 Ying Hao
Ying Hao Ningning Yang1†
Ningning Yang1† Xingjuan Chen
Xingjuan Chen- 1College of Sports, Northwest Normal University, Lanzhou, China
- 2Institute of Medical Research, Northwestern Polytechnical University, Xi’an, China
Osteoporosis, a systemic skeletal disorder marked by diminished bone mass and compromised bone microarchitecture, is becoming increasingly prevalent due to an aging population. The underlying pathophysiology of osteoporosis is attributed to an imbalance between osteoclast-mediated bone resorption and osteoblast-mediated bone formation. Osteoclasts play a crucial role in the development of osteoporosis through various molecular pathways, including the RANK/RANKL/OPG signaling axis, cytokines, and integrins. Notably, the calcium signaling pathway is pivotal in regulating osteoclast activation and function, influencing bone resorption activity. Disruption in calcium signaling can lead to increased osteoclast-mediated bone resorption, contributing to the progression of osteoporosis. Emerging research indicates that calcium-permeable channels on the cellular membrane play a critical role in bone metabolism by modulating these intracellular calcium pathways. Here, we provide an overview of current literature on the regulation of plasma membrane calcium channels in relation to bone metabolism with particular emphasis on their dysregulation during the progression of osteoporosis. Targeting these calcium channels may represent a potential therapeutic strategy for treating osteoporosis.
Introduction
Osteoporosis is a systemic skeletal disorder characterized by decreased bone mass and deterioration of bone microarchitecture, resulting in increased bone fragility and susceptibility to fractures (1, 2). According to a prevalence report encompassing 86 studies across five continents, the global prevalence rate of osteoporosis stands at 18.3%, with only 31-36% of individuals aged over 70 maintaining normal bone health. The remaining population exhibits varying degrees of osteopenia or osteoporosis (3). This number is expected to increase further due to the growing aging population. The incidence of osteoporotic fractures exceeds three to four times that of cardiovascular disease or cancer. A report in the US indicated that due to demographic changes, by 2040, approximately 25% of individuals over age 50 who have experienced hip fractures related to osteoporosis are expected to die within one year (4). The individual and societal impacts posed by osteoporosis and its complications present significant challenges for healthcare systems.
Primary osteoporosis includes postmenopausal and senile forms primarily attributed to declining estrogen levels and aging process (3, 5). Secondary osteoporosis is caused by underlying diseases or their treatments, including cardiovascular, neurological, endocrine disorders, and malignancies, as well as long-term glucocorticoid use, lifestyle factors, and major depression (6–9). Bone remodeling, a lifelong process, involves osteoclast-mediated bone resorption and osteoblast-driven bone formation (10–13). Additionally, osteocytes embedded within the bone matrix also play a role in this remodeling process, they are currently considered as the main source of molecules regulating the osteoclast and osteoblast activity, such as receptor activator of nuclear factor-κB ligand (RANKL) and sclerostin. Osteocytes detect and respond to mechanical and hormonal stimuli to coordinate both bone resorption and formation (14, 15). The primary pathological mechanism in osteoporosis is increased bone resorption due to abnormal osteoclast differentiation and proliferation (Figure 1) (16, 17). Consequently, treatment strategies focus on targeting osteoclast activity (18).
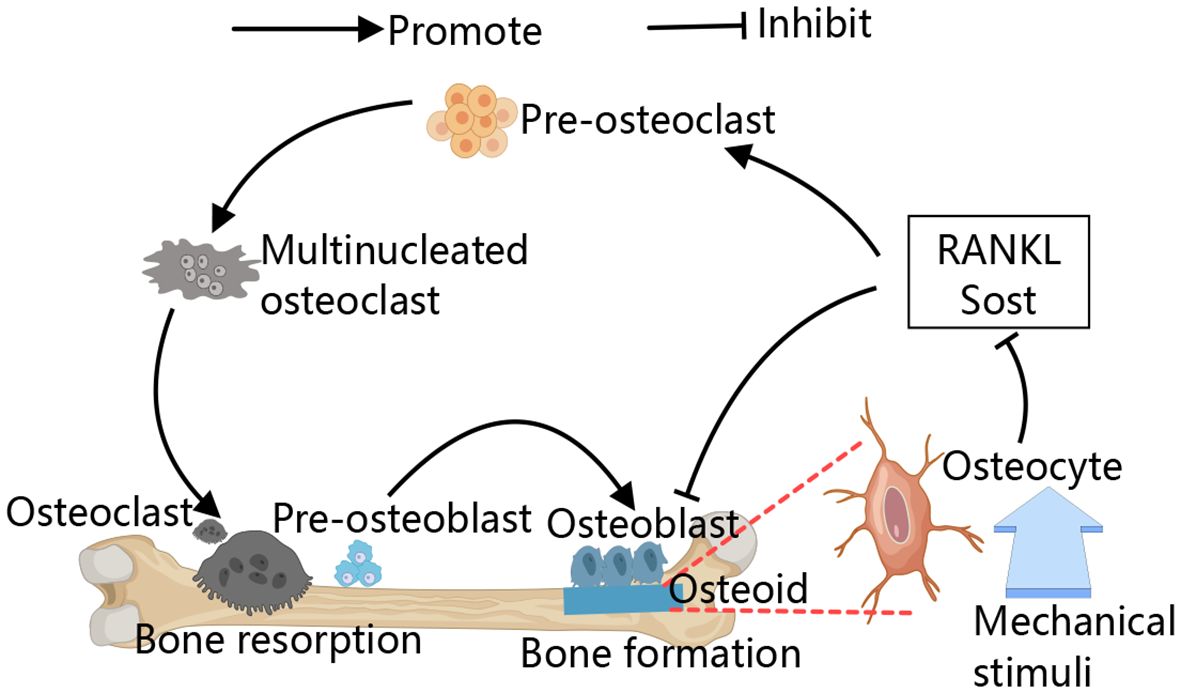
Figure 1 Schematic diagram of the bone remodeling. The process of bone remodeling begins with the recruitment of osteoclast precursors, which fuse to form multinucleated, active osteoclasts that mediate bone resorption. After resorption, osteoclasts leave the site, allowing osteoblasts to move in and cover the excavated area. Osteoblasts then initiate new bone formation by secreting osteoid. Osteocytes detect and respond to mechanical stimuli to regulate bone remodeling by regulating their secreted cytokines, for example, RANKL and sclerostin.
Additionally, recent studies suggest a correlation between bone microvessels and bone loss, with reduced trans-cortical vessels (TCV) observed in osteoporosis models and a positive correlation between TCV numbers and bone mass (19–21). Trans-cortical vessels are capillaries that run vertically through the cortical bone, connecting the endosteal and periosteal surfaces (20). Xiao CL et al. used old mice, ovariectomy mice, and db/db mice as osteoporosis models, finding reduced TCV in all models, which correlated positively with bone mass (20).
The regulation of osteoclasts formation and differentiation involves a complex interplay of cytokines, hormones, immune factors, gut microbiota, and cellular aging (22–25). Among the various cytokines and hormones that regulate osteoclasts formation and function, macrophage colony-stimulating factor (M-CSF) and RANKL are the most critical molecules. Senescent cells can produce senescence-associated secretory phenotypes (SASP), which exert deleterious paracrine and systemic effects, including the development of osteoporosis (26, 27). Comparative analysis between young mice (6 months old) and old mice (24 months old) revealed significantly higher expression levels of multiple SASP markers in osteocytes from the latter group (28). SASP has been reported to promote osteoclastogenesis by enhancing the survival of monocyte osteoclast progenitors. Moreover, inhibition of SASP using the JAK1/2 inhibitor Ruxolitinib has been shown to prevent age-related bone loss (29).
As a crucial intracellular second messenger, Ca2+ plays a significant role in the regulation of osteoclast differentiation and bone resorption (30). Many studies have shown that osteoclast dysfunction is often accompanied by increased intracellular calcium levels (17). Consequently, there has been a surge of interest in devising strategies to modulate the intracellular calcium system as a pivotal approach for regulating osteoclast function. Ca2+-permeable channels located on cell membranes are an essential component of the calcium signaling system, mediating the influx of extracellular Ca2+. Recently, accumulating evidence has highlighted the critical role these channels play in maintaining dynamic bone metabolism. In this review, we present a summary of existing research on the control of calcium channels in the plasma membrane concerning bone health, with specific focus on their irregularities during the development of osteoporosis. The exploration of these calcium channels as a potential therapeutic approach for managing osteoporosis is discussed.
Calcium signaling in the process of bone remodeling
Calcium signaling plays a crucial role in bone metabolism, with imbalanced Ca2+ homeostasis significantly affecting osteoblast activity and bone formation. Excessive Ca2+ loading in osteoblasts can limit differentiation by inducing apoptosis in mitochondria and the endoplasmic reticulum (ER) (31, 32). Mitochondria are essential for normal bone formation; however, elevated intracellular calcium levels can disrupt the function of the inner mitochondrial membrane, thereby impairing bone formation (33, 34). Similarly, high intracellular calcium levels can cause ER stress, leading to apoptosis in osteoblasts (35). Bone marrow mesenchymal stem cells (BMMSCs) differentiate into osteoblasts and regulate osteoclast activity through the secretion of RANKL and osteoprotegerin (OPG), maintaining bone metabolism balance. BMMSCs play a crucial role in bone remodeling by directly forming new bone and indirectly influencing bone resorption. Li et al. reported disrupted intracellular calcium homeostasis in bone samples from osteoporosis patients and mice, leading to impaired osteoblast differentiation and compromised bone formation (36).
In osteoclast differentiation and function, calcium signaling is key (37) (Figure 2). RANKL utilizes this pathway to activate NFATC, promoting osteoclast formation (38). RANKL/RANK signaling transactivates phospholipase C (PLC), producing inositol 1,4,5-trisphosphate (IP3), which binds to inositol 1,4,5-trisphosphate receptors (IP3Rs) on the ER, triggering Ca2+ oscillations. This leads to Ca2+ binding to calmodulin, activating calcineurin, which dephosphorylates NFATc1, thus promoting osteoclast differentiation (39–41). Additionally, Kim et al. have identified an upstream signaling pathway for RANKL-induced PLC activation and Ca2+ oscillations. The small G protein Rac1 is the most upstream component activated by RANKL, and its activation can induce long-term production of reactive oxygen species (ROS) and trigger calcium oscillation by activating PLC to promote osteoclast differentiation (42, 43). However, continuous supply of Ca2+ is required for Ca2+ oscillations to reload the stores between spikes. RANKL-induced ER Ca2+ release occurs through the activation of STIM1 on the ER membrane, leading to STIM1 aggregation into puncta at the ER-PM junction. This induces an influx of extracellular calcium via store-operated calcium entry (SOCE), facilitating osteoclast genesis (42–44). Both types of Ca2+ oscillations are terminated when extracellular Ca2+ removing or SOCE blocker Gd3+ are used (43). RANK-bound RANKL also activates TRAF6, leading to NF-κB activation and NFATc1 transcription (45, 46). Studies on (–)-Epicatechin 3-O-β-D-allopyranoside (ECAP) show it inhibits osteoclastogenesis by blocking NF-κB activation and reducing NFATc1 expression (47).
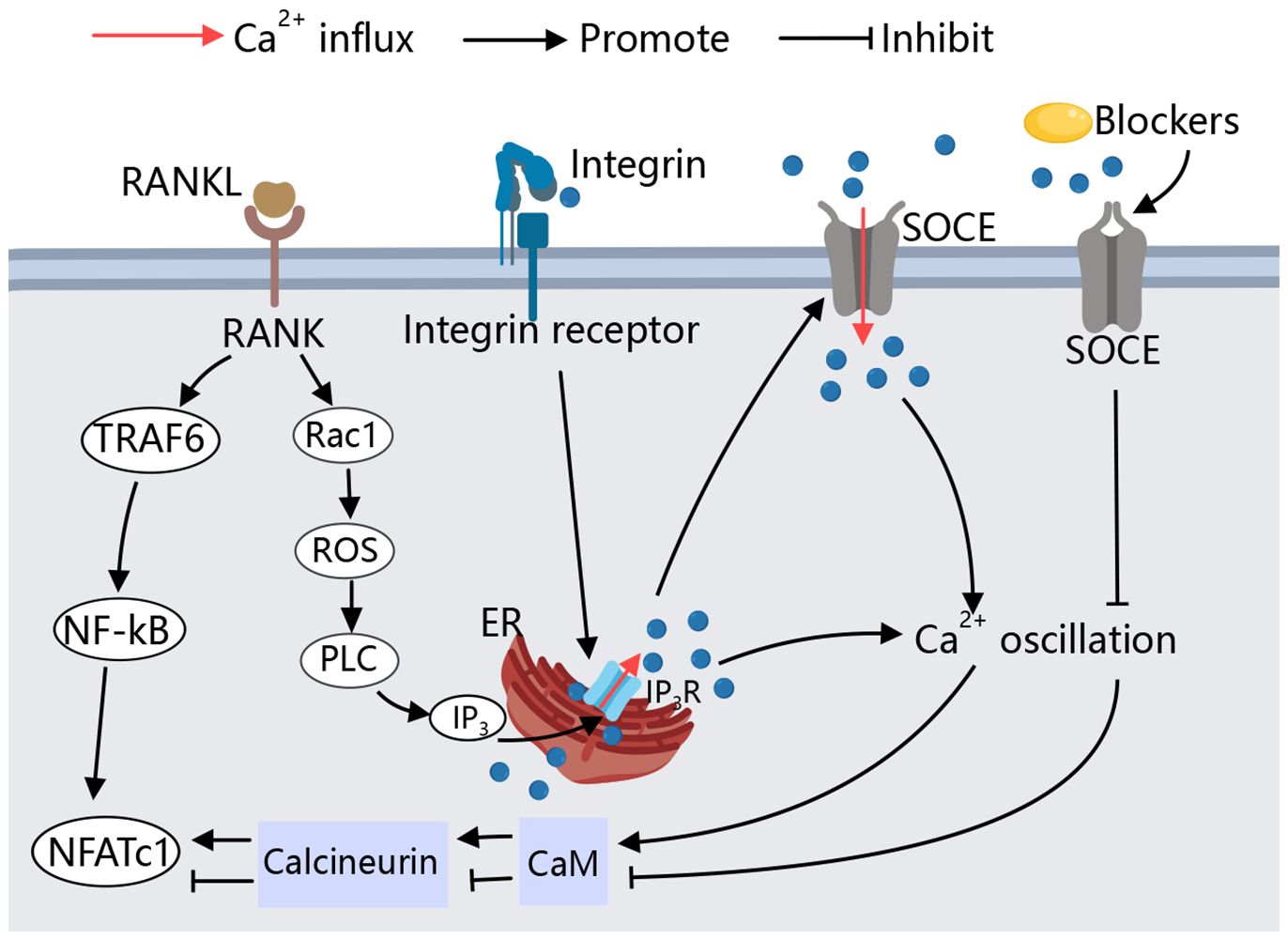
Figure 2 Calcium signaling in osteoclast. During RANKL-mediated osteoclastogenesis, cytoplasmic Ca2+ oscillation, Causing downstream related pathways to activate NFATC1,and thus inducing osteoclast differentiation. SOCE is involved in RANKL-induced Ca2+ oscillations and maintains the sustained production of Ca2+ oscillations, the Ca2+ oscillations induced by RANKL are blocked by SOCE blockers. Specific peptide activation of integrin receptors also causes a transient Ca2+ response, allowing osteoclasts to adhere to the bone matrix.
In the absence of cellular calcium Ca2+, integrins are also able to activate intracellular Ca2+ storage structures by binding to integrin receptors at the cell membrane, induced an increase in intracellular Ca2+ (48). IP3Rs, particularly IP3R2, regulate calcium release from the ER, playing a critical role in osteoclast genesis (49). In cultured osteoclasts and monocyte precursors, increased intracellular Ca2+ lead to decreased adhesion and reduced expression of podosomes, key to osteoclasts function (50).
Maintaining proper intracellular Ca2+ levels is essential for osteoclast differentiation and activity, making calcium signaling pathways potential therapeutic targets for osteoporosis. The calcium permeability channels in the cell membrane are critical for extracellular calcium influx, which is vital for bone metabolism. An imbalance in calcium homeostasis due to these channels can impact the function and differentiation of both osteoblasts and osteoclasts.
Calcium channels in osteoclasts
Calcium channels influence skeletal homeostasis by mediating processes that mediate the extracellular and intracellular Ca2+ balance, Osteoclasts differentiation and apoptosis are widely regulated by Ca2+ channels located in the cell membrane. The regulation of these Ca2+ channels on osteoclasts is detailed below (Table 1).
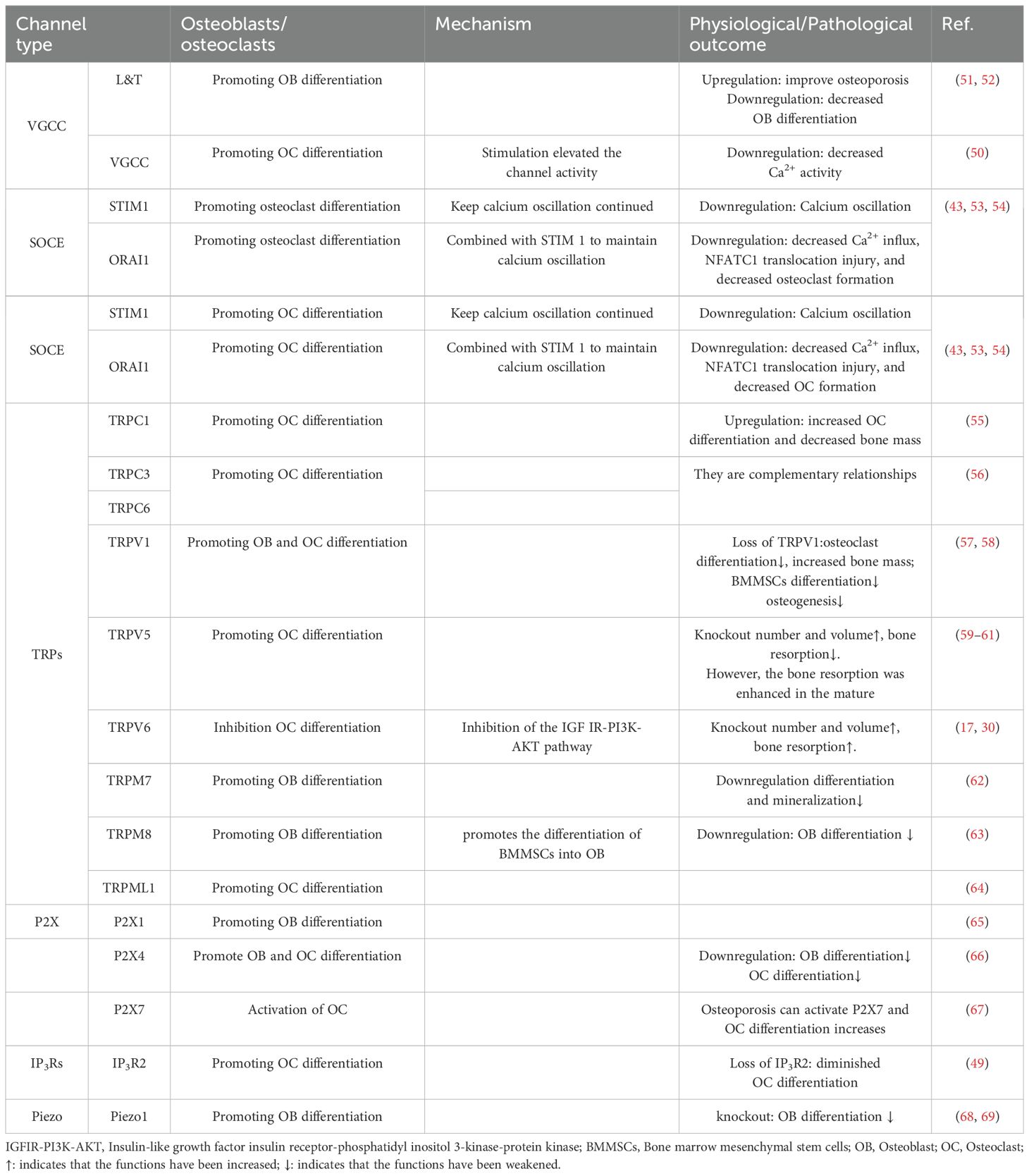
Table 1 Calcium channels and their functions in osteoblasts and osteoclasts.
Voltage-gated calcium channels
Most of the studies on VGCCs have focused on excitable cells such as neuro- and muscle cells. However, an increasing body of research has also demonstrated the involvement of VSCCs in nonexcitable cells, including bone cells and stem cells (70–72). These studies have highlighted the crucial role of VSCCs in bone remodeling. Specifically, disrupting VSCCs or using blockers can significantly impair osteogenesis and result in abnormal bone metabolism (73). The depolarization of the membrane potential activates VGCCs, mediating the influx of extracellular calcium (74). The Ca2+ influx of VGCCs was monitored in cultured osteoclasts of chickens by their depolarization to the membrane. Found that the enhanced activity of VGCCs in stimulated osteoclasts could affect cell adhesion and reduce bone resorption activity (50).
Store-operated calcium entry
SOCE is considered as the primary pathway for calcium release-activated calcium (CRAC) channel activation, involving the ORAI1 channel on the plasma membrane (PM) and the stromal interaction molecule 1 (STIM1) on the ER membrane. When ER calcium ion levels are high, STIM1 remains inactive. However, reduced ER Ca2+ levels activate both STIM1 and STIM2, triggering conformational changes in STIM1, its translocation to the PM, redistribution to ER-PM junctions, interaction with clustered Orai1 channels, and subsequent extracellular calcium influx into cells (75, 76). This process is crucial for maintaining intracellular calcium homeostasis during cellular activation by external stimuli. STIM1 knockout in precursor osteoclasts attenuates Ca2+ oscillations induced by RANKL (43), and ORAI1 knockdown impairs bone mineral resorption and leads to osteoclast deficiency, indicating that SOCE is essential for the Ca2+ oscillation/NFATc1-dependent signaling complex induced by RANKL (53, 54).
Transient receptor potential channels
TRP channels, comprising six subfamilies (TRPC, TRPV, TRPP, TRPM, TRPA, and TRPML), play significant roles in maintaining Ca2+ balance during bone homeostasis (77, 78). TRPC1 inactivation under physiological conditions in mice, and the subsequent upregulation of osteoclasts in MyoD family isoform “A” (I-mfa) knockout mice, suggests the role of TRPC1 in osteoclast differentiation (55). TRPC3 and TRPC6 are associated with osteoporosis, with TRPC3 promoting osteoclast differentiation and bone resorption in TRPC6-deficient phenotypes (56). TRPV channels are extensively documented in osteoblast and osteoclast differentiation. TRPV1 promotes osteoclast differentiation, with TRPV1-deficient mice showing reduced osteoclast numbers (57, 58). TRPV2 facilitates Ca2+ oscillation during osteoclast genesis (79), and TRPV4 is crucial during late osteoclast differentiation stages (80, 81). TRPV5 and TRPV6 regulate osteoclast size and number, with TRPV6 acting as a negative regulator (30, 59–61). Knockdown of TRPV6 resulting in a significant rise in bone resorption (17). Both knockdown and deletion of TRPML1 significantly reduced the differentiation of bone marrow-derived macrophages into osteoclasts (64), highlighting the complex roles of TRP channels in osteoclast function.
Others
Ryanodine receptors (RyRs) and IP3Rs mediate the release of Ca2+ from the endoplasmic reticulum (ER). The activation of PLC leads to the production of IP3, which subsequently binds to IP3Rs on the ER membrane, causing the release of Ca2+ stores. Gene knockout studies suggest that IP3R2 plays a critical role in calcium oscillation during osteoclastogenesis (49), with its absence resulting in a partial defect in osteoclast differentiation (82). RyRs calcium channels may contribute to the release of intracellular calcium stores, while plasma membrane-bound RyR2 potentially regulates osteoclast activity based on extracellular calcium concentration (83).
Purinoceptor 2X (P2X) receptors are ligand-gated ion channels primarily activated by ATP and exhibit significant permeability to Ca2+ (84). The P2X4 receptors are highly expressed in both osteoblasts and osteoclasts, and their inhibitors have been shown to dose-dependently inhibit osteoclastic and osteogenic differentiation (66). Studies examining OPG induced osteoclast adhesion structural changes mediated by mitogen-activated protein kinase (MAPK) signaling via P2X7 receptors indicate that loss of P2X7 receptors inhibits osteoclast activation (67).
Despite the large number of studies demonstrating the regulation of calcium channels in osteoclasts, Compared with mesenchymal osteoblasts and osteocytes, few studies regulate osteoclasts by VGCC, with unclear regulatory mechanisms likely due to the lower sensitivity of VGCCs in non-excitatory cells. Both ORAI1 and STIM1 have shown positive regulation of osteoclasts in the SOCE pathway. The regulation of osteoclast differentiation and activity by the TRP family, particularly TRPC and TRPV channels, is well-documented. Existing studies support the negative regulatory role of the TRPC family on osteoclasts. Additionally, TRPC3 has been found to be highly expressed in individuals with reduced bone mass (56), suggesting that TRPC3 may serve as an early warning signal for osteoporosis. The TRPV family is notably significant in regulating osteoclasts, with its six members influencing osteoclast differentiation and function. This family presents a potential target for osteoporosis treatment, though specific targets within the TRPV channels have yet to be identified. Future research should focus on discovering precise targets within TRPV channels and identifying new Ca2+ channels that could serve as therapeutic targets for osteoporosis.
Calcium channels in osteoblasts
The activation of calcium channels on the osteoblast membrane plays a crucial role in regulating cell differentiation and activity, with varying regulatory processes and effects observed across different types of calcium channels. The regulation of these Ca2+ channels on osteoclasts is outlined as follows (Figure 3).
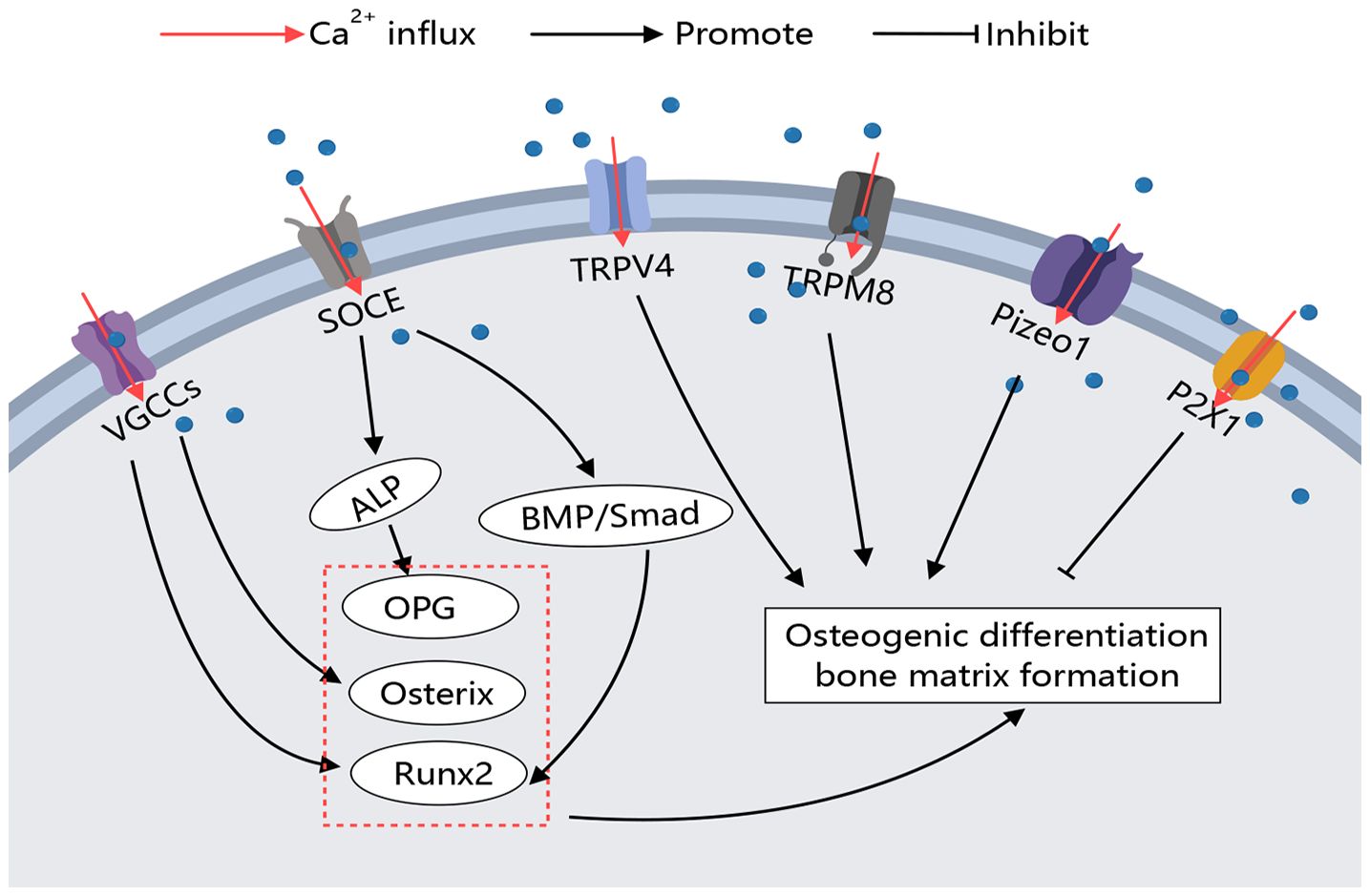
Figure 3 Calcium Channels in Osteoblasts. Various calcium channels on the osteoblast membrane play crucial roles in regulating osteoblast activity. Different types of calcium channels can modulate bone-forming factors, thereby promoting or inhibiting osteoblast differentiation and function through distinct signaling pathways.
VGCCs
The calcium increase caused by bone microinjury area induced the formation of bone matrix. The main mechanism is that extracellular high calcium stimulated cell membrane depolarization, which enabled Ca2+ to enter MC3T3-E1 cells in local areas through L-type and T-type VGCC and promoted bone matrix formation. When L and t-type blockers were used, the calcium inflow activity of cells in the bone micro-damage area was significantly reduced (51). Osteoblasts of the mouse skull were seeded on inactivated bovine bone wafers, and by inducing local diffuse damage near the cells, the cells on the damaged bone wafers showed significant increases in Runx2 and Osterix expression and synthesis of numerous osteocalcin and mineralized nodules. Moreover, when cells were treated with the nonselective VGCC inhibitor bepredil before loading, Runx2 and Osterix expression were significantly inhibited and osteocalcin and mineralized nodule formation was significantly reduced, indicating that the diffuse microlesion-induced Ca2+ efflux activates the anabolic response in osteoblasts by activating VGCCs (52). Functional mutated CaV1.2 mice exhibited elevated serum concentrations of OPG, while isolated BMMSCs displayed a reduced ratio of RANKL to OPG (85). Fei D et al. and Zhang Y et al. identified the downregulation of CaV1.2 in Zmpste24-mouse BMMSCs as a limiting factor for osteogenic differentiation.
In contrast, pharmacological upregulation of CaV1.2 activity alleviated osteoporosis in Zmpste24-/- mice (86). Bei Li et al. discovered that alkaline phosphatase (ALPL) regulates L-type calcium channel trafficking by binding to the α2δ subunit to maintain intracellular calcium homeostasis. Reduced intracellular calcium levels due to alkaline phosphatase deficiency lead to decreased osteogenic differentiation of BMMSCs, but ionomycin can improve the osteoporotic phenotype in alpl-/- mice and BMMSC-specific conditional alpl-/- mice by promoting L-type channel calcium flux (87).
SOCE
In the context of SOCE, ORAI1 deficiency leads to significant reductions in Ca2+ influx, alkaline phosphatase activity, substrate mineralization, and overall bone formation (51). Studies have indicated that long-term usage of lansoprazole (LPZ) induces calcium overload in osteoblasts and triggers apoptosis. This calcium release primarily occurs through the store-operated calcium entry (SOCE) influx at the cell membrane and via the IP3Rs located on the endoplasmic reticulum. Inhibition of IP3Rs and SOCE pathway using 2-APB improves osteoporosis condition (32). Furthermore, ORAI1 has been identified as a crucial mediator for enhancing the osteogenic potential of BMMSCs. The absence of ORAI1 results in limited phosphorylation of Smad 1/5/8 within the BMP signaling pathway; however, activation of BMP signaling can rescue the impaired osteogenic differentiation ability observed in ORAI1 BMMSCs. These findings suggest that targeting ORAI1-BMP signaling could be a potential therapeutic approach for treating bone formation defects (88).
TRPs
In vitro studies on TRPV1-deficient BMMSCs have demonstrated impaired osteoblast differentiation and mineralization (57). In vitro studies on TRPV1-deficient BMMSCs have demonstrated impaired osteoblast differentiation and mineralization (89). Although research on TRPM channels is limited, it has been shown that TRPM7 is upregulated during osteoblast differentiation, and its deficiency impairs osteoblast proliferation, differentiation, and mineralization (62). Notably, TRPM8 knockout mice exposed to cold temperatures exhibit reduced bone density and significant reductions in femoral size in males, along with lower vertebral bone microarchitectural parameters in females, suggesting TRPM8’s role in bone modeling and remodeling (90). Furthermore, TRPM8 promotes the differentiation of BMMSCs into osteoblasts. Treatments using TRPM8 agonists such as menthol or icilin enhance osteogenic differentiation, while the antagonist BCTC decreases it (63).
Others
P2X1 receptors negatively regulate osteoblast mineralization (65). Recently discovered mechanosensory ion channels Piezo1 and Piezo2 also play a regulatory role in osteoblasts (68). Targeted deletion of Piezo1 in osteoblasts resulted in severe osteoporosis and spontaneous fractures, highlighting Piezo1’s function in growth plate chondrocytes (69). Additionally, the activation of Sr2+ on the calcium-sensing receptor (CaR) exerts an anti-osteoporotic effect. Compared to Sr2+, CaR has a better sensitivity to Ca2+, and the combination of Ca2+ and CaR can better promote osteoclast apoptosis and osteoblast differentiation, thereby enhancing bone tissue (91).
The regulatory effect of calcium channels on osteoblasts primarily focuses on the positive regulation of osteogenesis by VGCCs. L-type and T-type VGCCs are widely present on osteoblasts and are activated by changes in the extracellular environment, such as high calcium levels and hormones, promoting osteogenic differentiation. Additionally, TRPM8 has been shown to promote BMMSC osteogenic differentiation, although the specific mechanisms require further study. Through these findings, it can be observed that the activation of VGCCs, TRPs, SOCE, and Piezo channels on the osteoblast membrane can reduce the symptoms of osteoporosis (Figure 3).
Calcium channels in osteocytes
Osteocytes are mechanical load-sensing cells that mediate bone formation, adaptation, and resorption in response to mechanical load (92). Various mechanical stimuli, including fluid shear stresses and matrix strains (e.g., compressive, tensile, and torsional loads), can activate osteocytes within the pericellular matrix (93). In the bone microenvironment, osteocytes are encased in a pericellular matrix at the interface between the cell membrane and the hard bone matrix. Small force stimuli generate fluid shear from the extracellular fluid flow due to spatial deformation, which activates mechanically stimulus-sensitive ion channels and integrins on the osteocyte membranes. This activation initiates a series of downstream pathways to regulate bone remodeling (Figure 4).
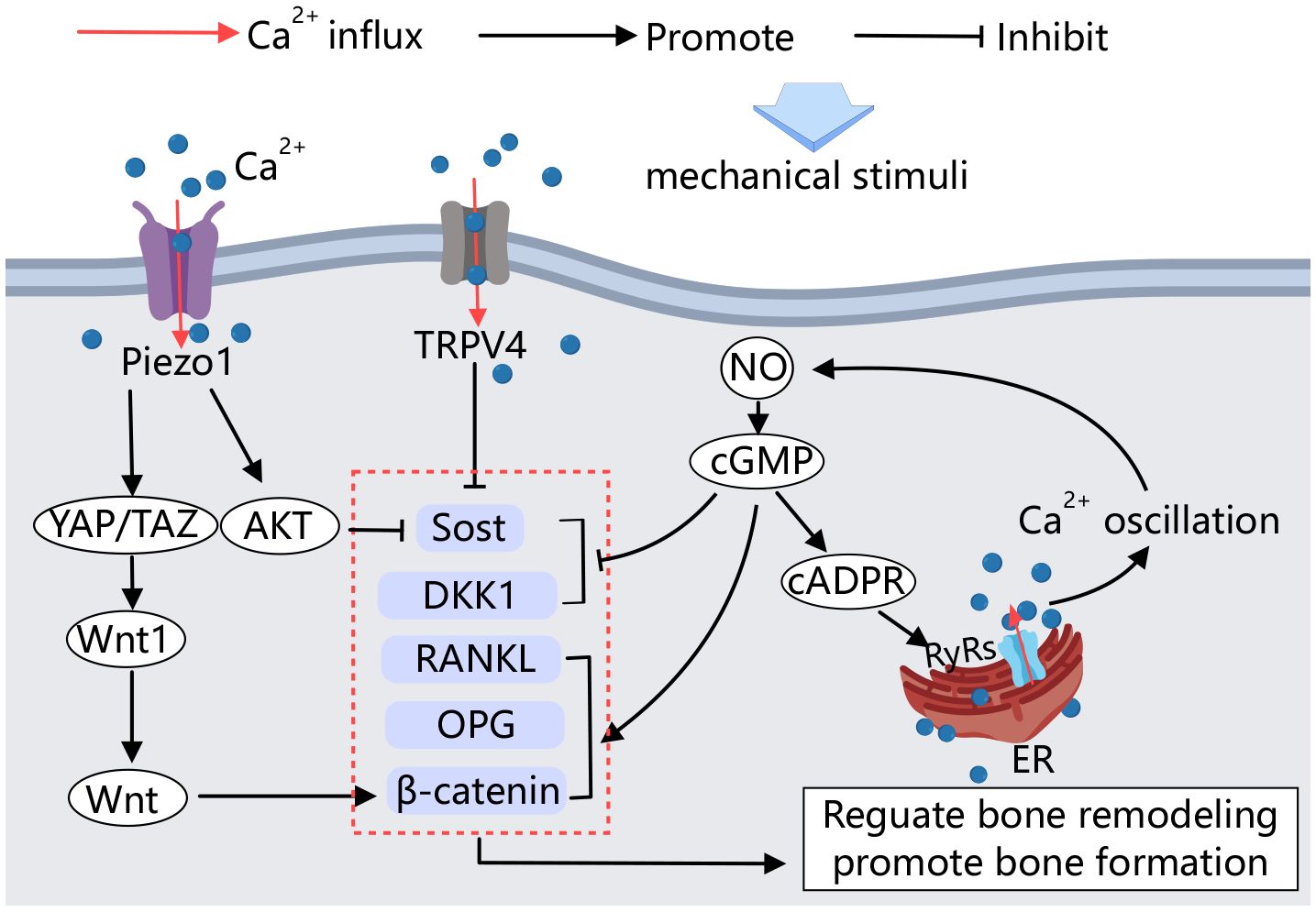
Figure 4 Calcium channels in osteocytes. Mechanical stimulation acting on osteocytes activates Piezo1 and TRPV4, influencing the activity of factors involved in bone formation and resorption, thereby regulating bone remodeling. Additionally, calcium signaling plays a crucial role in this process. Upon mechanical stimulation, an amplified positive feedback loop of Ca2+ signaling is established, further regulating bone remodeling.
Several studies have demonstrated that mechanosensitive calcium channels in the osteoblast membrane are involved in the sensing of mechanical signals by osteocytes. Calcium influx is an early response to mechanical stimulation of osteocytes both in vitro and in vivo (94). The Ca2+ channel Piezo1 mediates mechanical signaling in osteocytes. Fluid shear stress has been shown to increase the expression of Piezo1 on the MLO-Y4 cell membrane and elevate intracellular calcium concentration. Conditional knockdown of Piezo1 in osteocytes leads to significant reductions in bone mass and impaired bone structure and strength in mice (95). Educed Piezo1 expression weakens the MLO-Y4 response to external stress. Activation of the Piezo1 calcium channel by the Piezo1-specific agonist Yoda1 produces effects similar to those observed in mechanically stimulated osteocytes (96).
Moreover, YAP1/TAZ has been identified as key mediators of sensory transmission to mechanical signals in various cell types. Mechanical stimulation of Piezo1 on osteocyte membranes activates YAP1 and TAZ, increasing Wnt1 production by osteoblasts (96). Wnt1 production, in turn, activates the Wnt/β-catenin signaling pathway, promoting bone formation (97). In vitro cell culture studies have shown that Piezo1 can also regulate the biological behavior of osteocytes through the Akt/Sost pathway. Sost is a key of bone regulator produced by osteocytes, inhibits the classical Wnt signaling pathway, thereby regulating bone formation. This signaling pathway stimulates resorption (98). High expression of Piezo1 in osteocytes, stimulated by mechanical stretching, immediately induces calcium efflux and Akt phosphorylation, which inhibits Sost expression, promotes bone formation, and inhibits bone resorption (99).
TRPV4 calcium channels have also been shown to inhibit Sost expression in vitro by mechanically stimulating osteocytes (100). Mechanical stimulation can regulate the transcription of bone cytokines through the NO-Ca2+ positive feedback signaling pathway in osteocytes (101). NO is an important signaling molecule secreted by osteocytes under mechanical loading. It regulates the downstream cGMP signaling pathway, which influences the transcription of cellular factors such as RANKL, OPG, and DKK1, β-catenin, and causes intracellular Ca2+ oscillation through the NO-cGMP-cADPR-RyRs pathway. This Ca2+ oscillation further promotes NO formation through a positive feedback loop, thereby regulating bone remodeling and promoting osteoblast angiogenesis (101–103).
Conclusions and prospects
Osteoporosis, a serious age-related disease, poses a global challenge. Bone metabolism in osteoporosis is closely linked to the expression of Ca2+ channels in osteocytes, making their regulation crucial for managing the disease. TRP channels, known for their high calcium permeability, have several members significantly associated with osteoporosis. While research on TRPM8 has largely focused on tumors, cardiovascular diseases and pain (104, 105), its role in bone metabolism remains underexplored, particularly its impact on osteoclasts. Developing new therapeutic targets to address bone loss is essential, as current osteoporosis treatments often have side effects such as constipation, diarrhea, tumorigenesis, and cardiovascular disease. Further understanding of calcium channels in osteocytes, especially osteoclasts, could lead to healthier regulatory measures and new treatment ideas for osteoporosis.
Postmenopausal osteoporosis is the most common form of the disease, driven by significantly reduced estrogen levels, which lead to decreased bone mass, damaged bone microstructure, and higher fracture risk. Estrogen decline promotes osteoclast activity through mechanisms like upregulating RANKL and regulating microRNA-21 biogenesis, while also inhibiting osteogenic differentiation via increased TNF-α activity (106, 107). The specific interaction between estrogen and calcium ion channels remains unclear, presenting a promising direction for future research.
Author contributions
YH: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Resources, Project administration, Methodology, Funding acquisition, Data curation, Conceptualization. NY: Writing – review & editing, Writing – original draft, Visualization, Software, Resources, Project administration, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. MS: Writing – original draft, Visualization, Supervision, Software, Methodology, Investigation, Formal analysis, Data curation. SY: Writing – review & editing, Visualization, Supervision, Software, Data curation. XC: Writing – review & editing, Writing – original draft, Validation, Supervision, Resources, Project administration, Methodology, Funding acquisition, Conceptualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Social Science Fund of China (Grant No. 21BTY078).
Acknowledgments
We are extremely thankful to Yiwei An for revising the grammar of the manuscript. Figures were created by the online website MedPeer(https://user.medpeer.cn/).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gao Y, Patil S, Jia J. The development of molecular biology of osteoporosis. Int J Mol Sci. (2021) 22:8182. doi: 10.3390/ijms22158182
2. Chandra A, Rajawat J. Skeletal aging and osteoporosis: mechanisms and therapeutics. Int J Mol Sci. (2021) 22:3553. doi: 10.3390/ijms22073553
3. Salari N, Ghasemi H, Mohammadi L, Behzadi MH, Rabieenia E, Shohaimi S, et al. The global prevalence of osteoporosis in the world: a comprehensive systematic review and meta-analysis. J Orthop Surg Res. (2021) 16:609. doi: 10.1186/s13018-021-02772-0
4. Aibar-Almazán A, Voltes-Martínez A, Castellote-Caballero Y, Afanador-Restrepo DF, Carcelén-Fraile MDC, López-Ruiz E. Current status of the diagnosis and management of osteoporosis. Int J Mol Sci. (2022) 23:9465. doi: 10.3390/ijms23169465
5. Sobh MM, Abdalbary M, Elnagar S, Nagy E, Elshabrawy N, Abdelsalam M, et al. Secondary osteoporosis and metabolic bone diseases. J Clin Med. (2022) 11:2382. doi: 10.3390/jcm11092382
6. Heng MWY, Chan AWD, Man REK, Fenwick EK, Chew STH, Tay L, et al. Individual and combined associations of sarcopenia, osteoporosis and obesity with frailty in a multi-ethnic asian older adult population. BMC Geriatr. (2023) 23:802. doi: 10.1186/s12877-023-04500-1
7. Chuang TL, Koo M, Wang YF. The impact of diabetes, anemia, and renal function in the relationship between osteoporosis and fasting blood glucose among Taiwanese women: a cross-sectional study. BMC Womens Health. (2024) 24:23. doi: 10.1186/s12905-023-02851-w
8. Hu X, Ma S, Chen L, Tian C, Wang W. Association between osteoporosis and cardiovascular disease in elderly people: evidence from a retrospective study. PeerJ. (2023) 11:e16546. doi: 10.7717/peerj.16546
9. Jha SS, Kumar M, Agrawal PK, Thakur DK. Osteoporosis in asthma and COPD. Indian J Orthop. (2023) 57:200–8. doi: 10.1007/s43465-023-01048-5
10. Wang L, You X, Zhang L, Zhang C, Zou W. Mechanical regulation of bone remodeling. Bone Res. (2022) 10:16. doi: 10.1038/s41413-022-00190-4
11. Föger-Samwald U, Dovjak P, Azizi-Semrad U, Kerschan-Schindl K, Pietschmann P. Osteoporosis: Pathophysiology and therapeutic options. Excli J. (2020) 19:1017–37. doi: 10.17179/excli2020-2591
12. Du J, Yang J, He Z, Cui J, Yang Y, Xu M, et al. Osteoblast and osteoclast activity affect bone remodeling upon regulation by mechanical loading-induced leukemia inhibitory factor expression in osteocytes. Front Mol Biosci. (2020) 7:585056. doi: 10.3389/fmolb.2020.585056
13. Tang CH. Osteoporosis: from molecular mechanisms to therapies. Int J Mol Sci. (2020) 21:714. doi: 10.3390/ijms21030714
14. Nahian A, AlEssa AM. Histology, Osteocytes. Treasure Island (FL): StatPearls (2024). ineligible companies. Disclosure: Ahmed AlEssa declares no relevant financial relationships with ineligible companies.: StatPearls Publishing Copyright © 2024, StatPearls Publishing LLC.
15. McNamara LM. Osteocytes and estrogen deficiency. Curr Osteoporos Rep. (2021) 19:592–603. doi: 10.1007/s11914-021-00702-x
16. Da W, Tao L, Zhu Y. The role of osteoclast energy metabolism in the occurrence and development of osteoporosis. Front Endocrinol (Lausanne). (2021) 12:675385. doi: 10.3389/fendo.2021.675385
17. Ma J, Zhu L, Zhou Z, Song T, Yang L, Yan X, et al. The calcium channel TRPV6 is a novel regulator of RANKL-induced osteoclastic differentiation and bone absorption activity through the IGF-PI3K-AKT pathway. Cell Prolif. (2021) 54:e12955. doi: 10.1111/cpr.12955
18. Li H, Xiao Z, Quarles LD, Li W. Osteoporosis: mechanism, molecular target and current status on drug development. Curr Med Chem. (2021) 28:1489–507. doi: 10.2174/0929867327666200330142432
19. Shangguan WJ, Zhang YH, Li ZC, Tang LM, Shao J, Li H. Naringin inhibits vascular endothelial cell apoptosis via endoplasmic reticulum stress− and mitochondrial−mediated pathways and promotes intraosseous angiogenesis in ovariectomized rats. Int J Mol Med. (2017) 40:1741–9. doi: 10.3892/ijmm
20. Grüneboom A, Hawwari I, Weidner D, Culemann S, Müller S, Henneberg S, et al. A network of trans-cortical capillaries as mainstay for blood circulation in long bones. Nat Metab. (2019) 1:236–50. doi: 10.1038/s42255-018-0016-5
21. Xiao CL, Liu LL, Tang W, Liu WY, Wu LY, Zhao K. Reduction of the trans-cortical vessel was associated with bone loss, another underlying mechanism of osteoporosis. Microvasc Res. (2024) 152:104650. doi: 10.1016/j.mvr.2023.104650
22. Huang J, Lin D, Wei Z, Li Q, Zheng J, Zheng Q, et al. Parathyroid hormone derivative with reduced osteoclastic activity promoted bone regeneration via synergistic bone remodeling and angiogenesis. Small. (2020) 16:e1905876. doi: 10.1002/smll.201905876
23. Sjögren K, Engdahl C, Henning P, Lerner UH, Tremaroli V, Lagerquist MK, et al. The gut microbiota regulates bone mass in mice. J Bone Miner Res. (2012) 27:1357–67. doi: 10.1002/jbmr.1588
24. Yang Q, Wei Z, Wei X, Zhang J, Tang Y, Zhou X, et al. The age-related characteristics in bone microarchitecture, osteoclast distribution pattern, functional and transcriptomic alterations of BMSCs in mice. Mech Ageing Dev. (2023) 216:111877. doi: 10.1016/j.mad.2023.111877
25. Kurotaki D, Yoshida H, Tamura T. Epigenetic and transcriptional regulation of osteoclast differentiation. Bone. (2020) 138:115471. doi: 10.1016/j.bone.2020.115471
26. Basisty N, Kale A, Jeon OH, Kuehnemann C, Payne T, Rao C, et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. (2020) 18:e3000599. doi: 10.1371/journal.pbio.3000599
27. Farr JN, Rowsey JL, Eckhardt BA, Thicke BS, Fraser DG, Tchkonia T, et al. Independent roles of estrogen deficiency and cellular senescence in the pathogenesis of osteoporosis: evidence in young adult mice and older humans. J Bone Miner Res. (2019) 34:1407–18. doi: 10.1002/jbmr.3729
28. Farr JN, Fraser DG, Wang H, Jaehn K, Ogrodnik MB, Weivoda MM, et al. Identification of senescent cells in the bone microenvironment. J Bone Miner Res. (2016) 31:1920–9. doi: 10.1002/jbmr.2892
29. Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, Onken JL, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med. (2017) 23:1072–9. doi: 10.1038/nm.4385
30. Chen F, Ni B, Yang YO, Ye T, Chen A. Knockout of TRPV6 causes osteopenia in mice by increasing osteoclastic differentiation and activity. Cell Physiol Biochem. (2014) 33:796–809. doi: 10.1159/000358653
31. Zayzafoon M. Calcium/calmodulin signaling controls osteoblast growth and differentiation. J Cell Biochem. (2006) 97:56–70. doi: 10.1002/(ISSN)1097-4644
32. Cheng Z, Liu Y, Ma M, Sun S, Ma Z, Wang Y, et al. Lansoprazole-induced osteoporosis via the IP3R- and SOCE-mediated calcium signaling pathways. Mol Med. (2022) 28:21. doi: 10.1186/s10020-022-00448-x
33. Nie Z, Deng S, Zhang L, Chen S, Lu Q, Peng H. Crocin protects against dexamethasone−induced osteoblast apoptosis by inhibiting the ROS/Ca2+−mediated mitochondrial pathway. Mol Med Rep. (2019) 20:401–8. doi: 10.3892/mmr
34. Cai WJ, Chen Y, Shi LX, Cheng HR, Banda I, Ji YH, et al. AKT-GSK3β Signaling pathway regulates mitochondrial dysfunction-associated OPA1 cleavage contributing to osteoblast apoptosis: preventative effects of hydroxytyrosol. Oxid Med Cell Longev. (2019) 2019:4101738. doi: 10.1155/2019/4101738
35. Liu W, Xu C, Ran D, Wang Y, Zhao H, Gu J, et al. CaMKII mediates cadmium induced apoptosis in rat primary osteoblasts through MAPK activation and endoplasmic reticulum stress. Toxicology. (2018) 406–407:70–80. doi: 10.1016/j.tox.2018.06.002
36. Li J, Liu C, Li Y, Zheng Q, Xu Y, Liu B, et al. TMCO1-mediated Ca(2+) leak underlies osteoblast functions via CaMKII signaling. Nat Commun. (2019) 10:1589. doi: 10.1038/s41467-019-09653-5
37. Kim HJ, Lee J, Lee GR, Kim N, Lee HI, Kwon M, et al. Flunarizine inhibits osteoclastogenesis by regulating calcium signaling and promotes osteogenesis. J Cell Physiol. (2021) 236:8239–52. doi: 10.1002/jcp.30496
38. Zhang J, Xu H, Han Z, Chen P, Yu Q, Lei Y, et al. Pulsed electromagnetic field inhibits RANKL-dependent osteoclastic differentiation in RAW264.7 cells through the Ca(2+)-calcineurin-NFATc1 signaling pathway. Biochem Biophys Res Commun. (2017) 482:289–95. doi: 10.1016/j.bbrc.2016.11.056
39. Mao D, Epple H, Uthgenannt B, Novack DV, Faccio R. PLCgamma2 regulates osteoclastogenesis via its interaction with ITAM proteins and GAB2. J Clin Invest. (2006) 116:2869–79. doi: 10.1172/JCI28775
40. Yang Z, Kim S, Mahajan S, Zamani A, Faccio R. Phospholipase Cγ1 (PLCγ1) controls osteoclast numbers via colony-stimulating factor 1 (CSF-1)-dependent diacylglycerol/β-catenin/cyclinD1 pathway. J Biol Chem. (2017) 292:1178–86. doi: 10.1074/jbc.M116.764928
41. Park JH, Lee NK, Lee SY. Current understanding of RANK signaling in osteoclast differentiation and maturation. Mol Cells. (2017) 40:706–13. doi: 10.14348/molcells.2017.0225
42. Domazetovic V, Marcucci G, Iantomasi T, Brandi ML, Vincenzini MT. Oxidative stress in bone remodeling: role of antioxidants. Clin cases Miner Bone Metab. (2017) 14:209–16. doi: 10.11138/ccmbm/2017.14.1.209
43. Kim MS, Yang YM, Son A, Tian YS, Lee SI, Kang SW, et al. RANKL-mediated reactive oxygen species pathway that induces long lasting Ca2+ oscillations essential for osteoclastogenesis. J Biol Chem. (2010) 285:6913–21. doi: 10.1074/jbc.M109.051557
44. Shrestha N, Bacsa B, Ong HL, Scheruebel S, Bischof H, Malli R, et al. TRIC-A shapes oscillatory Ca2+ signals by interaction with STIM1/Orai1 complexes. PLoS Biol. (2020) 18:e3000700. doi: 10.1371/journal.pbio.3000700
45. Lorenzo J. The many ways of osteoclast activation. J Clin Invest. (2017) 127:2530–2. doi: 10.1172/JCI94606
46. Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. (2002) 3:889–901. doi: 10.1016/S1534-5807(02)00369-6
47. Hsiao HB, Wu JB, Lin WC. (-)-Epicatechin 3-O-β-D-allopyranoside prevent ovariectomy-induced bone loss in mice by suppressing RANKL-induced NF-κB and NFATc-1 signaling pathways. BMC Complement Altern Med. (2017) 17:245. doi: 10.1186/s12906-017-1737-9
48. Shankar G, Davison I, Helfrich MH, Mason WT, Horton MA. Integrin receptor-mediated mobilisation of intranuclear calcium in rat osteoclasts. J Cell Sci. (1993) 105:61–8. doi: 10.1242/jcs.105.1.61
49. Kuroda Y, Hisatsune C, Nakamura T, Matsuo K, Mikoshiba K. Osteoblasts induce Ca2+ oscillation-independent NFATc1 activation during osteoclastogenesis. Proc Natl Acad Sci U.S.A. (2008) 105:8643–8. doi: 10.1073/pnas.0800642105
50. Miyauchi A, Hruska KA, Greenfield EM, Duncan R, Alvarez J, Barattolo R, et al. Osteoclast cytosolic calcium, regulated by voltage-gated calcium channels and extracellular calcium, controls podosome assembly and bone resorption. J Cell Biol. (1990) 111:2543–52. doi: 10.1083/jcb.111.6.2543
51. Jung H, Best M, Akkus O. Microdamage induced calcium efflux from bone matrix activates intracellular calcium signaling in osteoblasts via L-type and T-type voltage-gated calcium channels. Bone. (2015) 76:88–96. doi: 10.1016/j.bone.2015.03.014
52. Jung H, Akkus O. Diffuse microdamage in bone activates anabolic response by osteoblasts via involvement of voltage-gated calcium channels. J Bone Miner Metab. (2020) 38:151–60. doi: 10.1007/s00774-019-01042-8
53. Robinson LJ, Blair HC, Barnett JB, Soboloff J. The roles of Orai and Stim in bone health and disease. Cell Calcium. (2019) 81:51–8. doi: 10.1016/j.ceca.2019.06.001
54. Hwang SY, Putney JW Jr. Calcium signaling in osteoclasts. Biochim Biophys Acta. (2011) 1813:979–83. doi: 10.1016/j.bbamcr.2010.11.002
55. Ong EC, Nesin V, Long CL, Bai CX, Guz JL, Ivanov IP, et al. A TRPC1 protein-dependent pathway regulates osteoclast formation and function. J Biol Chem. (2013) 288:22219–32. doi: 10.1074/jbc.M113.459826
56. Klein S, Mentrup B, Timmen M, Sherwood J, Lindemann O, Fobker M, et al. Modulation of transient receptor potential channels 3 and 6 regulates osteoclast function with impact on trabecular bone loss. Calcif Tissue Int. (2020) 106:655–64. doi: 10.1007/s00223-020-00673-8
57. He LH, Liu M, He Y, Xiao E, Zhao L, Zhang T, et al. TRPV1 deletion impaired fracture healing and inhibited osteoclast and osteoblast differentiation. Sci Rep. (2017) 7:42385. doi: 10.1038/srep42385
58. Idris AI, Landao-Bassonga E, Ralston SH. The TRPV1 ion channel antagonist capsazepine inhibits osteoclast and osteoblast differentiation in vitro and ovariectomy induced bone loss in vivo. Bone. (2010) 46:1089–99. doi: 10.1016/j.bone.2010.01.368
59. Chamoux E, Bisson M, Payet MD, Roux S. TRPV-5 mediates a receptor activator of NF-kappaB (RANK) ligand-induced increase in cytosolic Ca2+ in human osteoclasts and down-regulates bone resorption. J Biol Chem. (2010) 285:25354–62. doi: 10.1074/jbc.M109.075234
60. van der Eerden BC, Hoenderop JG, de Vries TJ, Schoenmaker T, Buurman CJ, Uitterlinden AG, et al. The epithelial Ca2+ channel TRPV5 is essential for proper osteoclastic bone resorption. Proc Natl Acad Sci U S A. (2005) 102:17507–12. doi: 10.1073/pnas.0505789102
61. Chen F, Ouyang Y, Ye T, Ni B, Chen A. Estrogen inhibits RANKL-induced osteoclastic differentiation by increasing the expression of TRPV5 channel. J Cell Biochem. (2014) 115:651–8. doi: 10.1002/jcb.24700
62. Abed E, Moreau R. Importance of melastatin-like transient receptor potential 7 and cations (magnesium, calcium) in human osteoblast-like cell proliferation. Cell Prolif. (2007) 40:849–65. doi: 10.1111/j.1365-2184.2007.00476.x
63. Henao JC, Grismaldo A, Barreto A, Rodríguez-Pardo VM, Mejía-Cruz CC, Leal-Garcia E, et al. TRPM8 channel promotes the osteogenic differentiation in human bone marrow mesenchymal stem cells. Front Cell Dev Biol. (2021) 9:592946. doi: 10.3389/fcell.2021.592946
64. Erkhembaatar M, Gu DR, Lee SH, Yang YM, Park S, Muallem S, et al. Lysosomal Ca(2+) signaling is essential for osteoclastogenesis and bone remodeling. J Bone Miner Res. (2017) 32:385–96. doi: 10.1002/jbmr.2986
65. Orriss IR, Key ML, Brandao-Burch A, Patel JJ, Burnstock G, Arnett TR. The regulation of osteoblast function and bone mineralisation by extracellular nucleotides: The role of p2x receptors. Bone. (2012) 51:389–400. doi: 10.1016/j.bone.2012.06.013
66. Orriss IR, Davies BK, Bourne LE, Arnett TR. Modulation of osteoblast differentiation and function by the P2X4 receptor. Purinergic Signal. (2023) 19:367–78. doi: 10.1007/s11302-022-09887-x
67. Ma Y, Shi X, Zhao H, Song R, Zou H, Zhu J, et al. Potential mechanisms of osteoprotegerin-induced damage to osteoclast adhesion structures via P2X7R-mediated MAPK signaling. Int J Mol Med. (2022) 49:59. doi: 10.3892/ijmm
68. Hendrickx G, Fischer V, Liedert A, von Kroge S, Haffner-Luntzer M, Brylka L, et al. Piezo1 inactivation in chondrocytes impairs trabecular bone formation. J Bone Miner Res. (2021) 36:369–84. doi: 10.1002/jbmr.4198
69. Nie X, Chung MK. Piezo channels for skeletal development and homeostasis: Insights from mouse genetic models. Differentiation. (2022) 126:10–5. doi: 10.1016/j.diff.2022.06.001
70. Dolphin AC. Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J Physiol. (2016) 594:5369–90. doi: 10.1113/JP272262
71. Kaestner L, Wang X, Hertz L, Bernhardt I. Voltage-activated ion channels in non-excitable cells-A viewpoint regarding their physiological justification. Front Physiol. (2018) 9:450. doi: 10.3389/fphys.2018.00450
72. Tan YZ, Fei DD, He XN, Dai JM, Xu RC, Xu XY, et al. L-type voltage-gated calcium channels in stem cells and tissue engineering. Cell Prolif. (2019) 52:e12623. doi: 10.1111/cpr.12623
73. Wright CS, Robling AG, Farach-Carson MC, Thompson WR. Skeletal functions of voltage sensitive calcium channels. Curr Osteoporos Rep. (2021) 19:206–21. doi: 10.1007/s11914-020-00647-7
74. Atlas D. Voltage-gated calcium channels function as Ca2+-activated signaling receptors. Trends Biochem Sci. (2014) 39:45–52. doi: 10.1016/j.tibs.2013.12.005
75. Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. (2006) 443:226–9. doi: 10.1038/nature05108
76. Soboloff J, Rothberg BS, Madesh M, Gill DL. STIM proteins: dynamic calcium signal transducers. Nat Rev Mol Cell Biol. (2012) 13:549–65. doi: 10.1038/nrm3414
77. Li H. TRP channel classification. Adv Exp Med Biol. (2017) 976:1–8. doi: 10.1007/978-94-024-1088-4_1
78. Lieben L, Carmeliet G. The involvement of TRP channels in bone homeostasis. Front Endocrinol (Lausanne). (2012) 3:99. doi: 10.3389/fendo.2012.00099
79. Kajiya H, Okamoto F, Nemoto T, Kimachi K, Toh-Goto K, Nakayana S, et al. RANKL-induced TRPV2 expression regulates osteoclastogenesis via calcium oscillations. Cell Calcium. (2010) 48:260–9. doi: 10.1016/j.ceca.2010.09.010
80. Masuyama R, Vriens J, Voets T, Karashima Y, Owsianik G, Vennekens R, et al. TRPV4-mediated calcium influx regulates terminal differentiation of osteoclasts. Cell Metab. (2008) 8:257–65. doi: 10.1016/j.cmet.2008.08.002
81. Shin SH, Lee EJ, Chun J, Hyun S, Kang SS. Phosphorylation on TRPV4 serine 824 regulates interaction with STIM1. Open Biochem J. (2015) 9:24–33. doi: 10.2174/1874091X01509010024
82. Zhang D, Lin L, Yang B, Meng Z, Zhang B. Knockdown of Tcirg1 inhibits large-osteoclast generation by down-regulating NFATc1 and IP3R2 expression. PLoS One. (2020) 15:e0237354. doi: 10.1371/journal.pone.0237354
83. Robinson LJ, Blair HC, Barnett JB, Zaidi M, Huang CL. Regulation of bone turnover by calcium-regulated calcium channels. Ann N Y Acad Sci. (2010) 1192:351–7. doi: 10.1111/j.1749-6632.2009.05219.x
84. Burnstock G, Kennedy C. P2X receptors in health and disease. Adv Pharmacol. (2011) 61:333–72. doi: 10.1016/B978-0-12-385526-8.00011-4
85. Cao C, Ren Y, Barnett AS, Mirando AJ, Rouse D, Mun SH, et al. Increased Ca2+ signaling through CaV1.2 promotes bone formation and prevents estrogen deficiency-induced bone loss. JCI Insight. (2017) 2:95512. doi: 10.1172/jci.insight.95512
86. Fei D, Zhang Y, Wu J, Zhang H, Liu A, He X, et al. Ca(v) 1.2 regulates osteogenesis of bone marrow-derived mesenchymal stem cells via canonical Wnt pathway in age-related osteoporosis. Aging Cell. (2019) 18:e12967. doi: 10.1111/acel.12967
87. Li B, He X, Dong Z, Xuan K, Sun W, Gao L, et al. Ionomycin ameliorates hypophosphatasia via rescuing alkaline phosphatase deficiency-mediated L-type Ca(2+) channel internalization in mesenchymal stem cells. Bone Res. (2020) 8:19. doi: 10.1038/s41413-020-0090-7
88. Lee SH, Park Y, Song M, Srikanth S, Kim S, Kang MK, et al. Orai1 mediates osteogenic differentiation via BMP signaling pathway in bone marrow mesenchymal stem cells. Biochem Biophys Res Commun. (2016) 473:1309–14. doi: 10.1016/j.bbrc.2016.04.068
89. Yoneda M, Suzuki H, Hatano N, Nakano S, Muraki Y, Miyazawa K, et al. PIEZO1 and TRPV4, which are distinct mechano-sensors in the osteoblastic MC3T3-E1 cells, modify cell-proliferation. Int J Mol Sci. (2019) 20:4960. doi: 10.3390/ijms20194960
90. Lelis Carvalho A, Treyball A, Brooks DJ, Costa S, Neilson RJ, Reagan MR, et al. TRPM8 modulates temperature regulation in a sex-dependent manner without affecting cold-induced bone loss. PLoS One. (2021) 16:e0231060. doi: 10.1371/journal.pone.0231060
91. Cheshmedzhieva D, Ilieva S, Permyakov EA, Permyakov SE, Dudev T. Ca(2+)/Sr(2+) selectivity in calcium-sensing receptor (CaSR): implications for strontium's anti-osteoporosis effect. Biomolecules. (2021) 11:1576. doi: 10.3390/biom11111576
92. Choi JUA, Kijas AW, Lauko J, Rowan AE. The mechanosensory role of osteocytes and implications for bone health and disease states. Front Cell Dev Biol. (2021) 9:770143. doi: 10.3389/fcell.2021.770143
93. Wittkowske C, Reilly GC, Lacroix D, Perrault CM. In vitro bone cell models: impact of fluid shear stress on bone formation. Front Bioeng Biotechnol. (2016) 4:87. doi: 10.3389/fbioe.2016.00087
94. Lu XL, Huo B, Park M, Guo XE. Calcium response in osteocytic networks under steady and oscillatory fluid flow. Bone. (2012) 51:466–73. doi: 10.1016/j.bone.2012.05.021
95. Wang L, You X, Lotinun S, Zhang L, Wu N, Zou W. Mechanical sensing protein PIEZO1 regulates bone homeostasis via osteoblast-osteoclast crosstalk. Nat Commun. (2020) 11:282. doi: 10.1038/s41467-019-14146-6
96. Li X, Han L, Nookaew I, Mannen E, Silva MJ, Almeida M, et al. Stimulation of Piezo1 by mechanical signals promotes bone anabolism. Elife. (2019) 8:49631. doi: 10.7554/eLife.49631
97. Joeng KS, Lee YC, Lim J, Chen Y, Jiang MM, Munivez E, et al. Osteocyte-specific WNT1 regulates osteoblast function during bone homeostasis. J Clin Invest. (2017) 127:2678–88. doi: 10.1172/JCI92617
98. Robling AG, Niziolek PJ, Baldridge LA, Condon KW, Allen MR, Alam I, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. (2008) 283:5866–75. doi: 10.1074/jbc.M705092200
99. Sasaki F, Hayashi M, Mouri Y, Nakamura S, Adachi T, Nakashima T. Mechanotransduction via the Piezo1-Akt pathway underlies Sost suppression in osteocytes. Biochem Biophys Res Commun. (2020) 521:806–13. doi: 10.1016/j.bbrc.2019.10.174
100. Lyons JS, Joca HC, Law RA, Williams KM, Kerr JP, Shi G, et al. Microtubules tune mechanotransduction through NOX2 and TRPV4 to decrease sclerostin abundance in osteocytes. Sci Signal. (2017) 10:5748. doi: 10.1126/scisignal.aan5748
101. Wang D, Cai J, Pei Q, Yan Z, Zhu F, Zhao Z, et al. Gut microbial alterations in arginine metabolism determine bone mechanical adaptation. Cell Metab. (2024) 36:1252–68.e8. doi: 10.1016/j.cmet.2024.04.004
102. Klein-Nulend J, van Oers RF, Bakker AD, Bacabac RG. Nitric oxide signaling in mechanical adaptation of bone. Osteoporos Int. (2014) 25:1427–37. doi: 10.1007/s00198-013-2590-4
103. Tan SD, Bakker AD, Semeins CM, Kuijpers-Jagtman AM, Klein-Nulend J. Inhibition of osteocyte apoptosis by fluid flow is mediated by nitric oxide. Biochem Biophys Res Commun. (2008) 369:1150–4. doi: 10.1016/j.bbrc.2008.03.007
104. Das UN. Potential role of TRPM8 in cold-induced hypertension and its clinical implications. Discovery Med. (2023) 35:451–7. doi: 10.24976/Discov.Med.202335177.46
105. Kong N, Li W, Zhang J, Wang X, Hu L, Xu Q. TRPM8 as a potential biomarker and therapeutic target for gastric cancer identified by a combination of text mining and RNA sequencing. Curr Gene Ther. (2023) 23:391–9. doi: 10.2174/1566523223666230529142423
106. Hu CH, Sui BD, Du FY, Shuai Y, Zheng CX, Zhao P, et al. miR-21 deficiency inhibits osteoclast function and prevents bone loss in mice. Sci Rep. (2017) 7:43191. doi: 10.1038/srep43191
Keywords: osteoporosis, calcium ion channels, bone metabolism, calcium signaling, therapeutic targeting
Citation: Hao Y, Yang N, Sun M, Yang S and Chen X (2024) The role of calcium channels in osteoporosis and their therapeutic potential. Front. Endocrinol. 15:1450328. doi: 10.3389/fendo.2024.1450328
Received: 17 June 2024; Accepted: 25 July 2024;
Published: 07 August 2024.
Edited by:
Satoru Otsuru, University of Maryland, United StatesReviewed by:
Daohua Xu, Guangdong Medical University, ChinaWenjun Xie, Xi’an Jiaotong University, China
Copyright © 2024 Hao, Yang, Sun, Yang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Hao, aGFveWluZ0Bud251LmVkdS5jbg==; Xingjuan Chen, eGpjaGVuQG53cHUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship