 Baizhou Tan
Baizhou Tan Beiyu Zhang
Beiyu Zhang Hongping Chen
Hongping Chen- 1Department of Histology and Embryology, School of Basic Medical Sciences, Jiangxi Medical College, Nanchang University, Nanchang, China
- 2Queen Mary School, Jiangxi Medical College, Nanchang University, Nanchang, China
- 3Jiangxi Key Laboratory of Experimental Animals, Nanchang University, Nanchang, China
The incidence of gastroenteropancreatic neuroendocrine neoplasms (GEP NEN) is increasing at a rapid pace and is becoming an increasingly important consideration in clinical care. Epidemiological data from multiple countries indicate that the incidence of gastroenteropancreatic neuroendocrine neoplasms (GEP NEN) exhibits regional, site-specific, and gender-based variations. While the genetics and pathogenesis of some GEP NEN, particularly pancreatic NENs, have been investigated, there are still many mechanisms that require further investigation. The management of GEP NEN is diverse, but surgery remains the primary option for most cases. Peptide receptor radionuclide therapy (PRRT) is an effective treatment, and several clinical trials are exploring the potential of immunotherapy and targeted therapy, as well as combination therapy.
1 Introduction
Neuroendocrine neoplasm (NEN) is a heterogeneous group of tumors originating from the diffuse neuroendocrine system (1). NEN can be present in any part of the body and can lead to various hormonal syndromes (2). The gastroenteropancreatic (GEP) NEN represents the most prevalent site of NEN, accounting for approximately 55% to 70% of the total number of NEN cases (3, 4). GEP NEN is primarily observed in the foregut, midgut, and hindgut (5). NEN can manifest in other locations, including the lungs, thymus, parathyroid, thyroid, adrenal glands, and pituitary glands (6). Furthermore, approximately 10% of NEN cases are associated with genetic syndromes, including multiple endocrine neoplasia type 1, von Hippel-Lindau and neurofibromatosis type 1 (7).
The occurrence of GEP NEN is rare (1). However, the global incidence of NENs has increased markedly over the past few decades, particularly in North America, and the clinical significance of NENs is becoming increasingly important (8, 9). The data from Surveillance, Epidemiology, and End Results (SEER) program show a 6.4-fold increase in the incidence of NEN in the United States between 1973 and 2012, and the trend is more pronounced for NEN than for other tumor types (4).In addition, the incidence of high grade GEP NEN increased approximately 5.3-fold between 1988 and 2010 (10). At the time of initial diagnosis, more than 50% of patients with NENs have already developed lymph node metastases (11). The liver was the primary site of metastasis, accounting for 82% of all cases, while the small intestine was identified as the primary source of NEN metastasis (12).
Malignant GEP neuroendocrine tumors were initially described by Oberndorfer as carcinoid due to their distinctive clinical characteristics, which have been observed for over a century (8). Nevertheless, the term carcinoid is still employed, occasionally resulting in some ambiguity.
NEN can be classified as either functional or non-functional, depending on whether it produces hormones. The percentage of functioning pancreatic NET (PNET) is estimated to be between 30 and 40% (13). Most tumors are non-functional, whereas functional NEN can produce hormones such as insulin, gastrin, serotonin, glucagon, etc., which can lead to different clinical symptoms (14–16). Tumor burden and the primary site of the tumor can also affect clinical signs and symptoms (17).
Over the past 10 years, methods and techniques for the classification, diagnosis and treatment of NENs have advanced significantly (4, 18). Currently, the main treatments for GEN NEN are surgery, peptide receptor radionuclide therapy, chemotherapy, and newer treatments such as immunotherapy and targeted therapy are still being developed, and combination therapies have become popular in the treatment of GEP NEN. This review focuses on the epidemiology, genetics, and treatment of GEP NEN.
2 2022 WHO classification
There has been confusion over the naming and classification of NEN (19). The World Health Organization (WHO) has established the most frequently used system for classifying NEN, which provides a new nomenclature for NEN (20). The most recent updates are the 2019 and 2022 versions.
The WHO classifies neuroendocrine neoplasms NEN into two categories: neuroendocrine tumors (NET) and neuroendocrine carcinomas (NEC). While NET is characterized by well-differentiated neuroendocrine cells, NECs exhibit less pronounced differentiation. These two types not only exhibit disparate pathological, morphological, and molecular characteristics but also manifest distinct epidemiological, clinical, therapeutic, and prognostic features.
In 2022, WHO published the latest version of the classification of GEP NEN (Table 1). Based on whether the tumor secretes hormones and causes characteristic clinical manifestations it is classified as non-functional and functional.
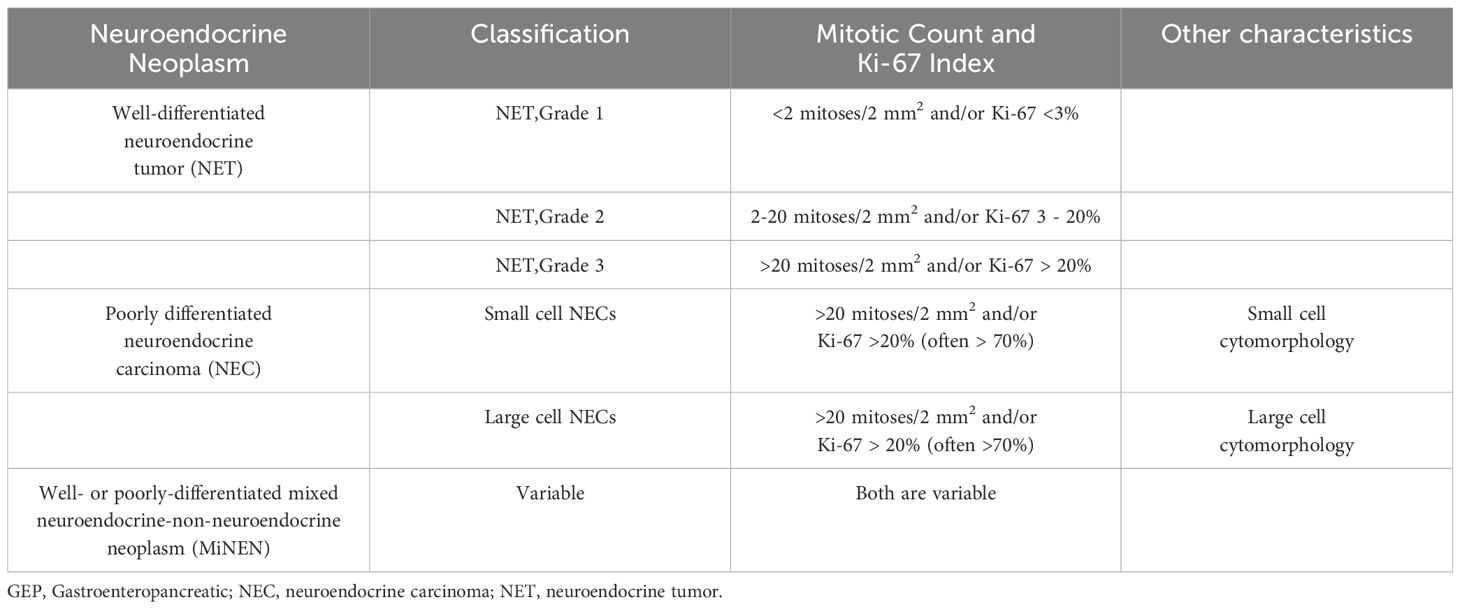
Table 1. The 2022 WHO epithelial neuroendocrine neoplasms classification for Gastrointestinal tract and pancreato-biliary tract (21).
NET is typically classified as low-grade (G1), intermediate-grade (G2), or high-grade (G3) based on their proliferation rate, as indicated by the Ki-67 index and mitotic count. NET G1 is characterized by a low proliferation rate, with a Ki-67 index typically below 3%, and is classified as the least aggressive among neuroendocrine neoplasms.NET G2 demonstrates a moderate proliferation rate, with a Ki-67 index ranging from 3% to 20%, and is more aggressive than G1 but less so than G3. NET G3 is highly aggressive, with a Ki-67 index exceeding 20%, reflecting a high rate of cell division and the most aggressive behavior within the NET.
In contrast, NECs are characterized by a high proliferative rate and a rapid growth pattern, and are classified as tumors. NEC can be categorized into small cell type and large cell based on the morphology of tumor cells. The WHO Classification of Endocrine Tumors, 5th edition, has adopted these classification principles (21).
However, some studies are still using outdated WHO classifications such as the 2010 version. Crucially, what is now graded as grade 3 NET are still considered NECs, since grade 3 was specifically designated as NECs in the 2010 WHO classification (22).
3 Epidemiology
As evidenced by pertinent research, the prevalence of GEP NET is on the rise globally, particularly in North America (23). The incidence of GEP NET exhibits considerable variation across countries and regions, with notable differences in the most common sites of cancer (Table 2) (Figure 1). Small intestinal NET (SiNET) and rectum NET are most common in North America (9). In Asia, the incidence of rectal and pancreatic NET is highest, and in Europe, the most common NET are small intestine and appendix (27–31) (Table 3). Overall survival rates for patients with GEP NET seem to be improving in recent years (23). The increased prevalence of GEP NEN is attributed to recent improvements in diagnostic techniques and histological classification, particularly in the rectum, stomach, and pancreas (31, 32). NET can occur at all ages, except for appendiceal tumors, which occur mainly after 50 years of age, and hereditary syndromes can develop earlier (16).
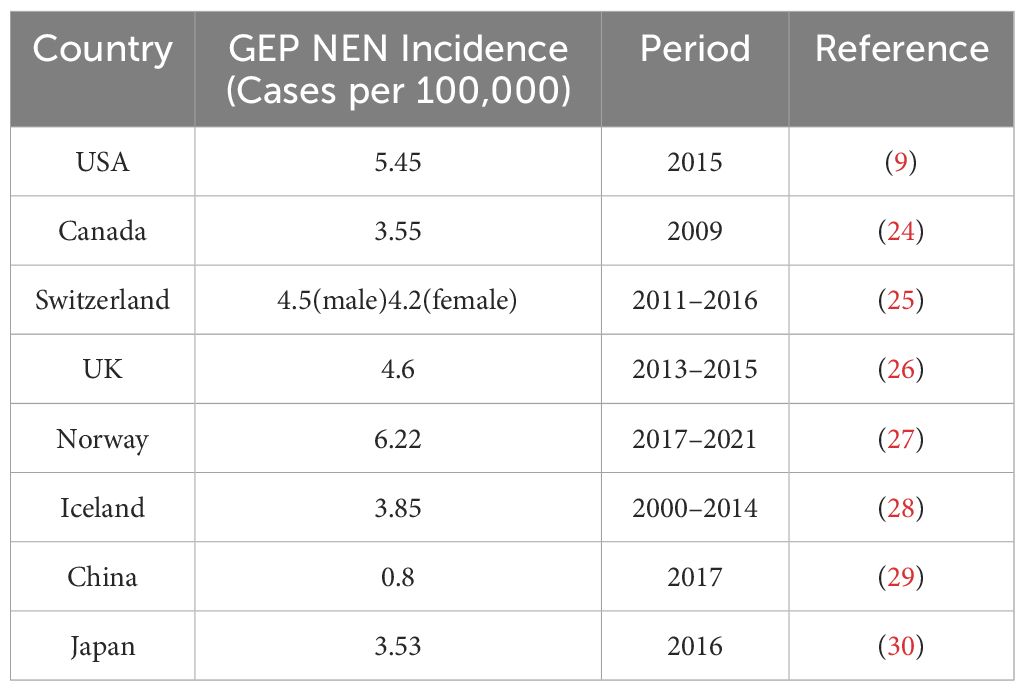
Table 2. The age-adjusted incidence of GEP NEN, by country.
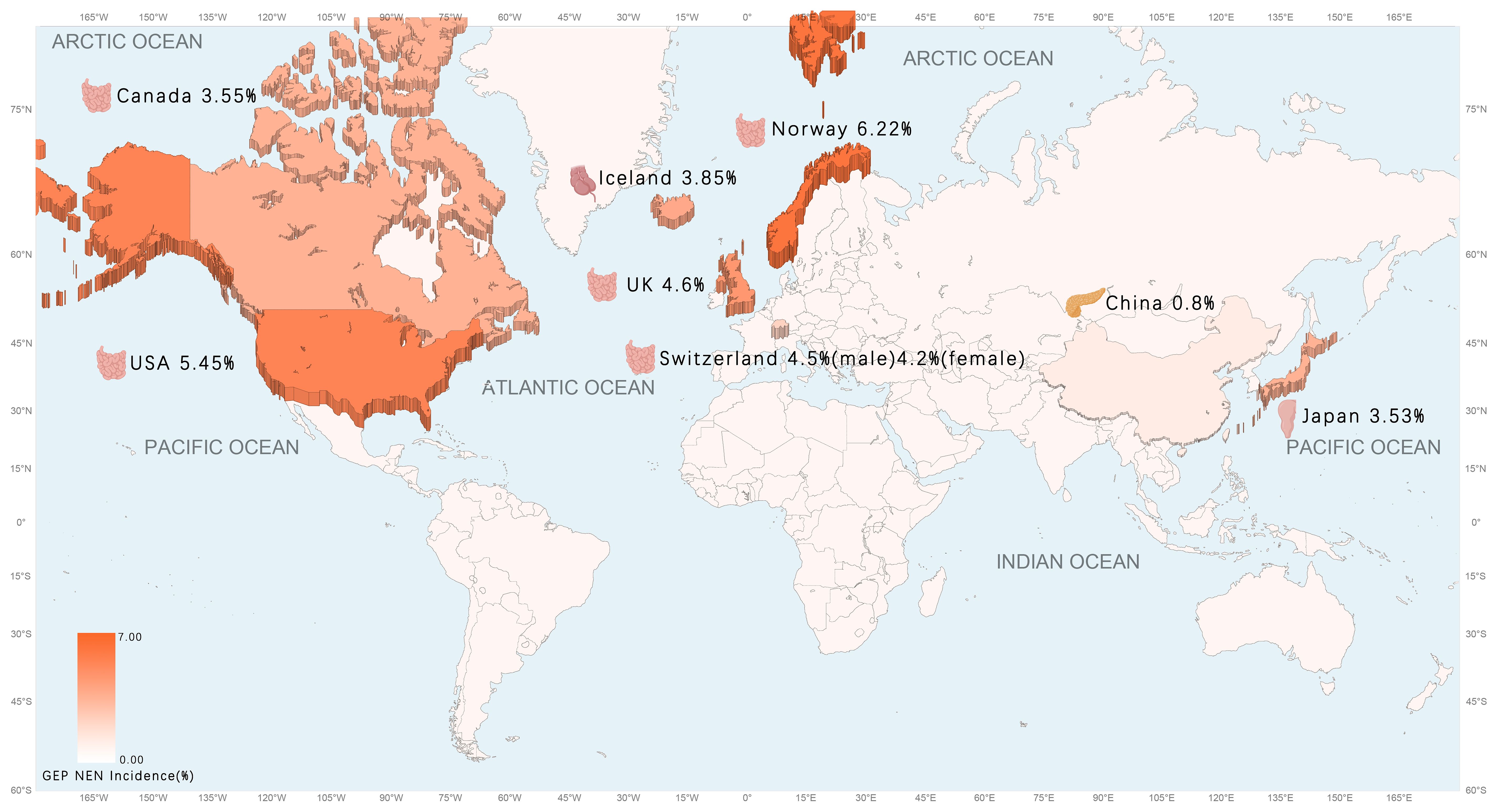
Figure 1. The age-adjusted incidence of GEP NEN in the world map.
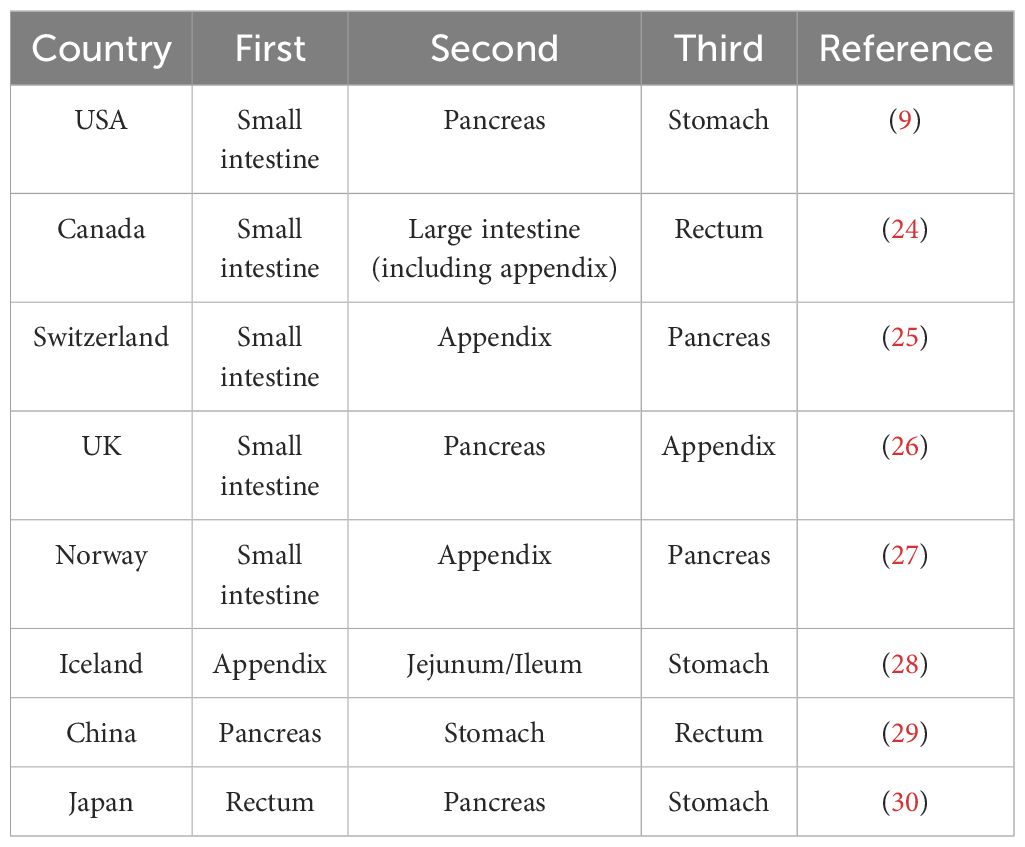
Table 3. Common primary sites of GEP NET, by country.
A gender-based disparity in GEP NET survival has been observed, with females exhibiting a higher survival rate than males, irrespective of staging and morphology. In most digestive system organs, females demonstrated enhanced survival rates, which reached statistical significance (33). Male pancreatic NET is larger than female pancreatic NET, and the rate of undifferentiated and poorly differentiated is higher, but there is no significant difference in distant metastasis rate. Overall survival differences are found in the early stage, but not in stage 3 or 4 disease (34). Another study demonstrated that women with GEP NET exhibited a higher prevalence of G1 stage tumors, yet comparable proliferation rates to men. There were no notable discrepancies in the metastatic rate or metastatic site, and no discernible differences in treatment by gender (35).
3.1 The United States
In a cohort study comprising over 40,000 patients with GEP NET, most patients had NEN in the rectum and small intestine. The age-adjusted incidence rate of GEP NET exhibited a notable upward trajectory from 1975 to 2015, with a ratio of 1.05 per 100,000 in 1975 and 5.45 per 100,000 in 2015. The most common primary tumor sites for GEP NET were the rectum (28.6%), the small intestine (28.1%), and the pancreas (16.4%) (9). A paper based on the SEER database indicates that the incidence of pancreatic neuroendocrine tumors (PNET) in the USA has increased with age from 1975 to 2018 (36). The highest rates of GEP NET are found in African Americans, with men having the highest incidence of high-grade tumors (37).
3.2 Canada
The incidence of GEP NET in Canada increased from 1.18 per 100,000 in 1994 to 3.55 per 100,000 in 2009. The most common sites of primary NET overall were the lungs (25.0%), the small intestine (18.1%), the large intestine (12.9%), and the rectum (12.3%) (24).
3.3 Switzerland
The incidence of GEP NET increased consistently between 1976 and 2016, with no significant gender differences, but the exact reason for the rise is unknown (25). The incidence of GEP NEN has increased for both genders. For males, the rate rose from 2.4 per 100,000 individuals between the periods of 1976-1980 to 4.5 per 100,000 individuals between 2011-2016. For females, the rate increased from 2.3 per 100,000 individuals between the periods of 1976-1980 to 4.2 per 100,000 individuals between 2011-2016. The majority of GEP NET are localized in the small intestine (33%), the appendix (30%), and the pancreas (12%). The most common site of GEP NEC is the pancreas (28%) (25).
3.4 Norway
The incidence of NEC of gastrointestinal tract in Norway increased over 2-fold between 1993-2021 and between 2017-2021, the most common NET tumors were small intestine (23%), lung (19%) appendix (13%) and pancreas (12%). The authors observed a stabilization or even a decrease in the incidence of several sites (e.g. stomach, rectum) during the last 5 years compared to the previous 5-year period (27).
3.5 Iceland
The total mean annual GEP NET incidence rate was 3.39 per 100,000 in 1985-1999 and 3.85 per 100,000 in 2000-2014. The most common primary tumor was found to be the appendix (32%), followed by the jejunum/ileum (24%) and the stomach (17%) (28).
3.6 The United Kingdom
As reported by NHS England, between 1995 and 2018, the age-adjusted NEN incidence rate increased by a factor of 3.7, from 2.35 cases per 100,000 people to 8.61 cases per 100,000 people. The incidence has increased significantly in the past 23 years. The most common sites were small intestine (1.46 per 100,000), pancreas (1.00 per 100,000) and appendix (0.95 per 100,000) (31). Another study showed a GEP NET incidence of 4.6 per 100,000 from 2013 to 2015 (26).
3.7 China
TIn 2017, the age-standardized incidence rate of neuroendocrine neoplasms in China was 1.14 per 100,000 individuals. The incidence rate was higher in males than in females (1.42 per 100,000 vs. 0.86 per 100,000), and higher in rural than in urban areas (1.28 per 100,000 vs. 1.05 per 100,000). The GEP NET incidence rate is 0.8 per 100,000 in 2017. The most common primary sites of GEP NET in China are the pancreas, stomach and rectum (29).
3.8 Japan
As indicated by data from the National Cancer Registry of Japan, the incidence of GEP NEN in Japan in 2016 was 3.53 per 100,000 individuals. Of these cases, rectal NEN constituted 53% of the total, followed by pancreas (20%) and stomach (30). There are large differences between Japanese and Western GEP NET, largely attributable to the prevalence of MEN-1 in non-functioning pancreatic endocrine tumors (38).
4 Genetics of GEP NEN
Two genetically distinct forms of GEP NEN have been identified: well-differentiated NET and poor differentiated NECs. NECs are characterized by TP53 and Rb1 inactivation, which is associated with a poor prognosis. In contrast, NET exhibit a wide range of molecular alterations. While GEP NET may originate from multiple sites, including the duodenum to the rectum, most molecular studies have primarily focused on two broad categories of tumors: PNET and SiNET.
4.1 Pancreatic NET(PNET)
4.1.1 MEN1
MEN1 is the most frequently mutated and studied gene in PNET. The germline mutations in this tumor suppressor gene cause multiple endocrine neoplasia type 1 (MEN 1). A review of the literature reveals that, by the age of 50, over 90% of MEN 1 patients have one or more forms of endocrine malignancies, with the majority of cases involving parathyroid tumors (>80%), pancreatic endocrine tumors (80%-100%), anterior pituitary tumors (54%-65%), and adrenal adenomas (27%-36%) (39). Age-related prevalence of MEN1 surpasses 50% at age 20 and 95% at age 40 in all clinical features (40). The observed increase in prevalence with age may be attributed to the ontogenesis of MEN1, which is consistent with the two-hit mutation hypothesis proposed by Knudson during his study of the Rb1 gene (41, 42).This hypothesis posits that the loss of the remaining wild-type copy at the mutated MEN1 allele at the disease locus is a key mechanism underlying the disease’s pathogenesis. Since loss of heterozygosity has been identified in 86% of macrotumors, 100% of microadenomas, and, surprisingly, 95% of monohormonal endocrine cells clusters, it has been studied as a potential marker for neoplastic growth (43). Given that MEN1 is an autosomal dominant disorder, it is not anticipated that there will be a significant difference in the incidence between the genders. However, there have been reports indicating a greater prevalence of female patients (44, 45). Further validation may be required to fully comprehend this phenomenon. The MEN1 mutations play a significant role in the development of hereditary tumors in patients with MEN1 syndrome. Additionally, this mutation is the most prevalent genetic event identified in sporadic PNET, occurring in more than 35% of sporadic PNET patients in a somatic way (43). Whole exome sequencing has demonstrated that MEN1 somatic mutations are present in 40-56% of sporadic PNET, representing a considerably higher frequency than that observed for any other single gene within these tumors (46). It is noteworthy that a recent study demonstrated that allelic deletions in MEN1 are two to three times more prevalent than mutations in MEN1, aneuploid individuals exhibit more significant roles than those with single-gene mutations (46, 47). This phenomenon can be attributed to the inactivation of the MEN1 gene or the deletion of other tumor-suppressor genes located on chromosome 11, band q13.
Menin is a 68 kDa protein encoded by MEN1 gene, serving as a crucial scaffold protein. In the nucleus, menin interacts with diverse proteins to regulate gene transcription and cellular signaling pathway. For example, menin interacts to JunD and inhibits its transcriptional activity, while it binds to Smad3 to enhance TGF-β and BMP signaling pathways, thereby showing their proliferating inhibitory effects (48, 49). Menin interacts with death-domain-associated protein (DAXX) to inhibit the proliferation of NET cells, enhancing the expression of membrane metallo-endopeptidase (MME) in a synergistic manner. The menin T429K mutation, however, disrupts binding to DAXX, eliminating its MME suppression effect and promoting NET cell proliferation (50). Menin also works together with Phosphatase and Tensin homolog (PTEN), which negatively controls PI3K-Akt-mTOR pathway to suppress tumorigenesis. Studies have indicated that, in contrast to mice with single gene deletions, mice with dual knockouts of both the MEN1 and PTEN genes exhibit a more rapid development of well-differentiated G1/G2 PNET (51). This observation underscores the significance of the Menin-PTEN crosstalk. Furthermore, MEN1 has been shown to inhibit mTOR signaling, which in turn promotes lipid peroxidation and ferroptosis. This process is known to be involved in a number of different cancers (52). Given its role in the mixed-lineage leukemia histone methyltransferase complex, it is unsurprising that menin plays a pivotal role in epigenetic regulation. Furthermore, research has demonstrated that hypermethylation of numerous potential tumor suppressor genes is a common occurrence in MEN1-associated PNET, including CDCA7L and RBM47 (53). Histone modification H3K4me3 is predominantly present in the promoter region in close proximity to the transcription start site, where it serves to trigger gene transcription. In endocrine pancreas, menin directly cooperate with the promoter of p27 and p18, known as cyclin-dependent kinase inhibitors. Study showed that menin increase the methylation of H3K4me3 and promote the expression of p27 and p18 to suppress cell proliferation (54). Menin can also enhance the H3K9me3 levels at the MME promoter to suppress PNET when cooperate with DAXX (51).
4.1.2 mTOR
The mammalian target of rapamycin(mTOR) is a serine threonine kinase that encoded by the mTOR gene. And mTOR locate downstream of PI3K/AKT signaling pathway, which mediate several basic cellular functions. The regulation of mTOR pathway involves upstream regulatory proteins (such as PTEN and PI3K) and downstream effectors (including MDM2, FOXO, and GSK-3β), which also regulated by diverse other signaling pathways (55). Dysregulation of PI3K/AKT signaling lead to tumorigenesis through increasing protein expression, cell migrating and promoting angiogenesis (56). For example, mutation in PTEN cause many cancers, such as breast, colon, lung, prostate (57).
In PNET, aberrant activating of PI3K-Akt-mTOR pathway involves both familial and sporadic PNET (Figure 2). Mutation in the TSC1 and TSC2 genes cause tuberous sclerosis (TS), characterized by benign hamartoma, cognitive impairment and epilepsy. There is also a higher risk in TSC patient for developing malignancies, including renal cell carcinoma, breast cancer and thyroid cancer (58). The development of PNET in patients with TSC is relatively common, primarily attributed to TSC2 mutations identified through genetic analysis (59, 60). The protein, tuberin, encoded by TSC2 gene, could facilitate Rheb GTP hydrolysis, leading to the inhibition of mTORC1 activation (61). It is well-established that aberrant activation of the mTOR pathway plays a significant role in sporadic PNET. The initial whole-exome sequencing (WES) study of 68 sporadic PNET cases revealed that 8.8% of cases had TSC2 mutation and 7.3% of cases had PTEN mutations (62). Additionally, a case with a PIK3CA missense mutation was also identified in this study (62). Furthermore, an inactivating mutation of DEPDC5, a tumor suppressor gene in the mTOR signaling pathway, has been recently discovered (63).
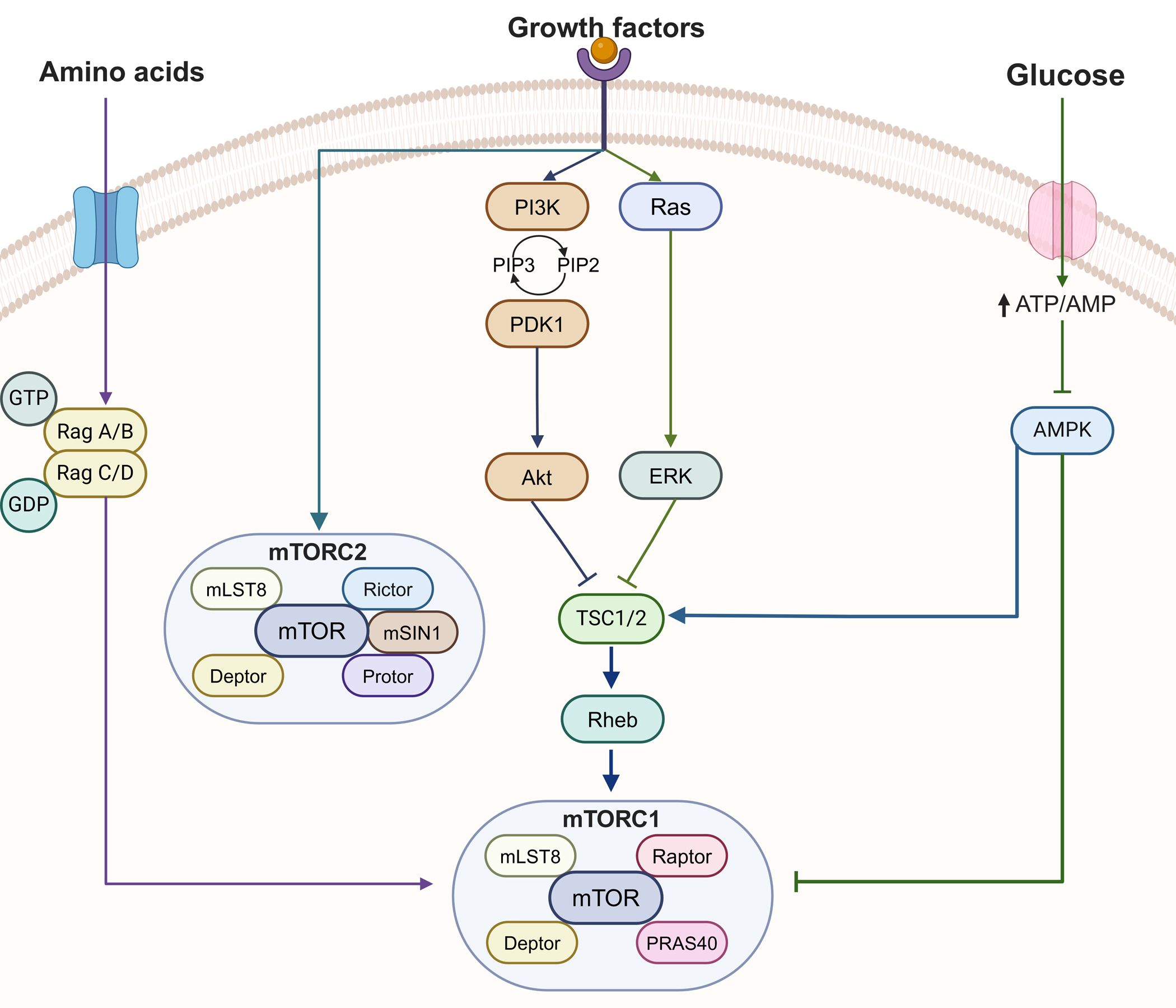
Figure 2. The mTOR signaling pathway. mTORC1 is linked to 3 input signals, whereas mTORC2 is controlled by a growth factor. AMPK, AMP-activated kinase; ERK, extra-cellular regulated kinase; deptor, DEP-domain-containing mTOR interacting protein; mLST8, mammalian lethal with Sec13 protein 8; mTORC, mammalian target of rapamycin complex; PDK1, phosphoinositide-dependent kinase 1; PI3K, phosphatidylinositol 3-kinase; PIP2, phosphatidylinositol bisphosphate; PIP3, phosphatidylinositol triphosphate; PRAS40, proline-rich Akt1 substrate 1; Rheb, Ras homolog enriched in brain; mSin1, stress activated protein kinase interaction protein 1; protor, protein observed with Rictor-1/; TSC1/2, tuberous sclerosis complex1/2.
In addition to mutations in mTOR-related genes, altered expression of pathway members is common in PNET patients. RT-PCR and immunohistochemistry results revealed that TSC2 and PTEN were downregulated in sporadic PNET, which was significantly related to disease progression and shorter overall survival (64). In normal islet cells, PTEN primarily exhibited in a nuclear pattern. However, a study showed that 19 of 23 sporadic PNET correlated with abnormal high cytoplasmic expression (65). Compare to normal tissues, miR-144/451 is significantly overexpressed in insulinomas, which, in turn, promotes β-cells proliferation by up-regulating the PTEN-Akt pathway (66). Overall, these results demonstrated that a significant proportion of PNET are naturally dysregulated in the PI3K/Akt/mTOR pathway.
4.1.3 VHL
Von Hippel-Lindau (VHL) syndrome, an autosomal dominant tumor syndrome, is caused by a germline mutation in VHL tumor suppressor gene. Patients with VHL are at an increased risk for developing hemangioblastoma of the central nervous system, renal angiomas, renal cell carcinoma, pheochromocytoma and pancreatic lessions (including PNET) (67). VHL encodes a 232-amino acid protein, pVHL. Under normoxic conditions, pVHL binds to hypoxia-inducible factor 1-alpha (HIF1-α), undergoing proteasomal degradation after polyubiquitination (68). During hypoxia, HIF 1-α translocates to the nucleus and interacts with HIF-1β to serve as a transcription factor, leading to increased proliferation (PDGFR and EGFR), angiogenesis (VEGF), glycolysis (CAIX and GLUT1) and Mesenchymal-epithelial transition (MET) (OCT4) (69).
Deletion of pVHL restricts the degradation of HIF-1α in patients with VHL syndrome. Consequently, HIF target genes are upregulated, leading to tumorigenesis. Research has demonstrated elevated expression levels of VEGF in PNET (70, 71). Immunostaining results have shown that the majority of PNET express another member of the VEGF protein family, VEGF-C, at moderate to high levels. The expression of VEGF-C in uncertain or low-grade malignant PNET is relatively higher compared to benign PNET, suggesting its role in mediating tumor progression (72). Scientists have also identified VEGFR-2 as the major VEGF-C receptor highly expressed in endothelial cells of all lesions examined, highlighting its role in angiogenesis in PNET (72). In addition, VEGF family protein can also transduce autocrine signals necessary for proliferation, survival and cell migration (73, 74). These examples illustrate significant roles in regulating one of the cancer-related pathways, the VEGF pathway (Figure 3). Another research revealed that patients with VHL gene promoter hypermethylated also showed active hypoxia signals and related to poor prognosis (75).
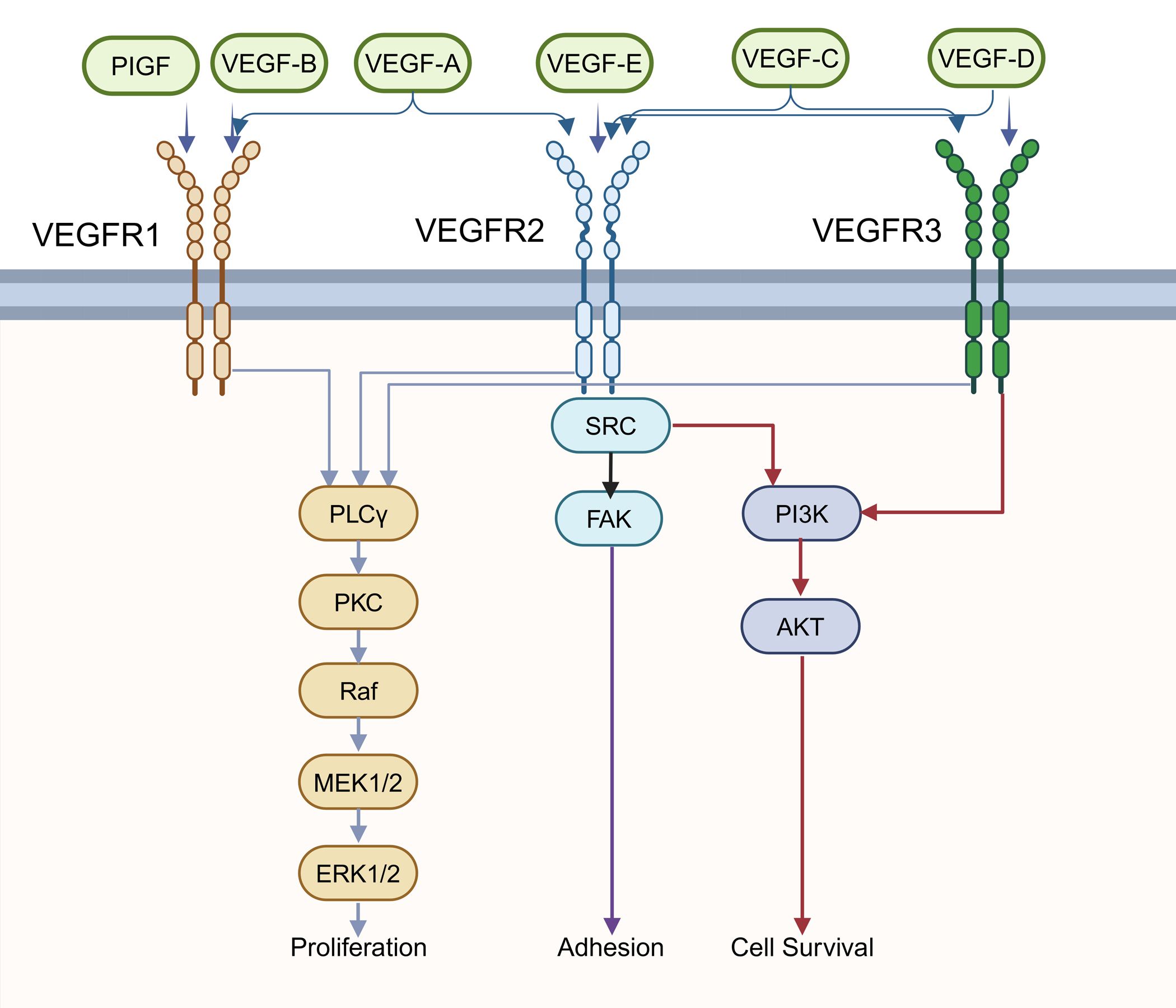
Figure 3. The VEGF signaling pathway. The Vascular endothelial growth factor (VEGF) family has 6 ligands (VEGF-A, B, C, D, E, placenta growth factor [PlGF]) that bind specifically to the VEGF receptor to activate different signaling pathways. The phospholipase C isoform-γ (PLCγ)–protein kinase C (PKC) pathway activates Raf protein kinase, and then the downstream Mitogen- activated protein kinase kinases 1 and 2 (MEK1/2), extra-cellular regulated kinase 1/2 (ERK1/2), will be activated sequentially, which can eventually control proliferation. Phosphoinositide 3-kinase (PI3K)/AKT signaling pathway regulates cell survival. In addition, adhesion is associated with focal adhesion kinase (FAK).
4.1.4 NF1 and RAS-MAPK pathway
Approximately 1/3000–4000 people worldwide are affected by neurofibromatosis type 1 (NF1), an autosomal dominant disorder. It is caused by NF1 germline mutation, which is a cancer suppressor located on chromosome 17q11.2. Patients with NF1 are predisposed to cancers of gastrointestinal tract, liver, lung, thyroid, ovary and breast. Scientists found that up to 10% NF1 individuals develop PNET, most commonly periampullary duodenal somatostainomas, pancreatic somatostinomas, gastrinomas or insulinomas (76). NF1 encodes a RAS GTPase-activating protein, neurofibromin, which function as a negative regulator of RAS/MAPK pathway and inhibitor of mTOR (77). Therefore, loss of neurofibromin results in aberrant activation of RAS/MAPK pathway, which mediate proliferation, survival, differentiation and metabolism in normal cell and often dysregulated in cancer.
Notably, regulated RAS/MAPK signaling is crucial for PNET cell survival and growth. Research revealed that 47 out of 422 cases exhibited copy number loss of HRAS (78). Additionally, KRAS was found to have a higher mutation rate in metastatic species compared to primary PNET, and is associated with metastasis and poor prognosis (78). It is interesting to find out that methylation of negative regulator of RAS/MAPK pathway, RASSF1A, is related to its low expression, thus decrease its roles in inhibiting cell growth.
4.1.5 DAXX and ATRX
In the first whole-exome study of PNET, researchers reported two novel frequently mutated genes, alpha-thalassemia X-linked intellectual disability syndrome (ATRX) and death-domain-associated protein (DAXX) (62). In 68 cases, Jiao et al. identified that 17.6% of patients had ATRX mutations and 25% had DAXX mutations (62). ATRX is a heterochromatin component that belongs to the SNF2 family of chromatin remodeling proteins, regulating gene expression through chromatin modification (79). DAXX is a histone chaperone that specially interacts with H3.3. DAXX is recruited by ATRX to form heterodimers and the ATRX/DAXX complex coordinates the deposition of histone H3.3 at the pericentromeric and telomeric heterochromatin, mediating chromatin remodeling and stabilize telomere length (80, 81). In most PNET, ATRX and DAXX mutations occurs in a mutually exclusive manner, consistent with their co-function in the same pathway (62). In addition, inactivation of ATRX/DAXX complex lead to chromosomal instability and the alternative lengthening of telomeres (ALT). Telomere-specific fluorescence in situ hybridization (FISH) was carried out in 41 PNET by Heaphy et al. Results demonstrated that PNET with ATRX/DAXX mutation displayed aberrant telomere signals, indicating telomerase independent telomere modification, known as ALT (82). Interestingly, several studies found that ATRX/DAXX also play crucial roles in methylation regulation in genes, such as RASSF1 and PTEN (83, 84). For example, DAXX is recruited by combination of p53 and RASSF1A to involve in RASSF1A methylation and inactivation, regulating stability of murine double minute 2 (MDM2). And the methylation level is strictly controlled by DAXX expression (83).
The significance of ARTX/DAXX mutation in prognosis also has been highlighted. Chan el at. found that ARTR/DAXX mutant PNET have the characteristic of alpha cells’ gene expression, indicating a worse outcome (85). Several studies showed that ARTX/DAXX loss is positively correlated with tumor grading and staging, disease recurrence, and survival rate (85–88). However, Jiao el at. hold the opposite opinion that patients with ATRX/DAXX mutation have better overall survival than patients with wild gene type. This difference may due to selection of patient pool, and additional study include larger and varied patient are necessary (62).
4.2 Small intestinal neuroendocrine tumor (SiNET)
Compared to PNET, mutation analysis of SiNET is less revealing. In exosome and genome sequencing, CDKN1B was identified as the most frequent recurrent mutations in 9% of SiNET patients (89). Another gene, APC, mutation was identified in 23% carried APC mutation in 30 SiNET patients, and recent research confirmed the finding as 8% cases carried APC mutation in 52 sporadic primary SiNET. Notably, although the modest frequency of genomic disturbances, 50% of SiNET hold diver mutations in common tumor suppressor gene and proto-oncogenes (90). However, mutation only account for less than 25% cases, although they can also exert effects on haplounder-deficient genes through without the need for a “second blow”. It seems that SiNET tumorigenesis is more dependent on chromosomal alterations and aberrant methylation instead of mutations.
The whole exome sequencing data demonstrated the duplicate loss of chromosomes 11 and 18, as well as the increase in chromosomes 4, 5, 14, and 20 (91, 92). The fact that loss of chromosome 18 is the most frequently events in SiNET led to further research into Smad2 and Smad4, which are tumor suppressor gene located on chromosome 18. However, scientists didn’t find relevant expression loss of Smad2 and Smad4 (92). Therefore, additional researches are necessary to explore the function of chromosome 18 loss in SiNET.
Enrichment analysis revealed overactivation of MAPK, mTOR and Wnt pathways. Notably, members in PI3K/mTOR play crucial roles, induced by amplification pf EGFR, HER2 or PDGFR (93). Methylation of RASSF 1A was identified in 32% SiNET cases and it can promote proliferation through cell cycle control and semaphorin 3 inactivation. Meanwhile, there are unexpected effects of SEMA 3F methylation and, thus, its product, semaphorin 3 loss. Researchers reported that semaphorin 3 loss can disinhibit PI3K and mTORC, representing a possible resistance mechanism to everolimus (94). Furthermore, the expression of semaphorin 3 is correlated with higher tumor stage (94). MiRNA upregulation and downregulation were identified in metastatic SiNET patients. The most consistent findings included the upregulation of miR-96, -182, -183, -196a and the downregulation of miR-1, -31, -129-5p, -133a, -215, miR-143-3p, and miR-375 (95–97). These differences in microRNA expression are clinically utilized as predictors of overall patient survival and miR-375 was identified as the strongest one (98).
5 Treatment
5.1 Surgery
Variation in the surgery of GEP NEN depends on the site and type of tumor. Major conferences and guidelines recommend surgery for most GEP NEN patients (99).
However, pancreatic neuroendocrine carcinoma (PNEC) has a poor prognosis and are not amenable to surgical resection (100). But even in those patients for whom there is no possibility of cure, surgery may be necessary in case of acute life-threatening complications (101).
Surgery is the first line of treatment for PNET and the only treatment for locally advanced NET, 70-90% of cases can be cured with surgery (102, 103).
PNET are clinically classified as either non-functional or functional, and functional PNET are supposed to be evaluated for surgery (104, 105). The NANETS guidelines recommend pancreatectomy for tumors larger than 2 cm for non-functional PNET, if there is localized functional PNET without distant metastases, resection is recommended (106).
The types of pancreatic surgery vary from “typical” to “atypical” resections, depending on the tumor burden, but laparoscopic resection is used most often (107–109). Patients in good general condition can undergo pancreas-sparing pancreatectomies, which can decrease the incidence of pancreatic insufficiency (104). The use of endoscopic ultrasound-guided therapies can be considered as an alternative treatment for patients for whom surgery is not available at the low-grade level of less than 20 mms PNEN (110–112). Minimally invasive pancreatectomy is technically feasible and safe and has advantages in terms of postoperative recovery (109, 113, 114). Furthermore, robotic distal pancreatectomy is also safe and effective (108).
Gastric NET can be divided into three subtypes based on clinical and histological features (115, 116). Treatments need to be selected based on the type of gastric NET. The treatment of type 2 gastric NET is generally similar to that of type 1. Survival of type 1 after endoscopic surveillance or surgical resection is high (117). Type 3 is more malignant and more likely to metastasise than other gastric NET types (118, 119). The choice of endoscopic mucosal resection, endoscopic submucosal dissection or surgery is based on the number of lesions and whether or not the invasion of the muscularis propria occurs (20, 120). The Nordic guidelines and the ENETS guidelines recommend surgical resection with lymphadenectomy similar to that performed for gastric adenocarcinoma (20, 120, 121). Chinese guidelines and a review point to endoscopic resection as an alternative for patients for who are not candidates for surgery, but there is a higher risk of lymph node spread (118, 122).
SiNET are frequently found in the ileum, small intestine resection with removal of lymph nodes is recommendable for SiNET (123, 124). ESMO guidelines recommend surgery for locally advanced SiNET because the presence of a large mesenteric mass can lead to intestinal obstruction and/or ischaemia (evidence level V, recommendation level B) (125). Duodenal neuroendocrine tumors(dNEN) have a high incidence of lymph node metastasis but have a positive surgery prognosis (126). In patients with colorectal NET who have predictive factors for lymph node metastasis, surgical resection with lymph node dissection is an option (127). Conventional imaging has been demonstrated to have a low detection rate of localized regional lymph nodes and micrometastases in preoperative dNEN, with understaged cases present in 38%. It is recommended that endoscopic ultrasound be included as a preoperative tool to obtain more accurate local staging (128).
In select cases of smaller colonic NET G1 (typically less than 10 mm), endoscopic resection may be a viable option, as recommended by the 2023 ENETS guidelines (129). In the treatment of rectal neuroendocrine tumors, endoscopic or surgical technique should be selected based on the size of the tumor. Endoscopic resection is the recommended course of action for lesions measuring 10 mm or less in diameter, as it has a relatively low recurrence rate (130–132). For lesions between 10-20 mm, a comprehensive imaging assessment should be conducted and deliberated by a multidisciplinary team (MDT) to ascertain the optimal course of action, which may entail endoscopic resection or surgical intervention (129). For lesions exceeding 20 mm, surgical resection, including low anterior resection or abdominal perineal resection, is advised following the exclusion of unresectable distant metastases (133).
Most appendiceal neuroendocrine neoplasms (aNEN) can be treated with simple appendectomy or right hemicolectomy. Patients with aNEN >2 cm are recommended for right hemicolectomy, while patients with aNEN <1cm could undergo appendectomy (134). The treatment of aNEN with diameters of 1-2 cm is controversial, but a recent study noted that right hemicolectomy is unadvisable after the complete removal of a 1-2 cm aNEN by appendectomy (135).A lot of people with NET develop liver metastases, cytoreduction can be an option when NET liver metastases are resectable in at least 70% of patients (136).
In advanced stages of NEN, surgical intervention has been shown to confer benefits to a subset of patients (101). The findings of the study indicate that surgical resection can be advantageous for patients with NEN G3 and mixed neuroendocrine-non-neuroendocrine neoplasms (MiNEN) (137).
However, a single-center study of 615 patients with SiNET revealed a significant risk of recurrence following intended radical surgery (138). In another study, 441 patients were included, of whom 224 had PNET and 217 had SiNET. The results demonstrated that approximately 30% of patients with enteropancreatic NET experienced recurrence within five years of radical surgery (139).
Surgical approaches to GEP NEN vary widely according to primary tumor site, tumor classification and lesion size, and each NEN requires dedicated assessment to determine the relevant characteristics of GEP NEN (140).
5.2 Somatostatin analogs (SSAs)
SSAs have high affinity for STTR2 and moderate affinity to SSTR5, which can be used to control the symptoms of hormone overproduction, especially in functional metastatic PNET (141–144). Five subtypes of SSTR receptors have been identified. SSTR receptors belong to the family of G protein-coupled receptors, and more than 70% of NET tumor cells overexpress SSTR type 2 and 5 (145–147). SSTR5 is expressed in somatostatinomas and SSTR2 is expressed in gastrinomas and glucagonomas in the functional PNET (147).
Positive somatostatin receptor imaging is required when applying SSAs for antiproliferative effects (148, 149). SSAs also have antiproliferative effects, cell cycle inhibition and an increase in apoptosis (150). The results of the PROMID, CLARINET and CLARINET FORTE clinical studies showed that for in midgut NET, SSAs could lengthen time to tumor progression, and in progressive GEP NETs could improve progression-free survival (143, 147, 151–154). Two SSAs are currently available that have been approved for GEP NET treatment in the United States, octreotide and lanreotide, with both being long-acting (144, 155). Both SSAs were well tolerated and had a very low-adverse reaction rate (149). No evidence exists to suggest that the two SSAs differ in controlling hormone secretion and tumor growth (142). The initial prospective study to assess the efficacy of growth inhibitor analogs in MEN1-related PNET revealed that lanreotide demonstrated efficacy as an antiproliferative therapy for MEN1-related PNET with a diameter of less than 2 cm (128).ESMO,NANETS,ASCO guidelines recommend SSAs as first-line treatment for functional NET or STTR-positive or metastatic high-differentiated GEP NET (125, 156, 157). However, patients can become resistant to SSAs treatment, and the exact mechanism is unclear (158).
Second generation SSAs is now available for the treatment of other diseases with a greater affinity for SSTR5, and may be used in the future for the treatment of GEP NEN (159, 160).
The most commonly reported adverse effects associated with SSAs are gastrointestinal events (diarrhea and constipation), abdominal discomfort, and the formation of gallstones (161).
5.3 Peptide receptor radionuclide therapy (PRRT)
The somatostatin receptors (SSTR) is commonly expressed in GEP NEN, especially in well-differentiated NET, whereas it is not expressed in normal tissues, and radioactive peptides can be used to label this receptor, which allows the radionuclides to enter the tumor tissue and eventually kill the tumor cells (17, 162–164). The use of highly active SSTR-binding ligands is now commonly referred to as peptide receptor radionuclide therapy (PRRT) (165). SSTRs receptor number is directly related to the therapeutic efficacy of PRRT (166). Somatostatin receptors are rarely expressed in NEC, making PRRT less suitable for NEC.PRRT is valid and secure in the treatment of NET (167–172). The results of the NETTER-1 trial showed that 177Lu-DOTATATE was more effective, and progression free survival was significantly improved compared to the use of high-dose octreotide (173). It is noteworthy that the NETTER-1trial exclusively included patients with SiNET and excluded those with PNET.
PRRT may also be effective in patients with G3 NET, but this needs to be validated with more evidence from clinical studies (174). A large retrospective study showed that PRRT treatment was effective in the treatment of NET G3, and in this study PRRT treatment showed promising response rate and disease control rate (175). The results of the NETTER-2 trial indicated that PRRT is an appropriate treatment option for patients with advanced Grade 2-3 GEP NET. The combination of 177Lu-Dotatate and octreotide 30 mg long-acting repeatable (LAR) demonstrated a 72% reduction in the risk of disease progression or mortality compared to high-dose octreotide 60 mg LAR (176).
Not all patients will benefit from PRRT, and it is necessary to investigate ways to improve the effectiveness of PRRT. Using alpha-emitters, PRRT in combination with chemotherapy, these methods may have more effective outcomes (177–180). However, attention should also be paid to the potential for greater toxicity associated with combination therapy (179) (Table 4).
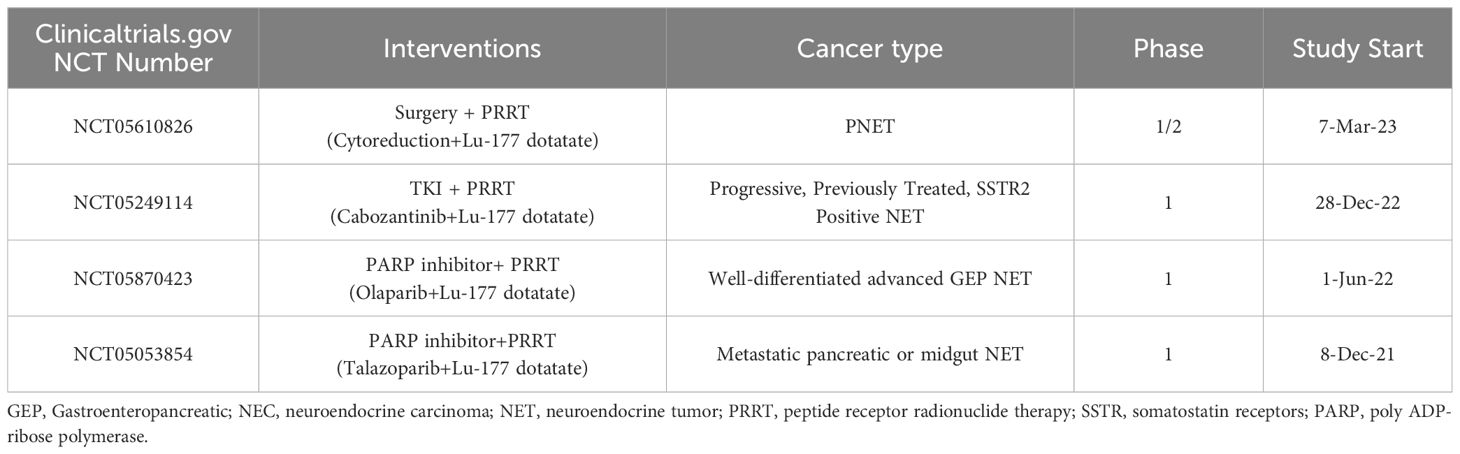
Table 4. Clinical trials investigating PRRT in combination with other treatment.
The first generation of radionuclide gamma-emitting 111Indium had a weak cytotoxic effect (181). Second-generation radionuclides include beta-emitting 90Yttrium (90Y) and 177Lutetium (177Lu), which have better therapeutic effects killing nearby tumor cells (170). The alpha-emitters have been the focus of current research due to their higher energy and greater tendency to cause DNA breaks. The alpha-emitters used in NEN patients are 213Bi, 225Ac, 212Pb (182). Current phase I clinical trials have shown [212Pb] Pb-DOTAMTATE to be well tolerated with fewer adverse effects (183).
The NANETS guidelines and ASCO guidelines recommend PRRT as a second-line treatment for metastatic intestinal NET G1/G2 if positive for SSTR expression (157, 180, 184). ESMO guidelines recommend PRRT as second-line treatment for midgut NET patients with progressive disease after SSA therapy [I, A] (125, 156). The delivery mode of PRRT also can change the therapeutic effect. PRRT arterial delivery to the hepatic artery may lead to improved results in the treatment of NEN with hepatic metastases (185). PRRT retreatment after initial PRRT did not negatively affect safety (186). The results of the meta-analysis demonstrated that Salvage PRRT exhibited a lower objective response rate (17.6% vs. 38.6%, P < 0.001) and a lower disease control rate (77.6% vs. 99.7%, P < 0.001) compared to the initial treatment (187).
A cohort study indicates that the use of upfront PRRT in patients with enteropancreatic NET who have experienced disease progression with SSA treatment was associated with improved progression-free survival outcomes compared with upfront chemotherapy or targeted therapy (in unmatched populations 2.5 years vs. 0.7 years; P < .001]) (188). Furthermore, a review of the literature indicates that patients with PNEN may benefit from PRRT as a neoadjuvant approach (189).
The side effects of PRRT are nausea, vomiting, myelosuppression and abdominal discomfort (125, 190–192). The incidence of Grade 3/4 toxicity in any of the blood counts was less than 15% in the treated patient (192). A retrospective study of 807 patients revealed that renal toxicity was observed in 35% of cases, with Grade 3/4 toxicity occurring in 1.5% (193).
Strategies may be employed to customize PRRT for each patient, including considerations such as patient selection, dosimetry, combination therapies, and modification of the therapeutic index (194).
5.4 Targeted therapy
Currently, VEGF and mTOR targeting drugs have been applied in the treatment of NET, while other targeting drugs such as Hypoxia-inducible factor (HIF) inhibitor, Cyclin dependent-kinase 4/6 inhibitors are under investigation (125). The use of targeted drugs in combination with immunotherapy in NET is also a hot topic of current research (195).
5.4.1 Mammalian target of rapamycin (mTOR) pathway
The mTOR inhibitor everolimus has been used in the treatment of NET. Guidelines recommend that everolimus can be used in SSTR-negative G1-G2 and advanced PNET (20, 157). The RADIANT-4 study demonstrated that treatment with everolimus improved the progression-free survival of GEP NET and was better tolerated, while also exhibiting strong antitumor activity (196). The combination of everolimus and SSA may be considered for routine treatment of patients with functional NET in the future (197). A new mTOR inhibitor, nab-Sirolimus, is under investigation. It has been found discovered that nab-Sirolimus has superior target inhibition when compared to oral mTOR inhibitors (198). A phase 2 clinical trial is now investigating the effect of nab-Sirolimus in the well-differentiated, advanced inoperable metastatic GEP NET(NCT05997056). Other mTOR inhibitors, such as sapanisertib, could play an important role in treating NEN and further studies are needed to assess the effect in PNET (199).
The most frequently observed adverse effects associated with everolimus therapy include stomatitis, fatigue, rash, diarrhea, hyperglycemia, and anemia (200–204). The most common Grade 3 or 4 drug-related adverse events were stomatitis (9%), diarrhea (7%), infections (7%), anemia (4%), and fatigue (4%) (196).
5.4.2 Vascular endothelial growth factor (VEGF) pathway
VEGF is one of the most widely studied biomarkers with the ability to induce abnormal angiogenesis in tumors, the ability of tumors to escape the immune system, invade and metastasise may be enhanced (205–207). VEGF binds to VEGF receptors (VEGFRs),VEGFR-1, VEGFR-2 and VEGFR-3 (208) (Figure 3).
Surufatinib can simultaneously target VEGFR-1, 2, 3 and fibroblast growth factor receptor 1, targeting multiple pathways simultaneously reduces tumor angiogenesis more effectively than targeting one pathway alone (209–211). Phase 3 clinical trials of surufatinib have demonstrated favorable therapeutic outcomes in both advanced pancreatic and extra-pancreatic NEN (212). Surufatinib has a controlled safety profile when combined with toripalimab immunotherapy and appears to have superior anti-tumor activity against NEC (213).
The most common adverse effects associated with surufatinib therapy encompass proteinuria, diarrhea, increased thyroid stimulating hormone in the blood and increased bilirubin in the blood, the most prevalent grade 3 or worse adverse events were hypertension (36%) (212).
Sunitinib is a multitarget tyrosine kinase inhibitor that targets VEGF and platelet-derived growth factor (214). Sunitinib demonstrates efficacy in patients with PNET and exhibits a favorable long-term safety profile (214, 215). Other drugs that target VEGFR and have potential include Cabozantinib, Lenvatinib, Nintedanib, Anlotinib (216, 217).
The most prevalent adverse effects associated with sunitinib therapy include fatigue, gastrointestinal intolerance, and dermatitis. Additionally, serious toxicities, classified as Grades 3 and 4, have been observed in 4 out of 41 cases (218).
In a hypoxic environment, HIF-1α induces VEGF expression, which may cause angiogenesis in tumors (219). Belzutifan is the first HIF inhibitor and the first FDA-approved systemic therapy for VHL-related tumors (219, 220). In VHL disease, the incidence of PNET ranges from 9% to 17% (221).The 2021 edition of the NCCN guidelines recommends the use of belzutifan for the treatment of G1 and G2 locoregional advanced metastatic progressive PNET if the VHL gene is mutated (222). Future drug combination strategies may lead to a better prognosis for patients, with simultaneous inhibition of MTOR and VEGF pathways showing promise in the treatment of NEN (216, 223).
The most commonly reported side effects of Belzutifan include anemia and hypoxia-related symptoms (224, 225). In a phase 1 trial include 43 patient A total of 31 patients (72%) experienced treatment-related adverse events of any grade, while 8 patients (19%) across all dose levels experienced treatment-related adverse events of grade 3 and 4 (226).
5.4.3 Cyclin dependent-kinase 4/6 inhibitors
Cyclin dependent-kinase 4/6 inhibitors, has promising potential for treatment in NEN and trials are already underway. However, a multicenter, phase II study reported that the combination of Ribociclib and Everolimus was not effective and may be toxic in well-differentiated foregut NET. One trial of the role of Abemaciclib’s anti-tumor activity in metastasized inoperable GEP NET is ongoing (NCT03891784).
Cyclin-dependent kinase 4/6 inhibitors have been identified as a promising avenue for treatment in NEN, with clinical trials already underway (227). However, a multicenter, phase II study reported that the combination of ribociclib and everolimus was not effective and may be toxic in well-differentiated foregut NET (228). One trial investigating the role of abemaciclib’s anti-tumor activity in metastasized inoperable gastrointestinal GEP NET is currently ongoing (NCT03891784).
5.5 Chemotherapy
Chemotherapy is primarily used to treat patients with G3 GEP NET and NEC. In contrast to G1,G2 GEP NET, chemotherapy is more important in the treatment of G3 GEP NET, as a high proliferative index indicates more effective chemotherapy (229). Platinum and etoposide chemotherapy is now the first-line choice for extrapulmonary NEC (156, 230). NANETS guidelines recommend the use of fluoropyrimidine in combination with platinum as second-line treatment for extrapulmonary NEC (156). ASCO guidelines recommend CAPTEM(Capecitabine and Temozolomide) chemotherapy in patients with SSTR-negative G1-G2 PNET (157). The effects of chemotherapy in combination with other treatments are being studied in clinical trials (Table 5). Moreover, the study by Tafuto et al. demonstrated that metronomic temozolomide (mTMZ) as monotherapy represents a viable treatment option for patients with advanced G2-G3 NET, particularly in those with an ECOG Performance Status (PS) score of 2, exhibiting good tolerability and clinical improvement (231).
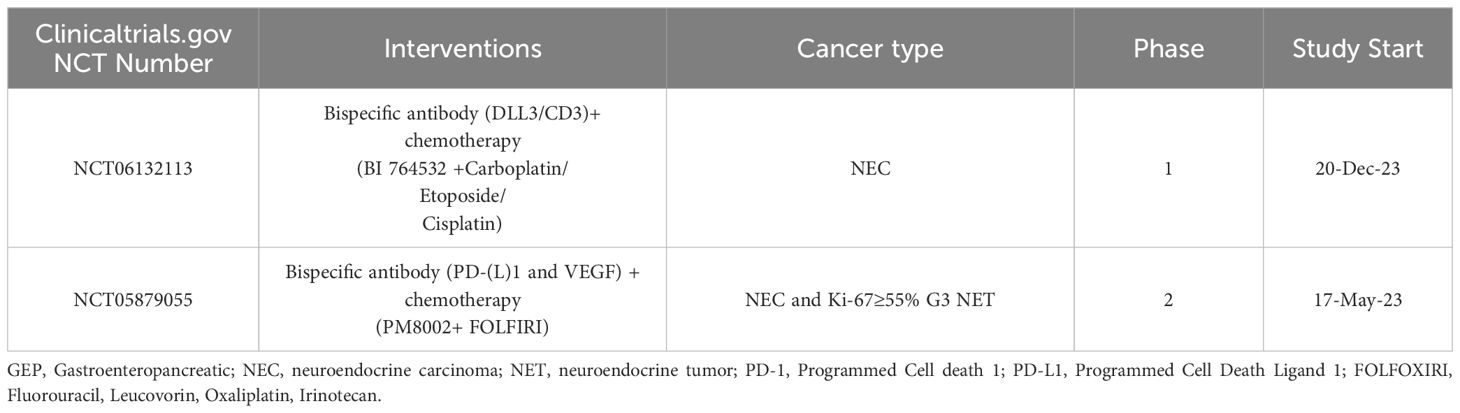
Table 5. Clinical trials investigating chemotherapy.
The side effects of temozolomide include anemia, platelet count decreased, neutrophil count decreased, fatigue, nausea, and constipation and vomiting, with Grade 3-4 toxicity rate of 22% when used alone (232).
5.6 Immunotherapy
Immune checkpoint inhibitors (ICIs) can be broadly classified into two groups, antibodies to programmed death 1 (PD-1) and its ligand (PD-L1) and antibodies targeting cytotoxic t-lymphocyte antigen 4 (CTLA-4), which can be monitored in many cancer types (233, 234). These have negative regulatory effects on t-cell immune function, and inhibition of these targets increases immune system activity (235). Immunotherapy has been developing rapidly recently and has promising applications in many tumors, however the efficacy of treatment in NEN is not well defined and more prospective studies are required to evaluate the value of ICI in GEP NEN therapy (236–239). However, ICIs may be a promising treatment option for high-grade, poorly differentiated NEN (240, 241).
One study indicates less PD-1/PD-L1 in the small intestine or pancreas, which may lead to poor use of ICI (242).However, one study noted that PD-1/PD-L1 expression is common in poorly differentiated NEC of the digestive system (243). One article shows limited potential for anti-PD-1/PD-L1 monotherapy for digestive NECs (244). A recent animal trial showed that the combination of PRRT and anti-PD1 provided the strongest response to NET, with an overall effect superior to that of ICI or PRRT alone (245, 246).
Pembrolizumab monotherapy has a proven safety profile in patients with advanced NEN, but has limited anti-tumor activity and further studies may be needed to identify effective combination safety profile in the treatment of high-grade NEN following first-line chemotherapy (247). The DART trial showed efficacy of ipilimumab in combination with nivolumab in non-pancreatic neuroendocrine tumors, particularly high-grade NEC (248). The primary adverse effects associated with pembrolizumab treatment include fatigue, arthralgia, edema, nausea, vomiting, and diarrhea (249, 250). Of the total 29 of cases, only 9 were classified as grade 3 events, and no grade 4 events were identified as potentially drug-related (249).
The NCCN guidelines classify the use of Pembrolizumab for the treatment of advanced or high TMB tumors as category 2B(lower level of evidence and NCCN considers the treatment to be appropriate) (222, 250). Treatment of patients with locally advanced or metastatic NET with the combination of lipilimumab and nivolumab is also recommended as an alternative to clinical trials (category 2B) (222, 251).
Investigating the microenvironment and immune mechanisms of NEN tumors can drive advances toward ICI and other combination therapies, which are critical for NEN immunotherapy (252) (Table 6).
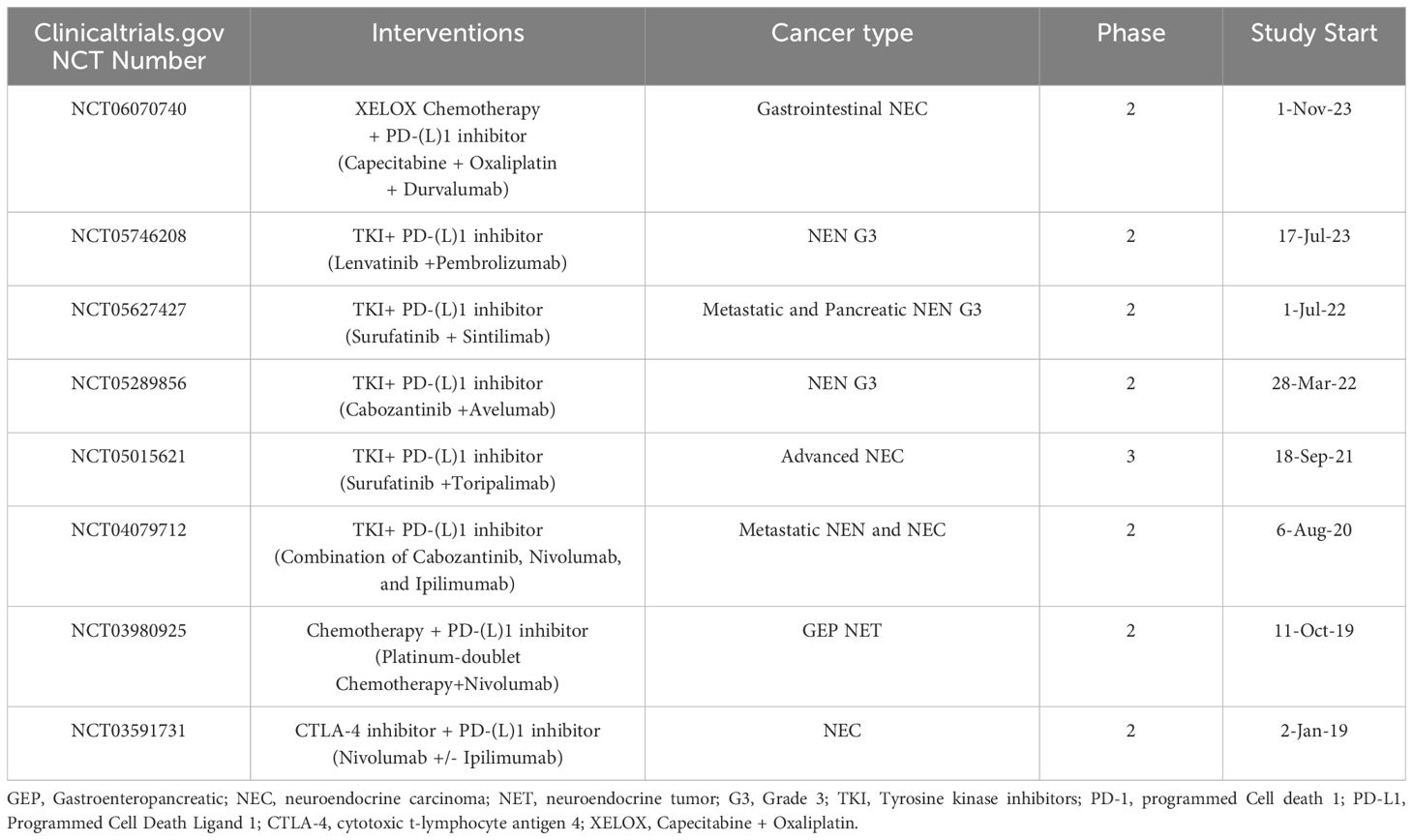
Table 6. Clinical trials investigating ICI in combination with other treatment.
5.7 Interferon-alpha (IFN-α)
Interferon-alpha (IFN-α) has been demonstrated to possess antiproliferative and pro-apoptotic properties in NET. IFN-α represents a second-line treatment option, employed primarily in the management of progressive or functional PNET (253, 254). In accordance with the ESMO guidelines, if a NET patient has a SSTR-negative status, IFN-α may be considered as an alternative to other treatments [IV, B] (125, 184). Treatment with IFN-α is a treatment for refractory carcinoid syndrome, but it is not well-tolerated (125, 255). The use of interferon in clinical practice has been constrained by the occurrence of adverse effects. The main adverse effects of IFN-α are fever(76%), anorexia(71%), arthralgia(52%),injection site pain(28%) and headache(14%) (256). Among these, the suppression of bone marrow function represents a significant concern associated with IFN-α (257). The incidence of side effects may be reduced with the use of weekly pegylated formulations (258). In a phase III trial, no significant difference in progression-free survival between the bevacizumab and IFN groups, indicating that these drugs exhibit comparable antitumor efficacy in patients with advanced NET (259). The combination of IFN-α with octreotide or 131I-MIBG was found to be ineffective and did not demonstrate any synergistic effects (260, 261).
5.8 External beam radiotherapy (EBRT)
Radiotherapy techniques have developed at a relatively rapid pace, from total abdominal irradiation to stereotactic approaches, with major improvements in side effects (262). EBRT is rarely employed for the treatment of GEP NET, and there is a paucity of published literature on this subject. Several studies have demonstrated that EBRT can be utilized as a treatment for inoperable PanNEN, resulting in favorable local control and is well tolerated (263–265). Stereotactic body radiotherapy enables the precise delivery of high doses of radiation to small targets (266).
6 Summary and prospects
NEN is a heterogeneous group of tumors, and GEP NEN is the most prevalent. The incidence and location of GEP NEN vary across different countries. As research into NEN has progressed, a number of genetic and pathogenic mechanisms associated with GEP NEN have been identified. These include the MEN1, mTOR, VHL, NF1, DAXX and ATRX pathways. A variety of treatment options are currently available for neuroendocrine neoplasms (NEN). Surgical intervention represents the sole treatment option for localized GEP NET, and it also plays a role in the management of metastatic NET and NEC. PRRT employs the targeted delivery of radionuclides through the utilization of the high expression of SSTR observed in GEP NEN. The current focus of research is on immunotherapy and targeted therapies. SSA has been demonstrated to be an effective agent for the control of hormone overproduction, while chemotherapy has been shown to be a valuable adjunctive treatment for G3 GEP NET and NEC. It is imperative that future research on the genetics of GEP NEN be intensified in order to identify new therapeutic targets and potentially alter treatment strategies. Combination therapy represents a promising avenue of research, but it is of the utmost importance that researchers take note of the incidence of adverse effects and drug toxicity. Accurate assessment of the patient’s condition and selection of the appropriate treatment modality can lead to personalized treatment that is more beneficial to the patient.
Author contributions
BT: Conceptualization, Writing – original draft, Writing – review & editing. BZ: Conceptualization, Writing – original draft, Writing – review & editing. HC: Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Acknowledgments
The authors thank Ziqiang Li, Hunan University, for his help of Figure 1. Figures 2, 3 are created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Modlin IM, Oberg K, Chung DC, Jensen RT, de Herder WW, Thakker RV, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. (2008) 9:61–72. doi: 10.1016/S1470-2045(07)70410-2
2. Garcia-Carbonero R, Capdevila J, Crespo-Herrero G, Díaz-Pérez JA, Martínez del Prado MP, Alonso Orduña V, et al. Incidence, patterns of care and prognostic factors for outcome of gastroenteropancreatic neuroendocrine tumors (Gep-nets): results from the national cancer registry of Spain (Rgetne). Ann Oncol. (2010) 21:1794–803. doi: 10.1093/annonc/mdq022
3. Klöppel G. Neuroendocrine neoplasms: dichotomy, origin and classifications. Visc Med. (2017) 33:324–30. doi: 10.1159/000481390
4. Dasari A, Shen C, Halperin D, Zhao B, Zhou S, Xu Y, et al. Trends in the incidence, prevalence, and survival outcomes in patients with neuroendocrine tumors in the United States. JAMA Oncol. (2017) 3:1335–42. doi: 10.1001/jamaoncol.2017.0589
5. Williams ED, Sandler M. The classification of carcinoid tumours. Lancet. (1963) 281:238–9. doi: 10.1016/S0140-6736(63)90951-6
6. Kulke MH, Benson AB, Bergsland E, Berlin JD, Blaszkowsky LS, Choti MA, et al. Neuroendocrine tumors. J Natl Compr Cancer Network J Natl Compr Canc Netw. (2012) 10:724–64. doi: 10.6004/jnccn.2012.0075
7. Ruggeri RM, Benevento E, De Cicco F, Fazzalari B, Guadagno E, Hasballa I, et al. Neuroendocrine neoplasms in the context of inherited tumor syndromes: A reappraisal focused on targeted therapies. J Endocrinological Invest. (2023) 46:213–34. doi: 10.1007/s40618-022-01905-4
8. Yao JC, Hassan M, Phan A, Dagohoy C, Leary C, Mares JE, et al. One hundred years after "Carcinoid": epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. (2008) 26:3063–72. doi: 10.1200/jco.2007.15.4377
9. Xu Z, Wang L, Dai S, Chen M, Li F, Sun J, et al. Epidemiologic trends of and factors associated with overall survival for patients with gastroenteropancreatic neuroendocrine tumors in the United States. JAMA Network Open. (2021) 4:e2124750–e. doi: 10.1001/jamanetworkopen.2021.24750
10. Mosquera C, Koutlas NJ, Fitzgerald TL. Localized high-grade gastroenteropancreatic neuroendocrine tumors: defining prognostic and therapeutic factors for a disease of increasing clinical significance. Eur J Surg Oncol (EJSO). (2016) 42:1471–7. doi: 10.1016/j.ejso.2016.07.137
11. Clift AK, Kidd M, Bodei L, Toumpanakis C, Baum RP, Oberg K, et al. Neuroendocrine neoplasms of the small bowel and pancreas. Neuroendocrinology. (2020) 110:444–76. doi: 10.1159/000503721
12. Riihimäki M, Hemminki A, Sundquist K, Sundquist J, Hemminki K. The epidemiology of metastases in neuroendocrine tumors. Int J Cancer. (2016) 139:2679–86. doi: 10.1002/ijc.30400
13. Alshareefy Y, Cummins S, Mazzoleni A, Sharma V, Guggilapu S, Leong AWY, et al. A review of functional pancreatic neuroendocrine tumors: exploring the molecular pathogenesis, diagnosis and treatment. Med (Baltimore). (2023) 102:e36094. doi: 10.1097/md.0000000000036094
14. van der Graaf WTA, Tesselaar MET, McVeigh TP, Oyen WJG, Fröhling S. Biology-guided precision medicine in rare cancers: lessons from sarcomas and neuroendocrine tumours. Semin Cancer Biol. (2022) 84:228–41. doi: 10.1016/j.semcancer.2022.05.011
15. Pobłocki J, Jasińska A, Syrenicz A, Andrysiak-Mamos E, Szczuko M. The neuroendocrine neoplasms of the digestive tract: diagnosis, treatment and nutrition. Nutrients. (2020) 12:1437. doi: 10.3390/nu12051437
16. Vinik AI, Chaya C. Clinical presentation and diagnosis of neuroendocrine tumors. Hematology/Oncology Clinics North America. (2016) 30:21–48. doi: 10.1016/j.hoc.2015.08.006
17. Sorbye H, Grande E, Pavel M, Tesselaar M, Fazio N, Reed NS, et al. European neuroendocrine tumor society (Enets) 2023 guidance paper for digestive neuroendocrine carcinoma. J Neuroendocrinol. (2023) 35:e13249. doi: 10.1111/jne.13249
18. Sultana Q, Kar J, Verma A, Sanghvi S, Kaka N, Patel N, et al. A comprehensive review on neuroendocrine neoplasms: presentation, pathophysiology and management. J Clin Med. (2023) 12:5138. doi: 10.3390/jcm12155138
19. Modlin IM, Shapiro MD, Kidd M. Siegfried oberndorfer: origins and perspectives of carcinoid tumors. Hum Pathol. (2004) 35:1440–51. doi: 10.1016/j.humpath.2004.09.018
20. Janson ET, Knigge U, Dam G, Federspiel B, Grønbaek H, Stålberg P, et al. Nordic guidelines 2021 for diagnosis and treatment of gastroenteropancreatic neuroendocrine neoplasms. Acta Oncol. (2021) 60:931–41. doi: 10.1080/0284186X.2021.1921262
21. Rindi G, Mete O, Uccella S, Basturk O, La Rosa S, Brosens LAA, et al. Overview of the 2022 who classification of neuroendocrine neoplasms. Endocr Pathol. (2022) 33:115–54. doi: 10.1007/s12022-022-09708-2
22. Helderman NC, Suerink M, Kilinç G, van den Berg JG, Nielsen M, Tesselaar MET. Relation between who classification and location- and functionality-based classifications of neuroendocrine neoplasms of the digestive tract. Neuroendocrinology. (2023) 114:120–33. doi: 10.1159/000534035
23. Das S, Dasari A. Epidemiology, incidence, and prevalence of neuroendocrine neoplasms: are there global differences? Curr Oncol Rep. (2021) 23:43. doi: 10.1007/s11912-021-01029-7
24. Hallet J, Law CHL, Cukier M, Saskin R, Liu N, Singh S. Exploring the rising incidence of neuroendocrine tumors: A population-based analysis of epidemiology, metastatic presentation, and outcomes. Cancer. (2015) 121:589–97. doi: 10.1002/cncr.29099
25. Alwan H, La Rosa S, Andreas Kopp P, Germann S, Maspoli-Conconi M, Sempoux C, et al. Incidence trends of lung and gastroenteropancreatic neuroendocrine neoplasms in Switzerland. Cancer Med. (2020) 9:9454–61. doi: 10.1002/cam4.3524
26. Genus TSE, Bouvier C, Wong KF, Srirajaskanthan R, Rous BA, Talbot DC, et al. Impact of neuroendocrine morphology on cancer outcomes and stage at diagnosis: A uk nationwide cohort study 2013-2015. Br J Cancer. (2019) 121:966–72. doi: 10.1038/s41416-019-0606-3
27. Thiis-Evensen E, Boyar Cetinkaya R. Incidence and prevalence of neuroendocrine neoplasms in Norway 1993–2021. J Neuroendocrinol. (2023) 35:e13264. doi: 10.1111/jne.13264
28. Gudmundsdottir H, Möller PH, Jonasson JG, Björnsson ES. Gastroenteropancreatic neuroendocrine tumors in Iceland: A population-based study. Scandinavian J Gastroenterol. (2019) 54:69–75. doi: 10.1080/00365521.2018.1553061
29. Zheng R, Zhao H, An L, Zhang S, Chen R, Wang S, et al. Incidence and survival of neuroendocrine neoplasms in China with comparison to the United States. Chin Med J (Engl). (2023) 136:1216–24. doi: 10.1097/cm9.0000000000002643
30. Masui T, Ito T, Komoto I, Uemoto S. Recent epidemiology of patients with gastro-entero-pancreatic neuroendocrine neoplasms (Gep-nen) in Japan: A population-based study. BMC Cancer. (2020) 20:1104. doi: 10.1186/s12885-020-07581-y
31. White BE, Rous B, Chandrakumaran K, Wong K, Bouvier C, Van Hemelrijck M, et al. Incidence and survival of neuroendocrine neoplasia in england 1995-2018: A retrospective, population-based study. Lancet Reg Health Eur. (2022) 23:100510. doi: 10.1016/j.lanepe.2022.100510
32. Takayanagi D, Cho H, Machida E, Kawamura A, Takashima A, Wada S, et al. Update on epidemiology, diagnosis, and biomarkers in gastroenteropancreatic neuroendocrine neoplasms. Cancers. (2022) 14:1119. doi: 10.3390/cancers14051119
33. White BE, Russell B, Remmers S, Rous B, Chandrakumaran K, Wong KF, et al. Sex differences in survival from neuroendocrine neoplasia in england 2012&Ndash;2018: A retrospective, population-based study. Cancers. (2023) 15(6):1863. doi: 10.3390/cancers15061863
34. Greenberg JA, Ivanov NA, Egan CE, Lee YJ, Zarnegar R, Fahey TJ, et al. Sex-based clinicopathologic and survival differences among patients with pancreatic neuroendocrine tumors. J Gastrointestinal Surg. (2022) 26:2321–9. doi: 10.1007/s11605-022-05345-6
35. Jann H, Krieg S, Krieg A, Eschrich J, Luedde T, Kostev K, et al. Analyses of sex-based clinicopathological differences among patients with gastrointestinal neuroendocrine neoplasms in europe. J Cancer Res Clin Oncol. (2023) 149:7557–63. doi: 10.1007/s00432-023-04711-4
36. Liu X, Chen B, Chen J, Su Z, Sun S. The incidence, prevalence, and survival analysis of pancreatic neuroendocrine tumors in the United States. J Endocrinological Invest. (2023) 46:1373–84. doi: 10.1007/s40618-022-01985-2
37. Leoncini E, Boffetta P, Shafir M, Aleksovska K, Boccia S, Rindi G. Increased incidence trend of low-grade and high-grade neuroendocrine neoplasms. Endocrine. (2017) 58:368–79. doi: 10.1007/s12020-017-1273-x
38. Ito T, Sasano H, Tanaka M, Osamura RY, Sasaki I, Kimura W, et al. Epidemiological study of gastroenteropancreatic neuroendocrine tumors in Japan. J Gastroenterol. (2010) 45:234–43. doi: 10.1007/s00535-009-0194-8
39. Jensen RT, Berna MJ, Bingham DB, Norton JA. Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer. (2008) 113:1807–43. doi: 10.1002/cncr.23648
40. Thakker RV, Newey PJ, Walls GV, Bilezikian J, Dralle H, Ebeling PR, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (Men1). J Clin Endocrinol Metab. (2012) 97:2990–3011. doi: 10.1210/jc.2012-1230
41. Toliat MR, Berger W, Ropers HH, Neuhaus P, Wiedenmann B. Mutations in the men I gene in sporadic neuroendocrine tumours of gastroenteropancreatic system. Lancet (London England). (1997) 350:1223. doi: 10.1016/s0140-6736(05)63453-8
42. Corbo V, Dalai I, Scardoni M, Barbi S, Beghelli S, Bersani S, et al. Men1 in pancreatic endocrine tumors: analysis of gene and protein status in 169 sporadic neoplasms reveals alterations in the vast majority of cases. Endocrine-related Cancer. (2010) 17:771–83. doi: 10.1677/erc-10-0028
43. Modica R, Liccardi A, Minotta R, Cannavale G, Benevento E, Colao A. Current understanding of pathogenetic mechanisms in neuroendocrine neoplasms. Expert Rev Endocrinol Metab. (2024) 19:49–61. doi: 10.1080/17446651.2023.2279540
44. Giusti F, Cianferotti L, Boaretto F, Cetani F, Cioppi F, Colao A, et al. Multiple endocrine neoplasia syndrome type 1: institution, management, and data analysis of a nationwide multicenter patient database. Endocrine. (2017) 58:349–59. doi: 10.1007/s12020-017-1234-4
45. Vezzosi D, Cardot-Bauters C, Bouscaren N, Lebras M, Bertholon-Grégoire M, Niccoli P, et al. Long-term results of the surgical management of insulinoma patients with men1: A groupe D'étude des tumeurs endocrines (Gte) retrospective study. Eur J Endocrinol. (2015) 172:309–19. doi: 10.1530/eje-14-0878
46. Görtz B, Roth J, Krähenmann A, de Krijger RR, Muletta-, Rütimann K, et al. Mutations and allelic deletions of the men1 gene are associated with a subset of sporadic endocrine pancreatic and neuroendocrine tumors and not restricted to foregut neoplasms. Am J Pathol. (1999) 154:429–36. doi: 10.1016/s0002-9440(10)65289-3
47. Lawrence B, Blenkiron C, Parker K, Tsai P, Fitzgerald S, Shields P, et al. Recurrent loss of heterozygosity correlates with clinical outcome in pancreatic neuroendocrine cancer. NPJ genomic Med. (2018) 3:18. doi: 10.1038/s41525-018-0058-3
48. Agarwal SK, Guru SC, Heppner C, Erdos MR, Collins RM, Park SY, et al. Menin interacts with the ap1 transcription factor jund and represses jund-activated transcription. Cell. (1999) 96:143–52. doi: 10.1016/s0092-8674(00)80967-8
49. Kaji H, Canaff L, Lebrun JJ, Goltzman D, Hendy GN. Inactivation of menin, a smad3-interacting protein, blocks transforming growth factor type beta signaling. Proc Natl Acad Sci United States America. (2001) 98:3837–42. doi: 10.1073/pnas.061358098
50. Feng Z, Wang L, Sun Y, Jiang Z, Domsic J, An C, et al. Menin and daxx interact to suppress neuroendocrine tumors through epigenetic control of the membrane metallo-endopeptidase. Cancer Res. (2017) 77:401–11. doi: 10.1158/0008-5472.Can-16-1567
51. Wong C, Tang LH, Davidson C, Vosburgh E, Chen W, Foran DJ, et al. Two well-differentiated pancreatic neuroendocrine tumor mouse models. Cell Death differentiation. (2020) 27:269–83. doi: 10.1038/s41418-019-0355-0
52. Ye Z, Chen H, Ji S, Hu Y, Lou X, Zhang W, et al. Men1 promotes ferroptosis by inhibiting mtor-scd1 axis in pancreatic neuroendocrine tumors. Acta Biochim Biophys Sin. (2022) 54:1599–609. doi: 10.3724/abbs.2022162
53. Tirosh A, Mukherjee S, Lack J, Gara SK, Wang S, Quezado MM, et al. Distinct genome-wide methylation patterns in sporadic and hereditary nonfunctioning pancreatic neuroendocrine tumors. Cancer. (2019) 125:1247–57. doi: 10.1002/cncr.31930
54. Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, et al. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding P27kip1 and P18ink4c. Proc Natl Acad Sci United States America. (2005) 102:14659–64. doi: 10.1073/pnas.0503484102
55. Ersahin T, Tuncbag N, Cetin-Atalay R. The pi3k/akt/mtor interactive pathway. Mol Biosyst. (2015) 11:1946–54. doi: 10.1039/c5mb00101c
56. Leslie NR, Yang X, Downes CP, Weijer CJ. The regulation of cell migration by pten. Biochem Soc Trans. (2005) 33:1507–8. doi: 10.1042/bst0331507
57. Álvarez-Garcia V, Tawil Y, Wise HM, Leslie NR. Mechanisms of pten loss in cancer: it's all about diversity. Semin Cancer Biol. (2019) 59:66–79. doi: 10.1016/j.semcancer.2019.02.001
58. Sauter M, Belousova E, Benedik MP, Carter T, Cottin V, Curatolo P, et al. Rare manifestations and Malignancies in tuberous sclerosis complex: findings from the tuberous sclerosis registry to increase disease awareness (Tosca). Orphanet J rare Dis. (2021) 16:301. doi: 10.1186/s13023-021-01917-y
59. Koc G, Sugimoto S, Kuperman R, Kammen BF, Karakas SP. Pancreatic tumors in children and young adults with tuberous sclerosis complex. Pediatr Radiol. (2017) 47:39–45. doi: 10.1007/s00247-016-3701-0
60. Sancak O, Nellist M, Goedbloed M, Elfferich P, Wouters C, Maat-Kievit A, et al. Mutational analysis of the tsc1 and tsc2 genes in a diagnostic setting: genotype–phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur J Hum genetics: EJHG. (2005) 13:731–41. doi: 10.1038/sj.ejhg.5201402
61. Castro AF, Rebhun JF, Clark GJ, Quilliam LA. Rheb binds tuberous sclerosis complex 2 (Tsc2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem. (2003) 278:32493–6. doi: 10.1074/jbc.C300226200
62. Jiao Y, Shi C, Edil BH, de Wilde RF, Klimstra DS, Maitra A, et al. Daxx/atrx, men1, and mtor pathway genes are frequently altered in pancreatic neuroendocrine tumors. Sci (New York NY). (2011) 331:1199–203. doi: 10.1126/science.1200609
63. Scarpa A, Chang DK, Nones K, Corbo V, Patch AM, Bailey P, et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature. (2017) 543:65–71. doi: 10.1038/nature21063
64. Missiaglia E, Dalai I, Barbi S, Beghelli S, Falconi M, della Peruta M, et al. Pancreatic endocrine tumors: expression profiling evidences a role for akt-mtor pathway. J Clin oncology: Off J Am Soc Clin Oncol. (2010) 28:245–55. doi: 10.1200/jco.2008.21.5988
65. Perren A, Komminoth P, Saremaslani P, Matter C, Feurer S, Lees JA, et al. Mutation and expression analyses reveal differential subcellular compartmentalization of pten in endocrine pancreatic tumors compared to normal islet cells. Am J Pathol. (2000) 157:1097–103. doi: 10.1016/s0002-9440(10)64624-x
66. Jiang X, Shan A, Su Y, Cheng Y, Gu W, Wang W, et al. Mir-144/451 promote cell proliferation via targeting pten/akt pathway in insulinomas. Endocrinology. (2015) 156:2429–39. doi: 10.1210/en.2014-1966
67. Nielsen SM, Rhodes L, Blanco I, Chung WK, Eng C, Maher ER, et al. Von hippel-lindau disease: genetics and role of genetic counseling in a multiple neoplasia syndrome. J Clin oncology: Off J Am Soc Clin Oncol. (2016) 34:2172–81. doi: 10.1200/jco.2015.65.6140
68. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein vhl targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. (1999) 399:271–5. doi: 10.1038/20459
69. Choudhry H, Harris AL. Advances in hypoxia-inducible factor biology. Cell Metab. (2018) 27:281–98. doi: 10.1016/j.cmet.2017.10.005
70. Zhang J, Jia Z, Li Q, Wang L, Rashid A, Zhu Z, et al. Elevated expression of vascular endothelial growth factor correlates with increased angiogenesis and decreased progression-free survival among patients with low-grade neuroendocrine tumors. Cancer. (2007) 109:1478–86. doi: 10.1002/cncr.22554
71. Terris B, Scoazec JY, Rubbia L, Bregeaud L, Pepper MS, Ruszniewski P, et al. Expression of vascular endothelial growth factor in digestive neuroendocrine tumours. Histopathology. (1998) 32:133–8. doi: 10.1046/j.1365-2559.1998.00321.x
72. Hansel DE, Rahman A, Hermans J, de Krijger RR, Ashfaq R, Yeo CJ, et al. Liver metastases arising from well-differentiated pancreatic endocrine neoplasms demonstrate increased vegf-C expression. Modern pathology: an Off J United States Can Acad Pathology Inc. (2003) 16:652–9. doi: 10.1097/01.Mp.0000077416.68489.50
73. Mercurio AM, Bachelder RE, Bates RC, Chung J. Autocrine signaling in carcinoma: vegf and the alpha6beta4 integrin. Semin Cancer Biol. (2004) 14:115–22. doi: 10.1016/j.semcancer.2003.09.016
74. Wiszniak S, Schwarz Q. Exploring the intracrine functions of vegf-A. Biomolecules. (2021) 11(1):128. doi: 10.3390/biom11010128
75. Schmitt AM, Schmid S, Rudolph T, Anlauf M, Prinz C, Klöppel G, et al. Vhl inactivation is an important pathway for the development of Malignant sporadic pancreatic endocrine tumors. Endocrine-related Cancer. (2009) 16:1219–27. doi: 10.1677/erc-08-0297
76. Geurts JL. Inherited syndromes involving pancreatic neuroendocrine tumors. J gastrointestinal Oncol. (2020) 11:559–66. doi: 10.21037/jgo.2020.03.09
77. Noë M, Pea A, Luchini C, Felsenstein M, Barbi S, Bhaijee F, et al. Whole-exome sequencing of duodenal neuroendocrine tumors in patients with neurofibromatosis type 1. Modern pathology: an Off J United States Can Acad Pathology Inc. (2018) 31:1532–8. doi: 10.1038/s41379-018-0082-y
78. Guo Y, Tian C, Cheng Z, Chen R, Li Y, Su F, et al. Molecular and functional heterogeneity of primary pancreatic neuroendocrine tumors and metastases. Neuroendocrinology. (2023) 113:943–56. doi: 10.1159/000530968
79. Tang J, Wu S, Liu H, Stratt R, Barak OG, Shiekhattar R, et al. A novel transcription regulatory complex containing death domain-associated protein and the atr-X syndrome protein. J Biol Chem. (2004) 279:20369–77. doi: 10.1074/jbc.M401321200
80. Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. Daxx is an H3.3-specific histone chaperone and cooperates with atrx in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci United States America. (2010) 107:14075–80. doi: 10.1073/pnas.1008850107
81. Drané P, Ouararhni K, Depaux A, Shuaib M, Hamiche A. The death-associated protein daxx is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. (2010) 24:1253–65. doi: 10.1101/gad.566910
82. Heaphy CM, de Wilde RF, Jiao Y, Klein AP, Edil BH, Shi C, et al. Altered telomeres in tumors with atrx and daxx mutations. Sci (New York NY). (2011) 333:425. doi: 10.1126/science.1207313
83. Zhang H, He J, Li J, Tian D, Gu L, Zhou M. Methylation of rassf1a gene promoter is regulated by P53 and daxx. FASEB journal: Off Publ Fed Am Societies Exp Biol. (2013) 27:232–42. doi: 10.1096/fj.12-215491
84. Benitez JA, Ma J, D'Antonio M, Boyer A, Camargo MF, Zanca C, et al. Pten regulates glioblastoma oncogenesis through chromatin-associated complexes of daxx and histone H3.3. Nat Commun. (2017) 8:15223. doi: 10.1038/ncomms15223
85. Chan CS, Laddha SV, Lewis PW, Koletsky MS, Robzyk K, Da Silva E, et al. Atrx, daxx or men1 mutant pancreatic neuroendocrine tumors are a distinct alpha-cell signature subgroup. Nat Commun. (2018) 9:4158. doi: 10.1038/s41467-018-06498-2
86. Marinoni I, Kurrer AS, Vassella E, Dettmer M, Rudolph T, Banz V, et al. Loss of daxx and atrx are associated with chromosome instability and reduced survival of patients with pancreatic neuroendocrine tumors. Gastroenterology. (2014) 146:453–60.e5. doi: 10.1053/j.gastro.2013.10.020
87. Heaphy CM, Singhi AD. Reprint of: the diagnostic and prognostic utility of incorporating daxx, atrx, and alternative lengthening of telomeres (Alt) to the evaluation of pancreatic neuroendocrine tumors (Pannets). Hum Pathol. (2023) 132:1–11. doi: 10.1016/j.humpath.2023.01.004
88. Hackeng WM, Brosens LAA, Kim JY, O'Sullivan R, Sung YN, Liu TC, et al. Non-functional pancreatic neuroendocrine tumours: atrx/daxx and alternative lengthening of telomeres (Alt) are prognostically independent from arx/pdx1 expression and tumour size. Gut. (2022) 71:961–73. doi: 10.1136/gutjnl-2020-322595
89. Francis JM, Kiezun A, Ramos AH, Serra S, Pedamallu CS, Qian ZR, et al. Somatic mutation of cdkn1b in small intestine neuroendocrine tumors. Nat Genet. (2013) 45:1483–6. doi: 10.1038/ng.2821
90. Samsom KG, Levy S, van Veenendaal LM, Roepman P, Kodach LL, Steeghs N, et al. Driver mutations occur frequently in metastases of well-differentiated small intestine neuroendocrine tumours. Histopathology. (2021) 78:556–66. doi: 10.1111/his.14252
91. Andersson E, Swärd C, Stenman G, Ahlman H, Nilsson O. High-resolution genomic profiling reveals gain of chromosome 14 as a predictor of poor outcome in ileal carcinoids. Endocrine-related Cancer. (2009) 16:953–66. doi: 10.1677/erc-09-0052
92. Nieser M, Henopp T, Brix J, Stoß L, Sitek B, Naboulsi W, et al. Loss of chromosome 18 in neuroendocrine tumors of the small intestine: the enigma remains. Neuroendocrinology. (2017) 104:302–12. doi: 10.1159/000446917
93. Karpathakis A, Dibra H, Pipinikas C, Feber A, Morris T, Francis J, et al. Progressive epigenetic dysregulation in neuroendocrine tumour liver metastases. Endocrine-related Cancer. (2017) 24:L21–l5. doi: 10.1530/erc-16-0419
94. Bollard J, Massoma P, Vercherat C, Blanc M, Lepinasse F, Gadot N, et al. The axon guidance molecule semaphorin 3f is a negative regulator of tumor progression and proliferation in ileal neuroendocrine tumors. Oncotarget. (2015) 6:36731–45. doi: 10.18632/oncotarget.5481
95. Li SC, Essaghir A, Martijn C, Lloyd RV, Demoulin JB, Oberg K, et al. Global microrna profiling of well-differentiated small intestinal neuroendocrine tumors. Modern pathology: an Off J United States Can Acad Pathology Inc. (2013) 26:685–96. doi: 10.1038/modpathol.2012.216
96. Miller HC, Frampton AE, Malczewska A, Ottaviani S, Stronach EA, Flora R, et al. Micrornas associated with small bowel neuroendocrine tumours and their metastases. Endocrine-related Cancer. (2016) 23:711–26. doi: 10.1530/erc-16-0044
97. Ruebel K, Leontovich AA, Stilling GA, Zhang S, Righi A, Jin L, et al. Microrna expression in ileal carcinoid tumors: downregulation of microrna-133a with tumor progression. Modern pathology: an Off J United States Can Acad Pathology Inc. (2010) 23:367–75. doi: 10.1038/modpathol.2009.161
98. Arvidsson Y, Rehammar A, Bergström A, Andersson E, Altiparmak G, Swärd C, et al. Mirna profiling of small intestinal neuroendocrine tumors defines novel molecular subtypes and identifies mir-375 as a biomarker of patient survival. Modern pathology: an Off J United States Can Acad Pathology Inc. (2018) 31:1302–17. doi: 10.1038/s41379-018-0010-1
99. Albers MB, Almquist M, Bergenfelz A, Nordenström E. Complications of surgery for gastro-entero-pancreatic neuroendocrine neoplasias. Langenbecks Arch Surg. (2020) 405:137–43. doi: 10.1007/s00423-020-01869-0
100. Pulvirenti A, Pea A, Chang DK, Jamieson NB. Clinical and molecular risk factors for recurrence following radical surgery of well-differentiated pancreatic neuroendocrine tumors. Front Med (Lausanne). (2020) 7:385. doi: 10.3389/fmed.2020.00385
101. Goretzki PE, Mogl MT, Akca A, Pratschke J. Curative and palliative surgery in patients with neuroendocrine tumors of the gastro-entero-pancreatic (Gep) tract. Rev Endocrine Metab Disord. (2018) 19:169–78. doi: 10.1007/s11154-018-9469-9
102. Cuthbertson DJ, Shankland R, Srirajaskanthan R. Diagnosis and management of neuroendocrine tumours. Clin Med. (2023) 23:119–24. doi: 10.7861/clinmed.2023-0044
103. Pulvirenti A, Javed AA, Michelakos T, Sekigami Y, Zheng J, Kalvin HL, et al. Recurring pancreatic neuroendocrine tumor: timing and pattern of recurrence and current treatment. Ann Surg. (2023) 278:e1063–e7. doi: 10.1097/sla.0000000000005809
104. Gharios J, Hain E, Dohan A, Prat F, Terris B, Bertherat J, et al. Pre- and intraoperative diagnostic requirements, benefits and risks of minimally invasive and robotic surgery for neuroendocrine tumors of the pancreas. Best Pract Res Clin Endocrinol Metab. (2019) 33:101294. doi: 10.1016/j.beem.2019.101294
105. Mansour JC, Chavin K, Morris-Stiff G, Warner SG, Cardona K, Fong ZV, et al. Management of asymptomatic, well-differentiated pnets: results of the delphi consensus process of the americas hepato-pancreato-biliary association. HPB. (2019) 21:515–23. doi: 10.1016/j.hpb.2018.09.020
106. Howe JR, Merchant NB, Conrad C, Keutgen XM, Hallet J, Drebin JA, et al. The north american neuroendocrine tumor society consensus paper on the surgical management of pancreatic neuroendocrine tumors. Pancreas. (2020) 49:1–33. doi: 10.1097/mpa.0000000000001454
107. Scott AT, Howe JR. Evaluation and management of neuroendocrine tumors of the pancreas. Surg Clin North Am. (2019) 99:793–814. doi: 10.1016/j.suc.2019.04.014
108. Zhang J, Jin J, Chen S, Gu J, Zhu Y, Qin K, et al. Minimally invasive distal pancreatectomy for pnets: laparoscopic or robotic approach? Oncotarget. (2017) 8:33872–83. doi: 10.18632/oncotarget.17513
109. Ferraro V, Tedeschi M, Laera L, Ammendola M, Riccelli U, Silvestris N, et al. The role of laparoscopic surgery in localized pancreatic neuroendocrine tumours. Curr Treat Options Oncol. (2021) 22:27. doi: 10.1007/s11864-021-00824-5
110. Zilli A, Arcidiacono PG, Conte D, Massironi S. Clinical impact of endoscopic ultrasonography on the management of neuroendocrine tumors: lights and shadows. Dig Liver Dis. (2018) 50:6–14. doi: 10.1016/j.dld.2017.10.007
111. Armellini E, Facciorusso A, Crinò SF. Efficacy and safety of endoscopic ultrasound-guided radiofrequency ablation for pancreatic neuroendocrine tumors: A systematic review and metanalysis. Medicina (Kaunas). (2023) 59(2). doi: 10.3390/medicina59020359
112. Khoury T, Sbeit W, Napoléon B. Endoscopic ultrasound guided radiofrequency ablation for pancreatic tumors: A critical review focusing on safety, efficacy and controversies. World J Gastroenterol. (2023) 29:157–70. doi: 10.3748/wjg.v29.i1.157
113. Szeliga J, Jackowski M. Pancreatic neuroendocrine neoplasms: A role of laparoscopy in surgical treatment: review. Surg Laparosc Endosc Percutan Tech. (2018) 28(3):147–52. doi: 10.1097/SLE.0000000000000523
114. Memeo R, Roselli S, Lupo L, Cherkaoui Z, Pessaux P. Laparoscopic management of neuroendocrine tumors: state-of-the-art. Trans Gastroenterol Hepatol. (2017) 2:109. doi: 10.21037/tgh.2017.11.19
115. Rindi G, Luinetti O, Cornaggia M, Capella C, Solcia E. Three subtypes of gastric argyrophil carcinoid and the gastric neuroendocrine carcinoma: A clinicopathologic study. Gastroenterology. (1993) 104:994–1006. doi: 10.1016/0016-5085(93)90266-F
116. Guo X, Zhao X, Huang G, Yu Y. Advances in endoscopic diagnosis and treatment of gastric neuroendocrine neoplasms. Digestive Dis Sci. (2024) 69:27–35. doi: 10.1007/s10620-023-08180-0
117. Gladdy RA, Strong VE, Coit D, Allen PJ, Gerdes H, Shia J, et al. Defining surgical indications for type I gastric carcinoid tumor. Ann Surg Oncol. (2009) 16:3154–60. doi: 10.1245/s10434-009-0687-y
118. Roberto GA, Rodrigues CMB, Peixoto RD, Younes RN. Gastric neuroendocrine tumor: A practical literature review. World J Gastrointest Oncol. (2020) 12:850–6. doi: 10.4251/wjgo.v12.i8.850
119. Putzer D, Schullian P, Jaschke W, Bale R. Nen: advancement in diagnosis and minimally invasive therapy. Rofo. (2020) 192:422–30. doi: 10.1055/a-1030-4631
120. Manfredi S, Walter T, Baudin E, Coriat R, Ruszniewski P, Lecomte T, et al. Management of gastric neuro-endocrine tumours in a large french national cohort (Gte). Endocrine. (2017) 57:504–11. doi: 10.1007/s12020-017-1355-9
121. Delle Fave G, O'Toole D, Sundin A, Taal B, Ferolla P, Ramage JK, et al. Enets consensus guidelines update for gastroduodenal neuroendocrine neoplasms. Neuroendocrinology. (2016) 103:119–24. doi: 10.1159/000443168
122. Jie C, Yongzhan N, Wenming W, Association. SoNNoCA-C. China anti-cancer association guideline for diagnosis and treatment of neuroendocrine neoplasm. China Oncol. (2022) 32:545–79. doi: 10.19401/j.cnki.1007-3639.2022.06.010
123. Howe JR, Cardona K, Fraker DL, Kebebew E, Untch BR, Wang Y-Z, et al. The surgical management of small bowel neuroendocrine tumors: consensus guidelines of the north american neuroendocrine tumor society. Pancreas. (2017) 46(6):715–31. doi: 10.1097/MPA.0000000000000846
124. Keck KJ, Maxwell JE, Utria AF, Bellizzi AM, Dillon JS, O'Dorisio TM, et al. The distal predilection of small bowel neuroendocrine tumors. Ann Surg Oncol. (2018) 25:3207–13. doi: 10.1245/s10434-018-6676-2
125. Pavel M, Öberg K, Falconi M, Krenning EP, Sundin A, Perren A, et al. Gastroenteropancreatic neuroendocrine neoplasms: esmo clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2020) 31:844–60. doi: 10.1016/j.annonc.2020.03.304
126. Zhang XF, Wu XN, Tsilimigras DI, Poultsides G, Rocha F, Abbott DE, et al. Duodenal neuroendocrine tumors: impact of tumor size and total number of lymph nodes examined. J Surg Oncol. (2019) 120:1302–10. doi: 10.1002/jso.25753
127. Yamaguchi T, Takahashi K, Yamada K, Bando H, Baba H, Ito M, et al. A nationwide, multi-institutional collaborative retrospective study of colorectal neuroendocrine tumors in Japan. Ann Gastroenterological Surg. (2021) 5:215–20. doi: 10.1002/ags3.12403
128. Rossi RE, Milanetto AC, Andreasi V, Campana D, Coppa J, Nappo G, et al. Risk of preoperative understaging of duodenal neuroendocrine neoplasms: A plea for caution in the treatment strategy. J Endocrinol Invest. (2021) 44:2227–34. doi: 10.1007/s40618-021-01528-1
129. Rinke A, Ambrosini V, Dromain C, Garcia-Carbonero R, Haji A, Koumarianou A, et al. European neuroendocrine tumor society (Enets) 2023 guidance paper for colorectal neuroendocrine tumours. J Neuroendocrinol. (2023) 35:e13309. doi: 10.1111/jne.13309
130. Ngamruengphong S, Kamal A, Akshintala V, Hajiyeva G, Hanada Y, Chen YI, et al. Prevalence of metastasis and survival of 788 patients with T1 rectal carcinoid tumors. Gastrointest Endosc. (2019) 89:602–6. doi: 10.1016/j.gie.2018.11.010
131. Kuiper T, van Oijen MGH, van Velthuysen MF, van Lelyveld N, van Leerdam ME, Vleggaar FD, et al. Endoscopically removed rectal nets: A nationwide cohort study. Int J Colorectal Dis. (2021) 36:535–41. doi: 10.1007/s00384-020-03801-w
132. Gonzalez RS. Diagnosis and management of gastrointestinal neuroendocrine neoplasms. Surg Pathol Clinics. (2020) 13:377–97. doi: 10.1016/j.path.2020.04.002
133. Fujii Y, Kobayashi K, Kimura S, Uehara S, Miyai H, Takiguchi S. Indications for lateral lymph node dissection in patients with rectal neuroendocrine tumors: A case report and review of the literature. Mol Clin Oncol. (2021) 14:80. doi: 10.3892/mco.2021.2242
134. Andrini E, Lamberti G, Alberici L, Ricci C, Campana D. An update on appendiceal neuroendocrine tumors. Curr Treat Options Oncol. (2023) 24:742–56. doi: 10.1007/s11864-023-01093-0
135. Nesti C, Bräutigam K, Benavent M, Bernal L, Boharoon H, Botling J, et al. Hemicolectomy versus appendectomy for patients with appendiceal neuroendocrine tumours 1–2 cm in size: A retrospective, europe-wide, pooled cohort study. Lancet Oncol. (2023) 24:187–94. doi: 10.1016/S1470-2045(22)00750-1
136. Tran CG, Sherman SK, Chandrasekharan C, Howe JR. Surgical management of neuroendocrine tumor liver metastases. Surg Oncol Clin N Am. (2021) 30:39–55. doi: 10.1016/j.soc.2020.08.001
137. Holmager P, Langer SW, Kjaer A, Ringholm L, Garbyal RS, Pommergaard H-C, et al. Surgery in patients with gastro-entero-pancreatic neuroendocrine carcinomas, neuroendocrine tumors G3 and high grade mixed neuroendocrine-non-neuroendocrine neoplasms. Curr Treat Options Oncol. (2022) 23:806–17. doi: 10.1007/s11864-022-00969-x
138. Slott C, Langer SW, Møller S, Krogh J, Klose M, Hansen CP, et al. Outlook for 615 small intestinal neuroendocrine tumor patients: recurrence risk after surgery and disease-specific survival in advanced disease. Cancers. (2024) 16(1):204. doi: 10.3390/cancers16010204
139. Merola E, Pascher A, Rinke A, Bartsch DK, Zerbi A, Nappo G, et al. Radical resection in entero-pancreatic neuroendocrine tumors: recurrence-free survival rate and definition of a risk score for recurrence. Ann Surg Oncol. (2022) 29:5568–77. doi: 10.1245/s10434-022-11837-1
140. Hallet J, Clarke CN. Aso practice guidelines series: surgical management of gastrointestinal (Midgut) neuroendocrine neoplasms. Ann Surg Oncol. (2024) 31:1704–13. doi: 10.1245/s10434-023-14802-8
141. Stueven AK, Kayser A, Wetz C, Amthauer H, Wree A, Tacke F, et al. Somatostatin analogues in the treatment of neuroendocrine tumors: past, present and future. Int J Mol Sci. (2019) 20:3049. doi: 10.3390/ijms20123049
142. Agarwal P, Mohamed A. Systemic therapy of advanced well-differentiated small bowel neuroendocrine tumors progressive on somatostatin analogues. Curr Treat Options Oncol. (2022) 23:1233–46. doi: 10.1007/s11864-022-00998-6
143. Caplin ME, Pavel M, Ćwikła JB, Phan AT, Raderer M, Sedláčková E, et al. Lanreotide in metastatic enteropancreatic neuroendocrine tumors. N Engl J Med. (2014) 371:224–33. doi: 10.1056/NEJMoa1316158
144. Palmieri L-J, Dermine S, Barré A, Dhooge M, Brezault C, Cottereau A-S, et al. Medical treatment of advanced pancreatic neuroendocrine neoplasms. J Clin Med. (2020) 9:1860. doi: 10.3390/jcm9061860
145. Srirajaskanthan R, Watkins J, Marelli L, Khan K, Caplin ME. Expression of somatostatin and dopamine 2 receptors in neuroendocrine tumours and the potential role for new biotherapies. Neuroendocrinology. (2009) 89:308–14. doi: 10.1159/000179899
146. Rai U, Thrimawithana TR, Valery C, Young SA. Therapeutic uses of somatostatin and its analogues: current view and potential applications. Pharmacol Ther. (2015) 152:98–110. doi: 10.1016/j.pharmthera.2015.05.007
147. Gomes-Porras M, Cárdenas-Salas J, Álvarez-Escolá C. Somatostatin analogs in clinical practice: A review. Int J Mol Sci. (2020) 21(5):1682. doi: 10.3390/ijms21051682
148. Pavel M, Valle JW, Eriksson B, Rinke A, Caplin M, Chen J, et al. Enets consensus guidelines for the standards of care in neuroendocrine neoplasms: systemic therapy - biotherapy and novel targeted agents. Neuroendocrinology. (2017) 105:266–80. doi: 10.1159/000471880
149. La Salvia A, Modica R, Rossi RE, Spada F, Rinzivillo M, Panzuto F, et al. Targeting neuroendocrine tumors with octreotide and lanreotide: key points for clinical practice from net specialists. Cancer Treat Rev. (2023) 117:102560. doi: 10.1016/j.ctrv.2023.102560
150. Mazziotti G, Mosca A, Frara S, Vitale G, Giustina A. Somatostatin analogs in the treatment of neuroendocrine tumors: current and emerging aspects. Expert Opin Pharmacotherapy. (2017) 18:1679–89. doi: 10.1080/14656566.2017.1391217
151. Rinke A, Wittenberg M, SChade-Brittinger C, Aminossadati B, Ronicke E, Gress TM, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide lar in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors (Promid): results of long-term survival. Neuroendocrinology. (2017) 104:26–32. doi: 10.1159/000443612
152. Caplin ME, Pavel M, Phan AT, Ćwikła JB, Sedláčková E, Thanh XT, et al. Lanreotide autogel/depot in advanced enteropancreatic neuroendocrine tumours: final results of the clarinet open-label extension study. Endocrine. (2021) 71:502–13. doi: 10.1007/s12020-020-02475-2
153. Pavel M, Ćwikła JB, Lombard-Bohas C, Borbath I, Shah T, Pape UF, et al. Efficacy and safety of high-dose lanreotide autogel in patients with progressive pancreatic or midgut neuroendocrine tumours: clarinet forte phase 2 study results. Eur J Cancer. (2021) 157:403–14. doi: 10.1016/j.ejca.2021.06.056
154. Rinke A, Müller H-H, SChade-Brittinger C, Klose K-J, Barth P, Wied M, et al. Placebo-controlled, double-blind, prospective, randomized study on the effect of octreotide lar in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the promid study group. J Clin Oncol. (2009) 27:4656–63. doi: 10.1200/jco.2009.22.8510
155. Chahid Y, Hashimi K, van de Garde EMW, Klümpen HJ, Hendrikse NH, Booij J, et al. The influence of long-acting somatostatin analogs on 68 ga-dotatate uptake in patients with neuroendocrine tumors. Clin Nucl Med. (2023) 48:757–62. doi: 10.1097/rlu.0000000000004776
156. Halfdanarson TR, Strosberg JR, Tang L, Bellizzi AM, Bergsland EK, O'Dorisio TM, et al. The north american neuroendocrine tumor society consensus guidelines for surveillance and medical management of pancreatic neuroendocrine tumors. Pancreas. (2020) 49:863–81. doi: 10.1097/mpa.0000000000001597
157. Rivero JD, Perez K, Kennedy EB, Mittra ES, Vijayvergia N, Arshad J, et al. Systemic therapy for tumor control in metastatic well-differentiated gastroenteropancreatic neuroendocrine tumors: asco guideline. J Clin Oncol. (2023) 41:5049–67. doi: 10.1200/jco.23.01529
158. Alonso-Gordoa T, Manneh R, Grande E, Molina-Cerrillo J. High-dose somatostatin analogs for the treatment of neuroendocrine neoplasms: where are we now? Curr Treat Options Oncol. (2022) 23:1001–13. doi: 10.1007/s11864-022-00983-z
159. Faggiano A. Long-acting somatostatin analogs and well differentiated neuroendocrine tumors: A 20-year-old story. J Endocrinol Invest. (2024) 47:35–46. doi: 10.1007/s40618-023-02170-9
160. Bolanowski M, Kałużny M, Witek P, Jawiarczyk-Przybyłowska A. Pasireotide-a Novel Somatostatin Receptor Ligand after 20 years of Use. Rev Endocr Metab Disord. (2022) 23:601–20. doi: 10.1007/s11154-022-09710-3
161. Marasco M, Dell'Unto E, Laviano A, Campana D, Panzuto F. Gastrointestinal side effects of somatostatin analogs in neuroendocrine tumors: A focused review. J Gastroenterol Hepatol. (2024) 39(9):1737–44. doi: 10.1111/jgh.16638
162. Lawhn-Heath C, Fidelman N, Chee B, Jivan S, Armstrong E, Zhang L, et al. Intraarterial peptide receptor radionuclide therapy using 90y-dotatoc for hepatic metastases of neuroendocrine tumors. J Nucl Med. (2021) 62:221–7. doi: 10.2967/jnumed.119.241273
163. Reubi JC, Schaer JC, Waser B, Mengod G. Expression and localization of somatostatin receptor sstr1, sstr2, and sstr3 messenger rnas in primary human tumors using in situ hybridization. Cancer Res. (1994) 54:3455–9.
164. Scott AT, Howe JR. Management of small bowel neuroendocrine tumors. Surg Oncol Clinics North America. (2020) 29:223–41. doi: 10.1016/j.soc.2019.11.006
165. Hicks RJ, Kwekkeboom DJ, Krenning E, Bodei L, Grozinsky-Glasberg S, Arnold R, et al. Enets consensus guidelines for the standards of care in neuroendocrine neoplasms: peptide receptor radionuclide therapy with radiolabelled somatostatin analogues. Neuroendocrinology. (2017) 105:295–309. doi: 10.1159/000475526
166. Thomas KEH, Voros BA, Boudreaux JP, Thiagarajan R, Woltering EA, Ramirez RA. Current treatment options in gastroenteropancreatic neuroendocrine carcinoma. Oncologist. (2019) 24:1076–88. doi: 10.1634/theoncologist.2018-0604
167. Saravana-Bawan B, Bajwa A, Paterson J, McEwan AJB, McMullen TPW. Efficacy of 177lu peptide receptor radionuclide therapy for the treatment of neuroendocrine tumors: A meta-analysis. Clin Nucl Med. (2019) 44(9):719–27. doi: 10.1097/RLU.0000000000002646
168. Strosberg J, El-Haddad G, Wolin E, Hendifar A, Yao J, Chasen B, et al. Phase 3 trial of 177lu-dotatate for midgut neuroendocrine tumors. New Engl J Med. (2017) 376:125–35. doi: 10.1056/NEJMoa1607427
169. Fröss-Baron K, Garske-Roman U, Welin S, Granberg D, Eriksson B, Khan T, et al. 177lu-dotatate therapy of advanced pancreatic neuroendocrine tumors heavily pretreated with chemotherapy: analysis of outcome, safety, and their determinants. Neuroendocrinology. (2020) 111:330–43. doi: 10.1159/000506746
170. Hauser H, Gerson DS, Reidy-Lagunes D, Raj N. Systemic therapies for metastatic pancreatic neuroendocrine tumors. Curr Treat Options Oncol. (2019) 20:87. doi: 10.1007/s11864-019-0690-x
171. Zhang-Yin J, Guilabert N, Kiffel T, Montravers F, Calais P, Lumbroso J, et al. Patient external dose rate after (177)Lu-dotatate therapy: factors affecting its decrease and predictive value. Int J Med Sci. (2021) 18:2725–35. doi: 10.7150/ijms.58680
172. Kim S-J, Pak K, Koo PJ, Kwak JJ, Chang S. The efficacy of 177lu-labelled peptide receptor radionuclide therapy in patients with neuroendocrine tumours: A meta-analysis. Eur J Nucl Med Mol Imaging. (2015) 42:1964–70. doi: 10.1007/s00259-015-3155-x
173. Strosberg JR, Caplin ME, Kunz PL, Ruszniewski PB, Bodei L, Hendifar A, et al. 177lu-dotatate plus long-acting octreotide versus high−Dose long-acting octreotide in patients with midgut neuroendocrine tumours (Netter-1): final overall survival and long-term safety results from an open-label, randomised, controlled, phase 3 trial. Lancet Oncol. (2021) 22:1752–63. doi: 10.1016/S1470-2045(21)00572-6
174. Sorbye H, Kong G, Grozinsky-Glasberg S. Prrt in high-grade gastroenteropancreatic neuroendocrine neoplasms (Who G3). Endocr Relat Cancer. (2020) 27:R67–r77. doi: 10.1530/erc-19-0400
175. Carlsen EA, Fazio N, Granberg D, Grozinsky-Glasberg S, Ahmadzadehfar H, Grana CM, et al. Peptide receptor radionuclide therapy in gastroenteropancreatic nen G3: A multicenter cohort study. Endocr Relat Cancer. (2019) 26:227–39. doi: 10.1530/erc-18-0424
176. Singh S, Halperin D, Myrehaug S, Herrmann K, Pavel M, Kunz PL, et al. (177)Lu]Lu-dota-tate plus long-acting octreotide versus high−Dose long-acting octreotide for the treatment of newly diagnosed, advanced grade 2-3, well-differentiated, gastroenteropancreatic neuroendocrine tumours (Netter-2): an open-label, randomised, phase 3 study. Lancet. (2024) 403:2807–17. doi: 10.1016/s0140-6736(24)00701-3
177. Ichikawa Y, Kobayashi N, Takano S, Kato I, Endo K, Inoue T. Neuroendocrine tumor theranostics. Cancer Sci. (2022) 113:1930–8. doi: 10.1111/cas.15327
178. Kunikowska J, Królicki L. Targeted Α-emitter therapy of neuroendocrine tumors. Semin Nucl Med. (2020) 50:171–6. doi: 10.1053/j.semnuclmed.2019.11.003
179. Garcia-Alvarez A, Hernando Cubero J, Capdevila J. Drug development in neuroendocrine tumors: what is on the horizon? Curr Treat Options Oncol. (2021) 22:43. doi: 10.1007/s11864-021-00834-3
180. Yordanova A, Ahrens H, Feldmann G, Brossart P, Gaertner FC, Fottner C, et al. Peptide receptor radionuclide therapy combined with chemotherapy in patients with neuroendocrine tumors. Clin Nucl Med. (2019) 44:e329–e35. doi: 10.1097/rlu.0000000000002532
181. Capello A, Krenning EP, Breeman WA, Bernard BF, de Jong M. Peptide receptor radionuclide therapy in vitro using [111in-dtpa0]Octreotide. J Nucl Med. (2003) 44:98–104.
182. Fortunati E, Bonazzi N, Zanoni L, Fanti S, Ambrosini V. Molecular imaging theranostics of neuroendocrine tumors. Semin Nucl Med. (2023) 53:539–54. doi: 10.1053/j.semnuclmed.2022.12.007
183. Delpassand ES, Tworowska I, Esfandiari R, Torgue J, Hurt J, Shafie A, et al. Targeted Α-emitter therapy with (212)Pb-dotamtate for the treatment of metastatic sstr-expressing neuroendocrine tumors: first-in-humans dose-escalation clinical trial. J Nucl Med. (2022) 63:1326–33. doi: 10.2967/jnumed.121.263230
184. Strosberg JR, Halfdanarson TR, Bellizzi AM, Chan JA, Dillon JS, Heaney AP, et al. The north american neuroendocrine tumor society consensus guidelines for surveillance and medical management of midgut neuroendocrine tumors. Pancreas. (2017) 46:707–14. doi: 10.1097/mpa.0000000000000850
185. Rabei R, Fidelman N. Liver-directed therapy for neuroendocrine tumor metastases in the era of peptide receptor radionuclide therapy. Curr Treat Options Oncol. (2023) 24:1994–2004. doi: 10.1007/s11864-023-01152-6
186. Strosberg J, Leeuwenkamp O, Siddiqui MK. Peptide receptor radiotherapy re-treatment in patients with progressive neuroendocrine tumors: A systematic review and meta-analysis. Cancer Treat Rev. (2021) 93:102141. doi: 10.1016/j.ctrv.2020.102141
187. Kim Y-i. Salvage peptide receptor radionuclide therapy in patients with progressive neuroendocrine tumors: A systematic review and meta-analysis. Nucl Med Commun. (2021) 42:451–8. doi: 10.1097/mnm.0000000000001350
188. Pusceddu S, Prinzi N, Tafuto S, Ibrahim T, Filice A, Brizzi MP, et al. Association of upfront peptide receptor radionuclide therapy with progression-free survival among patients with enteropancreatic neuroendocrine tumors. JAMA Netw Open. (2022) 5:e220290. doi: 10.1001/jamanetworkopen.2022.0290
189. Lania A, Ferraù F, Rubino M, Modica R, Colao A, Faggiano A. Neoadjuvant therapy for neuroendocrine neoplasms: recent progresses and future approaches. Front Endocrinol (Lausanne). (2021) 12:651438. doi: 10.3389/fendo.2021.651438
190. Tsoli M, Alexandraki K, Xanthopoulos C, Kassi E, Kaltsas G. Medical treatment of gastrointestinal neuroendocrine neoplasms. Horm Metab Res. (2020) 52:614–20. doi: 10.1055/a-1110-7251
191. Gordon S, Chan DLH, Bernard EJ, Eslick ME, Willowson KP, Roach PJ, et al. Single-centre experience with peptide receptor radionuclide therapy for neuroendocrine tumours (Nets): results using a theranostic molecular imaging-guided approach. J Cancer Res Clin Oncol. (2023) 149:7717–28. doi: 10.1007/s00432-023-04706-1
192. Brabander T, Teunissen JJM, Van Eijck CHJ, Franssen GJH, Feelders RA, de Herder WW, et al. Peptide receptor radionuclide therapy of neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab. (2016) 30:103–14. doi: 10.1016/j.beem.2015.10.005
193. Bodei L, Kidd M, Paganelli G, Grana CM, Drozdov I, Cremonesi M, et al. Long-term tolerability of prrt in 807 patients with neuroendocrine tumours: the value and limitations of clinical factors. Eur J Nucl Med Mol Imaging. (2015) 42:5–19. doi: 10.1007/s00259-014-2893-5
194. Sundlöv A, Sjögreen-Gleisner K. Peptide receptor radionuclide therapy – prospects for personalised treatment. Clin Oncol. (2021) 33:92–7. doi: 10.1016/j.clon.2020.10.020
195. Mosalem O, Sonbol MB, Halfdanarson TR, Starr JS. Tyrosine kinase inhibitors and immunotherapy updates in neuroendocrine neoplasms. Best Pract Res Clin Endocrinol Metab. (2023) 37:101796. doi: 10.1016/j.beem.2023.101796
196. Yao JC, Fazio N, Singh S, Buzzoni R, Carnaghi C, Wolin E, et al. Everolimus for the treatment of advanced, non-functional neuroendocrine tumours of the lung or gastrointestinal tract (Radiant-4): A randomised, placebo-controlled, phase 3 study. Lancet. (2016) 387:968–77. doi: 10.1016/s0140-6736(15)00817-x
197. Capdevila J, Teulé A, Barriuso J, Castellano D, Lopez C, Manzano JL, et al. Phase ii study of everolimus and octreotide lar in patients with nonfunctioning gastrointestinal neuroendocrine tumors: the getne1003_Everlar study. Oncologist. (2019) 24:38–46. doi: 10.1634/theoncologist.2017-0622
198. Wagner AJ, Ravi V, Riedel RF, Ganjoo K, Van Tine BA, Chugh R, et al. Nab-sirolimus for patients with Malignant perivascular epithelioid cell tumors. J Clin Oncol. (2021) 39:3660–70. doi: 10.1200/jco.21.01728
199. Lewis CS, Elnakat Thomas H, Orr-Asman MA, Green LC, Boody RE, Matiash K, et al. Mtor kinase inhibition reduces tissue factor expression and growth of pancreatic neuroendocrine tumors. J Thromb Haemost. (2019) 17:169–82. doi: 10.1111/jth.14342
200. Rugo HS, Hortobagyi GN, Yao J, Pavel M, Ravaud A, Franz D, et al. Meta-analysis of stomatitis in clinical studies of everolimus: incidence and relationship with efficacy. Ann Oncol. (2016) 27:519–25. doi: 10.1093/annonc/mdv595
201. Abdel-Rahman O, Fouad M. Risk of mucocutaneous toxicities in patients with solid tumors treated with everolimus; a systematic review and meta-analysis. Expert Rev Anticancer Ther. (2014) 14:1529–36. doi: 10.1586/14737140.2014.953936
202. Ravaud A, Urva SR, Grosch K, Cheung WK, Anak O, Sellami DB. Relationship between everolimus exposure and safety and efficacy: meta-analysis of clinical trials in oncology. Eur J Cancer. (2014) 50:486–95. doi: 10.1016/j.ejca.2013.11.022
203. Shameem R, Lacouture M, Wu S. Incidence and risk of rash to mtor inhibitors in cancer patients–a meta-analysis of randomized controlled trials. Acta Oncol. (2015) 54:124–32. doi: 10.3109/0284186x.2014.923583
204. Lee L, Ito T, Jensen RT. Everolimus in the treatment of neuroendocrine tumors: efficacy, side-effects, resistance, and factors affecting its place in the treatment sequence. Expert Opin Pharmacotherapy. (2018) 19:909–28. doi: 10.1080/14656566.2018.1476492
205. Abdel-Rahman O. Vascular endothelial growth factor (Vegf) pathway and neuroendocrine neoplasms (Nens): prognostic and therapeutic considerations. Tumour Biol. (2014) 35:10615–25. doi: 10.1007/s13277-014-2612-7
206. Ghalehbandi S, Yuzugulen J, Pranjol MZI, Pourgholami MH. The role of vegf in cancer-induced angiogenesis and research progress of drugs targeting vegf. Eur J Pharmacol. (2023) 949:175586. doi: 10.1016/j.ejphar.2023.175586
207. Sandra I, Cazacu IM, Croitoru VM, Mihaila M, Herlea V, Diculescu MM, et al. Circulating angiogenic markers in gastroenteropancreatic neuroendocrine neoplasms: A systematic review. Curr Issues Mol Biol. (2022) 44:4001–14. doi: 10.3390/cimb44090274
208. Melincovici CS, Boşca AB, Şuşman S, Mărginean M, Mihu C, Istrate M, et al. Vascular endothelial growth factor (Vegf) - key factor in normal and pathological angiogenesis. Rom J Morphol Embryol. (2018) 59:455–67.
209. Xu J, Shen L, Bai C, Wang W, Li J, Yu X, et al. Surufatinib in advanced pancreatic neuroendocrine tumours (Sanet-P): A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. (2020) 21:1489–99. doi: 10.1016/S1470-2045(20)30493-9
210. Zhao Y, Adjei AA. Targeting angiogenesis in cancer therapy: moving beyond vascular endothelial growth factor. Oncologist. (2015) 20:660–73. doi: 10.1634/theoncologist.2014-0465
211. Bertolini F, Marighetti P, Martin-Padura I, Mancuso P, Hu-Lowe DD, Shaked Y, et al. Anti-vegf and beyond: shaping a new generation of anti-angiogenic therapies for cancer. Drug Discovery Today. (2011) 16:1052–60. doi: 10.1016/j.drudis.2011.08.007
212. Xu J, Shen L, Zhou Z, Li J, Bai C, Chi Y, et al. Surufatinib in advanced extrapancreatic neuroendocrine tumours (Sanet-ep): A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. (2020) 21:1500–12. doi: 10.1016/S1470-2045(20)30496-4
213. Zhang P, Shi S, Xu J, Chen Z, Song L, Zhang X, et al. Surufatinib plus toripalimab in patients with advanced neuroendocrine tumours and neuroendocrine carcinomas: an open-label, single-arm, multi-cohort phase ii trial. Eur J Cancer. (2024) 199:113539. doi: 10.1016/j.ejca.2024.113539
214. Mizuno Y, Kudo A, Akashi T, Akahoshi K, Ogura T, Ogawa K, et al. Sunitinib shrinks net-G3 pancreatic neuroendocrine neoplasms. J Cancer Res Clin Oncol. (2018) 144:1155–63. doi: 10.1007/s00432-018-2636-2
215. Valle JW, Borbath I, Rosbrook B, Fernandez K, Raymond E. Sunitinib in patients with pancreatic neuroendocrine tumors: update of safety data. Future Oncol. (2019) 15:1219–30. doi: 10.2217/fon-2018-0882
216. Vitale G, Cozzolino A, Malandrino P, Minotta R, Puliani G, Saronni D, et al. Role of fgf system in neuroendocrine neoplasms: potential therapeutic applications. Front Endocrinol. (2021) 12:665631. doi: 10.3389/fendo.2021.665631
217. Corti F, Brizzi MP, Amoroso V, Giuffrida D, Panzuto F, Campana D, et al. Assessing the safety and activity of cabozantinib combined with lanreotide in gastroenteropancreatic and thoracic neuroendocrine tumors: rationale and protocol of the phase ii lola trial. BMC Cancer. (2023) 23:908. doi: 10.1186/s12885-023-11287-2
218. Daskalakis K, Tsoli M, Angelousi A, Kassi E, Alexandraki KI, Kolomodi D, et al. Anti-tumour activity of everolimus and sunitinib in neuroendocrine neoplasms. Endocrine Connections. (2019) 8:641–53. doi: 10.1530/ec-19-0134
219. Toledo RA, Jimenez C, Armaiz-Pena G, Arenillas C, Capdevila J, Dahia PLM. Hypoxia-inducible factor 2 alpha (Hif2α) inhibitors: targeting genetically driven tumor hypoxia. Endocr Rev. (2023) 44:312–22. doi: 10.1210/endrev/bnac025
220. Fallah J, Brave MH, Weinstock C, Mehta GU, Bradford D, Gittleman H, et al. Fda approval summary: belzutifan for von hippel-lindau disease-associated tumors. Clin Cancer Res. (2022) 28:4843–8. doi: 10.1158/1078-0432.Ccr-22-1054
221. Tirosh A, Sadowski SM, Linehan WM, Libutti SK, Patel D, Nilubol N, et al. Association of vhl genotype with pancreatic neuroendocrine tumor phenotype in patients with von hippel-lindau disease. JAMA Oncol. (2018) 4:124–6. doi: 10.1001/jamaoncol.2017.3428
222. Shah MH, Goldner WS, Benson AB, Bergsland E, Blaszkowsky LS, Brock P, et al. Neuroendocrine and adrenal tumors, version 2.2021, nccn clinical practice guidelines in oncology. J Natl Compr Cancer Network. (2021) 19:839–68. doi: 10.6004/jnccn.2021.0032
223. Cella CA, Minucci S, Spada F, Galdy S, Elgendy M, Ravenda PS, et al. Dual inhibition of mtor pathway and vegf signalling in neuroendocrine neoplasms: from bench to bedside. Cancer Treat Rev. (2015) 41:754–60. doi: 10.1016/j.ctrv.2015.06.008
224. Curry L, Soleimani M. Belzutifan: A novel therapeutic for the management of von hippel–lindau disease and beyond. Future Oncol. (2024) 20:1251–66. doi: 10.2217/fon-2023-0679
225. Choi WW, Boland JL, Kalola A, Lin J. Belzutifan (Mk-6482): biology and clinical development in solid tumors. Curr Oncol Rep. (2023) 25:123–9. doi: 10.1007/s11912-022-01354-5
226. Choueiri TK, Bauer TM, Papadopoulos KP, Plimack ER, Merchan JR, McDermott DF, et al. Inhibition of hypoxia-inducible factor-2α in renal cell carcinoma with belzutifan: A phase 1 trial and biomarker analysis. Nat Med. (2021) 27:802–5. doi: 10.1038/s41591-021-01324-7
227. de Sousa MJ, Gervaso L, Meneses-Medina MI, Spada F, Abdel-Rahman O, Fazio N. Cyclin-dependent kinases 4/6 inhibitors in neuroendocrine neoplasms: from bench to bedside. Curr Oncol Rep. (2022) 24:715–22. doi: 10.1007/s11912-022-01251-x
228. Raj N, Zheng Y, Hauser H, Chou J, Rafailov J, Bou-Ayache J, et al. Ribociclib and everolimus in well-differentiated foregut neuroendocrine tumors. Endocr Relat Cancer. (2021) 28:237–46. doi: 10.1530/erc-20-0446
229. Chan DL, Singh S. Current chemotherapy use in neuroendocrine tumors. Endocrinol Metab Clinics North America. (2018) 47:603–14. doi: 10.1016/j.ecl.2018.04.006
230. Bardasi C, Spallanzani A, Benatti S, Spada F, Laffi A, Antonuzzo L, et al. Irinotecan-based chemotherapy in extrapulmonary neuroendocrine carcinomas: survival and safety data from a multicentric italian experience. Endocrine. (2021) 74:707–13. doi: 10.1007/s12020-021-02813-y
231. Tafuto S, von Arx C, Capozzi M, Tatangelo F, Mura M, Modica R, et al. Safety and activity of metronomic temozolomide in second-line treatment of advanced neuroendocrine neoplasms. J Clin Med. (2019) 8(8):1224. doi: 10.3390/jcm8081224
232. Kunz PL, Graham NT, Catalano PJ, Nimeiri HS, Fisher GA, Longacre TA, et al. Randomized study of temozolomide or temozolomide and capecitabine in patients with advanced pancreatic neuroendocrine tumors (Ecog-acrin E2211). J Clin Oncol. (2023) 41:1359–69. doi: 10.1200/jco.22.01013
233. Fazio N, Abdel-Rahman O. Immunotherapy in neuroendocrine neoplasms: where are we now? Curr Treat Options Oncol. (2021) 22:19. doi: 10.1007/s11864-021-00817-4
234. Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. (2020) 20:651–68. doi: 10.1038/s41577-020-0306-5
235. Buchbinder EI, Desai A. Ctla-4 and pd-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. (2016) 39:98–106. doi: 10.1097/coc.0000000000000239
236. Albertelli M, Dotto A, Nista F, Veresani A, Patti L, Gay S, et al. Present and future of immunotherapy in neuroendocrine tumors. Rev Endocr Metab Disord. (2021) 22:615–36. doi: 10.1007/s11154-021-09647-z
237. Takkenkamp TJ, Jalving M, Hoogwater FJH, Walenkamp AME. The immune tumour microenvironment of neuroendocrine tumours and its implications for immune checkpoint inhibitors. Endocr Relat Cancer. (2020) 27:R329–r43. doi: 10.1530/erc-20-0113
238. Bösch F, Brüwer K, Altendorf-Hofmann A, Auernhammer CJ, Spitzweg C, Westphalen CB, et al. Immune checkpoint markers in gastroenteropancreatic neuroendocrine neoplasia. Endocrine-Related Cancer. (2019) 26:293–301. doi: 10.1530/erc-18-0494
239. Gile JJ, Liu AJ, McGarrah PW, Eiring RA, Hobday TJ, Starr JS, et al. Efficacy of checkpoint inhibitors in neuroendocrine neoplasms: mayo clinic experience. Pancreas. (2021) 50:500–5. doi: 10.1097/mpa.0000000000001794
240. Xu G, Wang Y, Zhang H, She X, Yang J. Immunotherapy and potential predictive biomarkers in the treatment of neuroendocrine neoplasia. Future Oncol. (2021) 17:1069–81. doi: 10.2217/fon-2020-0703
241. Kaur J, Vijayvergia N. Narrative review of immunotherapy in gastroentero-pancreatic neuroendocrine neoplasms. Curr Oncol. (2023) 30:8653–64. doi: 10.3390/curroncol30090627
242. da Silva A, Bowden M, Zhang S, Masugi Y, Thorner AR, Herbert ZT, et al. Characterization of the neuroendocrine tumor immune microenvironment. Pancreas. (2018) 47:1123–9. doi: 10.1097/mpa.0000000000001150
243. Roberts JA, Gonzalez RS, Das S, Berlin J, Shi C. Expression of pd-1 and pd-L1 in poorly differentiated neuroendocrine carcinomas of the digestive system: A potential target for anti–pd-1/pd-L1 therapy. Hum Pathol. (2017) 70:49–54. doi: 10.1016/j.humpath.2017.10.003
244. Xing J, Ying H, Li J, Gao Y, Sun Z, Li J, et al. Immune checkpoint markers in neuroendocrine carcinoma of the digestive system. Front Oncol. (2020) 10:132. doi: 10.3389/fonc.2020.00132
245. Esfahani SA, Ferreira CDA, Summer P, Mahmood U, Heidari P. Addition of peptide receptor radiotherapy to immune checkpoint inhibition therapy improves outcomes in neuroendocrine tumors. J Nucl Med. (2023) 64:1056–61. doi: 10.2967/jnumed.123.265391
246. Maggio I, Manuzzi L, Lamberti G, Ricci AD, Tober N, Campana D. Landscape and future perspectives of immunotherapy in neuroendocrine neoplasia. Cancers. (2020) 12(4):832. doi: 10.3390/cancers12040832
247. Fottner C, Apostolidis L, Ferrata M, Krug S, Michl P, SChad A, et al. A phase ii, open label, multicenter trial of avelumab in patients with advanced, metastatic high-grade neuroendocrine carcinomas nec G3 (Who 2010) progressive after first-line chemotherapy (Avenec). J Clin Oncol. (2019) 37:4103. doi: 10.1200/JCO.2019.37.15_suppl.4103
248. Patel SP, Othus M, Chae YK, Giles FJ, Hansel DE, Singh PP, et al. A phase ii basket trial of dual anti–ctla-4 and anti–pd-1 blockade in rare tumors (Dart swog 1609) in patients with nonpancreatic neuroendocrine tumors. Clin Cancer Res. (2020) 26:2290–6. doi: 10.1158/1078-0432.Ccr-19-3356
249. Vijayvergia N, Dasari A, Deng M, Litwin S, Al-Toubah T, Alpaugh RK, et al. Pembrolizumab monotherapy in patients with previously treated metastatic high-grade neuroendocrine neoplasms: joint analysis of two prospective, non-randomised trials. Br J Cancer. (2020) 122:1309–14. doi: 10.1038/s41416-020-0775-0
250. Strosberg J, Mizuno N, Doi T, Grande E, Delord JP, Shapira-Frommer R, et al. Efficacy and safety of pembrolizumab in previously treated advanced neuroendocrine tumors: results from the phase ii keynote-158 study. Clin Cancer Res. (2020) 26:2124–30. doi: 10.1158/1078-0432.Ccr-19-3014
251. Klein O, Kee D, Markman B, Michael M, Underhill C, Carlino MS, et al. Immunotherapy of ipilimumab and nivolumab in patients with advanced neuroendocrine tumors: A subgroup analysis of the ca209-538 clinical trial for rare cancers. Clin Cancer Res. (2020) 26:4454–9. doi: 10.1158/1078-0432.Ccr-20-0621
252. Gubbi S, Vijayvergia N, Yu JQ, Klubo-Gwiezdzinska J, Koch CA. Immune checkpoint inhibitor therapy in neuroendocrine tumors. Horm Metab Res. (2022) 54:795–812. doi: 10.1055/a-1908-7790
253. Herrera-Martínez AD, Hofland J, Hofland LJ, Brabander T, Eskens F, Gálvez Moreno MA, et al. Targeted systemic treatment of neuroendocrine tumors: current options and future perspectives. Drugs. (2019) 79:21–42. doi: 10.1007/s40265-018-1033-0
254. Pavel M, O''Toole D, Costa F, Capdevila J, Gross D, Kianmanesh R, et al. Enets consensus guidelines update for the management of distant metastatic disease of intestinal, pancreatic, bronchial neuroendocrine neoplasms (Nen) and nen of unknown primary site. Neuroendocrinology. (2016) 103:172–85. doi: 10.1159/000443167
255. Oberg K, Funa K, Alm G. Effects of leukocyte interferon on clinical symptoms and hormone levels in patients with mid-gut carcinoid tumors and carcinoid syndrome. N Engl J Med. (1983) 309:129–33. doi: 10.1056/nejm198307213090301
256. Frank M, Klose KJ, Wied M, Ishaque N, SChade-Brittinger C, Arnold R. Combination therapy with octreotide and Α-interferon. Off J Am Coll Gastroenterol | ACG. (1999) 94:1381–7. doi: 10.1111/j.1572-0241.1999.01090.x
257. Larsson G, Janson ET. Anemia in patients with midgut carcinoid, treated with alpha interferon: effects by erythropoietin treatment on the perceived quality of life. Eur J Cancer Care. (2008) 17:200–4. doi: 10.1111/j.1365-2354.2007.00844.x
258. Zandee Wouter T, de Herder Wouter W. The evolution of neuroendocrine tumor treatment reflected by enets guidelines. Neuroendocrinology. (2018) 106:357–65. doi: 10.1159/000486096
259. Yao JC, Guthrie KA, Moran C, Strosberg JR, Kulke MH, Chan JA, et al. Phase iii prospective randomized comparison trial of depot octreotide plus interferon alfa-2b versus depot octreotide plus bevacizumab in patients with advanced carcinoid tumors: swog S0518. J Clin Oncol. (2017) 35:1695–703. doi: 10.1200/jco.2016.70.4072
260. Arnold R, Rinke A, Klose K-J, Müller H-H, Wied M, Zamzow K, et al. Octreotide versus octreotide plus interferon-alpha in endocrine gastroenteropancreatic tumors: A randomized trial. Clin Gastroenterol Hepatol. (2005) 3:761–71. doi: 10.1016/S1542-3565(05)00481-7
261. Zuetenhorst JM, Valdes Olmos RA, Muller M, Hoefnagel CA, Taal BG. Interferon and meta-iodobenzylguanidin combinations in the treatment of metastatic carcinoid tumours. Endocrine-Related Cancer Endocr Relat Cancer. (2004) 11:553–61. doi: 10.1677/erc.1.00810
262. Chan DL, Thompson R, Lam M, Pavlakis N, Hallet J, Law C, et al. External beam radiotherapy in the treatment of gastroenteropancreatic neuroendocrine tumours: A systematic review. Clin Oncol. (2018) 30:400–8. doi: 10.1016/j.clon.2018.03.006
263. Maidment BW, Ellison T, Herman JM, Sharma NK, Laheru D, Regine W, et al. Radiation in the management of pancreatic neuroendocrine tumors. J Clin Oncol. (2012) 30:335. doi: 10.1200/jco.2012.30.4_suppl.335
264. Iwata T, Ueno H, Itami J, Ito Y, Inaba K, Morizane C, et al. Efficacy of radiotherapy for primary tumor in patients with unresectable pancreatic neuroendocrine tumors. Japanese J Clin Oncol. (2017) 47:826–31. doi: 10.1093/jjco/hyx081
265. Bellefkih FZ, Benchakroun N, Lalya I, Amaoui B, El Kacemi H, Acharki A, et al. Radiotherapy in the management of rare gastrointestinal cancers: A systematic review. Cancer Radiother. (2023) 27:622–37. doi: 10.1016/j.canrad.2023.06.010
Keywords: gastroenteropancreatic neuroendocrine neoplasm, epidemiology, treatment, neuroendocrine tumor, genetics
Citation: Tan B, Zhang B and Chen H (2024) Gastroenteropancreatic neuroendocrine neoplasms: epidemiology, genetics, and treatment. Front. Endocrinol. 15:1424839. doi: 10.3389/fendo.2024.1424839
Received: 15 May 2024; Accepted: 10 September 2024;
Published: 30 September 2024.
Edited by:
Roberta Elisa Rossi, Humanitas Research Hospital, ItalyReviewed by:
Alex Giakoustidis, Aristotle University of Thessaloniki, GreeceRoberta Modica, University of Naples Federico II, Italy
Pernille Holmager, University of Copenhagen, Denmark
Copyright © 2024 Tan, Zhang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongping Chen, anhjaHAyMDAwQDEyNi5jb20=
†These authors have contributed equally to this work