
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 27 September 2024
Sec. Clinical Diabetes
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1394102
This article is part of the Research Topic Improving Diagnosis and Management of Genetic Lipodystrophy View all 6 articles
 Reivla Marques Vasconcelos Soares1
Reivla Marques Vasconcelos Soares1 Monique Alvares da Silva2Julliane Tamara Araújo de Melo Campos2,3
Monique Alvares da Silva2Julliane Tamara Araújo de Melo Campos2,3 Josivan Gomes Lima1*
Josivan Gomes Lima1*The PPARG gene encodes a member of a nuclear receptor superfamily known as peroxisome proliferator-activated gamma (PPARγ). PPARγ plays an essential role in adipogenesis, stimulating the differentiation of preadipocytes into adipocytes. Loss-of-function pathogenic variants in PPARG reduce the activity of the PPARγ receptor and can lead to severe metabolic consequences associated with familial partial lipodystrophy type 3 (FPLD3). This review focuses on recent scientific data related to FPLD3, including the role of PPARγ in adipose tissue metabolism and the phenotypic and clinical consequences of loss-of-function variants in the PPARG gene. The clinical features of 41 PPARG pathogenic variants associated with FPLD3 patients were reviewed, highlighting the genetic and clinical heterogeneity observed among 91 patients. Most of them were female, and the average age at the onset and diagnosis of lipoatrophy was 21 years and 33 years, respectively. Considering the metabolic profile, hypertriglyceridemia (91.9% of cases), diabetes (77%), hypertension (59.5%), polycystic ovary syndrome (58.2% of women), and metabolic-dysfunction-associated fatty liver disease (87,5%). We also discuss the current treatment for FPLD3. This review provides new data concerning the genetic and clinical heterogeneity in FPLD3 and highlights the importance of further understanding the genetics of this rare disease.
Lipodystrophies are rare conditions resulting from disturbances in adipogenesis or lipid storage, culminating in a loss of adipose tissue without nutritional restriction or catabolic state. The estimated prevalence of these syndromes ranges from 1.3-4.7 cases per million (1). It can be classified based on etiology (congenital or acquired) or the extent of lipoatrophy (partial or generalized). Based on these classifications, there are four groups of lipodystrophies: Congenital Generalized Lipodystrophy (CGL), Familial Partial Lipodystrophy (FPLD), Acquired Generalized Lipodystrophy (AGL), and Acquired Partial Lipodystrophy (APL) (2). Acquired lipodystrophies are generally associated with HIV infection and its treatment or with autoimmune diseases (3).
Among the genetic lipodystrophies, CGL has autosomal recessive inheritance, characterized by an almost complete absence of subcutaneous white adipose tissue (sWAT). Patients usually have less than 6% total body fat (4). It is considered an ultra-rare disease with a prevalence of 0.96/million, with around 500 cases described in the literature (5). In some countries, such as Brazil, the CGL prevalence is exceptionally high (32.3/million inhabitants) (6). The scarcity of adipose tissue in these patients leads to serious metabolic consequences such as severe hypertriglyceridemia (HTG), diabetes, liver cirrhosis due to steatosis, and a higher predisposition to infections. These complications reduce life expectancy by 35 years for the affected patients (7).
FPLD was initially described in 1970 and is the most common form of genetic lipodystrophy, with an estimated prevalence of 1.7-2.8 cases/million (8). This disorder results from a selective loss of adipose tissue, usually affecting the buttocks and lower limbs, with fat accumulation in other regions, such as the abdomen and neck. The relative deficiency of adipose tissue and its lipid storage capacity impairment causes metabolic consequences such as HTG, insulin resistance (IR), diabetes, non-alcoholic hepatic steatosis, hypertension, and atherosclerosis (8, 9).
FPLD has considerable genetic and phenotypic variability and can be classified into different types based on specific DNA changes. FPLD type 1 - or Köbberling syndrome - is characterized by a loss of adipose tissue concentrated in the lower limbs, but no specific genes involved have been described. FPLD type 2 – or Dunnigan’s syndrome – results from mutations in the LMNA gene, responsible for encoding laminas A and C, whose mutation causes cell damage and premature apoptosis of adipocytes. Type 3 FPLD has been described as associated with mutations in the PPARG gene, which is involved in adipogenesis. FPLD2 and FPLD3 account for almost 50% of partial lipodystrophy cases (10). FPLD2 has been reported in over 500 patients, while FPLD3 has been documented in around 20 affected families (8). Type 4 FPLD occurs due to heterozygous pathogenic variants in the PLIN1 gene, responsible for encoding the lipid droplet-associated perilipin-1 protein (9). FPLD type 5 results from a homozygous variant in the CIDEC gene, involving the formation of a unique lipid droplet in white adipose cells (9). Initiated in adulthood, FPLD type 6 results from variants of LIPE, which encodes hormone-sensitive lipase (HSL). The HSL hydrolyzes adipocyte triglycerides, providing free fatty acids (FFA) and glycerol. Other types of FPLD are caused by pathogenic variants in MFN2 and AKT2 genes (9).
Autosomal dominant variants cause PPARG loss-of-function and consequent reduction in its receptor activity, leading to a severe metabolic phenotype characteristic of FPLD3 (9). These data reinforce the role of PPARγ as a fundamental regulator in adipose tissue metabolism.
This review highlights the most recent scientific evidence on FPLD3, including the role of PPARγ in adipose tissue metabolism and the phenotypic consequences of loss-of-function variants in the PPARG gene, emphasizing the genetic and clinical heterogeneity observed among FPLD3 patients.
The PPARG (peroxisome proliferator-activated receptor) gene located on the short arm of chromosome 3 (3p25.2) encodes a member of a superfamily of nuclear receptors called peroxisome proliferator-activated receptors that comprise three isoforms (PPARα, PPARγ, and PPARδ) with distinct tissue distribution and physiological roles (1). The PPARγ functions as a ligand-activated transcription factor, and it is expressed in white and brown adipose tissues (WAT and BAT, respectively), the large intestine, and the spleen. Nevertheless, its expression is higher in adipose tissue, which plays a central role in adipocyte differentiation and function (11). The focus on PPARγ as a master in adipose tissue metabolism began in the 1990s after the discovery of thiazolidinediones and their power to induce adipocyte differentiation and improve insulin sensitivity (12).
PPARG gene has sixteen PPARG splicing variants in humans, according to the RefSeq database from the NCBI (National Center for Biotechnology Information). They are produced by the differential combination of alternative promoters. The three main isoforms of PPARγ in humans are PPARγ1, PPARγ2, and PPARγ3 (13). The PPARγ1 (NM_001354666; NP_001341595.2) contains 475 amino acids and is expressed at low levels in multiple tissues, such as adipose tissue, skeletal muscle, macrophages, and intestinal epithelium (colon). The PPARγ2 (NM015869.5; NP_056953.2) contains 505 amino acids and is predominantly expressed in WAT, BAT, and liver (13, 14). PPARγ3 (NM_001330615.4; NP_001317544.2) is expressed in macrophages, adipose tissue, and large intestine epithelium and has 278 amino acids (15). Figures 1A, B highlight the PPARG1, PPARG2, and PPARG3 transcripts and PPARγ1, 2, and 3 isoform domains, respectively.
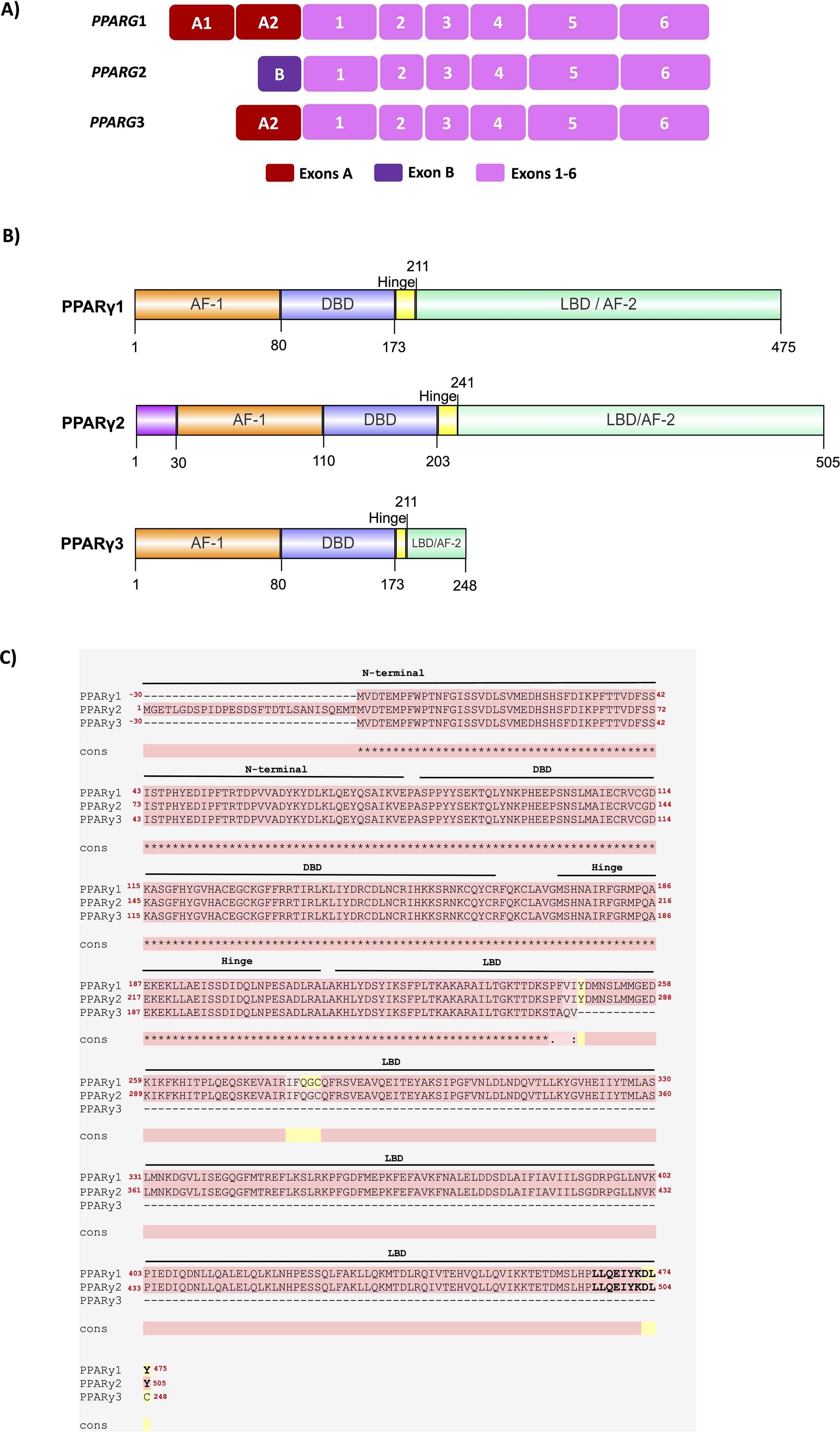
Figure 1. Schematic representation of the main human PPARG transcripts and its PPARγ protein isoforms. (A) The structure of the PPARG1, 2, and 3 transcripts is highlighted, showing exons 1 to 6, common to all PPARγ transcripts, while PPARG2 has the additional exon B and encodes the canonical and dominant PPARγ2 isoform. PPARG1 also presents exons A1 and A2, while PPARG3 also presents the A2 exon. (B) PPARγ1 has 475 amino acids and is expressed at low levels in adipose tissue, skeletal muscle, macrophages, and epithelium from the colon. PPARγ2 presents 30 additional amino acids (magenta) and is mainly found in WAT, BAT, and the liver. PPARγ3 has 248 aa and has higher expression levels in macrophages, adipose tissue, and large intestine epithelium. The main PPARγ isoforms 1, 2, and 3 are composed of 4 functional domains: N-terminus domain AF-1 (orange), DNA Binding Domain – DBD (blue), Hinge (yellow), and Ligand Binding Domain – LBD (green) in the C-terminus. The AF-1 domain and the Hinge region are poorly conserved, while the DBD, LBD, and AF-2 domains are highly conserved. The image was made using IBS 2.0 software. (C) Protein sequence alignment of the PPARγ1, 2, and 3 isoforms. PPARγ isoform sequences were aligned via T-Coffee. Pink represents identical alignments; yellow corresponds to similar alignments; and green regions show different alignments. ∗ corresponds to an equal match. Cons: consensus sequence. The PPARγ sequences used were: PPARγ1 (NM_001354666; NP_001341595.2), PPARγ2 (NM_015869.5; NP_056953.2), and PPARγ3 (NM_001330615.4; NP_001317544.2).
Both PPARγ1 and PPARγ2 isoforms have the intrinsic capacity to promote adipogenesis. However, recent studies show that PPARγ2 has a more decisive action on adipogenesis due to sensitivity to ligands and greater binding capacity to components of the mediator complex (16–18).
The PPARγ proteins have a molecular structure similar to that of other nuclear receptors and contain four domains: 1) N-terminus contains the Activation Function-1 (AF-1; also known as ligand-independent transactivation domain 1); 2) DNA binding domain (DBD); 3) HINGE, and 4) Ligand binding domain (LBD) located in the portion C-terminus. The AF-1 domain regulates ligand-independent transcriptional PPARγ activity, while the HINGE domain is involved in interactions with coactivators and corepressors. The DBD and LBD domains are the most essential and highly conserved among species. The DBD has the role of binding PPARγ to the promoter region of their target genes. The LBD domain is involved with ligand binding, the transactivation of many genes, and the transcriptional co-regulator interactions. The LBD is responsible for dimerization with the retinoid X receptor (RXR) and overlays the more powerful Activation Function-2 (AF-2) domain, which can be altered by the ligand binding (17, 19). AF-2 is the major transcriptional activation domain. It is essential for dimerization and regulates the ligand-dependent PPARγ transcriptional activity (1, 20–22). Figures 1B highlights the domains in PPARγ1, 2, and 3 isoforms. To better characterize the amino acid differences among the three main PPARγ isoforms, we performed an alignment to compare their protein sequences and domains, as shown in Figure 1C. These analyses were performed according to Alvares et al. (23). PPARγ2 has 30 additional amino acids in the N-terminus. Knockdown of Pparg in 3T3-L1 preadipocytes and Pparg null MEFs revealed that the PPARγ2 has a more potent role in adipocyte differentiation than the PPARγ1 isoform (17), indicating a crucial role of the longest PPARγ protein in the adipogenesis. However, how the N-terminus of PPARγ acts to promote adipogenesis remains an open question.
Despite being poorly conserved among species, the N-terminus portion showed an essential regulatory function in the action of PPARγ. The amino acids of the N-terminus domain have transcriptional activity when linked to a heterologous DNA-binding domain. Contradictorily, when this N-terminus region of PPARγ is deleted, this factor has greater transcriptional activity and more significant adipogenic action. This finding suggested that this N-terminus could also have some inhibitory function in the context of the holoreceptor, and a large part of this inhibitory action was linked to the phosphorylation of PPARγ by members of the MAP kinase family (20, 24). Furthermore, it was observed that the N-terminal domain influences the response to ligand binding of the LBD. Substitution of serine 112 by an aspartate residue inhibits ligand binding to the receptor (25).
To exert its biological action, PPARγ binds to members of the RXR family as an obligatory heterodimer at specific DNA binding sites, termed PPAR response elements (PPREs). The crystalline structure of the PPARγ-RXR heterodimer bounds to DNA in the presence of ligand results in a conformational change in the LBD and favors interaction with coactivator peptides (such as steroid receptor coactivators (SRCs), histone acetyltransferases (HATs), CBP and P300) and the Mediator complex, promoting transcription of PPARγ target genes, resulting in their physiological effects on adipogenesis and adipose tissue metabolism (12, 26).
The PPARγ participates in adipogenesis during the differentiation of preadipocytes into adipocytes, playing a central role in this process (20). The evidence that PPARγ is the master regulator of adipogenesis is well established. In vitro and in vivo studies show a lack of matured adipocytes without PPARγ (27).
During the adipocyte differentiation process, PPARγ participates in a transcriptional cascade. Its activation promotes the induction of a variety of differentiation-dependent target genes, which play an essential role in the uptake and storage of triglycerides in the adipocyte (18, 28, 29).
After ligand activation, PPARγ induces many target genes involved in lipogenesis and adipogenesis and activates the expression of C/EBPα. This transcription factor can bind directly to the CEBP site in the PPARγ promoter, creating a stable, self-reinforcing regulatory loop (30, 31). After activation, PPARγ stimulates regulatory regions of a large number of genes that have essential roles in lipogenesis and insulin sensitivity, including FABP4, PCK2, LPL, ADIPOQ, PLIN1, and SLC2A4 (which encode aP2, PEPCK, lipoprotein lipase, adiponectin, perilipin and Glut4 proteins, respectively), promoting the maturation of the adipocyte, which begins to capture and store lipids (Figure 2) (26, 28).
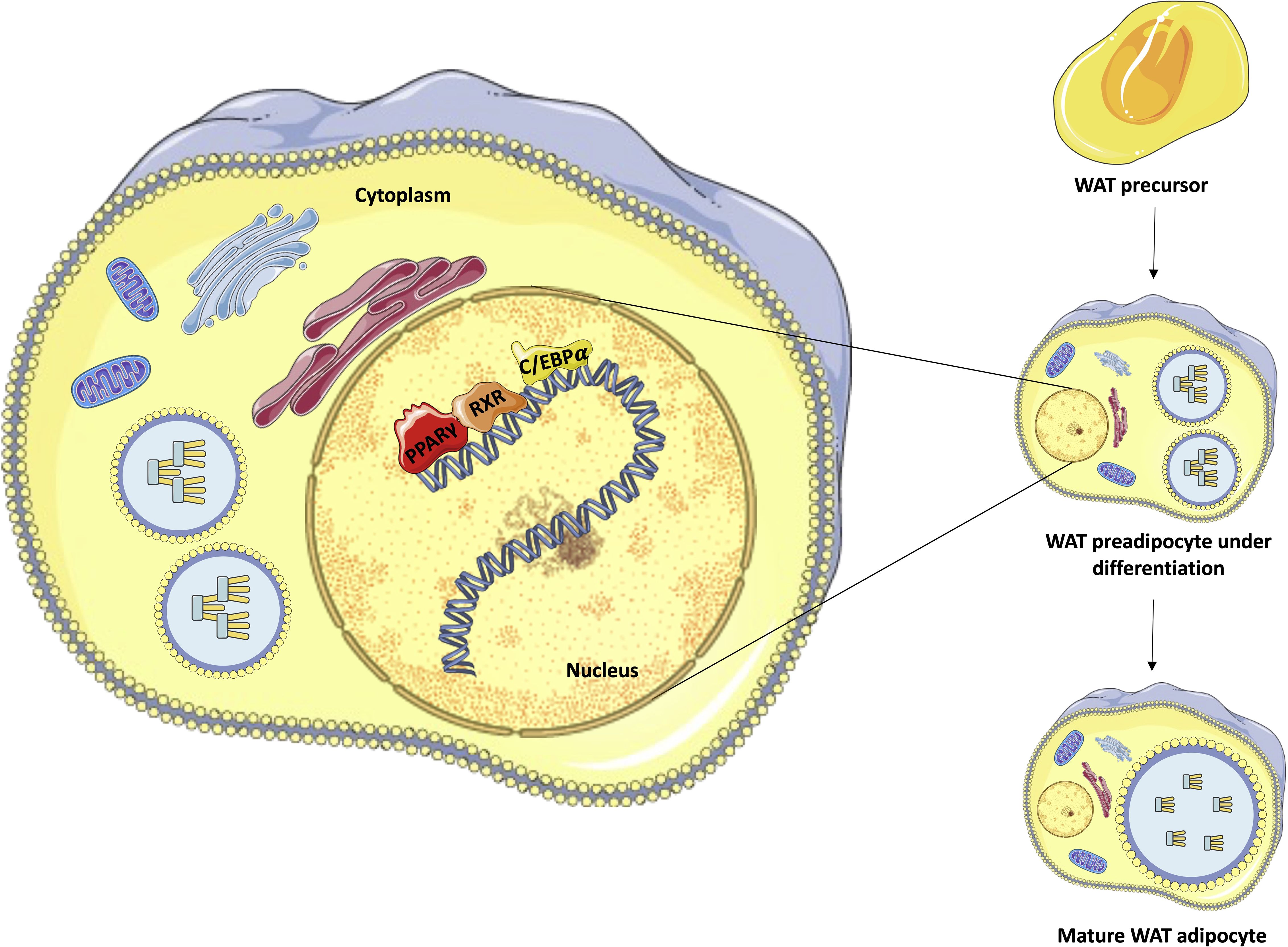
Figure 2. Representation of adipocyte differentiation control performed by PPARγ, C/EBP, and RXR. The figure emphasizes the process of differentiation of white adipocytes from pre-adipocytes, highlighting the transcriptional roles of PPARγ (red), C/EBP (orange), and RXR (yellow) in the nucleus of preadipocytes under differentiation to complete maturation. Own authorship using resources from SMART – Servier Medical Art.
Likewise, the PPARγ is essential for the development and function of BAT. PPARγ ligands induce terminal differentiation of the brown preadipocyte HIB-1B cell line and stimulate the expression of UCP-1, a mitochondrial proton transporter that confers thermogenic properties to BAT (32).
With the discovery of thiazolidinediones (TZDs) and their hypoglycemic action by improving IR, several studies were dedicated to understanding the role of PPARγ in insulin sensitivity. Activation of PPARγ by endogenous or synthetic ligands results in systemic insulin sensitization through complex mechanisms involving multiple organs. In adipose tissue, activated PPARγ promotes pre-adipocyte differentiation into insulin-sensitive adipocytes. This activation of PPARγ does not increase the size of adipocytes (hypertrophy); instead, it leads to the formation of smaller and more insulin-sensitive adipocytes, possibly due to de novo differentiation (12, 31). This process increases the capacity of WAT to store fatty acids (FA), reducing the ectopic concentration of FFA, whose accumulation leads to harmful effects on insulin action (33, 34). An additional but also important mechanism that favors insulin sensitivity is the functional improvement of adipose tissue after activation of PPARγ, which starts to produce more adiponectin, directly linked to insulin sensitivity (20). The adiponectin actions are already well established, improving muscle glucose uptake and reducing hepatic glucose production (Figure 3) (35). PPARγ activation by ligands in adipocytes is also associated with decreased levels of adipokines related to the IR onset, including tumor necrosis factor-alpha (TNFα) and resistin (36).
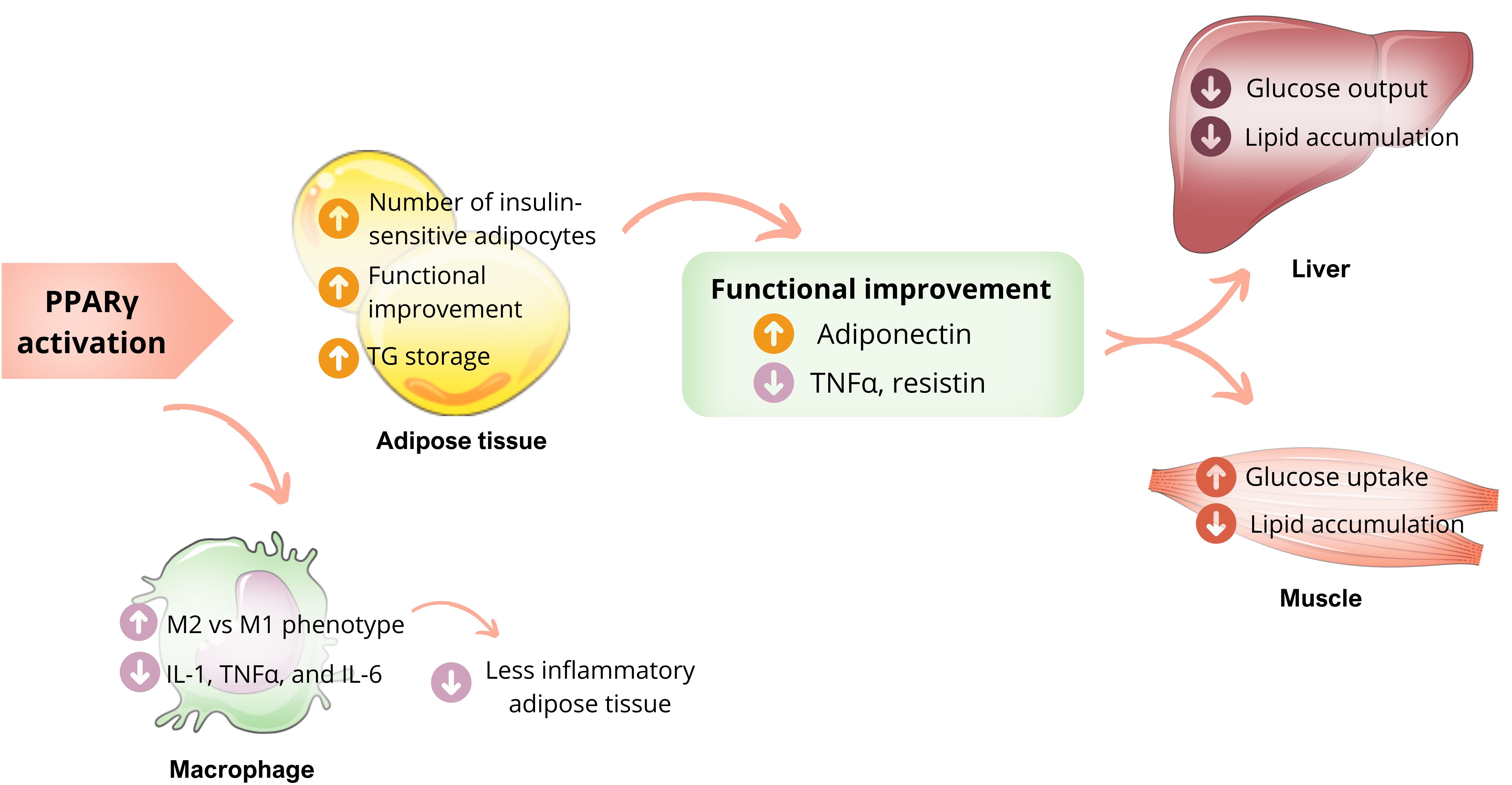
Figure 3. PPARγ actions and insulin sensibility. Activation of PPARγ in adipose tissue promotes the differentiation of pre-adipocytes into insulin-sensitive adipocytes, favoring the uptake of more lipids and influencing the production of adipokines, resulting in higher levels of adiponectin and reduced levels of TNF-α. These mechanisms benefit glucose metabolism, including less hepatic glucose production and more skeletal muscle glucose uptake, improving insulin sensibility. PPARγ activation also stimulates the transformation of macrophages into less inflammatory cells, thereby reducing macrophage infiltration into adipose tissue. Own authorship using resources from SMART – Servier Medical Art.
PPARγ plays a crucial role in promoting the alternative activation of macrophages, which results in less inflammatory adipose tissue (12). The relationship between macrophages and IR is already known. When macrophages infiltrate adipose tissue, they produce inflammatory cytokines such as IL-1, TNFα, and IL-6, which act on the insulin receptor. This causes the exchange of the phosphorylation residue to tyrosine by serine, resulting in less activation of the receptor and consequent IR. However, macrophages can also be activated alternatively (M2), producing arginase I (argl) and IL-10. These cytokines have less inflammatory power and less impact on the insulin receptor. PPARγ, especially the PPARγ3 isoform, plays a vital role in stimulating this alternative activation (M2), which configures the anti-inflammatory functions of this nuclear receptor (Figure 3) (15, 37).
Several pathogenic variants in the PPARG gene have been identified in the human population. Approximately 0.2% of the population presents missense variants of PPARG, but only 20% of these variants show functional impairments and are associated with metabolic commitments (38). The most common variant in the human PPARG gene is an alanine to proline substitution at position 12 in the PPARγ2 isoform (Pro12Ala) that has a variable physiological effect related to a decreased risk of type 2 diabetes mellitus (DM) (39).
In the general population, 1 in every 500 people carries a PPARG missense variant. However, only a small portion of them experience metabolic consequences that do not necessarily lead to FPLD3. After evaluating the FPLD3 clinical and phenotypic findings (Table 1), heterogeneity is observed among different PPARG pathogenic variants or members of the same family with similar pathogenic variants. This shows that the activity and expression of this gene are influenced by gene-gene and gene-environment interactions that determine the clinical consequences of genetic variants with significant genetic and phenotypic heterogeneity (38). Studies on mice indicate that a reduction in PPARγ activity up to 50% is still sufficient to maintain normal body composition. However, when PPARγ activity was reduced to 25%, it caused IR, decreased total body and fat mass, and dyslipidemia (66).
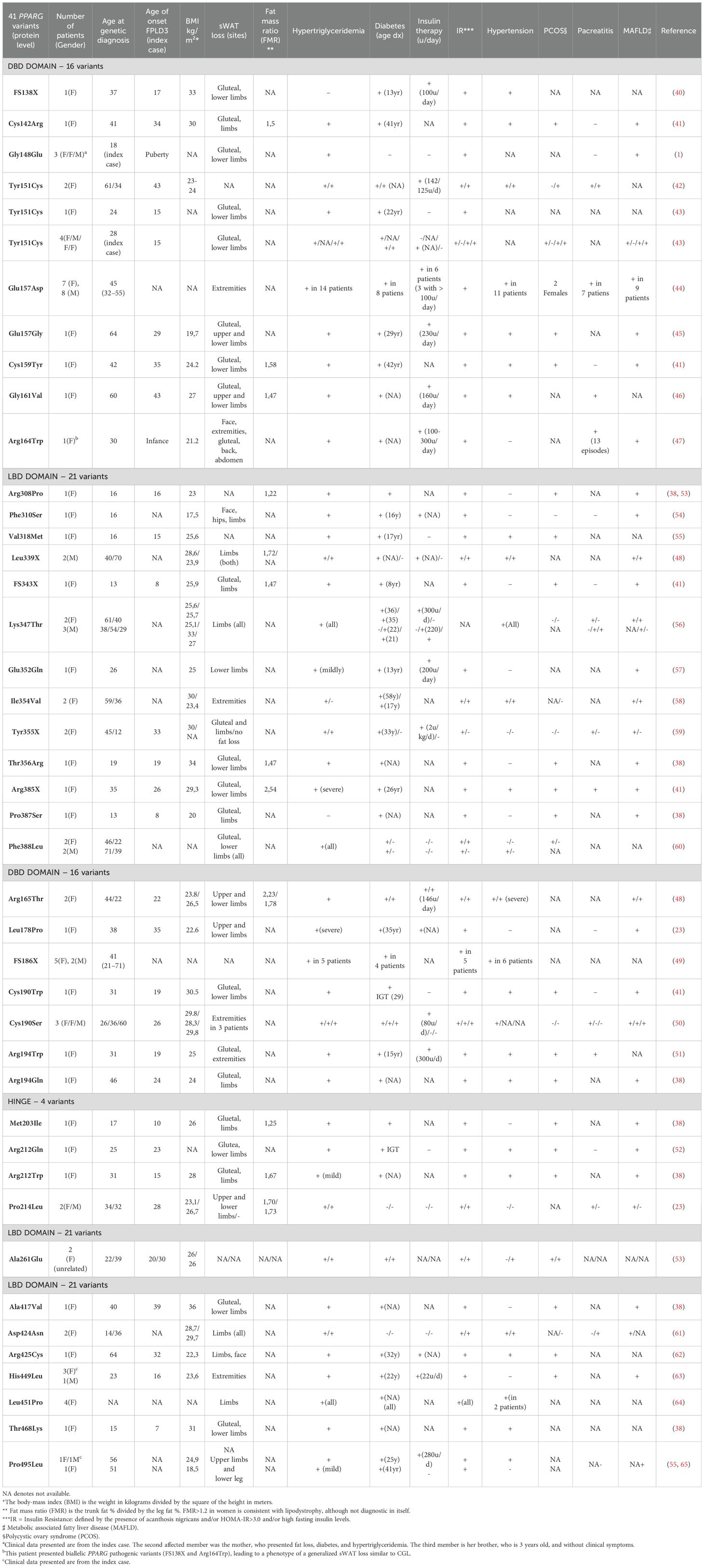
Table 1. Phenotype and clinical characteristics of the most frequent PPARG variants related to FPLD3.
Genetic lipodystrophies due to loss-of-function PPARG variants occur at a low frequency estimated at 1:100,000 individuals (67). These variants reduce the PPAR receptor activity and can lead to severe metabolic consequences associated with FPLD3. This disease has an autosomal dominant inheritance (all patients are heterozygous). It includes mainly amino acid substitutions (mainly in the DBD and LBD) and nonsense and frameshift mutations resulting in PPARγ inactivity (12).
As far as we know (based on the literature review carried out in PubMed, ClinVar, and The Human Gene Mutation Database), 44 PPARG pathogenic variants related to FPLD3 have been described until July 2024. These variants impair PPARγ transcriptional activity in several ways: some of the DBD variants show extreme dominant negative activity, suppressing the transcriptional activity of PPARγ; others cause an apparent impairment of transcriptional activity but do not show any dominant negative activity against the wild-type receptor. Likewise, variants affecting the LBD present dominant negative activity, even with little or no DNA binding activity (41). The clinical/phenotypic features of loss-of-function variants in the PPARG gene and FPLD3 will be discussed below.
FPLD3 certainly impacts the quality and expectations of individuals with this condition. Unfortunately, the exact natural history of these syndromes is not well documented, making their diagnosis difficult and creating an obstacle in developing specific therapies for this disease (43).
Patients generally present a loss of adipose tissue in the hips and lower limbs, severe IR, diabetes, hyperglyceridemia, hypertension, hepatic steatosis, and, in women, polycystic ovary syndrome (PCOS) with symptoms of hyperandrogenism were found. Classically, FPLD3 is characterized by a milder loss of adipose tissue and a more severe metabolic condition when compared to FPLD2 (Dunnigan disease) (58). One possible explanation for this paradoxical finding is that patients harboring PPARG pathogenic variants may have fewer small, insulin-sensitive adipocytes, with the preservation of large adipocytes. This could explain why individuals with FPLD3 experience less severe loss of fat tissue (lipoatrophy) despite having higher insulin resistance than those with FPLD2 (68).
We have reviewed the clinical features presented by the index case and the affected family members in 41 out of 44 PPARG variants associated with FPLD3. The summarized data is available in Table 2, while detailed information about each specific variant is provided in Table 1. The other three pathogenic variants (Pro387Ser, Lys395Arg, and Gln438Pro) were initially described by SekizKardes et al. However, clinical data on affected patients are not available (69). Since the majority of PPARG variants reviewed here were previously described without considering the Human Genome Variation Society (HGVS) recommendations, we classified all reviewed PPARG variants according to HGVS (70), and their pathogenicity classification was made according to the American College of Medical Genetics and Genomics (ACMG) (71). We also used the MutationTaster and Mutalyzer tools to confirm the HGVS nomenclature (72, 73). These data were inserted in Table 3. This analysis was challenging since most manuscripts reviewed here did not include detailed information concerning the variants at the coding DNA and protein levels. Therefore, to classify all PPARG variants according to ACMG guidelines, we first used the MutationTaster tool to obtain the correct PPARG variant position at genomic DNA, cDNA, and protein levels for missense, nonsense, and the frameshift variants FS138X and FS343X. Then, the Mutalyzer tool was used to confirm the correct HGVS nomenclature. For the FS186X variant, the correct PPARG variant nomenclature was obtained using only the Mutalyzer tool since this frameshift variant could not be analyzed by the MutationTaster tool. We used the GRCh37 genome reference for all Mutalyzer tool analyses. For manuscripts that informed only the variant nomenclature at the protein level, the transcript and protein sequences were obtained from the National Center of Biotechnology Information (NCBI). For all analyses, we used the PPARG transcript sequence that encodes the biggest PPARγ isoform 2 (NC_000003.11; NM_015869.5; NP_056953.2). Then, after obtaining all appropriate nomenclatures, in silico predictive algorithms were applied, as recommended by ACMG guidelines. Here, we evaluated the pathogenicity of all PPARG missense and nonsense variants reviewed using CADD (Combined Annotation Dependent Depletion) v.1.7 (74) and REVEL (rare exome variant ensemble learner) tools (75). After these steps, we classified all PPARG variants reviewed according to ACMG guidelines (Table 3). More details were inserted in the footnote of Table 3.
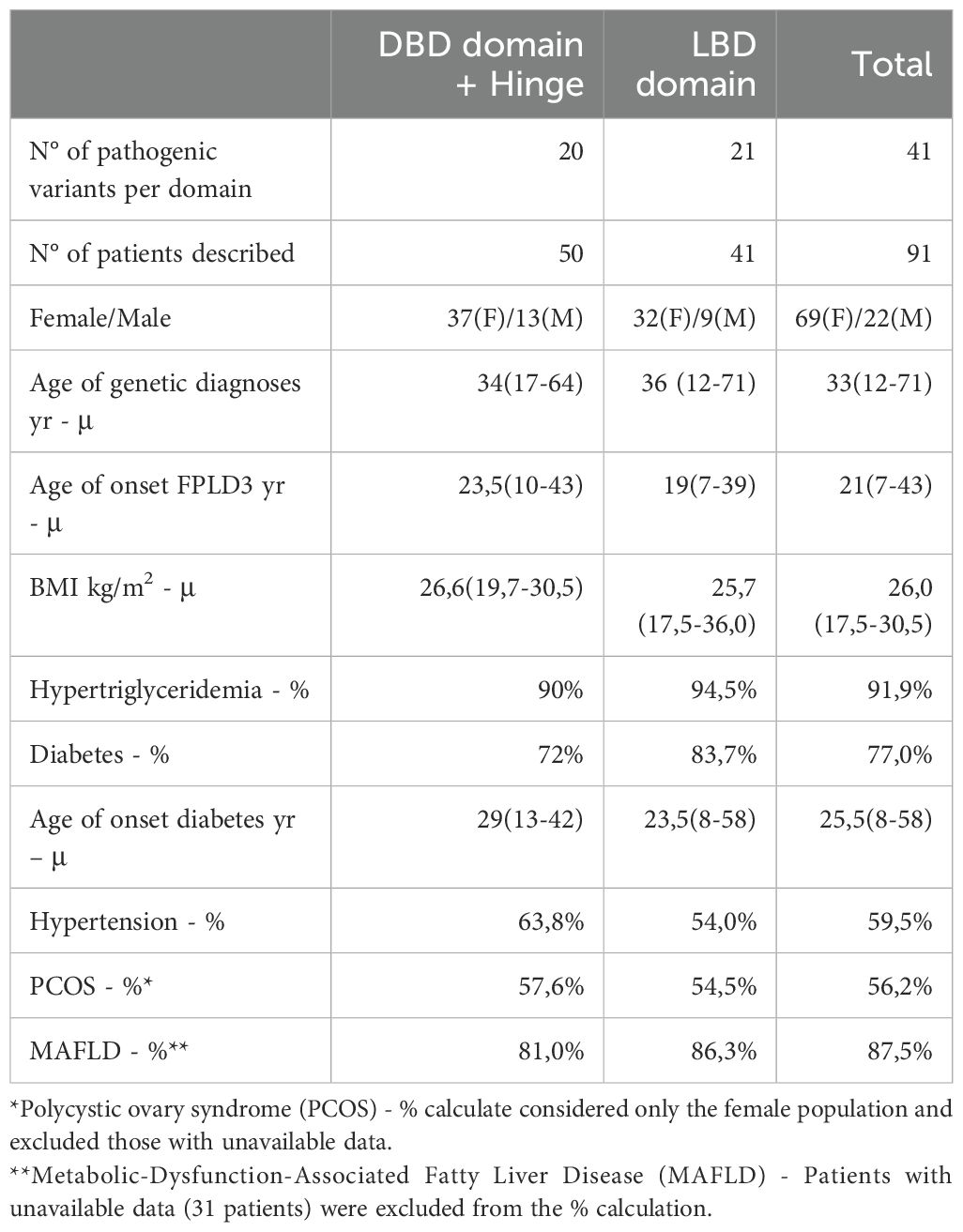
Table 2. Overview of FPLD3 clinical characteristics related to 41 PPARG pathogenic variants.
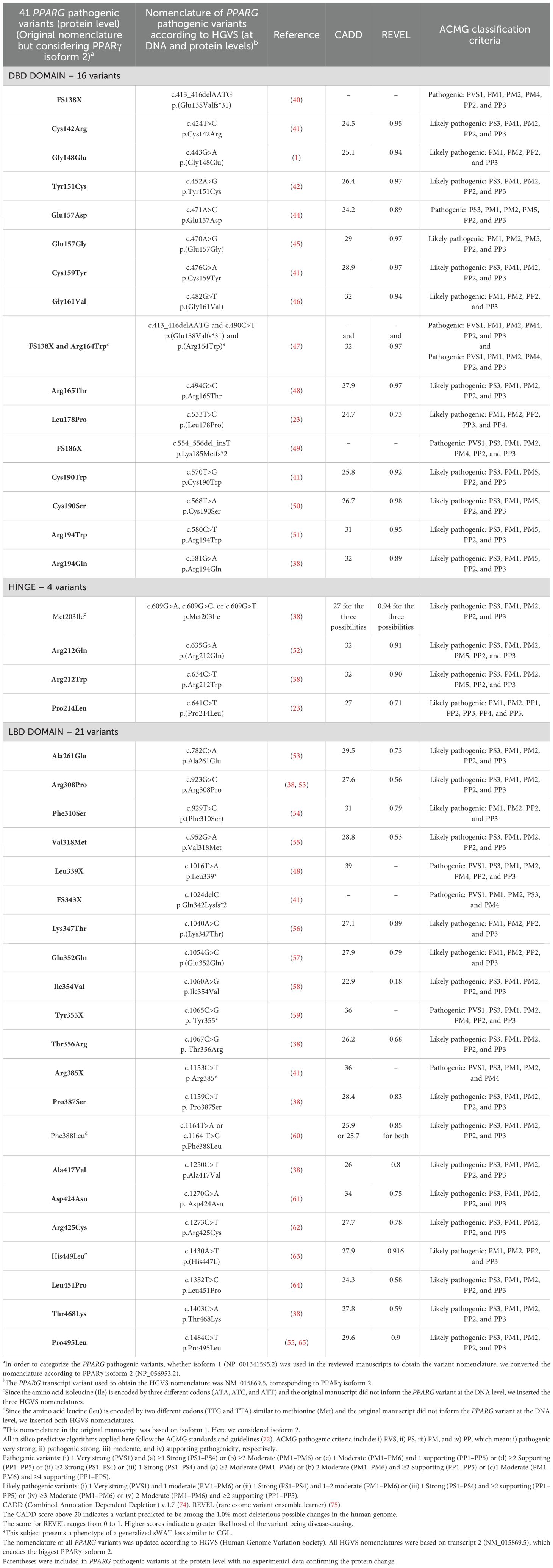
Table 3. Deleteriousness predictions of all 41 previously published PPARG variants according to the American College of Medical Genetics and Genomics (ACMG).
Cases of FPLD3 usually begin to show clinical manifestations at puberty or early adulthood, and females are most affected (1). Of the 91 cases described, 69 (75.8%) were women. The patients started experiencing symptoms around age 21 (Table 2). The average age of onset of FPLD3 symptoms is around 20 years, similar to other FPLD, but there is a significant variability in onset among the patients listed, ranging from 7 to 43 years. One limitation in analyzing these data is that 43 out of the 91 patients listed did not have information about the onset of symptoms or perception of lipoatrophy in the limbs. When analyzing data from patients with FPLD 2, it is observed that the onset of symptoms occurs during puberty in women and later in men, with few cases presenting in childhood or senescence (76).
The loss of WAT tends to be variable and mainly affects the lower limbs and gluteal, but it can also extend to other areas, such as the upper limbs and face. They also show a specific WAT accumulation in some areas, which is variable in these patients. Some sWAT accumulate fat in the face, trunk, back, and abdomen, while others do not (45, 54, 62). Some FPLD3 patients may exhibit a more subtle loss of sWAT, while others may not experience any reduction at all (59).
Biallelic PPARG variants (compound heterozygous) were related to a congenital generalized lipodystrophy phenotype, emphasizing the genetic heterogeneity of congenital lipodystrophies. This is evident when observing a 37-year-old woman carrying the FS138X pathogenic variant who presented a loss of adipose tissue in the gluteal region and lower limbs but accumulation in the back, periscapular region, abdomen, and visceral fat (40). On the other hand, a 30-year-old woman who carried biallelic pathogenic variants (Arg164Trp and FS138X) in heterozygosity showed a loss of WAT on the face, lower limbs, and buttocks since childhood. It progressed to the upper limbs, back, and abdomen in adulthood, resembling the phenotypic pattern of generalized congenital lipodystrophies (CGL) (47). This patient’s case was the first one correlating distinct heterozygous PPARG variants with the CGL phenotype, associated with a pathogenic variant that had not yet been reported in the literature (Arg164Trp).
Objective assessment of fat distribution can be done using Computed Tomography (CT), Magnetic Resonance Imaging (MRI), and Dual-Energy X-ray Absorptiometry (DXA). DXA is more clinically applicable among these due to its better accessibility and lower cost. The Fat Mass Ratio (FMR - the ratio between truncal and lower limb fat) can be evaluated by DXA and has been suggested as a tool for identifying partial lipodystrophy. An FMR value greater than 1.2 may indicate a lipoatrophic pattern suggestive of partial lipodystrophy, although it is not diagnostic (77). In our analysis, 12 FPLD3 patients had FMR values available, all of which were greater than 1.2 (ranging from 1.22 to 2.54), underlining the usefulness of this parameter as an additional measure for diagnosing FPLD3 (Table 1).
The development of metabolic disorders associated with lipodystrophies is linked to the inability to maintain adequate fat storage in sWAT and impaired postprandial lipid buffering (1). When the capacity for sWAT expansion is impaired, fat is relocated in non-adipose organs, such as the liver, skeletal muscle, and pancreas (78). The dysfunctional adipocytes develop lipotoxicity, macrophage infiltration, mitochondrial dysfunction, and oxidative stress (8). The lipotoxicity alters insulin receptor signaling pathways, causing severe IR and abnormal metabolism of lipids and glucose (79). Further, excessive levels of inflammatory adipokines and cytokines secreted by visceral AT induce the accumulation of TG and FFA in ectopic sites (78, 80).
FPLD3 subjects present a more severe IR, and the extent of the change in adipose tissue is greater. Insulin resistance can be identified through clinical signs such as acanthosis nigricans and acrochordons. These conditions occur due to high insulin levels in keratinocytes and fibroblasts from the skin, leading to hyperkeratosis and skin hyperpigmentation, especially in skin fold areas. It’s important to note that while acanthosis nigricans is common in patients with FPLD3, acrochordons are not typically described in this group. In contrast, this condition has been observed in other lipodystrophies, such as CGL, but only in a minority of cases (4). Fasting blood glucose and insulin levels can be considered to assess IR biochemically, along with calculating the Homeostasis Model Assessment-Insulin Resistance (HOMA-IR) index (1). In our review of 91 patients, 84 showed clinical or biochemical signs of insulin resistance, indicating its significant role in developing metabolic complications of FPLD3.
As a result of this process, patients tend to develop DM in early adulthood. These patients typically present with difficult-to-control hyperglycemia and require high-dose insulin therapy due to the severity of IR. Many patients suffer from microvascular and macrovascular complications due to poor glycemic control (40, 42, 45). Previous studies have shown that FPLD3 patients are more likely to develop DM than those with FPLD2, with 72% of the patients experiencing it, while the prevalence of diabetes in Dunnigan disease ranged from 28% to 51% (68, 76). Our analysis confirms this, as 77% of the patients we reviewed had DM. The median age at which this condition appeared was 25.5 years. A wide variation in the onset of diabetes was observed in this analysis (ranging from 8 to 58 years). In many cases, this data was unavailable (42 cases out of 67 patients with DM). Among the available data, 24 patients were using insulin therapy, the majority with doses greater than 100 U/day or 2U/kg/day, and some requiring up to 5U/kg/day.
HTG is a metabolic change that occurs early on and is considered a lipid indicator of ongoing lipodystrophy. When WAT deposits are reduced due to lipoatrophy, TG present in circulating lipoproteins, chylomicrons, and VLDL can only be partially stored in these deposits. Another contributing factor is increased VLDL synthesis due to hepatic steatosis, which is also found in these patients. These mechanisms likely lead to an increase in circulating TG levels (81).
HTG is generally severe and can be accompanied by eruptive xanthomas and lead to complications such as acute pancreatitis. Triglyceride levels can be two to three times higher in women with FPLD compared to men (1). Patients with FPLD3 typically have more severe HTG and a higher risk of acute pancreatitis than those with FPLD2 (1). Among patients with FPLD3, 91.9% had HTG, with only three (Arg212Trp, Glu352Gln, Pro496Leu) having slightly increased TG levels (Table 2). Although most patients with FPLD3 present with moderate or severe HTG, these two cases with mild HTG highlight the phenotypic heterogeneity observed in lipodystrophies. When comparing the pathogenic variants Arg212Trp and Arg212Gln (located in the same position of the gene), a phenotypic difference is observed concerning the intensity of HTG and the age at which fatty tissue loss begins (earlier in the Arg212Trp variant), which highlights the possibility of gene-gene interactions and gene-environment. Acute pancreatitis was observed in 16 patients, and one of them had 13 episodes of pancreatitis. Seven distinct variants presented a phenotype that included eruptive xanthomas (41, 42, 47, 52, 59).
Due to insulin resistance and HTG, patients often experience metabolic-associated fatty liver disease (MAFLD), which can cause hepatomegaly and liver cirrhosis (58, 72). In this review, 87,5% of the patients for whom data was available developed this condition. Thirty-one patients did not provide any information about MAFLD.
PPARγ regulates vascular tone and blood pressure and is expressed in many vascular system components (endothelial and smooth muscle cells). Loss-of-function variants are associated with hypertension, which is generally severe and has an early onset (82). Initially, two cases of distinct mutations in the PPARG gene (Val318Met and Pro495Leu) were described with severe and difficult-to-control arterial hypertension, which appeared around 30 years of age (55). Patients with different variants (Arg165Thr and Leu339X) had severe hypertension with no systemic renin-angiotensin system (RAS) alterations. However, components of the cellular RAS were markedly overexpressed and activated in fibroblasts and peripheral blood mononuclear cells (PBMCs) issued from 4 patients with this variant (48). Patients’ cells exhibited increased levels of angiotensin II receptor 1 (AT1R), renin, and angiotensinogen (AGT), with overactivation of angiotensin II signaling and oxidative stress and inflammation. These findings suggest that severe hypertension, which is a peculiar feature of patients with FPLD3, might be linked to tissue RAS overactivation resulting from PPARγ dysfunction (48).
It is important to note that hypertension, which is one of the typical clinical symptoms of FPLD3 that we have studied, can manifest differently among patients. In total, 59,5% of patients experience hypertension, with a higher percentage of patients affected among those with a PPARG variant in the DBD (63,8%) compared to those with a variant in the LDB (54,0%) (Table 2).
Women with FPLD3 often experience changes in their reproductive system, such as menstrual irregularity, hirsutism, infertility, and polycystic ovary syndrome (PCOS) (8). These changes are related to IR, which can cause high levels of circulating insulin that affect the ovaries. This can lead to overproduction of androgens by theca cells and interfere with follicular stimulation and ovulation (38, 43, 53). After analyzing 39 mutations, researchers found that PCOS was present in 56.2% of women, usually accompanied by irregular menstrual cycles and hirsutism.
Different PPARG pathogenic variants can result in similar physical and metabolic characteristics mentioned above, but this is not always true. Even though two patients have similar or distinct pathogenic variants in the PPARG gene, they can exhibit different physical characteristics and clinical symptoms. This is because gene-gene and gene-environment interactions can contribute to phenotypic and clinical heterogeneity. The specific clinical findings related to each PPARG pathogenic variant are detailed in Table 1. Therefore, being aware of the critical clinical presentations associated with FPLD can help to enhance understanding of the FPLD disease and prevent misdiagnosis (3).
Some patients with PPARG pathogenic variants have exhibited clinical findings that are not typically associated with FPLD3. For instance, a patient with a variant in the DBD domain (FS138X) had bilateral cataracts and bilateral hearing deficits (40). Additionally, two patients with different variants (Gly148Glu and Phe310Ser) reported hypothyroidism (1, 54). Furthermore, two men with the Leu339X variant experienced psoriasis (48). After reviewing the literature, it was observed that there is an association of PPAR with thyroid carcinoma but not with hypothyroidism (83).
To date, no association between FPLD3 and cataracts has been observed in the literature. There is only one reported case of congenital, autosomal dominant, partial lipodystrophy. It was associated with congenital cataracts and spinal cord and cerebellar dysfunction. However, the specific pathogenic variant associated with this phenotype was not described (variants in LMNA or BSCL2 genes were ruled out) (84).
Although many studies have evaluated the functional properties of PPARs in the eye and discovered fundamental PPAR mechanisms in the retina and cornea, PPARγ has not yet been associated with changes in the iris. PPARγ and PPARα are well established in their functions in ocular homeostasis regarding neuroprotection, neovascularization, and inflammation (85).
The literature contains no data supporting the connection between FPLD3 and psoriasis. However, some studies suggest the involvement of PPARγ in developing this skin condition, highlighting its direct effects on keratinocytes and immune cells. In psoriasis, the activation of PPARγ regulates the inflammatory response by reducing the expression and suppressing the genes of adhesion molecules. Additionally, the activation of PPARγ hinders the differentiation of Th CD4+ cells into Th17 cells. Some small-scale studies have shown improved skin symptoms after using pioglitazone in a limited number of patients (86).
Until now, there is no cure for FPLD3. Therapeutic approaches should be directed towards the associated comorbidities. This treatment is challenging and requires the combination of several strategies, such as lifestyle modifications and intensive treatment for DM and dyslipidemia (8). Lifestyle changes include physical exercise and a balanced diet containing approximately 50–60% of carbohydrates, 20–30% fat, and 20% protein (87). Due to the syndrome’s rarity, the evidence supporting pharmacological treatment is primarily based on expert opinion, case reports, or case series (88).
The initial management of hyperglycemia can benefit from insulin sensitizers like metformin and thiazolidinediones (TZDs), which are oral hypoglycemic options. TZDs medications can help manage partial lipodystrophies by stimulating the action of PPARγ to form sWAT and improve insulin sensitivity. There are reports of isolated cases using thiazolidinediones (TZD) in FPLD3 but with variable results. Pioglitazone showed favorable and sustained results in improving glycemic control and dyslipidemia in women carrying Tyr355X, His449Leu, Arg308Leu, and Phe310Ser pathogenic variants. These women initially had mild metabolic changes, except for the last two, who had more severe metabolic issues (53, 54, 59, 63). However, a different response was observed using rosiglitazone in patients with severe metabolic profiles who carried the PPARG pathogenic variants Pro495Leu, Val318Met, and Ala261Glu. In these cases, there was a slight and non-sustained improvement in blood glucose levels and serious adverse effects of the therapy. One possible explanation for this difference in response is the effectiveness of the medication. Pioglitazone is more effective than rosiglitazone in reducing the metabolic profile and improving fat distribution in animal models and clinical trials with DM2 patients (89–92). Another hypothesis suggests that the site of the PPARG pathogenic variant may interfere with its responsiveness to endogenous or synthetic ligands. In a study with structural modeling of PPARG pathogenic variants (Arg308Leu and Ala261Glu), it was found that the site of these variants interferes with the response to the endogenous ligand while maintaining a full transcriptional response to synthetic ligands, such as pioglitazone (Arg308Leu) and rosiglitazone (Ala261Glu). In the latter case, there was a metabolic improvement in clinical analysis, but serious adverse effects to the rosiglitazone were observed, resulting in treatment interruption (53, 93).
The sodium-glucose cotransporter 2 (SGLT2) inhibitors are essential for treating diabetes in lipodystrophic patients due to their insulin-independent effect and notable cardio-renal benefits. Bansal et al. analyzed the efficacy and safety of SGLT2 inhibitors in a cohort of 12 patients with partial lipodystrophy (4 with variants in LMNA, 1 in PPARG, 1 in PCYT1A, and 6 with unknown mutations). They found a significant reduction in HbA1c and blood pressure in patients using these medications. The most reported adverse effects included fungal infections, urinary infections, and limb pain. Serious adverse effects with diabetic ketoacidosis occurred in only one patient who was not compliant with insulin therapy (94).
Glucagon-like peptide 1 receptor agonists (GLP-1RA) have shown positive results in treating patients with FPLD. In a recent retrospective analysis, 13 patients with FPLD type 1 and 1 patient with FPLD type 2 were treated with GLP-1RA, and the metabolic effects of this treatment were observed by comparing the results before and six months after starting this drug. The treatment with GLP-1RA significantly reduced weight, BMI, HbA1c, and fasting glucose levels in FPLD patients. Additionally, triglyceride levels decreased from 334 ± 170 mg/dL before GLP-1RA treatment to 256 ± 82 mg/dL after six months (95).
Due to the severity of diabetes, insulin therapy is typically required for treating patients with FPLD. These patients often need high insulin doses, and in this situation, U500 insulins may be beneficial (8).
Metreleptin is a recombinant human leptin analog used as a specific therapy to manage human lipodystrophies. A study was conducted on seven patients with FPLD3 (Arg425Cys, Arg194Gln, Pro495Leu, Pro387Ser, Lys395Arg, and Gln438Pro pathogenic variants) who were treated with metreleptin for 13 months. The study showed that metreleptin improved glycemic control, as evidenced by decreased glycated hemoglobin and fasting glucose. The reduction in triglycerides in patients with PPARG pathogenic variants depended on the initial triglyceride value: four patients with baseline serum triglyceride levels >500 mg/dL were classified as metreleptin responders, whereas only one of three patients with baseline triglyceride levels <500 mg/dL was a responder (69).
Patients with PPARG loss-of-function variants display clinical symptoms that reflect the impact of this protein on the development and functioning of adipose tissue. PPARy is a significant regulator of adipogenesis and insulin response. The several pathogenic variants found in the PPARG gene leading to a classic pattern of FPLD3 show heterogeneity at the allelic level. Patients with the same pathogenic variant present some distinct clinical characteristics, suggesting heterogeneity at a clinical and phenotypic level. These interactions arise from gene-gene and gene-environment interactions. Understanding these characteristics will help in diagnosing this rare but underdiagnosed disease and can lead to more precise therapeutic interventions for patients.
RS: Conceptualization, Data curation, Writing – original draft. MdS: Writing – review & editing. JC: Supervision, Validation, Writing – review & editing. JL: Supervision, Validation, Writing – review & editing.
The authors declare that financial support was received for manuscript processing charges and the open access fee from Chiesi Global Rare Diseases (GRD). Chiesi GRD was not involved in the design, data collection, interpretation or decision to publish this manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Rutkowska L, Salachna D, Lewandowski K, Lewiński A, Gach A. Familial partial lipodystrophy—Literature review and report of a novel variant in PPARG expanding the spectrum of disease-causing alterations in FPLD3. Diagnostics. (2022) 12. doi: 10.3390/diagnostics12051122
2. Brown RJ, Araujo-Vilar D, Cheung T, Dunger D, Garg A, Jack M, et al. The diagnosis and management of lipodystrophy syndromes: A multi-society practice guideline. J Clin Endocrinol Metab. (2016) 101:4500–11. doi: 10.1210/jc.2016-2466
3. Broekema MF, Savage DB, Monajemi H, Kalkhoven E. Gene-gene and gene-environment interactions in lipodystrophy: Lessons learned from natural PPARγ mutants. Biochim Biophys Acta Mol Cell Biol Lipids. (2019) 1864:715–32. doi: 10.1016/J.BBALIP.2019.02.002
4. Lima JG, Nobrega LHC, De Lima NN, Do Nascimento Santos MG, Baracho MFP, Jeronimo SMB. Clinical and laboratory data of a large series of patients with congenital generalized lipodystrophy. Diabetol Metab Syndr. (2016) 8. doi: 10.1186/S13098-016-0140-X
5. Chiquette E, Oral EA, Garg A, Araújo-Vilar D, Dhankhar P. Estimating the prevalence of generalized and partial lipodystrophy: findings and challenges. Diabetes Metab Syndr Obes. (2017) 10:375–83. doi: 10.2147/DMSO.S130810
6. De Azevedo Medeiros LB, Cândido Dantas VK, Craveiro Sarmento AS, Agnez-Lima LF, Meireles AL, Xavier Nobre TT, et al. High prevalence of Berardinelli-Seip Congenital Lipodystrophy in Rio Grande do Norte State, Northeast Brazil. Diabetol Metab Syndr. (2017) 9. doi: 10.1186/S13098-017-0280-7
7. Lima JG, Nobrega LHC, Lima NN, dos Santos MCF, Silva PHD, Maria de Fatima PB, et al. Causes of death in patients with Berardinelli-Seip congenital generalized lipodystrophy. PloS One. (2018) 13:e0199052. doi: 10.1371/JOURNAL.PONE.0199052
8. Bagias C, Xiarchou A, Bargiota A, Tigas S. Familial partial lipodystrophy (FPLD): Recent insights. Diabetes Metab Syndrome Obes. (2020) 13:1531–44. doi: 10.2147/DMSO.S206053
9. Fernández-Pombo A, Sánchez-Iglesias S, Cobelo-Gómez S, Hermida-Ameijeiras Á, Araújo-Vilar D. Familial partial lipodystrophy syndromes. Presse Medicale. (2021) 50. doi: 10.1016/j.lpm.2021.104071
10. Jéru I, Vatier C, Araujo-Vilar D, Vigouroux C, Lascols O. Clinical Utility Gene Card for: Familial partial lipodystrophy. Eur J Hum Genet. (2016) 25. doi: 10.1038/ejhg.2016.102
11. Janani C, Ranjitha Kumari BD. PPAR gamma gene - A review. Diabetes Metab Syndrome: Clin Res Rev. (2015) 9:46–50. doi: 10.1016/j.dsx.2014.09.015
12. Astapova O, Leff T. PPARγ mutations, lipodystrophy and diabetes. Horm Mol Biol Clin Investig. (2014) 20:63–70. doi: 10.1515/hmbci-2014-0033
13. Fajas L, Auboeuf D, Raspé E, Schoonjans K, Lefebvre A-M, Saladin R, et al. The organization, promoter analysis, and expression of the human PPARg gene* (1997). Available online at: http://www.jbc.org (Accessed January 22, 2024).
14. Escher P, Braissant O, Basu-Modak S, Michalik L, Wahli W, Desvergne B. Rat PPARs: quantitative analysis in adult rat tissues and regulation in fasting and refeeding. Endocrinology. (2001) 142:4195–202. doi: 10.1210/ENDO.142.10.8458
15. Fajas L, Fruchart JC, Auwerx J. PPARgamma3 mRNA: a distinct PPARgamma mRNA subtype transcribed from an independent promoter. FEBS Lett. (1998) 438:55–60. doi: 10.1016/S0014-5793(98)01273-3
16. Ren D, Collingwood TN, Rebar EJ, Wolffe AP, Camp HS. PPARgamma knockdown by engineered transcription factors: exogenous PPARgamma2 but not PPARgamma1 reactivates adipogenesis. Genes Dev. (2002) 16:27–32. doi: 10.1101/GAD.953802
17. Aprile M, Cataldi S, Ambrosio MR, D’Esposito V, Lim K, Dietrich A, et al. PPARγΔ5, a naturally occurring dominant-negative splice isoform, impairs PPARγ Function and adipocyte differentiation. Cell Rep. (2018) 25:1577–1592.e6. doi: 10.1016/j.celrep.2018.10.035
18. Sarjeant K, Stephens JM. Adipogenesis. Cold Spring Harb Perspect Biol. (2012) 4. doi: 10.1101/cshperspect.a008417
19. Hernandez-Quiles M, Broekema MF, Kalkhoven E. PPARgamma in metabolism, immunity, and cancer: unified and diverse mechanisms of action. Front Endocrinol (Lausanne). (2021) 12:624112. doi: 10.3389/FENDO.2021.624112
20. Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. (2008) 77:289–312. doi: 10.1146/ANNUREV.BIOCHEM.77.061307.091829
21. Jeninga EH, Gurnell M, Kalkhoven E. Functional implications of genetic variation in human PPARgamma. Trends Endocrinol Metab. (2009) 20:380–7. doi: 10.1016/J.TEM.2009.04.005
22. Kroker AJ, Bruning JB. Review of the structural and dynamic mechanisms of PPARγ Partial agonism. PPAR Res. (2015) 2015. doi: 10.1155/2015/816856
23. da Silva MA, Soares RMV, de Oliveira Filho AF, Campos LRS, de Lima JG, de Melo Campos JTA. Case report: two novel PPARG pathogenic variants associated with type 3 familial partial lipodystrophy in Brazil. Diabetol Metab Syndrome. (2024) 16:1–13. doi: 10.1186/S13098-024-01387-9
24. Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. (1994) 79:1147–56. doi: 10.1016/0092-8674(94)90006-X
25. Shao D, Rangwala SM, Bailey ST, Krakow SL, Reginato MJ, Lazar MA. Interdomain communication regulating ligand binding by PPAR-gamma. Nature. (1998) 396:377–80. doi: 10.1038/24634
26. Lefterova MI, Haakonsson AK, Lazar MA, Mandrup S. PPARγ and the global map of adipogenesis and beyond. Trends Endocrinol Metab. (2014) 25:293–302. doi: 10.1016/j.tem.2014.04.001
27. Berger J, Patel HV, Woods J, Hayes NS, Parent SA, Clemas J, et al. A PPARgamma mutant serves as a dominant negative inhibitor of PPAR signaling and is localized in the nucleus. Mol Cell Endocrinol. (2000) 162:57–67. doi: 10.1016/S0303-7207(00)00211-2
28. Tavares V, Hirata MH, Crespo RD. Receptor Ativado por Proliferadores de Peroxissoma Gama (PPARγ): Estudo Molecular na Homeostase da Glicose, Metabolismo de Lipídeos e Abordagem Terapêutica. Arq Bras Endocrinol Metab. (2007). 51/4.
29. Kawai M. Adipose tissue and bone: Role of PPARγ in adipogenesis and osteogenesis. Horm Mol Biol Clin Investig. (2013) 15:105–13. doi: 10.1515/hmbci-2013-0036
30. Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. (2006) 444:847–53. doi: 10.1038/NATURE05483
31. Rosen ED, MacDougald OA. Adipocyte differentiation from the inside out. Nat Rev Mol Cell Biol. (2006) 7:885–96. doi: 10.1038/NRM2066
32. Sears IB, MacGinnitie MA, Kovacs LG, Graves RA. Differentiation-dependent expression of the brown adipocyte uncoupling protein gene: regulation by peroxisome proliferator-activated receptor gamma. Mol Cell Biol. (1996) 16:3410. doi: 10.1128/MCB.16.7.3410
33. Martin G, Schoonjans K, Lefebvre AM, Staels B, Auwerx J. Coordinate regulation of the expression of the fatty acid transport protein and acyl-CoA synthetase genes by PPARalpha and PPARgamma activators. J Biol Chem. (1997) 272:28210–7. doi: 10.1074/JBC.272.45.28210
34. Kishida K, Shimomura I, Nishizawa H, Maeda N, Kuriyama H, Kondo H, et al. Enhancement of the aquaporin adipose gene expression by a peroxisome proliferator-activated receptor gamma. J Biol Chem. (2001) 276:48572–9. doi: 10.1074/JBC.M108213200
35. Berg AH, Combs TP, Scherer PE. ACRP30/adiponectin: An adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metab. (2002) 13:84–9. doi: 10.1016/S1043-2760(01)00524-0
36. Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, et al. The hormone resistin links obesity to diabetes. Nature. (2001) 409:307–12. doi: 10.1038/35053000
37. Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, et al. Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature. (2007) 447:1116–20. doi: 10.1038/NATURE05894
38. Majithia AR, Tsuda B, Agostini M, Gnanapradeepan K, Rice R, Peloso G, et al. Prospective functional classification of all possible missense variants in PPARG. Nat Genet. (2016) 48:1570–5. doi: 10.1038/ng.3700
39. Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, Nemesh J, et al. The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet. (2000) 26:76–80. doi: 10.1038/79216
40. Hegele RA, Ur C, Ransom TP, Cao H. A frameshift mutation in peroxisome-proliferator-activated receptor-γ in familial partial lipodystrophy subtype 3 (FPLD3; MIM 604367). Clin Genet. (2006) 70:360–2. doi: 10.1111/j.1399-0004.2006.00674.x
41. Agostini M, Schoenmakers E, Mitchell C, Szatmari I, Savage D, Smith A, et al. Non-DNA binding, dominant-negative, human PPARgamma mutations cause lipodystrophic insulin resistance. Cell Metab. (2006) 4:303–11. doi: 10.1016/J.CMET.2006.09.003
42. Visser ME, Kropman E, Kranendonk ME, Koppen A, Hamers N, Stroes ES, et al. Characterisation of non-obese diabetic patients with marked insulin resistance identifies a novel familial partial lipodystrophy-associated PPARγ mutation (Y151C). Diabetologia. (2011) 54:1639–44. doi: 10.1007/s00125-011-2142-4
43. Akinci B, Onay H, Demir T, Savas-Erdeve Ş, Gen R, Simsir IY, et al. Clinical presentations, metabolic abnormalities and end-organ complications in patients with familial partial lipodystrophy. Metabolism. (2017) 72:109–19. doi: 10.1016/j.metabol.2017.04.010
44. Campeau PM, Astapova O, Martins R, Bergeron J, Couture P, Hegele RA, et al. Clinical and molecular characterization of a severe form of partial lipodystrophy expanding the phenotype of PPARγ deficiency. J Lipid Res. (2012) 53:1968–78. doi: 10.1194/jlr.P025437
45. Lambadiari V, Kountouri A, Maratou E, Liatis S, Dimitriadis GD, Karpe F. Case report: metreleptin treatment in a patient with a novel mutation for familial partial lipodystrophy type 3, presenting with uncontrolled diabetes and insulin resistance. Front Endocrinol (Lausanne). (2021) 12:684182. doi: 10.3389/fendo.2021.684182
46. Lau E, Carvalho D, Oliveira J, Fernandes S, Freitas P. Familial partial lipodystrophy type 3: a new mutation on the PPARG gene. Hormones. (2015) 14:317–20. doi: 10.14310/horm.2002.1585
47. Dyment DA, Gibson WT, Huang L, Bassyouni H, Hegele RA, Innes AM. Biallelic mutations at PPARG cause a congenital, generalized lipodystrophy similar to the Berardinelli-Seip syndrome. Eur J Med Genet. (2014) 57:524–6. doi: 10.1016/j.ejmg.2014.06.006
48. Auclair M, Vigouroux C, Boccara F, Capel E, Vigeral C, Guerci B, et al. Peroxisome proliferator-activated receptor-γ mutations responsible for lipodystrophy with severe hypertension activate the cellular renin-angiotensin system. Arterioscler Thromb Vasc Biol. (2013) 33:829–38. doi: 10.1161/ATVBAHA.112.300962
49. Savage DB, Agostini M, Barroso I, Gurnell M, Luan J, Meirhaeghe A, et al. Digenic inheritance of severe insulin resistance in a human pedigree. Nat Genet. (2002) 31:379–84. doi: 10.1038/ng926
50. Lüdtke A, Buettner J, Wu W, Muchir A, Schroeter A, Zinn-Justin S, et al. Peroxisome proliferator-activated receptor-gamma C190S mutation causes partial lipodystrophy. J Clin Endocrinol Metab. (2007) 92:2248–55. doi: 10.1210/JC.2005-2624
51. Monajemi H, Zhang L, Li G, Jeninga EH, Cao H, Maas M, et al. Clinical case seminar: Familial partial lipodystrophy phenotype resulting from a single-base mutation in deoxyribonucleic acid-binding domain of peroxisome proliferator-activated receptor-γ. J Clin Endocrinol Metab. Endocrine Soc. (2007) 92:1606–12. doi: 10.1210/jc.2006-1807
52. Sorkina EL, Kalashnikova MF, Likhodey NV, Koksharova EO, Ustyuzhanin DV, Mayorov AY, et al. Development of metabolic syndrome at a young age as a manifestation of familial partial lipodystrophy type 3 (PPARG mutation): The first description of its clinical case in Russia. Diabetes Mellitus. (2015) 18:99–105. doi: 10.14341/DM2015399-105
53. Agostini M, Schoenmakers E, Beig J, Fairall L, Szatmari I, Rajanayagam O, et al. A pharmacogenetic approach to the treatment of patients with PPARG mutations. Diabetes. (2018) 67:1086–92. doi: 10.2337/DB17-1236
54. Chen X, Ma Z, Chen P, Song X, Li W, Yu X, et al. Case report: A new peroxisome proliferator-activated receptor gamma mutation causes familial partial lipodystrophy type 3 in a Chinese patient. Front Endocrinol (Lausanne). (2022) 13:830708. doi: 10.3389/fendo.2022.830708
55. Barroso I, Gurnell M F, Crowley VE, Agostini M, Schwabek JW, Soos MA, et al. Dominant negative mutations in human PPARg associated with severe insulin resistance, diabetes mellitus and hypertension(1999). Available online at: www.nature.com. (Accessed: January 23, 2024).
56. Miehle K, Porrmann J, Mitter D, Stumvoll M, Glaser C, Fasshauer M, et al. Novel peroxisome proliferator-activated receptor gamma mutation in a family with familial partial lipodystrophy type 3. Clin Endocrinol (Oxf). (2016) 84:141–8. doi: 10.1111/cen.12837
57. Castell AL, Hiéronimus S, Lascols O, Fournier T, Fénichel P. Vascular placental abnormalities and newborn death in a pregnant diabetic woman with familial partial lipodystrophy type 3: a possible role for peroxisome proliferator-activated receptor γ. Diabetes Metab. (2012) 38:367–9. doi: 10.1016/J.DIABET.2012.02.012
58. Padova G, Prudente S, Vinciguerra F, Sudano D, Baratta R, Bellacchio E, et al. The novel loss of function Ile354Val mutation in PPARG causes familial partial lipodystrophy. Acta Diabetol. (2020) 57:589–96. doi: 10.1007/s00592-019-01462-y
59. Francis GA, Li G, Casey R, Wang J, Cao H, Leff T, et al. Peroxisomal proliferator activated receptor-γ deficiency in a Canadian kindred with familial partial lipodystrophy type 3 (FPLD3). BMC Med Genet. (2006) 7. doi: 10.1186/1471-2350-7-3
60. Hegele RA, Cao H, Frankowski C, Mathews ST, Leff T. Brief genetics report PPARG F388L, a transactivation-deficient mutant, in familial partial lipodystrophy. Diabetes. (2022) 51:3586–90. doi: 10.2337/diabetes.51.12.3586
61. Lüdtke A, Buettner J, Schmidt HHJ, Worman HJ. New PPARG mutation leads to lipodystrophy and loss of protein function that is partially restored by a synthetic ligand. J Med Genet. (2007) 44. doi: 10.1136/JMG.2007.050567
62. Agarwal AK, Garg A. A novel heterozygous mutation in peroxisome proliferator-activated receptor-gamma gene in a patient with familial partial lipodystrophy. J Clin Endocrinol Metab. (2002) 87:408–8. doi: 10.1210/JCEM.87.1.8290
63. Demir T, Onay H, Savage DB, Temeloglu E, Uzum AK, Kadioglu P, et al. Familial partial lipodystrophy linked to a novel peroxisome proliferator activator receptor -γ (PPARG) mutation, H449L: a comparison of people with this mutation and those with classic codon 482 Lamin A/C (LMNA) mutations. Diabetic Med. (2016) 33:1445–50. doi: 10.1111/dme.13061
64. Broekema MF, Massink MPG, Donato C, de Ligt J, Schaarschmidt J, Borgman A, et al. Natural helix 9 mutants of PPARγ differently affect its transcriptional activity. Mol Metab. (2019) 20:115–27. doi: 10.1016/J.MOLMET.2018.12.005
65. Iizaka T, Kodama E, Mikura K, Iida T, Imai H, Hashizume M, et al. Clinical characteristics and efficacy of pioglitazone in a Japanese patient with familial partial lipodystrophy due to peroxisome proliferator-activated receptor γ gene mutation. Endocr J. (2023) 70:69–76. doi: 10.1507/ENDOCRJ.EJ22-0140
66. Tsai YS, Tsai PJ, Jiang MJ, Chou TY, Pendse A, Kim HS, et al. Decreased PPARγ Expression compromises perigonadal-specific fat deposition and insulin sensitivity. Mol Endocrinol. (2009) 23:1787–98. doi: 10.1210/ME.2009-0073
67. Garg A, Peshock RM, Fleckenstein JL. Adipose tissue distribution pattern in patients with familial partial lipodystrophy (Dunnigan variety). J Clin Endocrinol Metab. (1999) 84:170–4. doi: 10.1210/JCEM.84.1.5383
68. Vasandani C, Li X, Sekizkardes H, Brown RJ, Garg A. Phenotypic differences among familial partial lipodystrophy due to LMNA or PPARG variants. J Endocr Soc. (2022) 6. doi: 10.1210/JENDSO/BVAC155
69. Sekizkardes H, Cochran E, Malandrino N, Garg A, Brown RJ. Efficacy of metreleptin treatment in familial partial lipodystrophy due to PPARG vs LMNA pathogenic variants. J Clin Endocrinol Metab. (2019) 104:3068–76. doi: 10.1210/JC.2018-02787
70. den Dunnen JT, Dalgleish R, Maglott DR, Hart RK, Greenblatt MS, Mcgowan-Jordan J, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. (2016) 37:564–9. doi: 10.1002/HUMU.22981
71. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/GIM.2015.30
72. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. (2014) 11:361–2. doi: 10.1038/NMETH.2890
73. Lefter M, Vis JK, Vermaat M, den Dunnen JT, Taschner PEM, Laros JFJ. Mutalyzer 2: next generation HGVS nomenclature checker. Bioinformatics. (2021) 37:2811–7. doi: 10.1093/BIOINFORMATICS/BTAB051
74. Schubach M, Maass T, Nazaretyan L, Röner S, Kircher M. CADD v1.7: using protein language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res. (2024) 52:D1143–54. doi: 10.1093/NAR/GKAD989
75. Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. (2016) 99:877–85. doi: 10.1016/J.AJHG.2016.08.016
76. Fernandez-Pombo A, Diaz-Lopez EJ, Castro AI, Sanchez-Iglesias S, Cobelo-Gomez S, Prado-Moraña T, et al. Clinical spectrum of LMNA-associated type 2 familial partial lipodystrophy: A systematic review. Cells. (2023) 12. doi: 10.3390/CELLS12050725
77. Valerio CM, Zajdenverg L, De Oliveira JEP, Mory PB, Moyses RS, Godoy-Matos AF. Body composition study by dual-energy x-ray absorptiometry in familial partial lipodystrophy: Finding new tools for an objective evaluation. Diabetol Metab Syndr. (2012) 4. doi: 10.1186/1758-5996-4-40
78. Mahyoub MA, Elhoumed M, Maqul AH, Almezgagi M, Abbas M, Jiao Y, et al. Fatty infiltration of the pancreas: a systematic concept analysis. Front Med (Lausanne). (2023) 10:1227188. doi: 10.3389/FMED.2023.1227188
79. Tan GD, Savage DB, Fielding BA, Collins J, Hodson L, Humphreys SM, et al. Fatty acid metabolism in patients with PPARγ mutations. J Clin Endocrinol Metab. (2008) 93:4462–70. doi: 10.1210/jc.2007-2356
80. Kawai T, Autieri MV, Scalia R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am J Physiol Cell Physiol. (2021) 320:C375–91. doi: 10.1152/AJPCELL.00379.2020/ASSET/IMAGES/LARGE/AJ-ACEL210014F002.JPEG
81. Capeau J, Magré J, Caron-Debarle M, Lagathu C, Antoine B, Béréziat V, et al. Human lipodystrophies: genetic and acquired diseases of adipose tissue. Endocr Dev Basel, Karger. (2010) 19:1–20. doi: 10.1159/000316893
82. Halabi CM, Beyer AM, de Lange WJ, Keen HL, Baumbach GL, Faraci FM, et al. Interference with PPARγ Function in smooth muscle causes vascular dysfunction and hypertension. Cell Metab. (2008) 7:215–26. doi: 10.1016/j.cmet.2007.12.008
83. Raman P, Koenig RJ. Pax-8-PPAR-γ fusion protein in thyroid carcinoma. Nat Rev Endocrinol. (2014) 10:616–23. doi: 10.1038/NRENDO.2014.115
84. Berger JR, Arioglu Oral E, Taylor SI. Familial lipodystrophy associated with neurodegeneration and congenital cataracts. Neurology. (2002) 58:43–7. doi: 10.1212/WNL.58.1.43
85. Escandon P, Vasini B, Whelchel AE, Nicholas SE, Matlock HG, Ma JX, et al. The role of peroxisome proliferator-activated receptors in healthy and diseased eyes. Exp Eye Res. (2021) 208:108617. doi: 10.1016/J.EXER.2021.108617
86. Sobolev VV, Tchepourina E, Korsunskaya IM, Geppe NA, Chebysheva SN, Soboleva AG, et al. The role of transcription factor PPAR-γ in the pathogenesis of psoriasis, skin cells, and immune cells. Int J Mol Sci. (2022) 23:9708. doi: 10.3390/IJMS23179708
87. Araújo-Vilar D, Santini F. Diagnosis and treatment of lipodystrophy: a step-by-step approach. J Endocrinol Invest. (2019) 42:61–73. doi: 10.1007/s40618-018-0887-z
88. Akinci B, Meral R, Oral EA. Update on therapeutic options in lipodystrophy. Curr Diabetes Rep. (2018) 18:1–12. doi: 10.1007/S11892-018-1100-7/METRICS
89. Yang KJ, Noh JR, Kim YH, Gang GT, Hwang JH, Yang SJ, et al. Differential modulatory effects of rosiglitazone and pioglitazone on white adipose tissue in db/db mice. Life Sci. (2010) 87:405–10. doi: 10.1016/J.LFS.2010.08.002
90. Beysen C, Murphy EJ, Nagaraja H, Decaris M, Riiff T, Fong A, et al. A pilot study of the effects of pioglitazone and rosiglitazone on de novo lipogenesis in type 2 diabetes. J Lipid Res. (2008) 49:2657–63. doi: 10.1194/jlr.M800165-JLR200
91. Goldberg RB, Kendall DM, Deeg MA, Buse JB, Zagar AJ, Pinaire JA, et al. A comparison of lipid and glycemic effects of pioglitazone and rosiglitazone in patients with type 2 diabetes and dyslipidemia. Diabetes Care. (2005) 28:1547–54. doi: 10.2337/DIACARE.28.7.1547
92. Olansky L, Marchetti A, Lau H. Multicenter retrospective assessment of thiazolidinedione monotherapy and combination therapy in patients with type 2 diabetes: Comparative subgroup analyses of glycemic control and blood lipid levels. Clin Ther. (2003) 25. doi: 10.1016/S0149-2918(03)80243-6
93. Savage DB, Tan GD, Acerini CL, Jebb SA, Agostini M, Gurnell M, et al. Human metabolic syndrome resulting from dominant-negative mutations in the nuclear receptor peroxisome proliferator-activated receptor. Available online at: http://diabetesjournals.org/diabetes/article-pdf/52/4/910/339498/db0403000910.pdf. (Accessed: February 01, 2024).
94. Bansal R, Cochran E, Startzell M, Brown RJ. Clinical effects of sodium-glucose transporter type 2 inhibitors in patients with partial lipodystrophy. Endocrine Pract. (2022) 28:610–4. doi: 10.1016/j.eprac.2022.03.006
Keywords: PPAR gamma, adipose tissue, genetic lipodystrophy, insulin resistance, diabetes mellitus
Citation: Soares RMV, da Silva MA, Campos JTAdM and Lima JG (2024) Familial partial lipodystrophy resulting from loss-of-function PPARγ pathogenic variants: phenotypic, clinical, and genetic features. Front. Endocrinol. 15:1394102. doi: 10.3389/fendo.2024.1394102
Received: 01 March 2024; Accepted: 10 September 2024;
Published: 27 September 2024.
Edited by:
Rinki Murphy, The University of Auckland, New ZealandReviewed by:
Ken Ebihara, Jichi Medical University, JapanCopyright © 2024 Soares, da Silva, Campos and Lima. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Josivan Gomes Lima, am9zaXZhbmxpbWFAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.