
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 30 August 2024
Sec. Adrenal Endocrinology
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1442691
 Bijun Lian1,2,3†
Bijun Lian1,2,3† Jun Lu3†
Jun Lu3† Xudong Fang1†Yiming Zhang1Wei Wang4Yi He5
Xudong Fang1†Yiming Zhang1Wei Wang4Yi He5 Hongyuan Yu3Feiping Li3Junwei Wang4Weiying Chen3*
Hongyuan Yu3Feiping Li3Junwei Wang4Weiying Chen3* Xiaoping Qi1,3*
Xiaoping Qi1,3*Objective: The aim of this study was to investigate the genotypic and clinical phenotypic characteristics of MAX germline mutation–associated pheochromocytoma (PCC) and paraganglioma (PGL).
Methods: We retrospectively analyzed the family investigation data and clinical genetic characteristics of six individuals from three independent families with PCC carrying MAX germline mutations from December 2005 to March 2024. A literature review was then conducted of the six carriers and another 103 carriers from the other 84 families with MAX germline mutations reported previously.
Results: There were 109 patients in 87 families with all five exons and 53 types of MAX germline mutations. p.R33* (c.97C>T; 21.1%), p.R75* (c.223C>T; 13.8%), and p.A67D (c.200C>A; 7.3%), which accounted for 42.2% of mutations detected, were the most common mutations. Moreover, 101 (92.7%) patients developed PCCs, including 59 bilateral PCCs and 42 unilateral PCCs, and 19 (18.8%) patients showed metastasis. The mean age at diagnosis was 32.8 ± 12.6 (13-80) years. The male-to-female ratio was 1.3:1. In 11 (10.9%) patients, the PCC was accompanied by chest or abdominal PGL, and one other patient had sole head and neck PGL. Nine (8.3%) patients also had functional pituitary adenomas, 11 (10.9%) developed other neuroendocrine tumors (NETs), and 7 (6.4%) presented with concomitant non-NET. Meanwhile, MAX-p.Q82Tfs*89 and p.E158A mutations are reported for the first time in this study.
Conclusion: MAX germline mutations may cause new types of multiple endocrine neoplasia. A comprehensive baseline assessment of neural crest cell–derived diseases is recommended for all individuals with MAX germline mutations. The risk of bilateral and metastatic PCCs should also be considered.
Pheochromocytoma (PCC) and paraganglioma (PGL) are collectively referred to as PPGL and they originate from the neural crest cells of the embryonic ectoderm. PPGL is a rare neuroendocrine tumor (NET) that often has endocrine functions (1, 2). Approximately 40% of PPGLs manifest as genetic syndromes involving at least 24 characteristic pathogenic germline driving genes (1–5). These gene mutations are divided into three main clusters based on the activation of a particular signaling pathway. Cluster 1 (C1, i.e., pseudohypoxia signaling cluster) would be implemented with VHL, FH, SDHA~D, SDHAF2, MDH2, EGLN2/PHD2, IDH1/2/3B, HIF2A/EPAS1, DLST, SUCLG2, SLC25A11 and IRP1 that controls cellular iron metabolism and negatively regulates HIF2a mRNA translation (not caused by hypoxia). Cluster 2 (C2, i.e., kinase signaling cluster) would mainly include RET, NF1, TMEM127, MAX, H-RAS, KIF1Bβ, MERTK, MET, MYCN and ATRX, which belongs to the SWI/SNF family of chromatin remodeling proteins, as their upregulation will activate the PI3K/AKT and RAS/MAPK signaling pathways resulting in tumor formation. Finally, cluster 3 (C3, i.e., Wnt signaling cluster) would be implemented with both CSDE1 and UBFT fusion at MAML3 related to somatic mutations and alterations of any of these genes will result in increase of target genes involved in Wnt receptor and Hedgehog signaling pathways, i.e., the Wnt-altered subtype (5). Classic PPGL-related genetic syndromes include multiple endocrine neoplasia type 2 (MEN 2), von Hippel–Lindau syndrome, neurofibromatosis 1, and PGL1-5, which are caused by pathogenic germline mutations in RET, VHL, NF1, SDHD, SDHAF2, SDHC, SDHB, and SDHA genes, respectively. In recent years, germline TMEM127 mutation–related familial PPGL; fumarate hydratase (FH) mutation–related hereditary leiomyomatosis and renal cell carcinoma syndrome (HLRCC); and MYC-associated factor X (MAX) mutation–related multiple endocrine tumors, including PPGL, have been discovered and reported (1–10). However, the genotype-clinical phenotype correlations of MAX mutation–related PPGL are not fully understood. We retrospectively analyzed clinical data from three different families with MAX germline mutation–related PCC, and performed a review of the literatures, to discuss the characteristics of MAX germline mutations, with the aim of improving the comprehensive ability to diagnose and treat MAX mutation–related PPGL.
Nineteen individuals from three independent PCC pedigree groups diagnosed and treated at the 903rd PLA Hospital, Taizhou Enze Medical Center (Group), People’s Hospital of Tiantai County, and the First Hospital of Jiaxing City from December 2005 to November 2022 were selected for investigation using pedigree analysis (Figure 1). Suspected PCC was evaluated based on the plasma-excreted amounts of catecholamines (epinephrine, norepinephrine, and dopamine) and metanephrine/normetanephrine. Other specific biochemical tests, including serum calcium, serum basal calcitonin, parathyroid hormone, growth hormone, prolactin concentrations and so on were tested. Imaging examinations, including Doppler B-ultrasound, computed tomography (CT), and magnetic resonance imaging (MRI) were conducted based on patients’ clinical symptoms, biochemical tests and so on. The diagnosis and metastasis criteria for PPGL were based on the World Health Organization classification of endocrine tumors (1, 2).

Figure 1. Pedigree investigation of the three MAX germline mutation-related families.
Peripheral blood was obtained from 19 individuals in the three PCC pedigree groups for genomic DNA extraction using a QIAamp DNA Blood Midi Kit (Qiagen, Hilden, Germany). Targeted next-generation sequencing of 24 genes associated with hereditary PCC, including RET, VHL, SDHA-D, SDHAF2, MAX, TMEM127, FH, and HIF2A/EPAS1, was performed. Online software Combined Annotation Dependent Depletion (CADD, https://cadd.gs.washington.edu/snv, accessed on 18 Jan 2024) and Rare Exome Variant Ensemble Learner (REVEL; https://sites.google.com/site/revelgenomics/, accessed on 18 Jan 2024) were used to predict pathogenic variants. Pathogenicity was classified according to the American College of Medical Genetics and Genomics criteria. Sanger sequencing was performed for identification and confirmation. In addition, AlphaFold 2 (V2.3.1) was used to predict the tertiary structures of mutant MAX proteins to validate mutations in specific protein domains.
Two authors independently searched the Cochrane Library, PubMed, Web of Science, and EMBASE up to March 2024. Literature retrieval strategies were as follows: #1 (“MYC associated factor X”[Mesh]) OR MAX [Title/Abstract]; #2 (“Pheochromocytoma” [Mesh]) OR Pheochromocytomas [Title/Abstract]); #3 (“Paraganglioma”[Mesh]) OR Paragangliomas [Title/Abstract]) OR Paragangliomata [Title/Abstract]) OR Paragangliomatas [Title/Abstract]; and #4 (#1 and #2 or #3). Full texts of the articles were evaluated for eligibility. Disputes were resolved by consensus among all the authors. A total of 27 publications were identified.
To determine whether there were any genotype-phenotype correlations, we further divided patients with MAX mutations into two groups: missense mutation (GM) and non-missense mutation (GN) groups. The GN group included patients with all other types of mutations, such as nonsense, frameshift, alternative splice site, deletion/insertion, duplication, and gene fusion mutations. Clinical parameters, including diagnostic age at presentation and the occurrence rate of PCC, PGL, PA, and other NETs, were compared between patients in the GM and GN groups.
SPSS Statistics for Windows, version 25.0 (IBM, Armonk, NY, USA) was used for data processing. Quantitative data were expressed as the mean ± standard deviation, and a Student’s t-test or non-parametric rank sum test was used for intergroup comparisons. The chi-square test or Fisher’s exact probability method was used for intergroup comparisons of categorical data. The significance level was set at P < 0.05.
The proband (Figures 1-FA-II-1) was a 24-year-old man who had been diagnosed as having “right PCC” in December 2005 with more than a 2-year history of elevated blood pressure and a right adrenal mass detected 2 weeks previously. His blood pressure was 200/110 mm Hg. B-ultrasound and CT revealed a right adrenal mass. Laparoscopic dissection of the right adrenal tumor was performed, and histopathological examination revealed right PCC. In August 2017, he was readmitted to the hospital after the detection of a left adrenal mass during a physical examination when he was 36 years old. His blood pressure fluctuated in the range of 120–130/75–90 mmHg, and his plasma norepinephrine concentration was elevated to 2,766.00ng/L (reference range, 0–600 ng/L). B-ultrasound and CT scanning revealed a 3.8 × 2.9 cm mass in the left adrenal gland. Biochemical and imaging examinations revealed no evidence of pituitary abnormalities or other related NET. The diagnosis was “left PCC after right PCC dissection.” After treatment with terazosin hydrochloride and volume expansion for 1 week, the patient underwent laparoscopic left adrenal tumor resection. The pathological results indicated a left PCC. The final diagnosis was a metachronous bilateral PCC. Pedigree investigations and genetic testing were performed in May 2020 (Figures 1-FA-I-1, I-2, II-1, III-1). Only the proband and his 13-year-old son (Figures 1-FA-III-1) carried a heterozygous nonsense mutation in exon 4 of MAX (p.R75*, c.223C>T; NM_002382.5; Figure 2A). Systematic screening of the pituitary gland, neck, chest, and abdomen showed no abnormalities.
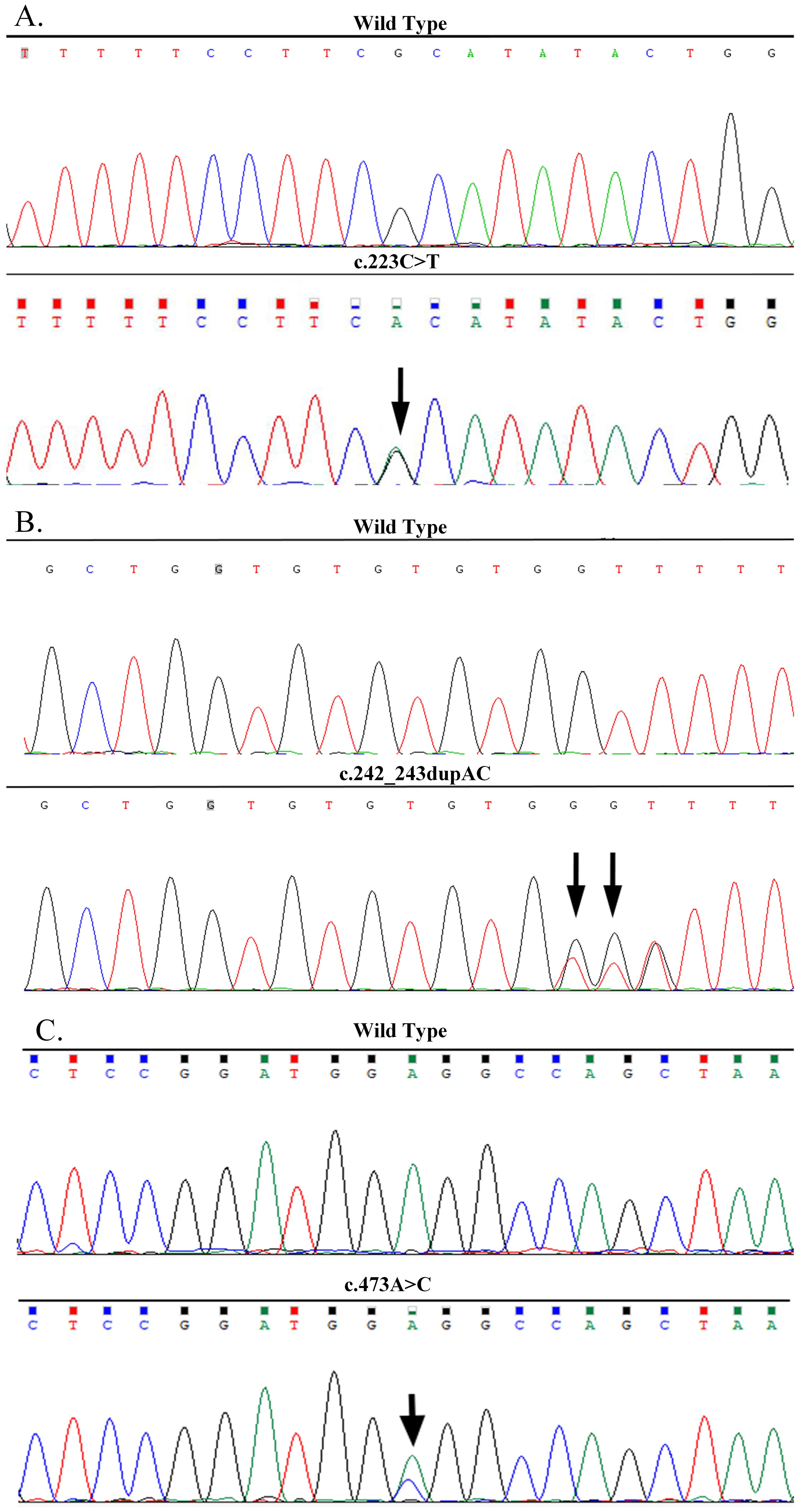
Figure 2. DNA sequencing confirmed the MAX mutations in the three families. (A) A heterozygous nonsense mutation of p.R75*(c.223C>T) in exon 4 of the MAX gene in the proband of Family A. (B) A heterozygous frameshift mutation of p.Q82Tfs*89 (c.242_243dupAC) in exon 4 of the MAX gene in the proband of Family B. (C) A heterozygous missense mutation of p.E158A (c.473A>C) in exon 5 of the MAX gene in the proband of Family C.
The proband (Figures 1-FB-II-1), a 34-year-old male, was admitted to the hospital in December 2021 with bilateral adrenal lesions. He had no symptoms, such as headache, palpitations, or excessive sweating. His blood pressure fluctuated in the range of 110–125/60–80 mmHg. B-ultrasound and CT examinations revealed bilateral adrenal masses (left, 2.8 cm; right, 2.2 cm). Plasma catecholamine, metanephrine, and normetanephrine levels were within normal limits. Imaging of the pituitary gland and neck and related biochemical examinations, such as serum calcium, calcitonin, parathyroid hormone, growth hormone, prolactin and so on, revealed no abnormalities. The patient was suspected to have “simultaneous bilateral PCC.” Subsequently, laparoscopic bilateral adrenal-sparing surgery was performed after treatment with terazosin hydrochloride and volume expansion for 1 week. Pathological results confirmed bilateral PCC. In January 2022, a family investigation and genetic testing (FB-II-1 and FB-II-1) were performed and the proband (FB-II-1) was found to carry a heterozygous frameshift mutation in exon 4 of MAX (p.Q82Tfs*89, c.242_243dupAC; NM_002382.5; Figure 2B). However, his parents (FB-I-1 and FB-I-2) showed no evidence of abnormalities in the pituitary gland, neck, chest, or abdomen. The proband refused genetic testing of his parents and his 3-year-old daughter (FB-III-1).
The proband (Figures 1-FC-II-1), a 64-year-old female, was admitted to our hospital in November 2022 with right adrenal lesions. She occasionally complained of paroxysmal headache. Her blood pressure fluctuated in the range of 100–130/66–90 mmHg. B-ultrasound and CT scanning revealed a 1.9 × 1.6 cm mass in the right adrenal gland. The plasma norepinephrine concentration was elevated to 2,243.7pmol/L. Imaging and biochemical examinations showed no evidence of pituitary, neck, or chest abnormalities. The diagnosis was “suspicion of right PCC.” After phenoxybenzamine and volume expansion treatment for 1 week, the patient underwent laparoscopic right adrenalectomy. A pathological report revealed a right PCC. In March 2024, genetic testing revealed a heterozygous missense mutation in exon 5 of MAX in the proband (p.E158A, c.473A>C; NM_002382.5; (Figure 2C). The mutation was detected in only one of her daughters and one of her sisters (Figures 1-FC-II-3, III-1), but their adrenal CT examination and blood biochemistry, such as plasma catecholamine, metanephrine, and normetanephrine, and so on, showed no abnormalities.
Six of 19 members of the three PPGL families with PCC harbored germline MAX variants (Figure 1, Table 1). Of these, only three (FA-II-1, FB-II-1, and FC-II-1) presented with PCC, including one with metachronous PCC, one with simultaneous bilateral PCC, and one with unilateral PCC. Their ages at the initial diagnosis were 24, 34, and 64 years, respectively. The remaining three (FA-III-1, and FC-II-3, FC-III-1), aged 13, 59, and 43 years, respectively, had no evidence of PPGL or clinical symptoms or biochemical or imaging findings of pituitary adenoma (PA) or other NET.
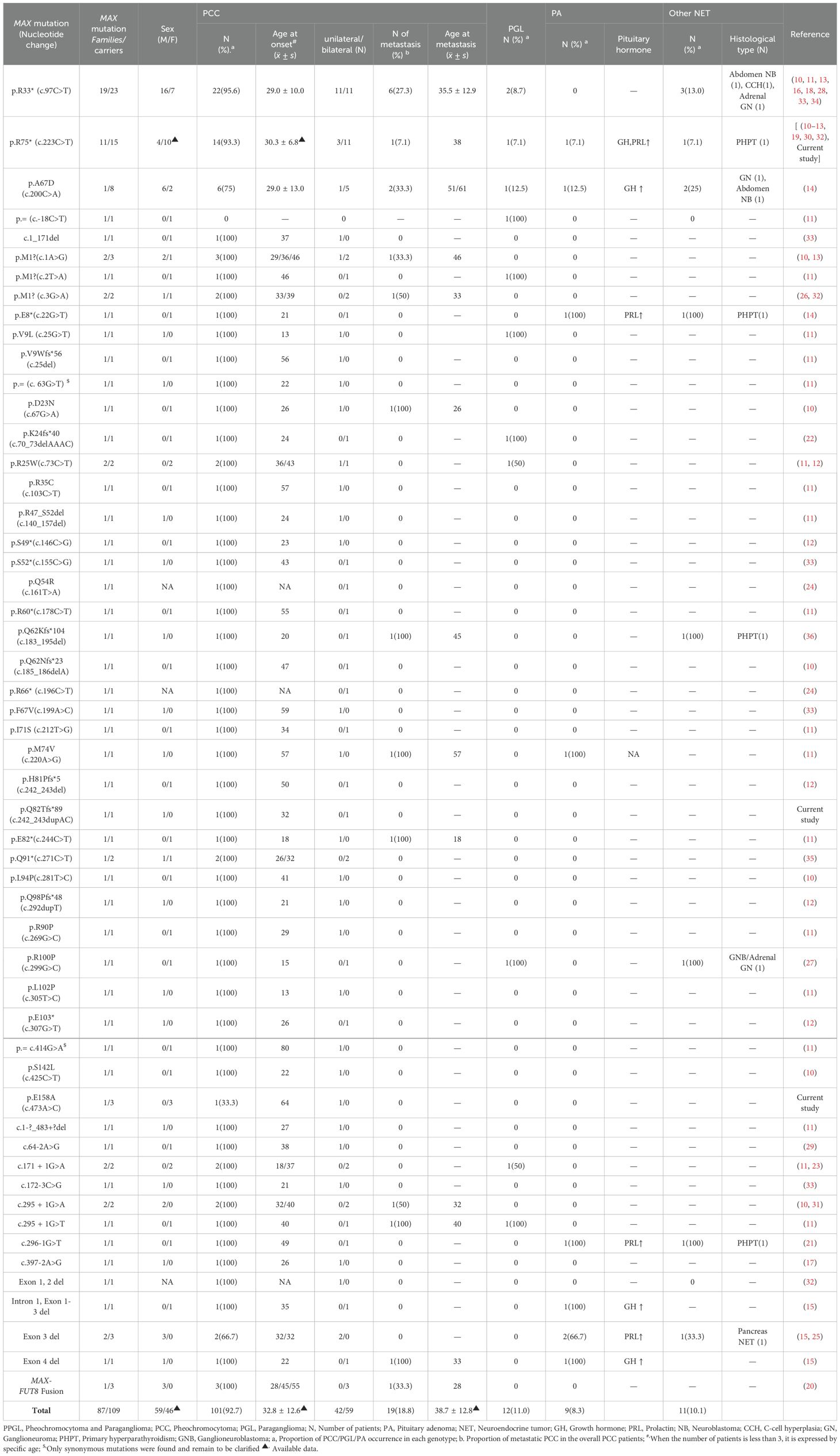
Table 1. Variation types and clinical characteristics of 87 MAX germline mutation-associated PPGL families.
The pathogenicity of the MAX variants was predicted using CADD and REVEL. High scores in the CADD (> 20) and REVEL (> 0.5; only evaluated for missense variants) implies a greater likelihood of the variant causing disease. The CADD values of the mutations c.223C>T and c.473A>C were 36 and 25, respectively, while the REVEL value of the mutation c.473A>C was 0.528, indicating that these two mutations are deleterious. Although the pathogenicity of the frameshift mutation c.242_243dupAC could not be predicted using the CADD and REVEL software, in combination with its clinical features, it is reasonable to consider it to be pathogenic (Table 2). In addition, the predicted 3D structure of the MAX protein indicated a structural change (Figure 3).
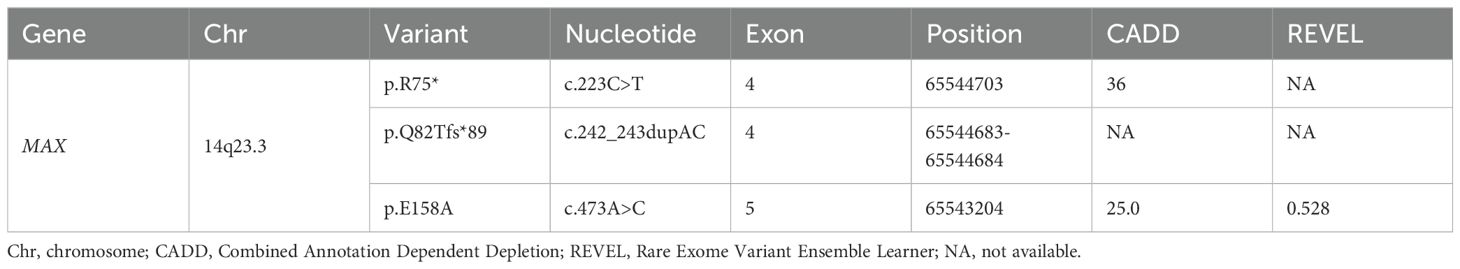
Table 2. The pathogenicity of MAX variants (NM_002382.5, hg38).
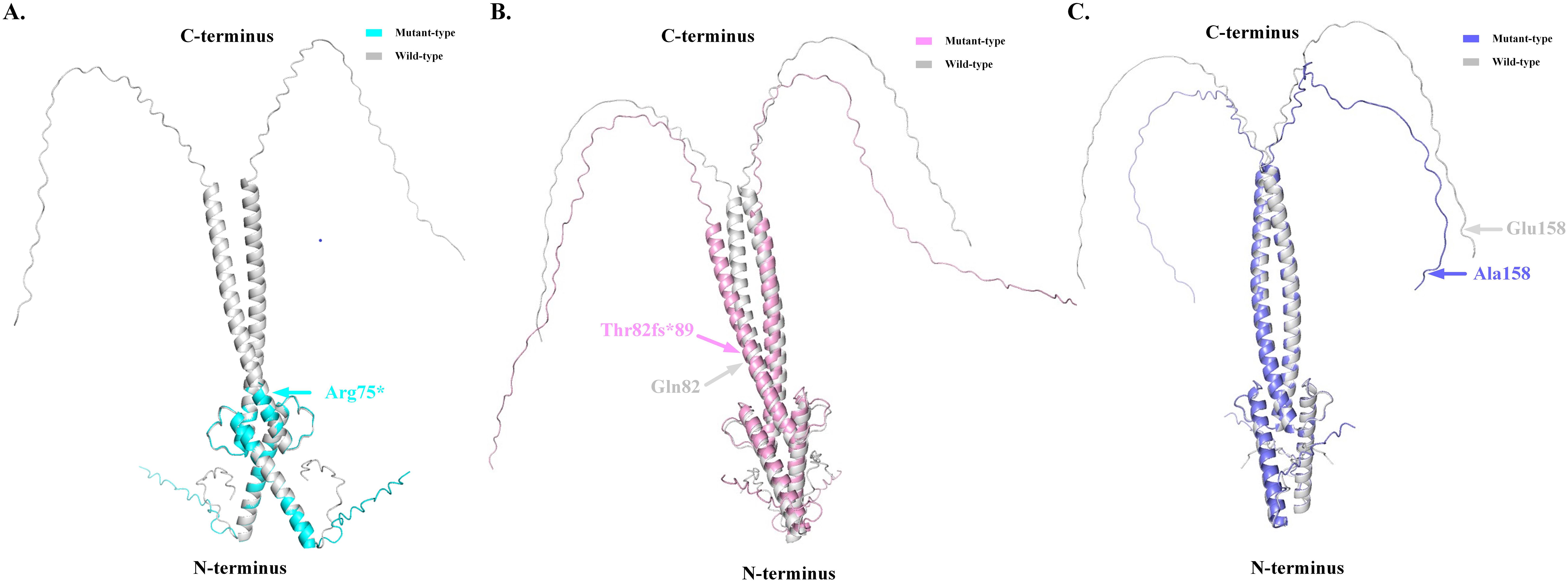
Figure 3. The predicted three-dimensional structure diagrams of the mutant-type MAX protein based on Alphafold 2 V2.3.1 (Bodies of grey represented as the wide type, and colored bodies represented as mutant types). (A). p.R75* mutation leads to the truncation of MAX protein. (B). Thr82fs89 alteration leads to the shortening of the C-terminal of MAX protein. (C). p.E158A mutation leads to the alteration in the Loop domain of MAX protein.
The clinical presentations of the six patients from the three families we reported and the remaining 103 patients from 84 families reported in the literature are shown in Table 1. Of the 53 MAX germline alterations identified, the most common mutations were nonsense mutations (47.7%), followed by missense mutations (23.8%), large insertions or deletions (14.8%), intronic variants (9.2%), synonymous mutations (1.8%), and gene fusion mutations (2.7%). The three most common mutations were p.R33* (c.97C>T; 21.1%), p.R75* (c.223C>T; 13.8%), and p.A67D (c.200C>A; 7.3%). The mutation rates in exons 1 to exon 5 were 8.3%, 0.1%, 29.4%, 34.9%, and 7.3%, respectively. In addition, another 10.1% of the alterations involved multiple exon deletions, and 9.2% of the variations occurred in introns.
Overall, 101 (92.7%) of the 109 patients developed PCC, with a mean age at diagnosis of 32.8 ± 12.6 years (range, 13–80), and the male-to-female ratio was 1.3:1. Of these, 59 (58.4%) developed bilateral PCC, with a mean diagnostic age of 31.7 ± 10.2 years (range, 14–55 years), of which 47 had simultaneous PCC and 12 had metachronous bilateral PCC with a mean interval time of 11.1 ± 6.7 years (range, 3–27 years). The remaining 42 (41.6%) were unilateral PCC with a mean diagnostic age of 34.3 ± 15.4 years (range, 13–80). Nineteen (18.8%) of the patients were diagnosed with metastatic PCC. Eleven (10.9%) had concomitant chest or abdominal PGL, and one presented with head and neck PGL without PCC. Additionally, the detection of the levels of blood/urine catecholamines and their metabolites were also reported in 63 of the 101 patients with PCC. The results showed that 61 (96.8%) were positive, of which 18 (29.5%) were positive for an increase in a single normetanephrine/norepinephrine and 43 (70.5%) were positive for a mixed increase in metanephrine/adrenaline, normetanephrine/norepinephrine, and/or dopamine.
Overall, nine (8.3%) patients were complicated with pituitary NET (PitNET) (16, 20, 24, 27). Among them, three, four, and one patient had a single increase in growth hormone, prolactin, and both hormone and prolactin concentrations, respectively, and were diagnosed with growth hormone tumors, prolactinomas, and mixed adenomas, showing acromegaly and/or prolactinoma symptoms. The hormone levels in the other patient were not reported. Four patients were diagnosed with PCC first, three with PA first, and two with PCC and PA simultaneously. Eleven (10.1%) developed other NETs, with an age at diagnosis ranging from 0.7 to 60 years, including seven combined with PCC: four primary hyperparathyroidism(PHPT) cases and each of one case in neuroblastoma, pelvic ganglioneuroblastoma/ganglionoma, and C-cell hyperplasia. Therein, the four PHPT patients’ mean diagnosis age was 41.8 ± 17.8 years(range, 18-60). Among them, one case showed multifocal parathyroid adenomas on thyroid ultrasound and was confirmed by postoperative pathology which typical of hereditary diseases. One case showed a single solid-appearing lower pole nodule of the left lobe without suspicious malignant features but did not receive surgical treatment. The other two patients’ thyroid ultrasound were not reported. In seven (6.4%) patients, PCC was accompanied by non-NETs, including one each of squamous cell carcinoma of the tongue, papillary thyroid carcinoma, prostate cancer, renal cell carcinoma, renal oncocytoma, breast cancer combined with renal oncocytoma, and chondrosarcoma of the chest combined with multifocal lung adenocarcinoma. In addition, eight (7.3%) patients with MAX mutations did not have PCC, but two had ganglioneomas, one had abdominal ganglioneuroblastoma, one had pancreatic NET, one head and neck PGL, and three were asymptomatic carriers. Among the 87 families, 22 (25.3%) had a family history of PPGL and eight (9.2%) had a clear paternal family genetic background. The ages at diagnosis of the different patient types are shown in Table 3. The occurrence rates of PCC, metastatic PCC, PGL, PA, other NET, and non-NET for the different mutation types (groups) are shown in Table 4. There was no significant difference in the age at diagnosis or the occurrence rate between Groups (all P > 0.05).
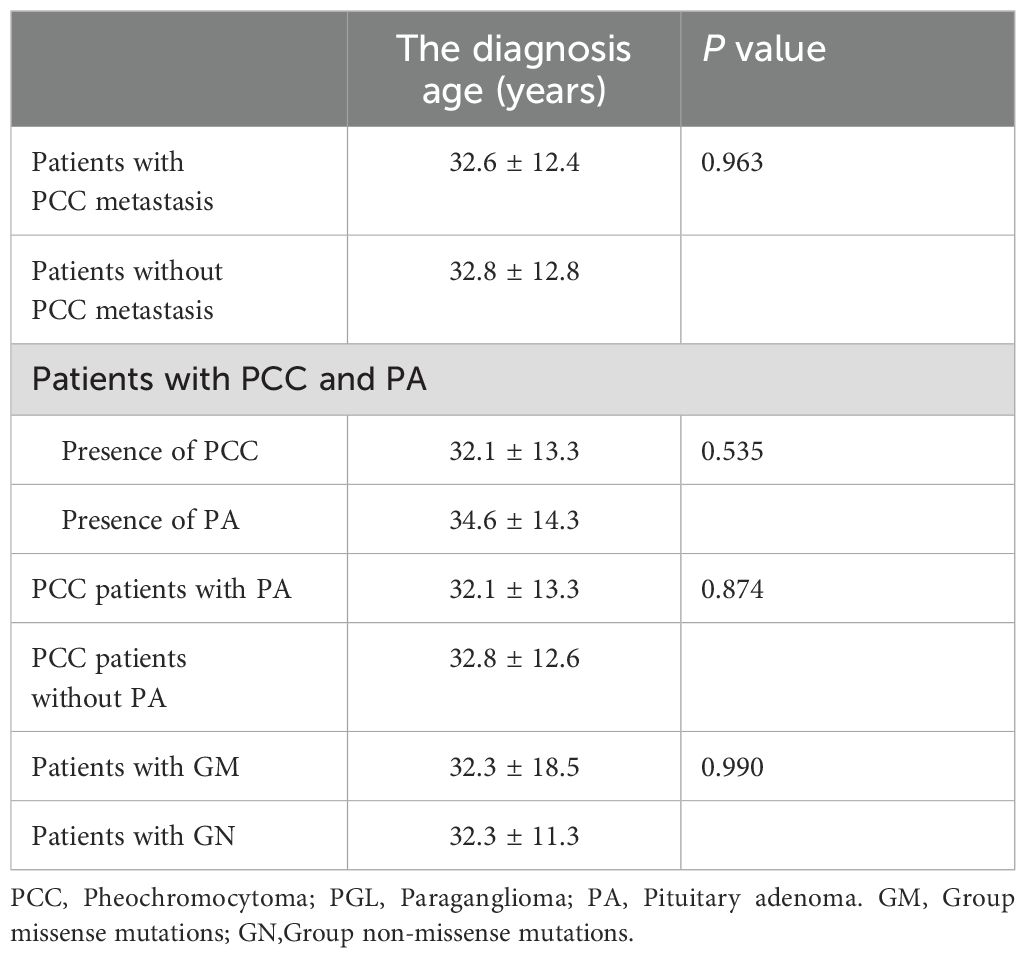
Table 3. The diagnosis age of different types (Groups) of patients.
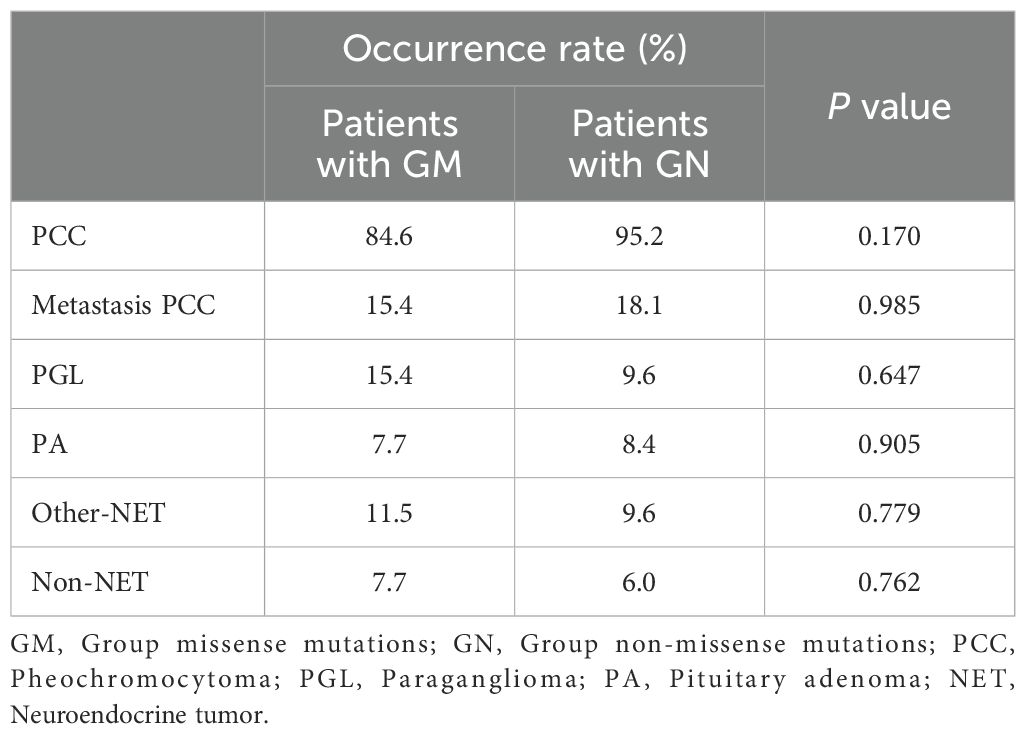
Table 4. The phenotypes’ occurrence rates of different mutation Groups.
The MAX germline mutations were first reported to be associated with hereditary PPGL in 2011 (10). The prevalence of MAX mutations in PPGL varies from 0.8%–1.9% (11–13). To date, almost all MAX mutations in PPGL have been reported in case reports or small family-based cohorts. In this study, we report six patients from three families with p.R75*, p.Q82Tfs*89, and p.E158A MAX mutations, the latter two of which have never been previously reported. Among them, two presented with bilateral PCC and one presented with unilateral PCC, whereas the other three were asymptomatic carriers without any signs of NET. Based on a literature review (10–36), 87 families with 109 MAX mutations have been discovered, with 53 different MAX mutation types involving all five exons. Most mutations were located in exons 3 and 4 of MAX. The mutation types were mainly nonsense, missense, and alternative splice-site mutations, with nonsense mutations p.R33 (21.1%) and p.R75* (13.8%) and the missense mutation p.A67D (7.3%) were the most common mutations, suggesting that there may be family and/or regional aggregation of MAX mutations (10–14). The MAX gene, located on chromosome 14q23.3, encodes a 160-amino acid MAX protein containing an N-terminal basic helix-loop-helix leucine zipper (bHLHLZip) domain and six C-terminal tyrosinase kinase II phosphorylation sites. MAX forms a specific DNA-binding protein complex with MYC or MAD, which recognize the core sequence 5’-CAC[GA]TG-3′ (E-box). MAX mutations result in an abnormal structure of the bHLHLZip domain, preventing MYC-MAX heterodimerization and MAX-MAD homodimerization, and inhibiting the suppression of E-box target DNA sequence transcription activity and/or somatic-level MAX allele loss of heterozygosity (LOH) (37, 38). However, pathogenic MAX mutations repressed the E-box-binding ability of MYC to a lesser extent (Figure 4) (39). Increased MYC expression levels appear to contribute to a more aggressive phenotype of chromaffin cells that develop from neural crest cells (40). MAX had a loss-of-function intolerance probability (pLI) score from the Genome Aggregation Database of 0.83, which was close to 1. A higher pLI score indicates that the gene is more intolerant to loss-of-function, implying that the mutation is more likely to be harmful. This explains the high pathogenicity and penetrance of MAX mutations. Although the MAX mutation is classified as type C2, including RET, NF1, TMEM127,H-RAS and ATRX, which belongs to the SWI/SNF family of chromatin remodeling proteins, as their upregulation will activate the PI3K/AKT and RAS/MAPK signaling pathways resulting in tumor formation (41). It’s clinical phenotype is similar to SDHD- and SDHAF2-related PPGL (C1 type), which is associated with metastatic PPGL and its underlying mechanism involves the “Warburg effect”, which means that genetic mutations lead to damage to aerobic glycolysis and oxidative phosphorylation functions, as well as the mechanism of the HIF-MYC/MAX interaction (1, 2).
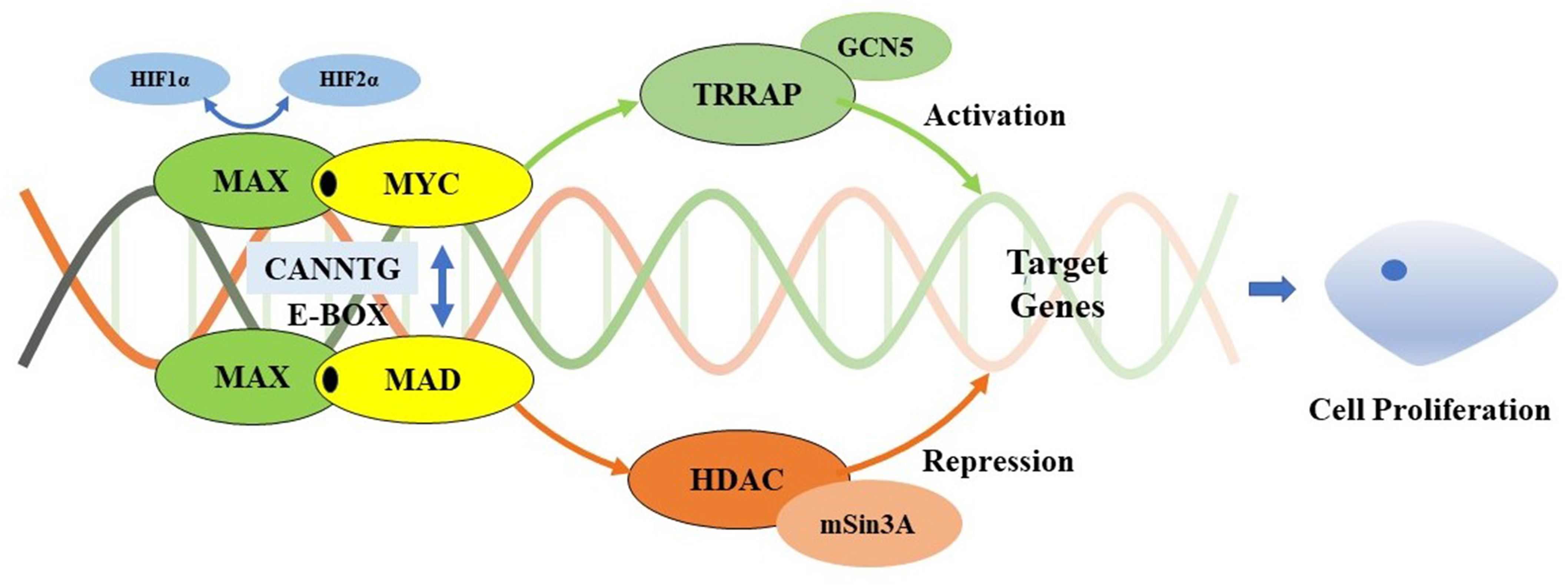
Figure 4. Myc/Max/Mad transcription module. MAX forms a specific DNA-binding protein complex with MYC or MAD which recognizes the core sequence 5’-CAC[GA] TG-3’(E-box). The MYC/MAX complex is a transcriptional activator, interacts with TRRAP that recruits the GCN5, which permitting transcription of target genes affecting cell proliferation. It can be regulated by HIF1α and HIF2α. HIF1α dimerizes with MAX and suppresses the binding to the E-box, while HIF2α leads to stabilization of the MYC/MAX complex to promote the activation of MYC target genes. Meanwhile, the MAD/MAX complex is a repressor which repress transcription by recruiting HDAC’s via mSin3A, and represses MYC transcriptional activity from E-box elements. MAX mutations result in preventing MYC-MAX heterodimerization and MAX-MAD homodimerization, inhibiting the suppression of E-boxs target DNA sequences transcription activity. However, pathogenic MAX mutations repress to a lesser extent MYC’s E-box binding ability. Increased expression of Myc appears to contribute to a more aggressive phenotype of chromaffin cells which develop from neural crest cells.
In all, 101 out of the 109 patients (92.7%) with MAX mutations developed PCC, of which bilateral PCC accounted for 58.4%, with a mean age at diagnosis of 31.7 ± 10.2 years (range, 14–55). Different from other classic PPGL genetic syndromes, which have a PCC incidence rate of 1%–50% (1, 2, 9), as well as sporadic PCC, which often occurs unilaterally, with an average age at diagnosis greater than 45 years (1, 9), MAX mutations always lead to a high incidence of bilateral PPGL and early onset In this study, 19 (18.8%) patients were diagnosed with metastatic PCC at the time of diagnosis, and the youngest age at diagnosis was 18 years, suggesting that patients with MAX mutations had a higher metastatic risk and a younger age at diagnosis than patients with non-metastatic PCC, although the difference was not statistically significant (P = 0.963). The PCC/PGL phenotype is most commonly observed in VHL-, FH-, and HIF2A/EPAS1-related syndromes (1, 2, 9, 14). In this study, 11 (10.9%) patients had PCC combined with thoracic or abdominal PGL, and the youngest age at diagnosis was 15 years. One other patient only presented with head and neck PGL, without PCC. This may be a characteristic manifestation of MAX mutations or it may be associated with the latent onset of PGL in the head and neck, which is often asymptomatic and can be easily misdiagnosed. Available data also suggest that MAX mutations are mostly characterized by a mixed phenotype of noradrenaline/adrenaline secretion rather than a single adrenaline secretion pattern (11, 14–23, 27–33), which may be related to MAX mutations leading to a decrease in phenylethanolamine N-methyltransferase expression levels, resulting in weakened conversion of noradrenaline to adrenaline (38). The positive rate of detecting blood/urine catecholamines and their metabolites metanephrine/normetanephrine in patients with MAX-mutation-related PPGL was 96.8% (11, 14–23, 26–35), which could benefit the preoperative qualitative diagnosis of such types of PPGL (1, 8, 9, 42).
Nine (8.3%) patients had PA, with the youngest age at diagnosis of 16 years. Most patients showed elevated levels of growth hormone only (3/9), prolactin only (4/9), or both (1/9). They manifested clinically as acromegaly and prolactinomas (14, 15, 21, 25, 30). The age at diagnosis was later for PA than for PCC, but the difference was not statistically significant (P = 0.535). PPGL with PA is also known as “3PA syndrome”, and was originally reported to be associated with pathogenic SDHA-D mutations, which was the most common pathogenic cause of familial 3PA syndrome (62.5% to 75%) (14, 43). Considering that the prevalence of PA is approximately 1/3,000, the high frequency of PitNET in MAX mutation carriers is not coincidental. MAX is a pathogenic susceptibility gene, although the direct mechanism of MAX in related PA remains to be elucidated (14).
Eleven (10.1%) patients had other NETs, diagnosed between 0.7 and 60 years old, including seven cases combined with PCC (14, 25, 28). Considering that these tumors (four parathyroid adenoma and one each of neuroblastoma, ganglioneuroblastoma/ganglionoma, and C-cell hyperplasia), PPGL, and PitNETs all originate from embryonic neural crest cells on histological examination, it is reasonable to assume that MAX mutations may trigger such tumors. In a cohort study of 372 neuroblastomas, it was found that 22% of tumor cells had LOH at chromosome 14q, especially at 14q23, including MAX. This further supports the close relationship between MAX mutations and the occurrence of these tumors (14, 44). The other four patients (3.7%) had single occurrences of ganglioneuroma (n = 2), abdominal neuroblastoma (n = 1), and pancreatic NET (n = 1) with the youngest age at diagnosis of 0.7 years (14, 25, 28). It is suspected that ancestral cells may overlap and have different expression spectra, and mature PPGL (type C2) originates from the chromaffin cell lineage, while mature neuroblastoma originates from the sympathetic neuroblastoma lineage. It may imply that MAX mutants have undergone a two-hit of single tumor stem cells on their second chromosome early in embryogenesis, before neural crest cells migrate and localize to the paraganglia or adrenal glands (39, 40). Two-hit hypothesis holds that certain genes (such as tumor suppressor genes) need to undergo “two hits”, that is, both alleles mutate or are inactivated, before tumors will occur. In hereditary tumors, the first hit is usually a germline variation of a certain tumor suppressor gene in germ cells, making the individual genetically susceptible; the second hit is a somatic variation of the other allele of this gene after birth, thereby promoting the occurrence and development of tumors. The MAX gene belongs to tumor suppressor genes, and its mutations may also follow the “two-hit” hypothesis (39, 40).
Seven (6.4%) patients had non-NETs (11, 14, 15, 20). The small number of cases and the involvement of six different systems and tissues (tongue, thyroid, kidney, prostate, breast, cartilage, and lung) may be an incidental or concomitant phenomenon; however, it is still difficult to exclude the possibility of a possible association with the MAX mutation. It should be noted, however, that of 109 patients with MAX mutations, three (2.8%) still had no evidence of a clinical phenotype.
Using MAX mutation genotype-phenotype analysis, we compared the occurrence rate of PCC (P = 0.170), metastatic PCC (P = 0.985), PGL (P = 0.647), PA (P = 0.905), other-NET (P = 0.779), and non-NET (P = 0.762), and the mean age at diagnosis at the presentation of PCC (P = 0.990) between patients with missense and non-missense mutations. The results indicated that the high penetrance of MAX mutation-related PCC and relatively high metastatic risk may not be related to MAX mutation types, or that the genotype-phenotype correlation is not strong. In addition, only 22 (25.3%) of the 87 MAX mutation pedigrees had a clear family history of PPGL (10–14, 20, 25–29, 33–35), similar to the case for RET mutations causing MEN2B (type C2) (42). Including the patient in pedigree II-1 of family A, MAX mutations may also have a very high de novo mutation rate (65.8%), and de novo MAX mutations may be a common pathogenic form. equations should be inserted in editable format from the equation editor.
In conclusion, MAX germline mutations may cause a new type of MEN originating from the neural crest, which may be called MEN 5 (14, 25, 45, 46). This suggests that all MAX germline mutation carriers should receive a comprehensive baseline assessment of the diversity of neurocrest-derived diseases, such as PPGL, PA, hyperparathyroidism, and ganglioneuroma/neuroblastoma. The appropriate frequency (interval) and extent of evaluation are optimized, but should take into consideration the risk preferences for bilateral and/or metastatic PCC, with lifelong intensive surveillance. Meanwhile, for patients with new diagnoses of PPGL, NGS is recommended to perform to identify potential mutations in all known susceptibility genes.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
The studies involving human participants were reviewed and approved by the Ethics Committee of the 903rd PLA Hospital, Taizhou Enze Medical Center (Group), Tiantai County People’s Hospital, and the First Hospital of Jiaxing. Written informed consent was obtained from all study subjects or their legal guardians.
BL: Data curation, Formal analysis, Writing – original draft. JL: Writing – original draft. XF: Data curation, Formal analysis, Writing – original draft. YZ: Writing – review & editing. WW: Data curation, Writing – review & editing. YH: Data curation, Writing – review & editing. HY: Writing – review & editing. FL: Writing – review & editing. JW: Writing – review & editing. WC: Writing – original draft. XQ: Conceptualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. National Natural Science Foundation of China (81472861), the Key Project of Zhejiang Province Science and Technology Plan (2014C03048-1), and Hangzhou Municipal Commission of Health and Family Planning Science and Technology Program (B20210355), Medical and Health Science and Technology Project of Zhejiang Province (2023KY1343, 2024KY1830); Taizhou Social Development Science and Technology Planning Project (23ywb56, 23ywb156).
We would like to thank Editage (www.editage.cn) for English language editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Garcia-Carbonero R, Matute Teresa F, Mercader-Cidoncha E, Mitjavila-Casanovas M, Robledo M, Tena I, et al. Multidisciplinary practice guidelines for the diagnosis, genetic counseling and treatment of pheochromocytomas and paragangliomas. Clin Transl Oncol. (2021) 23:1995–2019. doi: 10.1007/s12094-021-02622-9
2. Nölting S, Bechmann N, Taieb D, Beuschlein F, Fassnacht M, Kroiss M, et al. Personalized management of pheochromocytoma and paraganglioma. Endocr Rev. (2022) 43:199–239. doi: 10.1210/endrev/bnab019
3. Pillai S, Gopalan V, Smith RA, Lam AK. Updates on the genetics and the clinical impacts on phaeochromocytoma and paraganglioma in the new era. Crit Rev Oncol Hematol. (2016) 100:190–208. doi: 10.1016/j.critrevonc.2016.01.022
4. Lenders JWM, Kerstens MN, Amar L, Prejbisz A, Robledo M, Taieb D, et al. Genetics, diagnosis, management and future directions of research of phaeochromocytoma and paraganglioma: a position statement and consensus of the Working Group on Endocrine Hypertension of the European Society of Hypertension. J Hypertension. (2020) 38:1443–56. doi: 10.1097/hjh.0000000000002438
5. Jochmanova I, Pacak K. Genomic landscape of pheochromocytoma and paraganglioma. Trends Cancer. (2018) 4:6–9. doi: 10.1016/j.trecan.2017.11.001
6. Qi XP, Zhao JQ, Fang XD, Lian BJ, Li F, Wang HH, et al. Spectrum of Germline RET variants identified by targeted sequencing and associated Multiple Endocrine Neoplasia type 2 susceptibility in China. BMC Cancer. (2021) 21:369. doi: 10.1186/s12885-021-08116-9
7. Qi XP, Lian BJ, Fang XD, Dong F, Li F, Jin HY, et al. Simultaneous bilateral laparoscopic cortical-sparing adrenalectomy for bilateral pheochromocytomas in multiple endocrine neoplasia type 2. Front Surg. (2022) 9:1057821. doi: 10.3389/fsurg.2022.1057821
8. Geroula A, Deutschbein T, Langton K, Masjkur J, Pamporaki C, Peitzsch M, et al. Pheochromocytoma and paraganglioma: clinical feature-based disease probability in relation to catecholamine biochemistry and reason for disease suspicion. Eur J Endocrinol. (2019) 181:409–20. doi: 10.1530/eje-19-0159
9. Neumann HPH, Young WF Jr, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med. (2019) 381:552–65. doi: 10.1056/NEJMra1806651
10. Comino-Méndez I, Gracia-Aznárez FJ, Schiavi F, Landa I, Leandro-García LJ, Letón R, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet. (2011) 43:663–7. doi: 10.1038/ng.861
11. Burnichon N, Cascón A, Schiavi F, Morales NP, Comino-Méndez I, Abermil N, et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res. (2012) 18:2828–37. doi: 10.1158/1078-0432.Ccr-12-0160
12. Bausch B, Schiavi F, Ni Y, Welander J, Patocs A, Ngeow J, et al. Clinical characterization of the pheochromocytoma and paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for gene-informed prevention. JAMA Oncol. (2017) 3:1204–12. doi: 10.1001/jamaoncol.2017.0223
13. Ma X, Li M, Tong A, Wang F, Cui Y, Zhang X, et al. Genetic and clinical profiles of pheochromocytoma and paraganglioma: A single center study. Front Endocrinol (Lausanne). (2020) 11:574662. doi: 10.3389/fendo.2020.574662
14. Seabrook AJ, Harris JE, Velosa SB, Kim E, McInerney-Leo AM, Dwight T, et al. Multiple endocrine tumors associated with germline MAX mutations: Multiple endocrine neoplasia type 5? J Clin Endocrinol Metab. (2021) 106:1163–82. doi: 10.1210/clinem/dgaa957
15. Daly AF, Castermans E, Oudijk L, Guitelman MA, Beckers P, Potorac I, et al. A Novel Phenotype of Germline in three patients with MAX exon deletions. Endocr Relat Cancer. (2018) 25:L37–42. doi: 10.1530/erc-18-0065
16. Cascón A, Inglada-Pérez L, Comino-Méndez I, de Cubas AA, Letón R, Mora J, et al. Genetics of pheochromocytoma and paraganglioma in Spanish pediatric patients. Endocr Relat Cancer. (2013) 20:L1–6. doi: 10.1530/erc-12-0339
17. Urbini M, Nannini M, Astolfi A, Indio V, Vicennati V, De Luca M, et al. Whole exome sequencing uncovers germline variants of cancer-related genes in sporadic pheochromocytoma. Int J Genomics. (2018) 2018:6582014. doi: 10.1155/2018/6582014
18. Romanet P, Guerin C, Pedini P, Essamet W, Castinetti F, Sebag F, et al. Pathological and genetic characterization of bilateral adrenomedullary hyperplasia in a patient with germline MAX mutation. Endocr Pathol. (2017) 28:302–7. doi: 10.1007/s12022-016-9460-5
19. Pęczkowska M, Kowalska A, Sygut J, Waligórski D, Malinoc A, Janaszek-Sitkowska H, et al. Testing new susceptibility genes in the cohort of apparently sporadic phaeochromocytoma/paraganglioma patients with clinical characteristics of hereditary syndromes. Clin Endocrinol (Oxf). (2013) 79:817–23. doi: 10.1111/cen.12218
20. Korpershoek E, Koffy D, Eussen BH, Oudijk L, Papathomas TG, van Nederveen FH, et al. Complex MAX rearrangement in a family with Malignant pheochromocytoma, renal oncocytoma, and erythrocytosis. J Clin Endocrinol Metab. (2016) 101:453–60. doi: 10.1210/jc.2015-2592
21. Roszko KL, Blouch E, Blake M, Powers JF, Tischler AS, Hodin R, et al. Case report of a prolactinoma in a patient with a novel MAX mutation and bilateral pheochromocytomas. J Endocr Soc. (2017) 1:1401–7. doi: 10.1210/js.2017-00135
22. Shibata M, Inaishi T, Miyajima N, Adachi Y, Takano Y, Nakanishi K, et al. Synchronous bilateral pheochromocytomas and paraganglioma with novel germline mutation in MAX: a case report. Surg Case Rep. (2017) 3:131. doi: 10.1186/s40792-017-0408-x
23. Ferrara AM, Lombardi G, Pambuku A, Meringolo D, Bertorelle R, Nardin M, et al. Temozolomide treatment of a Malignant pheochromocytoma and an unresectable MAX-related paraganglioma. Anti-cancer Drugs. (2018) 29:102–5. doi: 10.1097/cad.0000000000000570
24. Albattal S, Alswailem M, Moria Y, Al-Hindi H, Dasouki M, Abouelhoda M, et al. Mutational profile and genotype/ phenotype correlation of non-familial pheochromocytoma and paraganglioma. Oncotarget. (2019) 10:5919–31. doi: 10.18632/oncotarget.27194
25. Petignot S, Daly AF, Castermans E, Korpershoek E, Scagnol I, Beckers P, et al. Pancreatic neuroendocrine neoplasm associated with a familial MAX deletion. Horm Metab Res. (2020) 52:784–7. doi: 10.1055/a-1186-0790
26. Choi H, Kim KJ, Hong N, Shin S, Choi JR, Kang SW, et al. Genetic analysis and clinical characteristics of hereditary pheochromocytoma and paraganglioma syndrome in korean population. Endocrinol Metab (Seoul). (2020) 35:858–72. doi: 10.3803/EnM.2020.683
27. Pozza C, Sesti F, Di Dato C, Sbardella E, Pofi R, Schiavi F, et al. A novel MAX gene mutation variant in a patient with multiple and "Composite" Neuroendocrine-neuroblastic tumors. Front Endocrinol (Lausanne). (2020) 11:234. doi: 10.3389/fendo.2020.00234
28. Chang X, Li Z, Ma X, Cui Y, Chen S, Tong A. A novel phenotype of germline pathogenic variants in MAX: concurrence of pheochromocytoma and ganglioneuroma in a chinese family and literature review. Front Endocrinol (Lausanne). (2020) 11:558. doi: 10.3389/fendo.2020.00558
29. Lam-Chung CE, Rodríguez LL, Vázquez JA, Chávarri-Guerra Y, Arízaga-Ramírez R, Antonio OF, et al. A novel, likely pathogenic MAX germline variant in a patient with unilateral pheochromocytoma. J Endocr Soc. (2021) 5:bvab085. doi: 10.1210/jendso/bvab085
30. Mamedova E, Vasilyev E, Petrov V, Buryakina S, Tiulpakov A, Belaya Z. Familial acromegaly and bilateral asynchronous pheochromocytomas in a female patient with a MAX mutation: A case report. Front Endocrinol (Lausanne). (2021) 12:683492. doi: 10.3389/fendo.2021.683492
31. Hata S, Asano M, Tominaga H, Hamaguchi M, Hongo F, Usui T, et al. Bilateral pheochromocytoma with germline MAX Variant without family history. Clin Pract. (2022) 12:299–305. doi: 10.3390/clinpract12030035
32. Parisien-La Salle S, Dumas N, Bédard K, Jolin J, Moramarco J, Lacroix A, et al. Genetic spectrum in a Canadian cohort of apparently sporadic pheochromocytomas and paragangliomas: New data on multigene panel retesting over time. Clin Endocrinol (Oxf). (2022) 96:803–11. doi: 10.1111/cen.14618
33. Taïeb D, Jha A, Guerin C, Pang Y, Adams KT, Chen CC, et al. 18F-FDOPA PET/CT imaging of MAX-related pheochromocytoma. J Clin Endocrinol Metab. (2018) 103:1574–82. doi: 10.1210/jc.2017-02324
34. Duarte DB, Ferreira L, Santos AP, Costa C, Lima J, Santos C, et al. Case report: pheochromocytoma and synchronous neuroblastoma in a family with hereditary pheochromocytoma associated with a MAX deleterious variant. Front Endocrinol (Lausanne). (2021) 12:609263. doi: 10.3389/fendo.2021.609263
35. Richter JE Jr, Hines S, Selvam P, Atwal H, Farres H, Caulfield TR, et al. Clinical description & molecular modeling of novel MAX pathogenic variant causing pheochromocytoma in family, supports paternal parent-of-origin effect. Cancer Genet. (2021) 252-253:107–10. doi: 10.1016/j.cancergen.2021.01.004
36. Charoenngam N, Mannstadt M. Primary hyperparathyroidism in a patient with bilateral pheochromocytoma and a mutation in the tumor suppressor MAX. JCEM Case Rep. (2023) 1:luad006. doi: 10.1210/jcemcr/luad006
37. Lüscher B, Larsson LG. The basic region/helix-loop-helix/leucine zipper domain of Myc proto-oncoproteins: function and regulation. Oncogene. (1999) 18:2955–66. doi: 10.1038/sj.onc.1202750
38. Grandori C, Cowley SM, James LP, Eisenman RN. The Myc/Max/Mad network and the transcriptional control of Cell behavior. Annu Rev Cell Dev Biol. (2000) 16:653–99. doi: 10.1146/annurev.cellbio.16.1.653
39. Comino-Méndez I, Leandro-García LJ, Montoya G, Inglada-Pérez L, de Cubas AA, Currás-Freixes M, et al. Functional and in silico assessment of MAX variants of unknown significance. J Mol Med (Berl). (2015) 93:1247–55. doi: 10.1007/s00109-015-1306-y
40. Bechmann N, Westermann F, Eisenhofer G. HIF and MYC signaling in adrenal neoplasms of the neural crest: implications for pediatrics. Front Endocrinol (Lausanne). (2023) 14:1022192. doi: 10.3389/fendo.2023.1022192
41. Koopman K, Gaal J, de Krijger RR. Pheochromocytomas and paragangliomas: new developments with regard to classification, genetics, and cell of origin. Cancers (Basel). (2019) 11:1070. doi: 10.3390/cancers11081070
42. Qi XP, Jin B. The trends in early precision diagnosis and treatment strategies of multiple endocrine neoplasia type 2. Zhonghua Wai Ke Za Zhi. (2022) 60:973–80. doi: 10.3760/cma.j.cn112139-20220714-00312
43. Xekouki P, Brennand A, Whitelaw B, Pacak K, Stratakis CA. The 3PAs: an Update on the association of pheochromocytomas, paragangliomas, and Pituitary Tumors. Horm Metab Res. (2019) 51:419–36. doi: 10.1055/a-0661-0341
44. Thompson PM, Seifried BA, Kyemba SK, Jensen SJ, Guo C, Maris JM, et al. Loss of heterozygosity for chromosome 14q in neuroblastoma. Med Pediatr Oncol. (2001) 36:28–31. doi: 10.1002/(ISSN)1096-911X
45. Singeisen H, Renzulli MM, Pavlicek V, Probst P, Hauswirth F, Muller MK, et al. Multiple endocrine neoplasia type 4: a new member of the MEN family. Endocr Connect. (2023) 12:e220411. doi: 10.1530/EC-22-0411
Keywords: multiple endocrine neoplasia, pheochromocytoma, paraganglioma, genealogy, MAX gene
Citation: Lian B, Lu J, Fang X, Zhang Y, Wang W, He Y, Yu H, Li F, Wang J, Chen W and Qi X (2024) Genotype and clinical phenotype characteristics of MAX germline mutation–associated pheochromocytoma/paraganglioma syndrome. Front. Endocrinol. 15:1442691. doi: 10.3389/fendo.2024.1442691
Received: 02 June 2024; Accepted: 15 August 2024;
Published: 30 August 2024.
Edited by:
Mirko Parasiliti-Caprino, University of Turin, ItalyCopyright © 2024 Lian, Lu, Fang, Zhang, Wang, He, Yu, Li, Wang, Chen and Qi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoping Qi, cXhwbG1kQDE2My5jb20=; Weiying Chen, Y2hlbnd5ODMzNUBlbnplbWVkLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.