 Saman Saedi1
Saman Saedi1 Yi Tan
Yi Tan Sara E. Watson
Sara E. Watson Kupper A. Wintergerst
Kupper A. Wintergerst Lu Cai
Lu Cai- 1Department of Animal Science, College of Agriculture, Shiraz University, Shiraz, Iran
- 2Pediatric Research Institute, Department of Pediatrics, University of Louisville School of Medicine, Louisville, KY, United States
- 3Wendy Novak Diabetes Institute, Norton Children’s Hospital, Louisville, KY, United States
- 4Department of Pharmacology and Toxicology, University of Louisville School of Medicine, Louisville, KY, United States
- 5Division of Endocrinology, Department of Pediatrics, University of Louisville, Norton Children’s Hospital, Louisville, KY, United States
- 6The Center for Integrative Environmental Health Sciences, University of Louisville School of Medicine, Louisville, KY, United States
- 7Department of Radiation Oncology, University of Louisville School of Medicine, Louisville, KY, United States
Diabetes and its complications are major diseases that affect human health. Diabetic cardiovascular complications such as cardiovascular diseases (CVDs) are the major complications of diabetes, which are associated with the loss of cardiovascular cells. Pathogenically the role of ferroptosis, an iron-dependent cell death, and cuproptosis, a copper-dependent cell death has recently been receiving attention for the pathogenesis of diabetes and its cardiovascular complications. How exposure to environmental metals affects these two metal-dependent cell deaths in cardiovascular pathogenesis under diabetic and nondiabetic conditions remains largely unknown. As an omnipresent environmental metal, cadmium exposure can cause oxidative stress in the diabetic cardiomyocytes, leading to iron accumulation, glutathione depletion, lipid peroxidation, and finally exacerbate ferroptosis and disrupt the cardiac. Moreover, cadmium-induced hyperglycemia can enhance the circulation of advanced glycation end products (AGEs). Excessive AGEs in diabetes promote the upregulation of copper importer solute carrier family 31 member 1 through activating transcription factor 3/transcription factor PU.1, thereby increasing intracellular Cu+ accumulation in cardiomyocytes and disturbing Cu+ homeostasis, leading to a decline of Fe–S cluster protein and reactive oxygen species accumulation in cardiomyocytes mitochondria. In this review, we summarize the available evidence and the most recent advances exploring the underlying mechanisms of ferroptosis and cuproptosis in CVDs and diabetic cardiovascular complications, to provide critical perspectives on the potential pathogenic roles of ferroptosis and cuproptosis in cadmium-induced or exacerbated cardiovascular complications in diabetic individuals.
1 Introduction
Diabetes is an endocrine system disease caused by a chronic hyperglycemic state. Indeed, diabetes is characterized by high blood glucose levels that result from insulin deficiency, in the context of β- cell dysfunction, insulin resistance or both (1). This chronic metabolic disorder is one of the most common and fastest-growing diseases worldwide, projected to affect 693 million adults by 2045 (2). Vascular complications of both the macrovascular system (cardiovascular disease (CVD) and microvascular system (diabetic kidney disease, diabetic retinopathy, and neuropathy) are the leading cause of morbidity and mortality in individuals with diabetes (3). Additionally, studies have shown that CVD is the major contributor to morbidity and mortality in individuals with type 2 diabetes (T2D), with a risk that is doubled in patients with diabetes compared with those without diabetes (4). These studies show diabetes acts as an independent risk factor for several forms of CVDs such as coronary artery diseases, heart failure (HF), cardiomyopathy, arrhythmia, and peripheral artery disease (5). To make matters worse, when patients with diabetes develop clinical CVDs, they sustain a worse prognosis for survival than do CVD patients without diabetes (6).
Diabetes and its cardiovascular complications are complex, multifactorial conditions with both main environmental and genetic components. Major risk factors for cardiovascular complications among individuals with diabetes include the presence of other microvascular complications, as well as BMI, age, sex, blood pressure, smoking status, and heavy metals (7–9). Cadmium is a toxic heavy metal that has been associated with hypertension, promotion of atherosclerosis, and impaired cardiac function (10). Experimental studies indicated that cadmium exposure is associated with increased risk of HF and damages the cardiomyocyte (11, 12). In addition, epidemiological studies suggested that higher cadmium exposure is associated with disruption of pancreatic islet function and an increased prevalence of diabetes (13–15). Although, it has been indicated that cadmium exacerbates cardiovascular complications of diabetic mice (9), the relationship between cadmium exposure to induce or exacerbate cardiovascular complications in individuals with diabetes is not clear.
Ferroptosis and cuproptosis are recently discovered form of regulated cell death triggered by excess iron (Fe) and copper (Cu2+), respectively (16, 17). Cuproptosis is highly related to cellular metabolism (18), and ferroptosis plays a critical role in cellular proliferation, and differentiation (16). Ferroptosis and cuproptosis have attracted significant interest due to their association with numerous pathological processes (19, 20). Recent studies have shown that impaired ferroptosis was implicated in the occurrence and development of various CVD (21). On the other hand, the essential role of Cu homeostasis has been shown in diabetic cardiomyopathy (DCM) (22).
Environmental toxicant exposures play a significant role in the development and progression of diabetic complications, particularly in T2D. The interaction between genetic predispositions and environmental factors can influence the clinical presentation of diabetes and its associated complications (23). Cadmium as an environmental toxin is highlighted due to its role in exacerbating complications in diabetic patients, particularly in comparison to other environmental toxins. Cadmium leads to oxidative stress and inflammation, which are known contributors to the progression of diabetic complications (24). Studies have demonstrated that cadmium exposure increases markers of oxidative stress, such as malondialdehyde, while decreasing antioxidant levels in diabetic subjects, thereby worsening diabetic complications (25). Besides, cadmium may disrupt trace element homeostasis such as zinc (Zn), magnesium (Mg), Cu and iron (26–29). These findings indicate that cadmium exposure may induce ferroptosis and cuproptosis mediated by change trace element homeostasis. Besides, previous studies have shown that ferroptosis and cuproptosis are associated with the risk of CVDs and diabetic cardiovascular complications (16, 30–32). However, potential roles of ferroptosis and cuproptosis in cadmium-induced or exacerbated cardiovascular complications in individuals with diabetes are still unknown. And whether there is a relationship between ferroptosis and cuproptosis with cadmium-induced cardiovascular complications in the individuals with diabetes remains unclear. In the current review, we present an overview on the most recent advances exploring the underlying mechanisms of ferroptosis and cuproptosis in CVDs and diabetic cardiovascular complications and provide critical perspectives on the potential pathogenic roles of ferroptosis and cuproptosis during cadmium exposure in cadmium-induced or exacerbated cardiovascular complications in the individuals with diabetes.
2 Cadmium and CVD in the individuals with diabetes
2.1 Cadmium
Cadmium is a toxic heavy metal harmful to human health, which is released by industrial activities such as leachate from combustion of fossil fuels, mining residues, and human activities including pesticides or naturally present in the environment (33). In the United States, approximately 600 tons of cadmium compounds are produced yearly and 150 tons are imported from other countries (34). Cadmium has a long biological half-life (10-30 years) and the average dietary intake of this metal ranges from 8–25 μg per day (35, 36). Cadmium can enter animals’ and humans’ bodies via breathing, food, and drinking water and accumulate in the organism, and cause severe damage to a variety of organs, including the lungs, liver, kidneys, testes, and brain (27, 37, 38). It has been reported that cadmium exposure is correlated with many health disorders such as neurotoxicity, osteoporosis, gastrointestinal injury, cancer, and diabetes (39–43).
2.2 Diabetes
Diabetes is a complex metabolic disorder characterized mainly by chronic hyperglycemia, a physiologically abnormal condition represented by continued enhanced blood glucose levels (1). In 2019, the number of adults with diabetes is estimated to be around 463 million, which corresponds to 9.3% of the total adult world population. By 2030, this number is estimated to rise to 578 million, representing 10.2% of the world’s total adult population, and further increase to 700 million by 2045, which represents 10.9% of the total world adult population (44). There are three main types of diabetes: type 1 diabetes (T1D), type 2 diabetes (T2D), and gestational diabetes during pregnancy. T1D is thought to be caused by an autoimmune reaction characterized by the destruction of the pancreatic islets of beta cells, leading to insufficient insulin secretion. Approximately 5-10% of the people who have diabetes have T1D. However, T2D accounts for more than 90% of all cases (45, 46). Diabetes and its complications are complex, and both major environmental and genetic factors participate in its etiopathogenesis (1). T2D has been more commonly associated with increasing age, family history of diabetes, obesity, physical inactivity, and adoption of modern lifestyle, but these factors are complex and remain largely unspecified. Nonetheless, unlike T1D, this disease is not associated with genes involved in the immune response including autoimmunity and consequently there is no immune-mediated destruction of pancreatic β-cells (47, 48).
2.3 Diabetic cardiovascular complications
Diabetes not only causes microangiopathy (associated with the three major diabetic complications, namely diabetic nephropathy, retinopathy, and neuropathy) but is also a major risk factor for macroangiopathy such as CVDs (49, 50). It has been indicated that diabetes can lead to multiple cardiovascular complications, including coronary artery disease, HF, and cardiac hypertrophy (51). CVDs are the leading cause of death worldwide, and they disproportionally affect people living in different communities and environments. Yearly, CVDs cause immense economic and health burdens in the United States and worldwide. In 2020, approximately 19 million deaths were attributed to CVDs, which was an increase of 18.7% since 2010 (52). Depending on the diabetes subtype and CVD complications for example, myocardial infarction, coronary heart disease, HF or stroke, individuals with diabetes have anywhere from a twofold to tenfold increased risk of CVD complications compared to individuals without diabetes (49, 53, 54). Despite the well-known increased risk of CVDs among individuals with diabetes, the pathophysiology linking the two conditions is poorly understood.
DCM is a specific diabetes-induced CVD that refers to a group of diseases that affect the heart muscle, leading to cardiac structure remodeling and dysfunction. Cell death, especially cardiomyocyte death, plays a critical role in the development of cardiomyopathy. When cardiomyocytes die, either by necrosis (uncontrolled cell death) or apoptosis (programmed cell death), there is a loss of contractile units, leading to impaired heart function. This occurs because cell death is followed by energy imbalances, chronic inflammation, and oxidative stress, all of which play crucial roles in the development and progression of cardiac dysfunction, i.e., cardiomyopathy (55–57). On the other hand, oxidative stress increases in patients with diabetes and animal models of diabetes (58). Indeed, oxidative stress and inflammation play a key role in the pathogenesis and progression of diabetes-induced CVD, where the enhanced expression of inflammatory proteins or cytokines such as oxidative stress-related proteins or C-reactive protein are shown to serve as biomarkers for the occurrence of cardiovascular complications (59).
2.4 Cadmium induces CVDs
Recent studies have highlighted the risks of chronic exposure to environmental toxicants, including heavy metals such as inorganic arsenic, lead, and cadmium in CVDs. Despite the fact that the exact effects of cadmium on the cardiovascular system are controversial, studies have shown that it can affect the cardiovascular system at extremely low doses. An in vitro study showed that a dose of cadmium less than toxic concentrations can trigger pathological changes in the vessel walls (60). Besides, chronic cadmium exposure has been associated with promotion of atherosclerosis, hypertension, and impaired cardiac function, as well as an increased risk of cardiovascular mortality (10, 61, 62). In addition, CVDs, stroke, coronary heart disease, and peripheral arterial disease increase due to cadmium exposure (63).
The underlying mechanisms by which cadmium can induce CVDs, cardiomyopathy, and coronary heart disease are still unclear, and further studies are needed to elucidate this issue. However, it has demonstrated that cadmium can disrupt endothelial cells integrity and induces cadmium-mediated endothelial cell death. The formation of gaps between the endothelial cells allows cadmium to diffuse from the bloodstream into the media layer (64). Cadmium is mainly retained in the smooth muscle cells after transport across the endothelial cells and can affect smooth muscle cells. Its effects on smooth muscle cells include cytotoxic effects, interactions with ion homeostasis and calcium ion flux, and stimulation of smooth muscle cell proliferation at low cadmium concentrations, leading to subsequent lipid accumulation in vessel walls and alteration of lipid profiles toward a more atherogenic state (65, 66). Cadmium accumulation also increases multiple important pro-inflammatory cytokines such as interleukin (IL)-6, IL-8, IL-1β, and tumor necrosis factor α (TNF-α) (67), which may play a critical role in atherosclerosis (68, 69). Moreover, exposure to cadmium alters lipid metabolism and decreases nitric oxide release from endothelial cells, leading to atherosclerosis, stroke, and hypertension (70).
3 Potential roles of cadmium-exacerbates cardiovascular complications in individuals with diabetes
Cardiovascular disease remains the leading cause of mortality and morbidity in individuals with diabetes. Recent studies have presented the risk of cardiotoxic effects of exposure to multiple metals particularly under diabetic or metabolic conditions (71–73). In one of these studies, Long et al. (72) demonstrated inverse associations between selenium and zinc incident CVD risk in patients with T2D. However, evidence for the association between heavy metal levels, especially cadmium and the risk of CVDs among individuals with T2D is limited. A recent study has presented that exposure to cadmium is significantly associated with higher risk of CVD mortality among patients with T2D (74). Animal studies have shown the effect of cadmium exposure on cardiac glucose metabolism and lipid accumulation in male Wistar rats. This study has indicated that cadmium exposure induces cardiac glucometabolic dysregulation and lipid accumulation independent of pyruvate dehydrogenase activity which could reduce cardiac efficiency (75). Besides, Xiuxiu Liu (9) has reported that cadmium exacerbates DCM development in mice. Altogether, potential roles of cadmium in exacerbating cardiovascular complications in the individuals with diabetes are described below (Figure 1).
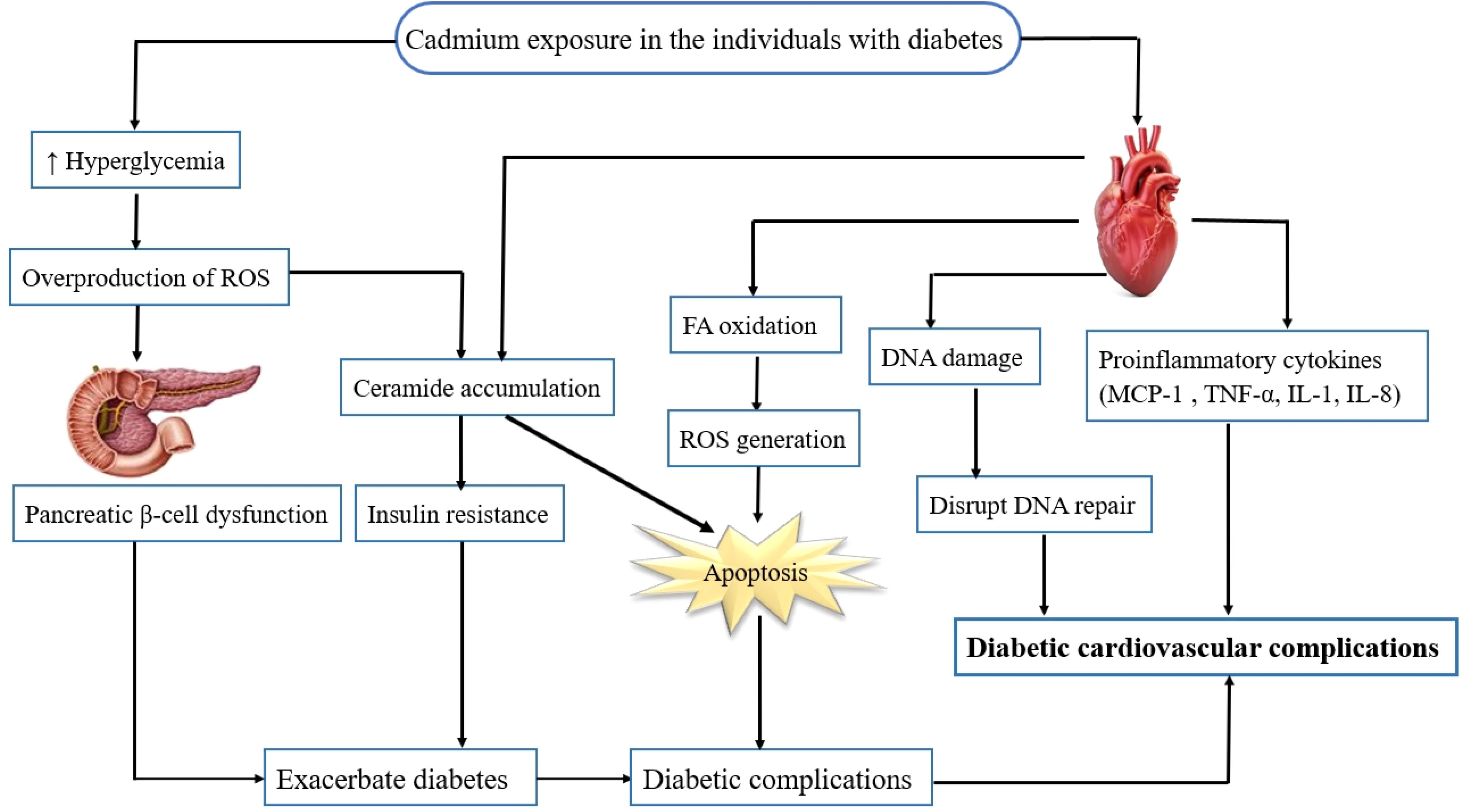
Figure 1. Potential role of cadmium in exacerbating cardiovascular complications in individuals with diabetes. Cadmium exposure in individuals with diabetes results in hyperglycemia, ceramide accumulation, and an increase in the FFA oxidation, proinflammatory cytokines as well as generation of ROS. The ROS activate several pathways involved in the pathogenesis of diabetic cardiovascular complications, including inflammation, disrupting DNA repair, oxidative stress, apoptosis, and cell death. FFA, free fatty acid; ROS, reactive oxygen species.
3.1 Oxidative stress
Based on the above discussion, diabetes is associated with cadmium exposure and cadmium is also a risk factor for cardiac oxidative damage. Cadmium-induced cardiac oxidative stress is mediated by free radicals generation and subsequent production of reactive oxygen species (ROS) such as superoxide anion free radical (O2.−), hydrogen peroxide (H2O2) as well as hydroxyl free radical (HO.) in the heart (76, 77). Enhanced production of ROS can overwhelm cells’ intrinsic antioxidant defenses, and result in oxidative stress, lipid oxidation, cell cycle progression, DNA damage, and apoptosis (78). Cadmium exposure provides cardiac oxidative damage; therefore, this heavy metal may also exacerbate cardiac dysfunction or CVDs in individuals with diabetes.
On the other hand, cadmium exposure is reported to induced hyperglycemia and oxidative status, leading to pancreatic β-cell dysfunction and progression of diabetes (79, 80). Hyperglycemia induces overproduction of some ROS like O2.− by the mitochondrial electron transport chain. It could be the first and key event in the activation of all other pathways involved in the pathogenesis of diabetic complications (81, 82) such as oxidative stress, inflammation, and CVDs (83). Additionally, studies have demonstrated that ROS generation under various pathological conditions such as cadmium exposure triggers ceramide accumulation in diabetic myocardium (83, 84). Ceramide accumulation has been proposed to be involved in insulin resistance, triggering cardiomyocyte apoptosis, cardiac arrhythmias, and CVDs (85, 86).
Generally, the heart uses fatty acid (60—80%) or glucose oxidation to produce adenosine triphosphate (ATP). Cadmium can further increase fatty acid (FA) oxidation and inhibit glucose oxidation in the heart under diabetic condition. FA oxidation decreases ATP and enhances ROS production in the mitochondria. A decrease in ATP and increased ROS production in the mitochondria due to cadmium exposure in individuals with diabetes leads to cardiovascular complications (77, 87).
3.2 Inflammation/signaling pathways changes
Cadmium disorders the arrangement of myocardial fibers and accelerates the fibrosis on myocardial fibers in diabetic mice. Furthermore, cadmium increases the level of transforming growth factor β1 (TGF-β1) in diabetic mice (9). TGF-β1 may contribute to cardiac dysfunction and induce DCM (88). On the other hand, cadmium exposure raises proinflammatory cytokines such as TNF-α, IL-1, and monocyte chemoattractant protein-1 (MCP-1) in diabetic mice (9). Rise of TNF-α, IL-1, and IL-8, levels in the serum and pancreas may affect inflammation in CVDs (89). MCP-1 plays a causative role in experimental DCM, and MCP-1 deficiency in animal models can attenuate HF (90–92). However, evidence to confirm the role of cadmium in exacerbating the risk of diabetic cardiovascular complications is limited, so more studies are needed to understand its underlying mechanisms.
3.3 Epigenetic effect
Other potential biological mechanisms for the effects of heavy metals, particularly cadmium, on the cardiovascular system include altered regulation of endocrine and endothelial vascular functions, as well as epigenetic pathways (93, 94). Some heavy metals can form covalent bonds with sulfhydryl groups of proteins due to their electron-sharing properties (95, 96). The binding to glutathione leads to depletion of its levels, resulting in an increase in the intracellular concentration of ROS. The consequences of this process include damage to cell membranes, promotion of lipid peroxidation, DNA damage, oxidation of amino acids in proteins leading to changes in their structure and function, and inactivation of enzymes (95). Increased oxidative stress and ROS generation may lead to the formation of gene products that cause cellular damage and loss of activity of DNA repair pathways, thereby contributing to the pathogenesis of diabetic cardiovascular complications (83, 97, 98).
3.4 Disturbing essential homeostasis
Interference with essential metal homeostasis, induction of oxidative stress and apoptosis by multifactorial mechanisms are considered as the most influential forms of cadmium toxicity in humans (94). Chronic exposure to cadmium results in several disorders through disrupt the homeostasis of trace elements such as Zn, Mg, Cu, and iron (26–29). For instance, cadmium displaces Zn in many sulfur-containing proteins, which leads to their dysfunction and the subsequent disruption of numerous biological processes (29). Animal study by (27) has demonstrated that early exposure to cadmium remarkably affected essential metals homeostasis such as Zn, Mg, Cu, and Fe levels in the kidney, liver, and heart in offspring. Cadmium can produce ROS by indirectly displacing an endogenous Fenton metal (e.g., Fe2+) from proteins, thus increasing the amount of free redox-active metals (99). Besides, excessive ROS production can lead to mitochondrial membrane depolarization, macromolecule oxidation, and apoptosis (99). Therefore, cadmium can disturb essential homeostasis by altering essential metals’ homeostasis and inducing and exacerbating different types of cell death such as apoptosis and necrosis. However, in the continuation of current review, we introduce other types of cell death such as ferroptosis and cuproptosis and provide potential pathogenic roles of ferroptosis and cuproptosis during cadmium exposure in cadmium-induced or exacerbated cardiovascular complications in the individuals with diabetes.
4 Ferroptosis
Ferroptosis is a novel non-apoptotic form of regulated cell death (100). Ferroptosis is induced by small molecules that inhibit glutathione (GSH) biosynthesis or glutathione peroxidase 4 (GPX4), and is characterized by iron-dependent accumulation of ROS and consumption of polyunsaturated fatty acids (PUFAs) in the plasma membrane (101). Ferroptosis differs from necroptosis, apoptosis, autophagy, and other types of cell death in terms of morphological characteristics and functions (102). The signaling pathway involved in ferroptosis includes multiple interconnected molecular events that key players in this pathway include iron metabolism, lipid metabolism, and antioxidant systems (30, 103). During the process of ferroptosis, the activity of cystine–glutamate antiporter (system Xc-) is inhibited, namely the amount of cystine entering into cells and glutamate transporting out of cells decreases. The lipid peroxide accumulates (103, 104). The production of lipid peroxides is iron-dependent. Iron accumulation in lipid oxidation facilitates the overproduction of lipid ROS. Then, reduced lipid peroxide scavenging due to inhibition of lipid antioxidants (e.g., GPX4) leads to ferroptosis (103, 105). Besides, other molecules and processes also contribute to the regulation of ferroptosis. The nicotinamide adenine dinucleotide phosphate oxidase (NOX) family of enzymes, such as NOX1 and NOX2, generate ROS that can contribute to lipid peroxidation and ferroptosis (106) as shown in Figure 2.
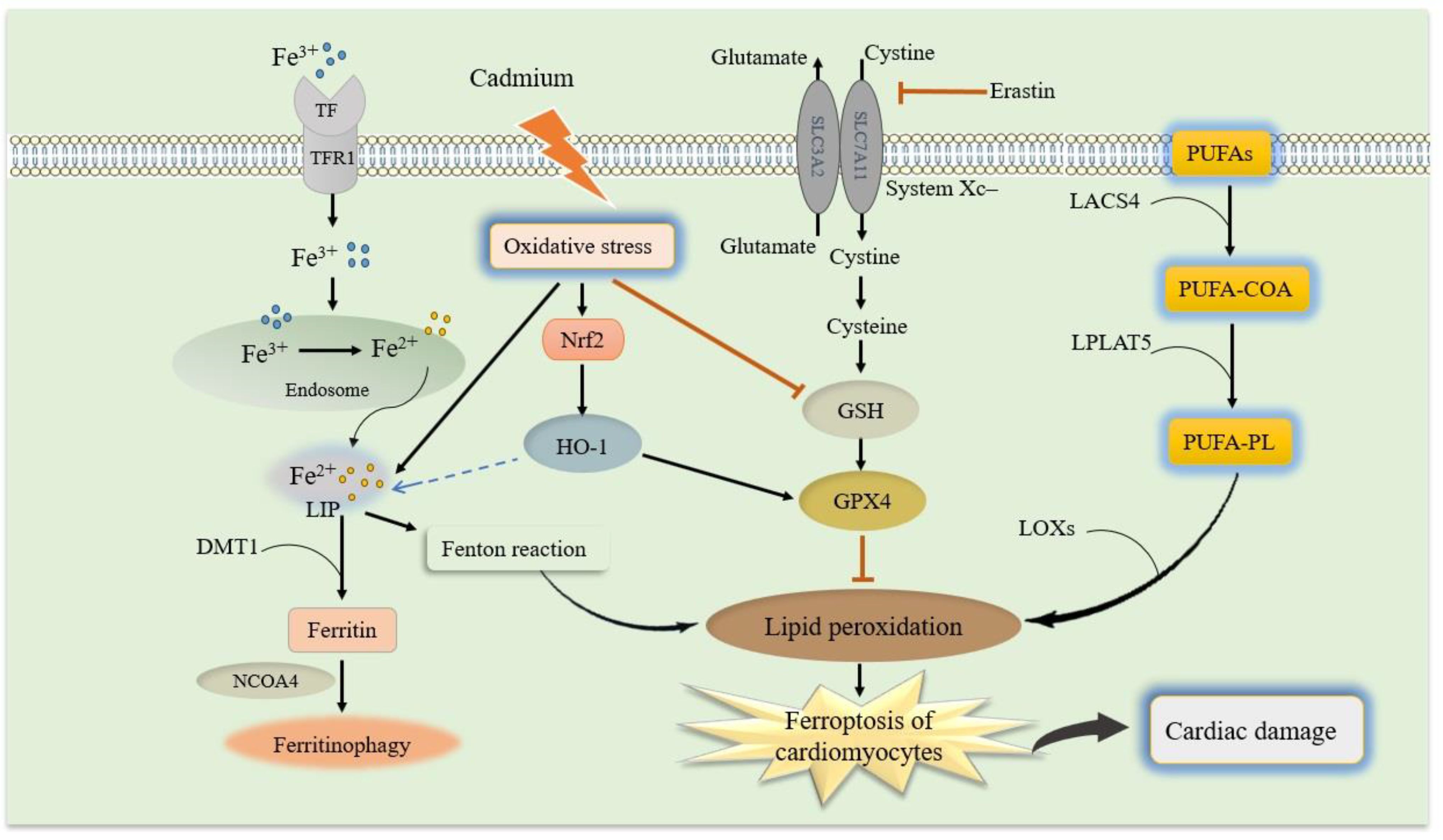
Figure 2. Schematic model of ferroptosis and molecular mechanisms of cadmium-exacerbated ferroptosis in diabetic cardiomyocytes. Molecular mechanisms of ferroptosis and related signaling pathways, which mainly include iron metabolism, GSH and GPX4, and lipid peroxidation. (1) Iron uptake via the transferrin receptor 1 (TfR1) or degradation of ferritin iron stores increases the labile iron pool, thereby cells are susceptible to ferroptosis via the formation of lipid hydroperoxides through the Fenton-like reaction. (2) Cystine-glutamate antiporter system Xc- regulates cysteine and GSH availability. GPX4 utilizes GSH to reduce lipid hydroperoxides, thereby preventing lipid peroxidation chain reactions. (3) The PUFA-PLs that are derived from PUFA-CoA by LPCAT3 and PUFA by ACSL4, respectively, are the main causative of lipid peroxidation. Cadmium exposure can cause oxidative stress in the diabetic cardiomyocytes, leading to iron accumulation, GSH depletion, lipid peroxidation, and finally exacerbate ferroptosis and disrupt the cardiac. In addition, Nrf2 up-regulation due to cadmium-induced oxidative stress can increase HO-1 expression to regulate iron hemostasis. GSH, reduced glutathione; GPX4, glutathione peroxidase 4; system Xc-, cystine–glutamate antiporter; PUFAs, polyunsaturated fatty acids; PUFA-CoA, PUFA coenzyme A; PUFA-PL, phospholipid PUFA; Nrf2, nuclear factor erythroid 2-related factor 2; HO-1, heme oxygenase.
4.1 Ferroptosis in diabetes
Ferroptosis has been implicated to play a pathogenic role in several disorders extending various systems, including liver disease, lung disease, cardiovascular disease, neurodegenerative disorders, cancers, kidney injury, gastrointestinal disease, and pancreas dysfunction (107). Since ferroptosis is regulated by the iron-dependent accumulation of lipid peroxides and subsequent oxidative damage, it has been suggested that ferroptosis may play a role in the pathogenesis of diabetes (108, 109). Excessive iron stores have been demonstrated to be associated with the development of diabetes, and ferritin levels are increased in T2D (110). Elevated ferritin in diabetes patients led to hyperglycemia (111). Besides, other iron metabolism indicators such as hepcidin could also be related to diabetic pathogenesis (110). Substantial researches have called that iron deficiency and excess may affect glucose regulation (112), and high glucose can cause a rise to iron overload (113), which is known to trigger ferroptosis. Meanwhile, under iron overload condition, oxidative stress can induce insulin resistance (114).
Pancreatic islet β-cells are susceptible to ferroptosis. Some studies indicate that pancreatic β-cells are susceptible to oxidative stress because they express a low level of antioxidant enzymes such as superoxide dismutase, GSH peroxidase, and catalase (115); therefore, ROS is prone to accumulate in the pancreatic islet β-cells. Accumulation of ROS in the pancreatic islet β-cells can trigger many forms of deterioration, including ferroptosis (105). Pancreatic islet β-cells function is closely associated with diabetes, and insufficient insulin secretion caused by pancreatic β-cells failure contributes to hyperglycemia. Hyperglycemia may inflict solute carrier family 7 member 11 and solute carrier family 3 member 2 impairment, leading to system Xc– dysfunction in the molecular mechanism of ferroptosis (116). Increased cellular iron alters the expression of genes involved in β-cell function and causes pancreatic β-cell dysfunction (117). Nutrient-deprivation autophagy factor-1 (NAF-1), is a class of iron-sulfur proteins that regulates mitochondrial iron levels (118). Interestingly, suppressed expression of NAF-1 in INS-1E pancreatic β-cell results in the appearance of ferroptosis-like features characterized by enhanced lipid peroxidation, and decreased expression of GPX4 (119). Krummel et al. have implicated that β-cells may have a high sensitivity to ferroptosis and confirmed that GPX4 distributes throughout the β-cell to a large extent, eliminating GPX4 induces ferroptosis (120). Altogether, ferroptosis features including upregulation in transferrin expression, elevated lipid peroxidation, and reduction in GPX4 have been observed in INS-1 E pancreatic β-cell, following the suppressed expression of NAF-1, which is a protein that regulates mitochondrial iron levels (107). A study showed remarkably reduced glucose-stimulated insulin secretion (GSIS) capacity in human islet β-cells when they were treated with the ferroptosis inducer erastin in vitro (121). Conversely, pretreatment with a ferroptosis inhibitor, Fer-1, rescued the damage to GSIS (121). On the other hand, inhibition of ferroptosis using specific inhibitor Fer-1 as the first ferroptosis inhibitor effectively improves the viability and functionality of pancreatic islets (122). These data indicate that ferroptosis as a new mechanism of cell death, plays an important role in maintaining homeostasis in the pancreatic islet cells, and may help to develop novel treatments for diabetes.
4.2 Ferroptosis in diabetic cardiovascular complications
Diabetes onset leads to a wide spectrum of macrovascular and microvascular disorders that affect the majority of the body. Diabetes is a high-risk factor to contribute CVDs, including atherosclerosis, myocardial ischemia/reperfusion injury in diabetes, DCM, and HF (123). Recent evidence illustrates that the development of diabetes complications particularly diabetic cardiovascular complications is closely related to ferroptosis (107). DCM is a group of myocardial diseases in patients with diabetes, which is distinguished by early onset of ventricular diastolic dysfunction and late onset of ventricular systolic dysfunction, cardiac hypertrophy, and fibrosis (124). The pathogenesis of DCM is primarily due to impaired cardiac energy metabolism and mitochondrial abnormalities, which consequently lead to ROS-mediated oxidative stress, and cardiac inflammation, all of which are associated with the same pathogenic risk factors for ferroptosis (108). Zang et al. demonstrated a potential increase in ferroptosis in the hearts of mice with T1D through increased iron deposition and cardiac 4-hydroxy-2-nonenal, a biomarker of lipid peroxidation for ferroptosis (125). Meanwhile, increased expression of cyclooxygenase 1 and fatty acid coenzyme A ligase proteins in the ferroptosis signaling pathway was observed in the hearts of rabbits with T1D, along with decreased GPX4 protein levels (126). Lipid peroxidation and reduced lipid peroxide scavenging due to inhibition of lipid antioxidants (e.g., GPX4) are pathogenic risk factors for ferroptosis.
Nuclear factor erythroid 2-related factor 2 (Nrf2) is a transcription factor controlling the expression of many ferroptosis-related genes such as GPX4 (127). Nrf2 and its target genes have an inhibitory effect on ferroptosis and act as a major regulator of cell redox state and detoxification to prevent high glucose-induced oxidative damage in DCM (128). Recent studies have shown that activation of Nrf2 reduced oxidative damage induced by high glucose in cultured cardiomyocytes and prevented the development of DCM in animal models (129, 130). Therefore, inactivating Nrf2 could mediate antioxidant defense and impair iron metabolism (125). Nevertheless, an interesting study demonstrated that Nrf2 has destructive effects on the heart by promoting ferroptosis during myocardial autophagy deficiency (125). However, targeting Nrf2 and its related targets remains a viable approach to prevent or treat DCM by regulating ferroptosis, which undoubtedly deserve further studies.
Myocardial ischemia is more likely to occur in those with diabetes, leading to myocardial vulnerability (131). Under diabetic conditions, oxidative stress and programmed cell death turn down AMP-activated protein kinase (AMPK) expression, leading to a higher level of NOX, whose main function is to supply ROS (132). Endoplasmic reticulum stress (ERS) is a cellular response to endoplasmic reticulum (ER) dysfunction and can be triggered by ROS, which is produced by the interaction between iron ions and NOX during ferroptosis (123). Erastin could aggravate ERS and cell injury by stimulating ferroptosis in high-glucose hypoxia reoxygenation group cell model. Meanwhile, inhibition of ERS could alleviate ferroptosis and cell injury. Also demonstrated is that suppressing ferroptosis may alleviate ERS, which was triggered by transcription factor 4- CCAAT-enhancer-binding protein homologous protein pathway and delay the progression of diabetic myocardial ischemia/reperfusion injury (133).
4.3 Cadmium and ferroptosis in diabetic cardiovascular complications
Several studies have shown that exposure to environmental pollutants such as arsenic and cadmium is closely associated with ferroptosis (134, 135). Cadmium exposure causes pancreatic β-cells dysfunction by disrupting lipid metabolism and inducing lipid accumulation in pancreatic β-cells (136). Cadmium can also cause abnormal iron metabolism and interfere with iron homeostasis (Figure 2). Hong et al. suggested that cadmium exposure can lead to iron accumulation, GSH depletion, GPX4 reduction, lipid peroxidation, mitochondrial membrane potential loss, and ultrastructural damage at the mitochondrial level by activating the Ager/Pkc/p65 pathway. Therefore, cadmium can induce ferroptosis of pancreatic β-cells by activating the GPX4/Ager/p65 pathway (137), exacerbating diabetes and its complications. On the other hand, high glucose levels in individuals with diabetes can induce ferroptosis in cardiomyocytes as a complication of diabetes (126).
Cardiomyocytes are rich in mitochondria and serve as the main source of ROS in the heart (138). Exposure to stressful conditions like heavy metals causes activation of lipid peroxidation in the heart (139, 140). Cadmium toxicity also inhibits mitochondrial function in cardiomyocytes by inactivating respiratory chain enzymes which results in the induction of ferroptosis (141). Moreover, cadmium interacts with endogenous and exogenous antioxidants and compromises the redox potential of the cells, which causes a series of complications such as lipid peroxidation of membranous structures, DNA damage–associated genotoxicity, and proteotoxicity associated ER-stress (142, 143). Therefore, this cardiac pathogen induces common conditions such as iron overload in cardiomyocytes, mitochondrial dysfunction, oxidative stress, and lipid peroxidation which lead to ferroptosis and CVDs (139).
Nrf2/heme oxygenase (HO-1) signaling pathway, as a critical signaling pathway in the oxidative stress response, is involved in anti-inflammatory, antioxidant, and other processes. Recent studies suggest that ferroptosis plays an indispensable role in DCM through the Nrf2/HO-1 pathway, and the activation and upregulation of Nrf2 can increase GPX4 and HO-1 expression to alleviate DCM (144). Another major feature of ferroptosis is an increase in intracellular free iron due to a disturbance in iron metabolism. Nrf2 regulates many ferroptosis-related proteins, such as ferritin, transferrin receptor (TfR), ATP-binding cassette subfamily B member 6, ferrochelatase. Nrf2 has also been shown to regulate the metabolism of heme to produce iron by regulating HO-1 and then regulating the concentration of iron in cells to reduce ferroptosis (145). The therapeutic potential of Nrf2 activation was well documented against pathological conditions associated with oxidative stress (146, 147). Additionally, when a cell was exposed to oxidative stress, the expression of the HO-1 mRNA and protein upregulated (145). Oxidative stress is one of the most important pathways of cadmium toxicity (148). Notably, cadmium exposure upregulates Nrf2 mRNA in H9c2 cardiomyocytes, which activate Nrf2 protein (149). Nrf-2-activation-mediated phase 2 enzymes were shown to be protective against toxic effects of cadmium in various cell lines and in mice (149). Cadmium induces cardiac oxidative stress and activates Nrf2 signaling pathway and activation of Nrf2 signaling pathway attenuates cadmium-induced cardiac oxidative stress (150) (Figure 2). Besides, activation of AMPK and Nrf2 plays an important role in preventing cardiac ferroptosis and consequently preserving cardiac function in T2D mice (151). Even though Nrf2 activation can be a novel ideal therapeutic strategy for alleviating cardiotoxicity and cardiac ferroptosis caused due to cadmium exposure in diabetic patients, further study is needed to confirm this.
5 Cuproptosis
The latest research in the field of cellular biology and disease research has discovered a new form of cell death, cuproptosis, which differs from pyroptosis, apoptosis, necroptosis, and ferroptosis (17). This process involves the activation of specific signaling cascades and is associated with changes in mitochondrial enzymes. In particular, the regulation of cuproptosis is based on copper overload, is dependent on mitochondrial respiration and adenosine triphosphate (ATP) production and is closely related to the tricarboxylic acid (TCA) cycle. Indeed, cuproptosis mainly occurs in mitochondria where its membrane oxidative damage can cause mitochondrial dysfunction and TCA cycle-related enzyme dysfunction, leading to cuproptosis (152). First, Elesclomol-Cu2+ transports Cu2+ into and out of cells and copper ion channels solute carrier family 31 member 1 (SLC31A1) and ATP7B also regulate the accumulation of copper ions by mediating the entry and exocytosis of copper ions, respectively (Cai, et al., 2024). Intracellular Cu targets and binds to lipoylated mitochondrial enzymes in the TCA cycle such as dihydrolipoamide S-acetyltransferase (DLAT), inducing the aggregation of these proteins (18). The aggregation of these Cu-bound lipoylated mitochondrial proteins and the subsequent reduction of Fe–S (iron–sulfur) clusters lead to proteotoxic stress and ultimately cell death. Ferredoxin 1 (FDX1) and lipoic acid synthetase (LIAS) are the critical regulators of protein lipoylation, facilitating the aggregation of mitochondrial proteins and loss of Fe–S clusters (17, 18). Interestingly, genetic knockout of either FDX1 or LIAS leads to an accumulation of pyruvate and α-ketoglutarate, reducing protein lipoylation and inhibiting Cu-induced cell death (18), as shown in Figure 3. However, the mechanisms that underlie Cu-induced cell death are poorly understood. Therefore, further research is needed to determine the specific molecular details and potential therapeutic interventions for cuproptosis (32, 153).
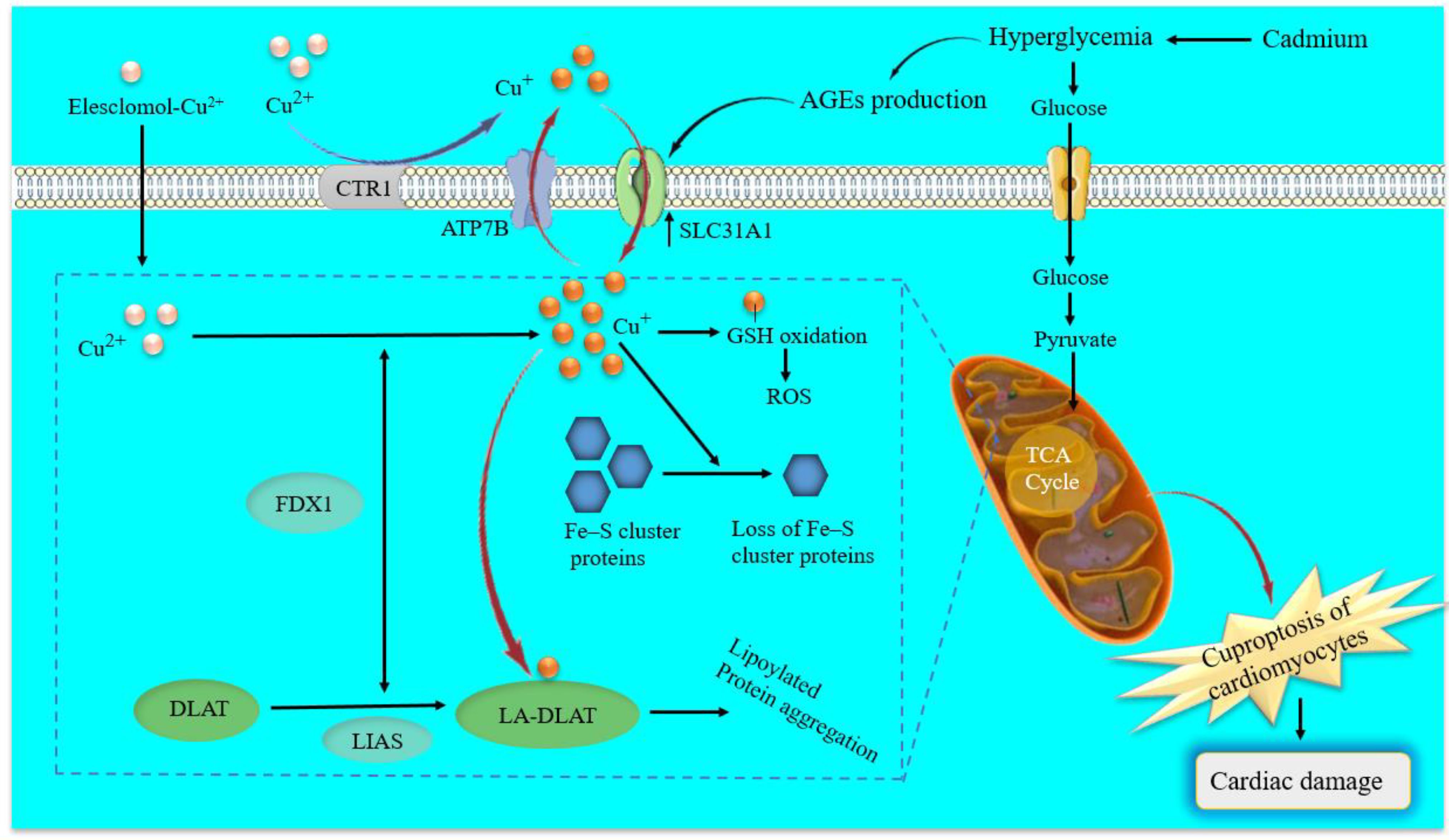
Figure 3. Schematic model of cuproptosis and molecular mechanisms of cadmium-induced cuproptosis in diabetic cardiomyocytes. Elesclomol binds extracellular copper (Cu2+) and transports it to intracellular compartments. Besides, copper ion channels SLC31A1 and ATP7B regulate the accumulation of Cu+ by mediating the entry and exocytosis of Cu+, respectively. Afterward, regulate cuproptosis sensitivity by affecting intracellular Cu+ levels. On the one hand, Cu+ binds to lipoylated mitochondrial enzymes in the TCA cycle such as DLAT, inducing the aggregation of these proteins. On the other hand, FDX1 as an upstream regulator of protein lipoylation reduces Cu2+ to Cu+, and with LIAS facilitates the aggregation of mitochondrial proteins and loss of Fe–S cluster proteins. Cadmium-induced hyperglycemia can enhance circulating AGEs. Excessive AGEs in diabetes promote the upregulation of copper importer SLC31A1 through ATF3/SPI1, thereby increasing intracellular Cu+ accumulation in cardiomyocytes and disturbing Cu+ homeostasis, leading to a decline of Fe–S cluster protein and ROS accumulation in cardiomyocytes mitochondria. ATP7B, ATPase copper transporting beta; SLC31A1, solute carrier family 31 member 1; Fe–S, iron-sulfur; FDX1, ferredoxin 1; TCA, tricarboxylic acid; DLAT, dihydrolipoamide S-acetyltransferase; LA-DLAT, lipoylated DLAT; LIAS, lipoyl synthase; AGEs, Advanced Glycosylation End Products.
5.1 Cuproptosis in diabetes and diabetic cardiovascular complications
Copper is an essential metal micronutrient, and abnormal copper metabolism is associated with multiple diseases, including Menkes disease, cardiovascular disease, Alzheimer’s disease, inflammation disorders, and cancers (32, 154–156). A high oral intake of Zn would affect copper absorption and can cause a decrease in copper absorption. Therefore, the Cu/Zn ratio is a key index to predict the risk of diabetes (157). Moreover, clinical studies have shown that plasma copper concentrations are positively associated with impaired glucose regulation in T1D and T2D, as well as all-cause mortality in patients with diabetes (158–160). Copper may induce diabetes via impaired glucose regulation and cuproptosis-induced oxidative stress and disrupted mitochondrial function.
Cuproptosis is caused by oxidative stress and impaired mitochondrial function, while diabetes alters the metabolism of cardiomyocytes, which also leads to oxidative stress that can result in increased copper levels in the cells (22). High levels of copper have been positively correlated with ROS generation (161). Copper promotes ROS generation, insulin resistance, and diabetes occurrence, leading to the development of atherosclerosis (162). On the one hand, accumulation of catalytically active Cu2+ in the cardiac extracellular myocardial induces copper toxicity, which is proposed as an important catalyst of cardiovascular damage in diabetes (163, 164). Copper toxicity leads to protein oxidation, GSH depletion, lipid damage, and redox imbalance (165, 166), which could cause impairment of heart function and exacerbate CVDs such as ischemic heart disease, arrhythmia, and heart hypertrophy. On the other hand, cardiomyocytes have the highest number of mitochondria. Therefore, it is reasonable to assume the potential induction of cuproptosis (108). This elevated copper accumulation can initiate a cascade of events that ultimately trigger cuproptosis (17) and its diabetic complications such as CVDs.
AGEs are formed by irreversible nonenzymatic reactions between reducing sugars, such as glucose, and amino groups in proteins, lipids, and nucleic acids during diabetes mellitus and could promote cardiomyocyte death through calcium overload, oxidative stress or excessive autophagy (167–169). Moreover, AGEs play an important role in the pathogenesis of diabetic complications, especially cardiomyopathy (170). AGEs promote the upregulation of high-affinity copper transporter-1 by upregulating the transcription factors activating transcription factor 3 (ATF3)/transcription factor PU.1 (SPI1)/SLC31A1, thus increasing intracellular copper accumulation and inducing cuproptosis in DCM (171). In addition, increased cuproptosis in the heart of streptozotocin-induced T1D and db/db T2D mice has been reported (171). Therefore, cuproptosis is closely related to the pathogenesis of DCM, which is expected to become another unique angle to explore the pathogenesis of diabetic cardiovascular complications, including DCM.
Besides, previous studies have suggested that changes in serum copper concentration and their effects may involve regulation at the genomic level (172, 173). A bioinformatics study recently showed that four key genes HSDL2, BCO2, CORIN, and SNORA80E are involved in the development of cardiomyocyte cuproptosis in patients with diabetes mellitus-associated HF through effects on the immune system (174). Previous studies also showed that hyperactivation of the PI3K/Akt/FoxO1/LOXL2 pathway promotes the development of myocardial fibrosis in the cardiomyocytes of patients with diabetes (175). This finding may enable us to quickly identify potentially relevant pathogenic factors. However, further cell or animal experiments are required to verify.
5.2 Cadmium and cuproptosis in diabetic cardiovascular complications
As cuproptosis is a novel kind of programmed cell death, recently reported by Tsvetkov et al. in March 2022 and its underlying mechanism and potential role of different environmental toxicants, including heavy metals such as inorganic arsenic, cadmium, and lead on cuproptosis are still not well known. Based on the results discussed in this review, heavy metals, especially cadmium, induce oxidative stress, pancreatic β-cell dysfunction, hyperglycemia, and disrupt insulin secretion, which induces and exacerbates T1D and T2D. Hyperglycemia induces oxidative stress, which causes mitochondrial dysfunction and AGE production (169). AGEs play an important role in the pathogenesis of diabetic complications, especially cardiomyopathy. Excessive AGEs in diabetes promote the upregulation of copper importer SLC31A1 through ATF3/SPI1, thereby increasing intracellular copper accumulation in cardiomyocytes and disturbing copper homeostasis. This encourages the decline of Fe–S cluster protein (FDX1, LIAS) and decreases lipoylation of DLAT-aggravated mitochondrial dysfunction in cardiomyocytes resulting in myocardial dysfunction (171). Moreover, excess copper-induced oxidative stress can lead to glutathione oxidation and decreased glutathione conjugation, ultimately resulting in cuproptosis and cardiotoxicity (176, 177) (Figure 3).
Although the direct effects of cadmium on hyperglycemia and the initiation of diabetes and the indirect effects of cadmium to exacerbate diabetic cardiovascular complications via ROS and AGE production have been demonstrated, further studies are undoubtedly needed to understand the interaction between cadmium and copper homeostasis in cardiac cuproptosis. Experimental evidence also has shown that inhibiting cuproptosis can attenuate mitochondrial dysfunction and cardiac damage in diabetic animal models. For instance, copper chelators like Trientine (triethylenetetramine, TETA) and Tetrathiomolybdate (TTM) can inhibit copper overload (178). However, further research is needed to fully understand the precise mechanisms linking cardiac cuproptosis in diabetic patients to explore potential therapeutic interventions targeting this pathway.
6 Conclusions and perspectives
Diabetes is characterized by high blood glucose levels that result from insulin deficiency, in the context of β- cell dysfunction, insulin resistance, or both (1). Oxidative stress and inflammation play a key role in the pathogenesis and progression of diabetes-induced CVD, where the enhanced expression of inflammatory proteins or cytokines such as oxidative stress-related proteins are shown to serve as biomarkers for the occurrence of cardiovascular complications (59). Oxidative stress and apoptosis by multifactorial mechanisms are considered as the most influential forms of cadmium toxicity in humans (94). The potential roles of cadmium in exacerbating cardiovascular complications in individuals with diabetes are described in the current review (Figure 1).
Cadmium exposure has been linked to cardiovascular complications in individuals with diabetes, potentially inducing ferroptosis and cuproptosis. The studies discussed above have provided preliminary, important evidence indicating the potential roles of ferroptosis and cuproptosis in cadmium-induced or exacerbated CVDs and diabetic cardiovascular complications. These processes involve disruption of trace element homeostasis, oxidative stress, GSH depletion, GPX4 reduction, and inflammation. Besides, cadmium induces common conditions such as iron overload in cardiomyocytes, mitochondrial dysfunction, oxidative stress, and lipid peroxidation, which lead to ferroptosis and cardiovascular complications in the individuals with diabetes (139). Furthermore, the present results are indicated that iron dysregulation by cadmium contributed to cadmium-induced ferroptosis, and induced or exacerbated cardiovascular complications in individuals with diabetes. Collectively, these results indicate that ferroptosis is involved in cadmium-induced cardiovascular complications in individuals with diabetes and is regulated by the Nrf2, HO-1, and GPX4 axis as well as promoting mitochondrial ROS generation (Figure 2).
Cuproptosis is the most recently described form of cell death triggered by copper accumulation and results in oxidative stress and mitochondrial dysfunction (22). This elevated copper accumulation can initiate a cascade of events that trigger cuproptosis (17) and its diabetic complications such as CVD. AGEs are formed by irreversible nonenzymatic reactions between reducing sugars, such as glucose, and amino groups in proteins, lipids, and nucleic acids during diabetes mellitus and could promote cardiomyocyte death through calcium overload, oxidative stress, or excessive autophagy (169). Cadmium-exacerbated hyperglycemia in diabetes patients can enhance AGEs, which excessive AGEs may promote the upregulation of copper importer SLC31A1 through ATF3/SPI1, thereby increasing intracellular copper accumulation in cardiomyocytes and induce myocardial cuproptosis (171). Overall, cuproptosis plays an essential role in cadmium-induced or exacerbated cardiovascular complications through copper importer SLC31A1 and mitochondrial accumulation of ROS in individuals with diabetes (Figure 3).
Little is known about the epigenetic effects of cadmium-exacerbates cardiovascular complications in the individuals with diabetes. Further research is needed to investigate the epigenetic effects of cadmium to exacerbate diabetic cardiovascular complications. Furthermore, the studies discussed above have provided preliminary, important evidence indicating the potential roles of ferroptosis and cuproptosis in cadmium-induced or exacerbated cardiovascular complications in individuals with diabetes. Nevertheless, the underlying mechanisms of ferroptosis and cuproptosis in cadmium-induced or exacerbated cardiovascular complications such as CVDs, cardiomyopathy, and coronary heart disease are still unclear, and further studies are needed to elucidate this issue. Activation of AMPK and Nrf2 plays an important role in preventing cardiac ferroptosis and preserving cardiac function in T2D mice (151). Even though Nrf2 activation can be a novel ideal therapeutic strategy for alleviating cardiotoxicity and cardiac ferroptosis caused due to cadmium exposure in diabetic patients, further study is needed to confirm this.
The direct effects of cadmium on hyperglycemia and the initiation of diabetes and the indirect effects of cadmium to exacerbate diabetic cardiovascular complications via ROS and AGE production have been demonstrated, further studies are undoubtedly needed to understand the interaction between cadmium and copper homeostasis in cardiac cuproptosis. Besides, experimental evidence has shown that inhibiting cuproptosis can attenuate mitochondrial dysfunction and cardiac damage in diabetic animal models. Copper chelators like TETA and TTM can inhibit copper overload (178). Further research is needed to fully understand the precise mechanisms linking cardiac cuproptosis in diabetic patients to explore potential therapeutic interventions targeting this pathway.
Author contributions
LC: Conceptualization, Supervision, Validation, Writing – original draft, Writing – review & editing. YT: Writing – review & editing. KW: Writing – review & editing. SW: Writing – review & editing. SS: Conceptualization, Methodology, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors of this review are supported in part by grants from the National Institute of Environmental Health Sciences (P30 ES030283 to KW, LC).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cole JB, Florez JC. Genetics of diabetes mellitus and diabetes complications. Nat Rev Nephrol. (2020) 16:377–90. doi: 10.1038/s41581-020-0278-5
2. Cho NH, Shaw JE, Karuranga S, Huang Y, da Rocha Fernandes JD, Ohlrogge AW, et al. IDF Diabetes Atlas: Global estimates of diabetes prevalence for 2017 and projections for 2045. Diabetes Res Clin Pract. (2018) 138:271–81. doi: 10.1016/j.diabres.2018.02.023
3. Morrish N, Wang S-L, Stevens L, Fuller J, Keen H, Group WMS. Mortality and causes of death in the WHO Multinational Study of Vascular Disease in Diabetes. Diabetologia. (2001) 44:S14–21. doi: 10.1007/PL00002934
4. Kannel WB, McGee DL. Diabetes and cardiovascular disease: the Framingham study. Jama. (1979) 241:2035–8. doi: 10.1001/jama.1979.03290450033020
5. Grundy SM, Benjamin IJ, Burke GL, Chait A, Eckel RH, Howard BV, et al. Diabetes and cardiovascular disease: a statement for healthcare professionals from the American Heart Association. Circulation. (1999) 100:1134–46. doi: 10.1161/01.CIR.100.10.1134
6. Singer DE, Moulton AW, Nathan DM. Diabetic myocardial infarction: interaction of diabetes with other preinfarction risk factors. Diabetes. (1989) 38:350–7. doi: 10.2337/diab.38.3.350
7. De Ferranti SD, De Boer IH, Fonseca V, Fox CS, Golden SH, Lavie CJ, et al. Type 1 diabetes mellitus and cardiovascular disease: a scientific statement from the American Heart Association and American Diabetes Association. Circulation. (2014) 130:1110–30. doi: 10.1161/CIR.0000000000000034
8. Turnbull F, Abraira C, Anderson R, Byington R, Chalmers J, Duckworth W, et al. Intensive glucose control and macrovascular outcomes in type 2 diabetes. Diabetologia. (2009) 52:2288–98. doi: 10.1007/s00125-009-1470-0
9. Liu X, Wang S, Li M, Li J, Sheng Z. Chronic cadmium exposure aggravates the cardiac dysfunction in type 2 diabetic mice by promoting inflammation and fibrosis. (2021). doi: 10.21203/rs.3.rs-639901/v1
10. Vallée A, Gabet A, Grave C, Blacher J, Olié V. Associations between urinary cadmium levels, blood pressure, and hypertension: the ESTEBAN survey. Environ Sci pollut Res. (2020) 27:10748–56. doi: 10.1007/s11356-019-07249-6
11. Everett CJ, Frithsen IL. Association of urinary cadmium and myocardial infarction. Environ Res. (2008) 106:284–6. doi: 10.1016/j.envres.2007.10.009
12. Turdi S, Sun W, Tan Y, Yang X, Cai L, Ren J. Inhibition of DNA methylation attenuates low-dose cadmium-induced cardiac contractile and intracellular Ca2+ anomalies. Clin Exp Pharmacol Physiol. (2013) 40:706–12. doi: 10.1111/cep.2013.40.issue-10
13. Li Y, Zhang Y, Wang W, Wu Y. Association of urinary cadmium with risk of diabetes: a meta-analysis. Environ Sci pollut Res. (2017) 24:10083–90. doi: 10.1007/s11356-017-8610-8
14. Han JC, Park SY, Hah BG, Choi GH, Kim YK, Kwon TH, et al. Cadmium induces impaired glucose tolerance in rat by down-regulating GLUT4 expression in adipocytes. Arch Biochem Biophys. (2003) 413:213–20. doi: 10.1016/S0003-9861(03)00120-6
15. Edwards J, Ackerman C. A review of diabetes mellitus and exposure to the environmental toxicant cadmium with an emphasis on likely mechanisms of action. Curr Diabetes Rev. (2016) 12:252–8. doi: 10.2174/1573399811666150812142922
16. Qin Y, Qiao Y, Wang D, Tang C, Yan G. Ferritinophagy and ferroptosis in cardiovascular disease: Mechanisms and potential applications. Biomed Pharmacother. (2021) 141:111872. doi: 10.1016/j.biopha.2021.111872
17. Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. (2022) 375:1254–61. doi: 10.1126/science.abf0529
18. Chen L, Min J, Wang F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct Target Ther. (2022) 7:378. doi: 10.1038/s41392-022-01229-y
19. Xie J, Yang Y, Gao Y, He J. Cuproptosis: mechanisms and links with cancers. Mol Cancer. (2023) 22:46. doi: 10.1186/s12943-023-01732-y
20. Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radical Biol Med. (2019) 131:356–69. doi: 10.1016/j.freeradbiomed.2018.12.011
21. Wang Y, Peng X, Zhang M, Jia Y, Yu B, Tian J. Revisiting tumors and the cardiovascular system: mechanistic intersections and divergences in ferroptosis. Oxid Med Cell Longev. (2020) 2020:738143. doi: 10.1155/2020/9738143
22. Cui X, Wang Y, Liu H, Shi M, Wang J, Wang Y. The molecular mechanisms of defective copper metabolism in diabetic cardiomyopathy. Oxid Med Cell Longev. (2022) 2022:5418376. doi: 10.1155/2022/5418376
23. Dawes K, Philibert W, Darbro B, Simons RL, Philibert R. Additive and interactive genetically contextual effects of HbA1c on cg19693031 methylation in type 2 diabetes. Genes. (2022) 13:683. doi: 10.3390/genes13040683
24. Zhang Y, Gong X, Li R, Gao W, Hu D, Yi X, et al. Exposure to cadmium and lead is associated with diabetic kidney disease in diabetic patients. Environ Health. (2024) 23:1. doi: 10.1186/s12940-023-01045-z
25. Samarghandian S, Borji A, Farkhondeh T, Majid A-S. Effect of cadmium on glucose, lipid profile and oxidative stress in streptozotocin-induced diabetic and non-diabetic rats. Toxicol Int. (2017) 24:9–16. doi: 10.22506/ti/2017/v24/i1/149027
26. Xiong L, Zhou B, Young JL, Wintergerst K, Cai L. Exposure to low-dose cadmium induces testicular ferroptosis. Ecotoxicol Environ Saf. (2022) 234:113373. doi: 10.1016/j.ecoenv.2022.113373
27. Young JL, Yan X, Xu J, Yin X, Zhang X, Arteel GE, et al. Cadmium and high-fat diet disrupt renal, cardiac and hepatic essential metals. Sci Rep. (2019) 9:14675. doi: 10.1038/s41598-019-50771-3
28. Miao X, Sun W, Fu Y, Miao L, Cai L. Zinc homeostasis in the metabolic syndrome and diabetes. Front Med. (2013) 7:31–52. doi: 10.1007/s11684-013-0251-9
29. Arnaud J, De Lorgeril M, Akbaraly T, Salen P, Arnout J, Cappuccio F, et al. Gender differences in copper, zinc and selenium status in diabetic-free metabolic syndrome European population–The IMMIDIET study. Nutrition Metab Cardiovasc Dis. (2012) 22:517–24. doi: 10.1016/j.numecd.2010.09.005
30. Fang X, Ardehali H, Min J, Wang F. The molecular and metabolic landscape of iron and ferroptosis in cardiovascular disease. Nat Rev Cardiol. (2023) 20:7–23. doi: 10.1038/s41569-022-00735-4
31. Li X, Ling J, Hu Q, Fang C, Mei K, Wu Y, et al. Association of serum copper (Cu) with cardiovascular mortality and all-cause mortality in a general population: a prospective cohort study. BMC Public Health. (2023) 23:2138. doi: 10.1186/s12889-023-17018-3
32. Chen X, Cai Q, Liang R, Zhang D, Liu X, Zhang M, et al. Copper homeostasis and copper-induced cell death in the pathogenesis of cardiovascular disease and therapeutic strategies. Cell Death Dis. (2023) 14:105. doi: 10.1038/s41419-023-05639-w
33. Kataranovski M, Janković S, Kataranovski D, Stošić J, Bogojević D. Gender differences in acute cadmium-induced systemi inflammation in rats. Biomed Environ Sci. (2009) 22:1–7. doi: 10.1016/S0895-3988(09)60014-3
34. Bernhoft RA. Cadmium toxicity and treatment. Sci World J. (2013) 2013:394652. doi: 10.1155/tswj.v2013.1
35. Berglund M, Larsson K, Grandér M, Casteleyn L, Kolossa-Gehring M, Schwedler G, et al. Exposure determinants of cadmium in European mothers and their children. Environ Res. (2015) 141:69–76. doi: 10.1016/j.envres.2014.09.042
36. Järup L, Åkesson A. Current status of cadmium as an environmental health problem. Toxicol Appl Pharmacol. (2009) 238:201–8. doi: 10.1016/j.taap.2009.04.020
37. Saedi S, Shirazi MRJ, Niazi A, Tahmasebi A, Ebrahimie E. Prepubertal exposure to high dose of cadmium induces hypothalamic injury through transcriptome profiling alteration and neuronal degeneration in female rats. Chemico-biological Interact. (2021) 337:109379. doi: 10.1016/j.cbi.2021.109379
38. Yan L-J, Allen DC. Cadmium-induced kidney injury: Oxidative damage as a unifying mechanism. Biomolecules. (2021) 11:1575. doi: 10.3390/biom11111575
39. Saedi S, Namavar MR, Shirazi MRJ, Rezazadeh FM, Tsutsui K. Exposure to cadmium alters the population of glial cell types and disrupts the regulatory mechanisms of the HPG axis in prepubertal female rats. Neurotoxicity Res. (2022) 40:1029–42. doi: 10.1007/s12640-022-00516-4
40. Ebrahimi M, Khalili N, Razi S, Keshavarz-Fathi M, Khalili N, Rezaei N. Effects of lead and cadmium on the immune system and cancer progression. J Environ Health Sci Eng. (2020) 18:335–43. doi: 10.1007/s40201-020-00455-2
41. Li M, Wang S, Liu X, Sheng Z, Li B, Li J, et al. Cadmium exposure decreases fasting blood glucose levels and exacerbates type-2 diabetes in a mouse model. Endocrine. (2022) 76:53–61. doi: 10.1007/s12020-021-02974-w
42. Ma Y, Ran D, Shi X, Zhao H, Liu Z. Cadmium toxicity: a role in bone cell function and teeth development. Sci Total Environ. (2021) 769:144646. doi: 10.1016/j.scitotenv.2020.144646
43. Tavakoli Pirzaman A, Ebrahimi P, Niknezhad S, Vahidi T, Hosseinzadeh D, Akrami S, et al. Toxic mechanisms of cadmium and exposure as a risk factor for oral and gastrointestinal carcinomas. Hum Exp Toxicol. (2023) 42:09603271231210262. doi: 10.1177/09603271231210262
44. Banday MZ, Sameer AS, Nissar S. Pathophysiology of diabetes: An overview. Avicenna J Med. (2020) 10:174–88. doi: 10.4103/ajm.ajm_53_20
45. Ozdemir M, Buyukbese M, Cetinkaya A, Ozdemir G. Risk factors for ocular surface disorders in patients with diabetes mellitus. Diabetes Res Clin Pract. (2003) 59:195–9. doi: 10.1016/S0168-8227(02)00244-9
47. Frayling TM. Genome–wide association studies provide new insights into type 2 diabetes aetiology. Nat Rev Genet. (2007) 8:657–62. doi: 10.1038/nrg2178
48. Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, et al. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. (2008) 40:638–45. doi: 10.1038/ng.120
49. Haffner SM, Lehto S, Rönnemaa T, Pyörälä K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med. (1998) 339:229–34. doi: 10.1056/NEJM199807233390404
50. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. (2001) 414:813–20. doi: 10.1038/414813a
51. Kannel WB, Hjortland M, Castelli WP. Role of diabetes in congestive heart failure: the Framingham study. Am J Cardiol. (1974) 34:29–34. doi: 10.1016/0002-9149(74)90089-7
52. Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, et al. Heart disease and stroke statistics—2022 update: a report from the American Heart Association. Circulation. (2022) 145:e153–639. doi: 10.1161/CIR.0000000000001052
53. Tuomilehto J, Borch-Johnsen K, Molarius A, Forsen T, Rastenyte D, Sarti C, et al. Incidence of cardiovascular disease in Type 1 (insulin-dependent) diabetic subjects with and without diabetic nephropathy in Finland. Diabetologia. (1998) 41:784–90. doi: 10.1007/s001250050988
54. Soedamah-Muthu SS, Fuller JH, Mulnier HE, Raleigh VS, Lawrenson RA, Colhoun HM. High risk of cardiovascular disease in patients with type 1 diabetes in the UK: a cohort study using the general practice research database. Diabetes Care. (2006) 29:798–804. doi: 10.2337/diacare.29.04.06.dc05-1433
55. Cai L, Kang YJ. Cell death and diabetic cardiomyopathy. Cardiovasc Toxicol. (2003) 3:219–28. doi: 10.1385/CT:3:3:219
56. Sheng S-y, Li J-m, Hu X-y, Wang Y. Regulated cell death pathways in cardiomyopathy. Acta Pharmacologica Sin. (2023) 44:1521–35. doi: 10.1038/s41401-023-01068-9
57. Cai L, Wang Y, Zhou G, Chen T, Song Y, Li X, et al. Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J Am Coll Cardiol. (2006) 48:1688–97. doi: 10.1016/j.jacc.2006.07.022
58. Kayama Y, Raaz U, Jagger A, Adam M, Schellinger IN, Sakamoto M, et al. Diabetic cardiovascular disease induced by oxidative stress. Int J Mol Sci. (2015) 16:25234–63. doi: 10.3390/ijms161025234
59. Braunwald E. Biomarkers in heart failure. N Engl J Med. (2008) 358:2148–59. doi: 10.1056/NEJMra0800239
60. Bernhard D, Rossmann A, Henderson B, Kind M, Seubert A, Wick G. Increased serum cadmium and strontium levels in young smokers: effects on arterial endothelial cell gene transcription. Arterioscler Thromb Vasc Biol. (2006) 26:833–8. doi: 10.1161/01.ATV.0000205616.70614.e5
61. Menke A, Muntner P, Silbergeld EK, Platz EA, Guallar E. Cadmium levels in urine and mortality among US adults. Environ Health Perspect. (2009) 117:190–6. doi: 10.1289/ehp.11236
62. Deering KE, Callan AC, Prince RL, Lim WH, Thompson PL, Lewis JR, et al. Low-level cadmium exposure and cardiovascular outcomes in elderly Australian women: a cohort study. Int J hygiene Environ Health. (2018) 221:347–54. doi: 10.1016/j.ijheh.2017.12.007
63. Tellez-Plaza M, Jones MR, Dominguez-Lucas A, Guallar E, Navas-Acien A. Cadmium exposure and clinical cardiovascular disease: a systematic review. Curr Atheroscl Rep. (2013) 15:1–15. doi: 10.1007/s11883-013-0356-2
64. Prozialeck WC, Edwards JR, Woods JM. The vascular endothelium as a target of cadmium toxicity. Life Sci. (2006) 79:1493–506. doi: 10.1016/j.lfs.2006.05.007
65. Prozialeck WC, Edwards JR, Nebert DW, Woods JM, Barchowsky A, Atchison WD. The vascular system as a target of metal toxicity. Toxicological Sci. (2008) 102:207–18. doi: 10.1093/toxsci/kfm263
66. Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. (1999) 340:115–26. doi: 10.1056/NEJM199901143400207
67. Hossein-Khannazer N, Azizi G, Eslami S, Alhassan Mohammed H, Fayyaz F, Hosseinzadeh R, et al. The effects of cadmium exposure in the induction of inflammation. Immunopharmacol Immunotoxicol. (2020) 42:1–8. doi: 10.1080/08923973.2019.1697284
68. Fatkhullina A, Peshkova I, Koltsova E. The role of cytokines in the development of atherosclerosis. Biochem (Moscow). (2016) 81:1358–70. doi: 10.1134/S0006297916110134
69. Bonaventura P, Lamboux A, Albarede F, Miossec P. Differential effects of TNF-α and IL-1β on the control of metal metabolism and cadmium-induced cell death in chronic inflammation. PloS One. (2018) 13:e0196285. doi: 10.1371/journal.pone.0196285
70. Refaie MM, El-Hussieny M, Bayoumi AM, Shehata S. Mechanisms mediating the cardioprotective effect of carvedilol in cadmium induced cardiotoxicity. Role of eNOS and HO1/Nrf2 pathway. Environ Toxicol Pharmacol. (2019) 70:103198. doi: 10.1016/j.etap.2019.103198
71. El-Kersh K, Hopkins CD, Wu X, Rai SN, Cave MC, Smith MR, et al. Metallomics in pulmonary arterial hypertension patients. Pulm Circ. (2023) 13:e12202. doi: 10.1002/pul2.12202
72. Huang J, El-Kersh K, Mann KK, James KA, Cai L. Overview of the cardiovascular effects of environmental metals: New preclinical and clinical insights. Toxicol Appl Pharmacol. (2022) 454:116247. doi: 10.1016/j.taap.2022.116247
73. Li Z, Wang R, Long T, Xu Y, Guo H, Zhang X, et al. Associations between plasma essential metals levels and the risks of all-cause mortality and cardiovascular disease mortality among individuals with type 2 diabetes. Nutrients. (2023) 15:1198. doi: 10.3390/nu15051198
74. Zhu K, Zhang Y, Lu Q, Geng T, Li R, Wan Z, et al. Associations of exposure to lead and cadmium with risk of all-cause and cardiovascular disease mortality among patients with type 2 diabetes. Environ Sci pollut Res. (2022) 29:76805–15. doi: 10.1007/s11356-022-21273-z
75. Oluranti OI, Agboola EA, Fubara NE, Ajayi MO, Michael OS. Cadmium exposure induces cardiac glucometabolic dysregulation and lipid accumulation independent of pyruvate dehydrogenase activity. Ann Med. (2021) 53:1109–18. doi: 10.1080/07853890.2021.1947519
76. Mitra E, Ghosh AK, Ghosh D, Mukherjee D, Chattopadhyay A, Dutta S, et al. Protective effect of aqueous Curry leaf (Murraya koenigii) extract against cadmium-induced oxidative stress in rat heart. Food Chem Toxicol. (2012) 50:1340–53. doi: 10.1016/j.fct.2012.01.048
77. Yazıhan N, Koçak MK, Akçıl E, Erdem O, Sayal A, Güven C, et al. Involvement of galectin-3 in cadmium-induced cardiac toxicity. Anatolian J Cardiology/Anadolu Kardiyoloji Dergisi. (2011) 11:479–84. doi: 10.5152/akd.2011.130
78. Kim HS, Kim YJ, Seo YR. An overview of carcinogenic heavy metal: molecular toxicity mechanism and prevention. J Cancer Prev. (2015) 20:232. doi: 10.15430/JCP.2015.20.4.232
79. Treviño S, Waalkes MP, Hernández JAF, León-Chavez BA, Aguilar-Alonso P, Brambila E. Chronic cadmium exposure in rats produces pancreatic impairment and insulin resistance in multiple peripheral tissues. Arch Biochem Biophys. (2015) 583:27–35. doi: 10.1016/j.abb.2015.07.010
80. Jacquet A, Arnaud J, Hininger-Favier I, Hazane-Puch F, Couturier K, Lénon M, et al. Impact of chronic and low cadmium exposure of rats: Sex specific disruption of glucose metabolism. Chemosphere. (2018) 207:764–73. doi: 10.1016/j.chemosphere.2018.05.099
81. Piconi L, Quagliaro L, Ceriello A. Oxidative stress in diabetes. Clin Chem Lab Med. (2003) 41(9):1144–9. doi: 10.1515/CCLM.2003.177
82. Rolo AP, Palmeira CM. Diabetes and mitochondrial function: role of hyperglycemia and oxidative stress. Toxicol Appl Pharmacol. (2006) 212:167–78. doi: 10.1016/j.taap.2006.01.003
83. Khullar M, Al-Shudiefat AA-RS, Ludke A, BiNepal G, Singal PK. Oxidative stress: a key contributor to diabetic cardiomyopathy. Can J Physiol Pharmacol. (2010) 88:233–40. doi: 10.1139/Y10-016
84. Lee W-K, Torchalski B, Thévenod F. Cadmium-induced ceramide formation triggers calpain-dependent apoptosis in cultured kidney proximal tubule cells. Am J Physiol-Cell Physiol. (2007) 293:C839–47. doi: 10.1152/ajpcell.00197.2007
85. Hayat SA, Patel B, Khattar RS, Malik RA. Diabetic cardiomyopathy: mechanisms, diagnosis and treatment. Clin Sci. (2004) 107:539–57. doi: 10.1042/CS20040057
86. Field BC, Gordillo R, Scherer PE. The role of ceramides in diabetes and cardiovascular disease regulation of ceramides by adipokines. Front Endocrinol. (2020) 11:569250. doi: 10.3389/fendo.2020.569250
87. El Hayek MS, Ernande L, Benitah J-P, Gomez A-M, Pereira L. The role of hyperglycaemia in the development of diabetic cardiomyopathy. Arch Cardiovasc Dis. (2021) 114:748–60. doi: 10.1016/j.acvd.2021.08.004
88. Yue Y, Meng K, Pu Y, Zhang X. Transforming growth factor beta (TGF-β) mediates cardiac fibrosis and induces diabetic cardiomyopathy. Diabetes Res Clin Pract. (2017) 133:124–30. doi: 10.1016/j.diabres.2017.08.018
89. Passino C, Barison A, Vergaro G, Gabutti A, Borrelli C, Emdin M, et al. Markers of fibrosis, inflammation, and remodeling pathways in heart failure. Clinica Chimica Acta. (2015) 443:29–38. doi: 10.1016/j.cca.2014.09.006
90. X-t W, Gong Y, Zhou B, J-j Y, Cheng Y, J-g Z, et al. Ursolic acid ameliorates oxidative stress, inflammation and fibrosis in diabetic cardiomyopathy rats. Biomed Pharmacother. (2018) 97:1461–7. doi: 10.1016/j.biopha.2017.11.032
91. Younce CW, Wang K, Kolattukudy PE. Hyperglycaemia-induced cardiomyocyte death is mediated via MCP-1 production and induction of a novel zinc-finger protein MCPIP. Cardiovasc Res. (2010) 87:665–74. doi: 10.1093/cvr/cvq102
92. Manna P, Sil PC. Impaired redox signaling and mitochondrial uncoupling contributes vascular inflammation and cardiac dysfunction in type 1 diabetes: protective role of arjunolic acid. Biochimie. (2012) 94:786–97. doi: 10.1016/j.biochi.2011.11.010
93. Yim G, Wang Y, Howe CG, Romano ME. Exposure to metal mixtures in association with cardiovascular risk factors and outcomes: a scoping review. Toxics. (2022) 10:116. doi: 10.3390/toxics10030116
94. Tinkov AA, Filippini T, Ajsuvakova OP, Skalnaya MG, Aaseth J, Bjørklund G, et al. Cadmium and atherosclerosis: A review of toxicological mechanisms and a meta-analysis of epidemiologic studies. Environ Res. (2018) 162:240–60. doi: 10.1016/j.envres.2018.01.008
95. Solenkova NV, Newman JD, Berger JS, Thurston G, Hochman JS, Lamas GA. Metal pollutants and cardiovascular disease: mechanisms and consequences of exposure. Am Heart J. (2014) 168:812–22. doi: 10.1016/j.ahj.2014.07.007
96. Mostafalou S, Baeeri M, Bahadar H, Soltany-Rezaee-Rad M, Gholami M, Abdollahi M. Molecular mechanisms involved in lead induced disruption of hepatic and pancreatic glucose metabolism. Environ Toxicol Pharmacol. (2015) 39:16–26. doi: 10.1016/j.etap.2014.11.001
97. Newsholme P, Haber E, Hirabara S, Rebelato E, Procopio J, Morgan D, et al. Diabetes associated cell stress and dysfunction: role of mitochondrial and non-mitochondrial ROS production and activity. J Physiol. (2007) 583:9–24. doi: 10.1113/jphysiol.2007.135871
98. Son SM. Role of vascular reactive oxygen species in development of vascular abnormalities in diabetes. Diabetes Res Clin Pract. (2007) 77:S65–70. doi: 10.1016/j.diabres.2007.01.036
99. Genchi G, Sinicropi MS, Lauria G, Carocci A, Catalano A. The effects of cadmium toxicity. Int J Environ Res Public Health. (2020) 17:3782. doi: 10.3390/ijerph17113782
100. Lei G, Mao C, Yan Y, Zhuang L, Gan B. Ferroptosis, radiotherapy, and combination therapeutic strategies. Protein Cell. (2021) 12:836–57. doi: 10.1007/s13238-021-00841-y
101. Zhou R-P, Chen Y, Wei X, Yu B, Xiong Z-G, Lu C, et al. Novel insights into ferroptosis: Implications for age-related diseases. Theranostics. (2020) 10:11976. doi: 10.7150/thno.50663
102. Stockwell BR, Angeli JPF, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
103. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
104. Sui X, Zhang R, Liu S, Duan T, Zhai L, Zhang M, et al. RSL3 drives ferroptosis through GPX4 inactivation and ROS production in colorectal cancer. Front Pharmacol. (2018) 9:1371. doi: 10.3389/fphar.2018.01371
105. Sha W, Hu F, Xi Y, Chu Y, Bu S. Mechanism of ferroptosis and its role in type 2 diabetes mellitus. J Diabetes Res. (2021) 2021:9999612. doi: 10.1155/2021/9999612
106. Yao W, Liao H, Pang M, Pan L, Guan Y, Huang X, et al. Inhibition of the NADPH oxidase pathway reduces ferroptosis during septic renal injury in diabetic mice. Oxid Med Cell Longev. (2022) 2022:1193734. doi: 10.1155/2022/1193734
107. Prasad MK, Mohandas S, Kunka Mohanram R. Role of ferroptosis inhibitors in the management of diabetes. BioFactors. (2023) 49:270–96. doi: 10.1002/biof.v49.2
108. Cai L, Tan Y, Holland B, Wintergerst K. Diabetic cardiomyopathy and cell death: focus on metal-mediated cell death. Cardiovasc Toxicol. (2024) 24:1–14. doi: 10.1007/s12012-024-09836-7
109. Li D, Jiang C, Mei G, Zhao Y, Chen L, Liu J, et al. Quercetin alleviates ferroptosis of pancreatic β cells in type 2 diabetes. Nutrients. (2020) 12:2954. doi: 10.3390/nu12102954
110. Liu J, Li Q, Yang Y, Ma L. Iron metabolism and type 2 diabetes mellitus: A meta-analysis and systematic review. J Diabetes Invest. (2020) 11:946–55. doi: 10.1111/jdi.13216
111. Gautam S, Alam F, Moin S, Noor N, Arif S. Role of ferritin and oxidative stress index in gestational diabetes mellitus. J Diabetes Metab Disord. (2021) 20:1615–9. doi: 10.1007/s40200-021-00911-2
112. Fernández-Real JM, McClain D, Manco M. Mechanisms linking glucose homeostasis and iron metabolism toward the onset and progression of type 2 diabetes. Diabetes Care. (2015) 38:2169–76. doi: 10.2337/dc14-3082
113. Shu T, Lv Z, Xie Y, Tang J, Mao X. Hepcidin as a key iron regulator mediates glucotoxicity-induced pancreatic β-cell dysfunction. Endocrine connections. (2019) 8:150–61. doi: 10.1530/EC-18-0516
114. Sung HK, Song E, Jahng JWS, Pantopoulos K, Sweeney G. Iron induces insulin resistance in cardiomyocytes via regulation of oxidative stress. Sci Rep. (2019) 9:4668. doi: 10.1038/s41598-019-41111-6
115. Wang J, Wang H. Oxidative stress in pancreatic beta cell regeneration. Oxid Med Cell Longev. (2017) 2017:1930261. doi: 10.1155/2017/1930261
116. Koppula P, Zhang Y, Shi J, Li W, Gan B. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J Biol Chem. (2017) 292:14240–9. doi: 10.1074/jbc.M117.798405
117. Hamad M, Mohammed AK, Hachim MY, Mukhopadhy D, Khalique A, Laham A, et al. Heme Oxygenase-1 (HMOX-1) and inhibitor of differentiation proteins (ID1, ID3) are key response mechanisms against iron-overload in pancreatic β-cells. Mol Cell Endocrinol. (2021) 538:111462. doi: 10.1016/j.mce.2021.111462
118. Mittler R, Darash-Yahana M, Sohn YS, Bai F, Song L, Cabantchik IZ, et al. NEET proteins: a new link between iron metabolism, reactive oxygen species, and cancer. Antioxidants Redox Signaling. (2019) 30:1083–95. doi: 10.1089/ars.2018.7502
119. Karmi O, Sohn Y-S, Marjault H-B, Israeli T, Leibowitz G, Ioannidis K, et al. A combined drug treatment that reduces mitochondrial iron and reactive oxygen levels recovers insulin secretion in NAF-1-deficient pancreatic cells. Antioxidants. (2021) 10:1160. doi: 10.3390/antiox10081160
120. Krümmel B, Plötz T, Jörns A, Lenzen S, Mehmeti I. The central role of glutathione peroxidase 4 in the regulation of ferroptosis and its implications for pro-inflammatory cytokine-mediated beta-cell death. Biochim Biophys Acta (BBA)-Molecular Basis Dis. (2021) 1867:166114. doi: 10.1016/j.bbadis.2021.166114
121. Bruni A, Pepper AR, Pawlick RL, Gala-Lopez B, Gamble AF, Kin T, et al. Ferroptosis-inducing agents compromise in vitro human islet viability and function. Cell Death Dis. (2018) 9:595. doi: 10.1038/s41419-018-0506-0
122. Li XY, Leung PS. Erastin-induced ferroptosis is a regulator for the growth and function of human pancreatic islet-like cell clusters. Cell Regeneration. (2020) 9:16. doi: 10.1186/s13619-020-00055-3
123. Duan J-Y, Lin X, Xu F, Shan S-K, Guo B, Li F-X-Z, et al. Ferroptosis and its potential role in metabolic diseases: a curse or revitalization? Front Cell Dev Biol. (2021) 9:701788. doi: 10.3389/fcell.2021.701788
124. Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia. (2014) 57:660–71. doi: 10.1007/s00125-014-3171-6
125. Zang H, Wu W, Qi L, Tan W, Nagarkatti P, Nagarkatti M, et al. Autophagy inhibition enables Nrf2 to exaggerate the progression of diabetic cardiomyopathy in mice. Diabetes. (2020) 69:2720–34. doi: 10.2337/db19-1176
126. Wei Z, Shaohuan Q, Pinfang K, Chao S. Curcumin attenuates ferroptosis-induced myocardial injury in diabetic cardiomyopathy through the Nrf2 pathway. Cardiovasc Ther. (2022) 2022:3159717. doi: 10.1155/2022/3159717
127. Jenkins T, Gouge J. Nrf2 in cancer, detoxifying enzymes and cell death programs. Antioxidants. (2021) 10:1030. doi: 10.3390/antiox10071030
128. Chen QM, Maltagliati AJ. Nrf2 at the heart of oxidative stress and cardiac protection. Physiol Genomics. (2018) 50:77–97. doi: 10.1152/physiolgenomics.00041.2017
129. Hu X, Rajesh M, Zhang J, Zhou S, Wang S, Sun J, et al. Protection by dimethyl fumarate against diabetic cardiomyopathy in type 1 diabetic mice likely via activation of nuclear factor erythroid-2 related factor 2. Toxicol Lett. (2018) 287:131–41. doi: 10.1016/j.toxlet.2018.01.020
130. Wang G, Song X, Zhao L, Li Z, Liu B. Resveratrol prevents diabetic cardiomyopathy by increasing Nrf2 expression and transcriptional activity. BioMed Res Int. (2018) 2018:2150218. doi: 10.1155/2018/2150218
131. Palomer X, Salvadó L, Barroso E, Vázquez-Carrera M. An overview of the crosstalk between inflammatory processes and metabolic dysregulation during diabetic cardiomyopathy. Int J Cardiol. (2013) 168:3160–72. doi: 10.1016/j.ijcard.2013.07.150
132. Wang C, Zhu L, Yuan W, Sun L, Xia Z, Zhang Z, et al. Diabetes aggravates myocardial ischaemia reperfusion injury via activating Nox2-related programmed cell death in an AMPK-dependent manner. J Cell Mol Med. (2020) 24:6670–9. doi: 10.1111/jcmm.v24.12
133. Li W, Li W, Leng Y, Xiong Y, Xia Z. Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol. (2020) 39:210–25. doi: 10.1089/dna.2019.5097
134. Li M-D, Fu L, Lv B-B, Xiang Y, Xiang H-X, Xu D-X, et al. Arsenic induces ferroptosis and acute lung injury through mtROS-mediated mitochondria-associated endoplasmic reticulum membrane dysfunction. Ecotoxicol Environ Saf. (2022) 238:113595. doi: 10.1016/j.ecoenv.2022.113595
135. Zhao C, Yu D, He Z, Bao L, Feng L, Chen L, et al. Endoplasmic reticulum stress-mediated autophagy activation is involved in cadmium-induced ferroptosis of renal tubular epithelial cells. Free Radical Biol Med. (2021) 175:236–48. doi: 10.1016/j.freeradbiomed.2021.09.008
136. Hong H, Xu Y, Xu J, Zhang J, Xi Y, Pi H, et al. Cadmium exposure impairs pancreatic β-cell function and exaggerates diabetes by disrupting lipid metabolism. Environ Int. (2021) 149:106406. doi: 10.1016/j.envint.2021.106406
137. Hong H, Lin X, Xu Y, Tong T, Zhang J, He H, et al. Cadmium induces ferroptosis mediated inflammation by activating Gpx4/Ager/p65 axis in pancreatic β-cells. Sci Total Environ. (2022) 849:157819. doi: 10.1016/j.scitotenv.2022.157819
138. Morales CR, Pedrozo Z, Lavandero S, Hill JA. Oxidative stress and autophagy in cardiovascular homeostasis. Antioxidants Redox Signaling. (2014) 20:507–18. doi: 10.1089/ars.2013.5359
139. Sahoo K, Sharma A. Understanding the mechanistic roles of environmental heavy metal stressors in regulating ferroptosis: adding new paradigms to the links with diseases. Apoptosis. (2023) 28:277–92. doi: 10.1007/s10495-022-01806-0
140. Meerson F, Kagan V, Kozlov YP, Belkina L, Arkhipenko YV. The role of lipid peroxidation in pathogenesis of ischemic damage and the antioxidant protection of the heart. Basic Res Cardiol. (1982) 77:465–85. doi: 10.1007/BF01907940
141. Hao R, Ge J, Song X, Li F, Sun-Waterhouse D, Li D. Cadmium induces ferroptosis and apoptosis by modulating miR-34a-5p/Sirt1axis in PC12 cells. Environ Toxicol. (2022) 37:41–51. doi: 10.1002/tox.23376
142. Belyaeva EA, Dymkowska D, Więckowski MR, Wojtczak L. Reactive oxygen species produced by the mitochondrial respiratory chain are involved in Cd2+-induced injury of rat ascites hepatoma AS-30D cells. Biochim Biophys Acta (BBA)-Bioenergetics. (2006) 1757:1568–74. doi: 10.1016/j.bbabio.2006.09.006
143. Zhang S, Hao S, Qiu Z, Wang Y, Zhao Y, Li Y, et al. Cadmium disrupts the DNA damage response by destabilizing RNF168. Food Chem Toxicol. (2019) 133:110745. doi: 10.1016/j.fct.2019.110745
144. Liu P, Zhang Z, Cai Y, Li Z, Zhou Q, Chen Q. Ferroptosis: mechanisms and role in diabetes mellitus and its complications. Ageing Res Rev. (2024) 94:102201. doi: 10.1016/j.arr.2024.102201
145. Deng X, Chu W, Zhang H, Peng Y. Nrf2 and ferroptosis: a new research direction for ischemic stroke. Cell Mol Neurobiol. (2023) 43:3885–96. doi: 10.1007/s10571-023-01411-y
146. Uruno A, Matsumaru D, Ryoke R, Saito R, Kadoguchi S, Saigusa D, et al. Nrf2 suppresses oxidative stress and inflammation in App knock-in Alzheimer’s disease model mice. Mol Cell Biol. (2020) 40:e00467–00419. doi: 10.1128/MCB.00467-19
147. Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. (2013) 53:401–26. doi: 10.1146/annurev-pharmtox-011112-140320
148. Ashrafizadeh M, Ahmadi Z, Farkhondeh T, Samarghandian S. Back to nucleus: combating with cadmium toxicity using Nrf2 signaling pathway as a promising therapeutic target. Biol Trace element Res. (2020) 197:52–62. doi: 10.1007/s12011-019-01980-4
149. Sasikumar S, Yuvraj S, Veilumuthu P, Godwin Christopher JS, Anandkumar P, Nagarajan T, et al. Ascorbic acid attenuates cadmium-induced myocardial hypertrophy and cardiomyocyte injury through Nrf2 signaling pathways comparable to resveratrol. 3 Biotech. (2023) 13:108. doi: 10.1007/s13205-023-03527-w
150. Guo K, Ge J, Zhang C, Lv M-W, Zhang Q, Talukder M, et al. Cadmium induced cardiac inflammation in chicken (Gallus gallus) via modulating cytochrome P450 systems and Nrf2 mediated antioxidant defense. Chemosphere. (2020) 249:125858. doi: 10.1016/j.chemosphere.2020.125858
151. Wang X, Chen X, Zhou W, Men H, Bao T, Sun Y, et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B. (2022) 12:708–22. doi: 10.1016/j.apsb.2021.10.005
152. Yuan H-J, Xue Y-T, Liu Y. Cuproptosis, the novel therapeutic mechanism for heart failure: a narrative review. Cardiovasc Diagnosis Ther. (2022) 12:681. doi: 10.21037/cdt-22-214
153. Wang D, Tian Z, Zhang P, Zhen L, Meng Q, Sun B, et al. The molecular mechanisms of cuproptosis and its relevance to cardiovascular disease. Biomed Pharmacother. (2023) 163:114830. doi: 10.1016/j.biopha.2023.114830
154. e Vairo FP, Chwal BC, Perini S, Ferreira MAP, de Freitas Lopes AC, Saute JAM. A systematic review and evidence-based guideline for diagnosis and treatment of Menkes disease. Mol Genet Metab. (2019) 126:6–13. doi: 10.1016/j.ymgme.2018.12.005
155. Sensi SL, Granzotto A, Siotto M, Squitti R. Copper and zinc dysregulation in Alzheimer’s disease. Trends Pharmacol Sci. (2018) 39:1049–63. doi: 10.1016/j.tips.2018.10.001
156. Shanbhag VC, Gudekar N, Jasmer K, Papageorgiou C, Singh K, Petris MJ. Copper metabolism as a unique vulnerability in cancer. Biochim Biophys Acta (BBA)-Molecular Cell Res. (2021) 1868:118893. doi: 10.1016/j.bbamcr.2020.118893
157. Chang W, Li P. Copper and diabetes: current research and prospect. Mol Nutr Food Res. (2023) 67:2300468. doi: 10.1002/mnfr.202300468
158. Yin J, Wang X, Li S, Zhu Y, Chen S, Li P, et al. Interactions between plasma copper concentrations and SOD1 gene polymorphism for impaired glucose regulation and type 2 diabetes. Redox Biol. (2019) 24:101172. doi: 10.1016/j.redox.2019.101172
159. Nie H, Hu H, Li Z, Wang R, He J, Li P, et al. Associations of plasma metal levels with type 2 diabetes and the mediating effects of microRNAs. Environ pollut. (2022) 292:118452. doi: 10.1016/j.envpol.2021.118452
160. Squitti R, Negrouk V, Perera M, Llabre MM, Ricordi C, Rongioletti MCA, et al. Serum copper profile in patients with type 1 diabetes in comparison to other metals. J Trace elements Med Biol. (2019) 56:156–61. doi: 10.1016/j.jtemb.2019.08.011
161. Masad A, Hayes L, Tabner BJ, Turnbull S, Cooper LJ, Fullwood NJ, et al. Copper-mediated formation of hydrogen peroxide from the amylin peptide: a novel mechanism for degeneration of islet cells in type-2 diabetes mellitus? FEBS Lett. (2007) 581:3489–93. doi: 10.1016/j.febslet.2007.06.061
162. Magenta A, Greco S, Capogrossi MC, Gaetano C, Martelli F. Nitric oxide, oxidative stress, and p66Shc interplay in diabetic endothelial dysfunction. BioMed Res Int. (2014) 2014:193095. doi: 10.1155/2014/193095
163. Cooper G, Young A, Gamble G, Occleshaw C, Dissanayake A, Cowan B, et al. A copper (II)-selective chelator ameliorates left-ventricular hypertrophy in type 2 diabetic patients: a randomised placebo-controlled study. Diabetologia. (2009) 52:715–22. doi: 10.1007/s00125-009-1265-3
164. Cooper GJ. Therapeutic potential of copper chelation with triethylenetetramine in managing diabetes mellitus and Alzheimer’s disease. Drugs. (2011) 71:1281–320. doi: 10.2165/11591370-000000000-00000
165. Tsang T, Davis CI, Brady DC. Copper biology. Curr Biol. (2021) 31:R421–7. doi: 10.1016/j.cub.2021.03.054
166. Fernández Rodarte BA, Soto Domínguez A, González Navarro A, Mellado Ayala M, Piña Mendoza E, Rodríguez Rocha H, et al. Copper induces damage, oxidative stress and cell death in endothelium of chronic intoxicated Wistar rats. Int J Morphol. (2022) 40:10–7. doi: 10.4067/S0717-95022022000100010
167. Rachek L. Glucose homeostatis and the pathogenesis of diabetes mellitus. Prog Mol Biol Transl Sci. (2014) 121:267–92. doi: 10.1016/B978-0-12-800101-1.00008-9
168. Hou X, Hu Z, Xu H, Xu J, Zhang S, Zhong Y, et al. Advanced glycation endproducts trigger autophagy in cadiomyocyte via RAGE/PI3K/AKT/mTOR pathway. Cardiovasc Diabetol. (2014) 13:1–8. doi: 10.1186/1475-2840-13-78
169. Yang Y-C, Tsai C-Y, Chen C-L, Kuo C-H, Hou C-W, Cheng S-Y, et al. Pkcδ activation is involved in ROS-mediated mitochondrial dysfunction and apoptosis in cardiomyocytes exposed to advanced glycation end products (ages). Aging Dis. (2018) 9:647. doi: 10.14336/AD.2017.0924
170. Bonanni LJ, Wittkopp S, Long C, Aleman JO, Newman JD. A review of air pollution as a driver of cardiovascular disease risk across the diabetes spectrum. Front Endocrinol. (2024) 15:1321323. doi: 10.3389/fendo.2024.1321323
171. Huo S, Wang Q, Shi W, Peng L, Jiang Y, Zhu M, et al. ATF3/SPI1/SLC31A1 signaling promotes cuproptosis induced by advanced glycosylation end products in diabetic myocardial injury. Int J Mol Sci. (2023) 24:1667. doi: 10.3390/ijms24021667
172. Zhao C, Zhang Z, Jing T. A novel signature of combing cuproptosis-with ferroptosis-related genes for prediction of prognosis, immunologic therapy responses and drug sensitivity in hepatocellular carcinoma. Front Oncol. (2022) 12:1000993. doi: 10.3389/fonc.2022.1000993
173. Zhang L, Shi J, Du D, Niu N, Liu S, Yang X, et al. Ketogenesis acts as an endogenous protective programme to restrain inflammatory macrophage activation during acute pancreatitis. EBioMedicine. (2022) 78:103959. doi: 10.1016/j.ebiom.2022.103959
174. Chen J, Yang X, Li W, Lin Y, Lin R, Cai X, et al. Potential molecular and cellular mechanisms of the effects of cuproptosis-related genes in the cardiomyocytes of patients with diabetic heart failure: a bioinformatics analysis. Front Endocrinol. (2024) 15:1370387. doi: 10.3389/fendo.2024.1370387
175. Zhou W-W, Dai C, Liu W-Z, Zhang C, Zhang Y, Yang G-S, et al. Gentianella acuta improves TAC-induced cardiac remodelling by regulating the Notch and PI3K/Akt/FOXO1/3 pathways. Biomed Pharmacother. (2022) 154:113564. doi: 10.1016/j.biopha.2022.113564
176. Yang Y, Feng Q, Luan Y, Liu H, Jiao Y, Hao H, et al. Exploring cuproptosis as a mechanism and potential intervention target in cardiovascular diseases. Front Pharmacol. (2023) 14:1229297. doi: 10.3389/fphar.2023.1229297
177. Alqarni MH, Muharram MM, Alshahrani SM, Labrou NE. Copper-induced oxidative cleavage of glutathione transferase F1-1 from Zea mays. Int J Biol Macromol. (2019) 128:493–8. doi: 10.1016/j.ijbiomac.2019.01.128
Keywords: cadmium, cuproptosis, cardiovascular diseases, diabetic cardiomyopathy, diabetes, ferroptosis
Citation: Saedi S, Tan Y, Watson SE, Wintergerst KA and Cai L (2024) Potential pathogenic roles of ferroptosis and cuproptosis in cadmium-induced or exacerbated cardiovascular complications in individuals with diabetes. Front. Endocrinol. 15:1461171. doi: 10.3389/fendo.2024.1461171
Received: 08 July 2024; Accepted: 16 September 2024;
Published: 02 October 2024.
Edited by:
Ying Xin, Jilin University, ChinaReviewed by:
Ji Li, University of Mississippi Medical Center, United StatesRongzhu Lu, Jiangsu University, China
Copyright © 2024 Saedi, Tan, Watson, Wintergerst and Cai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lu Cai, bHUuY2FpQGxvdWlzdmlsbGUuZWR1