
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Endocrinol. , 01 July 2024
Sec. Reproduction
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1396805
 Małgorzata Kałużna1†
Małgorzata Kałużna1† Bartłomiej Budny1†
Bartłomiej Budny1† Michał Rabijewski2
Michał Rabijewski2 Agnieszka Dubiel3
Agnieszka Dubiel3 Małgorzata Trofimiuk-Müldner3
Małgorzata Trofimiuk-Müldner3 Kosma Szutkowski4Adam Piotrowski5Elżbieta Wrotkowska1
Kosma Szutkowski4Adam Piotrowski5Elżbieta Wrotkowska1 Alicja Hubalewska-Dydejczyk3Marek Ruchała1
Alicja Hubalewska-Dydejczyk3Marek Ruchała1 Katarzyna Ziemnicka1*
Katarzyna Ziemnicka1*Introduction: Normosmic isolated hypogonadotropic hypogonadism (nIHH) is a clinically and genetically heterogeneous disorder. Deleterious variants in over 50 genes have been implicated in the etiology of IHH, which also indicates a possible role of digenicity and oligogenicity. Both classes of genes controlling GnRH neuron migration/development and hypothalamic/pituitary signaling and development are strongly implicated in nIHH pathogenesis. The study aimed to investigate the genetic background of nIHH and further expand the genotype–phenotype correlation.
Methods: A total of 67 patients with nIHH were enrolled in the study. NGS technology and a 38-gene panel were applied.
Results: Causative defects regarded as at least one pathogenic/likely pathogenic (P/LP) variant were found in 23 patients (34%). For another 30 individuals, variants of unknown significance (VUS) or benign (B) were evidenced (45%). The most frequently mutated genes presenting P/LP alterations were GNRHR (n = 5), TACR3 (n = 3), and CHD7, FGFR1, NSMF, BMP4, and NROB1 (n = 2 each). Monogenic variants with solid clinical significance (P/LP) were observed in 15% of subjects, whereas oligogenic defects were detected in 19% of patients. Regarding recurrence, 17 novel pathogenic variants affecting 10 genes were identified for 17 patients. The most recurrent pathogenic change was GNRHR:p.Arg139His, detected in four unrelated subjects. Another interesting observation is that P/LP defects were found more often in genes related to hypothalamic–pituitary pathways than those related to GnRH.
Conclusions: The growing importance of the neuroendocrine pathway and related genes is drawing increasing attention to nIHH. However, the underestimated potential of VUS variants in IHH etiology, particularly those presenting recurrence, should be further elucidated.
Isolated hypogonadotropic hypogonadism (IHH) is a rare genetic disorder comprising gonadoliberin (GnRH) deficiency with anosmia or hyposmia [Kallmann syndrome (KS)] and normal olfaction known as normosmic IHH (nIHH). The prevalence of IHH is approximately 1–10 in 100,000 live births, with approximately 40% of cases being normosmic (1). Both KS and nIHH are clinically and genetically heterogeneous. The diagnosis of IHH is challenging since patients with nIHH occasionally present a wide variety of nonreproductive defects, such as midline facial defects, renal anomalies, or limb malformations (2–4). Deficiency of GnRH secretion or action results in the impairment of pubertal development. The clinical spectrum of male nIHH includes absent or incomplete puberty, micropenis, cryptorchidism, impaired libido and potency, and infertility. Primary or secondary amenorrhea, partial breast development and infertility may occur in the course of female nIHH (2–4). Neonatal presentation of nIHH with a lack of mini-puberty and abnormal or underdeveloped external genitalia could be overlooked (5). Diagnostic difficulties in the population classified as nIHH may also be caused by nongenetic conditions that may affect GnRH secretion (6–9). The genetic background overlap seen for KS, nIHH, combined pituitary hormone deficiency (CPHD), and hypothalamic amenorrhea hinders routine genetic diagnostics for hypogonadism (10–13). The dysfunction of crucial components of the GnRH pathway underlies the pathogenesis of IHH (14, 15). Deleterious variants in over 50 genes contributing to neurodevelopmental or neuroendocrine activities have been implicated in the etiology of IHH (13, 15, 16). An abundance of genetic factors can impact heritability, leading to autosomal dominant and recessive transmissions and the occurrence of less common X-linked mode (17). Additionally, increasing evidence for non-Mendelian inheritance associated with the co-occurrence of more than one genetic factor (i.e., digenic and oligogenic), seen in both KS and nIHH, has been reported (15, 18, 19). The cumulative effect of multiple deleterious variants in various genes impacts their interactions and eventually draws a phenotypical picture of IHH patients (20). Both classes of genes controlling GnRH neuron migration, development, and function and coding neuroendocrine pathways are strongly implicated in nIHH pathogenesis (15, 19, 21). Therefore, the broad clinical spectrum may result from oligogenicity, variable expressivity, inheritance pattern, or incomplete penetrance related to particular genes (15). Gene defects have been detected in approximately 2%–21% of subjects with nIHH and in approximately 50% of all IHH cases (15, 20, 22, 23). Pathogenic mutations in the gonadotropin-releasing hormone receptor gene (GNRHR) constitute the foremost cause of nIHH, particularly in familial cases (22). However, owing to the introduction of massive sequencing technologies into routine diagnostics, the efficacy of genetic cause identification rose significantly, and the importance of new IHH genes was revealed (19).
This study aimed to investigate the genetic background of nIHH in adult patients. In addition, the study examined the rates of pathogenic/likely pathogenic (P/LP) variants and variants of uncertain significance (VUS) detected using the next-generation sequencing (NGS) approach. The study also looked for the recurrence of those variants among patients with nIHH. The study aimed to further delineate a genotype–phenotype correlation, with special consideration of adult-onset nIHH (AO-nIHH).
This study encompassed 67 unrelated Polish patients of Slavic origin (64 male and 3 female patients with an age range at genetic diagnosis of 16–46 years) diagnosed with nIHH. All patients underwent a complete physical and anthropometric examination, gynecological or andrological consultation with the sonographic assessment of the ovaries or testes, abdominal ultrasound examination, and pituitary magnetic resonance imaging (MRI). History of unilateral or bilateral cryptorchidism, micropenis, craniofacial anomalies (e.g., dental agenesis and cleft lip or palate), and hypogonadism in the family was obtained by a retrospective review of the patient’s medical records and from an interview with the patient. In the study group, there were 32 patients with AO-nIHH (48% of the tested subjects). Diagnosis of nIHH was based on the following criteria: (1) absent, delayed, or partial puberty or AO-nIHH; (2) reported and confirmed normal sense of smell; (3) clinical signs or symptoms of hypogonadism; (4) low or normal serum concentrations of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) (typically less than 4 IU/L) and serum testosterone level <3.5 nmol/L in male patients or serum estradiol level <20 pg/mL in female patients; (5) otherwise normal anterior pituitary function; (6) no previous lesions of a sellar region in MRI or surgery of sellar region; and (7) normal serum ferritin concentration (24–26). The ability to maintain an average adult testosterone serum concentration after 6 months of withdrawal of hormone replacement therapy was used to determine the reversibility of IHH in male subjects (27). Patients who completed pubertal development with normal gonad size but demonstrated erectile dysfunction with loss of spontaneous nocturnal erections in male patients, loss of libido, and infertility occurring with decreased testosterone and normal/low gonadotropins, with an absence of known acquired IHH cause (weight loss, intensive exercise, chronic severe illness, etc.), were classified as AO-nIHH (28).
The concentrations of LH and FSH, growth hormone (GH), adrenocorticotropic hormone (ACTH), thyrotropin (TSH), prolactin (PRL), testosterone, estradiol, morning cortisol, dehydroepiandrosterone sulfate (DHEA-S), insulin-like growth 1 (IGF-1) factor, free triiodothyronine (fT3), and free thyroxin (fT4) were assessed using the Cobas 6000 system (Roche Diagnostics, Switzerland) and dedicated electrochemiluminescence sandwich immunoassay (ECLIA) kits provided by the manufacturer. The free testosterone was measured using the following formula: (FTI) = 100 × (total testosterone/SHBG). Hemochromatosis was excluded by testing serum ferritin levels (ECLIA, Cobas 6000 system, CV < 4.4%).
The clinical, biochemical, and smell testing methodology was described in the authors’ previous publication reporting patients with KS (29).
NGS technology was used in the current study, and the gene panel was previously described and reported for KS patient cohorts (29). Briefly, the Ion Torrent Personal Genome Machine System (Ion PGM TM, Thermo Fisher Scientific, Inc. Waltham, MA, USA) was used, and a validated panel of 38 HH/CPHD genes was applied. Gene panel encompassed the following genes: ADAM7, ANOS1, BMP2, BMP4, CHD7, FGF8, FGF17, FGFR1, GLI2, GNRH1, GNRHR, HESX1, HS6ST1, IGSF10, KISS1, KISS1R, LEP, LEPR, LHB, LHX3, LHX4, LRRIQ3, NSMF, NR0B1, OTX1, OTX2, PCSK1, PITX1, PITX2, PROK2, PROKR2, PROP1, POU1F1, SEMA3A, SOX3, TAC3, TACR3, and WDR11. The sequences were primarily mapped to the human genome (GRCh37/hg19) using Torrent Suite™ software (version 4.0.2, Thermo Fisher Scientific, Waltham, MA, USA) and converted to GRCh38/hg38. Variants were assigned according to HGVS recommendations, and coding/genomic positions referred to the canonical transcripts. The pathogenicity estimation was made using Variant Effect Predictor (VEP) in Ensembl browser (19), MutationTaster2 (http://www.mutationtaster.org/), and PhenIX (http://compbio.charite.de/PhenIX/), and utilizing prediction from the following algorithms: FATHMM, CAD, PolyPhen2, SIFT, and MaxEntScan. Clinical databases HGMD (http://www.hgmd.cf.ac.uk) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) were also checked, and the frequency of variants was established using the GnomAD database (https://gnomad.broadinstitute.org/) with the minor allele frequency (MAF) of <1% as the filtering threshold. Alterations were classified according to updated ACMG guidelines [P/LP, VUS, and benign/likely benign (B/LB)] (30). Varsome (31) and InterVar (32) tools were used to determine the correctness of position estimation and HGVS nomenclature. Selected variants were subjected to in silico analysis to evaluate the effect of the change on protein architecture, considering its functionality and potential involvement in the manifestation of an abnormal phenotype.
Protein models were generated from FASTA files using homology modeling with Yasara 24.4.10 software (33), the PsiBLAST algorithm (34), and the Alphafold 3 server (35). The calculated structures, together with the model quality Z-scores, and PDB files were placed in the Supplementary Materials. Homology modeling in this version of the Yasara software uses, in addition to x-ray and NMR data (UniRef90H), the databases of three AI servers, i.e., Alphafold 2 (33), ESMFold (36), and OmegaFold. Models were aligned using the MUSTANG algorithm (37).
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the Institutional Ethics Committee of Poznan University of Medical Science (1002/13, 5 December 2013; 990/15, 5 November 2015, 567/16, 5 May 2016).
Informed consent was obtained from all subjects involved in the study.
In the study group, genetic defects were detected in 53 patients (79%). Four patients had cryptorchidism, three patients had micropenis, and one patient each had congenital adrenal hyperplasia (CAH), congenital adrenal hypoplasia, intellectual disability, arachnodactyly, facial bone dysplasia, and left renal agenesis. In the cohort of subjects presenting genetic abnormalities, 23 individuals were diagnosed with AO-nIHH, and the reversal form was found in only one patient. The majority of patients with AO-nIHH had no concomitant dysfunction or disorders. The detailed clinical data of the subjects were divided according to the onset and are presented in Tables 1 and 2 (childhood vs. adult-onset, AO). No genetic defects were found in 14 patients (13 men and 1 woman). The AO-nIHH was confirmed in seven patients, whereas the reversal form was found in two patients with nIHH and no genetic defect. One case of horseshoe kidney, one case of strabismus, and one case of unilateral cryptorchidism were noted in patients with no genetic defect.
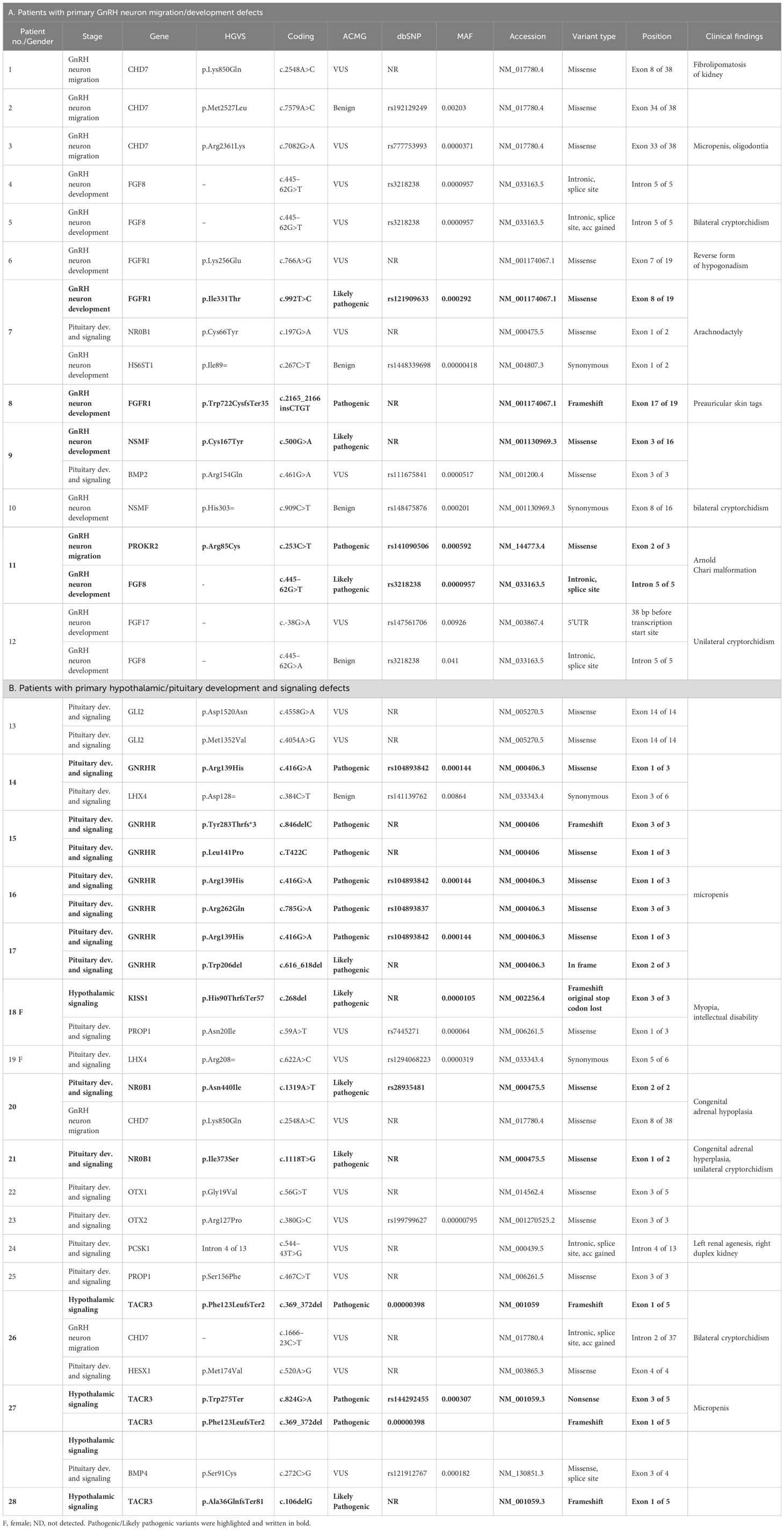
Table 1 Genetic test results and relevant clinical data in patients with childhood-onset nIHH with a genetic defect.
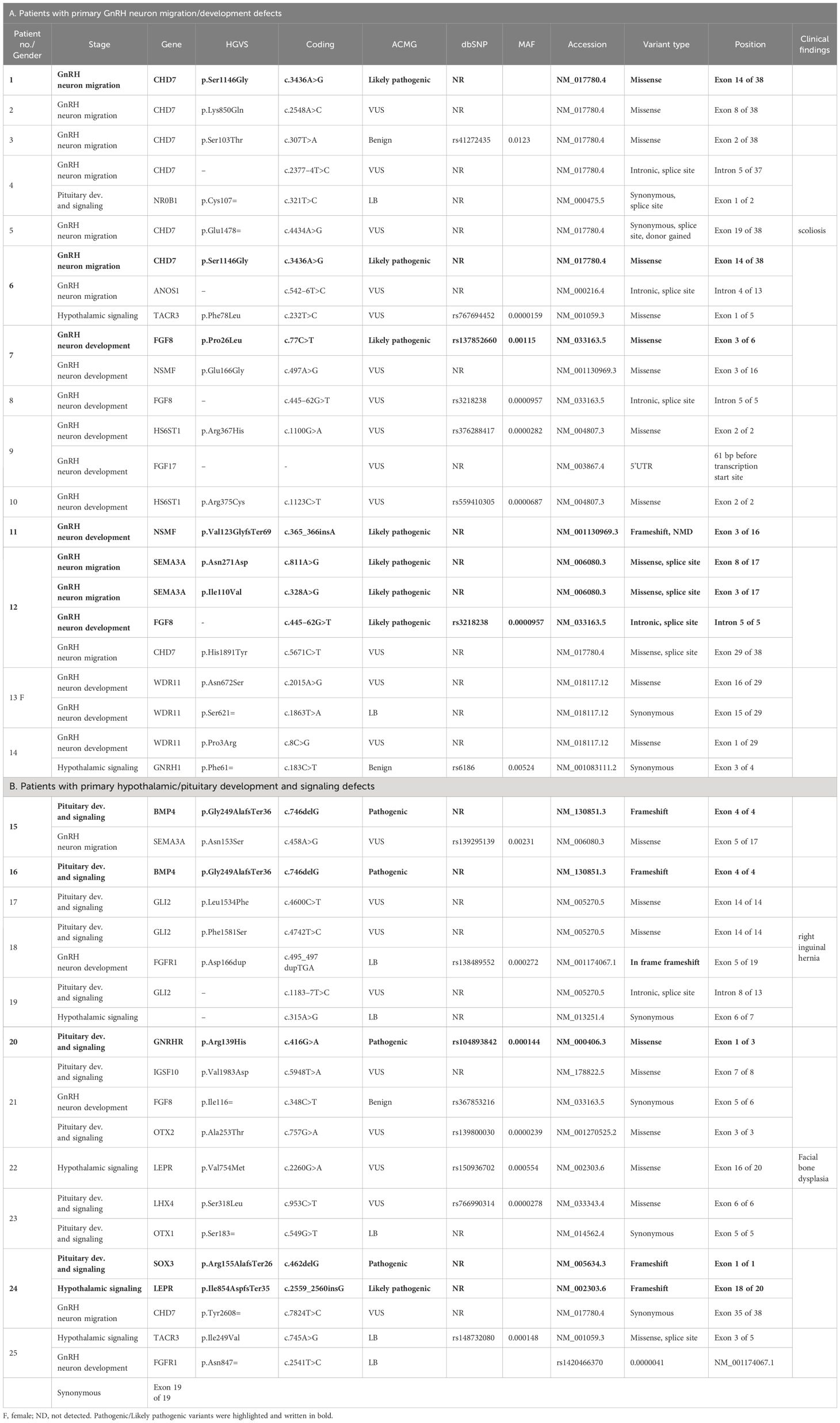
Table 2 Genetic test results and relevant clinical data in patients with adult-onset nIHH with a genetic defect.
In the current study, the authors used the same strategy as in their work on patients with KS (29). Clinically relevant, identified variants were classified as those related to GnRH neuron development and migration (Tables 1A, 2A) and those related to pituitary development and signaling (Tables 1B, 2B). In total, defects in 27 genes were found in 53 patients with nIHH (79%), with at least one P/LP variant detected in 23 patients (34%) and VUS/B in 30 individuals (45%). Of the total 88 identified variants, 15 were classified as pathogenic (P), 16 as likely pathogenic (LP), 42 as VUS, and 15 as B/LB. The most frequently mutated genes presenting P/LP alterations were GNRHR (n = 5, 3 compound heterozygotes, 2 heterozygotes, and 3 novel and not reported); TACR3 (n = 3; 1 compound heterozygote, 2 heterozygotes, and 3 novel), CHD7 (n = 2, 2 novel), FGFR1 (n = 2, 1 reported and 1 novel), NSMF (n = 2, 2 novel), BMP4 (n = 2, 2 novel), and NROB1 (n = 2, 1 reported and 1 novel). The hemizygous pathogenic variant was detected in the SOX3 (n = 1; novel) and VUS variant in ANOS1 gene (n = 1, novel). For 14 patients with nIHH, no causal or contributory variants were identified in the sequenced genes (21%). Finally, Table 1 shows the genetic defects detected in 28 out of 35 patients with childhood-onset nIHH recruited in the study, and Table 2 shows genetic variants in 25 out of 32 patients with AO-nIHH. In the childhood-onset nIHH group, significantly more P/LP variants were diagnosed (19) compared to the AO-nIHH group (11 defects). Monogenic P/LP variants were seen in 10 subjects (15%). Oligogenic defects with obligatory P/LP variant were detected in 13 patients (19%). In summary, 17 novel pathogenic variants, not previously reported for nIHH and affecting 10 genes (CHD7, FGFR1, NSMF, SEMA3A, BMP4, GNRHR, NROB1, SOX3, LEPR, and TACR3 were detected in 17 patients). Concerning the recurrence of pathogenic variants, heterozygous changes CHD7:p.Ser1146Gly, BMP4:p.Gly249AlafsTer36, and TACR3:p.Phe123LeufsTer2 were noted in more than one patient. For reported pathogenic variants, recurrence was found for GNRHR:p.Arg139His (four patients). Surprising results were obtained regarding VUS variants. The total number of identified VUS changes represented half of all identified changes, emphasizing their possible importance for the evaluated phenotype. The recurrence was found for intronic change FGF8:c.445–62G>T (five patients), CHD7:p.Lys850Gln (three patients, VUS), and p.Ser1146Gly (two patients, LP). The high frequency of presented VUS variants among patients might be considered as likely pathogenic onwards. The overall most mutated gene-bearing VUS alterations were CHD7 (nine patients), FGF8 (five patients), and GLI2 (five patients). Furthermore, those variants were intensively explored using the prediction of conformational changes, as presented in Figure 1. In summary, the applied approach allows identifying 40 variants in genes responsible for the development and migration of GnRH neurons and 48 alterations in genes contributing to the pituitary formation and pituitary/hypothalamic signaling. The histograms presenting the prevalence of mutations were classified by three metrics: severity of mutation (ACMG criteria: P/LP, VUS, and B/LB), oligogenicity, and novelty are shown in Figure 2.
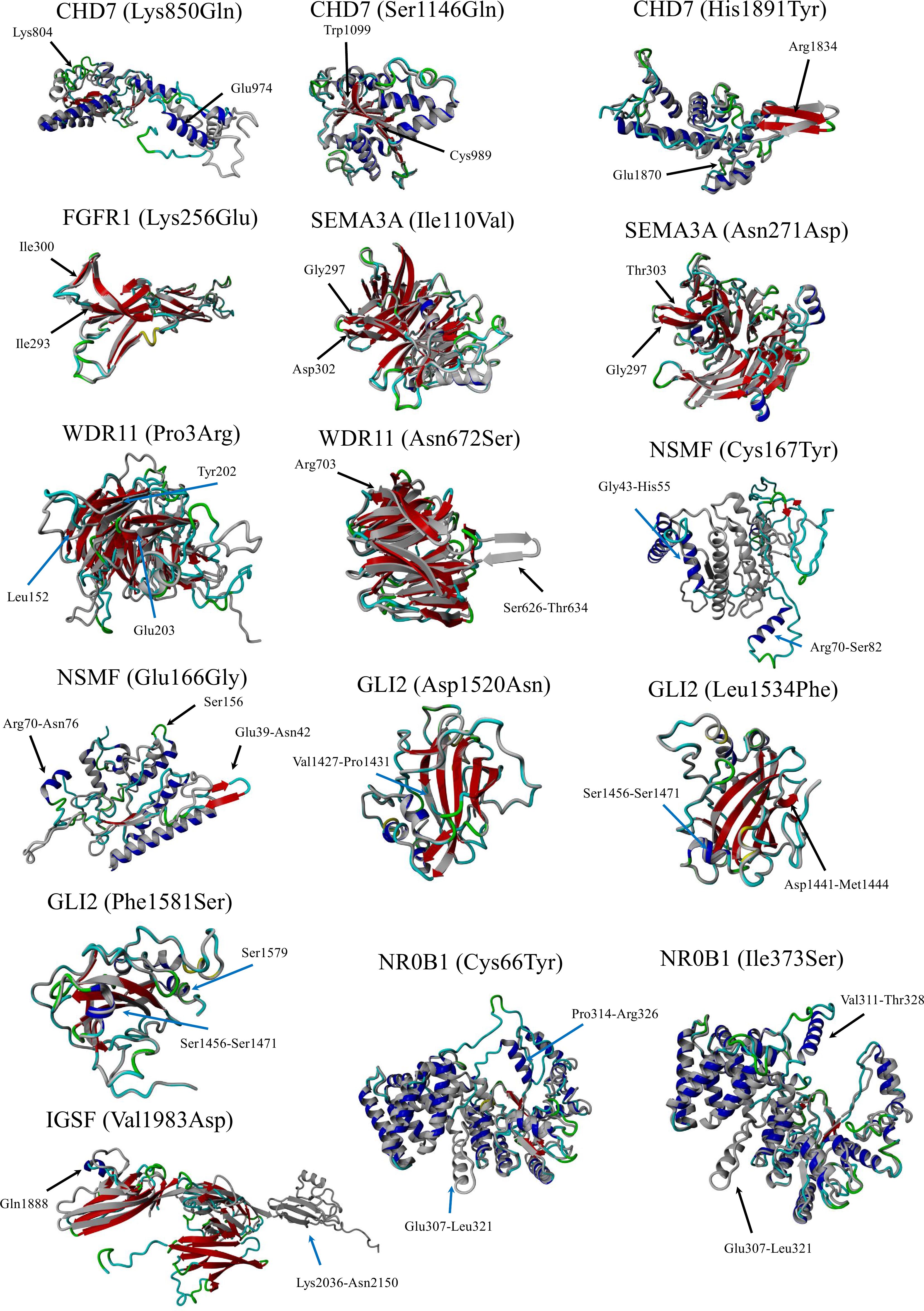
Figure 1 Simulated structures. The normal protein is colored gray, while the protein carrying a variant impacting structure is colored blue (coils) and red (β-pleated sheets). Protein models were generated using the Chimera v1.7 software.
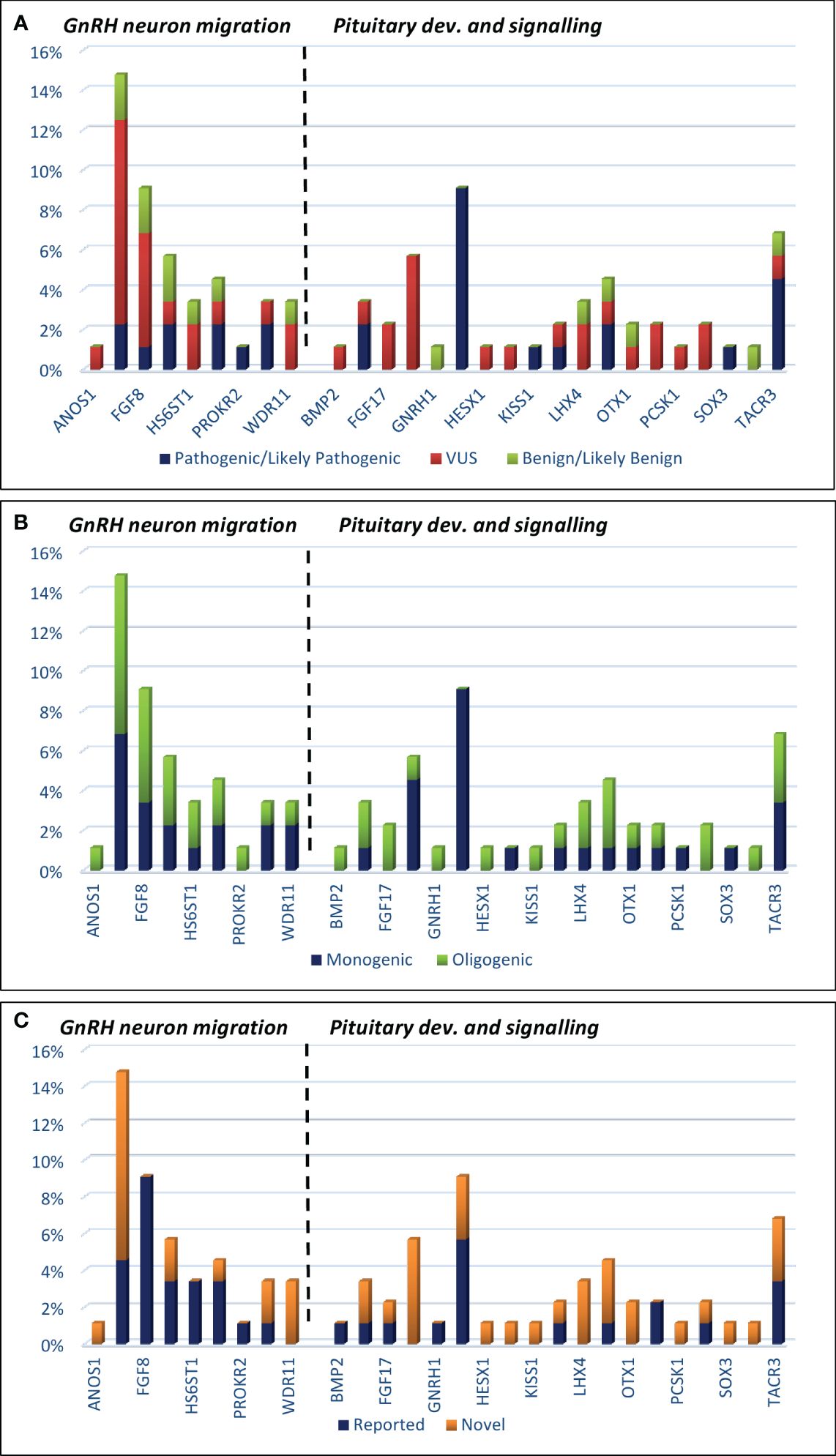
Figure 2 Histograms showing the prevalence of mutations among IHH patients, which are classified by three metrics: (A) mutation severity [ACMG criteria: pathogenic/likely pathogenic, VUS (variant of unknown significance), and benign/likely benign]; (B) oligogenicity; (C) novelty.
The simulated structures obtained from homology modeling are shown in Figure 1. The quality of models is represented by the Z-score parameter (Table 3). The accepted confidence scores were obtained for CHD7 750–1,000, 951–1,200, and 1,751–1,944 residues; FGFR1 (133–350); SEMA3A (31–514); NSMF (2–220, p.Cys167Tyr); IGSF10 (1,851–2,150); and NR0B1. The results were compared with the AlphaFold database and the confidence scores for the given residue range. While the CHD7 protein was not present in the AlphaFold database, the rest of the protein models obtained similar confidence scores. For the GLI2 domain 1,363–1,586, the Z-score was poor, although better than the AlphaFold structure for which only loops were present, and no structural information was achievable. The abbreviated structures were checked and presented using Chimera software. All calculated structures, including PDB structures, were placed in the Supplementary Materials.
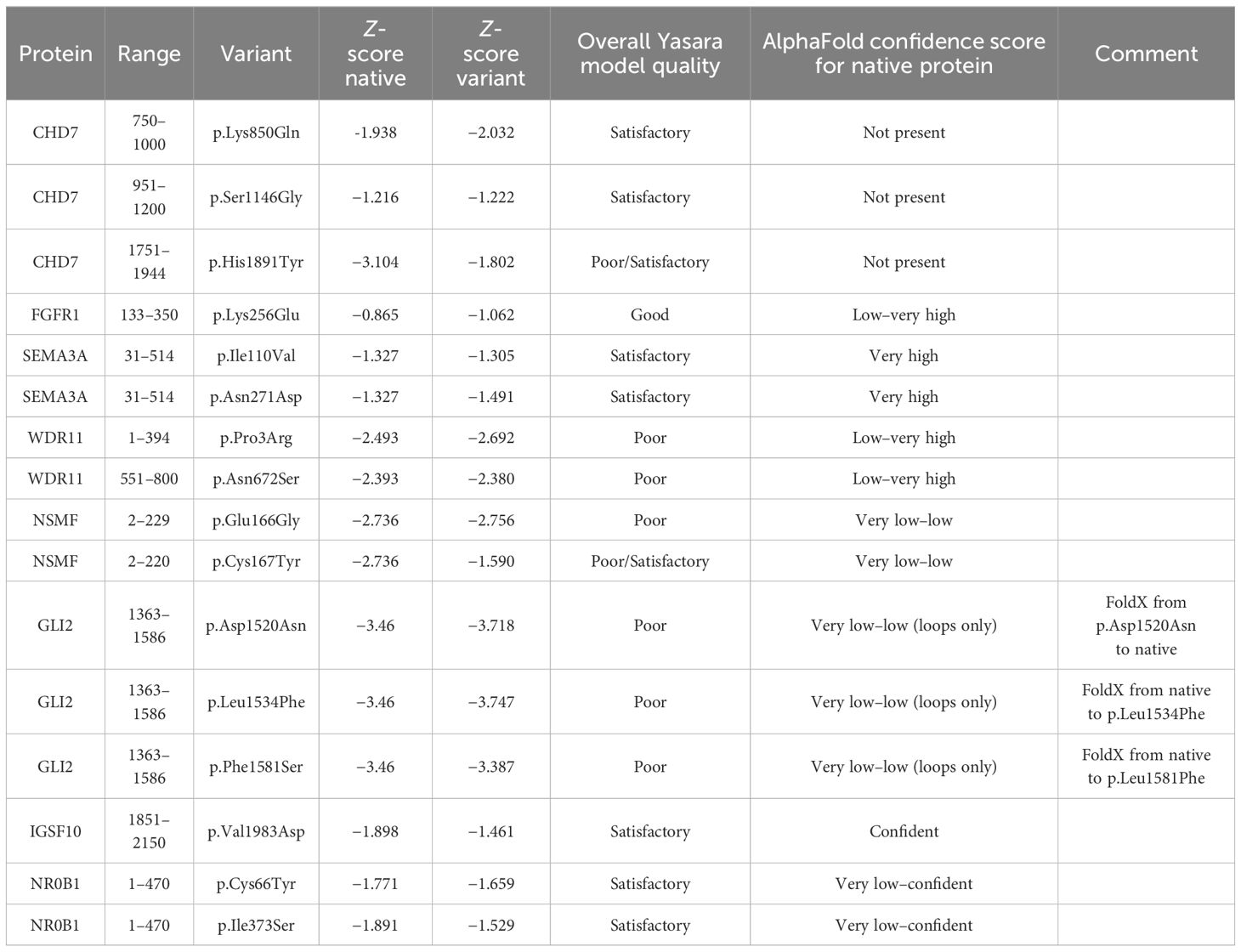
Table 3 Protein models’ overall Z-score parameters and comparison with AlphaFold database.
In patients with nIHH, P/LP defects were found more often in genes related to hypothalamic–pituitary signaling pathways than in GnRH neuron migration/development genes. Two patients with concomitant adrenal diseases (CAH and congenital adrenal hypoplasia) were found to have LP-type mutations in the NR0B1 gene. In the group of women studied (five patients), the LP variant was detected in one of them within the KISS1 gene. In addition to HH, she was also diagnosed with intellectual disability.
There was a greater diversity of genetic variants in patients with AO-nIHH than in the group with prepubertal or peri-pubertal timing of diagnosis. Only in the latter group were defects in the FGFR1, NR0B1, and TACR3 genes present, and the GNRHR defect was prevalent (Figure 2).
Unlike the KS form, the normosmic form of IHH is sporadically manifested with congenital nonreproductive abnormalities. Regarding the fact that multiple genes contribute to both forms, the reasonable explanation for phenotypical variability would be the nature and deleteriousness of variants and their impact on protein functionality. This study included extensive genetic diagnostics performed on the largest Polish nIHH patient group to date. Assuming a more complex and severe KS phenotype compared to nIHH, one could expect that stronger pathogenic alterations will play a key role in KS, and nIHH forms will result from milder, less harmful changes. This situation was well documented for CHD7 gene in patients with IHH and CHARGE syndrome (38). Unfortunately, strong pathogenic variants are seen for both KS and IHH. Therefore, their functional impact is difficult to estimate, while the action of extra-genetic factors cannot be excluded. The phenotypic complexity is further evidenced by new reports of oligogenicity and a growing role of causatives of accumulated milder variants in a single patient (19, 28, 39). A thorough bioinformatic analysis of data obtained in NGS and dedicated genetic panels (considering the clinical picture) is necessary. In patients with childhood-onset IHH, indicative of a more severe form, a greater number of LP/P defects were observed compared to AO-IHH. Patients with a more severe form of the disease, which manifests earlier, tend to exhibit more significant genetic defects (19, 28, 39).
The efficacy of setting up molecular diagnosis in patients with nIHH was higher than expected, approximately 20% according to literature and involved 34% of the examined cohort (P/LP variant identified in patients) (23). However, this number is promising since it is close to the rate of 40% for successful diagnosis for patients with KS (6, 7). The data confirm that known HH genes have an undisputable role in representing a solid foundation for comprehensive diagnostics. Focusing in detail on those genes and expanding analyses for their non-coding or regulatory sequences (i.e., splicing regulation) may benefit in leveraging mutation detection rate as evidenced by this study. The VUS call is made for the majority of clinically relevant variants since their direct deleterious effect on protein is predictive but difficult to prove (functional experiments required). Apart from that, VUS still meets important ACMG criteria (PM2 and BS2) of much higher frequency of such alterations in the patient group compared to the control or the combined general population. The contribution of VUS changes to the phenotype is further discussed in this manuscript.
Oligogenicity occurred in 19% of the patients in the current study, which is close to some data published so far (40). According to recent data, the molecular diagnosis was set for 34% of patients, which is more than expected for this cohort (23). However, it should be considered that publications differ in the number of genes studied. The most frequently mutated genes for P/LP variants were GNRHR (eight changes) and TACR3 (four changes), regarding genes contributing to pituitary development and signaling (a total of 19 alterations). In the case of genes playing a role in GnRH neuron migration, the frequencies were shared between multiple genes: CHD7, FGFR1, NSMF, and SEMA3A (two P/LP variants per gene, a total of 10 changes in this group). Therefore, the first striking conclusion that can be drawn is a higher contribution of genes related to the pituitary with a ratio of 1.5/1. Another notable finding of this research is the contribution of VUS variants. Compared to P/LP variants (29 identified), the overall frequency was higher (44 changes identified), and comparing both pathways (GnRH neuron migration and pituitary signaling), it was equally distributed (22/22). This fact raises the question of the clinical relevance of such variants, particularly regarding missing heritability in molecular diagnostics of patients with nIHH and obvious recurrence in the studied cohort. Recent data have confirmed the contribution of copy number variants (CNVs) in the etiopathogenesis of IHH (41). In the authors’ previous work on CPHD, they also confirmed the presence of such alterations in the genetic landscape of the disease (42). Therefore, the current study examined coding sequences of genes and sought long homozygous regions that would suggest potential allele deletions and prompt microarray scanning to be performed. Unfortunately, no such regions were found. It is agreed with Stamou et al. that these changes are possible but represent minor causes among patients with nIHH and present low prevalence (20). Taking into account all the issues mentioned, the authors took a closer look at selected genes where rare nucleotide variants were identified.
As expected, the authors noted the significant contribution of GNRHR variants, as this gene plays an essential role in IHH. Strikingly, all the identified changes were pathogenic and followed typical recessive transmission. The most familiar change was GNRHR: p.Arg139His (four patients) that appeared in four out of five patients presenting the nIHH phenotype. This variant is rare (MAF = 0.000114) and showed up in compound heterozygosity with other pathogenic changes: p.Trp206del and reported p.Arg262Gln (seven studies) (43). The mutation has so far been identified in 11 individuals with nIHH and may therefore represent a founder mutation in the Polish cohort. The three novel alterations among patients were truncating (p.Tyr283Thrfs*3; p.Trp206del) and missense (p.Leu141Pro). Surprisingly, no VUS variants were reported in this gene. Another critical gene in this cohort was TACR3. Similarly, as in GnRHR, biallelic loss-of-function mutations result in clinical consequences. In the study, three P/LP alterations (all truncating, two novel), one VUS and benign change (both missense and rare MAF = 0.0000159 and MAF = 0.000148, so far not reported for nIHH) were identified. The frameshift change p.Phe123LeufsTer2 was recurrent, and only one individual in the population database was found (MAF 0.00000398). Although the pathogenicity of detected variants seems convincing, biallelic deleterious changes were found not for all patients. Homozygosity or compound heterozygosity was tracked for three out of five patients regarding GnRHR and only one out of five for TACR3. Assuming AR inheritance, conclusive diagnostics for such patients cannot be made. These data suggest the need to expand sequencing for intronic alterations or focus more on existing rare VUS changes, which might have a decisive role in expressing phenotype.
Another justification for this strategy is the low oligogenicity rate and strong expectation for a typical monogenic background. One more mutated gene that did not escape the authors’ attention was transcription factor GLI2. Five novel VUS alterations were identified (four missense and one intronic) that were predicted to be deleterious according to applied algorithms, affecting the distal part of a gene, particularly in an evolutionarily conservative region in the vicinity of a carboxy-terminal domain responsible for transcriptional activation. The more detailed structure analysis the authors applied confirmed earlier predictions (Figure 1). Hitherto, the GLI2 gene was not linked to nIHH but was evidenced to impact pituitary function in hypopituitarism. The Gli2 protein contributes to Shh signaling in pituitary embryogenesis, expressed in the ventral diencephalon, where it induces Bmp4 and Fgf8 expression, and in the oral ectoderm, it induces pituitary progenitors (44). This finding encouraged the authors to look closely at coding sequences in the mentioned interacting genes. Pathogenic variants were also found for both, emphasizing the importance of pituitary development processes and their proper functioning. For BMP4, recurrent truncation p.Gly249AlafsTer36 (so far not reported) was identified. The second reported change was p.Ser91Cys (rs121912767, MAF = 0.000182). This change was evidenced as causative for kidney anomalies and supported by functional studies (45, 46), therefore meeting the criteria for a pathogenic call (PM2, PS3, PP3, and PP5), although the ClinVar database interpreted this variant as VUS due to conflicting reports. Since variants in the TACR3 gene were also shown in a patient with nIHH who had this defect, it is speculated that it had not had a direct impact on the disease, maintaining its uncertain significance.
Two different variants of the NR0B1 gene have been detected in patients with nIHH and concomitant adrenal dysfunction (congenital adrenal hypoplasia and CAH). The first one, missense p.Asn44Ile, had already been reported in X-linked primary adrenal insufficiency with associated IHH (47). Another novel missense variant, p.Ile373Ser, was present in a patient with nIHH and CAH. Single cases in the literature describe patients with NR0B1 and CAH (48).
A further gene whose expression is induced by GLI2 and turned out to be mutated in the nIHH cohort under study was FGF8. The protein belongs to a broad family of fibroblast growth factors and, as the ligand for the FGFR1 receptor, is directly involved in GnRH neuron migration (49). Data from the authors’ patients delivered pathogenic missense variant p.Pro26Leu (reported in patients with KS) and rare intronic variant c.445–62G>T (MAF = 0.0000957). This nucleotide position is conservative evolutionarily and was registered as a candidate for hypospadias (50) (HGMD CS072207) in its more common form c.445–62G>A (MAF = 0.04; in the cohort of the current study, only one patient). Surprisingly, five other patients with nIHH presented the G>T variant, which is predicted to affect splicing according to the NNSplice algorithm (SS acceptor gained). The alteration was estimated as VUS since interpretation relies on splice site prediction, and change might be population-specific. It is worth noting that all five patients with nIHH are sporadic, not related, and non-consanguineous. The authors also checked genomic data from testing other individuals (i.e., KS cohort, controls, and 300 chromosomes), and a variant was not found (29). The gene coding receptor for fibroblast growth factor (FGFR1) revealed abnormalities for five patients (two P/LP, one VUS, and two B/LB rare variants). The pathogenic variants p.Trp722CysfsTer35 and VUS p.Lys256Glu have not yet been reported. The missense change p.Ile331Thr was reported in patients with nIHH and is rare (MAF = 0.000292); however, in ClinVar, conflicting data on pathogenicity is assigned. This gene, in multiple studies, is regarded as the most frequently mutated and major IHH player affecting both KS and nIHH, sometimes even linked to the pleiotropic presentation (51). An evidenced contribution of other genes involved directly in the neurogenesis of the olfactory bulbs or neural crest cell guidance like PROKR2 and SEMA3A in nIHH phenotype was noted for one (PROKR2:p.Arg85Cys) and two patients, respectively (SEMA3A: p.[Asn271Asp]/[Ile110Val]; SEMA3A:p.Asn153Ser). These findings strengthen the importance of testing such genes routinely, even though significant variants showed higher frequency in the Asian population (52).
The unclear role of the NSMF gene (formerly NELF) and its co-occurrence with oligogenic cases is still disputable (53). The current study identified four NSMF variants (two P/LP, one VUS, and one B/LB), and three of them were novel. The pathogenic change p.Val123GlyfsTer69 was a single monogenic defect, whereas two others, p.Cys167Tyr (LP) and p.Glu166Gly (VUS), appeared in combination with variants in FGF8 and BMP2. Further studies are required to determine the importance of the NSMF gene in the pathogenesis of nIHH.
Finally, the gene that deserves detailed analysis is CHD7. Multiple families carrying CHD7 mutations in the AD form demonstrated a broad phenotypic variability, including CHARGE syndrome (OMIM 214800) and IHH (54). However, loss-of-function or truncating mutations in this gene are expected to be linked more frequently to the syndromic phenotype (CHARGE and KS) and lower frequency in nIHH (38, 55). Some authors even suggest that patients with milder phenotypes underwent insufficient exhaustive clinical examination (olfactometry, olfactory bulb MRI, deafness and outer-ear abnormalities, and cardiac problems), and milder syndromic symptoms might be simply overlooked (56, 57). Striking pleiotropy, like those observed in FGFR1, was noted for selected pedigrees as patients harboring identical mutation presented various phenotypical spectrums of KS, nIHH, or isolated anosmia (57). The assessment of pathogenicity for such genes as FGFR1 and CHD7 is therefore challenging, regarding their structural conformational complexity and the resulting abundance of involvement in multiple developmental processes. Its principal role includes neural crest cell migration impacting craniofacial bone formation or the development of a peripheral nervous system and cardiac structures (58).
Additionally, CHD7 is impacting phenotype indirectly via regulating multiple neurodevelopmental genes, i.e., SEMA3A, an important contributor to olfactory neuronal guidance. In the current study, pathogenic variants were identified in two patients (recurrent missense p.Ser1146Gly) and VUS variants were identified in another nine patients (three individuals with p.Lys850Gln recurrent missense, and missense/splice site intronic for others). Therefore, the observation of a higher frequency of non-truncating changes presenting partial loss of function or diminished residual activity of the protein, resulting in nIHH, seems to be confirmed in the cohort of the current study. However, the number of identified VUS alterations addresses an essential question of their clinical utility or relevance and further directives for molecular diagnostics in patients presenting with nIHH.
Multiple recent reports have presented robust, comprehensive, reasonably designed methodological approaches and testing strategies utilizing genomic tools like whole exome sequencing and whole genome copy number analysis (41). Although the applied methods have been proven to be efficient, they have not resulted in the identification of new genes that would explain missing heritability. The number of genes so far reported (~60) constitutes a core target for the nIHH phenotype, and it appears that it will not be raised dramatically. Despite some difficulties related to various gene disruption mechanisms (RSVs and CNVs), most researchers have applied a similar and efficient methodology. The definitive molecular diagnosis is still unsuccessful for over half of all patients with nIHH. In the current study, 44 VUS alterations were identified, and this number is higher than the total number of P/LP changes (28), which is rather expected and in agreement with other reports. Considering VUS for molecular diagnosis, the diagnostic yield would increase dramatically, but according to ACMG criteria and recommendations, that may also result in incorrect diagnosis and further improper genetic consultation. Therefore, all those criteria for pathogenicity estimation should comply rigorously. However, there are also several important reasons why it should be included in scientific reports. First, the striking observation is that most of the VUS found in the current study (55%) have not been reported. They usually met moderate ACMG principles: PM2 (absent from controls and population datasets), PM6 (assumed de novo, but without confirmation), sometimes for familial cases PP1 (cosegregation with disease among family members), PP2 (missense variants in genes as the common cause of disease), and finally PP3 (computing deleterious effects, conformation, conservation, splice, etc.). If the variant is identified and confirmed by others in a similar cohort of patients, it falls under the PS4 rule (strong evidence, prevalence in affected, and significantly increased compared to controls), which automatically shifts the estimation from VUS to likely pathogenic. Secondly, reporting VUS changes can prompt researchers to perform a functional evaluation in order to recapitulate the disease phenotype and encounter another strong ACMG rule, PS3 (evidence of damaging effect in functional studies). Missense variants often require individual evaluation because of unpredictable effects on protein function or a selected pathway (not necessarily a primary pathway) in case of multi-functional genes (i.e., CHD7). Recently, a comprehensive functional study on KS-linked genes FGFR1, HS6ST1, SOX9/10, and CHD7 was conducted. Using machine learning-based predictions and functional knock-outs, the authors evidenced convergence in signaling pathway and contribution to the same processes via various activities (59). Moreover, a significant burden is related to intronic changes since such variability is usually ignored due to a lack of convincing evidence (difficulties in estimation pathogenicity, limited access to algorithms predicting disruption of splice/regulatory sites, and their reliability). A similar problem is addressed in the estimation of oligogenicity consequences, where a synergistic effect is postulated. Here, it is even challenging to design proper functional studies, and the only possible line of evidence is the systematic reporting of genetic background in conclusive cohorts of patients.
Potential limitations of the study include its limited sample size and the use of NGS and a 38-gene targeted panel. Ideally, a whole genome sequencing approach and large, international, and interethnic groups should be used. The groups of patients with nIHH in other studies were of comparable size. Most studies have focused either holistically on patients with IHH or selectively on KS form. There are few studies on the genetic basis of nIHH that have such extensive NGS diagnostics and a dedicated gene panel. There are studies in which the groups of patients with hypogonadotropic hypogonadism without olfactory disorders are larger, but they probably included patients with hypogonadism secondary to, for example, obesity, among others. The selection of patients for the current study was very thorough and aimed at qualifying patients whose genetic background of hypogonadism was most likely, after excluding secondary causes.
Genetic screening should be offered in patients with nIHH, with special consideration for adult-onset forms. Careful qualification of the IHH patient for genetic testing seems to be crucial. In summary, the molecular diagnostics was successful for 34% of individuals with nIHH, and the applied strategy relying on clinically targeted sequencing is robust. The principal role of the neuroendocrine pathway and related genes like GNRHR1, TACR3, or NR0B1 is undisputed. The neurodevelopmental pathway, mainly associated with the migration of GnRH neurons, is becoming increasingly important. New insights into multifunction genes like CHD7, FGFR1, NSMF, SEMA3A, and FGF8 were made. A total of 29 pathogenic variants were added to the etiopathogenesis of IHH. The study also focused on the underestimated role of VUS variants, particularly those presenting recurrence, and genes that were found to be commonly affected by such variants as CHD7, FGF8, or GLI2.
Further analyses are needed to delineate the causes and mystery of the genetic landscape in patients with nIHH with unresolved disease backgrounds. It is important to have a proper interpretation of Mendelian and non-Mendelian inheritance patterns, as well as oligogenic interactions between genes, to gain an advanced understanding of the nature of nIHH and provide effective genetic counseling. Further development of the availability of targeted molecular diagnostics of nIHH is required.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving humans were approved by Institutional Ethics Committee of Poznan University of Medical Science (1002/13, 5th December 2013; 990/15, 5th November 2015, 567/16, 5th May 2016). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
MK: Conceptualization, Funding acquisition, Investigation, Writing – original draft, Writing – review & editing. BB: Conceptualization, Data curation, Formal Analysis, Writing – original draft, Writing – review & editing, Investigation, Methodology, Software. MRa: Investigation, Writing – review & editing. AD: Investigation, Writing – review & editing. MT-M: Writing – review & editing, Investigation. KS: Writing – review & editing, Investigation, Software, Visualization. AP: Writing – review & editing, Investigation, Software, Visualization. EW: Writing – review & editing, Investigation. AH-D: Writing – review & editing, Investigation. MRu: Writing – review & editing, Investigation, Supervision. KZ: Writing – original draft, Writing – review & editing, Investigation.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The research was created as a result of research project No. 2016/21/N/NZ5/00511, funded by the National Science Center. The APC was funded by the National Science Center and Poznan University of Medical Science.
The authors would like to thank Mark Jensen for editing the English version of the article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1396805/full#supplementary-material
1. Bianco SD, Kaiser UB. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol. (2009) 5:569–76. doi: 10.1038/nrendo.2009.177
2. Bhagavath B, Podolsky RH, Ozata M, Bolu E, Bick DP, Kulharya A, et al. Clinical and molecular characterization of a large sample of patients with hypogonadotropic hypogonadism. Fertil Steril. (2006) 85:706–13. doi: 10.1016/j.fertnstert.2005.08.044
3. Bonomi M, Vezzoli V, Krausz C, Guizzardi F, Vezzani S, Simoni M, et al. Characteristics of a nationwide cohort of patients presenting with isolated hypogonadotropic hypogonadism (Ihh). Eur J Endocrinol. (2018) 178:23–32. doi: 10.1530/EJE-17-0065
4. Brioude F, Bouligand J, Trabado S, Francou B, Salenave S, Kamenicky P, et al. Non-syndromic congenital hypogonadotropic hypogonadism: clinical presentation and genotype-phenotype relationships. Eur J Endocrinol. (2010) 162:835–51. doi: 10.1530/EJE-10-0083
5. Dwyer AA, Jayasena CN, Quinton R. Congenital hypogonadotropic hypogonadism: implications of absent mini-puberty. Minerva Endocrinol. (2016) 41:188–95.
6. Fernandez CJ, Chacko EC, Pappachan JM. Male obesity-related secondary hypogonadism - pathophysiology, clinical implications and management. Eur Endocrinol. (2019) 15:83–90. doi: 10.17925/EE.2019.15.2.83
7. Dwyer AA, Chavan NR, Lewkowitz-Shpuntoff H, Plummer L, Hayes FJ, Seminara SB, et al. Functional hypogonadotropic hypogonadism in men: underlying neuroendocrine mechanisms and natural history. J Clin Endocrinol Metab. (2019) 104:3403–14. doi: 10.1210/jc.2018-02697
8. Czarnywojtek A, Borowska M, Dyrka K, Moskal J, Koscinski J, Krela-Kazmierczak I, et al. The influence of various endocrine disruptors on the reproductive system. Endokrynol Pol. (2023) 74:221–33. doi: 10.5603/EP.a2023.0034
9. Rabijewski M. Male-specific consequences of obesity - functional hypogonadism and fertility disorders. Endokrynol Pol. (2023) 74:480–9. doi: 10.5603/ep.95626
10. Caronia LM, Martin C, Welt CK, Sykiotis GP, Quinton R, Thambundit A, et al. A genetic basis for functional hypothalamic amenorrhea. N Engl J Med. (2011) 364:215–25. doi: 10.1056/NEJMoa0911064
11. Budny B, Zemojtel T, Kaluzna M, Gut P, Niedziela M, Obara-Moszynska M, et al. Sema3a and igsf10 are novel contributors to combined pituitary hormone deficiency (Cphd). Front Endocrinol (Lausanne). (2020) 11:368. doi: 10.3389/fendo.2020.00368
12. Tusset C, Noel SD, Trarbach EB, Silveira LF, Jorge AA, Brito VN, et al. Mutational analysis of tac3 and tacr3 genes in patients with idiopathic central pubertal disorders. Arq Bras Endocrinol Metabol. (2012) 56:646–52.
13. Topaloglu AK, Turan I. Genetic etiology of idiopathic hypogonadotropic hypogonadism. Endocrines. (2022) 3:1–15.
14. Balasubramanian R, Dwyer A, Seminara SB, Pitteloud N, Kaiser UB, Crowley WF Jr. Human gnrh deficiency: A unique disease model to unravel the ontogeny of gnrh neurons. Neuroendocrinology. (2010) 92:81–99. doi: 10.1159/000314193
15. Butz H, Nyiro G, Kurucz PA, Liko I, Patocs A. Molecular genetic diagnostics of hypogonadotropic hypogonadism: from panel design towards result interpretation in clinical practice. Hum Genet. (2020). doi: 10.1007/s00439-020-02148-0
16. Stamou MI, Cox KH, Crowley WF Jr. Discovering genes essential to the hypothalamic regulation of human reproduction using a human disease model: adjusting to life in the "-omics" Era. Endocr Rev. (2015) 36:603–21. doi: 10.1210/er.2015-1045
17. Bonomi M, Libri DV, Guizzardi F, Guarducci E, Maiolo E, Pignatti E, et al. New understandings of the genetic basis of isolated idiopathic central hypogonadism. Asian J Androl. (2012) 14:49–56. doi: 10.1038/aja.2011.68
18. Sykiotis GP, Plummer L, Hughes VA, Au M, Durrani S, Nayak-Young S, et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc Natl Acad Sci U.S.A. (2010) 107:15140–4. doi: 10.1073/pnas.1009622107
19. Maione L, Dwyer AA, Francou B, Guiochon-Mantel A, Binart N, Bouligand J, et al. Genetics in endocrinology: genetic counseling for congenital hypogonadotropic hypogonadism and kallmann syndrome: new challenges in the era of oligogenism and next-generation sequencing. Eur J Endocrinol. (2018) 178:R55–80. doi: 10.1530/EJE-17-0749
20. Stamou MI, Georgopoulos NA. Kallmann syndrome: phenotype and genotype of hypogonadotropic hypogonadism. Metabolism. (2018) 86:124–34. doi: 10.1016/j.metabol.2017.10.012
21. Boehm U, Bouloux PM, Dattani MT, de Roux N, Dode C, Dunkel L, et al. Expert consensus document: european consensus statement on congenital hypogonadotropic hypogonadism–pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. (2015) 11:547–64. doi: 10.1038/nrendo.2015.112
22. Beate K, Joseph N, Nicolas de R, Wolfram K. Genetics of isolated hypogonadotropic hypogonadism: role of gnrh receptor and other genes. Int J Endocrinol. (2012) 2012:147893. doi: 10.1155/2012/147893
23. Amato LGL, Montenegro LR, Lerario AM, Jorge AAL, Guerra Junior G, Schnoll C, et al. New genetic findings in a large cohort of congenital hypogonadotropic hypogonadism. Eur J Endocrinol. (2019) 181:103–19. doi: 10.1530/eje-18-0764
24. Balasubramanian R, Crowley WF Jr. Isolated gnrh deficiency: A disease model serving as a unique prism into the systems biology of the gnrh neuronal network. Mol Cell Endocrinol. (2011) 346:4–12. doi: 10.1016/j.mce.2011.07.012
25. Lin J, Mao J, Wang X, Ma W, Hao M, Wu X. Optimal treatment for spermatogenesis in male patients with hypogonadotropic hypogonadism. Medicine. (2019) 98:e16616. doi: 10.1097/md.0000000000016616
26. Silveira LFG, Latronico AC. Approach to the patient with hypogonadotropic hypogonadism. J Clin Endocrinol Metab. (2013) 98:1781–8. doi: 10.1210/jc.2012-3550
27. Mao JF, Xu HL, Duan J, Chen RR, Li L, Li B, et al. Reversal of idiopathic hypogonadotropic hypogonadism: A cohort study in chinese patients. Asian J Androl. (2015) 17:497–502. doi: 10.4103/1008-682X.145072
28. Cangiano B, Duminuco P, Vezzoli V, Guizzardi F, Chiodini I, Corona G, et al. Evidence for a common genetic origin of classic and milder adult-onset forms of isolated hypogonadotropic hypogonadism. J Clin Med. (2019) 8:126. doi: 10.3390/jcm8010126
29. Kaluzna M, Budny B, Rabijewski M, Kaluzny J, Dubiel A, Trofimiuk-Muldner M, et al. Defects in gnrh neuron migration/development and hypothalamic-pituitary signaling impact clinical variability of kallmann syndrome. Genes (Basel). (2021) 12. doi: 10.3390/genes12060868
30. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the american college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
31. Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, Meyer R, et al. Varsome: the human genomic variant search engine. Bioinformatics. (2019) 35:1978–80. doi: 10.1093/bioinformatics/bty897
32. Li Q, Wang K. Intervar: clinical interpretation of genetic variants by the 2015 acmg-amp guidelines. Am J Hum Genet. (2017) 100:267–80. doi: 10.1016/j.ajhg.2017.01.004
33. Varadi M, Anyango S, Deshpande M, Nair S, Natassia C, Yordanova G, et al. Alphafold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. (2022) 50:D439–D44. doi: 10.1093/nar/gkab1061
34. Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. (1997) 25:3389–402. doi: 10.1093/nar/25.17.3389
35. Abramson J, Adler J, Dunger J, Evans R, Green T, Pritzel A, et al. Accurate structure prediction of biomolecular interactions with alphafold 3. Nature. (2024). doi: 10.1038/s41586-024-07487-w
36. Lin Z, Akin H, Rao R, Hie B, Zhu Z, Lu W, et al. Evolutionary-scale prediction of atomic-level protein structure with a language model. Science. (2023) 379:1123–30. doi: 10.1126/science.ade2574
37. Konagurthu AS, Whisstock JC, Stuckey PJ, Lesk AM. Mustang: A multiple structural alignment algorithm. Proteins. (2006) 64:559–74. doi: 10.1002/prot.20921
38. Balasubramanian R, Choi JH, Francescatto L, Willer J, Horton ER, Asimacopoulos EP, et al. Functionally compromised chd7 alleles in patients with isolated gnrh deficiency. Proc Natl Acad Sci U.S.A. (2014) 111:17953–8. doi: 10.1073/pnas.1417438111
39. Gach A, Pinkier I, Wysocka U, Sałacińska K, Salachna D, Szarras-Czapnik M, et al. New findings in oligogenic inheritance of congenital hypogonadotropic hypogonadism. Arch Med Sci. (2022) 18:353–64. doi: 10.5114/aoms.2020.98909
40. Cassatella D, Howard SR, Acierno JS, Xu C, Papadakis GE, Santoni FA, et al. Congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty have distinct genetic architectures. Eur J Endocrinol. (2018) 178:377–88. doi: 10.1530/eje-17-0568
41. Stamou MI, Brand H, Wang M, Wong I, Lippincott MF, Plummer L, et al. Prevalence and phenotypic effects of copy number variants in isolated hypogonadotropic hypogonadism. J Clin Endocrinol Metab. (2022) 107:2228–42. doi: 10.1210/clinem/dgac300
42. Budny B, Karmelita-Katulska K, Stajgis M, Zemojtel T, Ruchala M, Ziemnicka K. Copy number variants contributing to combined pituitary hormone deficiency. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21165757
43. Patil VA, Lila AR, Shah N, Arya S, Ekbote AV, Sarathi V, et al. Regional genotypic variations in normosmic congenital hypogonadotropic hypogonadism: our experience and systematic review. Pituitary. (2022) 25:444–53. doi: 10.1007/s11102-022-01209-z
44. Wang Y, Martin JF, Bai CB. Direct and indirect requirements of shh/gli signaling in early pituitary development. Dev Biol. (2010) 348:199–209. doi: 10.1016/j.ydbio.2010.09.024
45. Weber S, Taylor JC, Winyard P, Baker KF, Sullivan-Brown J, Schild R, et al. Six2 and bmp4 mutations associate with anomalous kidney development. J Am Soc Nephrol JASN. (2008) 19:891–903. doi: 10.1681/ASN.2006111282
46. Tabatabaeifar M, Schlingmann KP, Litwin M, Emre S, Bakkaloglu A, Mehls O, et al. Functional analysis of bmp4 mutations identified in pediatric cakut patients. Pediatr Nephrol. (2009) 24:2361–8. doi: 10.1007/s00467-009-1287-6
47. Schwartz M, Blichfeldt S, Müller J. X-linked adrenal hypoplasia in a large Greenlandic family. Detection of a missense mutation (N4401) in the dax-1 gene; implication for genetic counselling and carrier diagnosis. Hum Genet. (1997) 99:83–7. doi: 10.1007/s004390050316
48. Fan D-B, Li L, Zhang H-H. Nonsense variant of nr0b1 causes hormone disorders associated with congenital adrenal hyperplasia. Sci Rep. (2021) 11:16066. doi: 10.1038/s41598-021-95642-y
49. Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, et al. Decreased fgf8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest. (2008) 118:2822–31. doi: 10.1172/JCI34538
50. Beleza-Meireles A, Lundberg F, Lagerstedt K, Zhou X, Omrani D, Frisen L, et al. Fgfr2, fgf8, fgf10 and bmp7 as candidate genes for hypospadias. Eur J Hum Genet. (2007) 15:405–10. doi: 10.1038/sj.ejhg.5201777
51. Cangiano B, Swee DS, Quinton R, Bonomi M. Genetics of congenital hypogonadotropic hypogonadism: peculiarities and phenotype of an oligogenic disease. Hum Genet. (2021) 140:77–111. doi: 10.1007/s00439-020-02147-1
52. Zhou C, Niu Y, Xu H, Li Z, Wang T, Yang W, et al. Mutation profiles and clinical characteristics of chinese males with isolated hypogonadotropic hypogonadism. Fertil Steril. (2018) 110:486–95 e5. doi: 10.1016/j.fertnstert.2018.04.010
53. Spilker C, Grochowska KM, Kreutz MR. What do we learn from the murine jacob/nsmf gene knockout for human disease? Rare Dis. (2016) 4:e1241361. doi: 10.1080/21675511.2016.1241361
54. Li J-D, Wu J, Zhao Y, Wang X, Jiang F, Hou Q, et al. Phenotypic spectrum of idiopathic hypogonadotropic hypogonadism patients with chd7 variants from a large chinese cohort. J Clin Endocrinol Metab. (2019) 105:1515–26. doi: 10.1210/clinem/dgz182
55. Marcos S, Sarfati J, Leroy C, Fouveaut C, Parent P, Metz C, et al. The prevalence of chd7 missense versus truncating mutations is higher in patients with kallmann syndrome than in typical charge patients. J Clin Endocrinol Metab. (2014) 99:E2138–43. doi: 10.1210/jc.2014-2110
56. Bergman JE, Janssen N, Hoefsloot LH, Jongmans MC, Hofstra RM, van Ravenswaaij-Arts CM. Chd7 mutations and charge syndrome: the clinical implications of an expanding phenotype. J Med Genet. (2011) 48:334–42. doi: 10.1136/jmg.2010.087106
57. Xu C, Cassatella D, van der Sloot AM, Quinton R, Hauschild M, De Geyter C, et al. Evaluating charge syndrome in congenital hypogonadotropic hypogonadism patients harboring chd7 variants. Genet Med. (2018) 20:872–81. doi: 10.1038/gim.2017.197
58. Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, et al. Chd7 cooperates with pbaf to control multipotent neural crest formation. Nature. (2010) 463:958–62. doi: 10.1038/nature08733
Keywords: isolated hypogonadotropic hypogonadism, oligogenicity, next-generation sequencing (NGS), pathogenic/likely pathogenic (P/LP), normosmic
Citation: Kałużna M, Budny B, Rabijewski M, Dubiel A, Trofimiuk-Müldner M, Szutkowski K, Piotrowski A, Wrotkowska E, Hubalewska-Dydejczyk A, Ruchała M and Ziemnicka K (2024) Variety of genetic defects in GnRH and hypothalamic–pituitary signaling and development in normosmic patients with IHH. Front. Endocrinol. 15:1396805. doi: 10.3389/fendo.2024.1396805
Received: 06 March 2024; Accepted: 27 May 2024;
Published: 01 July 2024.
Edited by:
Pouneh K. Fazeli, University of Pittsburgh, United StatesReviewed by:
Vassos Neocleous, The Cyprus Institute of Neurology and Genetics, CyprusCopyright © 2024 Kałużna, Budny, Rabijewski, Dubiel, Trofimiuk-Müldner, Szutkowski, Piotrowski, Wrotkowska, Hubalewska-Dydejczyk, Ruchała and Ziemnicka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katarzyna Ziemnicka, a2F6aWVtQHVtcC5lZHUucGw=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.