 Yaqiong Liu
Yaqiong Liu Caomhán J. Lyons
Caomhán J. Lyons Christine Ayu
Christine Ayu Timothy O’Brien
Timothy O’Brien- Regenerative Medicine Institute (REMEDI), Biomedical Sciences Building, University of Galway, Galway, Ireland
Diabetes mellitus (DM) is a metabolic disease characterized by hyperglycemia, leading to various vascular complications. Accumulating evidence indicates that endothelial colony-forming cells (ECFCs) have attractive prospects for repairing and restoring blood vessels. Thus, ECFCs may be a novel therapeutic option for diabetic patients with vascular complications who require revascularization therapy. However, it has been reported that the function of ECFCs is impaired in DM, which poses challenges for the autologous transplantation of ECFCs. In this review, we summarize the molecular mechanisms that may be responsible for ECFC dysfunction and discuss potential strategies for improving the therapeutic efficacy of ECFCs derived from patients with DM. Finally, we discuss barriers to the use of ECFCs in human studies in light of the fact that there are no published reports using these cells in humans.
1 Introduction
Diabetes mellitus (DM) is a metabolic disease characterized by long-term hyperglycemia, which can be mainly classified into two subtypes: type 1 DM (T1DM) and type 2 DM (T2DM). T1DM is attributed to the immune-mediated destruction of pancreatic β-cells (1) and T2DM is caused by tissue resistance to insulin and a relative lack of insulin (2). It is estimated that approximately 537 million people are affected by DM worldwide in 2021, with the number expected to rise to 783 million by 2045 (3). DM can lead to macrovascular and microvascular complications, which imposes a significant health burden (4). The macrovascular complications of DM include coronary artery disease, peripheral arterial disease, and stroke. The primary microvascular complications encompass diabetic nephropathy, neuropathy, and retinopathy (5). For patients with severe vascular complications that need revascularization therapy, endothelial colony-forming cells (ECFCs) have emerged as a promising therapy due to their strong angiogenic ability. ECFCs are progenitors of endothelial cells, representing a cell population that has self-renewal ability and the capacity to form functional blood vessels (6). However, previous studies suggest that ECFCs isolated from diabetic patients exhibited functional defects, thereby limiting their therapeutic potential (Figure 1) (7, 8). Due to the difficulty in isolating ECFCs from peripheral blood (PB), most studies on ECFCs in diabetic patients have revolved around exploring the biology of ECFCs from gestational diabetes mellitus (GDM) patients. CB-ECFCs from GDM patients exhibit increased senescence, reduced proliferation, migration, and tube formation when compared with those from healthy donors (9–13). Besides, it has been reported that it was more difficult to isolate ECFCs from the PB of T2DM patients than from healthy donors (14). An altered function of PB-ECFCs was also found in diabetic patients with vascular complications. Tan et al. investigated the cell number and function of CD34+ CD45− ECFCs between healthy donors and patients with proliferative diabetic retinopathy (PDR) using a cross-sectional cohort design. Their results revealed that PB-ECFCs from patients with PDR had impaired capacity to migrate and were not able to form functional vascular tubes (15). Another study reported that PB-ECFCs isolated from patients with DM with neuroischemic (NI) or neuropathic (NP) foot ulcers exhibited reduced colony formation, proliferation, migration capability, and nitric oxide (NO) bioavailability (16). Hence, it is reasonable to suggest that optimizing the potency of ECFCs in DM by reversal of dysfunction may achieve the desired therapeutic potential. Several strategies have been developed to enhance the efficacy of ECFCs in DM, such as pretreatment with bioactive agents or chemical factors, genetic modification, and combination with other cell types such as mesenchymal stromal cells (MSCs). In this review, we aim to summarize the mechanisms that account for the reduced cell number and compromised function of ECFCs from DM and explore potential strategies to improve the therapeutic efficacy of ECFCs from patients with DM. Due to the dearth of clinical trials using these cells, we also discuss translational barriers.
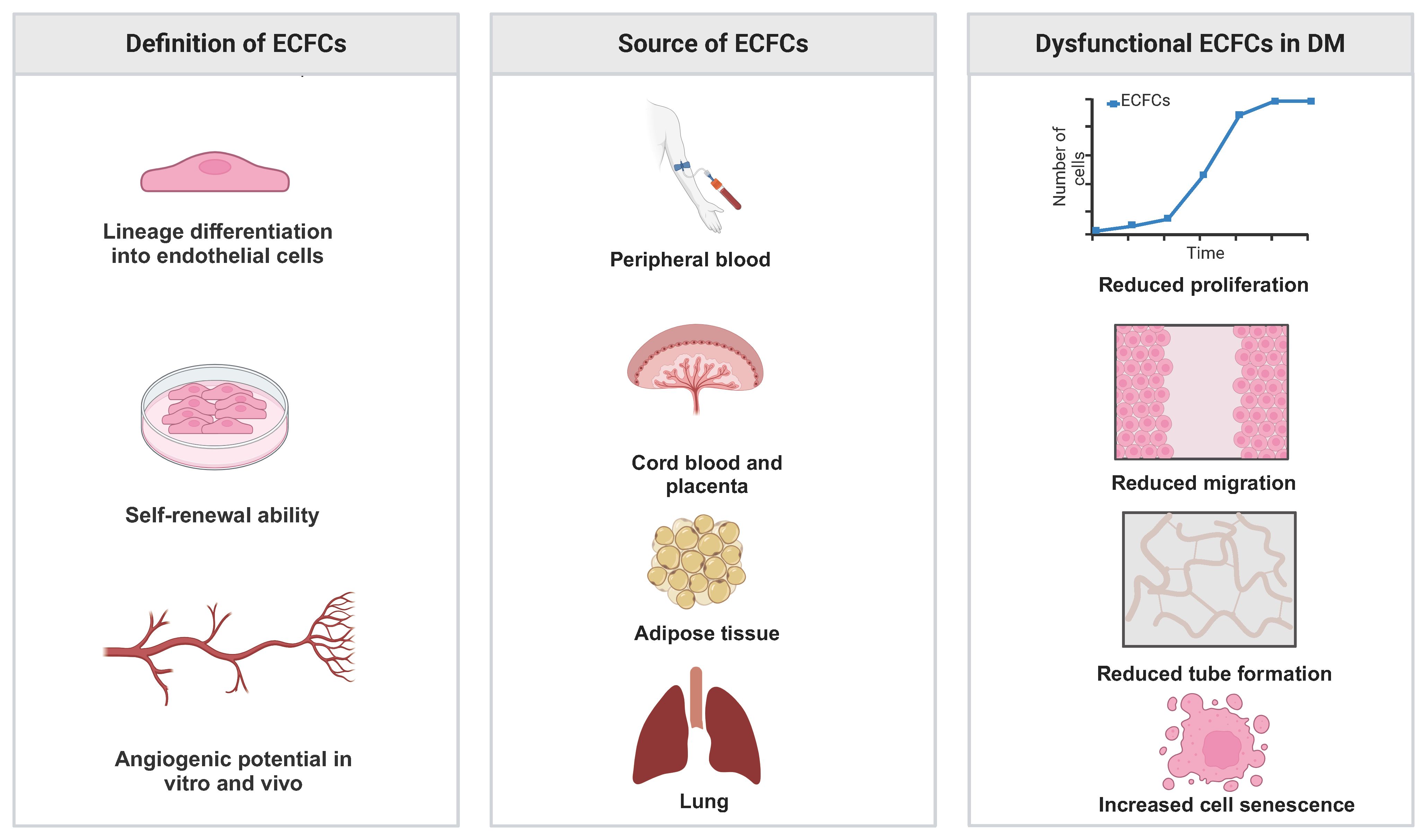
Figure 1 Definition, source of ECFCs and their phenotype in DM.
2 Definition and characteristics of human ECFCs
In 1997, Asahara et al. first isolated putative endothelial progenitor cells (EPCs) from peripheral blood mononuclear cells (PBMNCs). They reported that EPCs were able to integrate into regenerating host blood vessels within injured areas and contribute to vascular repair (17). Subsequent studies found that these EPCs promoted angiogenesis through a paracrine signaling mechanism, but they had limited proliferative ability and could not form blood vessels directly (18, 19). In 2004, Ingram et al. identified a new cell type termed ECFCs that exhibited superior clonal and angiogenic ability (20). In humans, ECFCs are defined using the following three features: (i) lineage differentiation into endothelial cells; (ii) self-renewal ability; and (iii) angiogenic potential in vitro and in vivo (21) (Figure 1). The first feature emphasizes that ECFCs exhibit the endothelial phenotype after in vitro culture, which is used to distinguish ECFCs from other blood outgrown cell types of hematopoietic lineages, such as myeloid angiogenic cells (MACs) (22). The second feature emphasizes the strong self-renewal potential of ECFCs. In some cases, a single ECFC is capable of expanding to a colony with >10,000 cells after 14 days in culture (20, 23). The third feature, angiogenic potential in vitro and vivo, is also essential for distinguishing ECFCs from MACs. MACs cannot form blood vessels themselves, but they support vascular repair by secreting angiogenic factors (22). To date, the origin of ECFCs is not well understood. Lin et al. have provided evidence for a bone marrow origin of ECFCs based on the analysis of blood samples from bone marrow transplant recipients who had received gender-mismatched transplants (24). Fujisawa et al. investigated the ECFCs isolated from PB of male patients after a sex mismatched allogeneic bone marrow transplant. They found that these ECFCs exhibited a XY phenotype instead of a XX phenotype, suggesting that ECFCs originated from blood vessels (25). Surprisingly, the two similar studies have reached very different conclusions. However, a recent study suggested that ECFCs originated from blood vessel walls through lineage tracing and single cell RNA sequence analysis (26). Although the origin of ECFCs is still controversial, ECFCs have been successfully isolated from various tissues (Figure 1). ECFCs are most frequently isolated from cord blood (CB) and peripheral blood (PB) (27) but can also be derived from other tissues (28–30) (Figure 1). CB-ECFCs and PB-ECFCs show differences in gene expression and pro-angiogenic capacity (31, 32). However, the frequency of ECFC is very low in CB and PB: approximately 50 CB-ECFCs per 1x108 cord blood mononuclear cells (CBMNCs) (33) and 1.7 PB-ECFCs per 1x108 PBMNCs (34). It is also important to point out that there is heterogeneity within ECFCs population. Based on the different proliferative potential, CB-ECFCs exhibit a hierarchy of three levels: (i) high proliferative potential ECFC (HPP-ECFC); (ii) low proliferative potential ECFC (LPP-ECFC); (iii) endothelial cell cluster (ECC). HPP-ECFC has the highest proliferative potential and forms colonies containing more than 2000 cells, which gives rise to LPP-ECFC and ECC. LPP-ECFC forms colonies with 50–2,000 cells and ECC forms colonies that contain less than 50 cells (20, 35). In 2019, the International Society on Thrombosis and Hemostasis Congress proposed a standardized protocol for the isolation and expansion of PB-ECFCs and CB-ECFCs without focusing particularly on their surface markers (36) (Figure 2). Generally, ECFCs are positive for CD34 (37), CD31, VEcadherin, von Willebrand factor (vWF), CD146, and VEGFR2 expression, whereas they are negative for hematopoietic markers such as CD14 and CD45 (6).
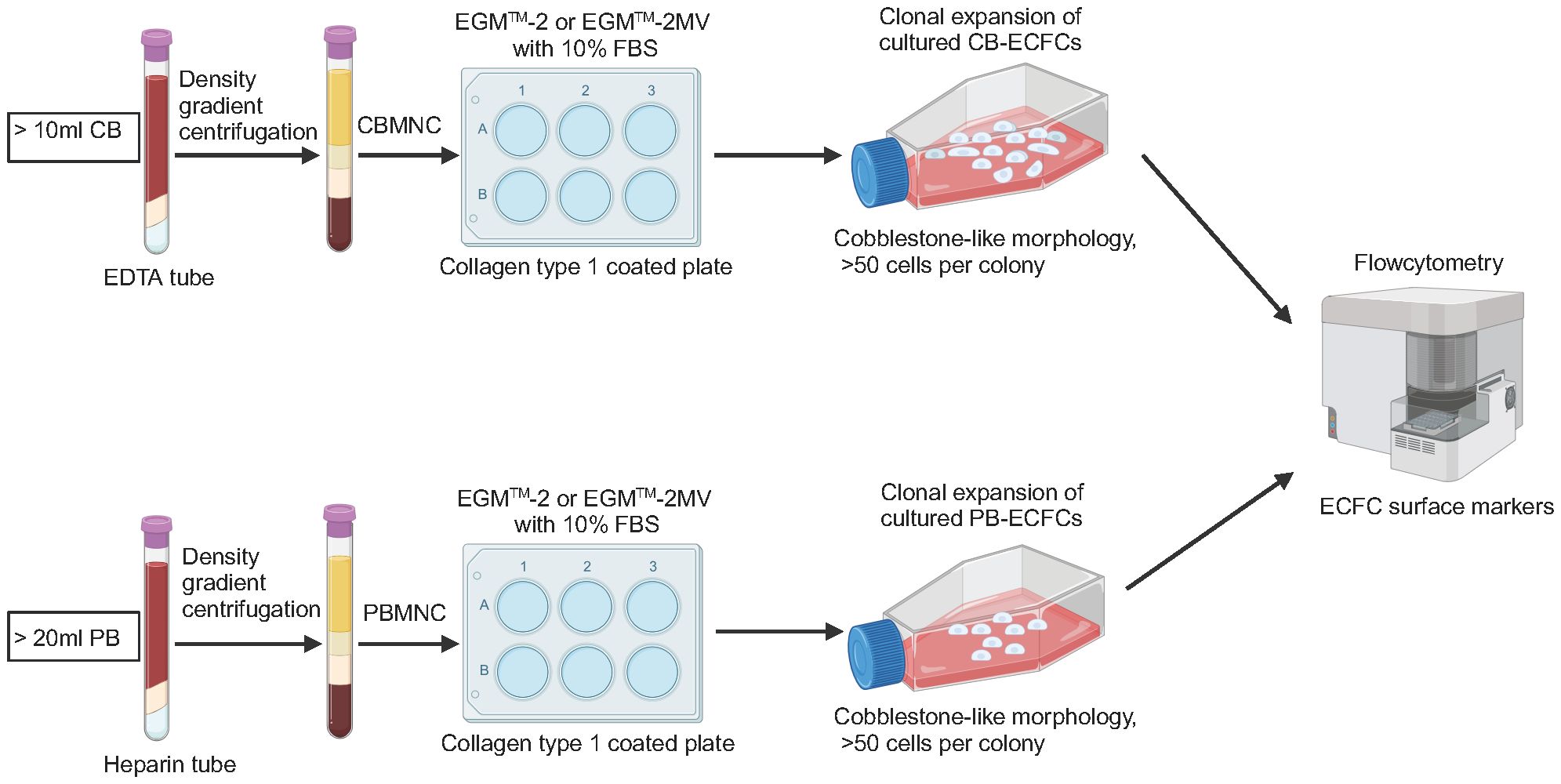
Figure 2 Schematic of isolation, culture, and characterization of ECFCs. CB, cord blood; EDTA, ethylene diamine tetra acetic acid; CBMNC, cord blood mononuclear cell; PB, peripheral blood; PBMNC, peripheral blood mononuclear cell.
3 Mechanisms leading to ECFCs dysfunction in DM
The triggering factors that lead to dysfunctional ECFCs in DM are still unclear, but hyperglycemia, oxidative stress, and inflammation have been hypothesized as key factors in ECFC dysfunction (Figure 3). These three triggering factors act independently and also interact with each other in a vicious cycle, which contributes to the altered function of ECFCs in DM.
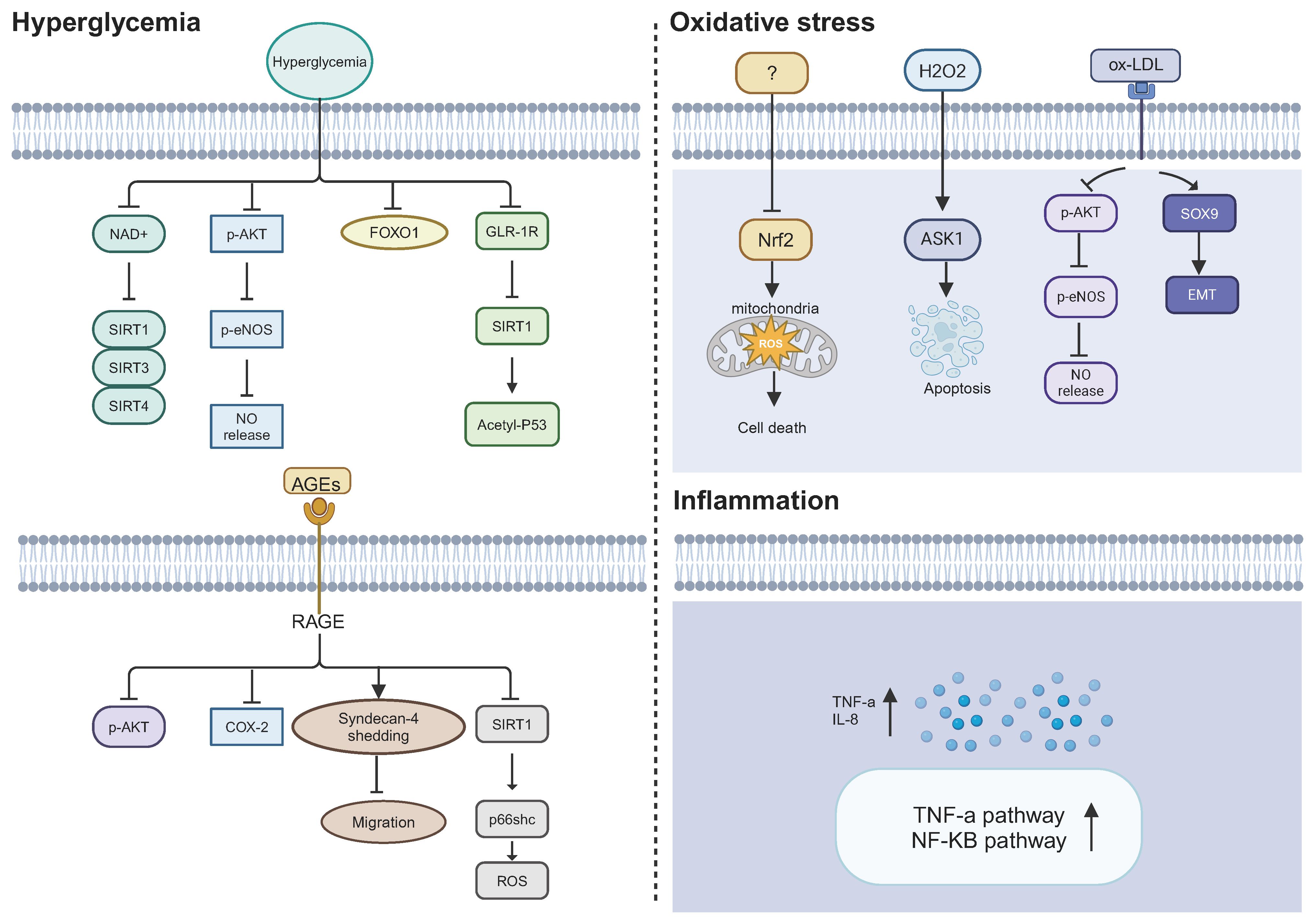
Figure 3 Potential mechanisms causing ECFC dysfunction in DM. Hyperglycemia, oxidative stress, and inflammation may lead to a defective phenotype of ECFCs in a diabetic state. NAD+, nicotinamide adenine dinucleotide; SIRT1/3/4, sirtuin 1; 3; and 4; p-AKT, phosphorylated protein kinase B; p-eNOS, phosphorylated endothelial nitric oxide synthase; NO, nitric oxide; FOXO1, fork head box O 1; GLP-1R, glucagon-like peptide 1 receptor; Acetyl-p53, acetylated p53; AGEs, advanced glycation end products; RAGE, receptor for AGEs; COX-2, cyclooxygenase-2; ROS, reactive oxygen species; Nrf2, nuclear factor erythroid 2-related factor 2; ASK1, apoptosis signal-regulating kinase 1; SOX9, SRY-box transcription factor 9; EMT, endothelial-to-mesenchymal transition; IL-8, Interluekin-8.
3.1 Hyperglycemia
Hyperglycemia, a hallmark of diabetes mellitus, may be one of the leading factors that result in ECFC dysfunction in DM. It has been reported that high glucose impaired the function of ECFCs, including colony formation, self-renewal capacity, and tube formation (38). The adverse effects of high glucose on ECFC have been suggested through affecting sirtuin (SIRT) family and nitric oxide (NO) bioavailability. The SIRT family are nicotinamide adenine dinucleotide (NAD)+-dependent histone deacetylases, which includes seven members (SIRT1–SIRT7) (39). SIRT1 is the most widely studied member of this family and shows anti-inflammatory and anti-oxidative stress effects and improves the mitochondrial function under stress (40). SIRT1 has been found to protect endothelial cells from stress-induced senescence and improve endothelium-dependent vasodilation by deacetylating endothelial nitric oxide synthase (eNOS) and increasing NO bioavailability (41). As for NO bioavailability, its decreased level is a key factor for endothelial cells dysfunction (42). There are several possible mechanisms for decreased NO bioavailability, including reduced eNOS mRNA or protein expression, endogenous competitive inhibitors of L-arginine, reduced tetrahydrobiopterin (BH4) level, and interaction between NO and superoxide (43). Due to the important role of SIRT1and NO bioavailability in endothelial cells, researchers have also investigated their alteration in ECFCs exposed to high glucose. Chen et al. demonstrated that high glucose (25 mM glucose) resulted in increased senescence and impaired proliferation, migration and tube formation ability of PB-ECFCs. Mechanistically, high glucose suppressed phosphorylation of Akt and eNOS, thereby reducing NO bioavailability. Besides, high glucose inhibited the activity of FOXO1 in PB-ECFCs. The negative effects of high glucose could be reversed by co-incubation with the NO donor sodium nitroprusside or p38 mitogen–activated protein kinase (MAPK) inhibitor. Conversely, the negative effects of high glucose could be enhanced by co-incubation with NOS inhibitor l-Ng-nitro-l-arginine methyl ester (l-NAME) or PI3K inhibitor LY294002. These results suggested that high glucose may impair ECFC function by modulating PI3K/Akt, NO, and p38 MAPK-related mechanisms (44). Likewise, Huang et al. reported that high glucose levels resulted in cellular senescence, defective migration, and tube formation of PB-ECFCs, as well as decreased phosphorylation of Akt and eNOS (45). Glucagon-like peptide-1 (GLP-1) plays a key role in regulating blood glucose homeostasis through GLP-1 receptor (GLP-1R) (46). Importantly, the activation of GLP-1R was able to increase NO production in endothelial cells and blunted high glucose-induced endothelial dysfunction (47). Therefore, Tu et al. investigated whether GLP-1R and SIRT1 led to PB-ECFCs dysfunction under high glucose conditions. They found that GLP-1R and SIRT1 were downregulated in PB-ECFCs under high glucose conditions (25 mM glucose). The knockdown of GLP-1R and SIRT1 aggravated the dysfunction of PB-ECFCs, including increased apoptosis, and impaired migration, adhesion and angiogenic abilities. Moreover, the upregulation of GLP-1R improved the dysfunctional ECFCs by regulating SIRT1 expression (48). Consistent with the aforementioned study, another group showed decreased transcription levels of SIRT1, SIRT3, and SIRT4 in CB-ECFCs after high glucose treatment (30 mM glucose) (49).
Persistent hyperglycemia increases the formation of advanced glycation end-products (AGEs) (50). AGEs are the products of non-enzymatic glycation of macromolecules (proteins, lipids, and nucleic acids) with monosaccharides such as glucose, glyceraldehyde, and fructose (51). AGEs are associated with increased oxidative stress (52) and inflammation (53). Chen et al. revealed that AGEs promoted the apoptosis of ECFCs and reduced the migration and tube formation of ECFCs. They investigated the effects of AGEs on Akt/eNOS and cyclooxygenase-2 (COX-2) protein expression as these proteins play an important role in endothelial cell homeostasis. The results showed that AGEs increased the expression of the receptor for AGE (RAGE) and decreased Akt and COX-2 protein expression. However, RAGE did not influence the mRNA nor protein levels of eNOS. These results provided evidence that Akt and COX-2 expression may be involved in the mechanism underlying this impairment induced by RAGE (54). Another study showed that AGEs accelerated the shedding of syndecan-4 (synd4, a ubiquitous heparan sulfate proteoglycan receptor), thereby impairing the migration of ECFCs. Besides, the lack of synd4 led to poor homing of ECFCs to the site of injury in lower limb ischemic mice (55). Additionally, a study reported that AGEs were able to promote oxidative stress via SIRT1/p66shc pathway and stimulate the upregulation of high mobility group box-1 protein (HMGB-1). The inhibition of HMGB-1 (a pro-inflammatory cytokine) attenuated oxidative stress induced by AGEs. Conversely, the effects of oxidative stress could be exacerbated by a positive feedback of which can bind to RAGEs, facilitating more ROS production (56).
Overall, mechanisms implicated in hyperglycemia-induced ECFCs dysfunction include: PI3K/Akt/eNOS pathway, the decreased SIRT1 level, and AGEs-RAGE axis. Previous studies have demonstrated that hyperglycemia causes endothelial dysfunction by four major mechanisms: (i) increased glucose flux into the polyol pathway; (ii) increased intracellular production of advanced glycation end-products (AGEs); (iii) activation of protein kinase C (PKC) isoforms; (iv) overactivity of the hexosamine pathway [see review (57)]. Therefore, it is worth exploring whether hyperglycemia could activate the polyol, hexosamine pathway, and PKC isoforms to cause ECFC damage (58).
3.2 Oxidative stress
Oxidative stress is caused by the imbalance between ROS production and ROS scavenging, which negatively affects the survival of cells (59). The important sources of ROS production include mitochondria, NADPH oxidases, xanthine oxidases, cyclooxygenases, and uncoupled endothelial (60). ROS are highly reactive oxidizing agents characterized by the presence of one or more unpaired electrons, such as superoxide anion (O2•−), hydroxyl radical (•OH), and H2O2 (61). ROS at low concentrations can serve as signaling molecules which transmit signals for normal physiological processes, such as cell growth and cellular adaptation responses (62). Excessive ROS would reduce NO bioavailability, activate inflammatory pathways mediated by TNF-α and NF-κB, leading to endothelial cell dysfunction (42). Notably, it has been reported that ROS was increased in ECFCs from patients with DM (63), which may play an important role in diabetes-induced ECFC dysfunction. But there is little evidence on how increased ROS levels disturb ECFC function in DM (64). A study found that increased ROS level in DM-ECFCs was mainly attributed to mitochondrial dysfunction. DM-ECFCs exhibited mitochondrial fragmentation and dysfunction accompanied by downregulation of nuclear factor erythroid 2-related factor 2 (Nrf2). Nrf2 is a key regulator of the cellular antioxidant system and the overexpression of Nrf2 improved the mitochondrial fragmentation and dysfunction by influencing protein associated with mitochondrial fission and fusion in DM-ECFCs (63). With the exception of decreased Nrf2 levels, other molecular mechanisms are involved in oxidative stress-mediated ECFCs dysfunction. Using the oxidative stress model induced by hydrogen peroxide (H2O2), Ingram et al. demonstrated that oxidative stress promoted apoptosis of ECFCs and diminished their tube formation in vitro and in vivo by activating apoptosis signal-regulating kinase 1 (ASK1) (35). ASK1 was shown previously to be negatively regulated by redox-sensitive binding partners (65). Furthermore, H2O2 treatment impaired the vessel formation of ECFCs in vivo (35). Another study showed that oxidative levels of five proteins (T-complex protein 1 subunit α, isoform A of prelamin-A/C, cofilin-1, peroxiredoxin-4, and actin) were upregulated in H2O2 induced ECFCs, which provided novel insight into the proteomic mechanisms of oxidative stress on ECFCs (66). Additionally, the generation of oxidized low-density lipoprotein (oxLDL) would increase under conditions of excessive oxidative stress (67). OxLDL has been shown to impair the growth and bioactivity of ECFCs, decrease Akt phosphorylation and eNOS protein expression in a dose-dependent manner, and increasing lectin-like OxLDL receptor protein expression (68). A very recent study found that ECFCs underwent endothelial-to-mesenchymal transition (EndoMT) and exhibited a loss in self-renewal and proliferative capacity after OxLDL stimulation (69). Collectively, oxidative stress impaired ECFC function by a variety of mechanisms, including causing mitochondrial fragmentation and dysfunction, increasing apoptosis, decreasing Akt phosphorylation and eNOS protein expression, and increased EndoMT. While oxidative stress has been recognized as a primary cause in endothelial cell dysfunction, its importance in ECFC dysfunction induced by diabetes requires further confirmation.
3.3 Inflammation
DM is associated with an inflammatory state, caused by complex factors, including increased levels of ROS, oxidized lipids, increased angiotensin II, free fatty acids, AGEs and reduced NO level (70). These factors lead to increased levels of plasma inflammatory cytokines in DM, including C-reactive protein (CRP) (71), interleukin-6 (IL-6), interleukin 1β (IL-1β), interleukin 8 (IL-8) (72), tumor necrosis factor-α (TNF-α), and so on (73). Theses inflammatory cytokines bind to various receptors that trigger a common pathway of mediators, like oxidative stress and NF-κB pathway, in endothelial cells, which eventually result in endothelial cell dysfunction (74). This paragraph aims to discuss the impact of inflammatory cytokines on ECFCs. Studies suggested that the inflammatory cytokines can lead to dual effects in ECFCs: (i)low level of inflammatory cytokines stimulated the proliferation of ECFCs (75); (ii) high level of inflammatory cytokines impaired the function of ECFCs (76). Two studies have reported that high concentrations of TNF-α (>10ng/ml) significantly inhibited the proliferation and tube formation of ECFCs and increased the apoptosis of CB-ECFCs (76, 77). But the mechanisms underlying the TNF-α-induced impairment of CB-ECFCs remains unclear. The interaction between TNF-α and transmembrane receptor 1 may be involved in the impairment of CB-ECFCs (78). Shen et al. provided additional evidence for inflammation induced ECFC dysfunction in DM. They compared the differentially expressed genes (DEGs) between healthy PB-ECFCs and diabetic PB-ECFCs using microarray data. Pathway analysis revealed that these DEGs were mainly associated with inflammatory pathways, such as “NF-κB signaling pathway” and “TNF-α signaling pathway”. Additionally, IL-8 (a pro-inflammatory chemokine) was upregulated in DM-ECFCs compared with healthy-ECFCs (79). However, another study has reported that increased plasma inflammatory cytokines in diabetic patients with chronic kidney disease did not influence the success rate of PB-ECFC isolation (80). Nevertheless, this study did not rule out the influence of inflammatory cytokines on other aspects of the biology of ECFCs.
Overall, inflammation could be caused by multiple factors, which leads to the increased inflammatory cytokines level in DM. These inflammatory cytokines could alter gene expression patterns of ECFCs and activate inflammatory pathways. These changes ultimately result in decreased tube formation and increased apoptosis of ECFCs.
4 Strategies for improving the therapeutic efficacy of ECFCs in DM
To achieve the desired therapeutic potential, several strategies have been proposed to improve the compromised function of ECFCs in DM. These strategies can be mainly divided into three categories: (i) pretreatment of ECFCs with biological compounds; (ii) genetic modification; (iii) co-injection with mesenchymal stem cells (MSCs) (Figure 4 and Table 1).
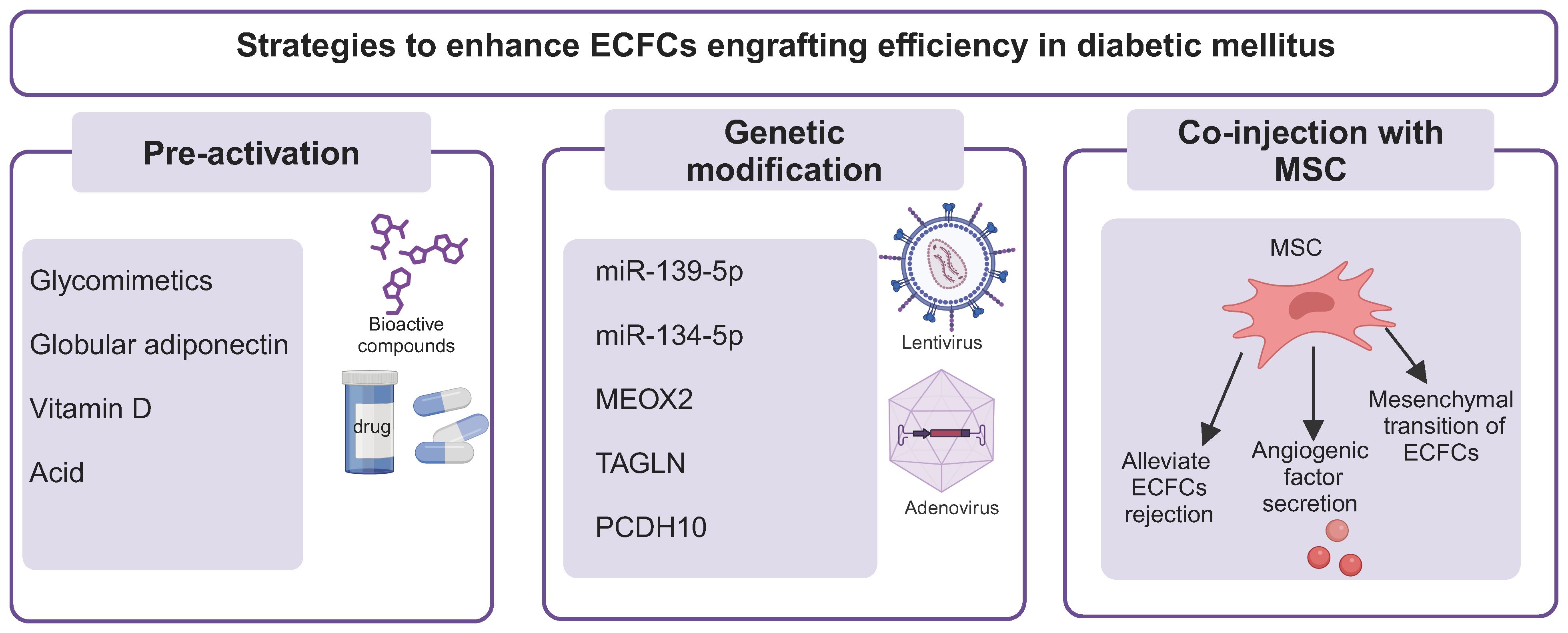
Figure 4 Strategies to enhance the therapeutic efficacy of ECFCs in DM. Several strategies have been used to improve the therapeutic efficiency of ECFCs in DM, including pretreatment of ECFCs with bioactive compounds or chemical factors (pH), genetic modification, and co-injection with MSCs. ECFCs, endothelial colony forming cells; MEOX2, mesenchyme homeobox 2; TAGLN, transgelin; PCDH10, protocadherin 10; MSC, mesenchymal stem cells; VEGFA, vascular endothelial growth factor A; FGF2, fibroblast growth factor-2.
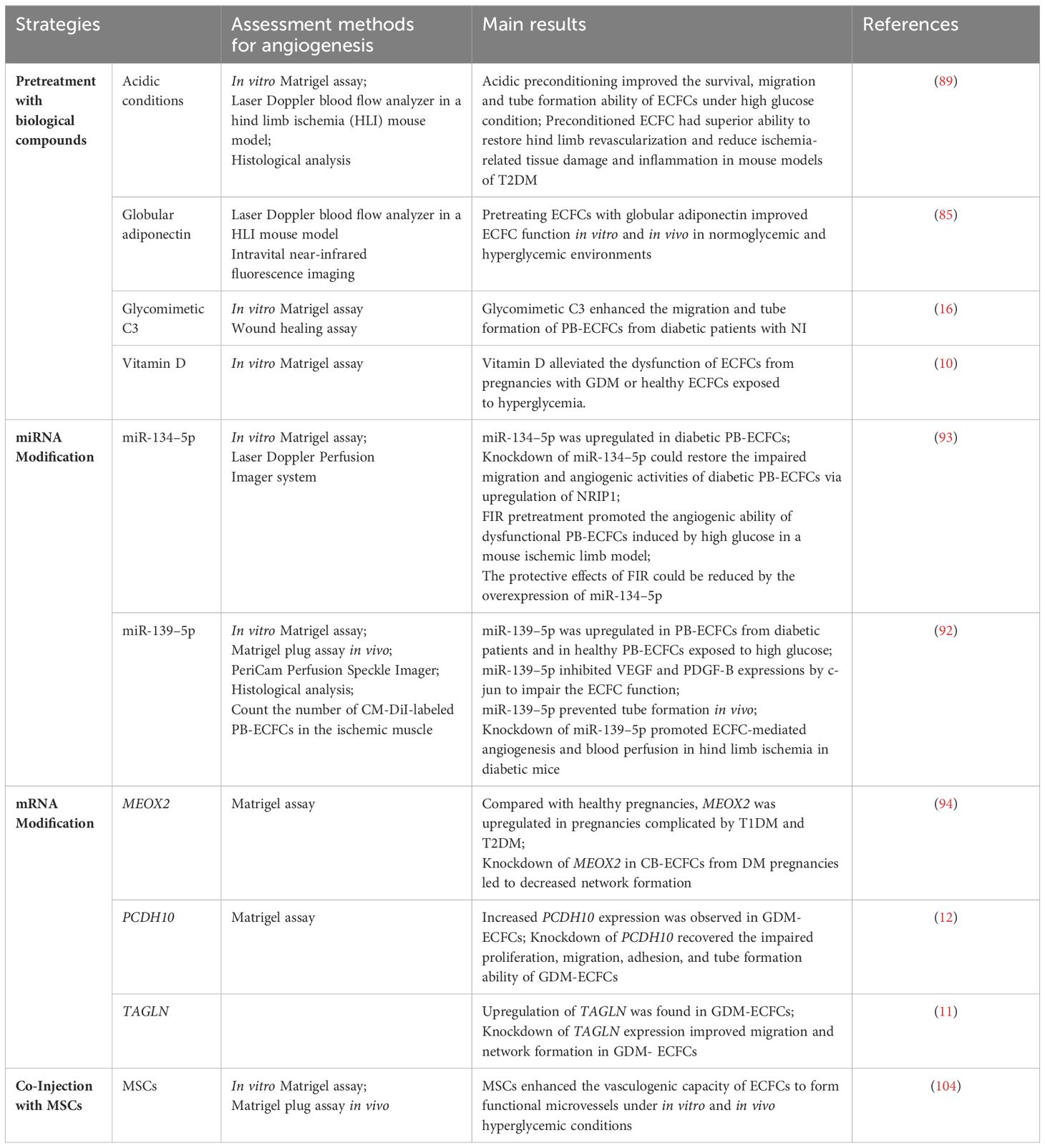
Table 1 Strategies for improving the therapeutic efficacy of ECFCs in DM.
4.1 Pretreatment of ECFCs with biological compounds
4.1.1 Pretreatment of ECFCs with glycomimetics
A previous study has demonstrated the defects in ECFCs from patients with diabetes with NI or NP ulcers (16). In an ex vivo study, it was suggested that glycomimetic C3 was able to enhance the migration ability of ECFCs from diabetic patients with NI and NP. Nevertheless, glycomimetic C3 only enhanced the tube formation in ECFCs from diabetic patients with NI. Although the mechanism by which glycomimetic C3 improved the function of ECFCs in diabetic patients was unclear, a previous study has shown that glycomimetic C3 exerted a protective effect in an endothelial model of lipid-induced oxidative stress through the Nrf2/ARE and Akt/eNOS signaling pathways (81).
4.1.2 Pretreatment of ECFCs with globular adiponectin
The literature has shown that individuals with higher serum adiponectin levels are less likely to develop insulin resistance and T2DM (82, 83). In addition, adiponectin exerts vasculoprotective actions by increasing NO production and inhibiting endothelial apoptosis (84). Leicht et al. investigated the effects of gAd (an active domain of adiponectin) on diabetic ECFCs and found that pre-treatment of diabetic ECFCs with gAd increased neovascularization in a diabetic hindlimb ischemia mouse model. Thus, preconditioning of DM-ECFCs with gAd may be a novel approach to counter their dysfunction in DM (85).
4.1.3 Pretreatment of ECFCs with vitamin D
Emerging evidence has suggested that vitamin D deficiency is associated with endothelial dysfunction (75). Vitamin D supplementation has antioxidant capacity and anti-inflammatory effects to rescue dysfunctional endothelial cells (76). Gui et al. showed that pre-activation of ECFCs with vitamin D improved the migration and tube formation of ECFCs from pregnancies with GDM (10). However, this study did not investigate the effects of vitamin D on diabetic ECFCs in vivo. Additionally, the effectiveness of vitamin D with a longer duration of supplementation and at different doses needs to be studied in the future to fully explore the potential of this therapy.
4.1.4 Pretreatment of ECFCs in acidic conditions
Reduced blood flow during tissue injury tends to drive cell metabolism into anaerobic glycolysis, which leads to lactate formation and pH reduction. Acidosis reduces cell proliferation and viability (86). Acidic preconditioning has been found to be a potential strategy to improve the cell viability in the host acidic environment (76). Mena et al. demonstrated that acid preconditioning enhanced the proliferation and tube formation of ECFCs under pro-inflammatory and high glucose condition. Compared with PBS treatment and non-preconditioned ECFCs, the transplantation of acidic preconditioned ECFCs increased the capillary density and diminished the inflammation score in the murine model of the diabetic ischemic limb (87). However, more experiments are needed to explore the different PH levels of cell culture and incubation time (88, 89) to obtain more beneficial effects and regenerative capacity of ECFCs.
4.2 Genetic modification
4.2.1 miRNAs
miRNAs are endogenous non-coding RNAs of 21–25 nucleotides that primarily affect the post-transcriptional regulation of target genes by binding to the 3’ untranslated region of mRNA (90). Luo et al. found that miR-139–5p, an anti-angiogenesis miRNA (91), was upregulated in diabetic ECFCs compared to ECFCs from healthy control patients. In vitro experiments showed that the inhibition of miR-139–5p can rescue the impaired migration and tube formation ability of diabetic ECFCs. Matrigel plug assays demonstrated that the inhibition of miR-139–5p facilitates restoration of the blood vessel-forming ability of diabetic ECFCs in vivo. Mechanistically, miR-139–5p was found to target the transcription factor c-jun, thereby decreasing the expression of VEGF/PDGF-B (92). Another study reported that miR-134–5p was more highly expressed in diabetic ECFCs compared to ECFCs from disease free controls, and was associated with impaired angiogenic activities of diabetic ECFCs. Knockdown of miR-134–5p restored the impaired migration and angiogenic activities of diabetic ECFCs via upregulation of the nuclear receptor-interacting protein 1 (NRIP1). Furthermore, the study suggested that far-infrared radiation (FIR) pretreatment promoted the angiogenic ability of dysfunctional ECFCs induced by high glucose in a mouse ischemic limb model. The protective effects of FIR could be attenuated by the overexpression of miR-134–5p (93). The study indicated that miR-134–5p may serve as a novel target for improving dysfunctional diabetic ECFCs. However, the limited sample size in combination with the lack of long-term monitoring makes it difficult to confirm the therapeutic effect of FIR.
4.2.2 Mesenchyme homeobox 2
Gohn et al. found that MEOX2 was upregulated in pregnancies complicated by T1DM and T2DM when compared with healthy pregnancies. MEOX2 knockdown can reduce the tube formation of GDM-ECFCs. This study suggested that the upregulation of MEOX2 might be a compensatory mechanism to alleviate the impaired migration and angiogenesis in GDM-ECFCs (94). But the other researchers have revealed that high MEOX2 expression resulted in increased senescence (95) and less tube formation (96) in human umbilical vein endothelial cells (HUVECs). Thus, the protective role of MEOX2 in GDM-ECFCs should be interpreted with caution.
4.2.3 Transgelin
TAGLN, a TGF-β inducible gene expressed in smooth muscle cells, has been reported to be involved in the angiogenesis of endothelial cells (97). Varberg et al. evaluated the function of TAGLN in GDM-ECFCs and found that decreasing TAGLN expression contributed to improved migration and tube formation in GDM-ECFCs (11). These results suggest that knockdown of TAGLN could be a novel strategy to improve the function of GDM-ECFCs. Interestingly, they developed a bioactive nanoparticle that can conjugate to the surface of ECFCs. The nanoparticles, loaded with SB-431542 (a TGF-β inhibitor), can stably normalize TAGLN expression in GDM-ECFCs. Not only did the bioactive nanoparticles improve migration of GDM-ECFCs, but also they augmented the vasculogenesis of GDM-ECFCs in vitro and in vivo (98).
4.2.4 Protocadherin 10
PCDH10, a member of the non-clustered protocadherins, is involved with adherens junctions (99). PCDH10 has been found to be upregulated in GDM-ECFCs compared to ECFCs from normal pregnancies. The upregulation of PCDH10 was due to the hypomethylation of the PCDH10 promoter. Knockdown of PCDH10 was able to rescue the defective proliferation, migration, cell adhesion, and angiogenic functions in GDM-ECFCs (12). Nevertheless, PCDH10 was identified as a tumor suppressor gene in various cancers (99). Thus, it is necessary to assess the safety of knockdown of PCDH10 in GDM-ECFCs.
4.3 Mesenchymal stem cells
In addition to specific biomolecules and genetic modification, co-injection with MSCs is an efficient strategy to boost the regenerative potential of ECFCs. MSCs are multipotent non-hematopoietic, fibroblast-like plastic adherent cells, which have the potential to differentiate into multiple cell types, such as chondrocytes, adipocytes, and osteocytes (100). MSCs have captured attention in vascular regeneration because these cells secrete large amounts of angiogenic factors, growth factors and cytokines (101). Several studies have demonstrated that combined transplantation of MSCs with healthy ECFCs exhibited stronger ability of vessel-like structures formation in mice when compared to translation of healthy ECFCs alone. The better effects of combined transplantation were due to the ability of MSCs to reduce the immune cell infiltration or their ability to produce paracrine factors (102, 103). However, there are very limited published data on the combination of ECFCs with MSCs in the context of DM. Lee et al. compared the vasculogenic capacity between ECFCs and ECFCs + MSCs under high glucose conditions (30 mM) in a diabetic immunodeficient mouse model. The results showed that ECFCs + MSCs potentiated the tube formation ability under hyperglycemic conditions compared to ECFCs alone. More importantly, the combination of ECFCs with MSCs was beneficial for forming tube functional microvessels in vitro (104). There are some advantages of combined transplantation of MSCs with ECFCs: (i) MSCs secrete a plethora of pro-angiogenic factors, such as VEGF-A and FGF2 (101); (ii) the immunomodulatory effects of MSCs protect against ECFCs rejection after transplantation (103); (iii) MSCs induce mesenchymal transition of ECFCs thorough NOTCH signaling, which improves engraftment and vasculogenic potential (105); (iv) MSCs bolster vasculogenic activity of ECFCs through the endoglin-mediated adhesion between MSCs and ECFCs (106).
4.4 Potential challenges and limitations of the above bioengineering strategies
Although the above strategies have shown promising results in vitro or in preclinical animal models, there are still some potential limitations that need to be considered. For pretreatment with biological compounds, this strategy is reliant on endogenous ECFC biology and its efficacy could be limited in defective ECFCs. More importantly, transient pretreatment may make it difficult to maintain the therapeutic effect for a much longer period. The use of genetically modified ECFCs will pose a biosafety concern. Viral transduction (adenovirus and lentivirus) is the most commonly used method for gene delivery into ECFCs because of its high and consistent transduction efficiency (107). Viral transduction with adenovirus and lentivirus has been associated with adverse effects such as toxicities, increased immunogenicity, and oncogenicity (108). As for co-injection with MSCs, it holds great potential for clinical translation because ECFCs and MSCs contribute to angiogenesis through different, yet complementary, mechanisms. However, the safety profile of combined therapy needs further research, as standardized approaches for combined therapy are not yet developed, including the optimal dosage, time, transplant type (autologous or allogenic), and route of administration.
5 Barriers to clinical translation of ECFCs and potential solutions
There is now significant evidence in pre-clinical models that ECFCs can be used as a therapy for diabetic vascular complications and that therapeutic effectiveness can be enhanced with modifications described above. However, it should be noted that there are no human clinical trials using ECFCs. The major challenges for clinical applications of ECFCs are divided into six different categories, including cell identity, cell dose, good manufacturing practice (GMP)-compliant cell manufacture, heterogeneity, efficacy, safety and cost (Table 2). First, it is necessary to establish unified surface markers for identifying ECFCs (37). Unified surface markers can improve the purity of ECFC populations, which may contribute to the production of a homogeneous cell population and facilitate superior comparison between studies using ECFCs. Single cell sequencing may further elucidate the characteristics of ECFCs and help to establish uniform surface markers. Uniform markers will also facilitate inter-study comparison and rapidly progress the ECFC field by reducing confusion and increasing knowledge. Second, the frequency of CB-ECFCs and PB-ECFCs is low (33, 34). ECFC large scale expansion strategies, e.g. bioreactor expansion, or enhancing ECFC potency is needed to achieve the desired clinical outcomes (109). Third, there is limited data in the literature describing GMP grade manufacture of ECFCs, with ECFC isolation and expansion being heavily reliant on high FBS content and the use of rat tail collagen for cell attachment (109). Collaboration between academic researchers, clinicians, and biotech companies can accelerate the development of GMP-compliant ECFCs. Fourth, ECFCs are an inherently heterogeneous cell population. The heterogeneity of ECFCs is affected by multiple factors, including the different donors (110), tissue origin (32, 111), and methods for isolation and culture of ECFCs (112). Therefore, it is difficult to reproducibly generate functionally equivalent ECFC populations. Fifth, disease status (113–115), cryopreservation (116), hypoxia and inflammatory microenvironment of transplantation sites (76) can lead to the loss of ECFC potency, which may impair therapeutic efficacy. The strategies mentioned in the previous paragraph should be explored to boost the potency of defective ECFCs. Lastly, the reagents (type I rat tail collagen and fetal bovine serum) for ECFC isolation and culture are not appropriate for clinical applications because they may carry animal-originated pathogens. The development of xeno-free medium might help to solve this issue, such as platelet lysate supplemented culture medium (109) and EC-Cult-XF ECFC medium (Stemcell Technologies, Canada). However, these xeno-free media are expensive to scale-up for clinical use currently. Collectively, there needs to be considerable attention paid to the good manufacturing practice (GMP) of human ECFCs keeping in mind the issues raised in this section on standardization, heterogeneity, optimal approaches for cell propagation, delivery and safety.
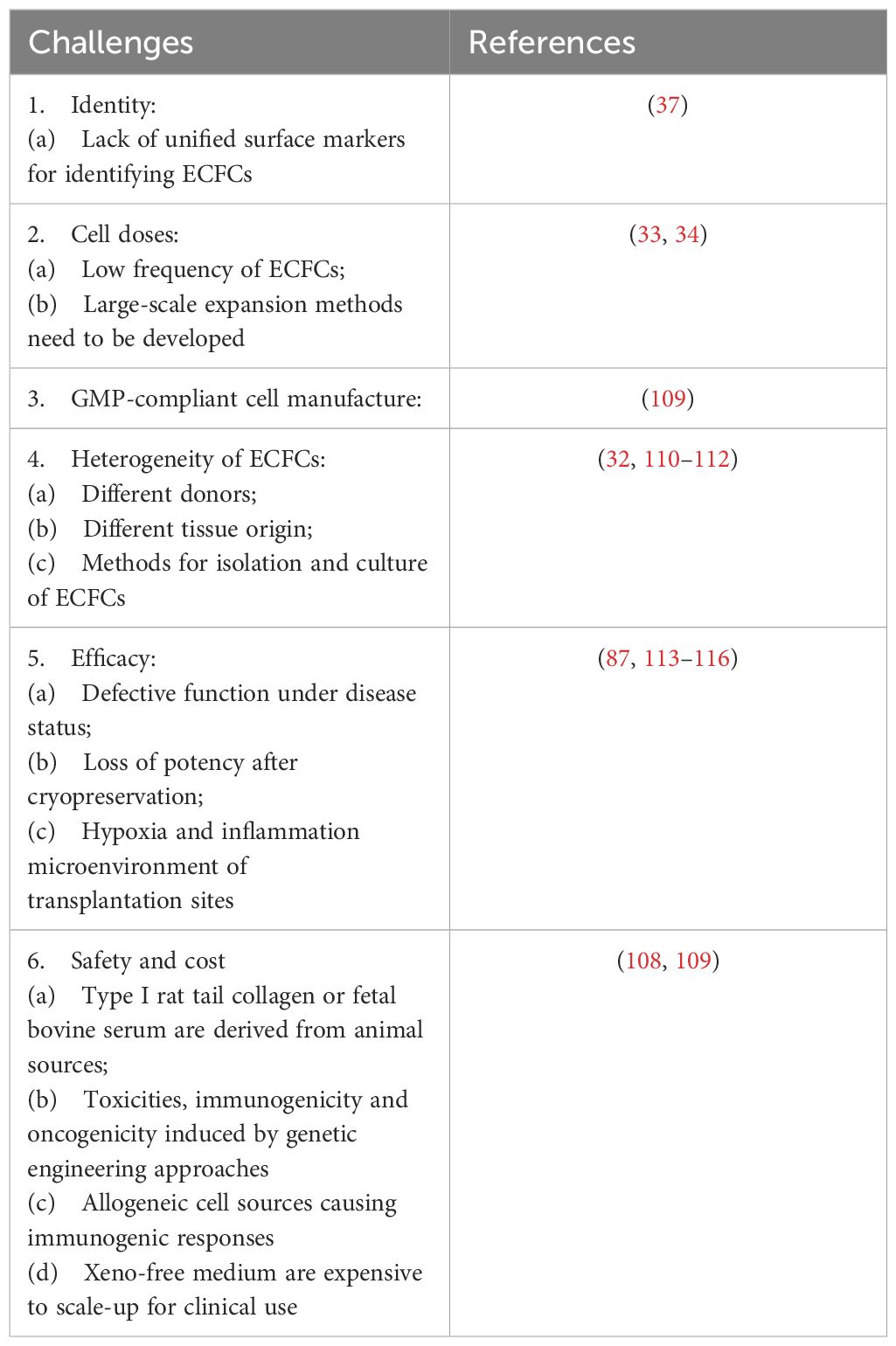
Table 2 Major Challenges for clinical applications of ECFCs.
6 Comparison of ECFCs with other possible cell or EV treatments in diabetic vascular complication
Due to the low frequency of PB-ECFCs and impaired function of ECFCs in DM, other possible sources have been explored for the treatment of diabetic vascular complication. Thus, we next compared the efficacy of ECFCs with other stem cell approaches or extracellular vesicles (EVs) derived from stem cells in diabetic vascular complications (Table 3).
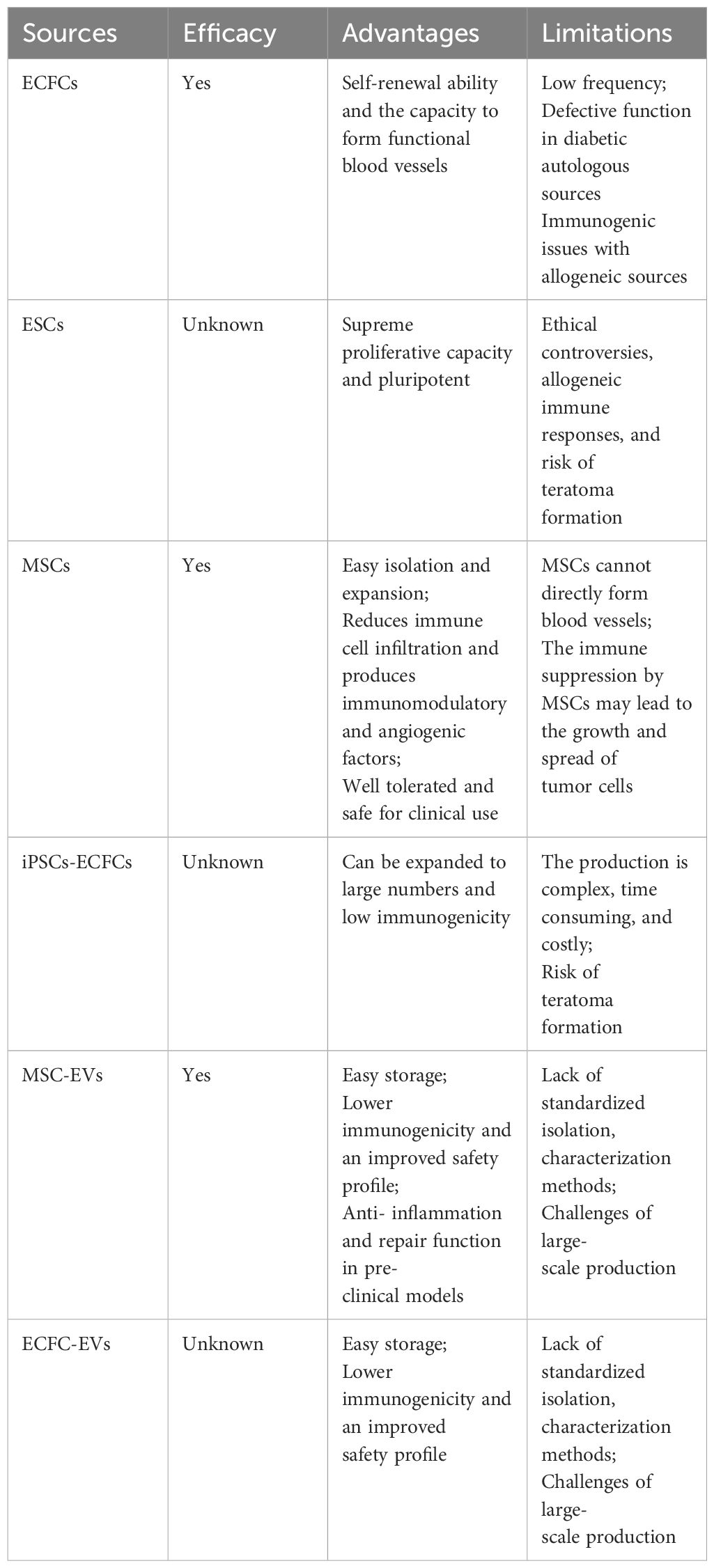
Table 3 Comparison of ECFCs with other possible cell or EV treatments in diabetic vascular complications.
Pluripotent embryonic stem cells (ESCs) originate from the inner cell mass of the blastocyst, which possess the capacity to differentiate into any cell type (117). Animal studies have suggested that ESCs-derived endothelial cells can incorporate into the vasculature of the ischemic limb and improve limb perfusion (118, 119). These studies indicated that ESCs can successfully treat ischemia, while evidence of their effects in diabetic vascular complications is still lacking. A significant potential advantage of differentiating ESCs into endothelial cells lies in the opportunity to induce tissue-specific EC phenotypes (120). However, although ESCs exhibit a greater proliferative capacity than adult stem cells, the application of ESCs in clinical settings is hindered by ethical controversies over human embryos, allogeneic immune responses post transplantation, and the risk of teratoma formation (121).
MSCs from different sources have been widely used in the treatment of diabetic vascular complications. Preclinical models and clinical trials have demonstrated a broad range of beneficial effects of MSCs in the treatment for diabetic complications, including vascular protective effects (122–124). For instance, Zhang et al. summarized the effects of umbilical cord mesenchymal stem cells (UC-MSCs) in the treatment of diabetic foot, including nine preclinical experiments and five clinical trials. These studies have demonstrated the impressive efficacy and safety of UC-MSCs in diabetic foot (125). Besides, researchers have suggested that UC-MSCs and adipose-derived MSCs could have superior clinical application prospects as they are more easily accessible and have better immunomodulatory properties than MSCs from bone marrow (126, 127). Nonetheless, MSCs promote vascular repair through paracrine and trophic mechanisms instead of forming blood vessels themselves (128). The combination between MSCs and ECFCs could be an exciting therapeutic prospect.
Induced pluripotent stem cells (iPSCs) are derived from mature cells like fibroblasts, which can be reprogrammed to differentiate into any of the three germ layers (129). One recent breakthrough was the observation that human induced pluripotent stem cells (iPSCs) can be differentiated into ECFCs. These iPSCs-ECFCs have been shown to possess high clonal proliferative potential, form capillary structures in Matrigel, and contribute to vascular repair in both oxygen-induced retinopathy and hind limb ischemia mice model. The iPSCs-ECFCs have similar capacity of promoting neovascularization when they were compared with CB-ECFCs (130). More importantly, iPSCs-ECFCs were derived from fibroblasts, which were easily obtained in large quantities and did not face the immunologic barriers associated with allogeneic cell therapies. However, differentiation of iPSCs into the ECFC lineage is complex, time consuming, and costly. Undifferentiated cells during the differentiation process could cause teratomas and the potential of iPSCs-ECFCs needs to be further investigated in the context of DM.
EVs have attracted considerable attention due to the therapeutic potential of bioactive molecules contained inside EVs, including peptides, proteins, lipids, and nucleic acids (microRNAs and mRNA) (131). An additional factor to consider is that EVs have low immunogenicity and an improved safety profile compared to cell-based therapies (132). EVs from MSC are considered as promising products for the treatment of diabetic vascular complications [see review (133)]. In particular, MSC-EVs have been reported to promote angiogenesis and wound healing in diabetic mice model (134). MSC-EVs exert their protective effects in diabetic vascular complications through multiple methods, such as reduction of inflammatory cytokines, activation of angiogenesis, and promotion of collagen synthesis (135). Despite promising in vitro and in vivo results, to date no clinical trials have been conducted to evaluate the efficacy and safety of MSC-EVs in diabetic vascular complications. As for ECFC-EVs, a few studies suggested that they had protective effects on preclinical mouse models of ischemia, such as ischemic retinopathy (136) and ischemic kidney injury (137). However, no studies have yet been performed to investigate the therapeutic effects of ECFC-EVs in ischemic conditions in the context of a diabetic environment. Moreover, it remains unknown whether tissue source, isolation protocol, and disease status influence the bioactivity of ECFC-EVs.
Therefore, MSCs are the most studied cell therapy in diabetic vascular complications ECFCs have shown promising outcomes in preclinical studies. The combination MSCs with ECFCs, iPSCs-ECFCs and EVs from MSC and ECFCs offer promising avenues for treating diabetic vascular complications.
7 Conclusion and future perspective
ECFCs enhance the formation of blood vessels and contribute to revascularization, which may benefit diabetic patients with severe vascular complications. However, ECFCs isolated from DM patients exhibited a dysfunctional phenotype. Although the involvement of hyperglycemia, oxidative stress, and inflammation has been discussed in this review, underlying the mechanisms of dysfunctional ECFCs in DM are not well understood. The role of noncoding RNAs and epigenetic regulation in DM-ECFCs requires further study.
Currently, combining ECFCs with biological compounds, gene modification, or MSC has shown efficacy in rescuing diabetes-mediated ECFC damage. However these strategies need to be evaluated in more studies and pre-clinical models in the future. Besides, regulatory issues related to cell modification and issues of ECFC expansion under diabetic conditions will need to be addressed to enhance the realization of the potential of this therapy. Moreover, the application of biomaterials, 3D bioprinting of vascular structures might represent potential future strategies to improve the properties and functional capabilities of ECFCs in DM. The development of ipSCs-ECFCs and ECFC-EVs could be promising alternative treatments for patients with DM.
Author contributions
YL: Conceptualization, Data curation, Funding acquisition, Investigation, Writing – original draft. CL: Methodology, Validation, Writing – review & editing. CA: Methodology, Validation, Writing – review & editing. TO: Conceptualization, Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors are grateful for the support of the China Scholarship Council (No. 202006370051) and Science Foundation Ireland (20/FFP-A/8794).
Acknowledgments
We would like to thank members of the O’Brien lab and Linyi Lai for constructive feedback. The figures were created with BioRender.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AGEs, Advanced glycation end-products; ASK1, Apoptosis signal-regulating kinase 1; CB, Cord blood; CBMNCs, Cord blood mononuclear cells; COX-2, Cycloxygenase-2; CRP, C-reactive protein; DEGs, Differently expressed genes; DM, Diabetes mellitus; ECFCs, Endothelial colony forming cells; ECC, Endothelial cell cluster; FIR, Far-infrared radiation; eNOS, Endothelial nitric oxide synthase; gAcrp, Globular adiponectin; GDM, Gestational diabetes mellitus; GLP-1R, Glucagonlike peptide-1 receptor; GMP, Good manufacturing practice; HA, Hyaluronic acid; HPP-ECFC, High proliferative potential ECFC; HUVECs, Human umbilical vein endothelial cells; IL-6, Interleukin 6; IL-1β, Interleukin 1β; IL-8, Interleukin 8; iPSCs, Induced pluripotent stem cells; LPP-ECFC, Low proliferative potential ECFC; MEOX2, Mesenchyme homeobox 2; MSCs, Mesenchymal stromal cells; NI, Neuroischemic; NO, Nitric oxide; NP, Neuropathic; NRIP1, Nuclear receptor-interacting protein 1; oxLDL, Oxidized low density lipoprotein; PB, Peripheral blood; PBMNCs, Peripheral blood mononuclear cells; PCDH10, Protocadherin 10; RAGE, Receptor for advanced glycation end-products; ROS, Reactive oxygen species; SIRT1, Sirtuin-1; TAGLN, Transgelin; TNF-a, Tumor necrosis factor-a; VWF, Von willebrand factor.
References
1. Atkinson MA, Eisenbarth GS, Michels AW. Type 1 diabetes. Lancet. (2014) 383:69–82. doi: 10.1016/S0140-6736(13)60591-7
2. Association AD. 2. Classification and diagnosis of diabetes: standards of medical care in diabetes—2020. Diabetes Care. (2020) 43:S14–31. doi: 10.2337/dc23-S002
3. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
4. Dal Canto E, Ceriello A, Rydén L, Ferrini M, Hansen TB, Schnell O, et al. Diabetes as a cardiovascular risk factor: An overview of global trends of macro and micro vascular complications. Eur J Prev Cardiol. (2019) 26:25–32. doi: 10.1177/2047487319878371
5. Fowler MJ. Microvascular and macrovascular complications of diabetes. Clin Diabetes. (2008) 26:77–82. doi: 10.2337/diaclin.26.2.77
6. Melero-Martin JM. Human endothelial colony-forming cells. Cold Spring Harbor Perspect Med. (2022) 12:a041154. doi: 10.1101/cshperspect.a041154
7. Lyons CJ, O’Brien T. The functionality of endothelial-colony-forming cells from patients with diabetes mellitus. Cells. (2020) 9:1731. doi: 10.3390/cells9071731
8. Wils J, Favre J, Bellien J. Modulating putative endothelial progenitor cells for the treatment of endothelial dysfunction and cardiovascular complications in diabetes. Pharmacol Ther. (2017) 170:98–115. doi: 10.1016/j.pharmthera.2016.10.014
9. Blue EK, DiGiuseppe R, Derr-Yellin E, Acosta JC, Pay SL, Hanenberg H, et al. Gestational diabetes induces alterations in the function of neonatal endothelial colony-forming cells. Pediatr Res. (2014) 75:266–72. doi: 10.1038/pr.2013.224
10. Gui J, Rohrbach A, Borns K, Hillemanns P, Feng L, Hubel C, et al. Vitamin D rescues dysfunction of fetal endothelial colony forming cells from individuals with gestational diabetes. Placenta. (2015) 36:410–8. doi: 10.1016/j.placenta.2015.01.195
11. Varberg KM, Garretson RO, Blue EK, Chu C, Gohn CR, Tu W, et al. Transgelin induces dysfunction of fetal endothelial colony-forming cells from gestational diabetic pregnancies. Am J Physiol-Cell Physiol. (2018) 315:C502–C15. doi: 10.1152/ajpcell.00137.2018
12. Kwon H, Jung YJ, Lee Y, Son G-H, Kim HO, Maeng Y-S, et al. Impaired angiogenic function of fetal endothelial progenitor cells via PCDH10 in gestational diabetes mellitus. Int J Mol Sci. (2023) 24:16082. doi: 10.3390/ijms242216082
13. Weiss E, Leopold-Posch B, Schrüfer A, Cvitic S, Hiden U. Fetal sex and maternal fasting glucose affect neonatal cord blood-derived endothelial progenitor cells. Pediatr Res. (2022) 92:1590–7. doi: 10.1038/s41390-022-01966-4
14. Jarajapu YP, Hazra S, Segal M, LiCalzi S, Jhadao C, Qian K, et al. Vasoreparative dysfunction of CD34+ cells in diabetic individuals involves hypoxic desensitization and impaired autocrine/paracrine mechanisms. PloS One. (2014) 9:e93965. doi: 10.1371/journal.pone.0093965
15. Tan K, Lessieur E, Cutler A, Nerone P, Vasanji A, Asosingh K, et al. Impaired function of circulating CD34+ CD45– cells in patients with proliferative diabetic retinopathy. Exp Eye Res. (2010) 91:229–37. doi: 10.1016/j.exer.2010.05.012
16. Langford-Smith AW, Hasan A, Weston R, Edwards N, Jones AM, Boulton AJ, et al. Diabetic endothelial colony forming cells have the potential for restoration with glycomimetics. Sci Rep. (2019) 9:2309. doi: 10.1038/s41598-019-38921-z
17. Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. (1997) 275:964–6. doi: 10.1126/science.275.5302.964
18. Rehman J, Li J, Orschell CM, March KL. Peripheral blood “endothelial progenitor cells” are derived from monocyte/macrophages and secrete angiogenic growth factors. Circulation. (2003) 107:1164–9. doi: 10.1161/01.CIR.0000058702.69484.A0
19. Medina RJ, O’Neill CL, O’Doherty TM, Knott H, Guduric-Fuchs J, Gardiner TA, et al. Myeloid angiogenic cells act as alternative M2 macrophages and modulate angiogenesis through interleukin-8. Mol Med. (2011) 17:1045–55. doi: 10.2119/molmed.2011.00129
20. Ingram DA, Mead LE, Tanaka H, Meade V, Fenoglio A, Mortell K, et al. Identification of a novel hierarchy of endothelial progenitor cells using human peripheral and umbilical cord blood. Blood. (2004) 104:2752–60. doi: 10.1182/blood-2004-04-1396
21. Medina RJ, Barber CL, Sabatier F, Dignat-George F, Melero-Martin JM, Khosrotehrani K, et al. Endothelial progenitors: a consensus statement on nomenclature. Stem Cells Trans Med. (2017) 6:1316–20. doi: 10.1002/sctm.16-0360
22. Pyšná A, Bém R, Němcová A, Fejfarová V, Jirkovská A, Hazdrová J, et al. Endothelial progenitor cells biology in diabetes mellitus and peripheral arterial disease and their therapeutic potential. Stem Cell Rev Rep. (2019) 15:157–65. doi: 10.1007/s12015-018-9863-4
23. Basile DP, Yoder MC. Circulating and tissue resident endothelial progenitor cells. J Cell Physiol. (2014) 229:10–6. doi: 10.1002/jcp.24423
24. Lin Y, Weisdorf DJ, Solovey A, Hebbel RP. Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest. (2000) 105:71–7. doi: 10.1172/JCI8071
25. Fujisawa T, Tura-Ceide O, Hunter A, Mitchell A, Vesey A, Medine C, et al. Endothelial progenitor cells do not originate from the bone marrow. Circulation. (2019) 140:1524–6. doi: 10.1161/CIRCULATIONAHA.119.042351
26. Lin Y, Banno K, Gil C-H, Myslinski J, Hato T, Shelley WC, et al. Origin, prospective identification, and function of circulating endothelial colony-forming cells in mice and humans. JCI Insight. (2023) 8:e164781. doi: 10.1172/jci.insight.164781
27. Mead LE, Prater D, Yoder MC, Ingram DA. Isolation and characterization of endothelial progenitor cells from human blood. Curr Protoc Stem Cell Biol. (2008) 6:2C. 1–2C. 1.27. doi: 10.1002/9780470151808.sc02c01s6
28. Lin R-Z, Moreno-Luna R, Muñoz-Hernandez R, Li D, Jaminet S-CS, Greene AK, et al. Human white adipose tissue vasculature contains endothelial colony-forming cells with robust in vivo vasculogenic potential. Angiogenesis. (2013) 16:735–44. doi: 10.1007/s10456-013-9350-0
29. Duong HT, Comhair SA, Aldred MA, Mavrakis L, Savasky BM, Erzurum SC, et al. Pulmonary artery endothelium resident endothelial colony-forming cells in pulmonary arterial hypertension. Pulmonary Circ. (2011) 1:475–86. doi: 10.4103/2045-8932.93547
30. Solomon I, O'Reilly M, Ionescu L, Alphonse RS, Rajabali S, Zhong S, et al. Functional differences between placental micro-and macrovascular endothelial colony-forming cells. Stem Cells Trans Med. (2016) 5:291–300. doi: 10.5966/sctm.2014-0162
31. Khoo CP, Roubelakis MG, Schrader JB, Tsaknakis G, Konietzny R, Kessler B, et al. miR-193a-3p interaction with HMGB1 downregulates human endothelial cell proliferation and migration. Sci Rep. (2017) 7:1–14. doi: 10.1038/srep44137
32. Ferratge S, Ha G, Carpentier G, Arouche N, Bascetin R, Muller L, et al. Initial clonogenic potential of human endothelial progenitor cells is predictive of their further properties and establishes a functional hierarchy related to immaturity. Stem Cell Res. (2017) 21:148–59. doi: 10.1016/j.scr.2017.04.009
33. Gumina DL, Black CP, Balasubramaniam V, Winn VD, Baker CD. Umbilical cord blood circulating progenitor cells and endothelial colony-forming cells are decreased in preeclampsia. Reprod Sci. (2017) 24:1088–96. doi: 10.1177/1933719116678692
34. Yoder MC, Mead LE, Prater D, Krier TR, Mroueh KN, Li F, et al. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood. (2007) 109:1801–9. doi: 10.1182/blood-2006-08-043471
35. Ingram DA, Krier TR, Mead LE, McGuire C, Prater DN, Bhavsar J, et al. Clonogenic endothelial progenitor cells are sensitive to oxidative stress. Stem Cells. (2007) 25:297–304. doi: 10.1634/stemcells.2006-0340
36. Blandinières A, Randi AM, Paschalaki KE, Guerin CL, Melero-Martin JM, Smadja DM. Results of an international survey about methods used to isolate human endothelial colony-forming cells: guidance from the SSC on Vascular Biology of the ISTH. J Thromb Hemostasis. (2023) 21:2611–9. doi: 10.1016/j.jtha.2023.06.014
37. Chambers SEJ, Pathak V, Pedrini E, Soret L, Gendron N, Guerin CL, et al. Current concepts on endothelial stem cells definition, location, and markers. Stem Cells Trans Med. (2021) 10:S54–61. doi: 10.1002/sctm.21-0022
38. Ingram DA, Lien IZ, Mead LE, Estes M, Prater DN, Derr-Yellin E, et al. In vitro hyperglycemia or a diabetic intrauterine environment reduces neonatal endothelial colony-forming cell numbers and function. Diabetes. (2008) 57:724–31. doi: 10.2337/db07-1507
39. Zhang H-n, Dai Y, Zhang C-h, Omondi AM, Ghosh A, Khanra I, et al. Sirtuins family as a target in endothelial cell dysfunction: implications for vascular ageing. Biogerontology. (2020) 21:495–516. doi: 10.1007/s10522-020-09873-z
40. Kitada M, Ogura Y, Monno I, Koya D. Sirtuins and type 2 diabetes: role in inflammation, oxidative stress, and mitochondrial function. Front Endocrinol. (2019) 10:187. doi: 10.3389/fendo.2019.00187
41. Mattagajasingh I, Kim C-S, Naqvi A, Yamamori T, Hoffman TA, Jung S-B, et al. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci. (2007) 104:14855–60. doi: 10.1073/pnas.0704329104
42. Magenta A, Greco S, Capogrossi MC, Gaetano C, Martelli F. Nitric oxide, oxidative stress, and p66Shc interplay in diabetic endothelial dysfunction. BioMed Res Int. (2014) 193095. doi: 10.1155/2014/193095
43. Chatterjee A, Catravas JD. Endothelial nitric oxide (NO) and its pathophysiologic regulation. Vasc Pharmacol. (2008) 49:134–40. doi: 10.1016/j.vph.2008.06.008
44. Chen Y-H, Lin S-J, Lin F-Y, Wu T-C, Tsao C-R, Huang P-H, et al. High glucose impairs early and late endothelial progenitor cells by modifying nitric oxide–related but not oxidative stress–mediated mechanisms. Diabetes. (2007) 56:1559–68. doi: 10.2337/db06-1103
45. Huang P-H, Chen J-S, Tsai H-Y, Chen Y-H, Lin F-Y, Leu H-B, et al. Globular adiponectin improves high glucose-suppressed endothelial progenitor cell function through endothelial nitric oxide synthase dependent mechanisms. J Mol Cell Cardiol. (2011) 51:109–19. doi: 10.1016/j.yjmcc.2011.03.008
46. Nadkarni P, Chepurny OG, Holz GG. Regulation of glucose homeostasis by GLP-1. Prog Mol Biol Trans Sci. (2014) 121:23–65. doi: 10.1016/B978-0-12-800101-1.00002-8
47. Koska J, Sands M, Burciu C, D’Souza KM, Raravikar K, Liu J, et al. Exenatide protects against glucose-and lipid-induced endothelial dysfunction: evidence for direct vasodilation effect of GLP-1 receptor agonists in humans. Diabetes. (2015) 64:2624–35. doi: 10.2337/db14-0976
48. Tu Q, Wang J-f, Xie H-q, Zhao Q, Fu J, Xu H-l, et al. Up-regulation of GLP-1R improved the dysfunction of late EPCs under hyperglycemia by regulating SIRT1 expression. Mol Cell Endocrinol. (2021) 538:111455. doi: 10.1016/j.mce.2021.111455
49. Gui J, Potthast A, Rohrbach A, Borns K, Das AM, von Versen-Hoeynck F. Gestational diabetes induces alterations of sirtuins in fetal endothelial cells. Pediatr Res. (2016) 79:788–98. doi: 10.1038/pr.2015.269
50. Goh S-Y, Cooper ME. The role of advanced glycation end products in progression and complications of diabetes. J Clin Endocrinol Metab. (2008) 93:1143–52. doi: 10.1210/jc.2007-1817
51. Ruiz HH, Ramasamy R, Schmidt AM. Advanced glycation end products: building on the concept of the “common soil” in metabolic disease. Endocrinology. (2020) 161:bqz006. doi: 10.1210/endocr/bqz006
52. Guimarães EL, Empsen C, Geerts A, van Grunsven LA. Advanced glycation end products induce production of reactive oxygen species via the activation of NADPH oxidase in murine hepatic stellate cells. J Hepatol. (2010) 52:389–97. doi: 10.1016/j.jhep.2009.12.007
53. Peyroux J, Sternberg M. Advanced glycation end products (AGEs): pharmacological inhibition in diabetes. Pathol Biol. (2006) 54:405–19. doi: 10.1016/j.patbio.2006.07.006
54. Chen Q, Dong L, Wang L, Kang L, Xu B. Advanced glycation end products impair function of late endothelial progenitor cells through effects on protein kinase Akt and cyclooxygenase-2. Biochem Biophys Res Commun. (2009) 381:192–7. doi: 10.1016/j.bbrc.2009.02.040
55. Xie J, Li R, Wu H, Chen J, Li G, Chen Q, et al. Advanced glycation end products impair endothelial progenitor cell migration and homing via syndecan 4 shedding. Stem Cells. (2017) 35:522–31. doi: 10.1002/stem.2506
56. Wu H, Li R, Wei Z-H, Zhang X-L, Chen J-Z, Dai Q, et al. Diabetes-induced oxidative stress in endothelial progenitor cells may be sustained by a positive feedback loop involving high mobility group box-1. Oxid Med Cell Longevity. (2016) 1943918. doi: 10.1155/2016/1943918
57. Maruhashi T, Higashi Y. Pathophysiological association between diabetes mellitus and endothelial dysfunction. Antioxidants. (2021) 10:1306. doi: 10.3390/antiox10081306
58. Sena CM, Pereira AM, Seiça R. Endothelial dysfunction—a major mediator of diabetic vascular disease. Biochim Biophys Acta (BBA)-Molecular Basis Dis. (2013) 1832:2216–31. doi: 10.1016/j.bbadis.2013.08.006
59. An Y, Xu B-t, Wan S-r, Ma X-m, Long Y, Xu Y, et al. The role of oxidative stress in diabetes mellitus-induced vascular endothelial dysfunction. Cardiovasc Diabetol. (2023) 22:237. doi: 10.1186/s12933-023-01965-7
60. Rochette L, Zeller M, Cottin Y, Vergely C. Diabetes, oxidative stress and therapeutic strategies. Biochim Biophys Acta (BBA)-General Subj. (2014) 1840:2709–29. doi: 10.1016/j.bbagen.2014.05.017
61. Fatehi-Hassanabad Z, Chan CB, Furman BL. Reactive oxygen species and endothelial function in diabetes. Eur J Pharmacol. (2010) 636:8–17. doi: 10.1016/j.ejphar.2010.03.048
62. van den Oever IA, Raterman HG, Nurmohamed MT, Simsek S. Endothelial dysfunction, inflammation, and apoptosis in diabetes mellitus. Mediators Inflammation. (2010) 792393. doi: 10.1155/2010/792393
63. Dai X, Wang K, Fan J, Liu H, Fan X, Lin Q, et al. Nrf2 transcriptional upregulation of IDH2 to tune mitochondrial dynamics and rescue angiogenic function of diabetic EPCs. Redox Biol. (2022) 56:102449. doi: 10.1016/j.redox.2022.102449
64. Guo Z, Wan X, Luo Y, Liang F, Jiang S, Yuan X, et al. The vicious circle of UHRF1 down-regulation and KEAP1/NRF2/HO-1 pathway impairment promotes oxidative stress-induced endothelial cell apoptosis in diabetes. Diabetic Med. (2023) 40:e15026. doi: 10.1111/dme.15026
65. Ichijo H, Nishida E, Irie K, Dijke PT, Saitoh M, Moriguchi T, et al. Induction of apoptosis by ASK1, a mammalian MAPKKK that activates SAPK/JNK and p38 signaling pathways. Science. (1997) 275:90–4. doi: 10.1126/science.275.5296.90
66. Wei J, Liu Y, Chang M, Sun C-L, Li D-W, Liu Z-Q, et al. Proteomic analysis of oxidative modification in endothelial colony-forming cells treated by hydrogen peroxide. Int J Mol Med. (2012) 29:1099–105. doi: 10.3892/ijmm.2012.944
67. Pennathur S, Heinecke JW. Mechanisms for oxidative stress in diabetic cardiovascular disease. Antioxid Redox Signaling. (2007) 9:955–69. doi: 10.1089/ars.2007.1595
68. Ma FX, Zhou B, Chen Z, Ren Q, Lu SH, Sawamura T, et al. Oxidized low density lipoprotein impairs endothelial progenitor cells by regulation of endothelial nitric oxide synthase. J Lipid Res. (2006) 47:1227–37. doi: 10.1194/jlr.M500507-JLR200
69. Zhao J, Sormani L, Jacquelin S, Li H, Styke C, Zhou C, et al. Distinct roles of SOX9 in self-renewal of progenitors and mesenchymal transition of the endothelium. Angiogenesis (2024) 1–16. doi: 10.1007/s10456-024-09927-7
70. Goldberg RB. Cytokine and cytokine-like inflammation markers, endothelial dysfunction, and imbalanced coagulation in development of diabetes and its complications. J Clin Endocrinol Metab. (2009) 94:3171–82. doi: 10.1210/jc.2008-2534
71. Wang X, Bao W, Liu J, OuYang Y-Y, Wang D, Rong S, et al. Inflammatory markers and risk of type 2 diabetes: a systematic review and meta-analysis. Diabetes Care. (2013) 36:166–75. doi: 10.2337/dc12-0702
72. Doganay S, Evereklioglu C, Er H, Türköz Y, Sevinc A, Mehmet N, et al. Comparison of serum NO, TNF-α, IL-1β, sIL-2R, IL-6 and IL-8 levels with grades of retinopathy in patients with diabetes mellitus. Eye. (2002) 16:163–70. doi: 10.1038/sj/eye/6700095
73. Liu C, Feng X, Li Q, Wang Y, Li Q, Hua M. Adiponectin, TNF-α and inflammatory cytokines and risk of type 2 diabetes: a systematic review and meta-analysis. cytokine. (2016) 86:100–9. doi: 10.1016/j.cyto.2016.06.028
74. Saad MI, Abdelkhalek TM, Saleh MM, Kamel MA, Youssef M, Tawfik SH, et al. Insights into the molecular mechanisms of diabetes-induced endothelial dysfunction: focus on oxidative stress and endothelial progenitor cells. Endocrine. (2015) 50:537–67. doi: 10.1007/s12020-015-0709-4
75. Green LA, Njoku V, Mund J, Case J, Yoder M, Murphy MP, et al. Endogenous transmembrane TNF-alpha protects against premature senescence in endothelial colony forming cells. Circ Res. (2016) 118:1512–24. doi: 10.1161/CIRCRESAHA.116.308332
76. Mena HA, Lokajczyk A, Dizier B, Strier SE, Voto LS, Boisson-Vidal C, et al. Acidic preconditioning improves the proangiogenic responses of endothelial colony forming cells. Angiogenesis. (2014) 17:867–79. doi: 10.1007/s10456-014-9434-5
77. Bian N, Du G, Ip MF, Ding J, Chang Q, Li Z. Pituitary adenylate cyclase-activating polypeptide attenuates tumor necrosis factor-α-induced apoptosis in endothelial colony-forming cells. Biomed Rep. (2017) 7:11–6. doi: 10.3892/br.2017.917
78. Naserian S, Abdelgawad ME, Afshar Bakshloo M, Ha G, Arouche N, Cohen JL, et al. The TNF/TNFR2 signaling pathway is a key regulatory factor in endothelial progenitor cell immunosuppressive effect. Cell Communication Signaling. (2020) 18:1–14. doi: 10.1186/s12964-020-00564-3
79. Shen Z, Chen Q, Ying H, Ma Z, Bi X, Li X, et al. Identification of differentially expressed genes in the endothelial precursor cells of patients with type 2 diabetes mellitus by bioinformatics analysis. Exp Ther Med. (2020) 19:499–510. doi: 10.3892/etm.2019.8239
80. Lapidos KA, Sprague SM, Ameer GA. Impact of serum source and inflammatory cytokines on the isolation of endothelial colony-forming cells from peripheral blood. J Tissue Eng Regenerative Med. (2014) 8:747–56. doi: 10.1002/term.v8.9
81. Mahmoud AM, Wilkinson FL, Jones AM, Wilkinson JA, Romero M, Duarte J, et al. A novel role for small molecule glycomimetics in the protection against lipid-induced endothelial dysfunction: involvement of Akt/eNOS and Nrf2/ARE signaling. Biochim Biophys Acta (BBA)-General Subj. (2017) 1861:3311–22. doi: 10.1016/j.bbagen.2016.08.013
82. Snehalatha C, Mukesh B, Simon M, Viswanathan V, Haffner SM, Ramachandran A. Plasma adiponectin is an independent predictor of type 2 diabetes in Asian Indians. Diabetes Care. (2003) 26:3226–9. doi: 10.2337/diacare.26.12.3226
83. Yamamoto Y, Hirose H, Saito I, Nishikai K, Saruta T. Adiponectin, an adipocyte-derived protein, predicts future insulin resistance: two-year follow-up study in Japanese population. J Clin Endocrinol Metab. (2004) 89:87–90. doi: 10.1210/jc.2003-031163
84. Achari AE, Jain SK. Adiponectin, a therapeutic target for obesity, diabetes, and endothelial dysfunction. Int J Mol Sci. (2017) 18:1321. doi: 10.3390/ijms18061321
85. Leicht SF, Schwarz TM, Hermann PC, Seissler J, Aicher A, Heeschen C. Adiponectin pretreatment counteracts the detrimental effect of a diabetic environment on endothelial progenitors. Diabetes. (2011) 60:652–61. doi: 10.2337/db10-0240
86. Moeinabadi-Bidgoli K, Babajani A, Yazdanpanah G, Farhadihosseinabadi B, Jamshidi E, Bahrami S, et al. Translational insights into stem cell preconditioning: from molecular mechanisms to preclinical applications. Biomed Pharmacother. (2021) 142:112026. doi: 10.1016/j.biopha.2021.112026
87. Mena HA, Zubiry PR, Dizier B, Schattner M, Boisson-Vidal C, Negrotto S. Acidic preconditioning of endothelial colony-forming cells (ECFC) promote vasculogenesis under proinflammatory and high glucose conditions in vitro and in vivo. Stem Cell Res Ther. (2018) 9:1–13. doi: 10.1186/s13287-018-0872-7
88. Cencioni C, Melchionna R, Straino S, Romani M, Cappuzzello C, Annese V, et al. Ex vivo acidic preconditioning enhances bone marrow ckit+ cell therapeutic potential via increased CXCR4 expression. Eur Heart J. (2013) 34:2007–16. doi: 10.1093/eurheartj/ehr219
89. Flacke J-P, Kumar S, Kostin S, Reusch HP, Ladilov Y. Acidic preconditioning protects endothelial cells against apoptosis through p38-and Akt-dependent Bcl-xL overexpression. Apoptosis. (2009) 14:90–6. doi: 10.1007/s10495-008-0287-5
90. Wahid F, Shehzad A, Khan T, Kim YY. MicroRNAs: synthesis, mechanism, function, and recent clinical trials. Biochim Biophys Acta (BBA)-Molecular Cell Res. (2010) 1803:1231–43. doi: 10.1016/j.bbamcr.2010.06.013
91. Lyttle BD, Vaughn AE, Bardill JR, Apte A, Gallagher LT, Zgheib C, et al. Effects of microRNAs on angiogenesis in diabetic wounds. Front Med. (2023) 10:1140979. doi: 10.3389/fmed.2023.1140979
92. Luo Y-F, Wan X-X, Zhao L-L, Guo Z, Shen R-T, Zeng P-Y, et al. MicroRNA-139–5p upregulation is associated with diabetic endothelial cell dysfunction by targeting c-jun. Aging (Albany NY). (2021) 13:1186. doi: 10.18632/aging.v13i1
93. Wang H-W, Su S-H, Wang Y-L, Chang S-T, Liao K-H, Lo H-H, et al. MicroRNA-134 contributes to glucose-induced endothelial cell dysfunction and this effect can be reversed by far-infrared irradiation. PloS One. (2016) 11:e0147067. doi: 10.1371/journal.pone.0147067
94. Gohn CR, Blue EK, Sheehan BM, Varberg KM, Haneline LS. Mesenchyme homeobox 2 enhances migration of endothelial colony forming cells exposed to intrauterine diabetes mellitus. J Cell Physiol. (2017) 232:1885–92. doi: 10.1002/jcp.25734
95. Douville JM, Cheung DY, Herbert KL, Moffatt T, Wigle JT. Mechanisms of MEOX1 and MEOX2 regulation of the cyclin dependent kinase inhibitors p21CIP1/WAF1 and p16INK4a in vascular endothelial cells. PloS One. (2011) 6:e29099. doi: 10.1371/journal.pone.0029099
96. Chen Y, Rabson AB, Gorski DH. MEOX2 regulates nuclear factor-κB activity in vascular endothelial cells through interactions with p65 and IκBβ. Cardiovasc Res. (2010) 87:723–31. doi: 10.1093/cvr/cvq117
97. Tsuji-Tamura K, Morino-Koga S, Suzuki S, Ogawa M. The canonical smooth muscle cell marker TAGLN is present in endothelial cells and is involved in angiogenesis. J Cell Sci. (2021) 134:jcs254920. doi: 10.1242/jcs.254920
98. Bui L, Edwards S, Hall E, Alderfer L, Round K, Owen M, et al. Engineering bioactive nanoparticles to rejuvenate vascular progenitor cells. Commun Biol. (2022) 5:635. doi: 10.1038/s42003-022-03578-4
99. Zhen Y, Pavez M, Li X. The role of Pcdh10 in neurological disease and cancer. J Cancer Res Clin Oncol. (2023) 149:8153–64. doi: 10.1007/s00432-023-04743-w
100. Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. Int Soc Cell Ther Position Statement Cytother. (2006) 8:315–7. doi: 10.1080/14653240600855905
101. Bronckaers A, Hilkens P, Martens W, Gervois P, Ratajczak J, Struys T, et al. Mesenchymal stem/stromal cells as a pharmacological and therapeutic approach to accelerate angiogenesis. Pharmacol Ther. (2014) 143:181–96. doi: 10.1016/j.pharmthera.2014.02.013
102. Souidi N, Stolk M, Rudeck J, Strunk D, Schallmoser K, Volk H-D, et al. Stromal cells act as guardians for endothelial progenitors by reducing their immunogenicity after co-transplantation. Stem Cells. (2017) 35:1233–45. doi: 10.1002/stem.2573
103. Shafiee A, Patel J, Lee JS, Hutmacher DW, Fisk NM, Khosrotehrani K. Mesenchymal stem/stromal cells enhance engraftment, vasculogenic and pro-angiogenic activities of endothelial colony forming cells in immunocompetent hosts. Sci Rep. (2017) 7:13558. doi: 10.1038/s41598-017-13971-3
104. Lee H, Huh Y-H, Kang K-T. Mesenchymal stem cells potentiate the vasculogenic capacity of endothelial colony-forming cells under hyperglycemic conditions. Life. (2022) 12:469. doi: 10.3390/life12040469
105. Shafiee A, Patel J, Yi Wong H, Donovan P, Hutmacher DW, Fisk NM, et al. Priming of endothelial colony-forming cells in a mesenchymal niche improves engraftment and vasculogenic potential by initiating mesenchymal transition orchestrated by NOTCH signaling. FASEB J. (2017) 31:610–24. doi: 10.1096/fj.201600937
106. Rossi E, Goyard C, Cras A, Dizier B, Bacha N, Lokajczyk A, et al. Co-injection of mesenchymal stem cells with endothelial progenitor cells accelerates muscle recovery in hind limb ischemia through an endoglin-dependent mechanism. Thromb Hemostasis. (2017) 117:1908–18. doi: 10.1160/TH17-01-0007
107. Faris P, Negri S, Perna A, Rosti V, Guerra G, Moccia F. Therapeutic potential of endothelial colony-forming cells in ischemic disease: strategies to improve their regenerative efficacy. Int J Mol Sci. (2020) 21:7406. doi: 10.3390/ijms21197406
108. Walther W, Stein U. Viral vectors for gene transfer: a review of their use in the treatment of human diseases. Drugs. (2000) 60:249–71. doi: 10.2165/00003495-200060020-00002
109. Reinisch A, Hofmann NA, Obenauf AC, Kashofer K, Rohde E, Schallmoser K, et al. Humanized large-scale expanded endothelial colony–forming cells function in vitro and in vivo. Blood J Am Soc Hematol. (2009) 113:6716–25. doi: 10.1182/blood-2008-09-181362
110. Tan CM, Lewandowski AJ, Williamson W, Huckstep OJ, Yu GZ, Fischer R, et al. Proteomic signature of dysfunctional circulating endothelial colony-forming cells of young adults. J Am Heart Assoc. (2021) 10:e021119. doi: 10.1161/JAHA.121.021119
111. Ha G, Ferratge S, Naserian S, Proust R, Ponsen A-C, Arouche N, et al. Circulating endothelial progenitors in vascular repair. J Cell Immunother. (2018) 4:13–7. doi: 10.1016/j.jocit.2018.09.004
112. Smadja DM, Melero-Martin JM, Eikenboom J, Bowman M, Sabatier F, Randi AM. Standardization of methods to quantify and culture endothelial colony-forming cells derived from peripheral blood. J Thromb Hemostasis. (2019) 17:1190–4. doi: 10.1111/jth.14462
113. Simoncini S, Toupance S, Labat C, Gautier S, Dumoulin C, Arnaud L, et al. Functional impairment of endothelial colony forming cells (ECFC) in patients with severe atherosclerotic cardiovascular disease (ASCVD). Int J Mol Sci. (2022) 23:8969. doi: 10.3390/ijms23168969
114. Paschalaki KE, Starke RD, Hu Y, Mercado N, Margariti A, Gorgoulis VG, et al. Dysfunction of endothelial progenitor cells from smokers and chronic obstructive pulmonary disease patients due to increased DNA damage and senescence. Stem Cells. (2013) 31:2813–26. doi: 10.1002/stem.1488
115. Vassallo PF, Simoncini S, Ligi I, Chateau A-L, Bachelier R, Robert S, et al. Accelerated senescence of cord blood endothelial progenitor cells in premature neonates is driven by SIRT1 decreased expression. Blood J Am Soc Hematol. (2014) 123:2116–26. doi: 10.1182/blood-2013-02-484956
116. Coldwell KE, Lee SJ, Kean J, Khoo CP, Tsaknakis G, Smythe J, et al. Effects of obstetric factors and storage temperatures on the yield of endothelial colony forming cells from umbilical cord blood. Angiogenesis. (2011) 14:381–92. doi: 10.1007/s10456-011-9222-4
117. Leeper NJ, Hunter AL, Cooke JP. Stem cell therapy for vascular regeneration: adult, embryonic, and induced pluripotent stem cells. Circulatio. (2010) 122:517–26. doi: 10.1161/CIRCULATIONAHA.109.881441
118. Huang NF, Niiyama H, Peter C, De A, Natkunam Y, Fleissner F, et al. Embryonic stem cell–derived endothelial cells engraft into the ischemic hindlimb and restore perfusion. Arteriosclerosis Thrombosis Vasc Biol. (2010) 30:984–91. doi: 10.1161/ATVBAHA.110.202796
119. Cho S-W, Moon S-H, Lee S-H, Kang S-W, Kim J, Lim JM, et al. Improvement of postnatal neovascularization by human embryonic stem cell–derived endothelial-like cell transplantation in a mouse model of hindlimb ischemia. Circulation. (2007) 116:2409–19. doi: 10.1161/CIRCULATIONAHA.106.687038
120. Aghazadeh Y, Khan ST, Nkennor B, Nunes SS. Cell-based therapies for vascular regeneration: past, present and future. Pharmacol Ther. (2022) 231:107976. doi: 10.1016/j.pharmthera.2021.107976
121. Fong CY, Gauthaman K, Bongso A. Teratomas from pluripotent stem cells: a clinical hurdle. J Cell Biochem. (2010) 111:769–81. doi: 10.1002/jcb.22775
122. Mikłosz A, Chabowski A. Efficacy of adipose-derived mesenchymal stem cell therapy in the treatment of chronic micro-and macrovascular complications of diabetes. Diabetes Obes Metab. (2024) 26:793–808. doi: 10.1111/dom.15375
123. Gomes A, Coelho P, Soares R, Costa R. Human umbilical cord mesenchymal stem cells in type 2 diabetes mellitus: the emerging therapeutic approach. Cell Tissue Res. (2021) 385:497–518. doi: 10.1007/s00441-021-03461-4
124. Zhu L, Wang S, Qu J, Hui Z, Kan C, Hou N, et al. The therapeutic potential of mesenchymal stem cells in the treatment of diabetes mellitus. Cell Reprogramming. (2022) 24:329–42. doi: 10.1089/cell.2022.0039
125. Zhang H, Gu Y, Zhang K, Tu Y, Ouyang C. Roles and mechanisms of umbilical cord mesenchymal stem cells in the treatment of diabetic foot: a review of preclinical and clinical studies. J Diabetes Complications. (2023) 38:108671. doi: 10.1016/j.jdiacomp.2023.108671
126. Melief SM, Zwaginga JJ, Fibbe WE, Roelofs H. Adipose tissue-derived multipotent stromal cells have a higher immunomodulatory capacity than their bone marrow-derived counterparts. Stem Cells Trans Med. (2013) 2:455–63. doi: 10.5966/sctm.2012-0184
127. Mattar P, Bieback K. Comparing the immunomodulatory properties of bone marrow, adipose tissue, and birth-associated tissue mesenchymal stromal cells. Front Immunol. (2015) 6:164869. doi: 10.3389/fimmu.2015.00560
128. Pankajakshan D, Agrawal DK. Mesenchymal stem cell paracrine factors in vascular repair and regeneration. J Biomed Technol Res. (2014) 1. doi: 10.19104/jbtr.2014.107
129. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. cell. (2006) 126:663–76. doi: 10.1016/j.cell.2006.07.024
130. Prasain N, Lee MR, Vemula S, Meador JL, Yoshimoto M, Ferkowicz MJ, et al. Differentiation of human pluripotent stem cells to cells similar to cord-blood endothelial colony–forming cells. Nat Biotechnol. (2014) 32:1151–7. doi: 10.1038/nbt.3048
131. Thakur A, Parra DC, Motallebnejad P, Brocchi M, Chen HJ. Exosomes: Small vesicles with big roles in cancer, vaccine development, and therapeutics. Bioactive Mater. (2022) 10:281–94. doi: 10.1016/j.bioactmat.2021.08.029
132. Elsharkasy OM, Nordin JZ, Hagey DW, de Jong OG, Schiffelers RM, Andaloussi SE, et al. Extracellular vesicles as drug delivery systems: Why and how? Adv Drug Deliv Rev. (2020) 159:332–43. doi: 10.1016/j.addr.2020.04.004
133. Li F-X-Z, Lin X, Xu F, Shan S-K, Guo B, Lei L-M, et al. The role of mesenchymal stromal cells-derived small extracellular vesicles in diabetes and its chronic complications. Front Endocrinol. (2021) 12:780974. doi: 10.3389/fendo.2021.780974
134. Born LJ, Chang KH, Shoureshi P, Lay F, Bengali S, Hsu ATW, et al. HOTAIR-loaded mesenchymal stem/stromal cell extracellular vesicles enhance angiogenesis and wound healing. Advanced Healthcare Mater. (2022) 11:2002070. doi: 10.1002/adhm.202002070
135. Hu W, Song X, Yu H, Sun J, Zhao Y. Therapeutic potentials of extracellular vesicles for the treatment of diabetes and diabetic complications. Int J Mol Sci. (2020) 21:5163. doi: 10.3390/ijms21145163
136. Dellett M, Brown ED, Guduric-Fuchs J, O'Connor A, Stitt AW, Medina RJ, et al. MicroRNA-containing extracellular vesicles released from endothelial colony-forming cells modulate angiogenesis during ischemic retinopathy. J Cell Mol Med. (2017) 21:3405–19. doi: 10.1111/jcmm.13251
Keywords: endothelial colony forming cells, diabetes mellitus, pharmacological conditioning, genetic modification, disease-related cellular dysfunction, clinical translation
Citation: Liu Y, Lyons CJ, Ayu C and O’Brien T (2024) Enhancing endothelial colony-forming cells for treating diabetic vascular complications: challenges and clinical prospects. Front. Endocrinol. 15:1396794. doi: 10.3389/fendo.2024.1396794
Received: 06 March 2024; Accepted: 14 June 2024;
Published: 15 July 2024.
Edited by:
Guifang Zhao, Mayo Clinic Florida, United StatesReviewed by:
Rongwei Lei, University of Houston, United StatesXiaodong Zou, The Chinese University of Hong Kong, China
Copyright © 2024 Liu, Lyons, Ayu and O’Brien. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Timothy O’Brien, dGltb3RoeS5vYnJpZW5AdW5pdmVyc2l0eW9mZ2Fsd2F5Lmll