 Chiara Bima1†
Chiara Bima1† Chiara Lopez1†
Chiara Lopez1† Gerdi Tuli2‡Jessica Munarin2‡Stefano Arata1Matteo Procopio1
Gerdi Tuli2‡Jessica Munarin2‡Stefano Arata1Matteo Procopio1 Martina Bollati1
Martina Bollati1 Mauro Maccario1
Mauro Maccario1 Luisa De Sanctis2§
Luisa De Sanctis2§ Mirko Parasiliti-Caprino1*§
Mirko Parasiliti-Caprino1*§- 1Endocrinology, Diabetes and Metabolism; Department of Medical Sciences; University of Turin, Turin, Italy
- 2Department of Sciences of Public Health and Pediatrics, University of Turin, Turin, Italy
Hypertensive crises in pediatric patients are rare conditions. However, determining their precise prevalence is more challenging than in adults due to the heterogeneity in the definition itself. These crises frequently occur without a prior diagnosis of hypertension and may indicate an underlying cause of secondary hypertension, including pheochromocytoma/paraganglioma (PPGL). The mechanisms of hypertensive crises in the pediatric population with PPGL are directly related to different types of catecholamine excess. Noradrenergic tumors typically present with sustained hypertension due to their predominant action on α1-adrenoceptors in the vasculature. Conversely, adrenergic tumors, through epinephrine binding to β2-adrenoceptors in addition to stimulation of α1- and α2-adrenoceptors, more frequently cause paroxysmal hypertension. Furthermore, the biochemical phenotype also reflects the tumor localization and the presence of a genetic mutation. Recent evidence suggests that more than 80% of PPGL in pediatric cases have a hereditary background. PPGL susceptibility mutations are categorized into three clusters; mutations in cluster 1 are more frequently associated with a noradrenergic phenotype, whereas those in cluster 2 are associated with an adrenergic phenotype. Consequently, the treatment of hypertensive crises in pediatric patients with PPGL, reflecting the underlying pathophysiology, requires first-line therapy with alpha-blockers, potentially in combination with beta-blockers only in the case of tachyarrhythmia after adequate alpha-blockade. The route of administration for treatment depends on the context, such as intraoperative or pre-surgical settings, and whether it presents as a hypertensive emergency (elevated blood pressure with acute target organ damage), where intravenous administration of antihypertensive drugs is mandatory. Conversely, in cases of hypertensive urgency, if children can tolerate oral therapy, intravenous administration may initially be avoided. However, managing these cases is complex and requires careful consideration of the selection and timing of therapy administration, particularly in pediatric patients. Therefore, facing these conditions in tertiary care centers through interdisciplinary collaboration is advisable to optimize therapeutic outcomes.
Introduction and epidemiology
The prevalence of arterial hypertension in pediatric patients varies from 0.5-1% to 5% depending on the studies considered (1, 2). However, determining the prevalence of hypertensive crises in children is more challenging due to the heterogeneity in its definition. Generally, a hypertensive crisis in pediatric patients is suspected when blood pressure exceeds the limits of stage II hypertension (3). Specifically, the European Society of Hypertension (ESH) guidelines define severe hypertensive crisis in children as blood pressure values 20% above the stage II hypertension limit (4). Hypertensive crises can be categorized as hypertensive urgency and emergency, with the latter defined by elevated blood pressure values associated with acute target organ damage (2).
In adults, hypertensive crises are often associated with poor compliance with antihypertensive treatment. While in children, they frequently occur without a known previous diagnosis of hypertension and may reflect an underlying cause of secondary hypertension, predominantly of renal origin (70-80% of cases), but also of an endocrine one, including the diagnosis of pheochromocytoma/paraganglioma (PPGL) (3, 5). PPGLs are rare neuroendocrine tumors that can secrete catecholamines (in about the 80% of cases), arising from chromaffin cells in the adrenal medulla or sympathetic paraganglia. The incidence of PPGL is approximately 1 in 300,000 per year, with only 10-20% occurring in pediatric age, with an estimated incidence of 0.5-2 per million children, with pheochromocytomas (PCC) representing 80-85% of cases compared to paragangliomas (PGL) (6–8). A retrospective study by Pamporaki et al. (9) showed a high percentage of hereditary (up to 70-80%), extra-adrenal, metastatic, multifocal, and recurrent disease in pediatric-onset tumors (10).
Among children with arterial hypertension, the incidence of PPGL is high, around 1.7%, compared to 0.2-0.6% in hypertensive adults (8, 10). Clinical presentation varies, with signs and symptoms of catecholamine hypersecretion often overlooked in children due to their high level of physical activity compared to adults. Children with PPGL are more likely to have sustained arterial hypertension, up to 60-90% of them, while adults often exhibit paroxysmal hypertension in about 50% of cases (5, 8, 10). The clinical presentation of PPGL reflects the underlying catecholamine secretory phenotype and genotype, with epinephrine-secreting tumors more associated with an acute and explosive presentation than norepinephrine and dopamine-secreting lesions (10).
Treating arterial hypertension, particularly hypertensive crises, in children presents significant challenges. Given the rarity of PPGL in pediatric patients and the limited data available in the literature, the objective of this review is to provide guidance for the clinical management of PPGL-induced hypertensive crises in children. This includes advocating for interdisciplinary collaboration among endocrinologists, pediatric intensivists, anesthesiologists, nephrologists, emergency physicians and surgeons to ensure comprehensive care.
Pathophysiology
The mechanisms of hypertensive crisis in pediatric population with PPGL is directly related to the different types of catecholamine excess (10, 11). From a physiological point of view, norepinephrine and epinephrine are released in different pattern from chromaffin cells tumors and show variable binding affinities for adrenoreceptors, leading to different clinical manifestations as the results of their effect on hemodynamics and metabolism (11). Approximately 50% of pheochromocytomas produce norepinephrine almost exclusively, whereas the other half secretes a combination of norepinephrine and epinephrine.
Adrenoreceptors are G-protein coupled receptor mediating the actions of epinephrine and norepinephrine. The major types of human adrenoreceptors are: α1, α2 and β, each having more subtypes. α1-adrenoceptors are mainly located in the vasculature and their stimulation induces vasoconstriction and increased peripheral vascular resistances, resulting in sustained hypertension. Instead, stimulation of presynaptic α2-adrenoceptors can reduce neuronal norepinephrine release, decreasing blood pressure, through a negative feedback mechanism. β1-adrenoceptors, localized on cardiomyocytes and the cardiac conduction system, mediate increase in blood pressure, through their action on heart rate and cardiac output. In contrast, stimulation of β2-adrenoceptors, present both on blood vessels and cardiomyocytes, could induce vasodilatation and reduced cardiac inotropy, and consequently hypotension (12). Norepinephrine exerts its cardiovascular effects by working mainly on α1-adrenoceptors in the vasculature, with low activity on cardiac β1-adrenoceptors. Instead, epinephrine is responsible for its hemodynamic and metabolic actions through its binding to β2-adrenoceptors, in addiction to stimulation of both α1- and α2-adrenoceptors (12).
Form the clinical point of view, the evaluation of catecholamine O-methylated metabolites is of primary importance because their production and release are continuous and independent from catecholamine secretion, making them reliable disease markers. The main metabolites are normetanephrine, metanephrine and 3-methoxytyramine, deriving from norepinephrine, epinephrine and dopamine, respectively (11, 13).
However, regardless of underlying etiology, hypertensive crisis is characterized by some common pathways that could contribute to the severity of hypertension itself and to end-stage organ damage (14). As shown by Harrison et al. (15), the initial stimulus to hypertension could involve several systems, determining the activation of renin-angiotensin-aldosterone system, oxidative stress and endothelial dysfunction. The consequent protein fragmentation and formation of neoantigens result in activation of T-cells and release of cytokines able to increase vasoconstriction and sodium and water retention. This theory thus recognizes a crucial role of inflammation in the pathogenesis of hypertensive crisis and possible organ damage (16).
The abovementioned pathophysiology of cathecolamine-induced hypertensive crises and related cardiovascular complications is depicted in Figure 1.
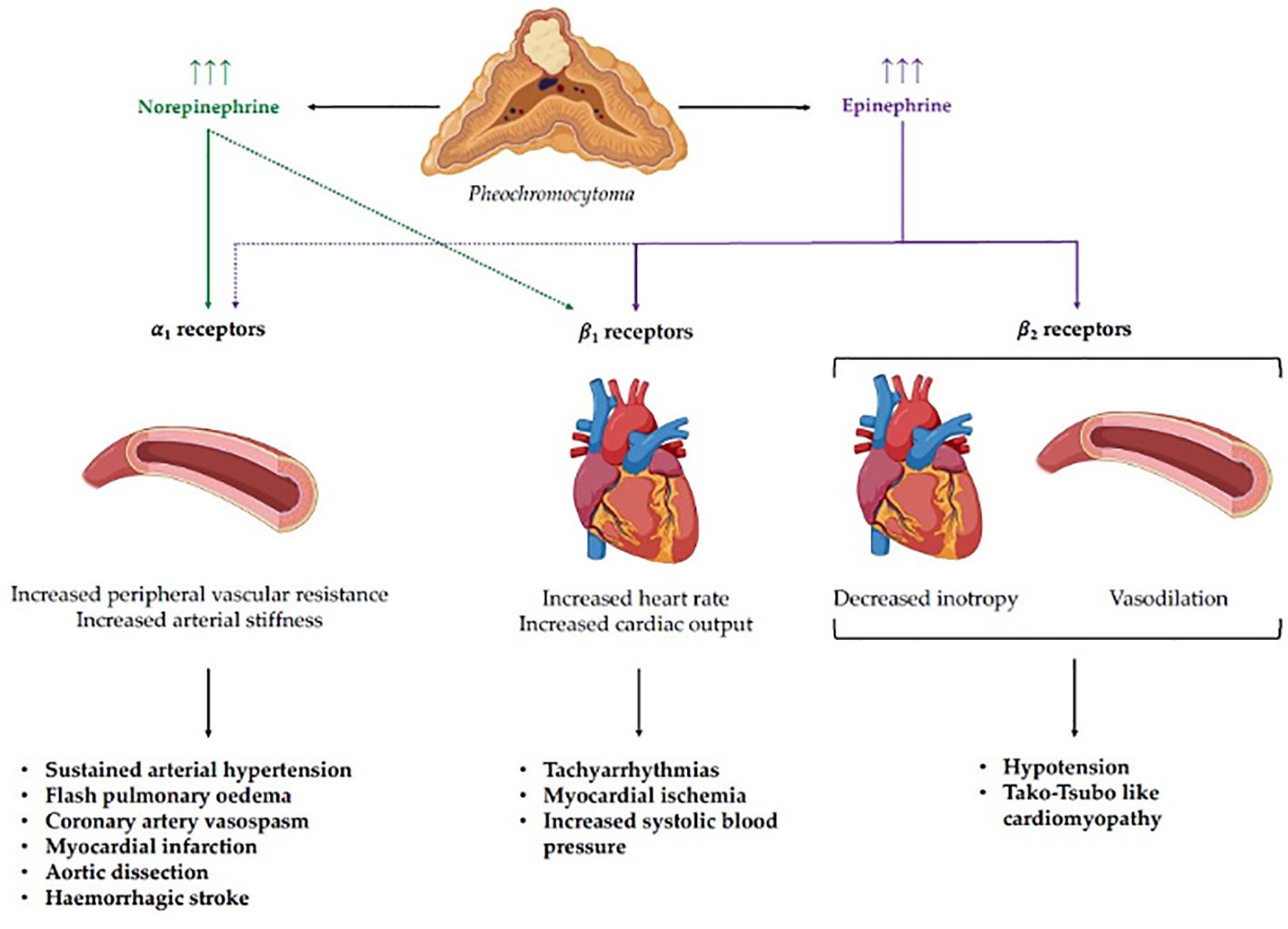
Figure 1 Pathophysiology of catecholamine-induced hypertensive crises and related cardiovascular complications. Norepinephrine and epinephrine display different binding affinities for adreno-receptors, leading to distinct clinical phenotypes according to the specific secreting pattern of the pheochromocytoma. Stimulation of α1 receptors, which show a higher affinity for norepinephrine, mediates an increase in peripheral vascular resistance, leading to sustained arterial hypertension and complications such as flash pulmonary edema, coronary artery vasospasm, myocardial infarction, aortic dissection, and hemorrhagic stroke. On the other hand, stimulation of cardiac β1 receptors, which have a greater affinity for epinephrine, leads to a positive chronotropic and inotropic effect, with the potential development of tachyarrhythmias, myocardial ischemia, and systolic hypertension. Finally, the stimulation of cardiac and vascular β2 receptors by epinephrine can lead to negative inotropism and vasodilation with subsequent hypotension and Tako-Tsubo like cardiomyopathy.
Clinical manifestations and biochemical phenotypes
In childhood functional PPGLs frequently manifest with sustained, rather than paroxysmal, hypertension present in approximately 60-90% of patients (5, 8). Furthermore, the classic “triad” including diaphoresis, headaches, and palpitations has been reported in up to 54% of children with PPGLs (8). In PPGL the cardiovascular complication of hypertensive crisis could derive, apart from marked and abrupt elevation in blood pressure values, also from catecholamine direct damage of the heart and vessels (17). However, the most common manifestations of hypertensive crisis in children are neurologic (14). Clinical signs and symptoms typically depend on type of hormonal secretion (11, 12). The specific secretion profile results in three different biochemical phenotypes: noradrenergic, adrenergic and dopaminergic, according to the predominant increase in normetanephrine, metanephrine and 3-methoxytyramine, respectively (12, 13). Moreover, there is a rare PPGL subtype that does not produce and release catecholamines, named biochemically silent (12). Noradrenergic tumors typically occur with sustained hypertension and possible consequences of excessive α1-adrenoceptors stimulation on cardiovascular system, such as flash pulmonary edema, coronary artery vasospasm and myocardial infarction, or aortic dissection. Instead, adrenergic tumors, in addition to the sequalae of α1-adrenoceptors stimulation, could also manifest more sequelae of excessive β-adrenoceptor stimulation, resulting in tachyarrhythmia, myocarditis, and demand myocardial ischemia or infarction (17, 18). Moreover, because of excessive β2-adrenoceptor binding, adrenergic tumors may also manifest with hypoglycemia, suppression of myocardial contractility (as in Takotsubo-like cardiomyopathy), orthostatic hypotension, and even hypotensive shock (8). Conversely, patients with dopamine-secreting tumors, could be diagnosed by mass effect symptoms, being longer normotensive and asymptomatic (5).
The biochemical phenotype also influences tumor localization. In fact, PPGL with noradrenergic phenotype is typically extra-adrenal instead adrenergic PPGLs are characterized by location within the adrenal glands (12).
Genetic background
Overall, PPGL are characterized by the highest rate of hereditary background compared to other human neoplasms. In fact, approximately 40–50% of cases are related to germline mutations in one of the known susceptibility genes and in 40-60% of sporadic cases a somatic mutation is found (19–21). Moreover, the hereditary rate rises to 70-80% in the pediatric setting (7, 9, 22). Baush et al. (7) reported an even higher 80% frequency of germline mutations among pediatric cases. Early onset of disease, bilateral multifocal, extra-adrenal, and malignant tumors are the clinical hallmarks of hereditary disease. Moreover, children usually presented symptomatic and potentially life-threatening disease (6).
Specific germline mutations have been attributed to at least 25 tumor-susceptibility genes (19, 20) that could be divided into three cluster groups based on transcriptomic profiles revealed by gene expression microarray analyses (23–26). This proportion of implicated genes will progressively increase as mutations impacting new genes are found (21).
Cluster 1 tumors comprehend those related to the following mutations: von Hippel-Lindau (VHL) suppressor, the four subunits of the succinate dehydrogenase complex (SDHA, SDHB, SDHC, and SDHD), and less commonly, the enzyme responsible for flavination of the SDHA subunit (SDHAF2), fumarate hydratase (FH), malate dehydrogenase 2 (MDH2), prolyl hydroxylase (PHD), and somatic gain-of-function pathogenic variants in the hypoxia-inducible factor 2 alpha gene (HIF2A or EPAS1) and some newly discovered genes that will be detailed in Table 1. Cluster 1 mutations result in stabilization of hypoxia-inducible factors and activation of the hypoxia signaling pathways (25, 26).
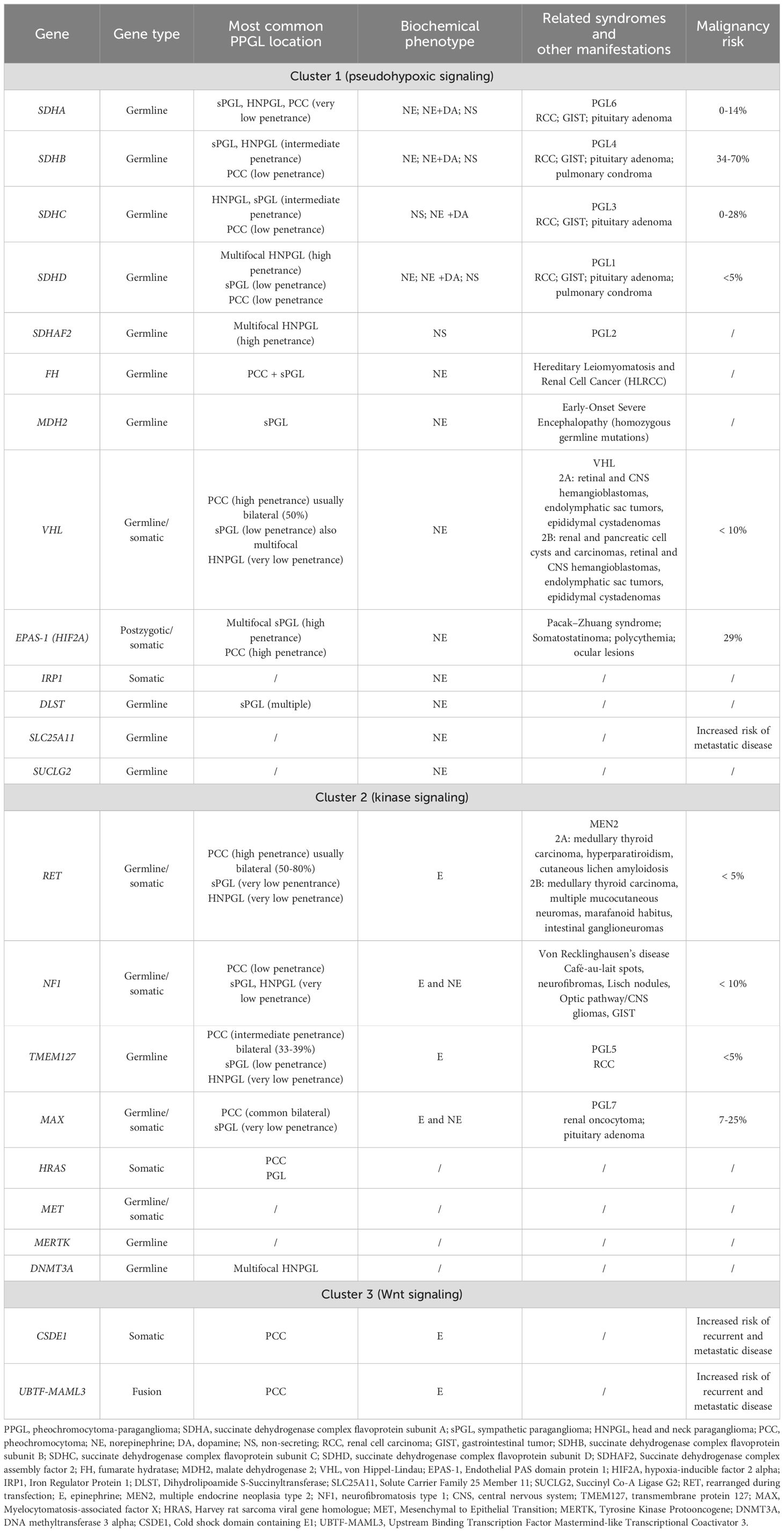
Instead, cluster 2 tumors include neoplasms due to mutations of the neurofibromatosis type 1 (NF1) tumor suppressor gene, the rearranged during transfection (RET) proto-oncogene, genes encoding transmembrane protein 127 (TMEM127), MYC-associated factor X (MAX) and HRAS. Mutations of cluster 2 genes regard activation of kinase receptor signaling pathways, translation initiation, protein synthesis, and pathways involved in maintenance of neural/neuroendocrine identity (26).
Cluster 3 comprehends somatic CSDE1 (Cold shock domain–containing E1 gene) mutations and MAML3 (a member of the Mastermind-like family of transcriptional co-activators) fusion variants implicated in Wnt-pathway signal alterations (25).
The specific genetic background influences the biochemical and clinical phenotypes because the underlying mutation determines a variable expression of biosynthetic enzymes involved in the synthesis of catecholamines by the tumors (28). In more detail, cluster 1 neoplasms are typically characterized by noradrenergic phenotype, on the contrary cluster 2 tumors by adrenergic phenotype (29). Literature data showed that noradrenergic PPGLs typically manifest in younger age that adrenergic ones (30). Based on these assumptions, a study performed by Pamporaki et al. (9) on large cohort of pediatric and adult patients with PPGLs demonstrated, not only a childhood predominance of extra-adrenal, multifocal, metastatic, recurrent, and hereditary PPGLs, but also the link between these phenotypic features to a higher prevalence of noradrenergic and related cluster 1 hereditary tumors in pediatric than adult patients.
As mentioned above, the biochemical properties of PPGL are related to the underlying genetic mutations able to influence differentiation of tumor progenitor cells and consequently the secretory pathways and the epigenetic remodeling profiles (21).
The complex relationship between genotype and phenotype in PPGLs and the characteristics of the main tumor-susceptibility genes and their related hereditary syndromes are summarized in Table 1.
Diagnostic work-up
Biochemical testing is recommended as the initial step in evaluation of suspected PPGL and should include plasma or 24-hours urinary free metanephrines measurement, performed using liquid chromatography assay (5, 8). Taking into account that 24-hour urine collection may not be feasible in young children, a plasma sample is often the initial biochemical test obtained in childhood. Indeed, two studies documented a high diagnostic accuracy of plasma free metanephrines for pediatric patients (31, 32). Pre-analytical considerations are specific challenges that impact upon the interpretation of biochemical tests in pediatric patients. In fact, sympathoadrenal activation triggered by upright posture, distress associated with venepuncture, emotional stress, as well as many medications (e.g. acetaminophen, tricyclic antidepressants, amphetamines,.) should be carefully evaluated. In general, normetanephrine or metanephrine values three-/four-fold or higher above the upper limit of the reference ranges require anatomical imaging for tumor localization and staging. Initial imaging studies include either contrasted enhanced computed tomography (CT) or magnetic resonance imaging (MRI), given their similar diagnostic performance (33). However, MRI is the preferred imaging modality for patients with head and neck PPGL and in those with metastatic disease and it is necessary if the initial imaging of the abdomen and pelvis does not identify the neoplasia. Moreover, functional imaging is a complementary technique useful for disease staging, detection of metastases or recurrent/multiple tumors. However, in childhood the indication for functional imaging must be carefully balanced against the radiation risk. The different functional imaging modalities include 68Ga-DOTATATE, 18F-fluorodopa (FDOPA), 18F-fluorodeoxyglucose (FDG) PET/TC and 123I-metaiodobenzylguanidine (123I-MIBG). The choice of the specific functional tracers should be guided by the known or suspected genetic mutation of the patient (34).
Prevention and treatment of PPGL-induced hypertensive crisis
The most important way to approach catecholamine-induced hypertensive crisis is the prevention of their onset. Therefore, the first part of this section is dedicated to the preoperative medical management.
Preoperative prevention of hypertensive crisis
To reduce the impact of hypertensive crises, potentially occurring during intraoperative manipulation of the tumor, adequate preoperative oral anti-hypertensive treatment is required.
From a pathophysiological perspective, a sequential approach with initial α-adrenergic blockade followed by β-adrenergic blockers is recommended to avoid reflex tachycardia (Table 2). Normalization of blood pressure and heart rate in pediatric patients requires longer treatment than in adults; therefore, compared with adults, it is recommended to start the treatment at least 14 days before surgery. This is probably related to the low-dose regimen and progressive titration of drugs to avoid side effects and to the fact that children are more sensitive to sympathetic overactivity (9, 11). In addition to antihypertensive therapy, adequate salt and water supplementation is required to avoid orthostatic hypotension related to volume contraction due to catecholamine release and drug side effects. After the initiation of α-blocker therapy, a supplementation of 6 to 10 grams per day of salt, depending on patient’s body surface area and an increase in fluid intake up to 1.5 times the weight-corrected levels are recommended.
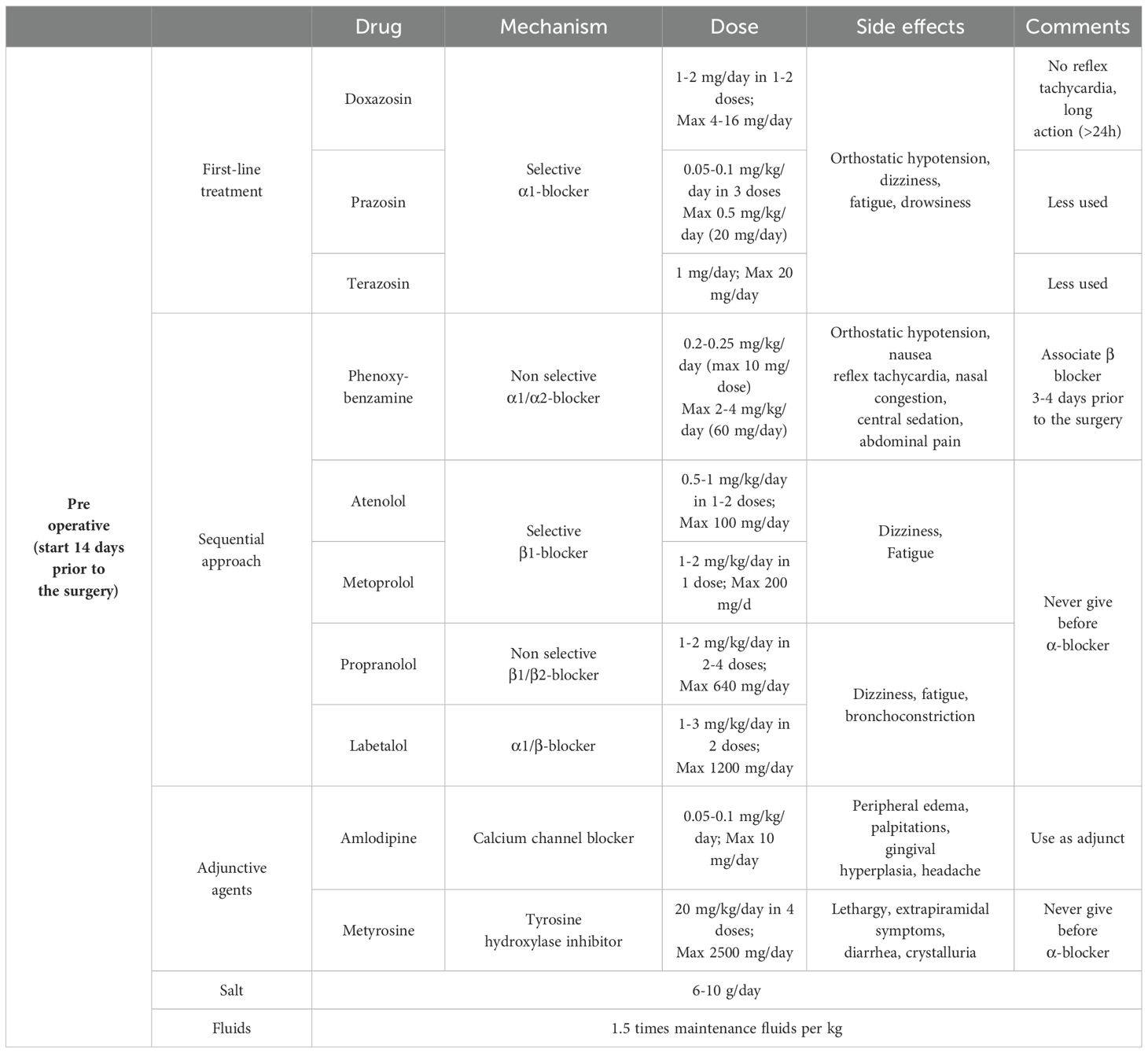
The goal is to achieve a blood pressure within the 50-90th percentile for age, gender and height, aiming to obtain values closer to the 50th percentile in the very last preoperative days.
The most used drugs are long-acting non-selective α1/α2 receptor blockers such as phenoxybenzamine or long-acting selective α1 receptor blockers such as doxazosin, prazosin or terazosin. The most frequent side effect is orthostatic hypotension. Non-selective α1/α2 receptor blockers additional side effects also include reflex tachycardia, central sedation, nasal congestion, nausea and abdominal pain. Selective α1 receptor blockers generally have a better side effect profile, with no reflex tachycardia and more prolonged activity (>24 hours). The risk of postoperative hypotension with selective blockers is also low, therefore they are used preferentially in many centers.
Second-line or adjunctive agents include calcium channel blockers, such as amlodipine or nifedipine and tyrosine hydroxylase inhibitors, such as metyrosine. The hypotensive effect of calcium channel blockers is minor, and their use is usually limited when blood pressure control with selective and nonselective α-blockers is ineffective or in case of severe adverse effects. Metyrosine is used for short periods before surgery in combination with α-blockers to provide better pre- and intraoperative blood pressure control. Side effect profile include extrapyramidal symptoms, diarrhea, orthostatic hypotension, drowsiness, xerostomia, and neuromuscular symptoms, as well as crystalluria, therefore its use in pediatric age is limited.
When nonselective α-blockers are used, β-adrenergic blockers are often added a few days before surgery to relieve α-blockers-related reflex tachycardia. β-adrenergic blockers should never be started before α-blockers because catecholamine-related vasoconstriction would trigger a hypertensive crisis. Selective β1-adrenergic blockers, such as atenolol and metoprolol, are preferred over nonselective β1/β2-blockers (propranolol) because of the risk of bronchoconstriction. Another nonselective β-blocker agent, with combined selective α1-blockade is labetalol. The latter should not be used alone for the high risk of hypertensive crisis (8–11).
General and intraoperative management of PPGL-induced hypertensive crisis
In case of hypertension urgency where children can tolerate oral therapy, intravenous administration may be initially avoided. However, hypertensive crises in children with PPGL occur predominantly during intraoperative manipulation of the tumor, even despite adequate preoperative treatment. Therefore, continuous invasive blood pressure monitoring by intra-arterial catheterization is essential during surgery to help anesthesiologists in assessing blood pressure fluctuations (8). In case of intraoperative hypertensive crisis, intravenous administration of short-acting antihypertensive drugs is mandatory. These indications can be also applied to the general management of hypertensive crisis out of the surgical setting.
From a pathophysiological perspective, as mentioned earlier, the drugs of choice to treat PPGL hypertensive crises are α-blockers, such as urapidil, a combined peripheral selective α1 receptor antagonist and central serotoninergic 1A receptor agonist, or phentolamine, a non-selective α1/α2 receptor antagonist. Urapidil is particularly useful, as it does not induce reflex tachycardia and is not associated with alterations of the renin-angiotensin-aldosterone system, thereby minimizing side effects. However, there are limited case series on the use of this drug in the pediatric population (14, 35). One of these is a multicentric Italian retrospective survey on treatment of hypertension in children with neuroblastoma. Intraoperative hypertension management was somewhat dissimilar among the participating centers, apart from a certain consistency in the intraoperative use of urapidil (36). Side effects associated with α-blockers in PPGL patients are rare and generally not severe. The main side effects reported in literature include excessive hypotension, tachycardia, dizziness, and nasal congestion (8, 10, 37).
In cases of hypertensive crises accompanied by tachyarrhythmias, when α-blockade is achieved, short-acting intravenous β-blockers may be utilized in combination, including labetalol, an α1-β blocker with a 1:7 ratio for intravenous administration, and esmolol, a selective β1-antagonist (10).
Although the majority of experience with esmolol, especially in little children younger than 6 years, is in management hypertension during and after intervention for aortic coarctation repair (38). Romero et al. (39) in a retrospective analysis of medical records of 10 children (from the age of 6 months to 18 years), that were diagnosed with a catecholamine secreting tumor from 2005–2013 and underwent surgical removal, showed how 80% of these patients experimented hypertension crises during surgery and most of them were treated with esmolol, labetalol, but also sodium nitroprusside. The disadvantage is the same as in all β-blockers, namely its negative inotropic effect and potential for bronchoconstriction. Consequently, it should be avoided in children with asthma or those suffering from decompensated or unstable congestive heart failure (14). The principal side effects of this class of drugs are dizziness, bradycardia and risk of atrio-ventricular block (10).
Second-third line treatments for hypertensive crises in children with PPGL include calcium-channel blockers, particularly second-generation dihydropyridines such as nicardipine, and nitroderivatives. Among these options, sodium nitroprusside, a preferential arterial vasodilation, is the drug of choice, because nitroglycerin, reducing cardiac preload by venous vasodilation, can potentially induce significant reflex tachycardia (10, 14). There is notable experience in using nicardipine for managing severe hypertension in children (40, 41). The main side effects observed were related to its vasodilatory effects, including tachycardia, flushing, palpitations, and hypotension. Sodium nitroprusside treatment provides additional benefits for controlling coronary vasospasm. However, it requires careful monitoring due to the potential risks of cyanide and thiocyanate toxicity. In addition, precautions must be taken to protect it from exposure to light (10, 14). Finally, in children with hypertensive crises due to PPGL, as an adjunctive therapy, treatment with dexmedetomidine, a central α2-agonist, and magnesium sulphate, a vasodilator that also inhibits catecholamine release from the adrenal medulla and sympathetic nerve endings, has been described (8, 10). In particular, magnesium sulphate is a valid and safe alternative for the pediatric population and is also suitable for use in pregnant women (42, 43). The main side effects of dexmedetomidine include respiratory depression and bradycardia. Conversely, magnesium sulphate should be used with caution in individuals with neuromuscular disorders due to the risk of paralysis (8, 10). Details about all these treatments are described in Table 3. In case of intraoperative hypotension, intravenous infusion of crystalloid or colloid fluids and administration of vasoactive agents may be necessary, with particular awareness in case of catecholamine-induced cardiomyopathy, as these patients are at risk for pulmonary edema secondary to volume overload.
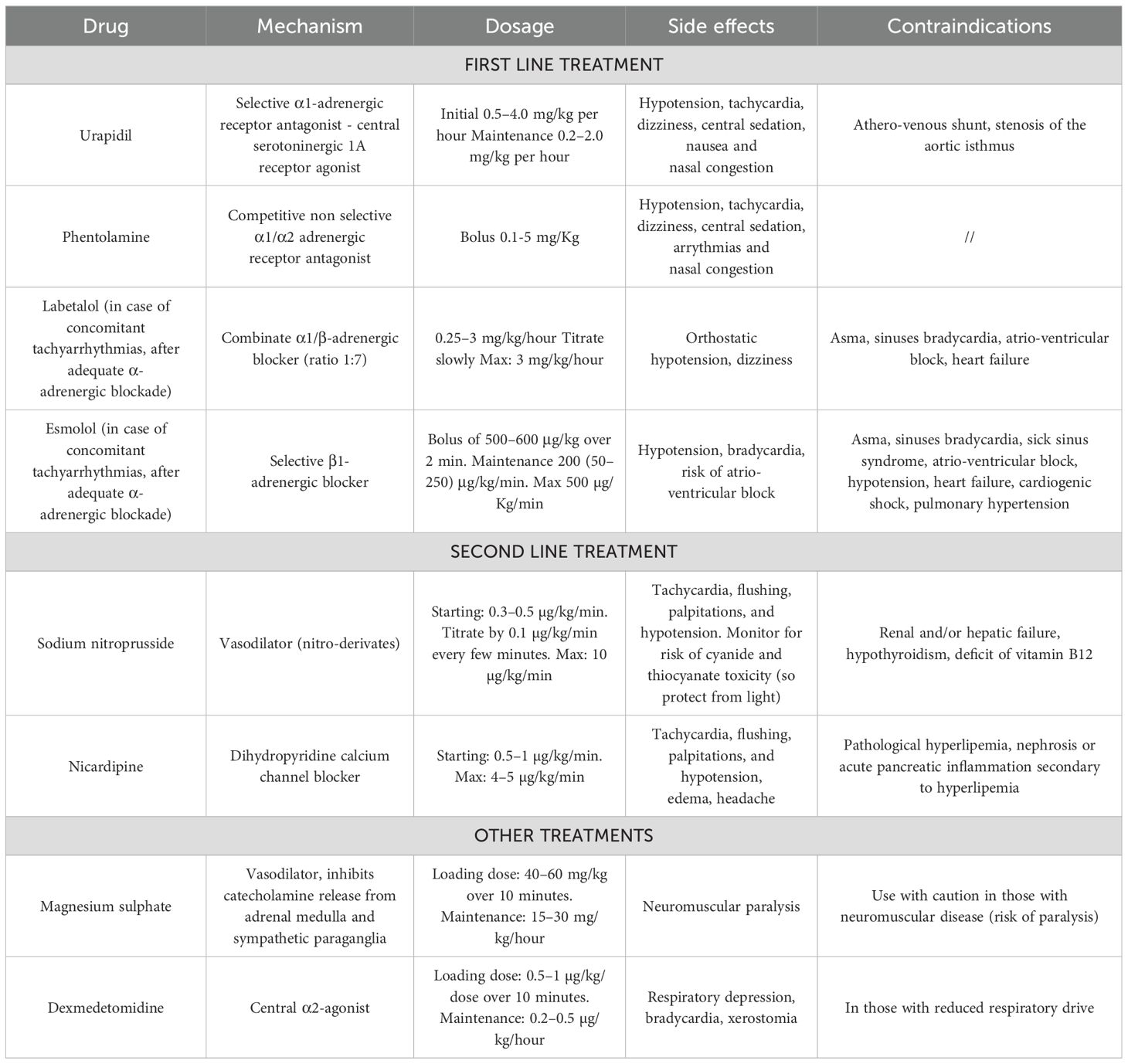
Table 3 Management of hypertensive crises induced by catecholamine secreting tumors in children (8, 10, 11, 14).
Postoperative complication management
Continuous monitoring in the first 48 hours after surgery is indicated for a high risk of hemodynamic instability. Multifactorial hypotension, responsive to colloid/crystalloid infusion, may occur due to the downregulation of adrenergic receptors and acute withdrawal of catecholamines after surgical removal of the mass, as well as the prolonged action of antihypertensive agents used in the preoperative period and the short-term effect of intraoperative management. Hypoglycemia may also occur postoperatively due to rebound hyperinsulinemia, which results from the loss of the inhibitory action of catecholamines on pancreatic β-cells. This condition can be treated with the infusion of glucose-containing fluids (8, 10).
Conclusions
Although hypertensive crises in children are rare conditions, the precise determination of their prevalence is more challenging than in adults, due to the heterogeneity in its definition. Catecholamine excess represents a rare cause of hypertensive crisis, the management of which is complex and requires careful selection and timing of therapy administration, even more in pediatric patients. Therefore, it would be advisable to manage these cases in tertiary care centers through interdisciplinary collaboration involving endocrinologists, pediatric intensivists, anesthesiologists, nephrologists, emergency physicians and surgeons to optimize therapeutic success (8–10).
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Yang WC, Lin MJ, Chen CY, Wu HP. Clinical overview of hypertensive crisis in children. World J Clin Cases. (2015) 3:510. doi: 10.12998/wjcc.v3.i6.510
2. Patel NH, Romero SK, Kaelber DC. Evaluation and management of pediatric hypertensive crises: Hypertensive urgency and hypertensive emergencies. Open Access Emergency Med. (2012) 4:85–92. doi: 10.2147/OAEM.S32809
3. Raina R, Mahajan Z, Sharma A, Chakraborty R, Mahajan S, Sethi SK, et al. Hypertensive crisis in pediatric patients: an overview. Front Pediatr. (2020) 8:588911. doi: 10.3389/fped.2020.588911
4. Lurbe E, Agabiti-Rosei E, Cruickshank JK, Dominiczak A, Erdine S, Hirth A, et al. 2016 European Society of Hypertension guidelines for the management of high blood pressure in children and adolescents. J Hypertens. (2016) 34:1887–920. doi: 10.1097/HJH.0000000000001039
5. Bholah R, Bunchman TE. Review of pediatric pheochromocytoma and paraganglioma. Front Pediatr. (2017) 5. doi: 10.3389/fped.2017.00155
6. Ciftci AO, Tanyel FC, Şenocak ME, Büyükpamukçu N. Pheochromocytoma in children. J Pediatr Surg. (2001) 36:447–52. doi: 10.1053/jpsu.2001.21612
7. Bausch B, Wellner U, Bausch D, Schiavi F, Barontini M, Sanso G, et al. Long-term prognosis of patients with pediatric pheochromocytoma. Endocr Relat Cancer. (2014) 21:17–25. doi: 10.1530/ERC-13-0415
8. Jain A, Baracco R, Kapur G. Pheochromocytoma and paraganglioma—an update on diagnosis, evaluation, and management. Pediatr Nephrol. (2020) 35:581–94. doi: 10.1007/s00467-018-4181-2
9. Pamporaki C, Hamplova B, Peitzsch M, Prejbisz A, Beuschlein F, Timmers HJLM, et al. Characteristics of pediatric vs adult pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. (2017) 102:1122–32. doi: 10.1210/jc.2016-3829
10. Seamon ML, Yamaguchi I. Hypertension in pheochromocytoma and paraganglioma: evaluation and management in pediatric patients. Curr Hypertens Rep. (2021) 23(5):32. doi: 10.1007/s11906-021-01150-9
11. Nazari MA, Hasan R, Haigney M, Maghsoudi A, Lenders JWM, Carey RM, et al. Catecholamine-induced hypertensive crises: current insights and management. Lancet Diabetes Endocrinol. (2023) 11:942–54. doi: 10.1016/S2213-8587(23)00256-5
12. Gupta G, Pacak K. Precision medicine: an update on genotype/biochemical phenotype relationships in pheochromocytoma/paraganglioma patients. Endocr Pract. (2017) 23:690–704. doi: 10.4158/EP161718.RA
13. Eisenhofer G, Pamporaki C, Lenders JWM. Biochemical assessment of pheochromocytoma and paraganglioma. Endocr Rev. (2023) 44:862–909. doi: 10.1210/endrev/bnad011
14. Seeman T, Hamdani G, Mitsnefes M. Hypertensive crisis in children and adolescents. Pediatr Nephrol. (2019) 34:2523–37. doi: 10.1007/s00467-018-4092-2
15. Harrison DG, Guzik TJ, Lob HE, Madhur MS, Marvar PJ, Thabet SR, et al. Inflammation, immunity, and hypertension. Hypertension. (2011) 57:132–40. doi: 10.1161/HYPERTENSIONAHA.110.163576
16. Derhaschnig U, Testori C, Riedmueller E, Aschauer S, Wolzt M, Jilma B. Hypertensive emergencies are associated with elevated markers of inflammation, coagulation, platelet activation and fibrinolysis. J Hum Hypertens. (2013) 27:368–73. doi: 10.1038/jhh.2012.53
17. Prejbisz A, Lenders JWM, Eisenhofer G, Januszewicz A. Cardiovascular manifestations of phaeochromocytoma. J Hypertens. (2011) 29:2049–60. doi: 10.1097/HJH.0b013e32834a4ce9
18. Petrák O, Rosa J, Holaj R, Štrauch B, Krátká Z, Kvasnička J, et al. Blood pressure profile, catecholamine phenotype, and target organ damage in pheochromocytoma/paraganglioma. J Clin Endocrinol Metab. (2019) 104:5170–80. doi: 10.1210/jc.2018-02644
19. Gimenez-Roqueplo AP, Robledo M, Dahia PLM. Update on the genetics of paragangliomas. Endocr Relat Cancer. (2023) 30:e220373. doi: 10.1530/ERC-22-0373
20. Lenders JWM, Kerstens MN, Amar L, Prejbisz A, Robledo M, Taieb D, et al. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: A position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J Hypertens. (2020) 38:1443–56. doi: 10.1097/HJH.0000000000002438
21. Jhawar S, Arakawa Y, Kumar S, Varghese D, Kim YS, Roper N, et al. New insights on the genetics of pheochromocytoma and paraganglioma and its clinical implications. Cancers (Basel). (2022) 14:594. doi: 10.3390/cancers14030594
22. Redlich A, Pamporaki C, Lessel L, Frühwald MC, Vorwerk P, Kuhlen M. Pseudohypoxic pheochromocytomas and paragangliomas dominate in children. Pediatr Blood Cancer. (2021) 68(7):e28981. doi: 10.1002/pbc.28981
23. Gimenez-Roqueplo AP, Dahia PL, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res. (2012) 44:328–33. doi: 10.1055/s-0031-1301302
24. Dahia PLM. Pheochromocytoma and paraganglioma pathogenesis: Learning from genetic heterogeneity. Nat Rev Cancer. (2014) 14:108–19. doi: 10.1038/nrc3648
25. Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell. (2017) 31:181–93. doi: 10.1016/j.ccell.2017.01.001
26. Jochmanova I, Pacak K. Genomic landscape of pheochromocytoma and paraganglioma. Trends Cancer. (2018) 4:6–9. doi: 10.1016/j.trecan.2017.11.001
27. Crona J, Taïeb D, Pacak K. New perspectives on pheochromocytoma and paraganglioma: Toward a molecular classification. Endocr Rev. (2017) 38:489–515. doi: 10.1210/er.2017-00062
28. Eisenhofer G, Huynh TT, Elkahloun A, Morris JC, Bratslavsky G, Marston Linehan W, et al. Differential expression of the regulated catecholamine secretory pathway in different hereditary forms of pheochromocytoma. Am J Physiol Endocrinol Metab. (2008) 295:E1223–33. doi: 10.1152/ajpendo.90591.2008
29. Qin N, De Cubas AA, Garcia-Martin R, Richter S, Peitzsch M, Menschikowski M, et al. Opposing effects of HIF1α and HIF2α on chromaffin cell phenotypic features and tumor cell proliferation: Insights from MYC-associated factor X. Int J Cancer. (2014) 135:2054–64. doi: 10.1002/ijc.28868
30. Eisenhofer G, Timmers HJ, Lenders JWM, Bornstein SR, Tiebel O, Mannelli M, et al. Age at diagnosis of pheochromocytoma differs according to catecholamine phenotype and tumor location. J Clin Endocrinol Metab. (2011) 96:375–84. doi: 10.1210/jc.2010-1588
31. Weise M, Merke DP, Pacak K, Walther MM, Eisenhofer G. Utility of plasma free metanephrines for detecting childhood pheochromocytoma. J Clin Endocrinol Metab. (2002) 87:1955–60. doi: 10.1210/jcem.87.5.8446
32. Sarathi V, Pandit R, Patil VK, Lia AR, Bandgar TR, Shah NS. Performance of plasma fractionated free metanephrines by enzyme immunoassay in the diagnosis of pheochromocytoma and paraganglioma in children. Endocrine Practice. (2012) 18:694–9. doi: 10.4158/EP12050.OR
33. Hanafy AK, Mujtaba B, Roman-Colon AM, Elsayes KM, Harrison D, Ramani NS, et al. Imaging features of adrenal gland masses in the pediatric population. Abdom Radiol (NY). (2020) 45:964–81. doi: 10.1007/s00261-019-02213-x
34. Taïeb D, Hicks RJ, Hindié E, Guillet BA, Avram A, Ghedini P, et al. European Association of Nuclear Medicine Practice Guideline/Society of Nuclear Medicine and Molecular Imaging Procedure Standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging. (2019) 46:2112–37. doi: 10.1007/s00259-019-04398-1
35. Schober JG, Pilossoff W, Biihlmeyer K. Pediatrics Urapidil therapy for acute hypertensive crises in infants and children. Eur J Eur J Pediatr. (1984) 143:87–91. doi: 10.1007/BF00445791
36. Pio L, Avanzini S, Mattioli G, Martucciello G, Sementa AR, Conte M, et al. Perioperative management of hypertensive neuroblastoma: A study from the Italian Group of Pediatric Surgical Oncologists (GICOP). J Pediatr Surg. (2017) 52:1633–6. doi: 10.1016/j.jpedsurg.2017.06.027
37. Eigelberger MS, Duh QY. Pheochromocytoma. Curr Treat Options Oncol. (2001) 2:321–9. doi: 10.1007/s11864-001-0025-5
38. Tabbutt S, Nicolson SC, Adamson PC, Zhang X, Hoffman ML, Wells W, et al. The safety, efficacy, and pharmacokinetics of esmolol for blood pressure control immediately after repair of coarctation of the aorta in infants and children: A multicenter, double-blind, randomized trial. J Thorac Cardiovasc Surgery. (2008) 136:321–8. doi: 10.1016/j.jtcvs.2007.09.086
39. Romero M, Kapur G, Baracco R, Valentini RP, Mattoo TK, Jain A. Treatment of hypertension in children with catecholamine-secreting tumors: A systematic approach. J Clin Hypertens. (2015) 17:720–5. doi: 10.1111/jch.12571
40. Flynn AL, Mottes TA, Brophy PD, Kershaw DB, Smoyer WE, Bunchman TE. Intravenous nicardipine for treatment of severe hypertension in children. J Pediatr. (2001) 139:38–43. doi: 10.1067/mpd.2001.114030
41. Michael J, Groshong T, Tobias JD. Nicardipine for hypertensive emergencies in children with renal disease. Pediatr Nephrol. (1998) 12:40–2. doi: 10.1007/s004670050400
42. Bryskin R, Weldon BC. Dexmedetomidine and magnesium sulfate in the perioperative management of a child undergoing laparoscopic resection of bilateral pheochromocytomas. J Clin Anesth. (2010) 22:126–9. doi: 10.1016/j.jclinane.2009.01.017
Keywords: juvenile hypertension, catecholamines, pheochromocytoma, paraganglioma, secondary hypertension, hypertensive crisis, children
Citation: Bima C, Lopez C, Tuli G, Munarin J, Arata S, Procopio M, Bollati M, Maccario M, De Sanctis L and Parasiliti-Caprino M (2024) Prevention and management of hypertensive crises in children with pheochromocytoma and paraganglioma. Front. Endocrinol. 15:1460320. doi: 10.3389/fendo.2024.1460320
Received: 05 July 2024; Accepted: 05 August 2024;
Published: 20 August 2024.
Edited by:
Rosario Ferrigno, AORN Santobono-Pausilipon, ItalyReviewed by:
Alejandro Gregorio Martinez-Aguayo, Pontificia Universidad Católica de Chile, ChileElsa Gabriela Sanso, Hospital General de Niños Ricardo Gutierrez, Argentina
Copyright © 2024 Bima, Lopez, Tuli, Munarin, Arata, Procopio, Bollati, Maccario, De Sanctis and Parasiliti-Caprino. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mirko Parasiliti-Caprino, bWlya28ucGFyYXNpbGl0aWNhcHJpbm9AdW5pdG8uaXQ=
†These authors share first authorship
‡These authors share second authorship
§These authors share last authorship