
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol. , 25 September 2024
Sec. Diabetes: Molecular Mechanisms
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1427413
This article is part of the Research Topic Advances in β-cell Development & Regeneration View all 5 articles
 Moustapha Ka1,2,3,4,5,6Eleanor Hawkins1,2,3,4,5,6Celio Pouponnot1,2,3,4,5,6
Moustapha Ka1,2,3,4,5,6Eleanor Hawkins1,2,3,4,5,6Celio Pouponnot1,2,3,4,5,6 Bertrand Duvillié1,2,3,4,5,6*
Bertrand Duvillié1,2,3,4,5,6*Diabetes is a complex metabolic disease which most commonly has a polygenic origin; however, in rare cases, diabetes may be monogenic. This is indeed the case in both Maturity Onset Diabetes of the Young (MODY) and neonatal diabetes. These disease subtypes are believed to be simpler than Type 1 (T1D) and Type 2 Diabetes (T2D), which allows for more precise modelling. During the three last decades, many studies have focused on rodent models. These investigations provided a wealth of knowledge on both pancreas development and beta cell function. In particular, they allowed the establishment of a hierarchy of the transcription factors and highlighted the role of microenvironmental factors in the control of progenitor cell proliferation and differentiation. Transgenic mice also offered the possibility to decipher the mechanisms that define the functional identity of the pancreatic beta cells. Despite such interest in transgenic mice, recent data have also indicated that important differences exist between mice and human. To overcome these limitations, new human models are necessary. In the present review, we describe these ex vivo models, which are created using stem cells and organoids, and represent an important step toward islet cell therapy and drug discovery.
Diabetes is a complex metabolic disease characterized by chronic hyperglycemia and categorized into two types: insulino-dependent (Type 1, T1D) and non-insulino-dependent diabetes (Type 2, T2D). T1D has a well-established auto-immune origin, while T2D is associated with insulin-resistance and overweight status. However, in the current context, this classification appears to be oversimplified due to the overlap between symptoms, and a sub-classification may be required. In 2018, Ahlqvist et al. performed a data-driven cluster analysis based on 6 parameters: glutamate decarboxylase antibodies, age at diagnosis, body mass index (BMI), Hba1c, beta-cell function, and insulin-resistance. This study identified 5 clusters of patients with diabetes, with distinct characteristics which may be associated with potential complications (1). T1D and T2D have a polygenic origin, making identification of the etiology of onset and progression of diabetes complicated. Conversely, some specific forms of diabetes, including maturity onset diabetes of the young (MODY) and neonatal diabetes, are monogenic. Analysis of these particular diseases may help to improve our understanding of the molecular events underlying pancreatic beta-cell dysfunction and the initiation of diabetes. Moreover, it should be noted that in recent decades, most studies in this field were performed using rodent models. One important advantage of rodent models is the ease of genetic manipulation, which allows the performance of metabolic analyses at the whole organism level. However, recent research has highlighted several important differences between mice and human. One example is the inter-species difference in the two transcription factors of the GATA family, GATA 4 and GATA 6. In humans, heterozygous mutations in GATA 6 lead to pancreas agenesis and neonatal diabetes (2, 3), while GATA 4 haploinsufficiency causes neonatal and childhood-onset diabetes (4, 5). Conversely, in mice, single mutations of GATA 4 or GATA 6 have no impact on pancreas development or glucose homeostasis (6, 7); however, co-existing mutations on three of the four alleles of GATA4/6 produce a phenotype similar to that observed in humans. These observations indicate a functional redundancy between GATA4 and GATA6 in mice that does not exist in human. Thus, such inter-species differences need to be considered not only for delineating the human-specific molecular pathways that contribute to the disease, but also when choosing the most appropriate treatment for patients. Recently, significant progress has been made in the generation of new ex vivo models to study human diabetes. These models aim to reproduce the physiological development of beta cells, their interaction with the microenvironment, and their biology. In the present review, we will describe how these innovative approaches can be used in research to help better understand and treat monogenic forms of diabetes.
Monogenic diabetes, also known as maturity onset diabetes of the young (MODY), is a clinically heterogenous disease characterized by nonketotic diabetes mellitus and defects in pancreatic beta cell function, with an autosomal dominant inheritance pattern. MODY generally develops before the age of 25 years, and is frequent during childhood and adolescence. MODY represents 3-5% of all cases of diabetes. Interestingly, some MODY genes have been also associated with T1D and T2D (8–11), indicating an overlap between the different types of diabetes. Thus far, causative mutations in at least 14 genes have been characterized. However, the fact that causative mutations remain unidentified in 15-20% of families with MODY (French AJD Association, “Aide aux Jeunes Diabétiques”) indicates that other MODY associated genes remains to be discovered. The known causative genes of MODY (Figure 1) include hepatocyte nuclear factor (HNF) 4a [MODY 1 (12)], the glycolytic enzyme glucokinase [GK, MODY2 (13, 14)], HNF1a [MODY3 (15)], insulin promoting factor 1 [IPF1, MODY 4 (16)], HNF1b [MODY 5 (17)], neurogenic differentiation factor 1 also named BETA2 [MODY 6 (18)], KLF transcription factor 11 [KLF11, MODY 7 (19, 20)], carboxyl ester lipase [CEL, MODY 8 (21)], the transcription factor Paired box 4 [PAX4, MODY9 (22)], Insulin [MODY10 (23)], the tyrosine protein kinase BLK [MODY11 (24)], the ATP binding cassette subfamily C member 8 ABCC8 [MODY 12 (25)], the Potassium Inwardly Rectifying Channel Subfamily J member 11 KCNJ11, which encodes for the Kir6.2 subunit of the ATP-sensitive potassium channel in the pancreatic beta cell [MODY 13 (26)], the Adaptor Protein Phosphotyrosine interacting with PH domain and Leucin Zipper 1 APPL1 [MODY14 (27)]. Further research has shown that neonatal diabetes and syndrome-associated diabetes are also caused by single gene mutations (28). These forms are commonly underdiagnosed, and require better characterization. A recently-discovered new candidate gene for MODY is v-Maf avian musculoaponeurotic fibrosarcoma oncogene homolog A (MAF-A). MAF-A is a transcription factor that controls glucose stimulated insulin gene expression (GSIS) and insulin secretion (29). Prior research using MafA knock-out mice has revealed alterations in GSIS and disruption of the architecture of the pancreatic islets (30). Recently, two different missense mutations in human MAFA, at p.Ser64Phe and p.Thr57Arg, were detected in three unrelated families (31, 32). Intriguingly, both mutations caused both insulinomatosis (predominantly in females) and a MODY-like diabetes mellitus (predominantly in males). Iacovazzo et al. also showed that the p.Ser64Phe mutation impaired the phosphorylation of MAF-A by GSK3 resulting in an enhanced transactivation activity and increased MAF-A protein stability according to our previous work (33). It is probable that the p.Thr57Arg mutation will have a similar effect, as this mutation affects one of the residues phosphorylated by GSK3 (34, 35).
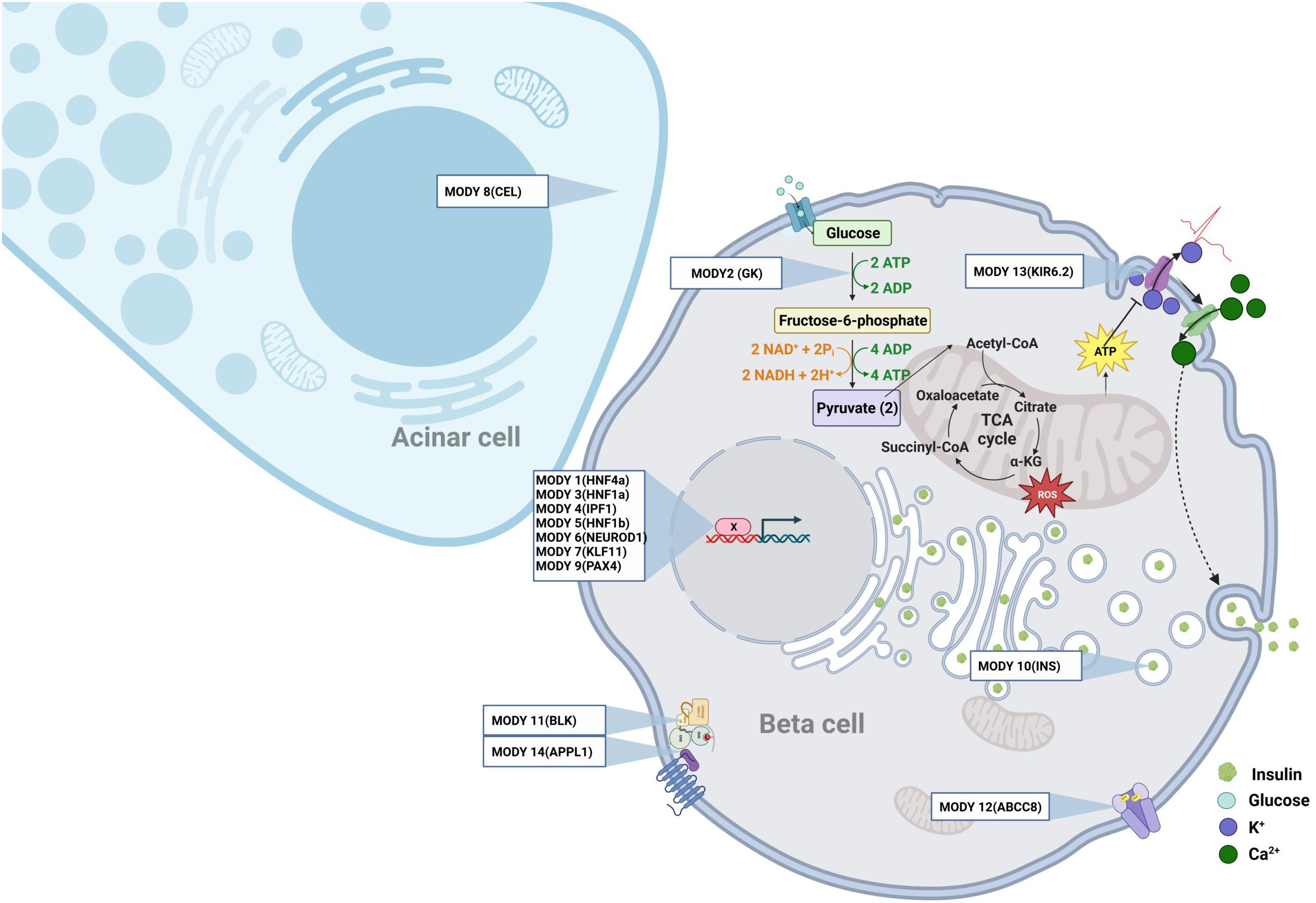
Figure 1. The monogenic diabetes. Representation at the cellular level of the different genes involved in Mody.
Recently, several new models have been developed to study MODY diabetes (Table 1). In particular, a variety of models can be used to study the biology of human beta cells, among which immortalized cells are commonly used. Several such cell lines have been produced and well characterized. For example, Blanchi et al. produced EndoC-bH5 human beta cells that show robust and highly-reproducible insulin secretion in response to glucose stimulation (36). These cells were initially produced by the integrative gene transfer of the immortalization genes hTERT and large T antigen to amplify the cells. Next, the transgenes were removed using a Cre recombinase system to facilitate physiological studies. However, this model cannot be used to investigate the process of differentiation. Adult pancreatic islets from cadaveric donors also represent a useful model, but these are scarce, difficult to genetically manipulate, and cannot be used to model beta cell development.
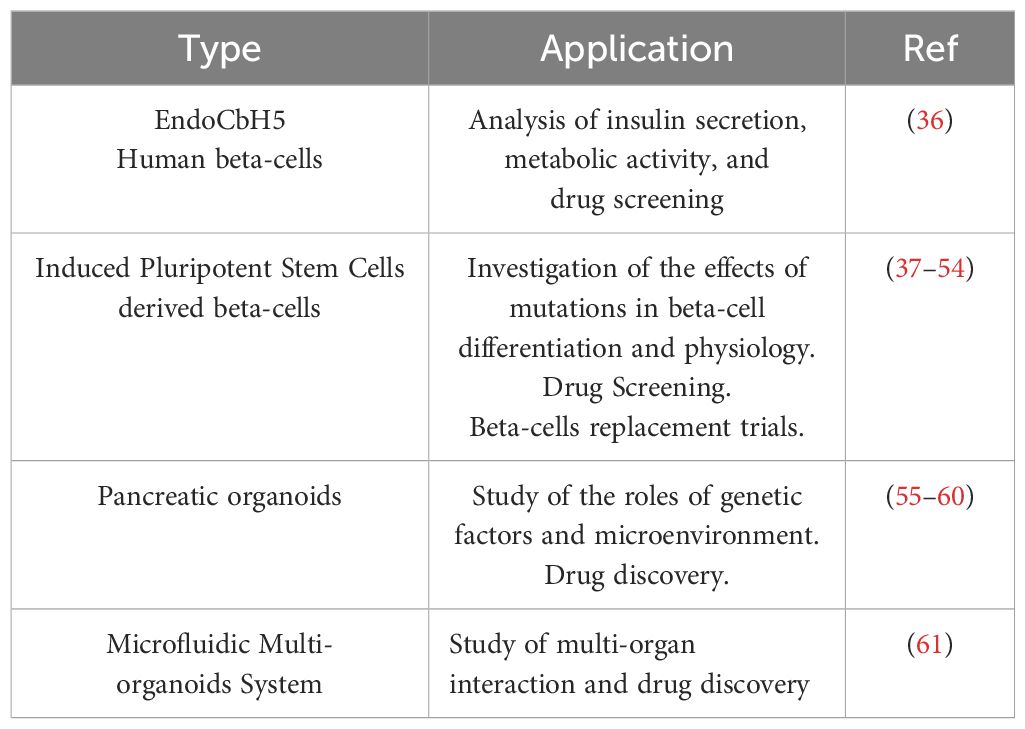
Table 1. The new human models for the study of MODY Diabetes.
More recently, the landscape of diabetes models has significantly progressed thanks to the introduction of human pluripotent stem cells (hPSC), including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). By studying the signals that govern the multiple steps of beta cell development in vivo, several protocols to generate insulin-secreting cells from iPS cells have been constantly improved. The first trials using hPSCs succeeded in generating pancreatic endoderms (62). Subsequently, pancreatic progenitor cells were obtained in vitro; however, transplantation into recipient mice was required to achieve complete endocrine differentiation (63). Finally, islet beta-like cells were produced from hPSCs in vitro, and were subsequently validated to reverse diabetes in mice (64, 65). Discoveries such as these have greatly increased interest in the field of cell therapy for diabetes. More recently, a number of studies have optimized the efficiency of terminal differentiation of beta cells, allowing more accurate investigation of their physiology (37–39). At present, research using stem cell islet technology has revealed several key features of human pancreas development and diabetes. Further, research combining disease modeling with gene editing and next generation sequencing has revealed the effects of mutations related to diabetes on multiple islet cell types (40) (see the modelization of monogenic diabetes using iPS cells in Table 2).
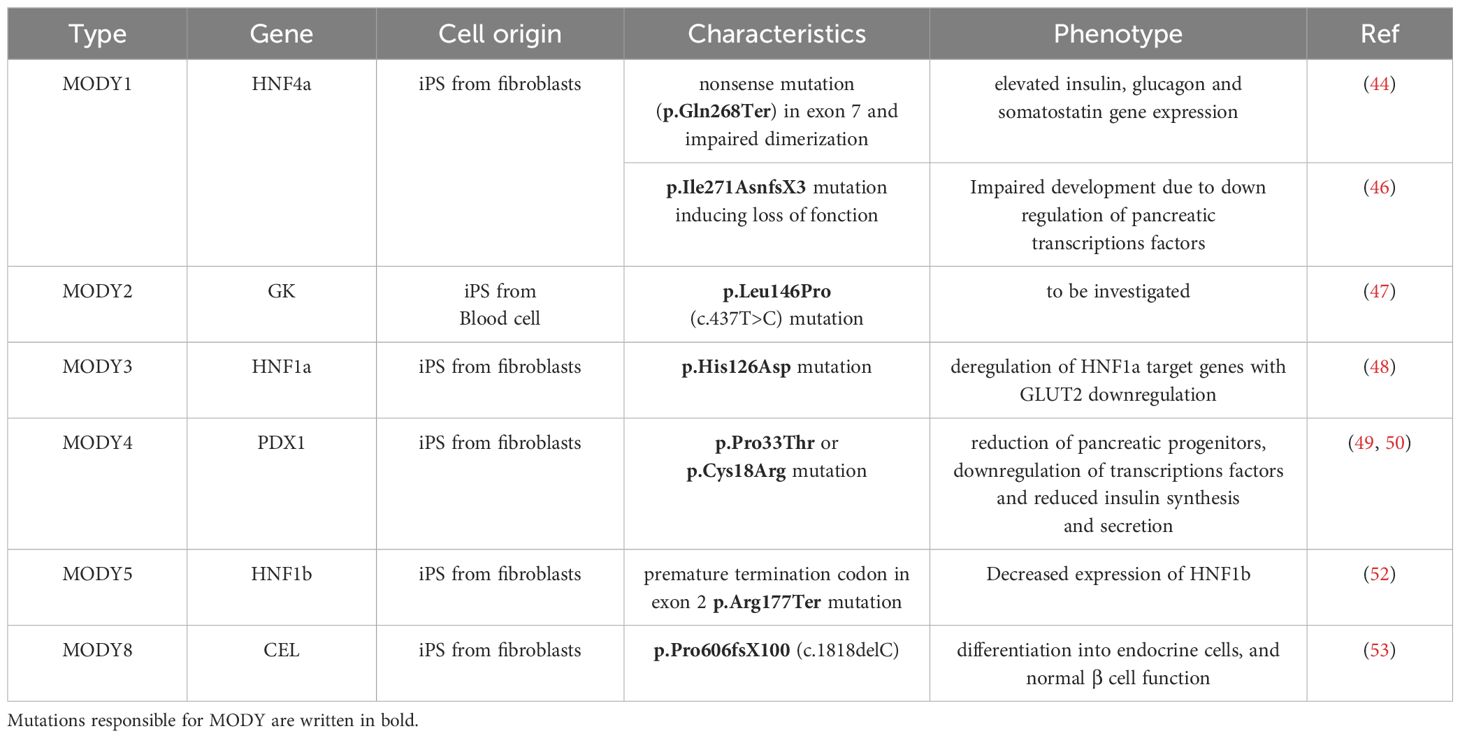
Table 2. Modelisation of monogenic diabetes using iPS cells.
MODY1 patients with HNF4α mutations have been shown to exhibit some alterations in beta cell function, but with normal insulin sensitivity (41, 42), concurrent with impairments in the secretion of glucagon and pancreatic polypeptide (41, 43). Treatments for MODY1 generally aim to increase insulin levels, while hypoglycemic drugs such as sulfonylureas can also be used (44). To establish a MODY 1 model, Braverman-Gross et al. used fibroblasts from two patients harboring a nonsense mutation in exon 7 of HNF4a (p.Gln268Ter) (44). This mutation impairs the dimerization of HNF4a and its transactivation domain. The authors subsequently analyzed the genetic signature of the pancreatic progenitors following induction of differentiation, revealing an increase in expression of stage specific transcription factors, as well as elevated expression of the insulin, glucagon, and somatostatin genes in MODY1 cells. This observation reflects the features of hyperinsulinemia observed in neonatal MODY1 patients (45), and may also correspond to a compensatory mechanism that favors pancreatic development in the presence of HNF4a heterozygosity. The differentiation of the primitive gut tube was also analyzed, revealing an enrichment of clusters for apolipoproteins, triglyceride catabolic process, lipoprotein metabolic process, lipid metabolic process, hormone biosynthesis and secretion in MODY 1 cells. This observation could be related to the dyslipidemia observed in MODY 1 patients. Moreover, NG et al. generated hepatopancreatic forgut endoderm cells, in addition to hepatic and beta-like cells, using hiPS cells carrying the p.Ile271AnsfsX3 mutation extracted from a MODY1 family (46). These authors showed that HNF4a haploinsufficiency alters foregut development, as well as both hepatic and pancreatic cell fates. Hepatic and beta-cell gene signatures were also impaired. This study therefore indicates that, in this model, foregut abnormalities further extend to the liver and the pancreas. For MODY2, a human iPS cell line, QBRli010-A, with a mutation in the GCK gene (pLeu146Pro, c.437T>C) was generated (47). This iPS cell line displays pluripotency characteristics and is able to produce the three germ layers. However, further studies will be necessary to generate beta cells and model diabetes in relation to MODY2.
Another study investigated MODY3, caused by mutation in HNF1a. Indeed, this disease is characterized by an alteration in insulin secretion; however, the specific molecular mechanisms in humans remain unclear. Su Jun Low et al. derived iPS cells carrying a p.His126Asp mutation in HNF1a from a MODY3 patient. Genome wide RNASeq and Chip Seq analysis on hiPS-derived endocrine progenitors showed that many HNF1a target genes were deregulated. Importantly, they also found a strong decrease in the expression of the glucose transporter, GLUT2, resulting in reduced glucose uptake and ATP production in MODY 3 hIPS derived beta-cells. Thus, these data demonstrate the role of HNF1a in the regulation of GLUT2 as well as several genes that regulate insulin secretion (48).
Pancreatic agenesis is caused by a homozygous mutation in the homeobox gene PDX1 (IPF1), while heterozygous mutations lead to MODY4 or T2D. Two iPS cell lines have previously been generated by episomal reprogramming of cells extracted from patients with missense coding mutations in the PDX1 gene. The first patient was a woman with a p.Cys18Arg mutation in PDX1 (49), and the second was a woman carrying a p.Pro33Thr mutation in the transactivation domain of PDX1 (50). These cell lines represent useful tools to delineate the molecular events that precede MODY4 (51). Isogenic cell lines carrying homozygous PDX1p.Cys18Arg/p.Cys18Arg and PDX1p.Pro33Thr/p.Pro33Thr mutations were also generated. Interestingly, the heterozygous PDX1p.Pro33Thr/+, PDX1p.Cys18Arg/+, and homozygous PDX1p.Pro33Thr/p.Pro33Thr and PDX1p.Cys18Arg/p.Cys18Arg mutations were found to alter beta-cell differentiation and function, while the PDX1p.Pro33Thr/p.Pro33Thr mutation also reduced the differentiation of pancreatic progenitors. This event is caused by the down-regulation of PDX1-bound genes. Together, these results demonstrate that all these mutations affect the endocrine lineages and participate in the development of diabetes.
Yabe et al. also analyzed MODY 5 using iPS cells from a Japanese patient to generate pancreatic beta cells (52). The iPS derived beta cells carried a MODY 5 mutation p.Arg177Ter, leading to a premature termination codon in exon 2 of HNF1b. The authors showed that the p.Arg177Ter mutant transcripts showed decreased expression compared to the wild type transcripts. They thus hypothesized that the mutant mRNA may be degraded by the nonsense-mediated decay pathway (NMD). Using cycloheximide to inhibit NMD, treatment increased the sequence signal of p.Arg177Ter mutant mRNA as compared to the controls, thus confirming their hypothesis.
More recently, Pelligrini et al. found a heterozygous pathogenic variant (p.Pro606fsX100, c.1818delC) in the CEL gene encoding carboxyl ester lipase (MODY8) (53). CEL is expressed in pancreatic acinar cells, and encodes a lipase secreted in the pancreatic juice. MODY8 is a rare disease leading to pancreatic exocrine dysfunction that precedes beta cell alterations (54). Pelligrini et al. derived three iPS clones from the patient’s skin fibroblasts, and used them to generate beta cells by following the developmental stages. These beta cells were found to show normal insulin secretion in response to glucose. Thus, this study appears to be useful not only for in vitro modelling of the disease, but also for beta cell replacement studies.
Organoids are 3D in vitro culture systems generated from stem or progenitor cells (55) which can mimic the function of some organs, including the pancreas. Organoid models can be used to study the roles of genetic factors, as well as the microenvironment in T1 and T2D (56). For example, some organoids can recapitulate organ development, thereby allowing evaluation of the impact of developmental defects. However, one limitation of such analyses is that they often represent a developmental stage rather than a mature organ (57). More recently, the use of organoids to study MODY3 has gained interest. Truncation of HNF1a (p.Pro291ProfsX25) is the most common mutation associated with MODY3 (58); however, while impaired HNF1a signaling is known to play a role in its development, the exact molecular mechanism remained unidentified. To explore this question, Cujba et al. generated CRISPR/CAS9 engineered HNF1ap.Pro291ProfsX25 cells from hiPS, which they used to generate 3D organoids. Using this model, they found a reduction of the number of progenitors as well as reduced beta-cell differentiation. At the molecular level, HNF1ap.Pro291ProfsX25 interacts with HNF1b and inhibits its function. In HNF1ap.Pro291ProfsX25 hiPS derived organoids, overexpression of HNF1b increased the PDX1+ progenitors. Similarly, overexpression of HNF1b in the HNF1ap.Pro291ProfsX25 hiPS cell line partially restored differentiation of the beta cells. Together, these data show that organoids can be used to model MODY3 and decipher the underlying intrinsic molecular mechanisms. To improve the representativeness of the human organoids, it is important to consider that T2D is a multi-organ metabolic disease, with a strong inter-relationship between organs. Interestingly, this is also the case for MODY 5, which displays a variable phenotype and age of onset, with interactions between several organs (66). Such consideration also has a strong impact on the pre-clinical steps of drug-therapy. To address this question, Tao et al. used a microfluidic multiorganoid system to reproduce the liver-islets axis (61). This technology allowed 30 days of 3D organoids co-culture under circulatory perfusion. A transcriptional analysis validated the activation of metabolically appropriate pathways. Moreover, under high glucose conditions, mitochondrial dysfunction and decreased glucose transport were detected both in the liver and organoid islets. Interestingly, this phenotype was rescued by metformin treatments. Thus, this new model has opened the door for the further investigation of multi-organ interaction and drug discovery.
The use of patient derived iPS cells for beta cell replacement is currently under investigation at the clinical level (67). The first clinical trial conducted on iPS cells was initiated in 2014. In this trial, differentiated Retinal Pigment Epithelial Cells were transplanted into a patient in Japan without any safety concerns. However, one major limit was the discovery of a genomic mutation in the derived iPS cells (68). More recently, experimentation using VX-880 cells has shown that stem-derived islet cell therapy could be applied to achieve insulin independence among individuals with T1D (American Diabetes Association, News Release June 23-26 2023, Vertex Press Release Jan7, 2024 and clinical trial NCT04786262). Moreover, in a new trial (VX-264), the same VX-880 cells were encapsulated in a device designed to eliminate the need for immunosuppressants. This study remains ongoing in multiple centers and countries (NCT05791201). Thus, these trials strongly suggest that beta-cell replacement is feasible in human. The same strategy could further be used to treat MODYs.
Furthermore, as indicated previously, Pellegrini et al. generated iPS cells by reprogramming somatic cells derived from a MODY8 patient with recurrent episodes of hyperglycemia without obesity. Interestingly, the authors were able to generate iPS-MODY8-derived beta cells completely devoid of functional alterations (53). Of note, these beta cells were able to secrete insulin following glucose stimulation. These experiments raise the possibility of autologous cell replacement therapy for MODY8.
In addition to cell therapy strategy, the use of iPS cells for drug discovery has also been investigated. One target investigated in this manner is dual-specificity tyrosine regulated kinase 1A (DYRK1A), which is ubiquitously expressed and has been implicated in brain development and function. DYRK1A haploinsufficiency in mice has been shown to lead to severe glucose intolerance, reduced beta cell mass, and diabetes (69). However, other studies have indicated that inhibition of DYRK1A stimulates beta cell proliferation in humans (70, 71). Recently, Barzowska et al. used a human organoid model to demonstrate that a set of DYRK1A small molecules inhibitors can enhance beta cell proliferation and long-term insulin secretion, in addition to balancing glucagon levels (59).
Moreover, Ilegems et al. recently identified the HIF1a inhibitor PX-478 as a good candidate to improve the function of beta cells (60). They first hypothesized that the beta cell dysfunction in T2D results from metabolic hypoxic stress. They further showed that administration of a HIF1a inhibitor could improve beta cell function in db/db mice and streptozotocin induced diabetes models. They further validated these results in pancreatic human organoids exposed to high glucose treatments.
In conclusion, the newly developed models based on human iPS cells discussed herein have considerably contributed to increasing our knowledge on the molecular mechanisms underlying diabetes in humans. The use of organoids and beta cells derived from patient iPS cells have paved the way for advances in drug discovery and regenerative medicine which may ultimately allow treatment of diabetes patients with autologous beta cell replacement. Moreover, these models have helped to translate data identified in mouse models in human models, thereby increasing the robustness of preclinical data.
MK: Conceptualization, Formal analysis, Investigation, Visualization, Writing – original draft, Writing – review & editing. EH: Writing – original draft, Writing – review & editing. CP: Writing – original draft, Writing – review & editing. BD: Conceptualization, Investigation, Supervision, Validation, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. BD was supported by Société Francophone du Diabète (SFD) (Grant number 26866).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Ahlqvist E, Storm P, Käräjämäki A, Martinell M, Dorkhan M, Carlsson A, et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. (2018) 6:361–9. doi: 10.1016/S2213-8587(18)30051-2
2. Catli G, Abaci A, Flanagan SE, De Franco E, Ellard S, Hattersley A, et al. A novel GATA6 mutation leading to congenital heart defects and permanent neonatal diabetes: A case report. Diabetes Metab. (2013) 39:370–4. doi: 10.1016/j.diabet.2013.01.005
3. Chao CS, McKnight KD, Cox KL, Chang AL, Kim SK, Feldman BJ. Novel GATA6 mutations in patients with pancreatic agenesis and congenital heart malformations. PloS One. (2015) 10:e0118449. doi: 10.1371/journal.pone.0118449
4. D’Amato E, Giacopelli F, Giannattasio A, D’Annunzio G, Bocciardi R, Musso M, et al. Genetic investigation in an Italian child with an unusual association of atrial septal defect, attributable to a new familial GATA4 gene mutation, and neonatal diabetes due to pancreatic agenesis: GATA4 mutation in ASD and pancreatic agenesis. Diabetic Med. (2010) 27:1195–200. doi: 10.1111/j.1464-5491.2010.03046.x
5. Shaw-Smith C, De Franco E, Lango Allen H, Batlle M, Flanagan SE, Borowiec M, et al. GATA4 mutations are a cause of neonatal and childhood-onset diabetes. Diabetes. (2014) 63:2888–94. doi: 10.2337/db14-0061
6. Carrasco M, Delgado I, Soria B, Martín F, Rojas A. GATA4 and GATA6 control mouse pancreas organogenesis. J Clin Invest. (2012) 122:3504–15. doi: 10.1172/JCI63240
7. Xuan S, Borok MJ, Decker KJ, Battle MA, Duncan SA, Hale MA, et al. Pancreas-specific deletion of mouse Gata4 and Gata6 causes pancreatic agenesis. J Clin Invest. (2012) 122:3516–28. doi: 10.1172/JCI63352
8. Noso S, Kataoka K, Kawabata Y, Babaya N, Hiromine Y, Yamaji K, et al. Insulin transactivator mafA regulates intrathymic expression of insulin and affects susceptibility to type 1 diabetes. Diabetes. (2010) 59:2579–87. doi: 10.2337/db10-0476
9. Cataldo LR, Singh T, Achanta K, Bsharat S, Prasad RB, Luan C, et al. MAFA and MAFB regulate exocytosis-related genes in human β-cells. Acta Physiologica. (2022) 234:e13761. doi: 10.1111/apha.13761
10. Li S-W, Koya V, Li Y, Donelan W, Lin P, Reeves WH, et al. Pancreatic duodenal homeobox 1 protein is a novel β-cell-specific autoantigen for type I diabetes. Lab Invest. (2010) 90:31–9. doi: 10.1038/labinvest.2009.116
11. Guo S, Dai C, Guo M, Taylor B, Harmon JS, Sander M, et al. Inactivation of specific β cell transcription factors in type 2 diabetes. J Clin Invest. (2013) 123:3305–16. doi: 10.1172/JCI65390
12. Yamagata K, Furuta H, Oda N, Kaisaki PJ, Menzel S, Cox NJ, et al. Mutations in the hepatocyte nuclear factor-4α gene in maturity-onset diabetes of the young (MODY1). Nature. (1996) 384:458–60. doi: 10.1038/384458a0
13. Miller SP, Anand GR, Karschnia EJ, Bell GI, LaPorte DC, Lange AJ. Characterization of glucokinase mutations associated with maturity-onset diabetes of the young type 2 (MODY-2): different glucokinase defects lead to a common phenotype. Diabetes. (1999) 48:1645–51. doi: 10.2337/diabetes.48.8.1645
14. Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S. Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia. (2010) 53:2504–8. doi: 10.1007/s00125-010-1799-4
15. Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, et al. Mutations in the hepatocyte nuclear factor-1α gene in maturity-onset diabetes of the young (MODY3). Nature. (1996) 384:455–8. doi: 10.1038/384455a0
16. Staffers DA, Ferrer J, Clarke WL, Habener JF. Early-onset type-ll diabetes mellitus (MODY4) linked to IPF1. Nat Genet. (1997) 17:138–9. doi: 10.1038/ng1097-138
17. Horikawa Y, Iwasaki N, Hara M, Furuta H, Hinokio Y, Cockburn BN, et al. Mutation in hepatocyte nuclear factor–1β gene (TCF2) associated with MODY. Nat Genet. (1997) 17:384–5. doi: 10.1038/ng1297-384
18. Malecki MT, Jhala US, Antonellis A, Fields L, Doria A, Orban T, et al. Mutations in NEUROD1 are associated with the development of type 2 diabetes mellitus. Nat Genet. (1999) 23:323–8. doi: 10.1038/15500
19. Fernandez-Zapico ME, Van Velkinburgh JC, Gutiérrez-Aguilar R, Neve B, Froguel P, Urrutia R, et al. MODY7 gene, KLF11, is a novel p300-dependent regulator of pdx-1 (MODY4) transcription in pancreatic islet β Cells. J Biol Chem. (2009) 284:36482–90. doi: 10.1074/jbc.M109.028852
20. Neve B, Fernandez-Zapico ME, Ashkenazi-Katalan V, Dina C, Hamid YH, Joly E, et al. Role of transcription factor KLF11 and its diabetes-associated gene variants in pancreatic beta cell function. Proc Natl Acad Sci USA. (2005) 102:4807–12. doi: 10.1073/pnas.0409177102
21. Johansson BB, Torsvik J, Bjørkhaug L, Vesterhus M, Ragvin A, Tjora E, et al. Diabetes and pancreatic exocrine dysfunction due to mutations in the carboxyl ester lipase gene-maturity onset diabetes of the young (CEL-MODY). J Biol Chem. (2011) 286:34593–605. doi: 10.1074/jbc.M111.222679
22. Plengvidhya N, Kooptiwut S, Songtawee N, Doi A, Furuta H, Nishi M, et al. PAX4 mutations in thais with maturity onset diabetes of the young. J Clin Endocrinol Metab. (2007) 92:2821–6. doi: 10.1210/jc.2006-1927
23. Meur G, Simon A, Harun N, Virally M, Dechaume A, Bonnefond A, et al. Insulin gene mutations resulting in early-onset diabetes: marked differences in clinical presentation, metabolic status, and pathogenic effect through endoplasmic reticulum retention. Diabetes. (2010) 59:653–61. doi: 10.2337/db09-1091
24. Borowiec M, Liew CW, Thompson R, Boonyasrisawat W, Hu J, Mlynarski WM, et al. Mutations at the BLK locus linked to maturity onset diabetes of the young and β-cell dysfunction. Proc Natl Acad Sci USA. (2009) 106:14460–5. doi: 10.1073/pnas.0906474106
25. Bowman P, Flanagan SE, Edghill EL, Damhuis A, Shepherd MH, Paisey R, et al. Heterozygous ABCC8 mutations are a cause of MODY. Diabetologia. (2012) 55:123–7. doi: 10.1007/s00125-011-2319-x
26. Liu L, Nagashima K, Yasuda T, Liu Y, Hu H, He G, et al. Mutations in KCNJ11 are associated with the development of autosomal dominant, early-onset type 2 diabetes. Diabetologia. (2013) 56:2609–18. doi: 10.1007/s00125-013-3031-9
27. Prudente S, Jungtrakoon P, Marucci A, Ludovico O, Buranasupkajorn P, Mazza T, et al. Loss-of-function mutations in APPL1 in familial diabetes mellitus. Am J Hum Genet. (2015) 97:177–85. doi: 10.1016/j.ajhg.2015.05.011
28. George MN, Leavens KF, Gadue P. Genome editing human pluripotent stem cells to model β-cell disease and unmask novel genetic modifiers. Front Endocrinol. (2021) 12:682625. doi: 10.3389/fendo.2021.682625
29. Aramata S, Han S, Kataoka K. Roles and regulation of transcription factor mafA in islet.BETA.-cells. Endocr J. (2007) 54:659–66. doi: 10.1507/endocrj.KR-101
30. Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol Cell Biol. (2005) 25:4969–76. doi: 10.1128/MCB.25.12.4969-4976.2005
31. Iacovazzo D, Flanagan SE, Walker E, Quezado R, De Sousa Barros FA, Caswell R, et al. MAFA missense mutation causes familial insulinomatosis and diabetes mellitus. Proc Natl Acad Sci USA. (2018) 115:1027–32. doi: 10.1073/pnas.1712262115
32. Fottner C, Sollfrank S, Ghiasi M, Adenaeuer A, Musholt T, SChad A, et al. Second MAFA variant causing a phosphorylation defect in the transactivation domain and familial insulinomatosis. Cancers. (2022) 14:1798. doi: 10.3390/cancers14071798
33. Rocques N, Abou Zeid N, Sii-Felice K, Lecoin L, Felder-Schmittbuhl M-P, Eychène A, et al. GSK-3-mediated phosphorylation enhances maf-transforming activity. Mol Cell. (2007) 28:584–97. doi: 10.1016/j.molcel.2007.11.009
34. Benkhelifa S, Provot S, Nabais E, Eychène A, Calothy G, Felder-Schmittbuhl M-P. Phosphorylation of mafA is essential for its transcriptional and biological properties. Mol Cell Biol. (2001) 21:4441–52. doi: 10.1128/MCB.21.14.4441-4452.2001
35. Guo S, Burnette R, Zhao L, Vanderford NL, Poitout V, Hagman DK, et al. The stability and transactivation potential of the mammalian mafA transcription factor are regulated by serine 65 phosphorylation. J Biol Chem. (2009) 284:759–65. doi: 10.1074/jbc.M806314200
36. Blanchi B, Taurand M, Colace C, Thomaidou S, Audeoud C, Fantuzzi F, et al. EndoC-βH5 cells are storable and ready-to-use human pancreatic beta cells with physiological insulin secretion. Mol Metab. (2023) 76:101772. doi: 10.1016/j.molmet.2023.101772
37. Velazco-Cruz L, Song J, Maxwell KG, Goedegebuure MM, Augsornworawat P, Hogrebe NJ, et al. Acquisition of dynamic function in human stem cell-derived β Cells. Stem Cell Rep. (2019) 12:351–65. doi: 10.1016/j.stemcr.2018.12.012
38. Nair GG, Liu JS, Russ HA, Tran S, Saxton MS, Chen R, et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived β cells. Nat Cell Biol. (2019) 21:263–74. doi: 10.1038/s41556-018-0271-4
39. Hogrebe NJ, Augsornworawat P, Maxwell KG, Velazco-Cruz L, Millman JR. Targeting the cytoskeleton to direct pancreatic differentiation of human pluripotent stem cells. Nat Biotechnol. (2020) 38:460–70. doi: 10.1038/s41587-020-0430-6
40. Maxwell KG, Millman JR. Applications of iPSC-derived beta cells from patients with diabetes. Cell Rep Med. (2021) 2:100238. doi: 10.1016/j.xcrm.2021.100238
41. Fajans SS, Bell GI, Polonsky KS. Molecular mechanisms and clinical pathophysiology of maturity-onset diabetes of the young. N Engl J Med. (2001) 345:971–80. doi: 10.1056/NEJMra002168
42. Winter WE. No title found. Rev Endocrine Metab Disord. (2003) 4:43–51. doi: 10.1023/A:1021823419393
43. Herman WH, Fajans SS, Smith MJ, Polonsky KS, Bell GI, Halter JB. Diminished insulin and glucagon secretory responses to arginine in nondiabetic subjects with a mutation in the hepatocyte nuclear factor–4α/MODY1 gene. Diabetes. (1997) 46:1749–54. doi: 10.2337/diab.46.11.1749
44. Braverman-Gross C, Nudel N, Ronen D, Beer NL, McCarthy MI, Benvenisty N. Derivation and molecular characterization of pancreatic differentiated MODY1-iPSCs. Stem Cell Res. (2018) 31:16–26. doi: 10.1016/j.scr.2018.06.013
45. Flanagan SE, Kapoor RR, Mali G, Cody D, Murphy N, Schwahn B, et al. Diazoxide-responsive hyperinsulinemic hypoglycemia caused by HNF4A gene mutations. Eur J Endocrinol. (2010) 162:987–92. doi: 10.1530/EJE-09-0861
46. Ng NHJ, Jasmen JB, Lim CS, Lau HH, Krishnan VG, Kadiwala J, et al. HNF4A haploinsufficiency in MODY1 abrogates liver and pancreas differentiation from patient-derived induced pluripotent stem cells. iScience. (2019) 16:192–205. doi: 10.1016/j.isci.2019.05.032
47. Aqel YWA, Ali G, Elsayed AK, Al-Khawaga S, Hussain K, Abdelalim EM. Generation of two human iPSC lines from patients with maturity-onset diabetes of the young type 2 (MODY2) and permanent neonatal diabetes due to mutations in the GCK gene. Stem Cell Res. (2020) 48:101991. doi: 10.1016/j.scr.2020.101991
48. Low BSJ, Lim CS, Ding SSL, Tan YS, Ng NHJ, Krishnan VG, et al. Decreased GLUT2 and glucose uptake contribute to insulin secretion defects in MODY3/HNF1A hiPSC-derived mutant β cells. Nat Commun. (2021) 12:3133. doi: 10.1038/s41467-021-22843-4
49. Wang X, Chen S, Burtscher I, Sterr M, Hieronimus A, Machicao F, et al. Generation of a human induced pluripotent stem cell (iPSC) line from a patient with family history of diabetes carrying a C18R mutation in the PDX1 gene. Stem Cell Res. (2016) 17:292–5. doi: 10.1016/j.scr.2016.08.005
50. Wang X, Chen S, Burtscher I, Sterr M, Hieronimus A, Machicao F, et al. Generation of a human induced pluripotent stem cell (iPSC) line from a patient carrying a P33T mutation in the PDX1 gene. Stem Cell Res. (2016) 17:273–6. doi: 10.1016/j.scr.2016.08.004
51. Wang X, Sterr M, Ansarullah, Burtscher I, Böttcher A, Beckenbauer J, et al. Point mutations in the PDX1 transactivation domain impair human β-cell development and function. Mol Metab. (2019) 24:80–97. doi: 10.1016/j.molmet.2019.03.006
52. Yabe SG, Iwasaki N, Yasuda K, Hamazaki TS, Konno M, Fukuda S, et al. Establishment of maturity-onset diabetes of the young-induced pluripotent stem cells from a Japanese patient. J Diabetes Invest. (2015) 6:543–7. doi: 10.1111/jdi.12334
53. Pellegrini S, Pipitone GB, Cospito A, Manenti F, Poggi G, Lombardo MT, et al. Generation of β Cells from iPSC of a MODY8 patient with a novel mutation in the carboxyl ester lipase (CEL) gene. J Clin Endocrinol Metab. (2021) 106:e2322–33. doi: 10.1210/clinem/dgaa986
54. Ræder H, Johansson S, Holm PI, Haldorsen IS, Mas E, Sbarra V, et al. Mutations in the CEL VNTR cause a syndrome of diabetes and pancreatic exocrine dysfunction. Nat Genet. (2006) 38:54–62. doi: 10.1038/ng1708
55. Scavuzzo MA, Teaw J, Yang D, Borowiak M. Generation of scaffold-free, three-dimensional insulin expressing pancreatoids from mouse pancreatic progenitors in vitro. J Vis Exp. (2018), 2(136):57599. doi: 10.3791/57599
56. Beydag-Tasöz BS, Yennek S, Grapin-Botton A. Towards a better understanding of diabetes mellitus using organoid models. Nat Rev Endocrinol. (2023) 19:232–48. doi: 10.1038/s41574-022-00797-x
57. Frum T, Spence JR. hPSC-derived organoids: models of human development and disease. J Mol Med. (2021) 99:463–73. doi: 10.1007/s00109-020-01969-w
58. Cujba A-M, Alvarez-Fallas ME, Pedraza-Arevalo S, Laddach A, Shepherd MH, Hattersley AT, et al. An HNF1α truncation associated with maturity-onset diabetes of the young impairs pancreatic progenitor differentiation by antagonizing HNF1β function. Cell Rep. (2022) 38:110425. doi: 10.1016/j.celrep.2022.110425
59. Barzowska A, Pucelik B, Pustelny K, Matsuda A, Martyniak A, Stępniewski J, et al. DYRK1A kinase inhibitors promote β-cell survival and insulin homeostasis. Cells. (2021) 10:2263. doi: 10.3390/cells10092263
60. Ilegems E, Bryzgalova G, Correia J, Yesildag B, Berra E, Ruas JL, et al. HIF-1α inhibitor PX-478 preserves pancreatic β cell function in diabetes. Sci Transl Med. (2022) 14:eaba9112. doi: 10.1126/scitranslmed.aba9112
61. Tao T, Deng P, Wang Y, Zhang X, Guo Y, Chen W, et al. Microengineered multi-organoid system from hiPSCs to recapitulate human liver-islet axis in normal and type 2 diabetes. Advanced Sci. (2022) 9:2103495. doi: 10.1002/advs.202103495
62. D’Amour KA, Agulnick AD, Eliazer S, Kelly OG, Kroon E, Baetge EE. Efficient differentiation of human embryonic stem cells to definitive endoderm. Nat Biotechnol. (2005) 23:1534–41. doi: 10.1038/nbt1163
63. D’Amour KA, Bang AG, Eliazer S, Kelly OG, Agulnick AD, Smart NG, et al. Production of pancreatic hormone–expressing endocrine cells from human embryonic stem cells. Nat Biotechnol. (2006) 24:1392–401. doi: 10.1038/nbt1259
64. Pagliuca FW, Millman JR, Gürtler M, Segel M, Van Dervort A, Ryu JH, et al. Generation of functional human pancreatic β Cells. In Vitro Cell. (2014) 159:428–39. doi: 10.1016/j.cell.2014.09.040
65. Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat Biotechnol. (2014) 32:1121–33. doi: 10.1038/nbt.3033
66. Ng N, Mijares Zamuner M, Siddique N, Kim J, Burke M, Byrne MM. Genotype–phenotype correlations and response to glucose lowering therapy in subjects with HNF1β associated diabetes. Acta Diabetol. (2022) 59:83–93. doi: 10.1007/s00592-021-01794-8
67. Millman JR, Pagliuca FW. Autologous pluripotent stem cell–derived β-like cells for diabetes cellular therapy. Diabetes. (2017) 66:1111–20. doi: 10.2337/db16-1406
68. Yoshihara M, Hayashizaki Y, Murakawa Y. Genomic instability of iPSCs: challenges towards their clinical applications. Stem Cell Rev Rep. (2017) 13:7–16. doi: 10.1007/s12015-016-9680-6
69. Rachdi L, Kariyawasam D, Guez F, Aïello V, Arbonés ML, Janel N, et al. Dyrk1a haploinsufficiency induces diabetes in mice through decreased pancreatic beta cell mass. Diabetologia. (2014) 57:960–9. doi: 10.1007/s00125-014-3174-3
70. Dirice E, Walpita D, Vetere A, Meier BC, Kahraman S, Hu J, et al. Inhibition of DYRK1A stimulates human β-cell proliferation. Diabetes. (2016) 65:1660–71. doi: 10.2337/db15-1127
Keywords: stem cells, organoids, pancreas, islets, diabetes, MODY
Citation: Ka M, Hawkins E, Pouponnot C and Duvillié B (2024) Modelling human diabetes ex vivo: a glance at maturity onset diabetes of the young. Front. Endocrinol. 15:1427413. doi: 10.3389/fendo.2024.1427413
Received: 23 July 2024; Accepted: 03 September 2024;
Published: 25 September 2024.
Edited by:
Simone Baltrusch, University Hospital Rostock, GermanyReviewed by:
Lise Bjørkhaug, Western Norway University of Applied Sciences, NorwayCopyright © 2024 Ka, Hawkins, Pouponnot and Duvillié. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bertrand Duvillié, YmVydHJhbmQuZHV2aWxsaWVAY3VyaWUuZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.