 Qiyuan Keith Liu
Qiyuan Keith Liu- MedStar Medical Group, MedStar Montgomery Medical Center, Olney, MD, United States
Glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) are two incretins that bind to their respective receptors and activate the downstream signaling in various tissues and organs. Both GIP and GLP-1 play roles in regulating food intake by stimulating neurons in the brain’s satiety center. They also stimulate insulin secretion in pancreatic β-cells, but their effects on glucagon production in pancreatic α-cells differ, with GIP having a glucagonotropic effect during hypoglycemia and GLP-1 exhibiting glucagonostatic effect during hyperglycemia. Additionally, GIP directly stimulates lipogenesis, while GLP-1 indirectly promotes lipolysis, collectively maintaining healthy adipocytes, reducing ectopic fat distribution, and increasing the production and secretion of adiponectin from adipocytes. Together, these two incretins contribute to metabolic homeostasis, preventing both hyperglycemia and hypoglycemia, mitigating dyslipidemia, and reducing the risk of cardiovascular diseases in individuals with type 2 diabetes and obesity. Several GLP-1 and dual GIP/GLP-1 receptor agonists have been developed to harness these pharmacological effects in the treatment of type 2 diabetes, with some demonstrating robust effectiveness in weight management and prevention of cardiovascular diseases. Elucidating the underlying cellular and molecular mechanisms could potentially usher in the development of new generations of incretin mimetics with enhanced efficacy and fewer adverse effects. The treatment guidelines are evolving based on clinical trial outcomes, shaping the management of metabolic and cardiovascular diseases.
1 Introduction
Glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) are two naturally occurring hormonal peptides produced in gastrointestinal tract, knowns as incretins. Together, they orchestrate a crucial hormonal regulation known as the incretin effect. The concept of incretin effect was first proposed by Creutzfeldt in the 1970s (1, 2), based on the early observations that insulin secretion was two to three times higher after oral glucose intake than that after an isocaloric intravenous glucose administration (3–5). The incretin effect was estimated to account for approximately 50% - 70% of the postprandial insulin responses in healthy individuals and may be substantially reduced to 20% – 30% in individuals with type 2 diabetes mellitus (T2DM) (6, 7), a complex disorder arising from inadequate compensation of insulin secretion by pancreas to counter peripheral insulin resistance. Consequently, researchers have devoted decades to studying incretins, postulating that incretin-based therapies could potentially reverse this diminished incretin effect and restore insulin secretion in patients with T2DM. This review presents a concise history of the discoveries of GIP and GLP-1, explores the physiology and pharmacology of incretins and their synthetic mimetics, and discusses the therapeutic applications of the USA FDA-approved GLP-1 and dual GIP/GLP-1 receptor agonists.
2 Physiology of incretins
2.1 Discoveries of GIP and GLP-1
The first incretin, GIP, was purified from canine intestinal extracts in the late 1960s (8) and initially named gastric inhibitory polypeptide (9) because the peptide inhibited gastrin-stimulated H+ secretion (10). In the early 1970s, Dupre et al. (11) discovered that infusion of GIP purified from porcine duodenojejunal mucosa, when combined with glucose, led to enhanced insulin secretion and improved glucose intolerance in humans. As a result, GIP was designated as the first incretin (1, 12). In normal subjects, both fat and carbohydrate stimulate GIP secretion from enteroendocrine K cells, which are dispersed in the upper portion of the gastrointestinal tract (duodenum and jejunum). Interestingly, fat appears to be a stronger stimulator of GIP secretion than carbohydrate (12, 13) [Figure 1 – Section A]. Most intestinal K cells secrete a biologically active form of GIP consisting of 42 amino acids, GIP (1–42), which is derived from a 153 amino acid preprohormone precursor distinct from preproglucagon (14). GIP is conserved across mammalian species (15); purification and sequencing of porcine and bovine GIP revealed only minor differences (two amino acids in porcine and one in bovine) compared to the human GIP peptide (16–18) [Figure 2].
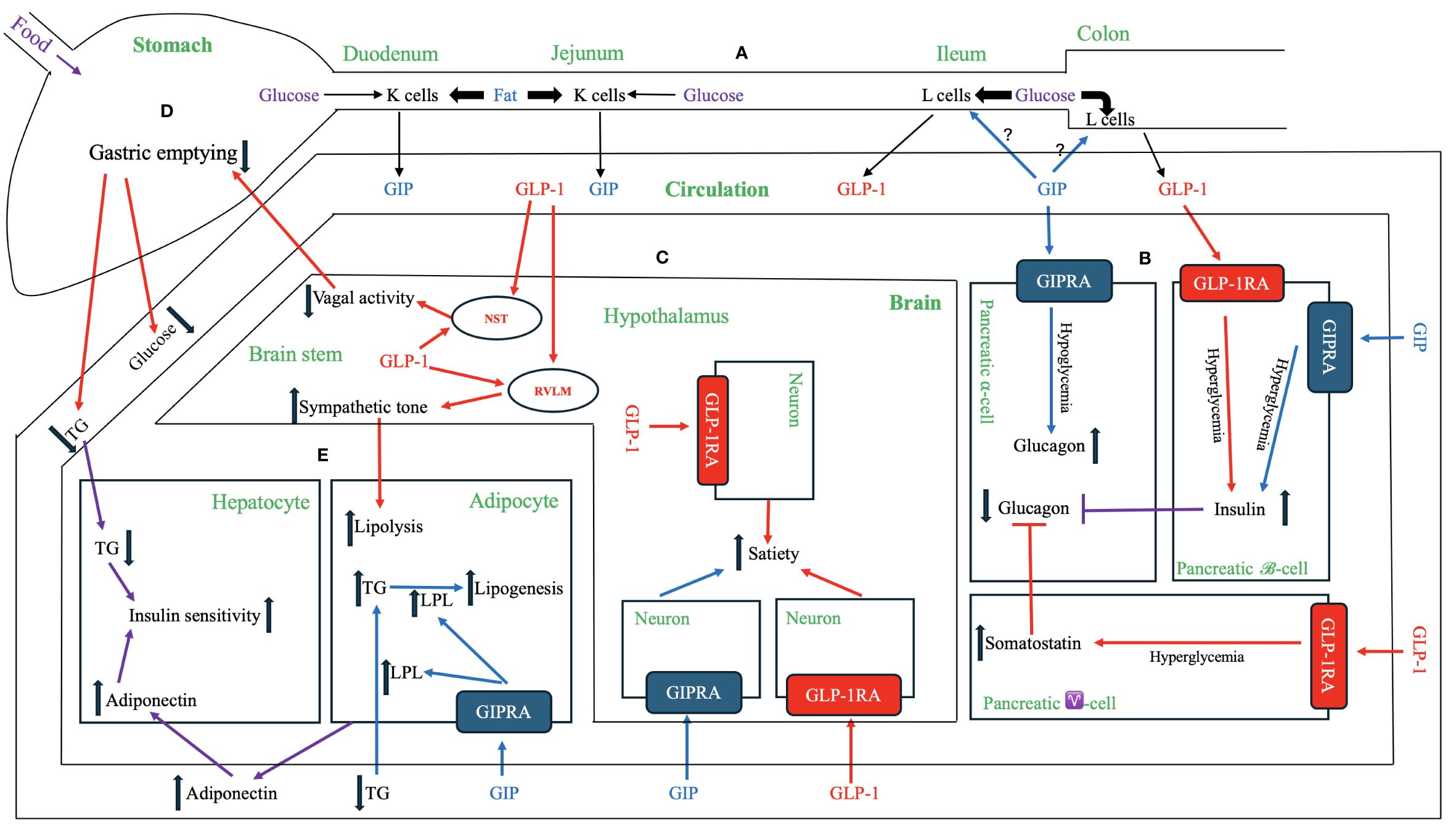
Figure 1 Physiological and Pharmacological Effects of GIP, GLP-1 and Their Receptor Agonists. (A) GIP and GLP-1 secretion. Enteroendocrine K cells in the upper gastrointestinal tract (duodenum and jejunum) and L cells in the distal gut (ileum and colon) produce and secret GIP and GLP-1, respectively, into the circulation. GIP peptide and the downstream signaling pathways are depicted in blue, while GLP-1 peptide and the downstream signaling pathways are represented in red. The signaling pathways regulated by both GIP and GLP-1 are shown in violet. (B) Effects on pancreatic endocrine functions. Both GIP and GLP-1 have insulinotropic effects on pancreatic β-cells during hyperglycemia. GIP has a glucagonotropic effect in pancreatic α-cells during hypoglycemia but no effect during hyperglycemia. GLP-1 has an indirect glucagonostatic effect during hyperglycemia but no effect during hypoglycemia. (C) Effects in the brain. Both GIP and GLP-1 activate their respective receptors in distinct neurons within central nervous system (e.g., hypothalamus) to increase the sense of satiety. Notably, GLP-1, but not GIP, is directly produced in the brain (e.g., nucleus of the solitary tract (NST) and hypothalamus), but both hormones can cross blood-brain barrier from systemic circulation. The central nervous system [e.g., rostral ventrolateral medulla (RVLM)] transmits GLP-1 signaling to increase sympathetic tone, which in turn enhances lipolysis in adipose tissue. The central nervous system (e.g., NST) also relays GLP-1 signaling between afferent and efferent vagal nerves to delay gastric emptying. (D) Effects on the stomach. GLP-1delays the gastric emptying, thereby reducing the postprandial lipid and carbohydrate surges. This effect of GLP-1 is at least partially mediated by the neuronal pathway, including afferent vagal nerve, brainstem (e.g., NST), and efferent vagal nerve. GIP does not have a similar mechanism for regulating gastric emptying. (E) Effects on adipose tissue and liver. GIP binds GIPR in adipose tissue and activates a signaling cascade that increases lipoprotein lipase (LPL) expression and secretion. LPL enhances triglyceride (TG) clearance from circulation, facilitates the transport of TG to adipocytes for lipogenesis, and attenuates ectopic fat accumulation in visceral organs. GLP-1 indirectly enhances lipolysis via increased sympathetic activity through the central nervous system. Both GIP and GLP-1 have indirect effects on the liver by increasing the secretion of adiponectin from adipose tissue and attenuating hypertriglyceridemia. These mechanisms reduce hepatic fat accumulation, ameliorate insulin resistance, and inhibits hepatic glucogenesis.
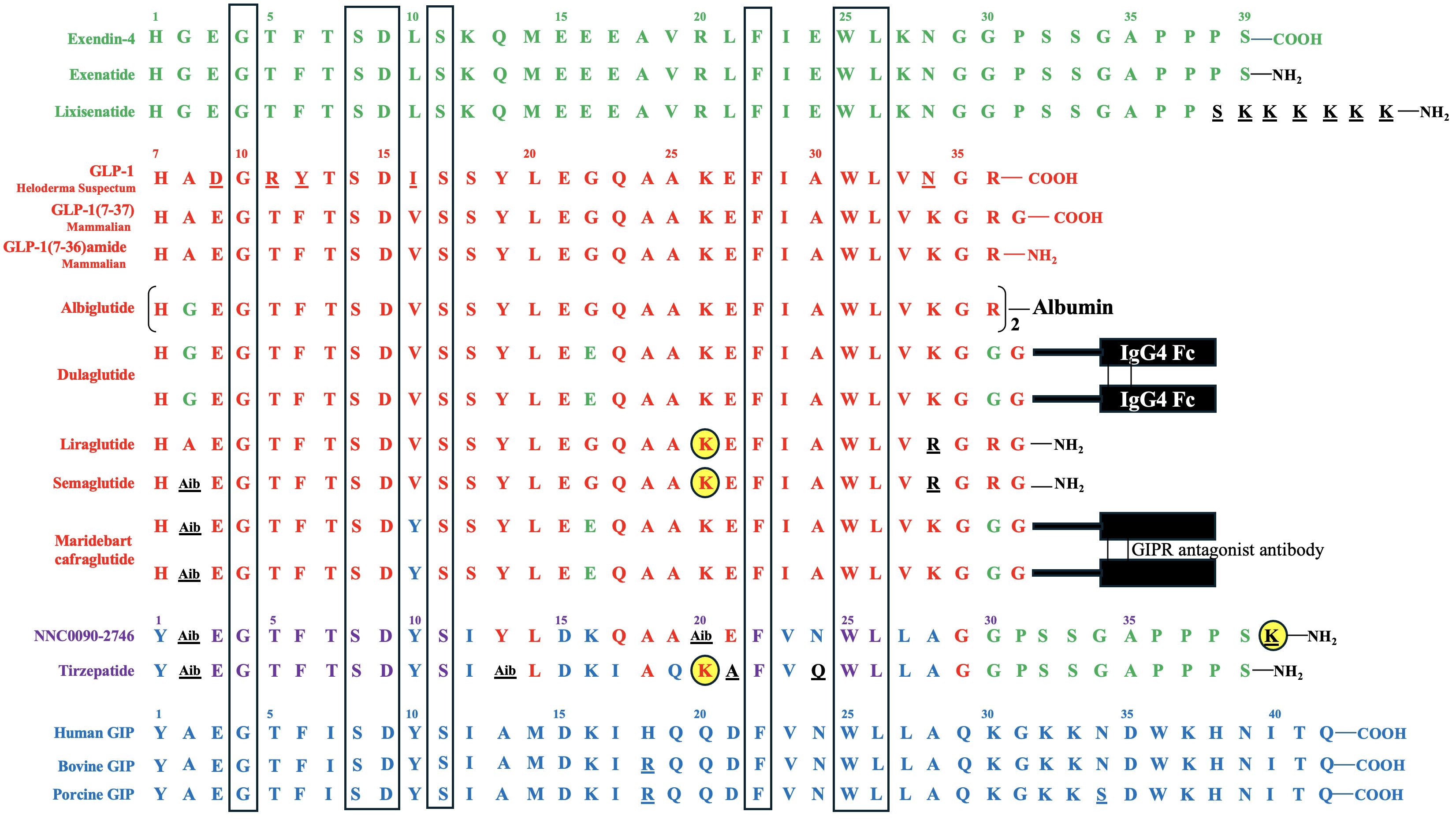
Figure 2 Amino Acid Sequences of Exendin-4, GLP-1, GIP, and Their Derivatives. Exendin-4 and its derivatives are depicted in green, while GLP-1 and its derivatives are represented in red. GLP-1 is conserved across mammalian species. Human GIP and some mammalian (porcine and bovine) GIP variants are shown in blue. The amino acids of the derivatives that differ from their backbone sequences are highlighted in black and underlined. The amino acids of bovine and porcine GIP that are not homologous to human GIP, as well as the amino acids of Heloderma Suspectum GLP-1 that are not homologous to human GLP-1, are also underlined. In NNC0090-2746 and tirzepatide, amino acids shared by both GIP and GLP-1 are displayed in violet, those unique to GIP are shown in blue, those exclusive to GLP-1 are shown in red, and those specific to exendin-4 are displayed in green, while those not identical to GIP, GLP-1 or exendin-4 are depicted in black and underlined. The acylated lysine residues in liraglutide, semaglutide, NNC0090-2746, and tirzepatide are indicated by yellow-filled circles. Seven amino acids conserved in all peptides are highlighted in the boxes.
The discovery of the second incretin, GLP-1, originated from observations in 1982 that anglerfish proglucagon mRNA contained coding sequences of glucagon-related peptide, flanked by pairs of basic amino acids characteristic of the sites cleaved during post-translational processing of prohormones (19). In 1983, Bell et al. reported the human and hamster GLP-1 gene and the deduced peptide sequences using cDNA hybridization technology and analysis of the human preproglucagon gene (20, 21). In 1986, Mojsov et al. (22) utilized rabbit anti-serum generated from synthetic peptide to identify GLP-1 peptide in both human pancreatic and intestinal tissues. Meanwhile, Holst et al. (23) employed hydrophobic gel permeation and HPLC technique to isolate GLP-1 from pig ileal mucosa. Subsequently, these two groups independently reported that the synthetic GLP-1 peptide acted as a potent stimulator of insulin secretion in isolated perfused rat pancreas (24) and isolated perfused pig pancreas (23), respectively, thus implicating GLP-1 as the second incretin. This second incretin is produced by enteroendocrine L cells, which are diffusedly distributed in the distal gut mucosa of ileum and colon (25, 26) [Figure 1 – Section A]. GLP-1 exists in two equipotent circulating peptides: GLP-1 (7–37) and GLP-1 (7–36) amide (27), with GLP-1 (7–36) amide being more abundant in the circulation after a meal (28). The complete conservation of GLP-1 across all mammalian species underscores its critical physiological role (29, 30) [Figure 2].
2.2 Molecular physiology of GIP and GLP-1
GIP and GLP-1 concentrations appear to be highly variable among individuals, both with and without T2DM. Interestingly, the mean values remain relatively normal in most T2DM groups (31–33), suggesting that the impaired incretin releases are not a typical prerequisite for the development of T2DM. The fasting plasma levels of GIP typically range between 10 – 20 pM with the peak values reaching around 80 – 150 pM after a meal (33–36). On the other hand, fasting plasma levels of bioactive GLP-1 typically fall within the ranges of 5 - 15 pM (33, 37, 38) and could increase 3 – 5 fold postprandially. Under physiological conditions, postprandial GIP levels are approximately 3 – 4 times higher in molar concentration compared to GLP-1, irrespective of diabetic status (28, 39).
Shortly after food intake, these incretins are released into body circulation and bind to their receptors. The physiological effects of GIP and GLP-1 are closely tied to the distribution of their respective receptors in various tissues and organs. Among the tissues and organs crucial in regulating glucose and lipid metabolism, pancreas, brain, and adipocytes express GIP receptors (GIPRs), while pancreas, brain, and gastrointestinal tract are rich in GLP-1 receptors (GLP-1Rs) (40). GIPR was first cloned in rats, identifying a 455-amino acid glycoprotein with a predicted molecular weight of approximately 59 kDa (41). Distinct from GIPR, GLP-1R consists of 463 amino acids and has a molecular weight of 62 kDa (42). Both GIPR and GLP-1R belong to the class B family of 7-transmembrane G protein-coupled receptors (GPCR) within the glucagon receptor superfamily (43). The C-terminal of hormone binds to the extracellular domain, while the N-terminal interacts with the transmembrane domain of the receptor (43). Notably, a study by Finan et al. (44) reported that the half-maximal effective concentrations (EC50) of GIP with GIPR and of GLP-1 with GLP-1R are 20 pM and 28 pM, respectively, without exhibiting cross-reactivity.
The hormonal binding at the extracellular domain is communicated to the intracellular receptor side, leading to G protein engagement and activation (45). The differential insulinotropic potency and other physiological effects of GIP and GLP-1 in both healthy individuals and those with T2DM may be linked to their distinct receptors and the downstream G proteins that transmit signals intracellularly. These pathways exhibit both overlapping function, such as stimulation of adenylate cyclase/cAMP pathway, and unique signaling transduction cascades. Notably, in murine pancreatic β-cells, GLP-1 can activate both G proteins Gαs and Gαq, whereas GIP selectively activates Gαs (45). Despite extensive research into the molecular mechanisms of GLP-1 and GIP actions in pancreatic β-cells, their effects in other cell types remain relatively unexplored.
2.3 Interaction of GIP and GLP-1 in the incretin effect
Studies in rodents (46) and canines (47) have delineated the existence of a proximal-distal incretin loop. Within this loop, nutrient stimulation of GIP secretion enhances GLP-1 release [Figure 1 – Section A]. However, a study by Nauck et al. (48) did not support the existence of this proximal-distal incretin loop in humans as intravenous injection of GIP into humans did not enhance GLP-1 secretion, pointing to the fact that the animal models may differ from humans in essential physiology. Interestingly, individuals with low levels of GIP may also have low levels of GLP-1 and vice versa (49). Once secreted, both endogenous GIP and GLP-1 undergo rapid degradation into the biologically inactive metabolites (50, 51). This degradation is catalyzed by the ubiquitous serum enzyme dipeptidyl peptidase 4 (DPP-4), which is produced both locally in the intestine and by circulating white blood cells. Both endogenous GIP (52) and GLP-1 (53) have very short half-lives, measured in minutes, resulting in only a small portion of these active hormones reaching the systemic circulation.
The effects of the incretins with respect to insulin secretion and postprandial glucose regulation have been extensively studied. In healthy individuals, studies indicate that the effects of these two hormones are additive (48, 54). However, the relative contributions of GIP and GLP-1 to the incretin effect and glycemic control under physiological and pharmacological conditions remain a topic of debate. When their respective postprandial concentrations were increased through exogenous infusion, GLP-1 elicited a more robust insulin response, suggesting its superior potency compared to GIP as an incretin (28, 55). Conversely, a study by Gasbjerg et al. (54) found that administering GIPR antagonist to healthy humans reduced postprandial insulin secretion and increased glucose excursions more than administering GLP-1R antagonist (GLP-1RA), indicating that endogenous GIP may play a more significant role in stimulating insulin secretion and reducing postprandial plasma glucose excursions compared to endogenous GLP-1. The relative importance of GIP and GLP-1 may vary depending on meal composition and diabetes stages. Notably, while both carbohydrate and fat ingestions induce considerable increase in GIP secretion, plasma GLP-1 levels primarily surge after glucose-rich meals (56) [Figure 1 – Section A].
3 Pharmacotherapeutic potential of GIP and GLP-1
3.1 Therapeutic potential in T2DM
Despite being the first identified incretin, GIP was initially disregarded as a viable therapeutic approach for T2DM. Earlier studies demonstrated that the incretin effect of GIP was severely impaired in patients with uncontrolled T2DM (57); even the pharmacological concentrations of GIP (> 1,000 pM) only marginally stimulate insulin secretion in these patients during hyperglycemic clamp experiments (58). When exposed to a hyperglycemic milieu, the GIPR was found to downregulate in a study of pancreatic islet cells (59) and the patients with T2DM might express a small amount of GIPR or defective GIPR (60, 61). Interestingly, research on clonal pancreatic β-cells revealed that fatty acid load stimulated GIPR expression via the activation of PPAR-α transcriptional factor under normoglycemia (99 mg/dL), but not during hyperglycemia (450 mg/dL) (59). These findings may explain the reduced responsiveness to GIP observed in individuals with T2DM who may have normal or even increased secretion of GIP.
In contrast, while the dose-response relationship between β-cell responsiveness to glucose (expressed as the slope of the linear relation between insulin secretion rate and the glucose concentration) and GLP-1 levels in circulation is partially impaired – a phenomenon referred as “GLP-1 resistance” (62) – a therapeutic infusion of GLP-1 at a pharmacological dose can restore β-cell insulin secretion responsiveness to glucose levels comparable to those observed in nondiabetic individuals (62). A seminal study by Zander et al. in patients with T2DM (38) found that continuous GLP-1 administration via a subcutaneous pump to raise serum GLP-1 levels from a mean of 9.7 pM at baseline to 282 pM over 6 weeks significantly reduced plasma glucose concentrations, improved glycated hemoglobin, and enhanced insulin sensitivity and β-cell function compared to placebo, with no significant limiting side effects reported. These findings provide compelling evidence for the therapeutic promise of GLP-1 in T2DM.
Furthermore, a preclinical study conducted by Finan et al. (44) reported that both GIPR agonist (GIPRA) and dual GIP/GLP-1 receptor agonist similarly improved glucose tolerance in GLP-1R knockout mice, but not in dual incretin receptor knockout mice. In addition, the insulinotropic effect of the dual GIP/GLP-1 receptor agonist was abrogated by GIP receptor antagonist (GIPRA) in GLP-1R null mice (63). These findings underscore the specific GIP action on its receptor in glucose metabolism. GLP-1 action may potentially sensitize GIP signaling, which is often compromised in individuals with uncontrolled T2DM. By facilitating GIP function at its full metabolic capacity alongside GLP-1, this interaction could effectively harness the full potential of incretin effect to enhance insulinotropic physiology and maintain euglycemia.
3.2 Therapeutic potential in obesity
Although the incretin effects of GIPR undoubtedly play a crucial role in preventing postprandial glucose excursion, there is still intensive debate on whether the GIPR should be activated or inhibited for the treatment of obesity. Genetic evidence, such as GWAS, has identified the single-nucleotide polymorphism (SNP) in GIPR, showing that lower function of GIPR is associated with lower BMI (64–66), allowing speculation that higher GIPR activity is obesity promoting. In a genetic preclinical study, embryonic GIPR knockout mice fed a high-fat diet were protected from obesity, supporting the role of GIPR antagonism as a method to promote weight loss and prevent weight gain (67). Based on these mouse and human genetic associations, GIPR antagonism has been explored as a therapy to treat obesity (68).
On the other hand, chronically elevating GIP levels in a transgenic mouse model exhibited reduced diet-induced obesity (69). Moreover, a study by Morz et al. (70) demonstrated that treatment with GIPRA led to dose-dependent reductions in body weight in both wild-type and GLP-1R knockout mice, and this effect was negated by co-administration of a GIPR antagonist. Additionally, the weight reduction effect of GIPRA was absent in mice deficient for the GIPR. These findings challenge the prior notion that GIP might be obesogenic and instead suggest that pharmacological activation of GIPR could offer therapeutic benefits for weight reduction. Consequently, these observations have sparked renewed discussions about the therapeutic potential of GIPRAs in the long-term treatment of obesity.
To reconcile the findings that both GIPR antagonists and GIPRAs reduce body weight, it is possible that physiological GIPR activity plays a permissive role in adipose tissue synthesis, while pharmacologically enhanced GIPR activity may reduce body weight through a different mechanism in the brain. The seemingly conflicting effects of GIP on body weight will be discussed further in later sections.
In contrast to the contrasting views on the impact of GIP on the weight, earlier studies unequivocally demonstrated the weight-reducing effects of GLP-1 and GLP-1RAs in both animals and humans. In rats, feeding activity and food intake decreased following intracerebroventricular injection of GLP-1, an effect counteracted by co-administration of a GLP-1 receptor antagonist (71, 72), suggesting central GLP-1 as a potent inhibitor of feeding. Additionally, intraperitoneal administration of GLP-1 at supraphysiological doses elicited an anti-adipogenic effect in rats (72), making GLP-1 a potential target for obesity treatment. A study involving patients with T2DM by Zander et al. (38) revealed that continuous GLP-1 infusion led to significant weight loss over a 6-week period, accompanied by increased sensation of satiety and fullness, along with reduced prospective food intake.
3.3 Development of unimolecular agents targeting both GIPR and GLP-1R
The anti-obesity effects of GIPR antagonism antibody, both alone and in combination with GLP-1RAs, have been demonstrated in preclinical models of mice and monkeys (68, 73). Maridebart cafraglutide (previously known as AMG133) is a bispecific molecule engineered by conjugating a fully human monoclonal anti-human GIPR antagonist antibody to two GLP-1 analogue agonist peptides using amino acid linkers. Its half maximal inhibitory concentration (IC50) for GIPR is 42.4 nM, and its EC50 for GLP-1R is 24.4 pM in a human cell-based functional assays (74). In a phase 1 clinical trial, maridebart cafraglutide effectively reduced body weight in participants with obesity (74). The phase 2 study to evaluate the efficacy, safety and tolerability of maridebart cafraglutide is currently ongoing (ClinicalTrials.gov number, NCT05669599).
On the other hand, preclinical studies in rodents have shown that co-administration of a long-acting GIPRA synergistically enhances the glucose-lowering and weight-reducing effects of GLP-1RA (44, 75). Unimolecular peptides with dual GIP/GLP-1 receptor agonism have been developed to improve metabolic efficacy and therapeutic index beyond what incretin mono-agonists can achieve (44, 63). The clinical studies have demonstrated the synergistic effects of GIP and GLP-1 agonism on glucose metabolism and body weight in these unimolecular dual agonists (63, 76), marking a significant achievement in the pharmaceutical development of incretins, which will be further discussed in the next section. Additionally, GLP-1RAs or dual GIP/GLP-1 receptor agonists have been combined with other nutrient-based hormone (e.g., glucagon and amylin) receptor agonists (77–82), a detailed discussion of which is beyond the scope of this review.
4 GLP-1 and dual GIP/GLP-1 receptor agonists
The efficacy of native GIP and GLP-1 in treating T2DM has been hampered by their very short half-lives. The current FDA-approved GLP-1RAs are developed as analogues of either exedin-4 or GLP-1. Exenatide and lixisenatide are exendin 4-based agents, while albiglutide, dulaglutide, liraglutide, and semaglutide are GLP-1-based agents. Tirzepatide, the only FDA-approved dual GIP/GLP-1 receptor agonist, is a peptide with potent and imbalanced co-agonism at both GIPR and GLP-1R. The pharmacokinetic differences between short-acting and long-acting analogues have profound implications for the mode of action, efficacy, and tolerability of these compounds. The development of these agents underscores the continuous improvement of the pharmacokinetic and pharmacodynamic profiles of these peptide agents [Table 1], expanding their applications beyond T2DM and obesity to encompass cardiovascular disease event reduction [Table 2].
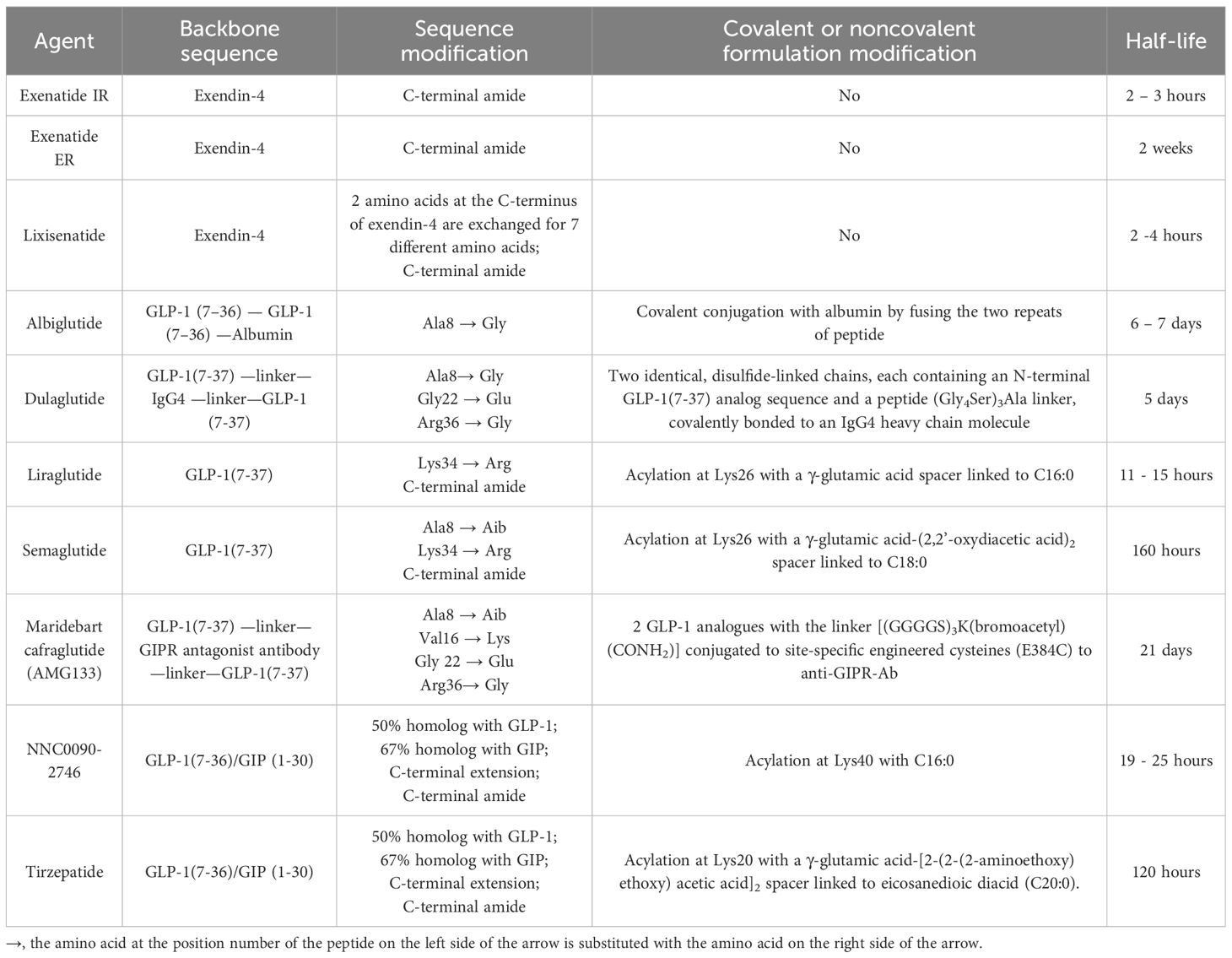
Table 1 The Peptide Sequences, Formulation Modifications, and Pharmacokinetics of GLP-1 and Dual GIP/GLP-1 Receptor Agonists.
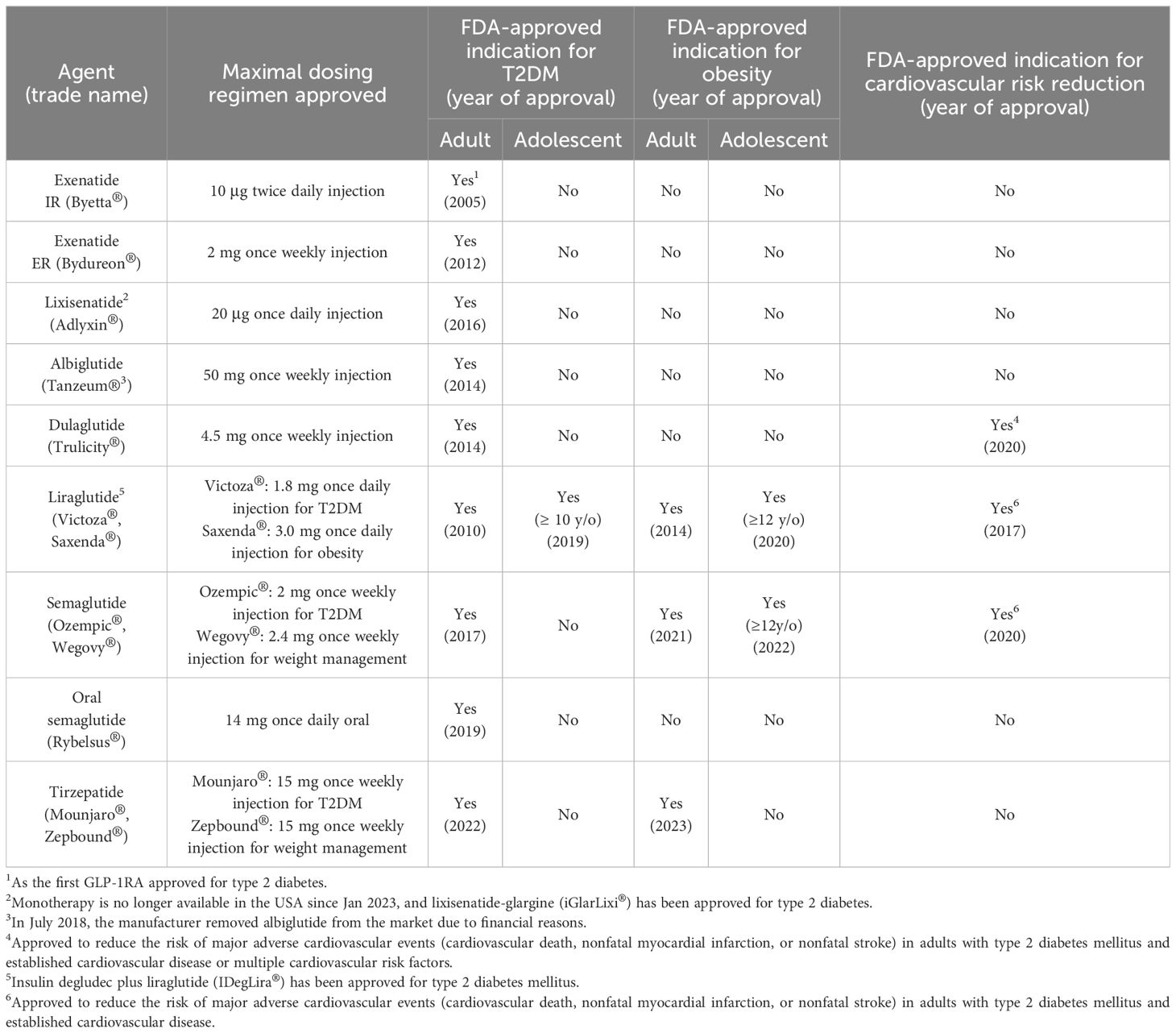
Table 2 FDA-Approved Dosages and Indications for GLP-1 and Dual GIP/GLP-1 Receptor Agonists.
4.1 Exendin 4-based agents
Exendin-4, a 39-amino acid peptide with 53% sequence homology to human GLP-1 [Figure 2], was originally isolated from Heloderma suspectum lizard venom by using an amino acid sequencing assay targeting peptides with an amino-terminal histidine residue (His1) (83). Although 16 of the 30 amino acids in its N-terminus are identical to human GLP-1 (7–36) amide, exendin-4 is not the lizard homolog of GLP-1. In comparison, lizard GLP-1 itself shares an 83% sequence identity to human GLP-1 (84) [Figure 2]. The shared biological properties of exendin-4 and human GLP-1 probably stem from their primary and secondary structures, but exendin-4 is naturally resistant to degradation by the DPP-4 enzyme, attributed to the presence of glycine at position 2, resulting in an intravenous half-life of approximately 30 minutes (85). In cellular assays, exendin-4 exhibits similar potency as human GLP-1 in binding to and activating GLP-1R (86). However, in murine models, exendin-4 demonstrates about 5,500-fold greater potency in improving glucose control, evidenced by the percentage fall in plasma glucose at 1 hour (87). The serendipitous discovery of exendin-4 led to the development of exenatide immediate-release (exenatide IR, Byetta®), a synthetic version of exendin-4 with C-terminal amidation to enhance its stability [Figure 2, Table 1], which became the first FDA-approved GLP-1RA in 2005 for the treatment of T2DM in adults [Table 2].
Lixisenatide is a 44 amino acid synthetic peptide derivative of exendin-4 [Figure 2, Table 1]. Binding studies in CHO-K1 cells expressing GLP-1R showed that lixisenatide is a potent and selective GLP-1RA, with a binding affinity to GLP-1R approximately four times greater than that of GLP-1 (88). Despite its relatively short half-life (2 – 4 hours), lixisenatide is recommended for once-daily dosing in the treatment of T2DM [Table 2]. However, as of 2023, lixisenatide is no longer available as a standalone agent in the USA; it is now only available in the combination formulation of insulin glargine plus lixisenatide (iGlarLixi ®).
A long-acting release form of exenatide (exenatide ER, Bydureon®) was approved by the FDA in 2012, becoming the first once-weekly injection of a GLP-1RA for adults with T2DM [Table 2]. The formulation of exenatide ER comprises the encapsulation of exenatide IR peptides within 0.06 mm-diameter injectable microspheres made of poly-(D,L lactide-co-glycolide) – a biodegradable medical polymer enabling controlled, gradual drug release over an extended period (89). As the polymer hydrolyzes, it steadily releases the encapsulated exenatide peptides over a maximal duration of 7 weeks. The breakdown products – lactic acid and glycolic acid – are subsequently eliminated from the body as carbon dioxide and water.
4.2 GLP-1-based agents
The focus of GLP-1RA development has predominantly been on designing GLP-1 derivatives resistant to DPP-4 enzymatic activity and exhibiting slow renal clearance through chemical modification and pharmaceutical formulation. These approaches are aimed at extending their pharmacological half-lives and attaining supraphysiological stimulation of GLP-1R.
Both albiglutide and dulaglutide are GLP-1 analogues with an alanine-to-glycine substitution at position 8, rendering the peptides resistant to DPP-4 enzymatic activity [Figure 2 and Table 1]. Albiglutide is generated from a genetic fusion of two modified recombinant GLP-1 (7–36) molecules linked in tandem to recombinant human albumin (90), while dulaglutide is created by conjugating two GLP-1 (7–37) analogues to the Fc fragment of a modified monoclonal antibody (IgG4) via peptide ((Gly4Ser)3Ala) linker (91). The large molecular entities of both albiglutide and dulaglutide delay their renal clearance (92, 93). Albiglutide was less effective in activating the GLP-1R compared to exendin-4 in an in vitro study of a BHK cell line expressing the rat GLP-1R (92). In another study, dulaglutide demonstrated full receptor activity in vitro and elicited insulinotropic effects in islets similar to GLP-1 (94). Both albiglutide and dulaglutide received FDA approval for the treatment of T2DM in 2014, with dulaglutide having an additional indication since 2020 to reduce the risk of major adverse cardiovascular events (MACE) in adults with T2DM and established cardiovascular disease or multiple cardiovascular risk factors [Table 2].
Once daily injectable liraglutide (marketed as Victoza® for T2DM and Saxenda® for obesity) and once weekly injectable semaglutide (marketed as Ozempic® for T2DM and Wegovy® for obesity) are two additional GLP-1 analogues, sharing 97% and 94% amino acid homology to GLP-1, respectively [Figure 2, Table 1]. Both employ fatty acid acylation of lysine 26 to facilitate serum albumin noncovalent binding to prolong their plasma half-lives (95). Semaglutide features a substitution of alanine residue with a non-coded amino acid, 2-aminoisobutyric acid (Aib) at position 8, shielding against DPP-4-mediated N-terminal proteolysis while preserving GLP-1R affinity (96). The crystal structures of un-acylated liraglutide and semaglutide are almost identical to GLP-1 (7–37). Pharmacokinetic data from a clinical trial involving adolescent participants treated with 3.0 mg liraglutide daily showed a median liraglutide concentration of 29.4 nM at week 8, declining to 17.7 nM at week 56 (97). In a population pharmacokinetic analysis of four clinical trials, the mean semaglutide plasma concentration was estimated at 15.3 nM with 0.5 mg/week of semaglutide and 30.6 nM with 1.0 mg/week of semaglutide in the blood samples collected from week 4 to week 56 of treatment (98). Liraglutide is equipotent to GLP-1 in activating GLP-1R, with an EC50 of 61 pM for liraglutide vs. 55 pM for GLP-1 in one cellular assay (99). In another study, semaglutide exhibited greater potency in activating GLP-1R compared to GLP-1, with an EC50 of 6.2 pM for semaglutide vs 16.2 pM for GLP-1 (95) (The variability in EC50 values among different studies for GLP-1 was not uncommon attributable to varying assay conditions). Both liraglutide and semaglutide have received FDA approval for the treatment of T2DM, obesity, and cardiovascular risk reduction [Table 2]. In addition, liraglutide combined with insulin degludec (IDegLira®) is a once-daily, fixed dual combination product approved for T2DM.
Peptide degradation in the gastrointestinal tract poses a significant challenge in the development of oral formulations of incretin analogues. An oral formulation of semaglutide (Rybelsus®) is the first oral GLP-1RA approved for treating T2DM [Table 2]. In this oral formula, semaglutide is non-covalently linked to sodium N-[8-(2-hydroxybenzoyl) aminocaprylate] (SNAC), which shields the peptide from enzymatic and acidic degradation in the stomach (100). The absorption is compound-specific and transcellular, facilitated by SNAC (100). Despite its oral bioavailability being less than 1%, this approach is therapeutically feasible because of the strong potency of semaglutide for GLP-1R activation (101). In a 10-week clinical trial, a daily dose of 20 mg oral semaglutide achieved a steady-state plasma concentration of 30 nM, and when daily dose of oral semaglutide was increased to 40 mg, the semaglutide plasma concentration reached about 60 nM (102), which was over a thousand times higher than the EC50 of semaglutide for GLP-1R activation (95). After absorption, the pharmacokinetic properties and effects of semaglutide are similar, irrespective of the route of administration (103). The half-life of oral semaglutide is approximately 1 week, aligning with subcutaneously administered form (102). The highest approved dose of oral semaglutide (14 mg/day) is currently indicated only for treating T2DM as an adjunct to diet and exercise. Higher doses of oral semaglutide (up to 50 mg/day) are under development for the treatment of both T2DM (104) and obesity (105), with effectiveness on par with 2.4 mg/week subcutaneous semaglutide approved for weight management.
4.3 Dual GIP/GLP-1 receptor agonists
The unimolecular dual incretin receptor agonist offers a more physiological approach to managing diseases associated with T2DM compared to GLP-1RAs alone. To date, clinical data have been reported for two dual GIP/GLP-1 receptor agonists. The first dual GIP/GLP-1 receptor agonist, NNC0090-2746 (also known as RG7697, RO6811135, or MAR709), is a 40-amino acid peptide acylated with a C16:0 fatty acid to lysine residue at position 40 [Figure 2, Table 1]. The modified peptide contains 2 Aib substitutions at positions 2 and 20 with a molecular weight of 4.5 kDa. It possesses in vitro balanced GIPR (EC50 = 3 pM) and GLP-1R (EC50 = 5 pM) agonism with relative activity seven times that of GIP to GIPR and five times that of GLP-1 to GLP-1R, respectively (44). Pharmacokinetics of NNC0090-2746 showed a Cmax at 5.4 nM with a single dose of 1.8 mg (106) and Cmax at 12 nM at the steady state with a daily dose of 2 mg in patients with T2DM (107).
The second dual GIP/GLP-1 receptor agonist, tirzepatide (previously known as LY3298176; marketed as Mounjaro® for T2DM and Zepbound® for obesity), is a 39-amino acid peptide acylated with a C20:0 fatty diacid moiety to lysine residue at position 20 with a molecular weight of 4.8 kDa [Figure 2, Table 1]. Tirzepatide is engineered as an imbalanced agonist in terms of its strong affinity and potency at the GIPR versus the GLP-1R. Preclinical data indicated that tirzepatide exhibited an affinity for GIPR equivalent to GIP binding, while its affinity for GLP-1R was approximately five times weaker compared to GLP-1 (63). In signaling studies using cell lines expressing GIPR or GLP-1R, tirzepatide demonstrated similar potency to GIP in activating GIPR (EC50 = 22.4 pM for tirzepatide vs. 33.4 pM for GIP) but approximately 13-fold weaker potency than GLP-1 in activating GLP-1R (EC50 = 934 pM for tirzepatide vs.70.5 pM for GLP-1). Pharmacokinetics of tirzepatide demonstrated dose proportionality over the wide dose range, with Cmax of 180 nM in individuals treated with one dose of 8 mg and a Cmax of 260 nM with 15 mg/week dose in the clinical study, exceeding the EC50 values for GIPR and GLP-1R activations by approximately 10,000-fold and 250-fold, respectively (63).
The acylation of lysine residues in NNC0090-2746 and tirzepatide enables noncovalent albumin binding and prolongs their renal clearance [Figure 2]. However, tirzepatide has significantly longer half-life than NNC0090-2746 in humans (120 hours vs. 19 – 25 hours) [Table 1]. NNC0090-2746 and tirzepatide also display unique agonism properties at their target receptors (108). In the cellular studies, NNC0090-2746 and tirzepatide showed comparable efficacy and potency at multiple signaling pathways connected to the GIPR. Tirzepatide, compared to both GLP-1 and NNC0090-2746, shows biased signaling at GLP-1R, favoring cAMP response over β-arrestin recruitment, which leads to low efficacy for GLP-1R internalization (108–110). It remains unclear whether the imbalanced pharmacology or the longer half-life of tirzepatide contributes to its superior efficacy in glucose and weight reduction compared to the more balanced dual agonist, NNC0090-2746.
Both NNC0090-2746 (107) and tirzepatide (63) delivered clinically meaningful improvements in glycemic control and body weight reduction in their phase 1 trials. In the phase 2b trial, however, the reductions in glycated hemoglobin and body weight in participants treated with NNC0090-2746 were similar to those in the group treated with liraglutide (76), and no phase 3 trial of NNC0090-2746 has been reported yet. Conversely, in another phase 2b study involving patients with T2DM, tirzepatide showed significantly better efficacy in glucose control and weight loss compared to both placebo and dulaglutide, with an acceptable safety and tolerability profile (111). The subsequent phase 3 trial of tirzepatide demonstrated more effective reductions in the glycated hemoglobin levels and body weight, along with greater overall improvements in the lipid profile compared to semaglutide in patients with T2DM, elevating incretin therapeutic agents to a new level (112). Tirzepatide became the first dual GLP/GLP-1 receptor agonist to gain FDA approval for treating T2DM in 2022 and obesity in 2023 [Table 2].
5 Pharmacological effects of GIP, GLP-1 and their mimetics
The pharmacological benefits of GIP, GLP-1, and their synthetic mimetics arise from their pleiotropic effects, encompassing at least five key functions: bolstering insulin secretion and survival of pancreatic β-cells and modulating glucagon release by the pancreatic α-cells in response to glycemic levels, acting on the satiety center in brain, slowing gastric emptying, regulating lipid and glucose metabolism through effects on adipose tissue and liver, and reducing systemic blood pressure.
5.1 GIP and GLP-1 effects on pancreas
5.1.1 Effects on pancreatic β-cell insulin secretion and survival
Activation of either GIPR or GLP-1R on pancreatic β-cells initiates distinct yet overlapping downstream signaling cascades that ultimately amplify glucose-stimulated insulin secretion. Previous physiological studies have shown that both GIP (113–116) and GLP-1 (117) act in concert with glucose to enhance insulin gene transcription, mRNA stability, insulin biosynthesis, and insulin secretion. However, a significant decrease in insulinotropic activity was observed with GIP in diabetic patient with hyperglycemia, whereas relatively preserved activity was noted with GLP-1 in the same patients (58), suggesting divergent effects of GIP and GLP-1 on β-cells (Figure 1 – Section B).
GIPRs are abundantly expressed on β-cells, and their activation induces a rise in cAMP and intracellular calcium levels, facilitating glucose-dependent insulin release from pancreatic β-cells (118). In a study involving 10 healthy male subjects, GIP infusion more than doubled the insulin secretion rate compared to the placebo group when the glucose was maintained at 216 mg/dL (35). Conversely, in another study with 10 healthy adults where glucose levels were maintained between 70 – 80 mg/dL, fat ingestion elicited a fivefold to sixfold rise in GIP levels without concomitant insulin secretion (119). These results suggest that mild to moderate hyperglycemia is required for the insulinotropic effect of GIP in nondiabetic subjects, acting as a mechanism to prevent reactive hypoglycemia following the consumption of fatty foods.
Similar to GIP, the stimulatory effects of GLP-1 on insulin expression and secretion were observed in the presence of high (450 mg/dL) but not normal (99 mg/dL) concentrations of glucose in the cultured cells, indicating the glucose-dependent insulinotropic effect of GLP-1 (117). The loss of GLP-1 effect on stimulating insulin secretion at basal plasma glucose concentrations limits the hypoglycemic risk even at high pharmacological concentrations of GLP-1 (28, 58). Marked insulinotropic effects have been observed in T2DM patients with a single injection of liraglutide (120) or acute infusion of exenatide (121). However, the long-term treatment with GLP-1RAs may reduce the insulin levels in both T2DM (122) and prediabetic patients (123), owing to the glycemic normalization and increased insulin sensitivity in these patients.
Pancreatic β-cell mass is significantly reduced by more than 50% in patients with T2DM, with lesser reductions already occurring in the prediabetes state (124). GIP and GLP-1 may exert long-term effects on β-cell survival and proliferation, potentially delaying the development of and even reversing T2DM. Studies conducted on various cellular and animal models have demonstrated that both GIP and GLP-1 exert marked anti-apoptotic effects in pancreas (125–127). GIP treatment has been shown to reduce glucolipotoxicity-induced cell death in C57 BL/6 and Bax-/- pancreatic islets, but not GIPR-/- pancreatic islets of the mouse models (114). A study by Wang et al. (128) in Wistar rats demonstrated that continuous subcutaneous infusion of GLP-1 reversed age-dependent decline in β-cell functions. In a study comparing the effects of GIPR and GLP-1R activation on β-cell survival in response to streptozotocin challenge in mice, Maida et al. (126) observed that GLP-1R signaling exerted more robust control of β-cell survival, relative to GIPR activation. Furthermore, exendin-4 has been found to stimulate β-cell mass expansion and pancreas regeneration in a partial pancreatectomy rat model of T2DM (129). However, the potential for delaying or preventing the progression of T2DM in patients treated with GIP and GLP-1 remains uncertain, as there is currently no definitive evidence from human studies demonstrating that enhanced incretin activity directly affects β-cell mass or alters the progressive loss of insulin secretory capacity in humans.
5.1.2 Effects on pancreatic α-cell glucagon release
In addition to their insulinotropic effects on β-cells, both GIP and GLP-1 play a role in regulating glucagon secretion of pancreatic α-cells, with GIP increasing and GLP-1 suppressing glucagon secretion, both in a glucose-dependent manner. GIPRs have been localized to human pancreatic α-cells, and intravenous GIP infusion activates GIPR and intracellular signaling in pancreatic α-cells, augmenting glucagonotropic effects in hypoglycemic conditions in humans (130). During hyperglycemia, GIP potentiates glucose-induced insulin secretion without exhibiting a glucagonotropic effect (35). Based on collective human data, a glycemic threshold of 99 – 108 mg/dL may exist, below which GIP primarily exerts glucagonotropic actions (35). Through its dual insulinotropic and glucagonotropic effects on stabilizing glucose levels, GIP could provide a buffer against hyperglycemia when large amounts of glucose are consumed and mitigate reactive hypoglycemia, especially after meals with high fat content. The safeguard against reactive hypoglycemia is evolutionally crucial, as central nervous tissue relies on stable blood glucose levels (Figure 1 – Section B).
On the contrary, GLP-1 suppresses glucagon secretion in both healthy individuals and those with T2DM under normoglycemia and hyperglycemia (58) but not during hypoglycemia (131), thereby reducing the potential for developing severe hyperglycemia or hypoglycemia. GLP-1’s inhibition of glucagon secretion appears to be indirectly triggered via paracrine effects in the islets. Evidence from experiments employing somatostatin receptor 2 (SSRT2) antagonists and SSRT2 knockout mice strongly suggests that GLP-1’s inhibitory actions on α-cells are indirect and partially mediated through somatostatin-dependent mechanisms (132). Somatostatin, secreted from pancreatic γ-cells, acts as an inhibitory paracrine to suppress glucagon release from pancreatic α-cells, and specific blockade of SSRT2 eliminates the inhibitory effect of GLP-1 on glucagon secretion. Insulin has also been suggested to play a significant role as a paracrine regulator of glucagon secretion (the intra-islet hypothesis), although the mechanism by which this inhibition occurs is not well elucidated (133). It has been demonstrated that the insulinotropic effect on β-cells and glucagonostatic effect on α-cells each contribute to about half of GLP-1’s blood glucose-lowering activity (134).
In an in vitro study, tirzepatide elicited a cAMP response in a human pancreatic β-cell line that was significantly higher than that observed for either GLP-1 or GIP alone, indicating a synergistic or additive activation of signaling in pancreatic β-cells by GIP and GLP-1 (63). A mechanism of action phase 1 study showed that the glycemic efficacy of tirzepatide in T2DM results from concurrent improvements in key components of diabetes pathophysiology, including β-cell function, insulin sensitivity, and glucagon secretion (135). These effects were substantial and help elucidate the remarkable glucose homeostasis abilities of tirzepatide observed in SURPASS-2 trial when compared with semaglutide (112).
5.2 GIP and GLP-1 effects on the central nervous system
The drive to forage and the regulation of satiety regarding nutrient intake are modulated by orexigenic and anorexigenic signaling pathways, respectively. These pathways are mainly located in arcuate nucleus of the hypothalamus (ARH). Neurons co-expressing orexigenic neuropeptide Y (NYP) and agouti-related peptide (AgRP) in the ARH activate melanocortin hormone- and orexin-expressing neurons in the lateral thalamus, leading to increase food intake. Conversely, satiety is primarily controlled by anorexigenic pro-opiomelanocortin (POMC) and cocaine- and amphetamine-regulated transcript (CART) neurons in the ARH that project to paraventricular nucleus neurons, resulting in the release α-melanocyte-stimulating hormone and a decrease of food intake. Modern foods, with their high caloric intensity, pose a challenge. The amount of oral intake needed to achieve gastric and intestinal fullness and signal satiety to the brain through the gut-brain axis can overwhelm the evolutionarily determined metabolic capacity of peripheral organs such as liver, pancreas, fat tissue, and muscles. This imbalance may lead to disruptions in glucose and lipid homeostasis, contributing to a range of metabolic diseases, including obesity. Incretins and their synthetic mimetics offer benefits by influencing the appetite and satiety centers in the brain, potentially resetting the body’s fat mass and body weight setpoints that are associated with the interplay between orexigenic and anorexigenic signaling pathways.
A study using in situ hybridization and polymerase chain reaction techniques did not detect GIP expression in the brain of adult male Sprague-Dawley rats (41). However, GIP immunoreactivity was identified in the adult rat brain using a specific monoclonal antibody against GIP (136), suggesting that while GIP is not expressed in the brain, peripheral GIP can reach the deep brain. GIPR was found in both neuronal and nonneuronal cells within the satiety and feeding centers of the hypothalamus in mice (137). Chronic central (intracerebroventricular) or peripheral (subcutaneous) infusion of GIP analogue in mice increased cFos neuronal activity in hypothalamic feeding center, resulting in reduced food intake and body weight. These effects were blunted in CNS-specific GIPR knockout mice, suggesting a key role of CNS GIPR in the control of energy metabolism (138) (Figure 1 – Section C).
Unlike GIP, the fully processed GLP-1 is produced by the neurons within the nucleus of the solitary tract (NST) in the brainstem of rats (139). The expression of pre-proglucagon gene and GLP-1 has been detected in the human brainstem and hypothalamus (140). GLP-1 is a well-characterized neurotransmitter involved in signaling satiety in the brain (71). Neurons producing GLP-1 project to targets in the hypothalamus, where GLP-1Rs are abundantly expressed. GLP-1Rs are also found in circumventricular organs (CVO), the hind brain, and the thalamus. The presence of GLP-1R in the phylogenetically oldest parts of the brain but not in the cortex emphasizes that GLP-1 is involved in the regulation of vital functions like feeding and satiety (141).
Brain GLP-1Rs, rather than peripheral GLP-1Rs, appear crucial for the weight-reducing effects of GLP-1RAs. Studies in mice with CNS neuron-specific GLP-1R knockout showed that liraglutide failed to induce weight loss but still improved glucose levels, suggesting that brain GLP-1Rs are necessary for the weight-lowering effect of GLP-1RAs but not essential for their glucose-lowering effects (142). The ability of peripherally circulating GLP-1 and GLP-1RAs to breach the blood-brain barrier remains uncertain; they may potentially do so via the relatively permeable CVO or through endocytosis and transcytosis of vascular endothelial cells to reach the deep brain (143–145). Salameh et al. (146) observed significant blood-to-brain influx for non-acylated GLP-1 mimetics like exendin-4 and lixisenatide, whereas acylated GLP-1 analogues such as liraglutide and semaglutide showed minimal blood-brain barrier penetration in mice. Interestingly, Gabery et al. (145) demonstrated that peripherally administered liraglutide and semaglutide accessed ARH directly without crossing the blood-brain barrier. However, semaglutide exerted broader effects, extending laterally and more deeply into the posterior ARH compared to liraglutide, potentially explaining its greater effectiveness in weight reduction (147). Emerging evidence suggests that GLP-1RAs may reduce food intake and body weight through diffuse brain effects rather than targeting a specific localized population of GLP-1Rs (145).
Notably, the amount of incretin mimetics entering the brain parenchyma does not correlate with their efficacy in weight reduction (146). There are other mechanisms by which circulating incretin mimetics can influence deep brain function without crossing the blood-brain barrier. Peripherally circulating GLP-1 (143) and some GLP-1RAs, such as liraglutide and semaglutide (145), can readily access the CVOs, including the subfornical organ and the area postrema, which express GLP-1R and have close neuroanatomical connections with hypothalamus involved in forage and satiety control. While both CVO and the hypothalamus are important, the hypothalamus is likely more central to the direct regulation of appetite and satiety through GLP-1 signaling. The role of CVO is crucial for sensing peripheral GLP-1 levels and relaying this information to the hypothalamus and other deep brain regions. Most pharmacokinetic studies assessing the brain uptake of incretin mimetics have been conducted in animals, and translating these findings to human subjects and clinical outcomes remains uncertain.
In the brain, the two incretins are not redundant and may complement each other in regulating orexigenic and anorexigenic pathways (148). Studies in mice suggest that GIP may potentiate the GLP-1-induced reduction of food intake and body weight by enhancing POMC expression and neuronal activation within distinct neuron populations in the hypothalamus (149). Importantly, preclinical studies have demonstrated that co-administration of GIP and GLP-1 intracerebroventricularly results in an additive enhancement in weight-lowering efficacy (138). The potential mechanisms through which GLP-1 and GIP/GLP-1 receptor agonists recalibrate and normalize the fat mass and body weight setpoint in patients with obesity may stem from their primary effects on incretin homeostasis. These effects include neurotrophic, neurogenic, neuroprotective, anti-inflammatory, and insulin sensitizing actions, a field currently under intensive investigation and not yet fully understood.
5.3 GIP and GLP-1 effects on gastric emptying
The secretion of endogenous GIP does not correlate with gastric half-emptying times (150). Neither acute nor chronic exposure to GIP or GIPRAs affects gastric emptying in healthy individuals or those with T2DM (151). Conversely, exogenous infusion of GLP-1 exhibits potent inhibitory effects of gastric emptying, indicating an enterogastrone mechanism. In a study with eight poorly controlled T2DM patients who received a liquid meal via a nasogastric tube and exogenous infusion of GLP-1 to achieve plasma levels of about 70 pM, gastric volume remained constant over 2 hours, and plasma glucose returned to normal fasting values within 3 – 4 hours (152). Counteraction of GLP-1 effect on gastric emptying by co-infusion with prokinetic drug erythromycin during a liquid test meal diminished GLP-1’s ability to decrease postprandial hyperglycemia in patients with T2DM (153). The dose-dependent effect of GLP-1 on decelerating gastric emptying plays a substantial role in improving postprandial glycemic concentrations to the extent that postprandial insulin secretion may no longer be stimulated or could even be reduced (154, 155) (Figure 1 – Section D).
Pharmacologically, only short-acting GLP-1RAs demonstrate a marked reduction in gastric emptying (156), while chronic activation of GLP-1R results in a waning effect on gastric emptying due to tachyphylaxis. This tachyphylaxis occurs with continuous infusion of GLP-1 (154, 157), long acting GLP-1RAs (156), or tirzepatide (151). Tachyphylaxis is contingent upon a full 24-hour exposure, as evidenced by a study in which rats treated with liraglutide (with half-life of 11 – 15 hours) twice daily for 14 days had markedly diminished gastric emptying while those treated with exenatide (with half-life of 2 – 3 hours) twice daily over the same period still displayed a profound reduction in gastric emptying, although both compounds had similar effects on body weight reduction (158). This phenomenon is further illustrated by results from a randomized, open-label mechanistic trial in patients with T2DM, which demonstrated that lixisenatide, with a half-life of 2 – 4 hours, had a significantly greater effect than liraglutide in delaying in gastric emptying and curtailing postprandial glucose excursions following a standardized solid breakfast (156).
GLP-1 has been shown to attenuate meal-induced gastric antral propagated contractions while increasing pyloric tone (159, 160). The experimental data from rats suggest that the inhibitory effect of GLP-1 on gastric motor function is mediated by the afferent vagal nerve, through interaction with GLP-1Rs located in vagal afferent fibers that transmit sensory signals to the brain (161). The brain regions (e.g., NST) responsible for mediating the inhibitory effect of GLP-1 on gastric emptying depend on intact vagal afferent input. In addition, the brain regions involved in mediating the effects of GLP-1 on gastric emptying and satiety differ, given that the threshold dose of GLP-1 required to inhibit gastric emptying is 3,000 to 10,000 times lower than the reported threshold doses necessary to suppress feeding behavior (161). GLP-1 may exert its effects locally via the vagus nerve before being degraded by DPP-4 in circulation. Studies in rats have identified GLP-1R expression in both the nodose ganglion (162) and vagal nerve terminals innervating the portal vein (163), and GLP-1 has been shown to increase impulse generation in both gastric and hepatic vagal afferent fibers (164, 165). Unlike GLP-1, GIP has not been found to possess a similar neural signaling mechanism for decelerating gastric emptying (54, 162).
The most common side effect of GLP-1RAs is nausea and vomiting, which are hypothesized to result from inhibition of gastric emptying. Long-acting agents seem to have a lower incidence of gastrointestinal adverse effects (166), likely due to tachyphylaxis in gastric emptying with sustained GLP-1R stimulation. Nonetheless, residual effects on gastric emptying could still produce significant gastrointestinal adverse effects in patients treated with long-acting GLP-RAs (167). The absence of GIP’s effect on gastric emptying underscores the physiological and pharmacological distinctions between GIP and GLP-1. Notably, the undesirable gastrointestinal effects associated with GLP-1RAs have not been observed with GIPRAs (168). Instead, GIP may counteract nausea and vomiting via direct modulation of the area postrema/NST emesis circuitry (169) and exert protective effects on unfavorable altered gut motility (44).
5.4 GIP and GLP-1 effects on lipid and glucose metabolism in adipose tissue and liver
Fat and liver are two organs that play key roles in lipid and glucose metabolism, working together in concert and complimentarily to maintain lipid and glucose homeostasis. Both GIP and GLP-1 exert direct or indirect effects on adipose and hepatic tissues to regulate lipid and glucose metabolism.
5.4.1 Effects on adipose tissue
GIPRs are abundant in adipose tissue (170). In a study utilizing a cultured mouse embryo preadipocyte cell line, GIP was shown to enhance the synthesis and release of lipoprotein lipase (LPL) into the medium, thereby facilitating the uptake of triglycerides into the adipocytes (171). Additionally, Ebert et al. (172) demonstrated that intravenous infusion of porcine GIP abolished plasma hypertriglyceridemia in rats during the fat load, while rats pretreated with GIP antiserum exhibited a significantly greater triglyceride increment late in the fat load time course. GIP promotes clearance of circulating triglycerides and free fatty acids and enhances adipose tissue triglyceride absorption, serving as a crucial hormonal regulator of postprandial triglyceride response in humans (173–175). Moreover, GIP has been found to boost glucose uptake by improving insulin sensitivity in adipocytes (176, 177), while tirzepatide has been demonstrated to enhance insulin sensitivity and promote glucose disposal in white adipose tissue in Glp-1r-null mice (178), further supporting a direct effect of GIP on adipose tissues. The anabolic effects of GIP enhance both the postprandial glucose and lipid buffering capacity of white adipose tissue and prevent ectopic fat deposition in visceral organs (179, 180). The anabolic effects of GIP may play a physiologically permissive role for adipose tissue synthesis, and GIP antagonist antibodies have been developed as pharmacological agents for obesity treatment (68, 74) (Figure 1 – Section E).
In contrast to GIPRs, GLP-1Rs are not expressed in adipose tissue (170). Existing data suggest that the effects of GLP-1 on adipose tissue are primarily indirect. GLP-1 may modulate lipid metabolism through increased sympathetic tone, leading to increased lipolysis, heightened triglyceride catabolism, and enhanced fatty acid oxidation (181). GLP-1Rs are densely expressed in the area postrema in rats (182). As the area postrema lacks the blood-brain barrier, it serves as a key site for peripheral GLP-1 to activate central autonomic regulatory sites. The targets of area postrema neuronal projections include the rostral ventrolateral medulla (RVLM), with the efferent projections from the RVLM innervating the sympathetic preganglionic neurons (183). These projections from the area postrema to the RVLM may constitute potential pathways activated by GLP-1 to elicit sympathetic responses. The sympathetic nerves release norepinephrine, which activates receptors on adipocytes, leading to the mobilization of stored triglycerides. Intriguingly, in patients with dumping syndrome, postprandial serum catecholamine rises correspond with increased serum GLP-1 levels (184). The increased sympathetic tone may explain a known GLP-1RA class effect of a modest but significant increase in heart rate, the clinical consequences of which are undetermined.
GIP promotes the proper storage of lipids in adipose tissue, whereas the robust fat oxidation mechanism of GLP-1 increases the utilization of these deposited lipids. The opposing effects of GIP and GLP-1 on lipogenesis and lipolysis, respectively, help maintain adipocytes in young and healthy state, fostering higher levels of adiponectin – a key adipokine known for its potential antidiabetic, anti-inflammatory, and antiatherogenic properties (185). Both GIP (186) and GLP-1 (187) enhance adiponectin expression and secretion in murine models. Elevated adiponectin concentrations have been noted in major clinical trials with GLP-1RAs (123, 188) and, more robustly, with tirzepatide (189). In a post hoc analysis of the SURPASS-1 trial involving patients with T2DM, the percentage increase in adiponectin levels from baseline ranged from 16% to 23% with tirzepatide compared to a decrease of 0.2% with placebo at week 40 (190).
5.4.2 Effects on liver
Despite the lack of expression of GIPR (41, 191) and GLP-1R (192, 193) in hepatocytes, both GIP and GLP-1 have indirect effects on hepatic lipid and glucose metabolism (194). The secretion of adiponectin from adipose tissue enhanced by both incretins elicits a signal akin to a fasting state in hepatocytes through the cAMP/pAMPK pathway. This signaling pathway upregulates enzymes such as carnitine palmitoyl transferase 1, a key enzyme in β-oxidation, while concurrently reducing the expression of lipogenesis-related genes, including SREBP-1c and FAS (195). Adiponectin also decreases hepatic de novo glucose production and reduces free fatty acid levels in the liver (196), thereby mitigating hepatic steatosis, enhancing insulin sensitivity, and improving the circulating lipid profile (Figure 1 – Section E).
Moreover, in addition to the direct action of GIP in adipose tissue to reduce serum triglyceride levels, GLP-1 may abolish the postprandial rise in triglyceride and free fatty acid concentrations by reducing intestinal absorption of dietary lipid, stemmed from the delayed gastric emptying (197, 198). Both GLP-1RAs and tirzepatide demonstrated efficacy in improving circulatory lipid profiles in the large clinical trials (199, 200). Intriguingly, a post hoc analysis of a phase 2 clinical trial revealed that dulaglutide treatment resulted in reductions in apoC-III and apoB levels without impacting serum LPL level, whereas treatment with tirzepatide showed a dose-dependent increase in serum LPL activity while concurrently reducing apoC-III and apoB levels (180).
Elevated levels of triglycerides and free fatty acids in circulation are major factors driving lipid accumulation in the liver, leading to metabolic dysfunction-associated steatohepatitis (MASH) and liver fibrosis. MASH contributes to worsening insulin resistance and is associated with an increased risk of cardiovascular disease. Clinical outcome studies have demonstrated the therapeutic potential of GLP-1RAs and tirzepatide in mitigating MASH. In small proof-of-concept phase 2 randomized controlled trials (RCTs) in patients with biopsy-confirmed MASH, liraglutide at 1.8 mg/day for 48 weeks (201), semaglutide at 0.4 mg/day for 72 weeks (202), and tirzepatide at 15 mg/week for 52 weeks (203) have been reported to achieve placebo-corrected percentages of participants meeting the criteria for resolution of MASH without worsening fibrosis by 30% (P = 0.019), 42% (P < 0.001), and 52% (P < 0.001), respectively. Additionally, the placebo-corrected percentages of participants with an improvement of at least one fibrosis stage without worsening MASH were 3% (P = 0.46), 10% (P = 0.48), and 21% (95% CI, 1 – 42), respectively. Treatment periods longer than 48 to 72 weeks may be needed to show substantial treatment effects on fibrosis with GLP-1 and dual GIP/GLP-1 receptor agonists. The effects of semaglutide on regression of fibrosis in patients with MASH are currently being investigated in the ongoing phase 3 ESSENCE trial (ClinicalTrials.gov number, NCT 04822181).
5.5 GLP-1 effects on systemic blood pressure
Intracerebroventricular GLP-1 injection stimulated the urinary excretion of water and sodium in a preclinical study of rats (72). GLP-1Rs have also been detected in kidney vasculature and tubular epithelial cells, potentially reducing the expression of fibrotic and inflammatory mediators in the kidney and decreasing sodium and fluid reabsorption in the proximal tubule (204). Liraglutide has been shown to enhance atrial natriuretic peptide secretion in mice (205), and this cardiac hormone reduces blood pressure mediated by natriuresis and vasodilatation. GLP-1RAs have been shown to increase natriuresis and diuresis in both healthy volunteers and individuals with obesity or diabetes. Exenatide has been demonstrated to diminish sodium intake in individuals with obesity without affecting salt craving (206). Gutzwiller et al. (207) reported that GLP-1 infusion evoked a dose-dependent increase in urinary sodium excretion in both healthy subjects and insulin-resistant men with obesity. Additionally, Lovshin et al. (208) reported that three weeks of liraglutide treatment significantly increased both 24-hour and nighttime urinary sodium excretion in hypertensive subjects with T2DM.
Consequently, chronic treatment with GLP-1RAs or tirzepatide improves both systolic and diastolic blood pressures, often occurring before significant weight loss is observed (209, 210). Data from a subset of 494 participants showed individuals receiving 10 mg/week of tirzepatide had lower placebo-adjusted systolic blood pressure and diastolic blood pressure by 10.6 mmHg and 2.9 mmHg, respectively, in ambulatory blood pressure monitoring at week 36 (211). Importantly, they do not reduce blood pressure in normotensive subjects, and hypotension has not been associated with GLP-1RAs and tirzepatide in clinical trials. Understanding how GLP-1R signaling coupled to blood pressure reduction is restricted or silenced in normotensive subjects is an important area for future investigation. On the other hand, the effects of GIP on blood pressure are not documented.
6 Clinical applications of GLP-1RAs and tirzepatide
Current treatment guidelines for T2DM have evolved from a narrow “glucose-centric” focus to addressing the underlying causes of disease and the associated detrimental health consequences, particularly adiposity and cardiovascular complications. Originally developed for the treatment of T2DM, GLP-1RAs and tirzepatide have shown promise in promoting weight loss and attenuating the risk of cardiovascular diseases.
6.1 Clinical applications in T2DM
In terms of glucose control effectiveness, most GLP-1 analogues outperform exendin-4 analogues [Supplementary Table]. Exendin-4-based agents, including exenatide IR (10 μg twice daily) (212, 213), exenatide ER (2 mg/week) (214), and lixisenatide (20 μg/day) (215, 216), all reduced glycated hemoglobin of less than 1% compared to placebo in the pivotal phase 3 RCTs conducted at their highest doses approved by the FDA. However, the placebo-adjusted reduction in glycated hemoglobin was 0.8% in the HARMONY 1 trial of albiglutide (30 mg/week) (217), 1.05% in the AWARD 1 trial of dulaglutide (1.5 mg/week) (218), 1.34% in the LEAD-1 SU trial of liraglutide (1.8 mg/day) (219), 1.5% in the SUSTAIN 1 trial of subcutaneous semaglutide (1.0 mg/week) (220), and 1.1% in the PIONEER 1 trial of oral semaglutide (14 mg daily) (221), respectively, when administered at doses of the upper limit approved by the FDA for the treatment of T2DM. Among GLP-1RAs, only liraglutide has been approved for use in adolescent population (age 10 or above) with T2DM [Table 2]. In the SURPASS-1 trial (222), the patients of T2DM treated with tirzepatide at 15 mg/week achieved a placebo-corrected glycated hemoglobin reduction by 2.11%, demonstrating superior efficacy compared to currently approved GLP-1RAs.
The reduction in glycated hemoglobin was primarily driven by lowering postprandial glucose with short-acting GLP-1RAs (e.g., exenatide-4 IR and lixisenatide), and a greater reduction in fasting blood glucose levels with a carryover effect on postprandial glucose following treatment with long-acting GLP-1RAs and tirzepatide (166, 223). In trials comparing the addition of injectable agents in individuals requiring further glucose lowering, glycemic efficacy of GLP-1RAs and tirzepatide was found to be similar or greater than that of basal insulin (224–226). GLP-1RAs and tirzepatide in these trials demonstrated a lower hazard of hypoglycemia, albeit with a higher incidence of gastrointestinal side effects. Consequently, these trials have led to the recommendation of GLP-1RAs and tirzepatide as the preferred agents for individuals in need of potency provided by an injectable therapy for glucose control (227).
6.2 Clinical applications in weight management
Weight loss was consistently observed in nearly all landmark clinical trials of GLP-1RAs and tirzepatide. Based on a series of comparison studies in patients with T2DM, the agents at their highest approved doses could be roughly ranked from least to most efficacy in terms of weight reduction as follows: albiglutide (50 mg/week) ≤ lixisenatide (20 μg/day) ≤ exenatide IR (10 μg twice a day) = exenatide ER (2 mg/week) ≤ dulaglutide (1.5 mg/week) < liraglutide (3.0 mg/day) < injectable semaglutide (2.4 mg/week) < tirzepatide (15 mg/week) [Supplementary Table]. The range of placebo-corrected weight loss varied from 0.83 kg (95%CI, -1.06 to -0.60) with albiglutide (50 mg/week) at 16 months in the Harmony Outcomes trial (228) to 11.6 kg with tirzepatide (15 mg/week) in the 72-week SURMOUNT-2 trial (229) in patients with T2DM. However, it’s important to approach indirect comparisons between trials with caution due to variations in study designs, durations, populations, and analysis methods. These studies ranged in duration from 24 weeks to 72 weeks, and it is unclear whether the relative efficacy of these agents would differ when used for longer durations.
Studies have indicated that albiglutide (228, 230) and dulaglutide (231) were not as effective as liraglutide and semaglutide in generating weight loss in head-to-head comparisons. These findings are consonant with the larger molecular structures of both albiglutide and dulaglutide, which limit their ability to reach the deep brain and thus have minimal effects on the brain’s satiety center. Only three injectable incretin analogues – liraglutide (dose up to 3.0 mg/day, marketed as Saxenda), injectable semaglutide (dose up to 2.4 mg/week, marketed as Wegovy), and tirzepatide (dose up to 15 mg/week, marketed as Zepbound) – are approved by the FDA as adjuncts to lifestyle modifications, including diet and exercise interventions, for weight loss. Additionally, both liraglutide and semaglutide are approved for indefinite use in weight management in adolescent aged 12 years and older (232) [Table 2].
A series of trials assessing the efficacy of GLP-1RAs and tirzepatide in reducing weight in patients, both with or without a diagnosis of T2DM, included the SCALE trials for liraglutide, the STEP trials for semaglutide, and the SURMOUNT trials for tirzepatide. Subcutaneous administration of liraglutide (3.0 mg/day) for 56 weeks, semaglutide (2.4 mg/week) for 68 weeks, or tirzepatide (15 mg/week) for 72 weeks resulted in placebo-corrected weight loss of 4.0% in the SCALE Diabetes study (122), 6.2% in the STEP 2 study (233), and 9.6% in the SURMOUNT-2 (229) study, respectively, in patients with T2DM. In contrast, they achieved more substantial weight loss of 6.0% in the SCALE Obesity and Prediabetes study (123), 12.4% in the STEP 1 study (234), and 17.8% in the SURMOUNT-1 study (200), respectively, in patients without diabetes. All three agents demonstrated reduced efficacy in individuals with T2DM, resulting in 30% to 50% less body weight reduction compared to those without diabetes. Moreover, in studies involving patients with T2DM, the magnitude of body-weight loss with GLP-1RAs diminished as baseline hyperglycemia worsened (235, 236). Based on these findings, it is hypothesized that the heightened insulin resistance might render patients more resistant to the weight-reducing effects of GLP-1RAs and tirzepatide.
Even modest reductions in body weight, ranging from 3 to 7%, can improve glycemic control and mitigate cardiometabolic risk factors, while more substantial weight loss (>10%) may lead to diabetes remission (237). Indeed, it has been suggested that agents capable of achieving an average weight reduction of about 15% in individuals with obesity represent a new generation of anti-obesity medications, as this level of weight loss is sufficient to combat or prevent a broader range of obesity-related complications (238). In the SURMOUNT-2 trial involving 938 adults with a BMI of 27 kg/m2 or higher and T2DM, up to 65%, 48%, and 31% of participants on tirzepatide 15 mg/week experienced body weight reductions of ≥ 10%, ≥ 15%, and ≥ 20%, respectively, by week 72, with 49% of those treated with tirzepatide achieving normoglycemia (glycated hemoglobin < 5.7%) compared to only 3% treated with placebo (229).
Participants treated with liraglutide (239), semaglutide (240), or tirzepatide (200) in the clinical trials had a percent reduction in fat mass approximately three times greater than the reduction in lean mass, leading to an overall improvement in body composition. The ratio of fat-mass loss to lean-mass loss was similar to that reported in lifestyle-based intervention in the Look AHEAD trial (241) and surgical treatment for obesity (242). However, loss of lean muscle mass could be potentially detrimental to patients, especially those who are older or have chronic illnesses. Ongoing research into nutritional and exercise interventions, as well as novel pharmacological agents – such as myostatin/activin pathway inhibitors (243) or agents targeting the apelin pathway (244) – holds promise for preserving lean mass and improving overall health in patients undergoing treatment with GLP-1 and dual GIP/GLP-1 receptor agonists.
In patients treated with GLP-1RAs and tirzepatide, gastrointestinal side effects such as nausea and vomiting are common but do not significantly contribute to weight loss. Although patients receiving exenatide IR and experiencing longer durations of nausea and vomiting tended to lose more weight, those in the trial group who did not experience the gastrointestinal symptoms (70%) generally lost weight as well (166, 224). A post hoc subgroup analysis assessing changes in BMI standard-deviation scores between participants with one or more gastrointestinal adverse events and those with none revealed that the effect of liraglutide in reducing BMI standard-deviation scores was independent of gastrointestinal adverse events (P = 0.82) (245). A previous mediation analysis of semaglutide in adults with overweight or obesity suggested that less than one percentage point of the body weight reduction achieved with subcutaneous semaglutide was attributable to gastrointestinal adverse events (246). The analysis of the SURPASS trials of tirzepatide revealed no association between weight reduction and gastrointestinal adverse events (229).
6.3 Clinical applications in cardiovascular disease prevention
Recent American College of Cardiology/American Heart Association guidelines advocate for the use of GLP-1RAs in patients with T2DM at high risk of atherosclerotic cardiovascular disease (ASCVD), irrespective of well-controlled glucose levels, based on the clinical trials mandated by FDA-issued industry guidelines in December 2008 to assess the cardiovascular effects of new diabetic therapies (247). As of 2022, GLP-1RAs have been recommended as first-line treatments, along with SGLT-2 inhibitors, for patients with T2DM and cardiovascular disease in American Diabetes Association’s Standards of Medical Care in Diabetes (248, 249). However, not all tested GLP-1RAs have consistently demonstrated reductions in cardiovascular events, and the impact on specific cardiovascular outcomes varied among the effective drugs.
Eight major RCTs of GLP-1RAs have investigated the cardiovascular outcomes, including the ELIXA trial of lixisenatide (250), the EXSCEL trial of exenatide ER (251), the LEARDER trial of liraglutide (252), the SUSTAIN-6 trial of subcutaneous semaglutide (253), the HARMONY trial of albiglutide (228), the REWIND trial of dulaglutide (254), the PIONEER trial of oral semaglutide (255), and the SELECT trial of subcutaneous semaglutide (199). They were all large-scale trials involving 3,000 to 18,000 patients who were either at high risk of or having established ASCVD, with trial durations ranging from 15 to 60 months. The first seven trials involved patients with T2DM and baseline glycated hemoglobin levels ranging from 7.3 to 8.7, whereas the most recent SELECT trial focused on nondiabetic patients with overweight or obesity and preexisting cardiovascular disease. All these trials were designed to evaluate the primary outcome of 3-MACE defined as cardiovascular death, nonfatal myocardial infarct, and nonfatal stroke, except for the ELIXA trial of exenatide ER, which included hospitalization for unstable angina as an additional component of the 4-MACE composite outcomes.
All eight reported studies have demonstrated the noninferiority of GLP-1RAs compared to placebo regarding MACE. Additionally, the superiority of GLP-1RAs compared to placebo in the primary endpoint of 3-MACE were found with liraglutide in the LEADER trial (HR 0.87; 95% CI 0.78 – 0.97; P = 0.01), injectable semaglutide in the SUSTAIN trial (HR 0.74; 95%CI 0.58 – 0.95; P = 0.02) and in SELECT trial (HR 0.80; 95% CI 0.72 – 0.90; P < 0.001), albiglutide in the HARMONY trial (HR 0.78; 95% CI 0.68 – 0.90; P = 0.0006), and dulaglutide in REWIND trial (HR 0.88; 95% CI 0.79 – 0.99; P = 0.026). The REWIND trial suggested that dulaglutide might be effective for both primary and secondary cardiovascular prevention in individuals with T2DM (254). Notably, significant reductions of death from any causes were shown in both the LEADER trial with liraglutide (HR 0.85; 95% CI 0.74 – 0.97; P = 0.02) (252) and the SELECT trial with semaglutide (HR 0.81; 95% CI 0.71 – 0.93) (199). A significant benefit in reducing death from cardiovascular causes was only shown in the LEADER trial with liraglutide (HR 0.78; 95% CI 0.66 – 0.93; P = 0.007). The recent FLOW trial with semaglutide in patients with chronic kidney disease and T2DM, with a composite of major kidney disease events as the primary outcome, showed significant reductions in both cardiovascular mortality (HR 0.71; 95% CI 0.56 – 0.89) and all-cause mortality (HR 0.80; 95% CI 0.67 – 0.95) in the confirmatory secondary outcomes, in addition to the reduced risk of the primary outcome events (HR 0.76; 95% CI 0.66 – 0.88; P = 0.0003) (256). Following the withdrawal of albiglutide from the market in 2018 by the manufacturer for financial reasons (257), liraglutide, injectable semaglutide, and dulaglutide should currently be the preferred choices within this class for reducing cardiovascular events until further data are available (248).
In the SUSTAIN-6 trial, injectable semaglutide demonstrated a notable effect in reducing the incidence of nonfatal stroke (hazard ratio [HR] 0.61; 95% CI 0.38 – 0.99; P = 0.04), a main contributor to the reduction of 3-MACE composite endpoint (253). Similarly, the REWIND trial showed a stronger impact of dulaglutide on nonfatal stroke (HR 0.76; 95% CI 0.61 – 0.98; P = 0.017) compared to nonfatal myocardial infarction (HR 0.96; 95% CI 0.79 – 1.16; P = 0.65) and cardiovascular death (0.91; 95% CI 0.78 – 1.067; P = 0.21) (254). On the other hand, the reports from the LEADER trial of liraglutide showed a less significant hazard ratio for nonfatal stroke (HR 0.89; 95% CI, 0.72 – 1.11; P = 0.30) (252), and the HARMONY Outcomes trial of albiglutide indicated a similar trend in reducing total stroke (HR 0.86; 95% CI 0.66 – 1.14; P = 0.30) (228). Whether the superior effects of semaglutide and dulaglutide on stroke reduction compared to other GLP-1RAs arise from inherent differences in drug properties or disparities in trial design remains uncertain. Confirmatory large-scale trials are essential to ascertain the potential efficacy of GLP-1RAs in specifically improving the stroke outcomes.
In addition to GLP-1RAs, pioglitazone and SGLT-2 inhibitors are two other classes of diabetic medications that have shown promise in reducing the risk of cardiovascular disease, albeit through different mechanisms. Pioglitazone represents another potentially important therapy for preventing stroke events (258). Unlike treatment with GLP-1RAs, which generally leads to body weight and fat mass reduction, pioglitazone dose-dependently increases body weight and fat mass. Paradoxically, this increase has been associated with improved all-cause mortality as well as cardiovascular mortality, according to a post hoc analysis of the PROactive trial (259). However, the benefit in reducing stroke events was not seen in the outcome studies of SGLT-2 inhibitors (260). Unlike GLP-1RAs, particularly dulaglutide (254), SGLT-2 inhibitors have not demonstrated cardiovascular benefits in primary prevention among individuals with multiple cardiovascular risk factors (261).
The SUPRPASS-4 trial (262), involving 2002 participants with high-risk cardiovascular profiles followed for up to 104 weeks, showed no difference in 4-MACE events (cardiovascular death, myocardial infarction, stroke, and hospitalization for unstable angina) with tirzepatide compared to insulin glargine (HR 0.74; 95% CI, 0.51 – 1.08), the latter having been demonstrated to have neutral effect on cardiovascular outcomes (263). A meta-analysis of seven SURPASS clinical trials in participants with T2DM showed the hazard ratios comparing tirzepatide versus controls of 0.80 (95% CI, 0.57 – 1.11) for 4-MACE, 0.90 (95% CI, 0.50 – 1.61) for cardiovascular death, and 0.80 (95% CI, 0.51 – 1.25) for all-cause mortality (264). The definitive impact of tirzepatide on cardiovascular disease will be addressed in the ongoing SURPASS-COVT study (ClinicalTrials.gov number, NCT04255433), which compares tirzepatide with dulaglutide in patients with T2DM and ASCVD. The study will provide a more comprehensive assessment of the cardiovascular outcomes of this dual GIP/GLP-1 receptor agonist.
The mechanisms underlying the favorable cardiovascular effects of GLP-1RAs remain incompletely understood. These benefits are independent of baseline glycemia and duration of diabetes and only minimally related to their glucose-lowering efficacy (254). Although the pathophysiological processes underlying the cardiovascular disease may improve with weight loss, weight loss alone is insufficient to fully account for the observed cardiovascular improvements. Clinical trials of GLP-1RAs have primarily focused on patients with obesity or overweight, yet the effectiveness in the cardiovascular protection appears independent of baseline BMI range (265). Furthermore, the cardiovascular benefits emerged early in treatment, preceding significant weight loss, suggesting that the cardiovascular effects may be mediated, at least partially, by other salutary processes in atherosclerotic protection. Treatment with GLP-1RAs leads to rapid reduction in blood pressure, which, according to a meta-analysis (266), could substantially lower the risk of cardiovascular disease and mortality. Additional factors such as increased release of adiponectin from adipose tissue, amelioration of insulin resistance in liver and peripheral tissues, and improved circulatory lipid profile all contribute to the reduced risk of ASCVD (267). Recent findings indicate that the anti-inflammatory effects of GLP-1RAs, mediated through GLP-1R in CNS neurons (268) and intrahepatic lymphocytes (269), play an important role in their antiatherogenic actions. The FLOW trial also highlights the renal protective effects of GLP-1RAs, suggesting a close link between renal and cardiovascular benefits, which warrants further mechanistic studies (256).
7 Conclusion and challenges
GLP-1RAs have been used to treat T2DM since 2005 and gained approval for weight management in 2014. In studies on cardiovascular diseases, GLP-1RAs have shown potential benefits. Notably, in high-quality large RCTs, tirzepatide demonstrated greater effectiveness than GLP-1R mono agonists in reducing serum glucose, fat mass, and body weight. Ongoing clinical trials will provide clarity on the presumed cardiovascular benefits of tirzepatide. However, the global supply of these medications remains limited, often resulting in a first-come, first-served distribution. To address this, there have been proposals to prioritize individuals at higher risk of premature death, aligning with ethical principles of equitable care (270). Nevertheless, it is crucial to consider the potential adverse effects of weight fluctuation after discontinuing these medications, particularly the increased risk of cardiovascular and all-cause mortality in patients with obesity or overweight.
For all their promise, GLP-1 and dual GIP/GLP-1 receptor agonists have raised more questions, particularly regarding their long-term safety implications. Clinical trials have provided limited insight into the long-term effects, as the longest trials spanned less than six years in adults and about one year in adolescents. Given the natural fluctuations in hormone levels and the intricate rhythms of human physiology throughout the day and over a person’s lifetime, there is concern about the uncertain health ramifications of sustained elevation of incretin activity over decades of treatment. Like virtually all medications, incretin mimetics come with side-effects, leading some patients to abandon treatment. Common adverse effects include nausea and vomiting, as well as increased hazards of gastroparesis and small bowel obstruction. The next generation of regimens may involve combining GLP-1 or dual GIP/GLP-1 receptor agonists with other nutrient-based hormone receptor agonists or antagonists that offer equal or greater effectiveness with fewer gastrointestinal adverse effects.
Author contributions
QL: Conceptualization, Data curation, Formal analysis, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
I thank Lily Lu, MD, PhD for her critical comments and David Liu and Erika Liu for their assistance with editing.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1431292/full#supplementary-material
References
1. Creutzfeldt W. The incretin concept today. Diabetologia. (1979) 16:75–85. doi: 10.1007/BF01225454
2. Creutzfeldt W. Gastrointestinal hormones and insulin secretion. N Engl J Med. (1973) 288:1238–9. doi: 10.1056/NEJM197306072882312
3. Elrick H, Stimmler L, Hlad CJ, Arai Y. Plasma insulin response to oral and intravenous glucose administration. J Clin Endocrinol Metab. (1964) 24:1076–82. doi: 10.1210/jcem-24-10-1076
4. Perley MJ, Kipnis DM. Plasma insulin responses to oral and intravenous glucose: studies in normal and diabetic sujbjects. J Clin Invest. (1967) 46:1954–62. doi: 10.1172/JCI105685
5. Mcintyre N, Holdsworth CD, Turner DS. New interpretation of oral glucose tolerance. Lancet. (1964) 2:20–1. doi: 10.1016/S0140-6736(64)90011-X
6. Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology. (2007) 132:2131–57. doi: 10.1053/j.gastro.2007.03.054
7. Holst JJ, Knop FK, Vilsbøll T, Krarup T, Madsbad S. Loss of incretin effect is a specific, important, and early characteristic of type 2 diabetes. Diabetes Care. (2011) 34 Suppl 2:S251–7. doi: 10.2337/dc11-s227
8. Brown JC, Pederson RA, Jorpes E, Mutt V. Preparation of highly active enterogastrone. Can J Physiol Pharmacol. (1969) 47:113–4. doi: 10.1139/y69-020
9. Brown JC. A gastric inhibitory polypeptide. I. The amino acid composition and the tryptic peptides. Can J Biochem. (1971) 49:255–61. doi: 10.1139/o71-037
10. Brown JC, Mutt V, Pederson RA. Further purification of a polypeptide demonstrating enterogastrone activity. J Physiol. (1970) 209:57–64. doi: 10.1113/jphysiol.1970.sp009155
11. Dupre J, Ross SA, Watson D, Brown JC. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metab. (1973) 37:826–8. doi: 10.1210/jcem-37-5-826
12. Creutzfeldt W, Ebert R, Willms B, Frerichs H, Brown JC. Gastric inhibitory polypeptide (GIP) and insulin in obesity: increased response to stimulation and defective feedback control of serum levels. Diabetologia. (1978) 14:15–24. doi: 10.1007/BF00429703
13. Buchan AM, Polak JM, Capella C, Solcia E, Pearse AG. Electronimmunocytochemical evidence for the K cell localization of gastric inhibitory polypeptide (GIP) in man. Histochemistry. (1978) 56:37–44. doi: 10.1007/BF00492251
14. Fujita Y, Asadi A, Yang GK, Kwok YN, Kieffer TJ. Differential processing of pro-glucose-dependent insulinotropic polypeptide in gut. Am J Physiol Gastrointest Liver Physiol. (2010) 298:G608–14. doi: 10.1152/ajpgi.00024.2010
15. Fehmann HC, Göke R, Göke B. Cell and molecular biology of the incretin hormones glucagon-like peptide-I and glucose-dependent insulin releasing polypeptide. Endocr Rev. (1995) 16:390–410. doi: 10.1210/edrv-16-3-390
16. Moody AJ, Thim L, Valverde I. The isolation and sequencing of human gastric inhibitory peptide (GIP). FEBS Lett. (1984) 172:142–8. doi: 10.1016/0014-5793(84)81114-X
17. Carlquist M, Maletti M, Jörnvall H, Mutt V. A novel form of gastric inhibitory polypeptide (GIP) isolated from bovine intestine using a radioreceptor assay. Fragmentation with staphylococcal protease results in GIP1-3 and GIP4-42, fragmentation with enterokinase in GIP1-16 and GIP17-42. Eur J Biochem. (1984) 145:573–7. doi: 10.1111/j.1432-1033.1984.tb08595.x
18. Jörnvall H, Carlquist M, Kwauk S, Otte SC, McIntosh CH, Brown JC, et al. Amino acid sequence and heterogeneity of gastric inhibitory polypeptide (GIP). FEBS Lett. (1981) 123:205–10. doi: 10.1016/0014-5793(81)80288-8
19. Lund PK, Goodman RH, Dee PC, Habener JF. Pancreatic preproglucagon cDNA contains two glucagon-related coding sequences arranged in tandem. Proc Natl Acad Sci U.S.A. (1982) 79:345–9.
20. Bell GI, Sanchez-Pescador R, Laybourn PJ, Najarian RC. Exon duplication and divergence in the human preproglucagon gene. Nature. (1983) 304:368–71. doi: 10.1038/304368a0
21. Bell GI, Santerre RF, Mullenbach GT. Hamster preproglucagon contains the sequence of glucagon and two related peptides. Nature. (1983) 302:716–8. doi: 10.1038/302716a0
22. Mojsov S, Heinrich G, Wilson IB, Ravazzola M, Orci L, Habener JF. Preproglucagon gene expression in pancreas and intestine diversifies at the level of post-translational processing. J Biol Chem. (1986) 261:11880–9. doi: 10.1016/S0021-9258(18)67324-7
23. Holst JJ, Orskov C, Nielsen OV, Schwartz TW. Truncated glucagon-like peptide I, an insulin-releasing hormone from the distal gut. FEBS Lett. (1987) 211:169–74. doi: 10.1016/0014-5793(87)81430-8
24. Mojsov S, Weir GC, Habener JF. Insulinotropin: glucagon-like peptide I (7-37) co-encoded in the glucagon gene is a potent stimulator of insulin release in the perfused rat pancreas. J Clin Invest. (1987) 79:616–9. doi: 10.1172/JCI112855
25. Orskov C, Holst JJ, Poulsen SS, Kirkegaard P. Pancreatic and intestinal processing of proglucagon in man. Diabetologia. (1987) 30:874–81. doi: 10.1007/BF00274797
26. Dhanvantari S, Seidah NG, Brubaker PL. Role of prohormone convertases in the tissue-specific processing of proglucagon. Mol Endocrinol. (1996) 10:342–55.
27. Nauck MA, Weber I, Bach I, Richter S, Orskov C, Holst JJ, et al. Normalization of fasting glycaemia by intravenous GLP-1 ([7-36 amide] or [7-37]) in type 2 diabetic patients. Diabetes Med. (1998) 15:937–45. doi: 10.1002/(ISSN)1096-9136
28. Kreymann B, Williams G, Ghatei MA, Bloom SR. Glucagon-like peptide-1 7-36: a physiological incretin in man. Lancet. (1987) 2:1300–4. doi: 10.1016/S0140-6736(87)91194-9
29. Orskov C. Glucagon-like peptide-1, a new hormone of the entero-insular axis. Diabetologia. (1992) 35:701–11. doi: 10.1007/BF00429088
30. Seino S, Welsh M, Bell GI, Chan SJ, Steiner DF. Mutations in the Guinea pig preproglucagon gene are restricted to a specific portion of the prohormone sequence. FEBS Lett. (1986) 203:25–30. doi: 10.1016/0014-5793(86)81429-6
31. Calanna S, Christensen M, Holst JJ, Laferrère B, Gluud LL, Vilsbøll T, et al. Secretion of glucagon-like peptide-1 in patients with type 2 diabetes mellitus: systematic review and meta-analyses of clinical studies. Diabetologia. (2013) 56:965–72. doi: 10.1007/s00125-013-2841-0
32. Calanna S, Christensen M, Holst JJ, Laferrère B, Gluud LL, Vilsbøll T, et al. Secretion of glucose-dependent insulinotropic polypeptide in patients with type 2 diabetes: systematic review and meta-analysis of clinical studies. Diabetes Care. (2013) 36:3346–52. doi: 10.2337/dc13-0465
33. Gutniak M, Orskov C, Holst JJ, Ahrén B, Efendic S. Antidiabetogenic effect of glucagon-like peptide-1 (7-36)amide in normal subjects and patients with diabetes mellitus. N Engl J Med. (1992) 326:1316–22. doi: 10.1056/NEJM199205143262003
34. Gjesing AP, Ekstrøm CT, Eiberg H, Urhammer SA, Holst JJ, Pedersen O, et al. Fasting and oral glucose-stimulated levels of glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP-1) are highly familial traits. Diabetologia. (2012) 55:1338–45. doi: 10.1007/s00125-012-2484-6
35. Christensen M, Vedtofte L, Holst JJ, Vilsbøll T, Knop FK. Glucose-dependent insulinotropic polypeptide: a bifunctional glucose-dependent regulator of glucagon and insulin secretion in humans. Diabetes. (2011) 60:3103–9. doi: 10.2337/db11-0979
36. Sarson DL, Wood SM, Kansal PC, Bloom SR. Glucose-dependent insulinotropic polypeptide augmentation of insulin. Physiology or pharmacology? Diabetes. (1984) 33:389–93.
37. Meier JJ, Nauck MA. Is the diminished incretin effect in type 2 diabetes just an epi-phenomenon of impaired beta-cell function? Diabetes. (2010) 59:1117–25.
38. Zander M, Madsbad S, Madsen JL, Holst JJ. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet. (2002) 359:824–30. doi: 10.1016/S0140-6736(02)07952-7
39. Vollmer K, Holst JJ, Baller B, Ellrichmann M, Nauck MA, Schmidt WE, et al. Predictors of incretin concentrations in subjects with normal, impaired, and diabetic glucose tolerance. Diabetes. (2008) 57:678–87. doi: 10.2337/db07-1124
40. Tan Q, Akindehin SE, Orsso CE, Waldner RC, DiMarchi RD, Müller TD, et al. Recent advances in incretin-based pharmacotherapies for the treatment of obesity and diabetes. Front Endocrinol (Lausanne). (2022) 13:838410. doi: 10.3389/fendo.2022.838410
41. Usdin TB, Mezey E, Button DC, Brownstein MJ, Bonner TI. Gastric inhibitory polypeptide receptor, a member of the secretin-vasoactive intestinal peptide receptor family, is widely distributed in peripheral organs and the brain. Endocrinology. (1993) 133:2861–70. doi: 10.1210/endo.133.6.8243312
42. Thorens B. Expression cloning of the pancreatic beta cell receptor for the gluco-incretin hormone glucagon-like peptide 1. Proc Natl Acad Sci U.S.A. (1992) 89:8641–5.
43. Zhang Y, Sun B, Feng D, Hu H, Chu M, Qu Q, et al. Cryo-EM structure of the activated GLP-1 receptor in complex with a G protein. Nature. (2017) 546:248–53. doi: 10.1038/nature22394
44. Finan B, Ma T, Ottaway N, Müller TD, Habegger KM, Heppner KM, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med. (2013) 5:209ra151. doi: 10.1126/scitranslmed.3007218
45. Oduori OS, Murao N, Shimomura K, Takahashi H, Zhang Q, Dou H, et al. Gs/Gq signaling switch in β cells defines incretin effectiveness in diabetes. J Clin Invest. (2020) 130:6639–55. doi: 10.1172/JCI140046
46. Roberge JN, Brubaker PL. Regulation of intestinal proglucagon-derived peptide secretion by glucose-dependent insulinotropic peptide in a novel enteroendocrine loop. Endocrinology. (1993) 133:233–40. doi: 10.1210/endo.133.1.8319572
47. Damholt AB, Buchan AM, Kofod H. Glucagon-like-peptide-1 secretion from canine L-cells is increased by glucose-dependent-insulinotropic peptide but unaffected by glucose. Endocrinology. (1998) 139:2085–91. doi: 10.1210/endo.139.4.5921
48. Nauck MA, Bartels E, Orskov C, Ebert R, Creutzfeldt W. Additive insulinotropic effects of exogenous synthetic human gastric inhibitory polypeptide and glucagon-like peptide-1-(7-36) amide infused at near-physiological insulinotropic hormone and glucose concentrations. J Clin Endocrinol Metab. (1993) 76:912–7.
49. Nauck MA, El-Ouaghlidi A, Gabrys B, Hücking K, Holst JJ, Deacon CF, et al. Secretion of incretin hormones (GIP and GLP-1) and incretin effect after oral glucose in first-degree relatives of patients with type 2 diabetes. Regul Pept. (2004) 122:209–17. doi: 10.1016/j.regpep.2004.06.020
50. Mentlein R, Gallwitz B, Schmidt WE. Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide, glucagon-like peptide-1(7-36)amide, peptide histidine methionine and is responsible for their degradation in human serum. Eur J Biochem. (1993) 214:829–35. doi: 10.1111/j.1432-1033.1993.tb17986.x
51. Kieffer TJ, McIntosh CH, Pederson RA. Degradation of glucose-dependent insulinotropic polypeptide and truncated glucagon-like peptide 1 in vitro and in vivo by dipeptidyl peptidase IV. Endocrinology. (1995) 136:3585–96. doi: 10.1210/endo.136.8.7628397
52. Vilsbøll T, Agersø H, Lauritsen T, Deacon CF, Aaboe K, Madsbad S, et al. The elimination rates of intact GIP as well as its primary metabolite, GIP 3-42, are similar in type 2 diabetic patients and healthy subjects. Regul Pept. (2006) 137:168–72. doi: 10.1016/j.regpep.2006.07.007
53. Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes. (1995) 44:1126–31. doi: 10.2337/diab.44.9.1126
54. Gasbjerg LS, Helsted MM, Hartmann B, Jensen MH, Gabe MBN, Sparre-Ulrich AH, et al. Separate and combined glucometabolic effects of endogenous glucose-dependent insulinotropic polypeptide and glucagon-like peptide 1 in healthy individuals. Diabetes. (2019) 68:906–17. doi: 10.2337/db18-1123
55. Mentis N, Vardarli I, Köthe LD, Holst JJ, Deacon CF, Theodorakis M, et al. GIP does not potentiate the antidiabetic effects of GLP-1 in hyperglycemic patients with type 2 diabetes. Diabetes. (2011) 60:1270–6. doi: 10.2337/db10-1332
56. Elliott RM, Morgan LM, Tredger JA, Deacon S, Wright J, Marks V. Glucagon-like peptide-1 (7-36)amide and glucose-dependent insulinotropic polypeptide secretion in response to nutrient ingestion in man: acute post-prandial and 24-h secretion patterns. J Endocrinol. (1993) 138:159–66. doi: 10.1677/joe.0.1380159
57. Krarup T, Saurbrey N, Moody AJ, Kühl C, Madsbad S. Effect of porcine gastric inhibitory polypeptide on beta-cell function in type I and type II diabetes mellitus. Metabolism. (1987) 36:677–82. doi: 10.1016/0026-0495(87)90153-3
58. Nauck MA, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest. (1993) 91:301–7. doi: 10.1172/JCI116186
59. Lynn FC, Thompson SA, Pospisilik JA, Ehses JA, Hinke SA, Pamir N, et al. A novel pathway for regulation of glucose-dependent insulinotropic polypeptide (GIP) receptor expression in beta cells. FASEB J. (2003) 17:91–3.
60. Holst JJ, Gromada J, Nauck MA. The pathogenesis of NIDDM involves a defective expression of the GIP receptor. Diabetologia. (1997) 40:984–6. doi: 10.1007/s001250050779
61. Kubota A, Yamada Y, Hayami T, Yasuda K, Someya Y, Ihara Y, et al. Identification of two missense mutations in the GIP receptor gene: a functional study and association analysis with NIDDM: no evidence of association with Japanese NIDDM subjects. Diabetes. (1996) 45:1701–5. doi: 10.2337/diab.45.12.1701
62. Kjems LL, Holst JJ, Vølund A, Madsbad S. The influence of GLP-1 on glucose-stimulated insulin secretion: effects on beta-cell sensitivity in type 2 and nondiabetic subjects. Diabetes. (2003) 52:380–6. doi: 10.2337/diabetes.52.2.380
63. Coskun T, Sloop KW, Loghin C, Alsina-Fernandez J, Urva S, Bokvist KB, et al. LY3298176, a novel dual GIP and GLP-1 receptor agonist for the treatment of type 2 diabetes mellitus: From discovery to clinical proof of concept. Mol Metab. (2018) 18:3–14. doi: 10.1016/j.molmet.2018.09.009
64. Berndt SI, Gustafsson S, Mägi R, Ganna A, Wheeler E, Feitosa MF, et al. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat Genet. (2013) 45:501–12. doi: 10.1038/ng.2606
65. Mohammad S, Patel RT, Bruno J, Panhwar MS, Wen J, McGraw TE. A naturally occurring GIP receptor variant undergoes enhanced agonist-induced desensitization, which impairs GIP control of adipose insulin sensitivity. Mol Cell Biol. (2014) 34:3618–29. doi: 10.1128/MCB.00256-14
66. Akbari P, Gilani A, Sosina O, Kosmicki JA, Khrimian L, Fang YY, et al. Sequencing of 640,000 exomes identifies. Science. (2021) 373.
67. Miyawaki K, Yamada Y, Ban N, Ihara Y, Tsukiyama K, Zhou H, et al. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nat Med. (2002) 8:738–42. doi: 10.1038/nm727
68. Killion EA, Wang J, Yie J, Shi SD, Bates D, Min X, et al. Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Sci Transl Med. (2018) 10. doi: 10.1126/scitranslmed.aat3392
69. Kim SJ, Nian C, Karunakaran S, Clee SM, Isales CM, McIntosh CH. GIP-overexpressing mice demonstrate reduced diet-induced obesity and steatosis, and improved glucose homeostasis. PloS One. (2012) 7:e40156. doi: 10.1371/journal.pone.0040156
70. Mroz PA, Finan B, Gelfanov V, Yang B, Tschöp MH, DiMarchi RD, et al. Optimized GIP analogs promote body weight lowering in mice through GIPR agonism not antagonism. Mol Metab. (2019) 20:51–62. doi: 10.1016/j.molmet.2018.12.001
71. Turton MD, O'Shea D, Gunn I, Beak SA, Edwards CM, Meeran K, et al. A role for glucagon-like peptide-1 in the central regulation of feeding. Nature. (1996) 379:69–72. doi: 10.1038/379069a0
72. Tang-Christensen M, Larsen PJ, Göke R, Fink-Jensen A, Jessop DS, Møller M, et al. Central administration of GLP-1-(7-36) amide inhibits food and water intake in rats. Am J Physiol. (1996) 271:R848–56. doi: 10.1152/ajpregu.1996.271.4.R848
73. Lu SC, Chen M, Atangan L, Killion EA, Komorowski R, Cheng Y, et al. GIPR antagonist antibodies conjugated to GLP-1 peptide are bispecific molecules that decrease weight in obese mice and monkeys. Cell Rep Med. (2021) 2:100263. doi: 10.1016/j.xcrm.2021.100263
74. Véniant MM, Lu SC, Atangan L, Komorowski R, Stanislaus S, Cheng Y, et al. A GIPR antagonist conjugated to GLP-1 analogues promotes weight loss with improved metabolic parameters in preclinical and phase 1 settings. Nat Metab. (2024) 6:290–303. doi: 10.1038/s42255-023-00966-w
75. Nørregaard PK, Deryabina MA, Tofteng Shelton P, Fog JU, Daugaard JR, Eriksson PO, et al. A novel GIP analogue, ZP4165, enhances glucagon-like peptide-1-induced body weight loss and improves glycaemic control in rodents. Diabetes Obes Metab. (2018) 20:60–8.
76. Frias JP, Bastyr EJ, Vignati L, Tschöp MH, Schmitt C, Owen K, et al. The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090-2746, in patients with type 2 diabetes. Cell Metab. (2017) 26:343–52.e2. doi: 10.1016/j.cmet.2017.07.011
77. Day JW, Ottaway N, Patterson JT, Gelfanov V, Smiley D, Gidda J, et al. A new glucagon and GLP-1 co-agonist eliminates obesity in rodents. Nat Chem Biol. (2009) 5:749–57. doi: 10.1038/nchembio.209
78. Finan B, Yang B, Ottaway N, Smiley DL, Ma T, Clemmensen C, et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat Med. (2015) 21:27–36. doi: 10.1038/nm.3761
79. Coskun T, Urva S, Roell WC, Qu H, Loghin C, Moyers JS, et al. LY3437943, a novel triple glucagon, GIP, and GLP-1 receptor agonist for glycemic control and weight loss: From discovery to clinical proof of concept. Cell Metab. (2022) 34:1234–47.e9. doi: 10.1016/j.cmet.2022.07.013
80. le Roux CW, Steen O, Lucas KJ, Startseva E, Unseld A, Hennige AM. Glucagon and GLP-1 receptor dual agonist survodutide for obesity: a randomised, double-blind, placebo-controlled, dose-finding phase 2 trial. Lancet Diabetes Endocrinol. (2024) 12:162–73. doi: 10.1016/S2213-8587(23)00356-X
81. Apovian CM, McDonnell ME. CagriSema and the link between obesity and type 2 diabetes. Lancet. (2023) 402:671–3. doi: 10.1016/S0140-6736(23)01291-6
82. Ambery P, Parker VE, Stumvoll M, Posch MG, Heise T, Plum-Moerschel L, et al. MEDI0382, a GLP-1 and glucagon receptor dual agonist, in obese or overweight patients with type 2 diabetes: a randomised, controlled, double-blind, ascending dose and phase 2a study. Lancet. (2018) 391:2607–18. doi: 10.1016/S0140-6736(18)30726-8
83. Eng J, Kleinman WA, Singh L, Singh G, Raufman JP. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from Guinea pig pancreas. J Biol Chem. (1992) 267:7402–5. doi: 10.1016/S0021-9258(18)42531-8
84. Chen YE, Drucker DJ. Tissue-specific expression of unique mRNAs that encode proglucagon-derived peptides or exendin 4 in the lizard. J Biol Chem. (1997) 272:4108–15. doi: 10.1074/jbc.272.7.4108
85. Kolterman OG, Kim DD, Shen L, Ruggles JA, Nielsen LL, Fineman MS, et al. Pharmacokinetics, pharmacodynamics, and safety of exenatide in patients with type 2 diabetes mellitus. Am J Health Syst Pharm. (2005) 62:173–81. doi: 10.1093/ajhp/62.2.173
86. Göke R, Fehmann HC, Linn T, Schmidt H, Krause M, Eng J, et al. Exendin-4 is a high potency agonist and truncated exendin-(9-39)-amide an antagonist at the glucagon-like peptide 1-(7-36)-amide receptor of insulin-secreting beta-cells. J Biol Chem. (1993) 268:19650–5. doi: 10.1016/S0021-9258(19)36565-2
87. Young AA, Gedulin BR, Bhavsar S, Bodkin N, Jodka C, Hansen B, et al. Glucose-lowering and insulin-sensitizing actions of exendin-4: studies in obese diabetic (ob/ob, db/db) mice, diabetic fatty Zucker rats, and diabetic rhesus monkeys (Macaca mulatta). Diabetes. (1999) 48:1026–34. doi: 10.2337/diabetes.48.5.1026
88. Thorkildsen C, Neve S, Larsen BD, Meier E, Petersen JS. Glucagon-like peptide 1 receptor agonist ZP10A increases insulin mRNA expression and prevents diabetic progression in db/db mice. J Pharmacol Exp Ther. (2003) 307:490–6. doi: 10.1124/jpet.103.051987
89. DeYoung MB, MacConell L, Sarin V, Trautmann M, Herbert P. Encapsulation of exenatide in poly-(D,L-lactide-co-glycolide) microspheres produced an investigational long-acting once-weekly formulation for type 2 diabetes. Diabetes Technol Ther. (2011) 13:1145–54. doi: 10.1089/dia.2011.0050
90. Bush MA, Matthews JE, De Boever EH, Dobbins RL, Hodge RJ, Walker SE, et al. Safety, tolerability, pharmacodynamics and pharmacokinetics of albiglutide, a long-acting glucagon-like peptide-1 mimetic, in healthy subjects. Diabetes Obes Metab. (2009) 11:498–505. doi: 10.1111/j.1463-1326.2008.00992.x
91. Kim DM, Chu SH, Kim S, Park YW, Kim SS. Fc fusion to glucagon-like peptide-1 inhibits degradation by human DPP-IV, increasing its half-life in serum and inducing a potent activity for human GLP-1 receptor activation. BMB Rep. (2009) 42:212–6. doi: 10.5483/BMBRep.2009.42.4.212
92. Baggio LL, Huang Q, Brown TJ, Drucker DJ. A recombinant human glucagon-like peptide (GLP)-1-albumin protein (albugon) mimics peptidergic activation of GLP-1 receptor-dependent pathways coupled with satiety, gastrointestinal motility, and glucose homeostasis. Diabetes. (2004) 53:2492–500. doi: 10.2337/diabetes.53.9.2492
93. Madsbad S. Review of head-to-head comparisons of glucagon-like peptide-1 receptor agonists. Diabetes Obes Metab. (2016) 18:317–32. doi: 10.1111/dom.12596
94. Glaesner W, Vick AM, Millican R, Ellis B, Tschang SH, Tian Y, et al. Engineering and characterization of the long-acting glucagon-like peptide-1 analogue LY2189265, an Fc fusion protein. Diabetes Metab Res Rev. (2010) 26:287–96. doi: 10.1002/dmrr.1080
95. Lau J, Bloch P, Schäffer L, Pettersson I, Spetzler J, Kofoed J, et al. Discovery of the once-weekly glucagon-like peptide-1 (GLP-1) analogue semaglutide. J Med Chem. (2015) 58:7370–80. doi: 10.1021/acs.jmedchem.5b00726
96. Deacon CF, Knudsen LB, Madsen K, Wiberg FC, Jacobsen O, Holst JJ. Dipeptidyl peptidase IV resistant analogues of glucagon-like peptide-1 which have extended metabolic stability and improved biological activity. Diabetologia. (1998) 41:271–8. doi: 10.1007/s001250050903
97. Kelly AS, Auerbach P, Barrientos-Perez M, Gies I, Hale PM, Marcus C, et al. A randomized, controlled trial of liraglutide for adolescents with obesity. N Engl J Med. (2020) 382:2117–28. doi: 10.1056/NEJMoa1916038
98. Petri KCC, Ingwersen SH, Flint A, Zacho J, Overgaard RV. Exposure-response analysis for evaluation of semaglutide dose levels in type 2 diabetes. Diabetes Obes Metab. (2018) 20:2238–45. doi: 10.1111/dom.13358
99. Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, Madsen K, Pedersen FZ, et al. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem. (2000) 43:1664–9. doi: 10.1021/jm9909645
100. Buckley ST, Bækdal TA, Vegge A, Maarbjerg SJ, Pyke C, Ahnfelt-Rønne J, et al. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci Transl Med. (2018) 10. doi: 10.1126/scitranslmed.aar7047
101. FDA U. Rybelsus prescribing information. USA: FDA (2020). Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/213051s000lbl.pdf.
102. Granhall C, Donsmark M, Blicher TM, Golor G, Søndergaard FL, Thomsen M, et al. Safety and pharmacokinetics of single and multiple ascending doses of the novel oral human GLP-1 analogue, oral semaglutide, in healthy subjects and subjects with type 2 diabetes. Clin Pharmacokinet. (2019) 58:781–91. doi: 10.1007/s40262-018-0728-4
103. Overgaard RV, Hertz CL, Ingwersen SH, Navarria A, Drucker DJ. Levels of circulating semaglutide determine reductions in HbA1c and body weight in people with type 2 diabetes. Cell Rep Med. (2021) 2:100387. doi: 10.1016/j.xcrm.2021.100387
104. Aroda VR, Aberle J, Bardtrum L, Christiansen E, Knop FK, Gabery S, et al. Efficacy and safety of once-daily oral semaglutide 25 mg and 50 mg compared with 14 mg in adults with type 2 diabetes (PIONEER PLUS): a multicentre, randomised, phase 3b trial. Lancet. (2023) 402:693–704. doi: 10.1016/S0140-6736(23)01127-3
105. Knop FK, Aroda VR, do Vale RD, Holst-Hansen T, Laursen PN, Rosenstock J, et al. Oral semaglutide 50 mg taken once per day in adults with overweight or obesity (OASIS 1): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2023) 402:705–19. doi: 10.1016/S0140-6736(23)01185-6
106. Portron A, Jadidi S, Sarkar N, DiMarchi R, Schmitt C. Pharmacodynamics, pharmacokinetics, safety and tolerability of the novel dual glucose-dependent insulinotropic polypeptide/glucagon-like peptide-1 agonist RG7697 after single subcutaneous administration in healthy subjects. Diabetes Obes Metab. (2017) 19:1446–53. doi: 10.1111/dom.13025
107. Schmitt C, Portron A, Jadidi S, Sarkar N, DiMarchi R. Pharmacodynamics, pharmacokinetics and safety of multiple ascending doses of the novel dual glucose-dependent insulinotropic polypeptide/glucagon-like peptide-1 agonist RG7697 in people with type 2 diabetes mellitus. Diabetes Obes Metab. (2017) 19:1436–45. doi: 10.1111/dom.13024
108. Novikoff A, O'Brien SL, Bernecker M, Grandl G, Kleinert M, Knerr PJ, et al. Spatiotemporal GLP-1 and GIP receptor signaling and trafficking/recycling dynamics induced by selected receptor mono- and dual-agonists. Mol Metab. (2021) 49:101181. doi: 10.1016/j.molmet.2021.101181
109. Willard FS, Douros JD, Gabe MB, Showalter AD, Wainscott DB, Suter TM, et al. Tirzepatide is an imbalanced and biased dual GIP and GLP-1 receptor agonist. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.140532
110. Zhao F, Zhou Q, Cong Z, Hang K, Zou X, Zhang C, et al. Structural insights into multiplexed pharmacological actions of tirzepatide and peptide 20 at the GIP, GLP-1 or glucagon receptors. Nat Commun. (2022) 13:1057. doi: 10.1038/s41467-022-28683-0
111. Frias JP, Nauck MA, Van J, Kutner ME, Cui X, Benson C, et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet. (2018) 392:2180–93. doi: 10.1016/S0140-6736(18)32260-8
112. Frías JP, Davies MJ, Rosenstock J, Pérez Manghi FC, Fernández Landó L, Bergman BK, et al. Tirzepatide versus semaglutide once weekly in patients with type 2 diabetes. N Engl J Med. (2021) 385:503–15. doi: 10.1056/NEJMoa2107519
113. Ehses JA, Casilla VR, Doty T, Pospisilik JA, Winter KD, Demuth HU, et al. Glucose-dependent insulinotropic polypeptide promotes beta-(INS-1) cell survival via cyclic adenosine monophosphate-mediated caspase-3 inhibition and regulation of p38 mitogen-activated protein kinase. Endocrinology. (2003) 144:4433–45. doi: 10.1210/en.2002-0068
114. Kim SJ, Winter K, Nian C, Tsuneoka M, Koda Y, McIntosh CH. Glucose-dependent insulinotropic polypeptide (GIP) stimulation of pancreatic beta-cell survival is dependent upon phosphatidylinositol 3-kinase (PI3K)/protein kinase B (PKB) signaling, inactivation of the forkhead transcription factor Foxo1, and down-regulation of bax expression. J Biol Chem. (2005) 280:22297–307. doi: 10.1074/jbc.M500540200
115. Trümper A, Trümper K, Trusheim H, Arnold R, Göke B, Hörsch D. Glucose-dependent insulinotropic polypeptide is a growth factor for beta (INS-1) cells by pleiotropic signaling. Mol Endocrinol. (2001) 15:1559–70.
116. Trümper A, Trümper K, Hörsch D. Mechanisms of mitogenic and anti-apoptotic signaling by glucose-dependent insulinotropic polypeptide in beta(INS-1)-cells. J Endocrinol. (2002) 174:233–46. doi: 10.1677/joe.0.1740233
117. Drucker DJ, Philippe J, Mojsov S, Chick WL, Habener JF. Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc Natl Acad Sci U.S.A. (1987) 84:3434–8.
118. Volz A, Göke R, Lankat-Buttgereit B, Fehmann HC, Bode HP, Göke B. Molecular cloning, functional expression, and signal transduction of the GIP-receptor cloned from a human insulinoma. FEBS Lett. (1995) 373:23–9. doi: 10.1016/0014-5793(95)01006-Z
119. Falko JM, Crockett SE, Cataland S, Mazzaferri EL. Gastric inhibitory polypeptide (GIP) stimulated by fat ingestion in man. J Clin Endocrinol Metab. (1975) 41:260–5. doi: 10.1210/jcem-41-2-260
120. Chang AM, Jakobsen G, Sturis J, Smith MJ, Bloem CJ, An B, et al. The GLP-1 derivative NN2211 restores beta-cell sensitivity to glucose in type 2 diabetic patients after a single dose. Diabetes. (2003) 52:1786–91. doi: 10.2337/diabetes.52.7.1786
121. Fehse F, Trautmann M, Holst JJ, Halseth AE, Nanayakkara N, Nielsen LL, et al. Exenatide augments first- and second-phase insulin secretion in response to intravenous glucose in subjects with type 2 diabetes. J Clin Endocrinol Metab. (2005) 90:5991–7. doi: 10.1210/jc.2005-1093
122. Davies MJ, Bergenstal R, Bode B, Kushner RF, Lewin A, Skjøth TV, et al. Efficacy of liraglutide for weight loss among patients with type 2 diabetes: the SCALE diabetes randomized clinical trial. JAMA. (2015) 314:687–99. doi: 10.1001/jama.2015.9676
123. Pi-Sunyer X, Astrup A, Fujioka K, Greenway F, Halpern A, Krempf M, et al. A randomized, controlled trial of 3.0 mg of liraglutide in weight management. N Engl J Med. (2015) 373:11–22. doi: 10.1056/NEJMoa1411892
124. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. (2003) 52:102–10. doi: 10.2337/diabetes.52.1.102
125. Farilla L, Hui H, Bertolotto C, Kang E, Bulotta A, Di Mario U, et al. Glucagon-like peptide-1 promotes islet cell growth and inhibits apoptosis in Zucker diabetic rats. Endocrinology. (2002) 143:4397–408. doi: 10.1210/en.2002-220405
126. Maida A, Hansotia T, Longuet C, Seino Y, Drucker DJ. Differential importance of glucose-dependent insulinotropic polypeptide vs glucagon-like peptide 1 receptor signaling for beta cell survival in mice. Gastroenterology. (2009) 137:2146–57. doi: 10.1053/j.gastro.2009.09.004
127. Widenmaier SB, Kim SJ, Yang GK, De Los Reyes T, Nian C, Asadi A, et al. A GIP receptor agonist exhibits beta-cell anti-apoptotic actions in rat models of diabetes resulting in improved beta-cell function and glycemic control. PloS One. (2010) 5:e9590.
128. Wang Y, Perfetti R, Greig NH, Holloway HW, DeOre KA, Montrose-Rafizadeh C, et al. Glucagon-like peptide-1 can reverse the age-related decline in glucose tolerance in rats. J Clin Invest. (1997) 99:2883–9. doi: 10.1172/JCI119482
129. Xu G, Stoffers DA, Habener JF, Bonner-Weir S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes. (1999) 48:2270–6. doi: 10.2337/diabetes.48.12.2270
130. Chia CW, Carlson OD, Kim W, Shin YK, Charles CP, Kim HS, et al. Exogenous glucose-dependent insulinotropic polypeptide worsens post prandial hyperglycemia in type 2 diabetes. Diabetes. (2009) 58:1342–9. doi: 10.2337/db08-0958
131. Nauck MA, Heimesaat MM, Behle K, Holst JJ, Nauck MS, Ritzel R, et al. Effects of glucagon-like peptide 1 on counterregulatory hormone responses, cognitive functions, and insulin secretion during hyperinsulinemic, stepped hypoglycemic clamp experiments in healthy volunteers. J Clin Endocrinol Metab. (2002) 87:1239–46. doi: 10.1210/jcem.87.3.8355
132. de Heer J, Rasmussen C, Coy DH, Holst JJ. Glucagon-like peptide-1, but not glucose-dependent insulinotropic peptide, inhibits glucagon secretion via somatostatin (receptor subtype 2) in the perfused rat pancreas. Diabetologia. (2008) 51:2263–70. doi: 10.1007/s00125-008-1149-y
133. Gromada J, Franklin I, Wollheim CB. Alpha-cells of the endocrine pancreas: 35 years of research but the enigma remains. Endocr Rev. (2007) 28:84–116. doi: 10.1210/er.2006-0007
134. Ørgaard A, Holst JJ. The role of somatostatin in GLP-1-induced inhibition of glucagon secretion in mice. Diabetologia. (2017) 60:1731–9. doi: 10.1007/s00125-017-4315-2
135. Heise T, Mari A, DeVries JH, Urva S, Li J, Pratt EJ, et al. Effects of subcutaneous tirzepatide versus placebo or semaglutide on pancreatic islet function and insulin sensitivity in adults with type 2 diabetes: a multicentre, randomised, double-blind, parallel-arm, phase 1 clinical trial. Lancet Diabetes Endocrinol. (2022) 10:418–29. doi: 10.1016/S2213-8587(22)00085-7
136. Nyberg J, Jacobsson C, Anderson MF, Eriksson PS. Immunohistochemical distribution of glucose-dependent insulinotropic polypeptide in the adult rat brain. J Neurosci Res. (2007) 85:2099–119. doi: 10.1002/jnr.21349
137. Adriaenssens AE, Biggs EK, Darwish T, Tadross J, Sukthankar T, Girish M, et al. Glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus regulate food intake. Cell Metab. (2019) 30:987–96.e6. doi: 10.1016/j.cmet.2019.07.013
138. Zhang Q, Delessa CT, Augustin R, Bakhti M, Colldén G, Drucker DJ, et al. The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling. Cell Metab. (2021) 33:833–44.e5. doi: 10.1016/j.cmet.2021.01.015
139. Larsen PJ, Tang-Christensen M, Holst JJ, Orskov C. Distribution of glucagon-like peptide-1 and other preproglucagon-derived peptides in the rat hypothalamus and brainstem. Neuroscience. (1997) 77:257–70. doi: 10.1016/S0306-4522(96)00434-4
140. Drucker DJ, Asa S. Glucagon gene expression in vertebrate brain. J Biol Chem. (1988) 263:13475–8. doi: 10.1016/S0021-9258(18)68261-4
141. Göke R, Larsen PJ, Mikkelsen JD, Sheikh SP. Distribution of GLP-1 binding sites in the rat brain: evidence that exendin-4 is a ligand of brain GLP-1 binding sites. Eur J Neurosci. (1995) 7:2294–300.
142. Sisley S, Gutierrez-Aguilar R, Scott M, D'Alessio DA, Sandoval DA, Seeley RJ. Neuronal GLP1R mediates liraglutide's anorectic but not glucose-lowering effect. J Clin Invest. (2014) 124:2456–63. doi: 10.1172/JCI72434
143. Orskov C, Poulsen SS, Møller M, Holst JJ. Glucagon-like peptide I receptors in the subfornical organ and the area postrema are accessible to circulating glucagon-like peptide I. Diabetes. (1996) 45:832–5. doi: 10.2337/diab.45.6.832
144. Hunter K, Hölscher C. Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis. BMC Neurosci. (2012) 13:33. doi: 10.1186/1471-2202-13-33
145. Gabery S, Salinas CG, Paulsen SJ, Ahnfelt-Rønne J, Alanentalo T, Baquero AF, et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.133429
146. Salameh TS, Rhea EM, Talbot K, Banks WA. Brain uptake pharmacokinetics of incretin receptor agonists showing promise as Alzheimer's and Parkinson's disease therapeutics. Biochem Pharmacol. (2020) 180:114187. doi: 10.1016/j.bcp.2020.114187
147. Capehorn MS, Catarig AM, Furberg JK, Janez A, Price HC, Tadayon S, et al. Efficacy and safety of once-weekly semaglutide 1.0mg vs once-daily liraglutide 1.2mg as add-on to 1-3 oral antidiabetic drugs in subjects with type 2 diabetes (SUSTAIN 10). Diabetes Metab. (2020) 46:100–9. doi: 10.1016/j.diabet.2019.101117
148. Campbell JE, Ussher JR, Mulvihill EE, Kolic J, Baggio LL, Cao X, et al. TCF1 links GIPR signaling to the control of beta cell function and survival. Nat Med. (2016) 22:84–90. doi: 10.1038/nm.3997
149. NamKoong C, Kim MS, Jang BT, Lee YH, Cho YM, Choi HJ. Central administration of GLP-1 and GIP decreases feeding in mice. Biochem Biophys Res Commun. (2017) 490:247–52. doi: 10.1016/j.bbrc.2017.06.031
150. Meier JJ, Goetze O, Anstipp J, Hagemann D, Holst JJ, Schmidt WE, et al. Gastric inhibitory polypeptide does not inhibit gastric emptying in humans. Am J Physiol Endocrinol Metab. (2004) 286:E621–5. doi: 10.1152/ajpendo.00499.2003
151. Urva S, Coskun T, Loghin C, Cui X, Beebe E, O'Farrell L, et al. The novel dual glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1 (GLP-1) receptor agonist tirzepatide transiently delays gastric emptying similarly to selective long-acting GLP-1 receptor agonists. Diabetes Obes Metab. (2020) 22:1886–91. doi: 10.1111/dom.14110
152. Willms B, Werner J, Holst JJ, Orskov C, Creutzfeldt W, Nauck MA. Gastric emptying, glucose responses, and insulin secretion after a liquid test meal: effects of exogenous glucagon-like peptide-1 (GLP-1)-(7-36) amide in type 2 (noninsulin-dependent) diabetic patients. J Clin Endocrinol Metab. (1996) 81:327–32.
153. Meier JJ, Kemmeries G, Holst JJ, Nauck MA. Erythromycin antagonizes the deceleration of gastric emptying by glucagon-like peptide 1 and unmasks its insulinotropic effect in healthy subjects. Diabetes. (2005) 54:2212–8. doi: 10.2337/diabetes.54.7.2212
154. Meier JJ, Gallwitz B, Salmen S, Goetze O, Holst JJ, Schmidt WE, et al. Normalization of glucose concentrations and deceleration of gastric emptying after solid meals during intravenous glucagon-like peptide 1 in patients with type 2 diabetes. J Clin Endocrinol Metab. (2003) 88:2719–25. doi: 10.1210/jc.2003-030049
155. Nauck MA, Niedereichholz U, Ettler R, Holst JJ, Orskov C, Ritzel R, et al. Glucagon-like peptide 1 inhibition of gastric emptying outweighs its insulinotropic effects in healthy humans. Am J Physiol. (1997) 273:E981–8. doi: 10.1152/ajpendo.1997.273.5.E981
156. Meier JJ, Rosenstock J, Hincelin-Méry A, Roy-Duval C, Delfolie A, Coester HV, et al. Contrasting effects of lixisenatide and liraglutide on postprandial glycemic control, gastric emptying, and safety parameters in patients with type 2 diabetes on optimized insulin glargine with or without metformin: A randomized, open-label trial. Diabetes Care. (2015) 38:1263–73. doi: 10.2337/dc14-1984
157. Nauck MA, Kemmeries G, Holst JJ, Meier JJ. Rapid tachyphylaxis of the glucagon-like peptide 1-induced deceleration of gastric emptying in humans. Diabetes. (2011) 60:1561–5. doi: 10.2337/db10-0474
158. Jelsing J, Vrang N, Hansen G, Raun K, Tang-Christensen M, Knudsen LB. Liraglutide: short-lived effect on gastric emptying – long lasting effects on body weight. Diabetes Obes Metab. (2012) 14:531–8. doi: 10.1111/j.1463-1326.2012.01557.x
159. Anvari M, Paterson CA, Daniel EE, McDonald TJ. Effects of GLP-1 on gastric emptying, antropyloric motility, and transpyloric flow in response to a nonnutrient liquid. Dig Dis Sci. (1998) 43:1133–40. doi: 10.1023/A:1018863716749
160. Schirra J, Houck P, Wank U, Arnold R, Göke B, Katschinski M. Effects of glucagon-like peptide-1(7-36)amide on antro-pyloro-duodenal motility in the interdigestive state and with duodenal lipid perfusion in humans. Gut. (2000) 46:622–31. doi: 10.1136/gut.46.5.622
161. Imeryüz N, Yeğen BC, Bozkurt A, Coşkun T, Villanueva-Peñacarrillo ML, Ulusoy NB. Glucagon-like peptide-1 inhibits gastric emptying via vagal afferent-mediated central mechanisms. Am J Physiol. (1997) 273:G920–7.
162. Nakagawa A, Satake H, Nakabayashi H, Nishizawa M, Furuya K, Nakano S, et al. Receptor gene expression of glucagon-like peptide-1, but not glucose-dependent insulinotropic polypeptide, in rat nodose ganglion cells. Auton Neurosci. (2004) 110:36–43. doi: 10.1016/j.autneu.2003.11.001
163. Vahl TP, Tauchi M, Durler TS, Elfers EE, Fernandes TM, Bitner RD, et al. Glucagon-like peptide-1 (GLP-1) receptors expressed on nerve terminals in the portal vein mediate the effects of endogenous GLP-1 on glucose tolerance in rats. Endocrinology. (2007) 148:4965–73. doi: 10.1210/en.2006-0153
164. Bucinskaite V, Tolessa T, Pedersen J, Rydqvist B, Zerihun L, Holst JJ, et al. Receptor-mediated activation of gastric vagal afferents by glucagon-like peptide-1 in the rat. Neurogastroenterol Motil. (2009) 21:978–e78. doi: 10.1111/j.1365-2982.2009.01317.x
165. Nishizawa M, Nakabayashi H, Uchida K, Nakagawa A, Niijima A. The hepatic vagal nerve is receptive to incretin hormone glucagon-like peptide-1, but not to glucose-dependent insulinotropic polypeptide, in the portal vein. J Auton Nerv Syst. (1996) 61:149–54. doi: 10.1016/S0165-1838(96)00071-9
166. Drucker DJ, Buse JB, Taylor K, Kendall DM, Trautmann M, Zhuang D, et al. Exenatide once weekly versus twice daily for the treatment of type 2 diabetes: a randomised, open-label, non-inferiority study. Lancet. (2008) 372:1240–50. doi: 10.1016/S0140-6736(08)61206-4
167. Umapathysivam MM, Lee MY, Jones KL, Annink CE, Cousins CE, Trahair LG, et al. Comparative effects of prolonged and intermittent stimulation of the glucagon-like peptide 1 receptor on gastric emptying and glycemia. Diabetes. (2014) 63:785–90. doi: 10.2337/db13-0893
168. Bergmann NC, Gasbjerg LS, Heimbürger SM, Krogh LSL, Dela F, Hartmann B, et al. No acute effects of exogenous glucose-dependent insulinotropic polypeptide on energy intake, appetite, or energy expenditure when added to treatment with a long-acting glucagon-like peptide 1 receptor agonist in men with type 2 diabetes. Diabetes Care. (2020) 43:588–96. doi: 10.2337/dc19-0578
169. Borner T, De Jonghe BC, Hayes MR. The antiemetic actions of GIP receptor agonism. Am J Physiol Endocrinol Metab. (2024) 326:E528–E36. doi: 10.1152/ajpendo.00330.2023
170. Nauck MA, Meier JJ. Incretin hormones: Their role in health and disease. Diabetes Obes Metab. (2018) 20 Suppl 1:5–21.
171. Eckel RH, Fujimoto WY, Brunzell JD. Gastric inhibitory polypeptide enhanced lipoprotein lipase activity in cultured preadipocytes. Diabetes. (1979) 28:1141–2. doi: 10.2337/diab.28.12.1141
172. Ebert R, Nauck M, Creutzfeldt W. Effect of exogenous or endogenous gastric inhibitory polypeptide (GIP) on plasma triglyceride responses in rats. Horm Metab Res. (1991) 23:517–21. doi: 10.1055/s-2007-1003745
173. Thondam SK, Daousi C, Wilding JP, Holst JJ, Ameen GI, Yang C, et al. Glucose-dependent insulinotropic polypeptide promotes lipid deposition in subcutaneous adipocytes in obese type 2 diabetes patients: a maladaptive response. Am J Physiol Endocrinol Metab. (2017) 312:E224–E33. doi: 10.1152/ajpendo.00347.2016
174. Asmar M, Asmar A, Simonsen L, Gasbjerg LS, Sparre-Ulrich AH, Rosenkilde MM, et al. The gluco- and liporegulatory and vasodilatory effects of glucose-dependent insulinotropic polypeptide (GIP) are abolished by an antagonist of the human GIP receptor. Diabetes. (2017) 66:2363–71. doi: 10.2337/db17-0480
175. Asmar M, Simonsen L, Madsbad S, Stallknecht B, Holst JJ, Bülow J. Glucose-dependent insulinotropic polypeptide may enhance fatty acid re-esterification in subcutaneous abdominal adipose tissue in lean humans. Diabetes. (2010) 59:2160–3. doi: 10.2337/db10-0098
176. Starich GH, Bar RS, Mazzaferri EL. GIP increases insulin receptor affinity and cellular sensitivity in adipocytes. Am J Physiol. (1985) 249:E603–7. doi: 10.1152/ajpendo.1985.249.6.E603
177. Hauner H, Glatting G, Kaminska D, Pfeiffer EF. Effects of gastric inhibitory polypeptide on glucose and lipid metabolism of isolated rat adipocytes. Ann Nutr Metab. (1988) 32. doi: 10.1159/000177467
178. Samms RJ, Christe ME, Collins KA, Pirro V, Droz BA, Holland AK, et al. GIPR agonism mediates weight-independent insulin sensitization by tirzepatide in obese mice. J Clin Invest. (2021) 131. doi: 10.1172/JCI146353
179. Samms RJ, Coghlan MP, Sloop KW. How may GIP enhance the therapeutic efficacy of GLP-1? Trends Endocrinol Metab. (2020) 31:410–21.
180. Wilson JM, Nikooienejad A, Robins DA, Roell WC, Riesmeyer JS, Haupt A, et al. The dual glucose-dependent insulinotropic peptide and glucagon-like peptide-1 receptor agonist, tirzepatide, improves lipoprotein biomarkers associated with insulin resistance and cardiovascular risk in patients with type 2 diabetes. Diabetes Obes Metab. (2020) 22:2451–9. doi: 10.1111/dom.14174
181. Nogueiras R, Pérez-Tilve D, Veyrat-Durebex C, Morgan DA, Varela L, Haynes WG, et al. Direct control of peripheral lipid deposition by CNS GLP-1 receptor signaling is mediated by the sympathetic nervous system and blunted in diet-induced obesity. J Neurosci. (2009) 29:5916–25. doi: 10.1523/JNEUROSCI.5977-08.2009
182. Merchenthaler I, Lane M, Shughrue P. Distribution of pre-pro-glucagon and glucagon-like peptide-1 receptor messenger RNAs in the rat central nervous system. J Comp Neurol. (1999) 403:261–80. doi: 10.1002/(ISSN)1096-9861
183. Ross CA, Ruggiero DA, Joh TH, Park DH, Reis DJ. Rostral ventrolateral medulla: selective projections to the thoracic autonomic cell column from the region containing C1 adrenaline neurons. J Comp Neurol. (1984) 228:168–85. doi: 10.1002/cne.902280204
184. Gebhard B, Holst JJ, Biegelmayer C, Miholic J. Postprandial GLP-1, norepinephrine, and reactive hypoglycemia in dumping syndrome. Dig Dis Sci. (2001) 46:1915–23. doi: 10.1023/A:1010635131228
185. Li X, Zhang D, Vatner DF, Goedeke L, Hirabara SM, Zhang Y, et al. Mechanisms by which adiponectin reverses high fat diet-induced insulin resistance in mice. Proc Natl Acad Sci U.S.A. (2020) 117:32584–93.
186. Varol C, Zvibel I, Spektor L, Mantelmacher FD, Vugman M, Thurm T, et al. Long-acting glucose-dependent insulinotropic polypeptide ameliorates obesity-induced adipose tissue inflammation. J Immunol. (2014) 193:4002–9. doi: 10.4049/jimmunol.1401149
187. Xiong X, Lu W, Qin X, Luo Q, Zhou W. Downregulation of the GLP-1/CREB/adiponectin pathway is partially responsible for diabetes-induced dysregulated vascular tone and VSMC dysfunction. BioMed Pharmacother. (2020) 127:110218. doi: 10.1016/j.biopha.2020.110218
188. Simental-Mendía LE, Sánchez-García A, Linden-Torres E, Simental-Mendía M. Impact of glucagon-like peptide-1 receptor agonists on adiponectin concentrations: A meta-analysis of randomized controlled trials. Br J Clin Pharmacol. (2021) 87:4140–9.
189. Thomas MK, Nikooienejad A, Bray R, Cui X, Wilson J, Duffin K, et al. Dual GIP and GLP-1 receptor agonist tirzepatide improves beta-cell function and insulin sensitivity in type 2 diabetes. J Clin Endocrinol Metab. (2021) 106:388–96. doi: 10.1210/clinem/dgaa863
190. Lee CJ, Mao H, Thieu VT, Landó LF, Thomas MK. Tirzepatide as monotherapy improved markers of beta-cell function and insulin sensitivity in type 2 diabetes (SURPASS-1). J Endocr Soc. (2023) 7:bvad056. doi: 10.1210/jendso/bvad056
191. Góralska J, Raźny U, Polus A, Dziewońska A, Gruca A, Zdzienicka A, et al. Enhanced GIP secretion in obesity is associated with biochemical alteration and miRNA contribution to the development of liver steatosis. Nutrients. (2020) 12. doi: 10.3390/nu12020476
192. Pyke C, Heller RS, Kirk RK, Ørskov C, Reedtz-Runge S, Kaastrup P, et al. GLP-1 receptor localization in monkey and human tissue: novel distribution revealed with extensively validated monoclonal antibody. Endocrinology. (2014) 155:1280–90. doi: 10.1210/en.2013-1934
193. Panjwani N, Mulvihill EE, Longuet C, Yusta B, Campbell JE, Brown TJ, et al. GLP-1 receptor activation indirectly reduces hepatic lipid accumulation but does not attenuate development of atherosclerosis in diabetic male ApoE(-/-) mice. Endocrinology. (2013) 154:127–39. doi: 10.1210/en.2012-1937
194. Rakipovski G, Rolin B, Nøhr J, Klewe I, Frederiksen KS, Augustin R, et al. The GLP-1 analogs liraglutide and semaglutide reduce atherosclerosis in apoE. JACC Basic Transl Sci. (2018) 3:844–57. doi: 10.1016/j.jacbts.2018.09.004
195. Ben-Shlomo S, Zvibel I, Shnell M, Shlomai A, Chepurko E, Halpern Z, et al. Glucagon-like peptide-1 reduces hepatic lipogenesis via activation of AMP-activated protein kinase. J Hepatol. (2011) 54:1214–23. doi: 10.1016/j.jhep.2010.09.032
196. Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, et al. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab. (2001) 86:1930–5. doi: 10.1210/jcem.86.5.7463
197. Sposito AC, Berwanger O, de Carvalho LSF, Saraiva JFK. GLP-1RAs in type 2 diabetes: mechanisms that underlie cardiovascular effects and overview of cardiovascular outcome data. Cardiovasc Diabetol. (2018) 17:157. doi: 10.1186/s12933-018-0800-2
198. Meier JJ, Gethmann A, Götze O, Gallwitz B, Holst JJ, Schmidt WE, et al. Glucagon-like peptide 1 abolishes the postprandial rise in triglyceride concentrations and lowers levels of non-esterified fatty acids in humans. Diabetologia. (2006) 49:452–8. doi: 10.1007/s00125-005-0126-y
199. Lincoff AM, Brown-Frandsen K, Colhoun HM, Deanfield J, Emerson SS, Esbjerg S, et al. Semaglutide and cardiovascular outcomes in obesity without diabetes. N Engl J Med. (2023) 389:2221–32. doi: 10.1056/NEJMoa2307563
200. Jastreboff AM, Aronne LJ, Ahmad NN, Wharton S, Connery L, Alves B, et al. Tirzepatide once weekly for the treatment of obesity. N Engl J Med. (2022) 387:205–16. doi: 10.1056/NEJMoa2206038
201. Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, et al. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. (2016) 387:679–90. doi: 10.1016/S0140-6736(15)00803-X
202. Newsome PN, Buchholtz K, Cusi K, Linder M, Okanoue T, Ratziu V, et al. A placebo-controlled trial of subcutaneous semaglutide in nonalcoholic steatohepatitis. N Engl J Med. (2021) 384:1113–24. doi: 10.1056/NEJMoa2028395
203. Loomba R, Hartman ML, Lawitz EJ, Vuppalanchi R, Boursier J, Bugianesi E, et al. Tirzepatide for metabolic dysfunction-associated steatohepatitis with liver fibrosis. N Engl J Med. (2024). doi: 10.1056/NEJMoa2401943
204. Skov J, Dejgaard A, Frøkiær J, Holst JJ, Jonassen T, Rittig S, et al. Glucagon-like peptide-1 (GLP-1): effect on kidney hemodynamics and renin-angiotensin-aldosterone system in healthy men. J Clin Endocrinol Metab. (2013) 98:E664–71. doi: 10.1210/jc.2012-3855
205. Kim M, Platt MJ, Shibasaki T, Quaggin SE, Backx PH, Seino S, et al. GLP-1 receptor activation and Epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nat Med. (2013) 19:567–75. doi: 10.1038/nm.3128
206. Smits MM, Ten Kulve JS, van Bloemendaal L, Tonneijck L, Muskiet MHA, Kramer MHH, et al. GLP-1 receptor agonists do not affect sodium intake: Exploratory analyses from two randomized clinical trials. Nutrition. (2019) 67-68:110524. doi: 10.1016/j.nut.2019.06.005
207. Gutzwiller JP, Tschopp S, Bock A, Zehnder CE, Huber AR, Kreyenbuehl M, et al. Glucagon-like peptide 1 induces natriuresis in healthy subjects and in insulin-resistant obese men. J Clin Endocrinol Metab. (2004) 89:3055–61. doi: 10.1210/jc.2003-031403
208. Lovshin JA, Barnie A, DeAlmeida A, Logan A, Zinman B, Drucker DJ. Liraglutide promotes natriuresis but does not increase circulating levels of atrial natriuretic peptide in hypertensive subjects with type 2 diabetes. Diabetes Care. (2015) 38:132–9. doi: 10.2337/dc14-1958
209. Gallwitz B, Vaag A, Falahati A, Madsbad S. Adding liraglutide to oral antidiabetic drug therapy: onset of treatment effects over time. Int J Clin Pract. (2010) 64:267–76. doi: 10.1111/j.1742-1241.2009.02265.x
210. Yang W, Chen L, Ji Q, Liu X, Ma J, Tandon N, et al. Liraglutide provides similar glycaemic control as glimepiride (both in combination with metformin) and reduces body weight and systolic blood pressure in Asian population with type 2 diabetes from China, South Korea and India: a 16-week, randomized, double-blind, active control trial(*). Diabetes Obes Metab. (2011) 13:81–8. doi: 10.1111/dom.2011.13.issue-1
211. de Lemos JA, Linetzky B, le Roux CW, Laffin LJ, Vongpatanasin W, Fan L, et al. Tirzepatide reduces 24-hour ambulatory blood pressure in adults with body mass index ≥27 kg/m. Hypertension. (2024).
212. Buse JB, Henry RR, Han J, Kim DD, Fineman MS, Baron AD, et al. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care. (2004) 27:2628–35. doi: 10.2337/diacare.27.11.2628
213. DeFronzo RA, Ratner RE, Han J, Kim DD, Fineman MS, Baron AD. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care. (2005) 28:1092–100. doi: 10.2337/diacare.28.5.1092
214. Guja C, Frías JP, Somogyi A, Jabbour S, Wang H, Hardy E, et al. Effect of exenatide QW or placebo, both added to titrated insulin glargine, in uncontrolled type 2 diabetes: The DURATION-7 randomized study. Diabetes Obes Metab. (2018) 20:1602–14. doi: 10.1111/dom.13266
215. Bolli GB, Munteanu M, Dotsenko S, Niemoeller E, Boka G, Wu Y, et al. Efficacy and safety of lixisenatide once daily vs. placebo in people with Type 2 diabetes insufficiently controlled on metformin (GetGoal-F1). Diabetes Med. (2014) 31:176–84. doi: 10.1111/dme.12328
216. Riddle MC, Aronson R, Home P, Marre M, Niemoeller E, Miossec P, et al. Adding once-daily lixisenatide for type 2 diabetes inadequately controlled by established basal insulin: a 24-week, randomized, placebo-controlled comparison (GetGoal-L). Diabetes Care. (2013) 36:2489–96. doi: 10.2337/dc12-2454
217. Reusch J, Stewart MW, Perkins CM, Cirkel DT, Ye J, Perry CR, et al. Efficacy and safety of once-weekly glucagon-like peptide 1 receptor agonist albiglutide (HARMONY 1 trial): 52-week primary endpoint results from a randomized, double-blind, placebo-controlled trial in patients with type 2 diabetes mellitus not controlled on pioglitazone, with or without metformin. Diabetes Obes Metab. (2014) 16:1257–64. doi: 10.1111/dom.12382
218. Wysham C, Blevins T, Arakaki R, Colon G, Garcia P, Atisso C, et al. Efficacy and safety of dulaglutide added onto pioglitazone and metformin versus exenatide in type 2 diabetes in a randomized controlled trial (AWARD-1). Diabetes Care. (2014) 37(8):2159–67. doi: 10.2337/dc13-2760
219. Marre M, Shaw J, Brändle M, Bebakar WM, Kamaruddin NA, Strand J, et al. Liraglutide, a once-daily human GLP-1 analogue, added to a sulphonylurea over 26 weeks produces greater improvements in glycaemic and weight control compared with adding rosiglitazone or placebo in subjects with Type 2 diabetes (LEAD-1 SU). Diabetes Med. (2009) 26:268–78. doi: 10.1111/j.1464-5491.2009.02666.x
220. Sorli C, Harashima SI, Tsoukas GM, Unger J, Karsbøl JD, Hansen T, et al. Efficacy and safety of once-weekly semaglutide monotherapy versus placebo in patients with type 2 diabetes (SUSTAIN 1): a double-blind, randomised, placebo-controlled, parallel-group, multinational, multicentre phase 3a trial. Lancet Diabetes Endocrinol. (2017) 5:251–60. doi: 10.1016/S2213-8587(17)30013-X
221. Aroda VR, Rosenstock J, Terauchi Y, Altuntas Y, Lalic NM, Morales Villegas EC, et al. PIONEER 1: randomized clinical trial of the efficacy and safety of oral semaglutide monotherapy in comparison with placebo in patients with type 2 diabetes. Diabetes Care. (2019) 42:1724–32. doi: 10.2337/dc19-0749
222. Rosenstock J, Wysham C, Frías JP, Kaneko S, Lee CJ, Fernández Landó L, et al. Efficacy and safety of a novel dual GIP and GLP-1 receptor agonist tirzepatide in patients with type 2 diabetes (SURPASS-1): a double-blind, randomised, phase 3 trial. Lancet. (2021) 398:143–55. doi: 10.1016/S0140-6736(21)01324-6
223. Cefalu WT, Kaul S, Gerstein HC, Holman RR, Zinman B, Skyler JS, et al. Cardiovascular outcomes trials in type 2 diabetes: where do we go from here? Reflections from a. Diabetes Care. (2018) 41:14–31. doi: 10.2337/dci17-0057
224. Heine RJ, Van Gaal LF, Johns D, Mihm MJ, Widel MH, Brodows RG, et al. Exenatide versus insulin glargine in patients with suboptimally controlled type 2 diabetes: a randomized trial. Ann Intern Med. (2005) 143:559–69. doi: 10.7326/0003-4819-143-8-200510180-00006
225. Henry RR, Buse JB, Sesti G, Davies MJ, Jensen KH, Brett J, et al. Efficacy of antihyperglycemic therapies and the influence of baseline hemoglobin A(1C): a meta-analysis of the liraglutide development program. Endocr Pract. (2011) 17:906–13. doi: 10.4158/EP.17.6.906
226. Ludvik B, Giorgino F, Jódar E, Frias JP, Fernández Landó L, Brown K, et al. Once-weekly tirzepatide versus once-daily insulin degludec as add-on to metformin with or without SGLT2 inhibitors in patients with type 2 diabetes (SURPASS-3): a randomised, open-label, parallel-group, phase 3 trial. Lancet. (2021) 398:583–98. doi: 10.1016/S0140-6736(21)01443-4
227. Committee ADAPP. 9. Pharmacologic approaches to glycemic treatment: standards of care in diabetes-2024. Diabetes Care. (2024) 47:S158–S78.
228. Hernandez AF, Green JB, Janmohamed S, D'Agostino RB, Granger CB, Jones NP, et al. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (Harmony Outcomes): a double-blind, randomised placebo-controlled trial. Lancet. (2018) 392:1519–29. doi: 10.1016/S0140-6736(18)32261-X
229. Garvey WT, Frias JP, Jastreboff AM, le Roux CW, Sattar N, Aizenberg D, et al. Tirzepatide once weekly for the treatment of obesity in people with type 2 diabetes (SURMOUNT-2): a double-blind, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet. (2023) 402:613–26. doi: 10.1016/S0140-6736(23)01200-X
230. Pratley RE, Nauck MA, Barnett AH, Feinglos MN, Ovalle F, Harman-Boehm I, et al. Once-weekly albiglutide versus once-daily liraglutide in patients with type 2 diabetes inadequately controlled on oral drugs (HARMONY 7): a randomised, open-label, multicentre, non-inferiority phase 3 study. Lancet Diabetes Endocrinol. (2014) 2:289–97. doi: 10.1016/S2213-8587(13)70214-6
231. Dungan KM, Povedano ST, Forst T, González JG, Atisso C, Sealls W, et al. Once-weekly dulaglutide versus once-daily liraglutide in metformin-treated patients with type 2 diabetes (AWARD-6): a randomised, open-label, phase 3, non-inferiority trial. Lancet. (2014) 384:1349–57. doi: 10.1016/S0140-6736(14)60976-4
232. Hannon TS, Arslanian SA. Obesity in adolescents. N Engl J Med. (2023) 389:251–61. doi: 10.1056/NEJMcp2102062
233. Davies M, Færch L, Jeppesen OK, Pakseresht A, Pedersen SD, Perreault L, et al. Semaglutide 2·4 mg once a week in adults with overweight or obesity, and type 2 diabetes (STEP 2): a randomised, double-blind, double-dummy, placebo-controlled, phase 3 trial. Lancet. (2021) 397:971–84. doi: 10.1016/S0140-6736(21)00213-0
234. Wilding JPH, Batterham RL, Calanna S, Davies M, Van Gaal LF, Lingvay I, et al. Once-weekly semaglutide in adults with overweight or obesity. N Engl J Med. (2021) 384:989–1002. doi: 10.1056/NEJMoa2032183
235. Dahlqvist S, Ahlén E, Filipsson K, Gustafsson T, Hirsch IB, Tuomilehto J, et al. Variables associated with HbA1c and weight reductions when adding liraglutide to multiple daily insulin injections in persons with type 2 diabetes (MDI Liraglutide trial 3). BMJ Open Diabetes Res Care. (2018) 6:e000464. doi: 10.1136/bmjdrc-2017-000464
236. Bonora E, Frias JP, Tinahones FJ, Van J, Malik RE, Yu Z, et al. Effect of dulaglutide 3.0 and 4.5 mg on weight in patients with type 2 diabetes: Exploratory analyses of AWARD-11. Diabetes Obes Metab. (2021) 23:2242–50. doi: 10.1111/dom.14465
237. ElSayed NA, Aleppo G, Aroda VR, Bannuru RR, Brown FM, Bruemmer D, et al. 8. Obesity and weight management for the prevention and treatment of type 2 diabetes: standards of care in diabetes-2023. Diabetes Care. (2023) 46:S128–S39.
238. Garvey WT. New horizons. A new paradigm for treating to target with second-generation obesity medications. J Clin Endocrinol Metab. (2022) 107:e1339–e47.
239. Astrup A, Carraro R, Finer N, Harper A, Kunesova M, Lean ME, et al. Safety, tolerability and sustained weight loss over 2 years with the once-daily human GLP-1 analog, liraglutide. Int J Obes (Lond). (2012) 36:843–54. doi: 10.1038/ijo.2011.158
240. Blundell J, Finlayson G, Axelsen M, Flint A, Gibbons C, Kvist T, et al. Effects of once-weekly semaglutide on appetite, energy intake, control of eating, food preference and body weight in subjects with obesity. Diabetes Obes Metab. (2017) 19:1242–51. doi: 10.1111/dom.12932
241. Wing RR, Bolin P, Brancati FL, Bray GA, Clark JM, Coday M, et al. Cardiovascular effects of intensive lifestyle intervention in type 2 diabetes. N Engl J Med. (2013) 369:145–54. doi: 10.1056/NEJMoa1212914
242. Pownall HJ, Schwartz AV, Bray GA, Berkowitz RI, Lewis CE, Boyko EJ, et al. Changes in regional body composition over 8 years in a randomized lifestyle trial: The look AHEAD study. Obes (Silver Spring). (2016) 24:1899–905. doi: 10.1002/oby.21577
243. Nunn E, Jaiswal N, Gavin M, Uehara K, Stefkovich M, Drareni K, et al. Antibody blockade of activin type II receptors preserves skeletal muscle mass and enhances fat loss during GLP-1 receptor agonism. Mol Metab. (2024) 80:101880. doi: 10.1016/j.molmet.2024.101880
244. Re Cecconi AD, Barone M, Forti M, Lunardi M, Cagnotto A, Salmona M, et al. Apelin resistance contributes to muscle loss during cancer cachexia in mice. Cancers (Basel). (2022) 14. doi: 10.3390/cancers14071814
245. Kelly AS, Hale PM. Liraglutide for adolescents with obesity. Reply N Engl J Med. (2020) 383:1193–4.
246. Wharton S, Calanna S, Davies M, Dicker D, Goldman B, Lingvay I, et al. Gastrointestinal tolerability of once-weekly semaglutide 2.4 mg in adults with overweight or obesity, and the relationship between gastrointestinal adverse events and weight loss. Diabetes Obes Metab. (2022) 24:94–105. doi: 10.1111/dom.14551
247. Arnett DK, Blumenthal RS, Albert MA, Buroker AB, Goldberger ZD, Hahn EJ, et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: executive summary: A report of the american college of cardiology/american heart association task force on clinical practice guidelines. J Am Coll Cardiol. (2019) 74:1376–414.
248. Committee ADAPP. 10. Cardiovascular disease and risk management: standards of care in diabetes-2024. Diabetes Care. (2024) 47:S179–218.
249. Draznin B, Aroda VR, Bakris G, Benson G, Brown FM, Freeman R, et al. 9. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes-2022. Diabetes Care. (2022) 45:S125–S43.
250. Pfeffer MA, Claggett B, Diaz R, Dickstein K, Gerstein HC, Køber LV, et al. Lixisenatide in patients with type 2 diabetes and acute coronary syndrome. N Engl J Med. (2015) 373:2247–57. doi: 10.1056/NEJMoa1509225
251. Holman RR, Bethel MA, Mentz RJ, Thompson VP, Lokhnygina Y, Buse JB, et al. Effects of once-weekly exenatide on cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2017) 377:1228–39. doi: 10.1056/NEJMoa1612917
252. Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2016) 375:311–22. doi: 10.1056/NEJMoa1603827
253. Marso SP, Bain SC, Consoli A, Eliaschewitz FG, Jódar E, Leiter LA, et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. (2016) 375:1834–44. doi: 10.1056/NEJMoa1607141
254. Gerstein HC, Colhoun HM, Dagenais GR, Diaz R, Lakshmanan M, Pais P, et al. Dulaglutide and cardiovascular outcomes in type 2 diabetes (REWIND): a double-blind, randomised placebo-controlled trial. Lancet. (2019) 394:121–30. doi: 10.1016/S0140-6736(19)31149-3
255. Husain M, Birkenfeld AL, Donsmark M, Dungan K, Eliaschewitz FG, Franco DR, et al. Oral semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N Engl J Med. (2019) 381:841–51. doi: 10.1056/NEJMoa1901118
256. Perkovic V, Tuttle KR, Rossing P, Mahaffey KW, Mann JFE, Bakris G, et al. Effects of semaglutide on chronic kidney disease in patients with type 2 diabetes. N Engl J Med. (2024) 391(2):109–21. doi: 10.1056/NEJMoa2403347
257. Wilcox T, De Block C, Schwartzbard AZ, Newman JD. Diabetic agents, from metformin to SGLT2 inhibitors and GLP1 receptor agonists: JACC focus seminar. J Am Coll Cardiol. (2020) 75:1956–74. doi: 10.1016/j.jacc.2020.02.056
258. Kernan WN, Viscoli CM, Furie KL, Young LH, Inzucchi SE, Gorman M, et al. Pioglitazone after ischemic stroke or transient ischemic attack. N Engl J Med. (2016) 374:1321–31. doi: 10.1056/NEJMoa1506930
259. Doehner W, Erdmann E, Cairns R, Clark AL, Dormandy JA, Ferrannini E, et al. Inverse relation of body weight and weight change with mortality and morbidity in patients with type 2 diabetes and cardiovascular co-morbidity: an analysis of the PROactive study population. Int J Cardiol. (2012) 162:20–6. doi: 10.1016/j.ijcard.2011.09.039
260. McGuire DK, Shih WJ, Cosentino F, Charbonnel B, Cherney DZI, Dagogo-Jack S, et al. Association of SGLT2 inhibitors with cardiovascular and kidney outcomes in patients with type 2 diabetes: A meta-analysis. JAMA Cardiol. (2021) 6:148–58. doi: 10.1001/jamacardio.2020.4511
261. Wyne KL, Litwin SE, Cusi K, Guyton JR. JCL roundtable. Obesity, diabetes, and liver disease in relation to cardiovascular risk. J Clin Lipidol. (2022) 16:115–27. doi: 10.1016/j.jacl.2022.03.007
262. Del Prato S, Kahn SE, Pavo I, Weerakkody GJ, Yang Z, Doupis J, et al. Tirzepatide versus insulin glargine in type 2 diabetes and increased cardiovascular risk (SURPASS-4): a randomised, open-label, parallel-group, multicentre, phase 3 trial. Lancet. (2021) 398:1811–24. doi: 10.1016/S0140-6736(21)02188-7
263. Gerstein HC, Bosch J, Dagenais GR, Díaz R, Jung H, Maggioni AP, et al. Basal insulin and cardiovascular and other outcomes in dysglycemia. N Engl J Med. (2012) 367:319–28. doi: 10.1056/NEJMoa1203858
264. Sattar N, McGuire DK, Pavo I, Weerakkody GJ, Nishiyama H, Wiese RJ, et al. Tirzepatide cardiovascular event risk assessment: a pre-specified meta-analysis. Nat Med. (2022) 28:591–8. doi: 10.1038/s41591-022-01707-4
265. Khera A, Powell-Wiley TM. SELECTing treatments for cardiovascular disease - obesity in the spotlight. N Engl J Med. (2023) 389:2287–8. doi: 10.1056/NEJMe2312646
266. Lewington S, Clarke R, Qizilbash N, Peto R, Collins R, Collaboration PS. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet. (2002) 360:1903–13. doi: 10.1016/S0140-6736(02)11911-8
267. Liu QK. Triglyceride-lowering and anti-inflammatory mechanisms of omega-3 polyunsaturated fatty acids for atherosclerotic cardiovascular risk reduction. J Clin Lipidol. (2021) 15:556–68. doi: 10.1016/j.jacl.2021.05.007
268. Wong CK, McLean BA, Baggio LL, Koehler JA, Hammoud R, Rittig N, et al. Central glucagon-like peptide 1 receptor activation inhibits Toll-like receptor agonist-induced inflammation. Cell Metab. (2024) 36:130–43.e5. doi: 10.1016/j.cmet.2023.11.009
269. McLean BA, Wong CK, Kaur KD, Seeley RJ, Drucker DJ. Differential importance of endothelial and hematopoietic cell GLP-1Rs for cardiometabolic versus hepatic actions of semaglutide. JCI Insight. (2021) 6. doi: 10.1172/jci.insight.153732
Keywords: GIP, GLP-1, GLP-1RA, tirzepatide, diabetes, obesity, cardiovascular disease
Citation: Liu QK (2024) Mechanisms of action and therapeutic applications of GLP-1 and dual GIP/GLP-1 receptor agonists. Front. Endocrinol. 15:1431292. doi: 10.3389/fendo.2024.1431292
Received: 11 May 2024; Accepted: 08 July 2024;
Published: 24 July 2024.
Edited by:
Hisamitsu Ishihara, Nihon University, JapanReviewed by:
Alessandro Pocai, Janssen Research and Development, United StatesHiroshi Nomoto, Hokkaido University Hospital, Japan
Copyright © 2024 Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qiyuan Keith Liu, UWl5dWFuLmxpdUBtZWRzdGFyLm5ldA==; bWVkcDgwNjRAaG90bWFpbC5jb20=