
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 10 June 2024
Sec. Diabetes: Molecular Mechanisms
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1414447
This article is part of the Research Topic Pancreatic Beta-Cell Dedifferentiation View all 5 articles
 Sumit Patel1
Sumit Patel1 Maria S. Remedi1,2,3*
Maria S. Remedi1,2,3*Type 2 diabetes (T2D) is a polygenic metabolic disorder characterized by insulin resistance in peripheral tissues and impaired insulin secretion by the pancreas. While the decline in insulin production and secretion was previously attributed to apoptosis of insulin-producing β-cells, recent studies indicate that β-cell apoptosis rates are relatively low in diabetes. Instead, β-cells primarily undergo dedifferentiation, a process where they lose their specialized identity and transition into non-functional endocrine progenitor-like cells, ultimately leading to β-cell failure. The underlying mechanisms driving β-cell dedifferentiation remain elusive due to the intricate interplay of genetic factors and cellular stress. Understanding these mechanisms holds the potential to inform innovative therapeutic approaches aimed at reversing β-cell dedifferentiation in T2D. This review explores the proposed drivers of β-cell dedifferentiation leading to β-cell failure, and discusses current interventions capable of reversing this process, thus restoring β-cell identity and function.
The international diabetes federation (IDF) determined that approximately 643 million people will have diabetes by the end of 2030 (1). Type 1 diabetes (T1D) is an autoimmune disorder leading to the destruction of the insulin producing β-cells, triggering a dysregulation of glucose homeostasis (2). Type 2 diabetes (T2D) is the commonest form of diabetes accounting for the 90% of the cases. T2D is a metabolic disorder driven by polygenes and environmental risk factors, which is characterized by impaired insulin secretion arising from progressive pancreatic β-cell dysfunction and loss of β-cell mass and insulin resistance in the target peripheral tissues, resulting in multiple long-term health complications (3–5). It had been widely accepted that the progressive decline in β-cell mass in T2D was due to increased apoptosis (6), however, apoptosis alone is not sufficient to explain the marked loss of functional β-cell mass (7–9), suggesting other causes. It is now known that chronic metabolic stress in T2D can lead to loss of β-cell identity with β-cell dedifferentiation, transdifferentiation or degranulation ultimately resulting in β-cell dysfunction (10–12). In this review, we discuss the findings regarding β-cell dedifferentiation and transdifferentiation in diabetes, and evidence supporting prevention and even reversion of these processes leading to restoration of β-cell identity and function.
Beta-cell dedifferentiation involves loss of mature β-cell identity and expression of markers of islet progenitors and other normally repressed (“disallowed”) genes, which may be one of the driving factors of loss of functional β-cell mass in diabetes. Disallowed genes such as lactate dehydrogenase (LdhA) and monocarboxylate transporter-1 (Mct1) that are highly expressed in other tissues for non-oxidative glucose metabolism are weakly expressed in the β-cells to prevent abnormal insulin secretion (13, 14). Upregulation of LdhA and Mct isoforms has been shown in the islets of rodent models of hyperglycemia and diabetes (15–20). Acyl-CoA thioesterase (Acot7) that encodes for the enzyme that catalyzes the hydrolysis of long-chain acyl-CoA esters into free fatty acids and coenzyme A, is another β-cell disallowed gene (14, 21). Increased expression levels of Acot7 have been shown in laser microdissected β-cell–enriched tissue from patients with T2D (22) and in Zucker diabetic fatty rat islets (23). Overexpression of mitochondrial Acot7 in β-cells of adult mice impaired insulin secretion worsening glucose tolerance (14). Therefore, altered metabolism through altered expression of forbidden genes in diabetes also drives β-cell dysfunction.
The notion that β-cell identity, rather than β-cell apoptosis, may be compromised was first shown in rats exposed to chronic hyperglycemia (15). Additional studies in rodent, non-human primates and human have furthered supported the notion that loss of β-cell identity and dedifferentiation underlies β-cell failure in diabetes (10, 20, 24–34). Although human diabetes studies are limited to terminal endpoints and are reliant on immunofluorescence techniques, β-cell dedifferentiation is more evident than apoptosis (24, 27, 29). Loss of β-cell identity in human T2D samples was shown by reduction of mRNA and protein levels of key β-cell identity markers: MAF BZIP Transcription Factor A (MafA) and NKX6 Homeobox 1 (Nkx6.1) and pancreatic and duodenal homeobox 1 (Pdx1) (27). These findings were further supported by another study that showed that isolated human T2D islets exhibited a similar reduction of mRNA levels of β-cell identity markers: Forkhead Box O1 (FoxO1), MafA and Nkx6.1. Further, immunostaining of T2D pancreata showed β-cells with increased Aldehyde dehydrogenase 1A3 (ALDH1A3) reactivity, indicative of dedifferentiation (24). Single-cell RNA seq (scRNA-Seq) studies from islets of non-diabetic and T2D individuals revealed that α- and β-cells from T2D exhibit similar transcriptomic profiles to islets from juvenile donors (35, 36). Polycomb repressive complex 2 (PRC2), has been identified as an essential chromatin regulatory complex involved in defining cell fate trajectories by repressing transcription and maintenance of β-cell identity. Loss of PRC2 function is observed in islets from human T2D. Elimination of PRC2 in mouse β-cells triggered progressive β-cell dedifferentiation (37). This suggests that disruption of the global gene silencing machinery in β-cells drives β-cell dedifferentiation in diabetes.
Animal models overcome the limitation of lineage-tracing in human studies, and have shown that β-cells do not undergo apoptosis but rather lose their mature β-cell identity and revert to progenitor-like state through dedifferentiation (10, 26, 28, 32–34). Early studies in mice deficient of FoxO1 in β-cells under chronic metabolic stress exhibited a marked decrease in β-cell mass due to loss of β cell identity markers such as MafA and Pdx1, accompanied by dedifferentiation of β-cells into progenitor-like cells expressing Neurogenin3 (Ngn3), Oct4, Nanog, and L-Myc (10). Loss of β-cell identity and dedifferentiation was also evident in leptin receptor deficient (db/db) and insulin-resistant diabetic (GIRKO) mice, suggesting β-cell dedifferentiation as a cause of β-cell dysfunction in diabetes (10, 38) (Figure 1). Beta-cell-specific inactivation of NKX6.1, another marker of β-cell identity, in mice also showed increased expression of Ngn3 (39). Loss of Urocortin 3 (Ucn3), signifying an early trigger of dedifferentiation, was seen in pancreata from obese diabetic (ob/ob and db/db) mice and from insulin-dependent diabetic mice (Ins2Akita) (33). Interestingly, islets from ob/ob, db/db and Ins2Akita diabetic mice treated with a TGFβ pathway inhibitor Alk5 inhibitor II, demonstrated increased mRNA levels of Ucn3, MafA, Nkx6.1 and Pdx1. β-cell dedifferentiation has been reported in non-obese diabetic (NOD) mouse model of T1D (40, 41). A subpopulation of β-cells (~15%) in NOD mice that evaded early immune attack exhibited decreased mRNA levels of β-cell identity markers: Ins1, Ins2, Glut2, FoxO1, Nkx6.1 and Pdx1, and increased levels of dedifferentiation markers Ngn3 and ALDH (40). IRE1α is a kinase involved in triggering unfolded protein response (UPR) due to elevated endoplasmic reticulum stress. NOD mice exhibit elevated UPR response, and β-cell specific deletion of IRE1α in NOD mice resulted in decreased mRNA and protein expression levels of β cell maturity markers MafA and Ucn3. It was further shown from scRNA-seq and bulk RNA-seq that β cell specific deletion of IRE1α increased expression of Aldh1a3, Gastrin (Gast), and Ngn3, indicative of dedifferentiation as a protection against immune detection (41). Treating human β-cells with polyinosinic-polycytidylic acid (PolyI:C), which mimics viral infections contributing to the development of T1D, resulted in decreased expression of β cell–specific genes such as Ins, MafA, and Slc30A8, along with a marked increase in progenitor markers such as Sox9, Hes1, and Myc (42).
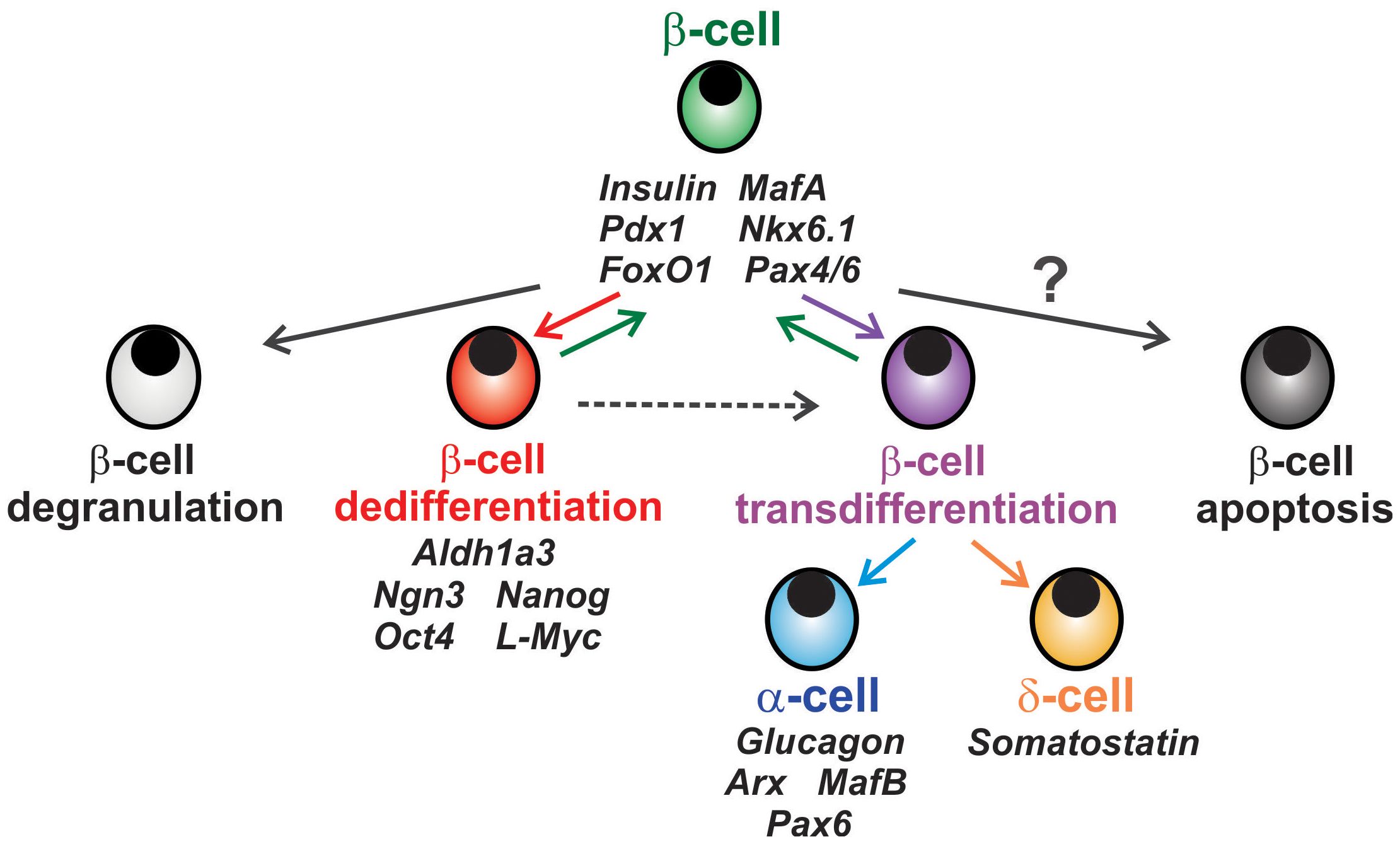
Figure 1 Fates of β-cell in diabetes. Chronic metabolic stress in diabetes can lead to loss of β-cell insulin granules known as β-cell degranulation (light grey cell). β-cells can also incur loss or reduction of mature β-cell identity markers and convert to an endocrine or pancreatic progenitor-like state known as β-cell dedifferentiation (red cell), a process which can be reversible. β-cells can also transition to a different endocrine cell subtype in a process called β-cell transdifferentiation (purple cell) to α- or δ-cells (blue and orange, respectively). The notion that β-cells can undergo apoptosis (dark grey cell) remains controversial, as β-cell apoptosis remains relatively low.
ALDH1A3 is an enzyme primarily involved in the catalysis of the oxidation of all-trans retinal to retinoic acid (RA) during RA synthesis (43). ALDH1A3 is a marker that is abnormally expressed in various cancers (44). Murine and human progenitor cells demonstrated increase in ALDH levels in comparison to other hematopoietic cells (45). As previously mentioned, dedifferentiated β-cells also exhibit a progenitor-like state that is similar to the differentiation observed in tumor progression (24, 26). ALDH1A3 was recently shown as a marker of β-cell dedifferentiation, with increased levels in failing β-cells from β-cell-specific FoxO1 knockout mice, with ALDH1A3 positive cells being less glucose responsive and demonstrating increased markers of uncommitted endocrine progenitors (Pax6, Rfx6, Rfx7, and Mlxipl) and decreased levels of mature β-cell markers (Glucokinase and MafA) (25). It was also demonstrated that while ALDH1A3 positive cells were almost undetectable in islets from pancreatic organ donors without diabetes, it was three-fold higher in islets from T2D individuals despite of adequate glucose control (24, 46). Db/db mice exhibit increased number of β-cells expressing ALDH1A3, compared to controls (30). In addition, ALDH1A3 protein expression was significantly elevated in β-cells of C57BL/6J mice subjected to high fat diet (HFD) for 13 weeks compared to 6 weeks on HFD (47).
Remarkably, mice lacking SUR1 (Abcc8) subunit of the KATP channels in β-cells showed increased ALDH1A3 expression, indicative of β-cell dedifferentiation potentially caused by chronically elevated intracellular Ca2+ (48). Moreover, Abcc8 knockout mice subjected to HFD exhibited upregulation of Aldh1a3 (49), suggesting that chronic β-cell depolarization coupled with overnutrition drives β-cell dedifferentiation. KATP-GOF mouse model of human neonatal diabetes also demonstrated increased ALDH1A3 expression in β-cells, potentially due to hyperstimulated glucose metabolism (50). Indeed, decreasing glucose metabolism by genetic reduction of glucokinase (Gck), reduced β-cell ALDH1A3 to control levels (50). Moreover, β-cell specific deletion of microRNA-483 (miR-483) in mice subjected to HFD led to impaired glucose homeostasis and increased β-cell ALDH1A3 expression (51), suggesting that microRNAs may play a critical role in protecting β-cell function by repressing dedifferentiation. Increased ALDH1A3 expression levels accompanied by decreased expression of CHGA (Chromogranin A) and PDX1 was also reported in β-cells of HFD/STZ (streptozotocin)-induced T2D and db/db mice. ScRNA-seq of β-cells from multiple-low-dose model of STZ-induced diabetic mice revealed a subset of β-cells with low expression of β-cell identity transcription factors such as Pdx1, Nkx2.2, Nkx6.1, Pax6, Isl1 and NeuroD1, and increased Aldh1a3 (52).
Early introduction of intensive insulin therapy in patients newly diagnosed with T2D has showed attainment of long-term remission in approximately 50% of patients, indicating a rescue of β-cell function from glucotoxicity (53). Moreover, a recent report has shown that early short-term insulin intervention coupled with metformin (biguanide) in newly diagnosed T2D patients improved β-cell function with superior and longer lasting glycemic and lipid control compared to glimepiride (sulfonylurea) coupled with metformin (54). Further, transgenic mouse models of monogenic neonatal diabetes with activated KATP channels resulting in hypoinsulinemia and hyperglycemia also showed loss of β-cell identity and dedifferentiation, evidenced by decreased in Ins, Nkx6.1, Pdx1 and a marked increase in Ngn3 expression (20, 55). Interestingly, these mice not only exhibited normalization of blood glucose levels but also redifferentiation of the same dedifferentiated NGN3-positive cells into insulin-positive mature β-cells upon intensive insulin therapy (20). Notably, neither treatment with insulin, phloridzin nor rosiglitazone could reduce β-cell ALDH1A3 expression, but calorie restriction was able to curtail β-cell dedifferentiation in db/db mice (30). Interestingly, infusions of human umbilical cord-derived MSCs (UC-MSCs) in HFD/STZ and db/db mice at an early stage of diabetes prevented β-cell dedifferentiation and protected β-cell function by increasing expression levels of CHGA and PDX1 (56). ALDH1A3 protein and mRNA levels were upregulated in islets upon disrupting the activity of CNOT3 in β-cells, an important post-transcriptional regulator of β-cell maturation and identity (57). Furthermore, db/db mice with specific deletion of Aldh1a3 in β-cells showed improved glucose homeostasis, enhanced glucose tolerance paralleled by improved β-cell function, and increased expression levels of PDX1, NKX6.1, E-Cadherin and MAFA (58). To further show that β-cell dedifferentiation can be curtailed, a recent report demonstrated enhanced β-cell function, enhanced glucose tolerance and increased expression of Insulin and PDX1 in db/db mice treated with an ALDH1A3 inhibitor (KOTX1) (34). Moreover, recent studies demonstrated loss of β-cell identity with reduction of NKX6.1 and PDX1 expression, and increased ALDH1A3 gene and protein levels in KK and KKAy polygenic mouse models of T2D. Strikingly, KK and KKAy diabetic mice subjected to intermittent fasting for 16 weeks demonstrated enhanced NKX6.1 and PDX1 expression, and decreased ALDH1A3, suggesting protection from loss of β-cell identity in T2D by intermittent fasting (20). Intermittent fasting also showed improved glucose tolerance especially in the severe polygenic KKAy model of T2D. Together, these findings further strengthen that β-cell dedifferentiation is a reversible process.
In addition to dedifferentiation, β-cells have also been shown to transdifferentiate into α-, δ- or PP cells. Increased α/β-cell ratio has been reported in human T2D studies (59–61), with the increased attributed to decreased β-cell mass with unchanged α-cell mass (61–63). Ex vivo studies of human islets showed that β-cells after undergoing degranulation can transdifferentiate into α-cells, with expression of PDX1 and NKX6.1 (11). Moreover, conversion of β-cells to α-cells can be curtailed by knockdown of Aristaless-related homeobox (Arx), an α-cell lineage marker. The presence of NKX6.1+GCG+INS− cells in macaques and humans with diabetes was indicative of that loss of functional β-cell mass could partly be due to conversion of β-cells to α-cells (64).
Beta-cell specific deletion of FoxO1 led to β-cell dedifferentiation as discussed above, but also transdifferentiation into α- and δ-cells (10). Overexpression of Paired Box 4 (PAX4) in mouse α-cells led to their transdifferentiation into β-cells (65), and inactivation of Arx in mouse α-cells led to conversion of α-cells to β-cells expressing key β-cell identity markers such as Pdx1, MafA and Glut2 (66, 67). Mutation of the NK2 homeobox 2 (Nkx2.2) tinman (TN) domain or deletion of DNA-methyltransferase 3A (DNMT3a) in mouse β-cells caused Arx-dependent β-to-α-cell transdifferentiation (68). Deletion of Nkx2.2 in adult β-cells also demonstrated transdifferentiation into α- or δ-cells (69). Simultaneous deletion of Arx and Dmnt1 (DNA-methyltransferase 1) in mouse α-cells promoted conversion into functional β-cells (70). Meanwhile, ectopic expression of Arx in embryonic and adult β-cells led to their transdifferentiation into α- and PP cells (71). Recent findings have shown that X-box binding protein 1 (XBP1), an important regulator of the ER stress response in β-cells also plays a role in maintaining β-cell identity, with inactivation of Xbp1 in adult mouse β-cells leading to β-cell dedifferentiation, β-to α-cell transdifferentiation and increased α cell mass (72). Therefore, based on the findings, β-cell transdifferentiation may also play an important role in β-cell dysfunction in diabetes.
Compelling evidence of β-cell dedifferentiation and transdifferentiation in diabetes demonstrates plasticity of β-cells and that these processes can be circumvented to restore functional β-cell mass. Recent findings of converting dedifferentiated cells into functional β-cells, either genetically or pharmacologically, provide insights into restoring β-cell identity and function during progression of diabetes. Future studies with advances in omics exploring the differences between dedifferentiated β-cells (ALDH1A3+ cells) and endocrine progenitor cells will help to identify potential mechanisms and new targets for preventing β-cell dedifferentiation. This will further enhance our understanding of how ALDH1A3 suppresses β-cell identity through epigenetic and transcriptional regulation. Moreover, identifying subpopulations of β-cells that are susceptible to metabolic stress and vulnerable to dedifferentiation and/or transdifferentiation, versus β-cells “resistant” to metabolic stress could be critical for preventing or even reverting loss of β-cell identity and function in diabetes. A major strength of this review is the comprehensive discussion of the latest studies in rodents demonstrating loss of β-cell identity, dedifferentiation, and transdifferentiation in diabetes. However, a significant limitation stems from the scarcity of human studies addressing loss of β-cell identity and dedifferentiation, mainly due to the limited availability of human pancreatic samples. Consequently, only a handful of reports using human samples are covered in this review.
SP: Writing – original draft, Writing – review & editing. MR: Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Work in Remedi lab related to the topic of this article was supported by National Institutes of Health grant R01 DK123163 (to MR).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. International Diabetes Federation. (2021). Available online at: https://diabetesatlas.org/atlas/tenth-edition/ (Accessed March 15, 2024).
2. Powers AC. Type 1 diabetes mellitus: much progress, many opportunities. J Clin Invest. (2021) 131. doi: 10.1172/JCI142242
3. Weir GC, Bonner-Weir S. Islet beta cell mass in diabetes and how it relates to function, birth, and death. Ann N Y Acad Sci. (2013) 1281:92–105. doi: 10.1111/nyas.12031
4. Cantley J, Ashcroft FM. Q&A: insulin secretion and type 2 diabetes: why do Β-cells fail? BMC Biol. (2015) 13. doi: 10.1186/s12915–015-0140–6
5. Nolan CJ, Damm P, Prentki M. Type 2 diabetes across generations: from pathophysiology to prevention and management. Lancet. (2011) 378:169–81. doi: 10.1016/S0140–6736(11)60614–4
6. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. (2003) 52:102–10. doi: 10.2337/diabetes.52.1.102
7. Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab. (2008) 10 Suppl 4:32–42. doi: 10.1111/j.1463-1326.2008.00969.x
8. Kahn SE, Zraika S, Utzschneider KM, Hull RL. The beta cell lesion in type 2 diabetes: there has to be a primary functional abnormality. Diabetologia. (2009) 52:1003–12. doi: 10.1007/s00125-009-1321-z
9. Dai C, Kayton NS, Shostak A, Poffenberger G, Cyphert HA, Aramandla R, et al. Stress-impaired transcription factor expression and insulin secretion in transplanted human islets. J Clin Invest. (2016) 126:1857–70. doi: 10.1172/JCI83657
10. Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic Β Cell dedifferentiation as a mechanism of diabetic Β Cell failure. Cell. (2012) 150:1223–34. doi: 10.1016/j.cell.2012.07.029
11. Spijker HS, Ravelli RBG, Mommaas-Kienhuis AM, van Apeldoorn AA, Engelse MA, Zaldumbide A, et al. Conversion of mature human Β-cells into glucagon-producing Α-cells. Diabetes. (2013) 62:2471–80. doi: 10.2337/db12–1001
12. Butler AE, Dhawan S, Hoang J, Cory M, Zeng K, Fritsch H, et al. Β-cell deficit in obese type 2 diabetes, a minor role of Β-cell dedifferentiation and degranulation. J Clin Endocrinol Metab. (2016) 101:523–32. doi: 10.1210/jc.2015–3566
13. Sekine N, Cirulli V, Regazzi R, Brown LJ, Gine E, Tamarit-Rodriguez J, et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential Role in Nutrient Sensing. J Biol Chem. (1994) 269:4895–902. doi: 10.1016/S0021-9258(17)37629-9
14. Martinez-Sanchez A, Pullen TJ, Chabosseau P, Zhang Q, Haythorne E, Cane MC, et al. Disallowance of acot7 in Β-cells is required for normal glucose tolerance and insulin secretion. Diabetes. (2016) 65:1268–82. doi: 10.2337/db15–1240
15. Jonas JC, Sharma A, Hasenkamp W, Ilkova H, Patanè G, Laybutt R, et al. Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J Biol Chem. (1999) 274:14112–21. doi: 10.1074/jbc.274.20.14112
16. Laybutt D, Sharma A, Sgroi D, Gaudet J, Bonner-Weir S, Weir G. Genetic regulation of metabolic pathways in beta-cells disrupted by hyperglycemia. J Biol Chem. (2002) 277:10912–21. doi: 10.1074/jbc.M111751200
17. Laybutt D, Hawkins Y, Lock J, Lebet J, Sharma A, Bonner-Weir S, et al. Influence of diabetes on the loss of beta cell differentiation after islet transplantation in rats. Diabetologia. (2007) 50:2117–25. doi: 10.1007/s00125–007-0749–2
18. Homo-Delarche F, Calderari S, Irminger J, Gangnerau M, Coulaud J, Rickenbach K, et al. Islet inflammation and fibrosis in a spontaneous model of type 2 diabetes, the GK rat. Diabetes. (2006) 55:1625–33. doi: 10.2337/db05–1526
19. Li X, Zhang L, Meshinchi S, Dias-Leme C, Raffin D, Johnson J, et al. Islet microvasculature in islet hyperplasia and failure in a model of type 2 diabetes. Diabetes. (2006) 55:2965–73. doi: 10.2337/db06–0733
20. Patel S, Yan Z, Remedi MS. Intermittent fasting protects Β-cell identity and function in a type-2 diabetes model. Metabolism. (2024) 153:155813. doi: 10.1016/j.metabol.2024.155813
21. Pullen TJ, Khan AM, Barton G, Butcher SA, Sun G, Rutter GA. Identification of genes selectively disallowed in the pancreatic islet. Islets. (2010) 2:89–95. doi: 10.4161/isl.2.2.11025
22. Marselli L, Thorne J, Dahiya S, Sgroi DC, Sharma A, Bonner-Weir S, et al. Gene expression profiles of beta-cell enriched tissue obtained by laser capture microdissection from subjects with type 2 diabetes. PloS One. (2010) 5:e11499. doi: 10.1371/journal.pone.0011499
23. Parton LE, McMillen PJ, Shen Y, Docherty E, Sharpe E, Diraison F, et al. Limited role for srebp-1c in defective glucose-induced insulin secretion from zucker diabetic fatty rat islets: A functional and gene profiling analysis. Am J Physiol Endocrinol Metab. (2006) 291:E982–94. doi: 10.1152/ajpendo.00067.2006
24. Cinti F, Bouchi R, Kim-Muller JY, Ohmura Y, Sandoval PR, Masini M, et al. Evidence of Β-cell dedifferentiation in human type 2 diabetes. J Clin Endocrinol Metab. (2016) 101:1044–54. doi: 10.1210/jc.2015–2860
25. Kim-Muller JY, Fan J, Kim YJ, Lee SA, Ishida E, Blaner WS, et al. Aldehyde dehydrogenase 1a3 defines a subset of failing pancreatic Β Cells in diabetic mice. Nat Commun. (2016) 7:12631. doi: 10.1038/ncomms12631
26. Wang Z, York NW, Nichols CG, Remedi MS. Pancreatic Β Cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. (2014) 19:872–82. doi: 10.1016/j.cmet.2014.03.010
27. Guo S, Dai C, Guo M, Taylor B, Harmon JS, Sander M, et al. Inactivation of specific Β Cell transcription factors in type 2 diabetes. J Clin Invest. (2013) 123:3305–16. doi: 10.1172/JCI65390
28. Fiori JL, Shin YK, Kim W, Krzysik-Walker SM, González-Mariscal I, Carlson OD, et al. Resveratrol prevents Β-cell dedifferentiation in nonhuman primates given a high-fat/high-sugar diet. Diabetes. (2013) 62:3500–13. doi: 10.2337/db13–0266
29. Amo-Shiinoki K, Tanabe K, Hoshii Y, Matsui H, Harano R, Fukuda T, et al. Islet cell dedifferentiation is a pathologic mechanism of long-standing progression of type 2 diabetes. JCI Insight. (2021) 6. doi: 10.1172/jci.insight.143791
30. Ishida E, Kim-Muller JY, Accili D. Pair feeding, but not insulin, phloridzin, or rosiglitazone treatment, curtails markers of Β-cell dedifferentiation in db/db mice. Diabetes. (2017) 66:2092–101. doi: 10.2337/db16–1213
31. Yamamoto Y, Miyatsuka T, Sasaki S, Miyashita K, Kubo F, Shimo N, et al. Preserving expression of Pdx1 improves Β-cell failure in diabetic mice. Biochem Biophys Res Commun. (2017) 483:418–24. doi: 10.1016/j.bbrc.2016.12.128
32. Sheng C, Li F, Lin Z, Zhang M, Yang P, Bu L, et al. Reversibility of Β-cell-specific transcript factors expression by long-term caloric restriction in db/db mouse. J Diabetes Res. (2016) 2016:6035046. doi: 10.1155/2016/6035046
33. Blum B, Roose AN, Barrandon O, Maehr R, Arvanites AC, Davidow LS, et al. Reversal of Β Cell de-differentiation by a small molecule inhibitor of the TGFβ Pathway. eLife. (2014) 3:e02809. doi: 10.7554/eLife.02809
34. Son J, Du W, Esposito M, Shariati K, Ding H, Kang Y, et al. Genetic and pharmacologic inhibition of ALDH1A3 as a treatment of Β-cell failure. Nat Commun. (2023) 14:558. doi: 10.1038/s41467–023-36315–4
35. Wang YJ, Schug J, Won K-J, Liu C, Naji A, Avrahami D, et al. Single-cell transcriptomics of the human endocrine pancreas. Diabetes. (2016) 65:3028–38. doi: 10.2337/db16–0405
36. Avrahami D, Wang YJ, Schug J, Feleke E, Gao L, Liu C, et al. Single-cell transcriptomics of human islet ontogeny defines the molecular basis of Β-cell dedifferentiation in T2d. Mol Metab. (2020) 42:101057. doi: 10.1016/j.molmet.2020.101057
37. Lu TT-H, Heyne S, Dror E, Casas E, Leonhardt L, Boenke T, et al. The polycomb-dependent epigenome controls Β Cell dysfunction, dedifferentiation, and diabetes. Cell Metab. (2018) 27:1294–308.e7. doi: 10.1016/j.cmet.2018.04.013
38. Lin HV, Ren H, Samuel VT, Lee H-Y, Lu TY, Shulman GI, et al. Diabetes in mice with selective impairment of insulin action in glut4-expressing tissues. Diabetes. (2011) 60:700–9. doi: 10.2337/db10–1056
39. Taylor Brandon L, Liu F-F, Sander M. Nkx6.1 is essential for maintaining the functional state of pancreatic beta cells. Cell Rep. (2013) 4:1262–75. doi: 10.1016/j.celrep.2013.08.010
40. Rui J, Deng S, Arazi A, Perdigoto AL, Liu Z, Herold KC. Β Cells that resist immunological attack develop during progression of autoimmune diabetes in nod mice. Cell Metab. (2017) 25:727–38. doi: 10.1016/j.cmet.2017.01.005
41. Lee H, Lee Y-S, Harenda Q, Pietrzak S, Oktay HZ, Schreiber S, et al. Beta cell dedifferentiation induced by IRE1α Deletion prevents type 1 diabetes. Cell Metab. (2020) 31:822–36.e5. doi: 10.1016/j.cmet.2020.03.002
42. Oshima M, Knoch KP, Diedisheim M, Petzold A, Cattan P, Bugliani M, et al. Virus-like infection induces human Β Cell dedifferentiation. JCI Insight. (2018) 3. doi: 10.1172/jci.insight.97732
43. Grün F, Hirose Y, Kawauchi S, Ogura T, Umesono K. Aldehyde dehydrogenase 6, a cytosolic retinaldehyde dehydrogenase prominently expressed in sensory neuroepithelia during development. J Biol Chem. (2000) 275:41210–8. doi: 10.1074/jbc.M007376200
44. Duan JJ, Cai J, Guo YF, Bian XW, Yu SC. ALDH1A3, a metabolic target for cancer diagnosis and therapy. Int J Cancer. (2016) 139:965–75. doi: 10.1002/ijc.30091
45. Pearce DJ, Taussig D, Simpson C, Allen K, Rohatiner AZ, Lister TA, et al. Characterization of cells with a high aldehyde dehydrogenase activity from cord blood and acute myeloid leukemia samples. Stem Cells. (2005) 23:752–60. doi: 10.1634/stemcells.2004–0292
46. Sun J, Ni Q, Xie J, Xu M, Zhang J, Kuang J, et al. Β-cell dedifferentiation in patients with T2d with adequate glucose control and nondiabetic chronic pancreatitis. J Clin Endocrinol Metab. (2019) 104:83–94. doi: 10.1210/jc.2018–00968
47. Tersey SA, Levasseur EM, Syed F, Farb TB, Orr KS, Nelson JB, et al. Episodic Β-cell death and dedifferentiation during diet-induced obesity and dysglycemia in male mice. FASEB J. (2018) 32:fj201800150RR. doi: 10.1096/fj.201800150RR
48. Stancill JS, Cartailler J-P, Clayton HW, O’Connor JT, Dickerson MT, Dadi PK, et al. Chronic Β-cell depolarization impairs Β-cell identity by disrupting a network of Ca2+-regulated genes. Diabetes. (2017) 66:2175–87. doi: 10.2337/db16–1355
49. Osipovich AB, Stancill JS, Cartailler J-P, Dudek KD, Magnuson MA. Excitotoxicity and overnutrition additively impair metabolic function and identity of pancreatic Β-cells. Diabetes. (2020) 69:1476–91. doi: 10.2337/db19–1145
50. Yan Z, Fortunato M, Shyr ZA, Clark AL, Fuess M, Nichols CG, et al. Genetic reduction of glucose metabolism preserves functional Β-cell mass in KATP-induced neonatal diabetes. Diabetes. (2022) 71:1233–45. doi: 10.2337/db21–0992
51. Wang Z, Mohan R, Chen X, Matson K, Waugh J, Mao Y, et al. Microrna-483 protects pancreatic Β-cells by targeting ALDH1A3. Endocrinology. (2021) 162. doi: 10.1210/endocr/bqab031
52. Sachs S, Bastidas-Ponce A, Tritschler S, Bakhti M, Böttcher A, Sánchez-Garrido MA, et al. Targeted pharmacological therapy restores Β-cell function for diabetes remission. Nat Metab. (2020) 2:192–209. doi: 10.1038/s42255–020-0171–3
53. Weng J, Li Y, Xu W, Shi L, Zhang Q, Zhu D, et al. Effect of intensive insulin therapy on beta-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: A multicentre randomised parallel-group trial. Lancet. (2008) 371:1753–60. doi: 10.1016/S0140-6736(08)60762-X
54. Stojanovic J, Andjelic-Jelic M, Vuksanovic M, Marjanovic-Petkovic M, Jojic B, Stojanovic M, et al. The effects of early short-term insulin treatment vs. Glimepiride on beta cell function in newly diagnosed type 2 diabetes with hba1c above 9. Turk J Med Sci. (2023) 53:552–62. doi: 10.55730/1300–0144.5616
55. Brereton MF, Iberl M, Shimomura K, Zhang Q, Adriaenssens AE, Proks P, et al. Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nat Commun. (2014) 5:4639. doi: 10.1038/ncomms5639
56. Li B, Cheng Y, Yin Y, Xue J, Yu S, Gao J, et al. Reversion of early- and late-stage Β-cell dedifferentiation by human umbilical cord-derived mesenchymal stem cells in type 2 diabetic mice. Cytotherapy. (2021) 23:510–20. doi: 10.1016/j.jcyt.2021.01.005
57. Mostafa D, Yanagiya A, Georgiadou E, Wu Y, Stylianides T, Rutter GA, et al. Loss of Β-cell identity and diabetic phenotype in mice caused by disruption of CNOT3-dependent mRNA deadenylation. Commun Biol. (2020) 3:476. doi: 10.1038/s42003–020-01201-y
58. Son J, Accili D. Reversing pancreatic Β-cell dedifferentiation in the treatment of type 2 diabetes. Exp Mol Med. (2023) 55:1652–58. doi: 10.1038/s12276–023-01043–8
59. Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, et al. Islet amyloid, increased a-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res. (1988) 9:151–9.
60. Deng S, Vatamaniuk M, Huang X, Doliba N, Lian M-M, Frank A, et al. Structural and functional abnormalities in the islets isolated from type 2 diabetic subjects. Diabetes. (2004) 53:624–32. doi: 10.2337/diabetes.53.3.624
61. Henquin JC, Rahier J. Pancreatic alpha cell mass in European subjects with type 2 diabetes. Diabetologia. (2011) 54:1720–5. doi: 10.1007/s00125–011-2118–4
62. Inaishi J, Saisho Y, Sato S, Kou K, Murakami R, Watanabe Y, et al. Effects of obesity and diabetes on Α- and Β-cell mass in surgically resected human pancreas. J Clin Endocrinol Metab. (2016) 101:2874–82. doi: 10.1210/jc.2016–1374
63. Henquin J-C, Ibrahim MM, Rahier J. Insulin, glucagon and somatostatin stores in the pancreas of subjects with type-2 diabetes and their lean and obese non-diabetic controls. Sci Rep. (2017) 7:11015. doi: 10.1038/s41598-017-10296-z
64. Spijker HS, Song H, Ellenbroek JH, Roefs MM, Engelse MA, Bos E, et al. Loss of Β-cell identity occurs in type 2 diabetes and is associated with islet amyloid deposits. Diabetes. (2015) 64:2928–38. doi: 10.2337/db14–1752
65. Collombat P, Xu X, Ravassard P, Sosa-Pineda B, Dussaud S, Billestrup N, et al. The ectopic expression of pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell. (2009) 138:449–62. doi: 10.1016/j.cell.2009.05.035
66. Wilcox CL, Terry NA, Walp ER, Lee RA, May CL. Pancreatic Α-cell specific deletion of mouse Arx leads to Α-cell identity loss. PloS One. (2013) 8:e66214. doi: 10.1371/journal.pone.0066214
67. Courtney M, Gjernes E, Druelle N, Ravaud C, Vieira A, Ben-Othman N, et al. The inactivation of Arx in pancreatic Α-cells triggers their neogenesis and conversion into functional Β-like cells. PloS Genet. (2013) 9:e1003934. doi: 10.1371/journal.pgen.1003934
68. Papizan JB, Singer RA, Tschen SI, Dhawan S, Friel JM, Hipkens SB, et al. Nkx2.2 repressor complex regulates islet Β-cell specification and prevents Β-to-Α-cell reprogramming. Genes Dev. (2011) 25:2291–305. doi: 10.1101/gad.173039.111
69. Gutiérrez GD, Bender AS, Cirulli V, Mastracci TL, Kelly SM, Tsirigos A, et al. Pancreatic Β Cell identity requires continual repression of non-Β Cell programs. J Clin Invest. (2017) 127:244–59. doi: 10.1172/jci88017
70. Chakravarthy H, Gu X, Enge M, Dai X, Wang Y, Damond N, et al. Converting adult pancreatic islet Α Cells into Β Cells by targeting both Dnmt1 and Arx. Cell Metab. (2017) 25:622–34. doi: 10.1016/j.cmet.2017.01.009
71. Collombat P, Hecksher-Sørensen J, Krull J, Berger J, Riedel D, Herrera PL, et al. Embryonic endocrine pancreas and mature beta cells acquire alpha and PP cell phenotypes upon Arx misexpression. J Clin Invest. (2007) 117:961–70. doi: 10.1172/jci29115
Keywords: diabetes, type 2 diabetes (T2D), β-cell, identity, dysfunction, dedifferentiation, transdifferentiation, apoptosis
Citation: Patel S and Remedi MS (2024) Loss of β-cell identity and dedifferentiation, not an irreversible process? Front. Endocrinol. 15:1414447. doi: 10.3389/fendo.2024.1414447
Received: 08 April 2024; Accepted: 27 May 2024;
Published: 10 June 2024.
Edited by:
Taiyi Diana Kuo, Davis, United StatesReviewed by:
Krishna Prasadan, University of Pittsburgh, United StatesCopyright © 2024 Patel and Remedi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria S. Remedi, bXJlbWVkaUB3dXN0bC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.