska A. Sildenafil can affect innate and adaptive immune system in both experimental animals and patients. J Immunol Res. (2017) 2017:4541958. doi: 10.1155/2017/4541958
81. Karakhanova S, Yang Y, Link J, Soltek S, von Ahn K, Umansky V, et al. Gender-specific immunological effects of the phosphodiesterase 5 inhibitor sildenafil in healthy mice. Mol Immunol. (2013) 56:649–59. doi: 10.1016/j.molimm.2013.06.021
82. Nunes AK, Rapôso C, Luna RL, Cruz-Höfling MA, Peixoto CA. Sildenafil (Viagra®) down regulates cytokines and prevents demyelination in a cuprizone-induced MS mouse model. Cytokine. (2012) 60:540–51. doi: 10.1016/j.cyto.2012.06.011
83. Pifarré P, Gutierrez-Mecinas M, Prado J, Usero L, Roura-Mir C, Giralt M, et al. Phosphodiesterase 5 inhibition at disease onset prevents experimental autoimmune encephalomyelitis progression through immunoregulatory and neuroprotective actions. Exp Neurol. (2014) 251:58–71. doi: 10.1016/j.expneurol.2013.10.021
84. Jerzak M, Kniotek M, Mrozek J, Górski A, Baranowski W. Sildenafil citrate decreased natural killer cell activity and enhanced chance of successful pregnancy in women with a history of recurrent miscarriage. Fertil Steril. (2008) 90:1848–53. doi: 10.1016/j.fertnstert.2007.08.043
85. Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. (2000) 87:840–4. doi: 10.1161/01.RES.87.10.840
86. Gerassimou C, Kotanidou A, Zhou Z, Simoes DC, Roussos C, Papapetropoulos A. Regulation of the expression of soluble guanylyl cyclase by reactive oxygen species. Br J Pharmacol. (2007) 150:1084–91. doi: 10.1038/sj.bjp.0707179
87. Kemp-Harper BK, Velagic A, Paolocci N, Horowitz JD, Ritchie RH. Cardiovascular therapeutic potential of the redox siblings, nitric oxide (NO•) and nitroxyl (HNO), in the setting of reactive oxygen species dysregulation. Handb Exp Pharmacol. (2021) 264:311–37. doi: 10.1007/164_2020_389
88. Strieter RM, Polverini PJ, Arenberg DA, Kunkel SL. The role of CXC chemokines as regulators of angiogenesis. Shock. (1995) 4:155–60. doi: 10.1097/00024382-199509000-00001
89. Szentes V, Gazdag M, Szokodi I, Dézsi CA. The role of CXCR3 and associated chemokines in the development of atherosclerosis and during myocardial infarction. Front Immunol. (2018) 9:1932. doi: 10.3389/fimmu.2018.01932
90. Giannattasio S, Corinaldesi C, Colletti M, Di Luigi L, Antinozzi C, Filardi T, et al. The phosphodiesterase 5 inhibitor sildenafil decreases the proinflammatory chemokine IL-8 in diabetic cardiomyopathy: in vivo and in vitro evidence. J Endocrinol Invest. (2019) 42:715–25. doi: 10.1007/s40618-018-0977-y
91. Filardi T, Ghinassi B, Di Baldassarre A, Tanzilli G, Morano S, Lenzi A, et al. Cardiomyopathy associated with diabetes: the central role of the cardiomyocyte. Int J Mol Sci. (2019) 20(13):3299. doi: 10.3390/ijms20133299
92. Tecchio C, Cassatella MA. Neutrophil-derived cytokines involved in physiological and pathological angiogenesis. Chem Immunol Allergy. (2014) 99:123–37. doi: 10.1159/000353358
93. Mandosi E, Giannetta E, Filardi T, Lococo M, Bertolini C, Fallarino M, et al. Endothelial dysfunction markers as a therapeutic target for Sildenafil treatment and effects on metabolic control in type 2 diabetes. Expert Opin Ther Targets. (2015) 19:1617–22. doi: 10.1517/14728222.2015.1066337
94. Giannetta E, Isidori AM, Galea N, Carbone I, Mandosi E, Vizza CD, et al. Chronic Inhibition of cGMP phosphodiesterase 5A improves diabetic cardiomyopathy: a randomized, controlled clinical trial using magnetic resonance imaging with myocardial tagging. Circulation. (2012) 125:2323–33. doi: 10.1161/CIRCULATIONAHA.111.063412
95. Apostolakis S, Vogiatzi K, Amanatidou V, Spandidos DA. Interleukin 8 and cardiovascular disease. Cardiovasc Res. (2009) 84:353–60. doi: 10.1093/cvr/cvp241
96. Katkenov N, Mukhatayev Z, Kozhakhmetov S, Sailybayeva A, Bekbossynova M, Kushugulova A. Systematic review on the role of IL-6 and IL-1β in cardiovascular diseases. J Cardiovasc Dev Dis. (2024) 11(7):206. doi: 10.3390/jcdd11070206
97. Lorenzatti AJ. Anti-inflammatory treatment and cardiovascular outcomes: results of clinical trials. Eur Cardiol. (2021) 16:e15. doi: 10.15420/ecr
98. Crescioli C, Buonamano A, Scolletta S, Sottili M, Francalanci M, Giomarelli P, et al. Predictive role of pretransplant serum CXCL10 for cardiac acute rejection. Transplantation. (2009) 87:249–55. doi: 10.1097/TP.0b013e3181919f5d
99. Li H, Chen C, Wang DW. Inflammatory cytokines, immune cells, and organ interactions in heart failure. Front Physiol. (2021) 12:695047. doi: 10.3389/fphys.2021.695047
100. Prapiadou S, Živković L, Thorand B, George MJ, van der Laan SW, Malik R, et al. Proteogenomic data integration reveals CXCL10 as a potentially downstream causal mediator for IL-6 signaling on atherosclerosis. Circulation. (2024) 149:669–83. doi: 10.1161/CIRCULATIONAHA.123.064974
101. Seta Y, Shan K, Bozkurt B, Oral H, Mann DL. Basic mechanisms in heart failure: the cytokine hypothesis. J Card Fail. (1996) 2:243–9. doi: 10.1016/S1071-9164(96)80047-9
102. Bauer JW, Baechler EC, Petri M, Batliwalla FM, Crawford D, Ortmann WA, et al. Elevated serum levels of interferon-regulated chemokines are biomarkers for active human systemic lupus erythematosus. PloS Med. (2006) 3:e491. doi: 10.1371/journal.pmed.0030491
103. Spinelli FR, Berti R, Farina G, Ceccarelli F, Conti F, Crescioli C. Exercise-induced modulation of Interferon-signature: a therapeutic route toward management of Systemic Lupus Erythematosus. Autoimmun Rev. (2023) 22:103412. doi: 10.1016/j.autrev.2023.103412
104. Lee EY, Lee ZH, Song YW. CXCL10 and autoimmune diseases. Autoimmun Rev. (2009) 8:379–83. doi: 10.1016/j.autrev.2008.12.002
105. Matucci-Cerinic M, Kahaleh B, Wigley FM. Review: evidence that systemic sclerosis is a vascular disease. Arthritis Rheum. (2013) 65:1953–62. doi: 10.1002/art.37988
106. Crescioli C, Corinaldesi C, Riccieri V, Raparelli V, Vasile M, Del Galdo F, et al. Association of circulating CXCL10 and CXCL11 with systemic sclerosis. Ann Rheum Dis. (2018) 77:1845–6. doi: 10.1136/annrheumdis-2018-213257
107. Maltez N, Maxwell LJ, Rirash F, Tanjong Ghogomu E, Harding SE, Tingey PC, et al. Phosphodiesterase 5 inhibitors (PDE5i) for the treatment of Raynaud’s phenomenon. Cochrane Database Syst Rev. (2023) 11:CD014089. doi: 10.1002/14651858.CD014089
108. Pope JE, Denton CP, Johnson SR, Fernandez-Codina A, Hudson M, Nevskaya T. State-of-the-art evidence in the treatment of systemic sclerosis. Nat Rev Rheumatol. (2023) 19:212–26. doi: 10.1038/s41584-023-00909-5
109. Corinaldesi C, Ross RL, Abignano G, Antinozzi C, Marampon F, di Luigi L, et al. Muscle damage in systemic sclerosis and CXCL10: the potential therapeutic role of PDE5 inhibition. Int J Mol Sci. (2021) 22(6):2894. doi: 10.3390/ijms22062894
110. Wharton J, Strange JW, Møller GM, Growcott EJ, Ren X, Franklyn AP, et al. Antiproliferative effects of phosphodiesterase type 5 inhibition in human pulmonary artery cells. Am J Respir Crit Care Med. (2005) 172:105–13. doi: 10.1164/rccm.200411-1587OC
111. Lerner A, Epstein PM. Cyclic nucleotide phosphodiesterases as targets for treatment of haematological Malignancies. Biochem J. (2006) 393:21–41. doi: 10.1042/BJ20051368
112. Zhu B, Strada S, Stevens T. Cyclic GMP-specific phosphodiesterase 5 regulates growth and apoptosis in pulmonary endothelial cells. Am J Physiol Lung Cell Mol Physiol. (2005) 289:L196–206. doi: 10.1152/ajplung.00433.2004
113. Zhu B, Strada SJ. The novel functions of cGMP-specific phosphodiesterase 5 and its inhibitors in carcinoma cells and pulmonary/cardiovascular vessels. Curr Top Med Chem. (2007) 7:437–54. doi: 10.2174/156802607779941198
114. Shi Z, Tiwari AK, Shukla S, Robey RW, Singh S, Kim IW, et al. Sildenafil reverses ABCB1- and ABCG2-mediated chemotherapeutic drug resistance. Cancer Res. (2011) 71:3029–41. doi: 10.1158/0008-5472.CAN-10-3820
115. Chang JF, Hsu JL, Sheng YH, Leu WJ, Yu CC, Chan SH, et al. Phosphodiesterase type 5 (PDE5) inhibitors sensitize topoisomerase II inhibitors in killing prostate cancer through PDE5-independent impairment of HR and NHEJ DNA repair systems. Front Oncol. (2018) 8:681. doi: 10.3389/fonc.2018.00681
116. Das A, Durrant D, Mitchell C, Mayton E, Hoke NN, Salloum FN, et al. Sildenafil increases chemotherapeutic efficacy of doxorubicin in prostate cancer and ameliorates cardiac dysfunction. Proc Natl Acad Sci U.S.A. (2010) 107:18202–7. doi: 10.1073/pnas.1006965107
117. Hamilton TK, Hu N, Kolomitro K, Bell EN, Maurice DH, Graham CH, et al. Potential therapeutic applications of phosphodiesterase inhibition in prostate cancer. World J Urol. (2013) 31:325–30. doi: 10.1007/s00345-012-0848-7
118. Liu N, Mei L, Fan X, Tang C, Ji X, Hu X, et al. Phosphodiesterase 5/protein kinase G signal governs stemness of prostate cancer stem cells through Hippo pathway. Cancer Lett. (2016) 378:38–50. doi: 10.1016/j.canlet.2016.05.010
119. Zhou F, Gao S, Han D, Han W, Chen S, Patalano S, et al. TMPRSS2-ERG activates NO-cGMP signaling in prostate cancer cells. Oncogene. (2019) 38:4397–411. doi: 10.1038/s41388-019-0730-9
120. Hsu JL, Leu WJ, Hsu LC, Ho CH, Liu SP, Guh JH. Phosphodiesterase type 5 inhibitors synergize vincristine in killing castration-resistant prostate cancer through amplifying mitotic arrest signaling. Front Oncol. (2020) 10:1274. doi: 10.3389/fonc.2020.01274
121. Pusztai L, Zhen JH, Arun B, Rivera E, Whitehead C, Thompson WJ, et al. Phase I and II study of exisulind in combination with capecitabine in patients with metastatic breast cancer. J Clin Oncol. (2003) 21:3454–61. doi: 10.1200/JCO.2003.02.114
122. Karami-Tehrani F, Moeinifard M, Aghaei M, Atri M. Evaluation of PDE5 and PDE9 expression in benign and Malignant breast tumors. Arch Med Res. (2012) 43:470–5. doi: 10.1016/j.arcmed.2012.08.006
123. Catalano S, Campana A, Giordano C, Győrffy B, Tarallo R, Rinaldi A, et al. Expression and function of phosphodiesterase type 5 in human breast cancer cell lines and tissues: implications for targeted therapy. Clin Cancer Res. (2016) 22:2271–82. doi: 10.1158/1078-0432.CCR-15-1900
124. Tinsley HN, Gary BD, Keeton AB, Zhang W, Abadi AH, Reynolds RC, et al. Sulindac sulfide selectively inhibits growth and induces apoptosis of human breast tumor cells by phosphodiesterase 5 inhibition, elevation of cyclic GMP, and activation of protein kinase G. Mol Cancer Ther. (2009) 8:3331–40. doi: 10.1158/1535-7163.MCT-09-0758
125. Tinsley HN, Gary BD, Keeton AB, Lu W, Li Y, Piazza GA. Inhibition of PDE5 by sulindac sulfide selectively induces apoptosis and attenuates oncogenic Wnt/β-catenin-mediated transcription in human breast tumor cells. Cancer Prev Res (Phila). (2011) 4:1275–84. doi: 10.1158/1940-6207.CAPR-11-0095
126. Chen L, Liu Y, Becher A, Diepold K, Schmid E, Fehn A, et al. Sildenafil triggers tumor lethality through altered expression of HSP90 and degradation of PKD2. Carcinogenesis. (2020) 41:1421–31. doi: 10.1093/carcin/bgaa001
127. Klutzny S, Anurin A, Nicke B, Regan JL, Lange M, Schulze L, et al. PDE5 inhibition eliminates cancer stem cells via induction of PKA signaling. Cell Death Dis. (2018) 9:192. doi: 10.1038/s41419-017-0202-5
128. Baravalle R, Valetti F, Catucci G, Gambarotta G, Chiesa M, Maurelli S, et al. Effect of sildenafil on human aromatase activity: From in vitro structural analysis to catalysis and inhibition in cells. J Steroid Biochem Mol Biol. (2017) 165:438–47. doi: 10.1016/j.jsbmb.2016.09.003
129. Greish K, Fateel M, Abdelghany S, Rachel N, Alimoradi H, Bakhiet M, et al. Sildenafil citrate improves the delivery and anticancer activity of doxorubicin formulations in a mouse model of breast cancer. J Drug Target. (2018) 26:610–5. doi: 10.1080/1061186X.2017.1405427
130. Poklepovic A, Qu Y, Dickinson M, Kontos MC, Kmieciak M, Schultz E, et al. Randomized study of doxorubicin-based chemotherapy regimens, with and without sildenafil, with analysis of intermediate cardiac markers. Cardiooncology. (2018) 4:7. doi: 10.1186/s40959-018-0033-2
131. Islam BN, Sharman SK, Hou Y, Bridges AE, Singh N, Kim S, et al. Sildenafil suppresses inflammation-driven colorectal cancer in mice. Cancer Prev Res (Phila). (2017) 10:377–88. doi: 10.1158/1940-6207.CAPR-17-0015
132. Stoner GD, Budd GT, Ganapathi R, DeYoung B, Kresty LA, Nitert M, et al. Sulindac sulfone induced regression of rectal polyps in patients with familial adenomatous polyposis. Adv Exp Med Biol. (1999) 470:45–53. doi: 10.1007/978-1-4615-4149-3
133. van Stolk R, Stoner G, Hayton WL, Chan K, DeYoung B, Kresty L, et al. Phase I trial of exisulind (sulindac sulfone, FGN-1) as a chemopreventive agent in patients with familial adenomatous polyposis. Clin Cancer Res. (2000) 6:78–89.
134. Thompson WJ, Piazza GA, Li H, Liu L, Fetter J, Zhu B, et al. Exisulind induction of apoptosis involves guanosine 3’,5’-cyclic monophosphate phosphodiesterase inhibition, protein kinase G activation, and attenuated beta-catenin. Cancer Res. (2000) 60:3338–42.
135. Liu L, Li H, Underwood T, Lloyd M, David M, Sperl G, et al. Cyclic GMP-dependent protein kinase activation and induction by exisulind and CP461 in colon tumor cells. J Pharmacol Exp Ther. (2001) 299:583–92.
136. Whitt JD, Li N, Tinsley HN, Chen X, Zhang W, Li Y, et al. A novel sulindac derivative that potently suppresses colon tumor cell growth by inhibiting cGMP phosphodiesterase and β-catenin transcriptional activity. Cancer Prev Res (Phila). (2012) 5:822–33. doi: 10.1158/1940-6207.CAPR-11-0559
137. Mei XL, Yang Y, Zhang YJ, Li Y, Zhao JM, Qiu JG, et al. Sildenafil inhibits the growth of human colorectal cancer in vitro and in vivo. Am J Cancer Res. (2015) 5:3311–24.
138. Domvri K, Zarogoulidis K, Zogas N, Zarogoulidis P, Petanidis S, Porpodis K, et al. Potential synergistic effect of phosphodiesterase inhibitors with chemotherapy in lung cancer. J Cancer. (2017) 8:3648–56. doi: 10.7150/jca.21783
139. Li Q, Shu Y. Pharmacological modulation of cytotoxicity and cellular uptake of anti-cancer drugs by PDE5 inhibitors in lung cancer cells. Pharm Res. (2014) 31:86–96. doi: 10.1007/s11095-013-1134-0
140. Zhang Md J, Zhang Md L, Yang Md Y, Liu Md Q, Ma Md H, Huang Md A, et al. Polymorphonuclear-MDSCs facilitate tumor regrowth after radiation by suppressing CD8. Int J Radiat Oncol Biol Phys. (2021) 109:1533–46. doi: 10.1016/j.ijrobp.2020.11.038
141. Giordano D, Giorgi M, Sette C, Biagioni S, Augusti-Tocco G. cAMP-dependent induction of PDE5 expression in murine neuroblastoma cell differentiation. FEBS Lett. (1999) 446:218–22. doi: 10.1016/S0014-5793(99)00227-6
142. Black KL, Yin D, Ong JM, Hu J, Konda BM, Wang X, et al. PDE5 inhibitors enhance tumor permeability and efficacy of chemotherapy in a rat brain tumor model. Brain Res. (2008) 1230:290–302. doi: 10.1016/j.brainres.2008.06.122
143. Booth L, Roberts JL, Tavallai M, Nourbakhsh A, Chuckalovcak J, Carter J, et al. OSU-03012 and viagra treatment inhibits the activity of multiple chaperone proteins and disrupts the blood-brain barrier: implications for anti-cancer therapies. J Cell Physiol. (2015) 230:1982–98. doi: 10.1002/jcp.24977
144. Hu J, Ljubimova JY, Inoue S, Konda B, Patil R, Ding H, et al. Phosphodiesterase type 5 inhibitors increase Herceptin transport and treatment efficacy in mouse metastatic brain tumor models. PloS One. (2010) 5:e10108. doi: 10.1371/journal.pone.0010108
145. Wang R, Chen W, Zhang Q, Liu Y, Qiao X, Meng K, et al. Phosphodiesterase type 5 inhibitor Tadalafil increases Rituximab treatment efficacy in a mouse brain lymphoma model. J Neurooncol. (2015) 122:35–42. doi: 10.1007/s11060-014-1690-0
146. Cesarini V, Martini M, Vitiani LR, Gravina GL, Di Agostino S, Graziani G, et al. Type 5 phosphodiesterase regulates glioblastoma multiforme aggressiveness and clinical outcome. Oncotarget. (2017) 8:13223–39. doi: 10.18632/oncotarget.v8i8
147. Lang F, Liu Y, Chou FJ, Yang C. Genotoxic therapy and resistance mechanism in gliomas. Pharmacol Ther. (2021) 228:107922. doi: 10.1016/j.pharmthera.2021.107922
148. Poklepovic AS, Shah P, Tombes MB, Shrader E, Bandyopadhyay D, Deng X, et al. Phase 2 study of sorafenib, valproic acid, and sildenafil in the treatment of recurrent high-grade glioma. medRxiv. (2024). doi: 10.1101/2024.04.23.24304634
149. Zhang G, Park MA, Mitchell C, Hamed H, Rahmani M, Martin AP, et al. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clin Cancer Res. (2008) 14:5385–99. doi: 10.1158/1078-0432.CCR-08-0469
150. Tang Y, Yacoub A, Hamed HA, Poklepovic A, Tye G, Grant S, et al. Sorafenib and HDAC inhibitors synergize to kill CNS tumor cells. Cancer Biol Ther. (2012) 13:567–74. doi: 10.4161/cbt.19771
151. Agarwal S, Sane R, Ohlfest JR, Elmquist WF. The role of the breast cancer resistance protein (ABCG2) in the distribution of sorafenib to the brain. J Pharmacol Exp Ther. (2011) 336:223–33. doi: 10.1124/jpet.110.175034
152. Dhaliwal A, Gupta M. PDE5 inhibitors. In: StatPearls. S. Publishing, Treasure Island (FL (2023).
153. Booth L, Roberts JL, Rais R, Cutler RE, Diala I, Lalani AS, et al. Neratinib augments the lethality of [regorafenib + sildenafil. J Cell Physiol. (2019) 234:4874–87. doi: 10.1002/jcp.27276
154. Danial C, Tichy AL, Tariq U, Swetman GL, Khuu P, Leung TH, et al. An open-label study to evaluate sildenafil for the treatment of lymphatic malformations. J Am Acad Dermatol. (2014) 70:1050–7. doi: 10.1016/j.jaad.2014.02.005
Keywords: PDE5i, cGMP, inflammation, cardiac diseases, cancer
Citation: Paronetto MP and Crescioli C (2024) Rethinking of phosphodiesterase 5 inhibition: the old, the new and the perspective in human health. Front. Endocrinol. 15:1461642. doi: 10.3389/fendo.2024.1461642
Received: 08 July 2024; Accepted: 28 August 2024;
Published: 17 September 2024.
Edited by:
Federica Campolo, Sapienza University of Rome, ItalyReviewed by:
Enio Setsuo Arakaki Pacini, Federal University of São Paulo, BrazilAndré Leite Moreira, University of Porto, Portugal
Francisco O. Silva, University of Texas Southwestern Medical Center, United States
Nobuaki Fukuma, Columbia University, United States
Copyright © 2024 Paronetto and Crescioli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Clara Crescioli, Y2xhcmEuY3Jlc2Npb2xpQHVuaXJvbWE0Lml0