 Senthil Sivakumar
Senthil Sivakumar Dechen Lama
Dechen Lama Nabil Rabhi
Nabil Rabhi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 09 July 2024
Sec. Obesity
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1393250
This article is part of the Research Topic Insights in Obesity: 2023 View all 9 articles
The prevalence of obesity and its associated comorbidities has surged dramatically in recent decades. Especially concerning is the increased rate of childhood obesity, resulting in diseases traditionally associated only with adulthood. While obesity fundamentally arises from energy imbalance, emerging evidence over the past decade has revealed the involvement of additional factors. Epidemiological and murine studies have provided extensive evidence linking parental obesity to increased offspring weight and subsequent cardiometabolic complications in adulthood. Offspring exposed to an obese environment during conception, pregnancy, and/or lactation often exhibit increased body weight and long-term metabolic health issues, suggesting a transgenerational inheritance of disease susceptibility through epigenetic mechanisms rather than solely classic genetic mutations. In this review, we explore the current understanding of the mechanisms mediating transgenerational and intergenerational transmission of obesity. We delve into recent findings regarding both paternal and maternal obesity, shedding light on the underlying mechanisms and potential sex differences in offspring outcomes. A deeper understanding of the mechanisms behind obesity inheritance holds promise for enhancing clinical management strategies in offspring and breaking the cycle of increased metabolic risk across generations.
The global rise in obesity presents a pressing public health challenge that demands thorough exploration to understand its complex origins and extensive impacts. According to the World Health Organization (WHO) in 2016, a staggering 39% of adults aged 18 years and over were classified as overweight, amounting to 1.9 billion individuals, with an alarming 13% falling into the obese category (World Health Organization, 2016). Projections by the World Obesity Federation suggest that if current trends persist, more than half (51%) of the global population will be affected by overweight or obesity by 2035 (1). Overweight and obesity are distinguished by Body Mass Index (BMI), with a BMI of ≥ 25 kg/m² indicating overweight and ≥ 30 kg/m² indicating obesity (WHO). This epidemiological trend carries significant implications, as obesity is closely linked to a myriad of health conditions, including cardiovascular disease (CVD), gastrointestinal disorders, type 2 diabetes (T2D), musculoskeletal disorders, respiratory complications, nonalcoholic fatty liver disease (NAFLD), chronic kidney disease (CKD), cancer, and psychiatric disorders (1).
The childhood obesity epidemic is equally worrisome, demanding urgent attention. In 2020, an alarming 39 million children below the age of 5 were affected by overweight or obesity, while over 340 million children and adolescents aged 5-19 faced similar challenges in 2016 (World Health Organization). What’s particularly concerning is the steady rise in the age-standardized mean BMI of children aged 5 to 18, which has increased globally by 0.32 kg/m² per decade from 1975 to 2016 . The consequences of childhood obesity are profound, encompassing an increased risk of lifelong obesity, the onset of chronic illnesses, and various psychosocial impacts (2–7). Notably, the challenges posed by the COVID-19 pandemic, including lockdowns and disruptions to routines, have further exacerbated the issue, leading to a troubling surge in childhood obesity rates (8). At its core, obesity arises from a complex interplay of imbalanced energy intake and expenditure, influenced by various factors such as changing lifestyles and dietary patterns (9, 10). Total energy expenditure includes basal metabolic rate, energy expended at rest, and physical activity expenditure. Recent trends underscore a societal transition toward a more sedentary way of life, contributing to diminished energy expenditure. Additionally, the obesogenic environment, characterized by easy access to high-calorie foods and limited physical activity opportunities, has fueled the prevalence of obesity. However, over the past decade, it has become clear that additional factors are involved (11).
Indeed, analysis of dietary data from the National Health and Nutrition Survey (NHANES) during the period from 1998 to 2006 found a noteworthy average increase of 2.3 kg/m² in BMI within the USA, even when accounting for dietary intake and exercise levels (12). These findings imply that factors beyond dietary habits and exercise participation warrant thorough investigation. Several plausible etiological factors for obesity have been posited, encompassing chronic sleep deprivation and circadian misalignment (13), alterations in the gut microbiome (14), and specific pharmaceutical agents known to induce weight gain (11, 15). Additionally, a multitude of genome-wide association studies (GWAS) have been conducted to elucidate genetically mediated heightened susceptibility to obesity (16). It is noteworthy, however, that genetic variants associated with obesity exhibit limited predictive capacity, collectively accounting for a mere ~3% of the variance in BMI (17). Finally, several lines of evidence demonstrated that environmental factors and alterations in nutritional conditions in-utero can exert direct influences on the epigenome of ancestral germline cells consequently imparting susceptibility to obesity (18–23). This mechanism is recognized as transgenerational or intergenerational inheritance of obesity. Notably, a recent meta-analyses, including 23 clinical studies, indicate a significant link between parental obesity and childhood obesity, showing that children with overweight or obese parents are nearly twice as likely to be obese compared to those with normal-weight parents (24). The risk escalates considerably when both parents are obese (odds ratio: 12.0), and even more so when both parents are severely obese (odds ratio: 22.3), independent of age, gender, socioeconomic status, and ethnicity (25). Moreover, studies predicting the risk of childhood or adolescent obesity have found that parental BMI and socioeconomic factors are better predictors of childhood obesity than genetic scores (26, 27). While both maternal and paternal obesity significantly contribute to the risk of childhood obesity, findings regarding the relative contributions of each parent vary across studies. Some studies suggest that maternal obesity has a stronger impact (25, 28, 29), while others indicate a greater influence from paternal obesity (26, 30, 31), highlighting the complexity and the need for further research to fully understand these dynamics.
In this review, we delve into the current evidence and molecular mechanisms underlying the inheritance of obesity, with a particular emphasis on inter- and trans-generational transmission. To ensure a thorough exploration of the topic, we conducted a comprehensive literature search, meticulously gathering relevant studies that shed light on the inheritance patterns of obesity. Our search encompassed a wide range of sources, including data on familial clustering of obesity, genetic studies elucidating predisposing factors, and animal models providing insights into transgenerational inheritance phenomena. The criteria for selecting studies were rigorously defined, prioritizing relevance to inter- and trans-generational inheritance and ensuring the robustness of review designs employed. By adopting this strategic approach, we aimed to provide a comprehensive overview of the inheritance mechanisms contributing to the obesity epidemic, thereby advancing our understanding of this complex and pressing public health issue.
The terms transgenerational and intergenerational are often used interchangeably when obesity-related effects are discussed hence before we continue it is opportune to clarify the definitions as previously established and used in this manuscript (Figure 1). The notion of ‘transgenerational effects’ encompasses phenomena exclusively attributed to factors that cannot be ascribed to the direct impact of a particular trigger on the affected organism. For instance, an environmental stimulus can directly influence a developing embryo, including the already-formed oocytes within a female embryo in mammals (32–34). Consequently, only modified phenotypes that emerge in the second generation (in the case of male transmission) or the third generation (in the case of female transmission) subsequent to a trigger can be accurately characterized as transgenerational inheritance in the context of obesity. In contrast, effects manifesting over shorter temporal intervals are categorized as ‘parental’ or ‘intergenerational’ in nature. It is important to note that while these intergenerational effects span fewer generations, they may share underlying mechanistic pathways with transgenerational effects.
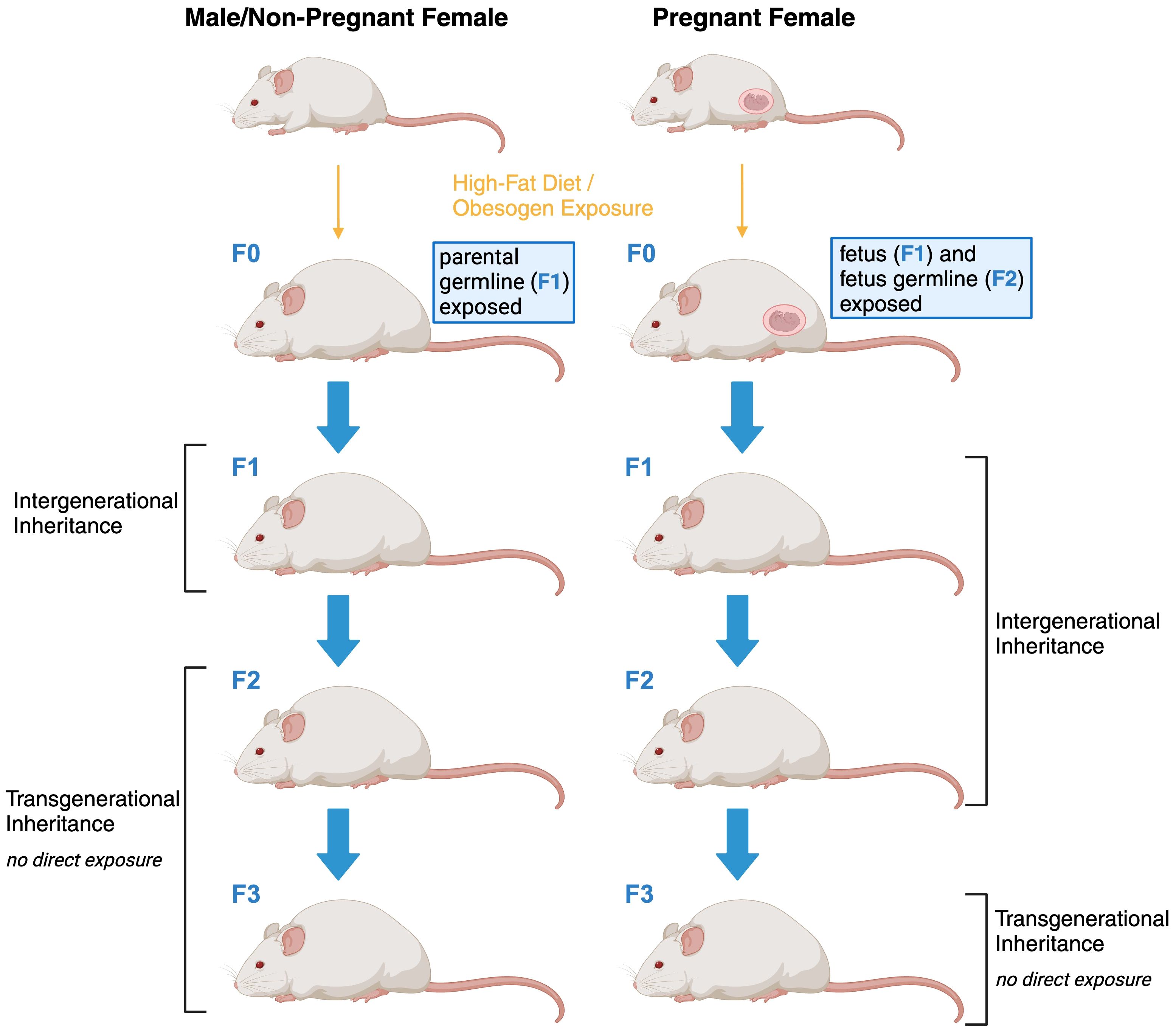
Figure 1 Schematic representation of intergenerational and transgenerational inheritance of obesity.
The heritability of obesity is estimated to range from between 40% and 75% (35). Genetic forms of obesity manifest along a continuum of clinical features traditionally classified into three overarching categories: Mendelian (monogenic) syndromic obesity, Mendelian non-syndromic obesity, and polygenic obesity (36).
Mendelian obesity forms result from rare chromosomal abnormalities and pathogenic gene variants that impact critical proteins involved in regulating energy balance. They are typically rare, early-onset and adhere to a Mendelian inheritance pattern and can be either autosomal or X-linked (37, 38). Syndromic forms of Mendelian obesity, also referred to as pleiotropic syndromes, are relatively uncommon within the general population. Syndromic obesity is characterized by obesity alongside additional distinctive features such as intellectual disabilities, dysmorphic traits, and congenital anomalies affecting specific organ systems. About 79 syndromes have been associated with obesity; notable examples include Albright hereditary osteodystrophy, Alström, Bardet-Biedl, and Prader-Willi syndrome (39). Those have been reviewed recently by Kaur et al. and will not be further discussed here (39).
On the other hand, non-syndromic Mendelian obesity forms identified thus far are primarily associated with genetic defects in the leptin/melanocortin pathway, leading to hyperphagic obesity (40, 41). These encompass mutations in genes encoding key components such as leptin, the leptin receptor, prohormone convertase 1, pro-opiomelanocortin, or melanocortin 4 receptor (Figure 2).
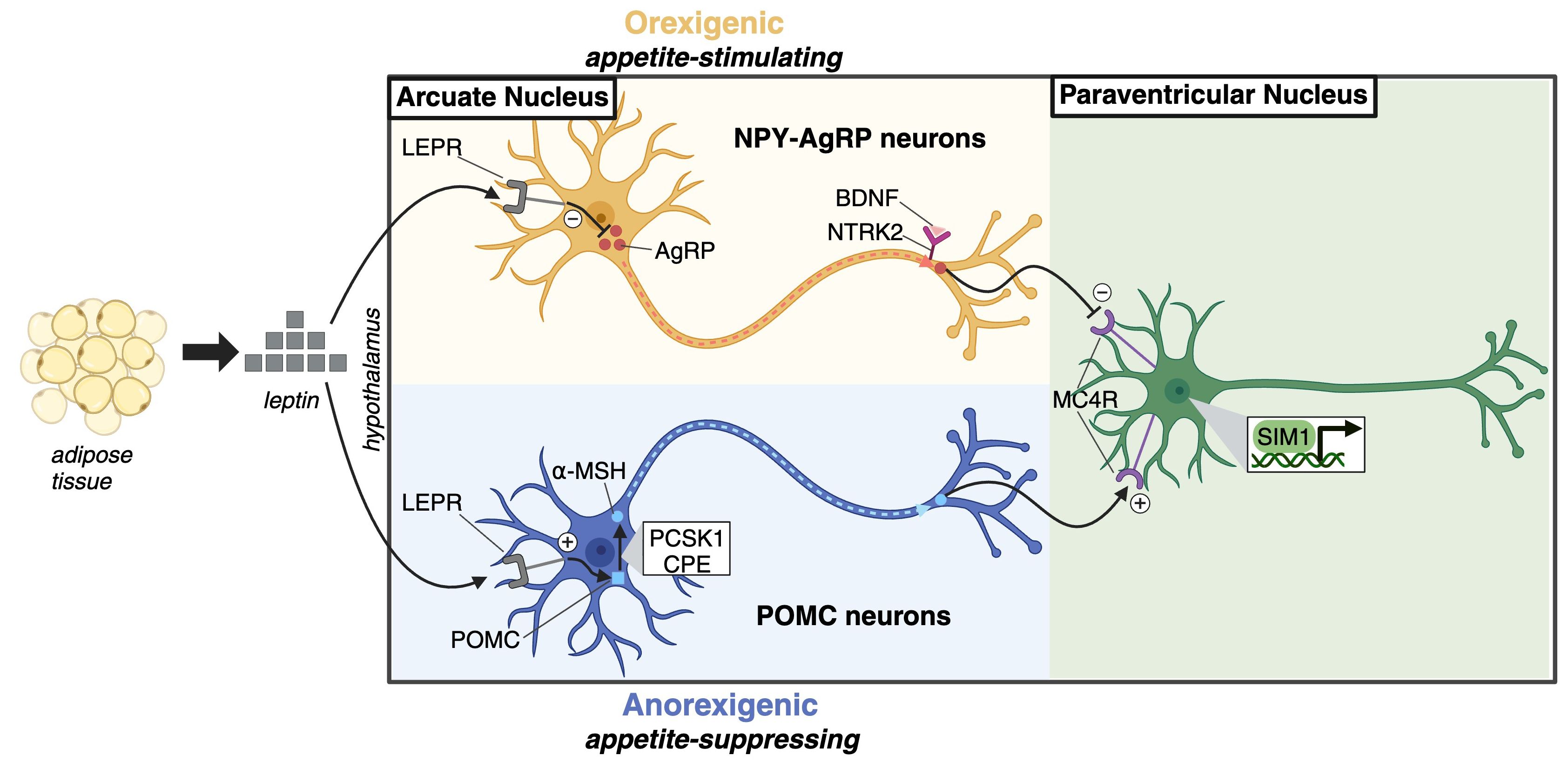
Figure 2 Leptin-melanocortin pathway. Leptin, primarily secreted by adipocytes, acts as a satiety factor in the hypothalamic arcuate nucleus. It binds to the leptin receptor (LEPR) on two neuron populations: neuropeptide Y (NPY)–agouti-related protein (AgRP) neurons and pro-opiomelanocortin (POMC) neurons. This binding downregulates orexigenic AgRP production in NPY-AgRP neurons and upregulates anorexigenic α-melanocyte-stimulating hormone (α-MSH) in POMC neurons. α-MSH acts as an agonist, while AgRP serves as a reverse agonist of MC4R on neurons in the paraventricular nucleus, signaling satiety and limiting food intake. Brain-derived neurotrophic factor (BDNF) modulates leptin-mediated synaptic plasticity through its receptor neurotrophic receptor tyrosine kinase 2 (NTRK2), while Single-minded homologue 1 (SIM1) is a transcription factor essential for the development of paraventricular nucleus neurons.
In contrast to monogenic obesity, polygenic obesity does not stem from a single gene with a significant impact on obesity development. Polygenic obesity is believed to be determined by the cumulative influence of numerous common genetic variants, each exerting modest effect. For instance, common variants located within intron 1 of the fat mass and obesity-associated gene (FTO) are among the most prominent contributors to polygenic obesity, accounting for approximately 1% of the variance in BMI within the general population.
Polygenic obesity, commonly known as common obesity, differs markedly from monogenic obesity, as it does not arise from a singular gene exerting a substantial impact on obesity development. Instead, polygenic obesity is shaped by the collective influence of numerous common genetic variants, each exerting modest effects (42–44). For example, common variants located within intron 1 of the fat mass and obesity-associated gene (FTO) are among the most prominent contributors to polygenic obesity, while accounting for only 1% of the variance in BMI within the general population (43, 44). This multifactorial condition aligns with heritability patterns observed in complex traits and diseases and shares a foundational biological framework with monogenic obesity. Specifically, both forms of obesity implicate the central nervous system (CNS) and the neural pathways governing food intake’s pleasurable aspects as crucial determinants of body weight regulation (45). Furthermore, emerging evidence suggests that mutations responsible for monogenic obesity may, to some extent, be influenced by an individual’s polygenic predisposition to obesity (46).
Genome-wide association studies (GWAS) have accelerated the identification of loci linked to polygenic obesity. Obesity-associated gene (FTO) was the first GWAS-identified obesity gene. The FTO locus has a well-established correlation with obesity but the specific mechanisms connecting FTO polymorphisms to obesity risks still need to be researched. Several loci discovered by GWAS are near genes that are also associated with monogenic obesity, including MC4R, BDNF, SH2B1, POMC, LEP, LEPR, NPY, SIM1, NTRK2, PCSK1 and KSR2 (45) (Table 1).
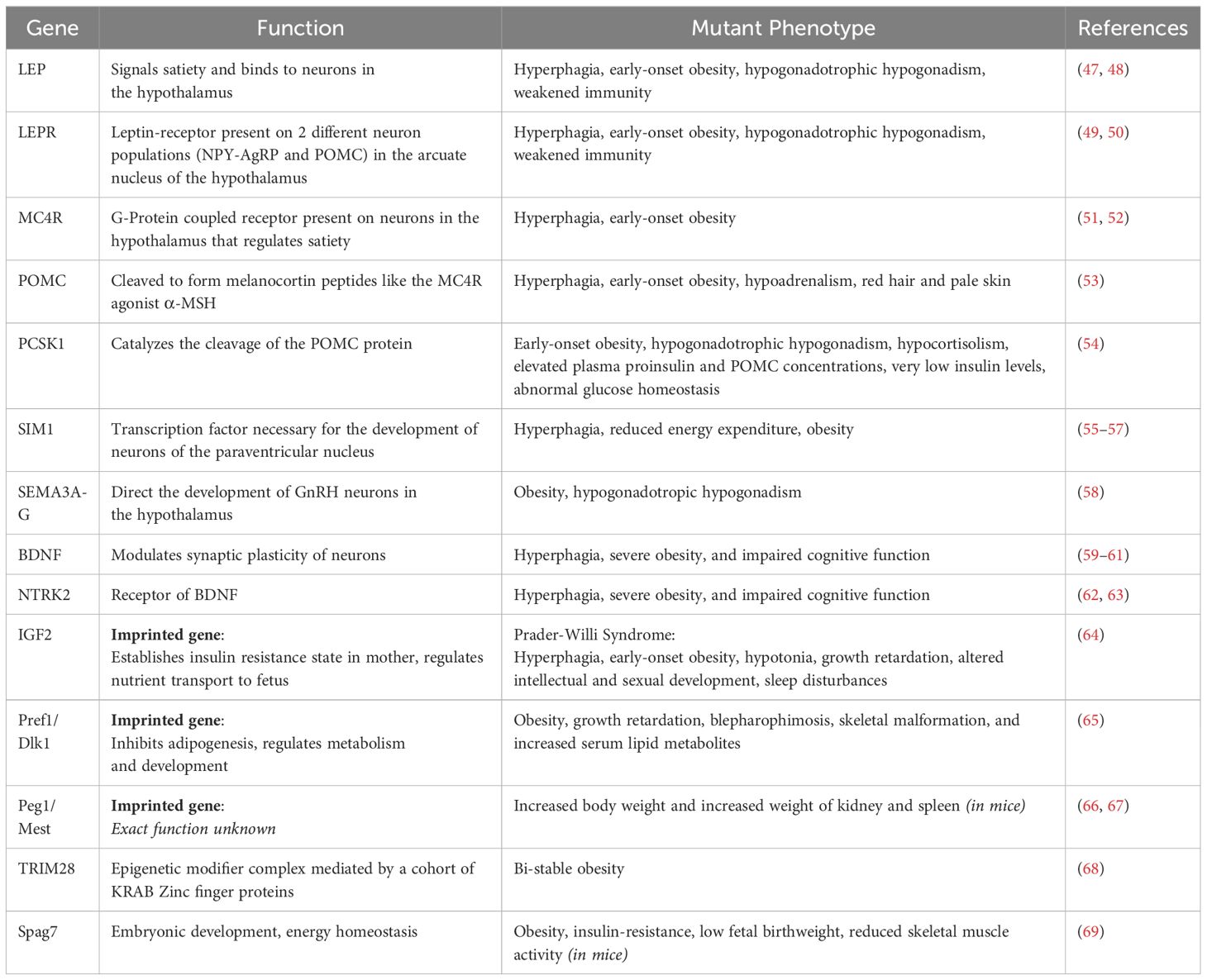
Table 1 List of genes found to control obesity inheritance in mice models and their functions.
Intergenerational phenotypes encompass alterations that arise from changes in ancestral generations’ phenotypes, which are distinct from primary DNA sequence modifications. These alterations involve various epigenetic modifications, including DNA methylation, histone modifications, and non-coding RNAs. The epigenome is remarkably sensitive to environmental influences such as lifestyle, dietary patterns, gut microbiota, and other factors (70, 71). Exposure early in life or in utero to those can alter metabolic outcomes through developmental epigenetic reprogramming (72). Understanding the mechanisms underlying intergenerational inheritance remains a complex challenge, as maternal lineage is influenced by various factors, including the intrauterine environment, placental function, and germ cell epigenetics. While firmly establishing germ cell-dependent epigenetic inheritance mechanisms presents difficulties, this section aims to provide insights into the current understanding of the roles of epigenetic marks in male and female germ cells.
In mammals, the predominant site for DNA methylation is observed on cytosines that are positioned before a guanine, referred to as CpG sites. This process involves the methylation of the 5’ carbon atom within the cytosine molecule, facilitated by enzymes known as DNA methyltransferases (Dnmts). CpG islands represent extended DNA sequences, typically around 1000 base pairs in length, characterized by a higher density of CpG dinucleotides compared to the rest of the genome. Remarkably, approximately 50% of CpG islands encompass well-established transcription start sites. The methylation of CpG islands plays a pivotal role in the stable silencing of gene expression. However, it is important to note that the majority of CpG islands remain unmethylated (73). The analysis of DNA methylation primarily finds their focus in population-based investigations. This preference arises from the relative stability and convenience associated with high-throughput, array-based assays, making DNA methylation a prominent subject of research in understanding epigenetic mechanisms.
Histones are pivotal proteins that play a crucial role in organizing the chromatin. Post-translationally, histones undergo modifications that exert a profound influence on the compaction state of chromatin, consequently influencing gene expression. This intricate regulation gives rise to two distinctive chromatin states: euchromatin, characterized by a more relaxed and open structure, facilitating high transcriptional activity; and heterochromatin, characterized by a tightly compacted configuration that renders it transcriptionally silent.
Numerous histone modifications contribute to the dynamic orchestration of chromatin structure and gene regulation, encompassing processes such as methylation, acetylation, phosphorylation, and ubiquitination (74). These modifications constitute fundamental components of the epigenetic machinery, finely tuning gene expression patterns.
Regulatory non-coding RNAs can mainly be divided into two categories based on size: short-chain non-coding RNAs (including siRNAs, miRNAs, and piRNAs) and long non-coding RNA (lncRNAs) (75).
Fathers play a crucial role in the genetic inheritance of their offspring, contributing approximately half of the nuclear DNA. This inheritance not only affects the genetic predisposition to diseases but also encompasses the transmission of genetic information from the paternal side. At specific loci known as paternally derived imprinted loci, the genetic information inherited by offspring is solely from the paternal allele. This exclusivity arises because DNA methylation effectively silences the maternal allele, allowing only the paternal allele to be expressed (76).
Nonetheless, there exists compelling evidence demonstrating that factors such as paternal age and environmental exposures possess the capacity to exert a direct influence on the genetic makeup of offspring, consequently shaping their subsequent traits. This influence arises from the induction of DNA damage and the generation of de novo genetic mutations within the male germline (77–79). Another viable hypothesis is that the paternal environment may play a role in the selection of specific haploid genomes. This could occur through mechanisms such as altering the genotype distribution within the ejaculate or inducing mutations that subsequently impact sperm function and the probability of reproductive success (80). For instance, studies in both humans and rodents showed that male obesity impairs sperm count, motility and morphology (77, 81–85). While these genetic defects may contribute to the paternal influence on offspring phenotype, it is important to clarify that paternal transgenerational obesity is defined as alterations that manifest independently of any variations in the offspring’s genotype. This implies that changes in offspring phenotype are not contingent upon variations in their genetic makeup. This distinction makes clinical and epidemiological data interpretation difficult (86–89). In mice, however, studies have clearly demonstrated that paternal high-fat diet (HFD) feeding has adverse consequences that manifest in the subsequent generation independently from the genotype (90–95). Paternal obesity in rodents has been linked to adverse effects on offspring health, including lower mitochondrial activity, smaller offspring size, altered carbohydrate metabolism, delayed cell cycle progression, decreased blastocyst number, and decreased blastulation rate (89–92). Fullston et al. have reported that paternal effects exhibit sex-specific transmission patterns to the next generation, affecting both first-generations of paternal and maternal lines (90). Intriguingly, even when both generations of offspring were maintained on a standard diet, the authors found that F1 males pass on a predisposition to obesity and insulin resistance to their F2 female offspring. Conversely, F1 females transmit a propensity for obesity, impaired metabolic health, and insulin resistance to their F2 male descendants. It is worth noting that these intergenerational effects originating from grand-paternal HFD exposure are most pronounced in F2 male offspring born to F1 females (90). Interestingly, male neonatal overnutrition has been shown to be sufficient to promote obesity, glucose intolerance and insulin resistance later in life. Most of the metabolic disorders excluding obesity were found to be inherited to at least the two next generations through the male lineage (96). Nevertheless, all the studies that examined paternal inheritance of metabolic disorders in successive generations found that those faded in severity with each generation (90, 96, 97). Finally, although in-utero exposure has a stronger transgenerational transmissibility, paternal HFD exposure has been found to have an additive metabolic effect for two generations (97).
All the studies to date overwhelmingly support the idea that paternal transmission is influenced by the inheritance of epigenetic modifications transmitted across successive generations. Remarkably, both human and rodent studies have consistently revealed a robust association between dietary habits and the epigenetic alterations present in sperm (93, 97–102). For example, paternal low protein diet (LPD) feeding has been shown to promote sperm hypomethylation leading to increased adiposity, glucose intolerance, altered gut bacteria profile, and hepatic liver function resembling fatty liver disease in offspring (103). These epigenetic modifications were detected in crucial metabolic genes, including adiponectin and leptin, as well as imprinted genes such as Igf2, Peg3, Cdkn1c, and Gnas (97, 98, 104–106). Pepin et al. found that obesity induced alterations in sperm H3K4me3 profile and suggested it as a metabolic sensor of paternal obesity and the inheritance of metabolic dysfunction (107). Further pathway analysis of genes with altered H3K4me3 modification revealed an enrichment of metabolic, inflammatory, and developmental processes (107). These processes were found to correlate with offspring metabolic dysfunction and corresponded to genes enriched for H3K4me3 in embryos, which also overlapped with embryonic and placental gene expression profiles (107). In rats, paternal obesity was found to downregulate histone marks H3K4me3, H3K9me3, and H4ac, while upregulating H3K27me3 and H3ac were in placentas derived from obese male rats.
Finally, the DNA methylation pattern of SETD2 a histone methyltransferase, which methylates H3K36me2 to generate H3K36me3, has been shown to be altered in paternal HFD sperm suggesting a potential role of SETD2 as an epigenetic carrier for paternal intergenerational and transgenerational inheritance (93). It is likely that dietary habits alter multiple epigenetic modifiers’ expression, activity and function which in turn synergically promote developmental programming and it transmitted across successive generations.
While both parents contribute equally to the genetic makeup of offspring, it is important to consider mothers separately in the context of genetic predispositions to diseases. Mothers have the unique ability to influence offspring phenotype during gestation and lactation, making this a critical window during which maternal factors can significantly impact offspring metabolic outcomes (108–110). Notably, maternal BMI has emerged as a significant determinant of offspring health, with some studies suggesting a stronger influence than paternal BMI (25, 28, 29). However, other studies using the Northern Finland Birth Cohort found a greater effect of paternal obesity, suggesting that the mechanisms and extents of their influences may differ, underscoring the need to consider both parental roles in obesity-related research (30, 31). Nerveless, the effect of maternal obesity is rooted in developmental programming by maternal obesity, although the precise mechanisms driving this transgenerational phenomenon remain poorly defined, with existing studies proposing varying mechanisms (111, 112). Research underscores the pivotal role of nutrient availability in the uterus in determining offspring obesity risk. Both fetal undernutrition and overnutrition have been implicated in shaping altered metabolic phenotypes, indicating the involvement of multiple mechanisms in the interaction between maternal nutrition and the transgenerationally transmitted phenotype (113, 114). Understanding these intricate pathways is crucial for unraveling the complexities of maternal influence on offspring health and developing effective interventions to mitigate the risk of obesity across generations.
The thrifty phenotype hypothesis, proposed by James Neel, offers an explanation for the observed associations between poor fetal nutrition and the subsequent development of metabolic disorders (115). According to this hypothesis, individuals with a thrifty phenotype are predisposed to store energy as body fat to aid survival during times of famine or food scarcity. However, in environments abundant with nutrition, this adaptive trait can increase the risk of obesity and associated metabolic syndromes. Supporting evidence for this hypothesis comes from studies of the Dutch hunger winter, which found that individuals born during the famine were more prone to developing obesity, diabetes, and other metabolic diseases compared to those born before the famine, with a more pronounced risk observed in males (116). Furthermore, infants exposed to famine during their mother’s first trimester faced a higher risk of metabolic diseases, underscoring the heightened susceptibility of the early developmental stage to environmental influences.
In the Dutch hunger winter study, Heijmans et al. (117) identified insulin-like growth factor II (IGF2) methylation as a potential epigenetic marker distinguishing individuals exposed to famine in utero. IGF2, which is maternally imprinted, exhibits relatively stable methylation patterns up to middle age, enabling the detection of in utero epigenetic changes later in life. Their findings indicated that individuals exposed to famine early in gestation displayed lower levels of IGF2 methylation compared to those unexposed, highlighting the lasting impact of temporary environmental influences on epigenetic modifications, which may contribute to adult disease risk. Hypomethylation of Igf2 in cord blood has also been associated with an increased risk of early childhood obesity (118, 119). The imprinted gene Igf2 plays a crucial role in fetal metabolism by regulating nutrient transport to the fetus and inducing insulin resistance in the mother. Fetuses lacking placental Igf2 were found to be growth-restricted and hypoglycemic, which may further elucidate the hypomethylation of Igf2 observed in famine conditions (64).
Recent metabolic studies have revealed that individuals with a thrifty metabolism exhibit decreased activation of brown adipose tissue (BAT) in response to cold exposure, potentially contributing to their increased susceptibility to weight gain (120). This observation is supported by a recent investigation linking sperm associated antigen 7 (SPAG7) deficiency to intrauterine growth restriction, which subsequently manifests as reduced energy expenditure, obesity and insulin resistance in adulthood (69). The authors found that SPAG7-deficient mice were born underweight but developed obesity later in life and identified reduced energy expenditure as a key driver for the onset of obesity and metabolic syndrome in these mice (69). Although the underlying mechanisms remain to be fully elucidated, these studies collectively support the concept of a ‘thrifty’ phenotype, suggesting the existence of genetic or epigenetic factors predisposing offspring to increased risks of metabolic diseases.
Maternal obesity has also been recognized as a major contributor to offspring obesity, with extensive epidemiological evidence linking pre-pregnancy BMI to increased offspring weight and subsequent cardiometabolic complications in adulthood (109, 121–130). Notably, offspring born after maternal bariatric gastrointestinal bypass surgery (AMS) exhibit lower birth weights and reduced obesity compared to siblings born before maternal surgery (131, 132). Moreover, alterations in maternal hormones, including resistin, insulin-like growth factor binding protein-1 (IGFBP-1), adiponectin, visfatin, and kisspeptin-1, have been correlated with variations in offspring birth weight (133–136). However, despite the correlations observed in these studies, deciphering the causal mechanisms driving maternal transgenerational obesity inheritance remains a formidable challenge, primarily due to constraints in accessing human specimens and acquiring comprehensive data from maternal-offspring cohorts.
One hypothesis suggests the transgenerational inheritance of epigenetic modifications from obese mothers. Early studies have identified correlations between retinoid X receptor alpha (RXRA) promoter methylation and later childhood adiposity across independent cohorts (137). Leveraging advancements in Targeted Bisulfite Sequencing technology, recent studies have conducted comprehensive genome-wide analyses of fetal umbilical cord blood from offspring of obese mothers. This scrutiny unveiled a notable reduction in methylated cytosines within both CpG islands and promoter regions compared to control groups. Intriguingly, these epigenetic alterations were found to be enriched in genes associated with pathways linked to an elevated susceptibility to metabolic disorders, cancer, and cardiomyopathy (138). Replication of these findings in sibling-offspring cohorts born BMS and AMS suggests potential reversibility through surgery-induced epigenetic reprogramming (132, 139). Further investigations in sibling offspring cohorts have revealed that maternal surgical intervention induced alterations in DNA methylation and transcriptional profiles of genes implicated in insulin and leptin signaling, as well as pro-inflammatory genes. These findings underscore the capacity for maternal metabolic health improvements to modulate offspring epigenetic profiles (132).
However, deciphering the mechanisms underlying maternal obesity inheritance in human settings remains challenging. Consequently, studies using established obesity models in controlled environments have been pivotal in advancing our understanding. For instance, in-depth investigations using HFD in C57BL/6 mice for three consecutive generations have revealed a progressive exacerbation of obesity across generations, with the severity increasing from F0 to F2 (140). Moreover, the F2 generation exhibited severe glucose intolerance and insulin resistance, accompanied by increased hepatic steatosis and elevated serum levels of triglycerides, insulin, and leptin (140). These metabolic alterations were concomitant with a gradual increase in hepatic lipogenesis and endoplasmic reticulum stress genes across generations. Interestingly, the F2 generation displayed a significant reduction in accumulation of methylated histones in LXRα and ERO1-α gene promoters (140). Nevertheless, elucidating the precise contributions of maternal versus paternal transmission of obesity remains challenging due to the intricate experimental designs employed in these studies. Using a targeted experimental approach to examine the specific repercussions of maternal obesity, Huang et al. unveiled compelling insights. Their study revealed that maternal HFD not only promoted glucose intolerance and insulin resistance in the F1 generation but also led to a diminution in embryonic developmental potential. Furthermore, HFD was associated with elevated levels of reactive oxygen species (ROS) and γH2AX, coupled with a decline in mitochondrial membrane potential (MMP) within oocytes, thus instigating significant oxidative stress and DNA damage (141). Additionally, the investigation identified an elevation in Rap1-interacting factor 1 (RIF1) levels in the oocytes of HFD-fed females associated with aberrations in DNA methylation and histone modification patterns during zygotic genome activation in obese mice (141). Furthermore, RIF1 knockdown experiments using Trim-Away methods revealed that degradation of RIF1 altered the enrichment of H3K4me3 and H3K9me3, subsequently triggering the transcriptional activation of the zygotic genome activation marker Murine Endogenous Retrovirus-Leucine (MuERV-L). In a recent investigation into fetal BAT from obese females, it was found that maternal obesity triggers an increase in the expression of Dio3, encoding deiodinase 3 (D3), thereby leading to the catabolization of triiodothyronine (T3). Simultaneously, the authors uncovered a suppression of the maternally imprinted long noncoding RNA, Dio3 antisense RNA (Dio3os), resulting in intracellular T3 deficiency and subsequent inhibition of BAT development. Furthermore, the investigators noted a higher degree of methylation in the Dio3os promoter region in the oocytes of obese mothers, a modification that persisted in the offspring (142). Conversely, Wang et al. demonstrated that maternal obesity suppressed genes associated with myogenesis and brown adipogenesis while promoting white adipogenesis during fetal BAT development by enhancing miR-204-5p expression, consequently leading to the suppression of PGC1α and Sirt1 (143). Collectively, these findings suggest a complex interplay of epigenetic modifications underlying the inheritance of maternal obesity, potentially persisting across generations.
It is noteworthy that sexual dimorphism in transgenerational inheritance of obesity has been reported, indicating a more complex mechanism than initially presumed. For instance, oocytes exposed to obesity have been previously found to accumulate and transmit dysfunctional mitochondria to offspring due to an impaired ability to activate mitophagy (144). Subsequent studies across generation have demonstrated the transmission of dysfunctional mitochondria to the second and third generations through the female germline (145). Furthermore, recent research has shown that maternal obesity induced by a HFD disrupts genomic methylation in oocytes, with at least some of the altered methylation transmitted to F2 oocytes and livers via females. Interestingly, the involvement of melatonin in regulating the hyper-methylation of HFD oocytes has been identified, with melatonin increasing the expression of DNMT3a and DNMT1 mediated by the cAMP/PKA/CREB pathway (146). These findings highlight the importance of considering significant distinctions in the mechanisms of maternal inheritance of obesity between female and male offspring in future studies.
The complex interplay occurring at the maternal-fetal interface exerts a profound influence on long-term fetal health outcomes, underscoring the need to delve into placental genomic regulations and nutrient sensing pathways as potential contributors to disrupted metabolic phenotypes in offspring (147). HFD consumption before and during pregnancy has been found to enhance nutrient transport and fetal overgrowth in both human and murine studies (148–151). These findings have been linked to the upregulated expression of the mammalian target of rapamycin (mTOR) complex 1, which modulates nutrient transporter expression. Interestingly, vasoactive intestinal peptide (VIP) has been identified as a regulator of glucose and amino acid uptake, exerting its effects by increasing GLUT1 and mTOR gene expression (152). Notably, offspring of VIP-deficient mothers exhibit a marked reduction in body weight (153). Maternal inheritance of obesity has also been associated with dysregulation of circulating steroid hormones during pregnancy. In addition to steroid hormone dysregulation, maternal obesity is associated with alterations in a wide range of hormones, growth factors, and cytokines, which can significantly affect pregnancy outcomes. Page L et al. provide an extensive review of these dysregulations, highlighting the complex endocrine environment in obese pregnancies and its potential implications for both maternal and fetal health (154). One study in rodents demonstrated that maternal obesity resulted in low fetal weight in the F1 generation, accompanied by modified DNA methylation and altered expression of the nuclear hormone receptor RXRα in a sex-dependent manner (155). Methylation of RXRα has also been linked to childhood adiposity (137). Examination of placentas from obese women reveals diminished mitochondrial β-oxidation of fatty acids (FA) and lipid accumulation in late pregnancy, fostering a lipotoxic environment (156). The authors found a significant reduction in genes associated with FA oxidation, uptake, synthesis, and storage, with pronounced effects notably observed in placentas of male fetuses (156).. Furthermore, alterations in placental genes implicated in modulating offspring glucose and insulin metabolism under the stress of maternal obesogenic conditions have been documented (152). Taken together, these findings suggest that maternal obesity-induced alterations in nutritional uptake may constitute critical determinants of maternal inheritance of obesity. Notably, evidence suggests that the action of epigenetic modifiers is sensitive to changes in dietary components and cellular metabolism intermediates, linking nutrition and energy metabolism to gene expression plasticity (157).
Considering the inheritability and genetic basis of obesity, it’s crucial to also examine gene-environment interactions as potential contributors to the mechanism of inheritance Studies have implicated exposure to environmental toxicants in the transgenerational inheritance of obesity and the development of DNA methylation sperm epimutations. Notable among these chemicals are plastics such as BPA, phthalates DEHP and DBP, bisphenol S, the herbicide glyphosate, insecticides like DDT and methoxychlor, the biocide tributyltin, the combustion byproduct benzo[a]pyrene, and hydrocarbons from jet fuel (158–165). Most of these chemicals are classified as endocrine disruptors and have been labeled as “obesogens,” capable of promoting obesity by increasing fat cell count, fat storage, and affecting appetite mechanisms. For instance, exposure to the estrogenic endocrine disruptor BPA has been linked to deregulated genomic methylation and hydroxymethylation (166, 167). Prenatal exposure to BPA in humans has been associated with early childhood obesity and methylation changes at the mesoderm-specific transcript homologue (MEST) locus (168). Studies on BPA-treated mouse spermatogonia showed reduced expression of Dnmt1, while exposure during oocyte maturation altered histone modifications due to oxidative stress (169, 170). BPA exposure has also been found to affect TET enzyme expression and function, leading to altered levels of 5hmC at several imprinted loci (167). This suggests a potential mechanism through which environmental toxicants can disrupt long-term imprinted gene regulation, ultimately contributing to obesity.
Moreover, some studies suggest that the effects of obesogens can be inherited across multiple generation. A transgenerational mouse study showed that grandparental exposure to tributyltin (TBT) resulted in increased fat depots in offspring extending to the F3 generation (171). Additionally, environmental factors such as paternal cold exposure have been implicated in influencing offspring obesity. Human studies have shown that individuals conceived in colder months exhibit higher BAT activation. Corresponding mouse studies demonstrated that paternal cold exposure affected sperm epigenetic programming and enhanced BAT activity in offspring (172).
Another environmental factor of interest is the maternal gut microbiome. Maternal diet influences the maternal gastrointestinal tract (GIT) microbiota, vaginal microbiota, and breast milk composition, which in turn influence the colonization of the fetus’ GIT either in utero or postnatally. The fetal microbiome plays a crucial role in GIT mucosa development and may be linked to obesity (173). Vaginal delivery is considered vital for infants to acquire bacterial communities resembling their mothers’, while cesarean delivery disrupts this process, leading to changes in offspring immune and metabolic programming (174). Indeed, studies have reported that infants born via cesarean delivery are more predisposed to obesity (175).
Treatments plans ranging from as simple as regulating the diet to as complex as surgery are available for the management of obesity. However, few methods are currently recommended for obese women during pregnancy or in the early stages of offspring development. Nonpharmacological interventions include dietary adjustments and increased physical activity, both of which been shown to confer benefits mediated through epigenetic mechanisms (176, 177). Notably, exercise during pregnancy has been shown to prevent the reduction in placental vascularization and fetal overgrowth associated with maternal obesity (178). Mechanistically, maternal exercise was found to downregulated mTOR protein expression mTOR and amino acid transporters promoting a healthier fetal outcome (179). Furthermore, maternal exercise during pregnancy as also found to improve fetal metabolic health through a vitamin D receptor-mediated increase in placental of superoxide dismutase 3 (SOD3), which in turn enhances liver function, and improves glucose tolerance in offspring (180). Extensive reviews have highlighted the effects of maternal and paternal exercise on offspring metabolism (181–184), underscoring the potential of regular exercise to break the cycle of increased metabolic risk across generations. However, the translation of these findings to the human population remains to be addressed.
Pharmacological treatment options typically target patients with a BMI ranging from 27 to 30 kg/m², with discontinuation recommended if less than 5% of the target weight is lost within three months of starting the medication (185). Currently, there are seven FDA-approved drugs for long-term weight loss, including Semaglutide, Setmelanotide, Gelesis100, Liraglutide, Bupropion-naltrexone, Phentermine-topiramate, and Orlistat (Table 2) (185). Of these, semaglutide and liraglutide, which are glucagon-like peptide 1 (GLP-1) agonists, show the most promising results, with semaglutide demonstrating the highest placebo-subtracted weight loss percentage (186, 187). Off-label drugs for weight loss include Bupropion, Metformin, Pramlintide, Sodium glucose cotransporter 2 (SGLT-2) inhibitors, Topiramate, and Zonisamide (185). A summary of the most common FDA-approved drugs for long-term weight loss, along with those currently in clinical trials, is provided in Table 2. However, none of these drugs have been tested in the context of maternal obesity and its impact on future offspring.
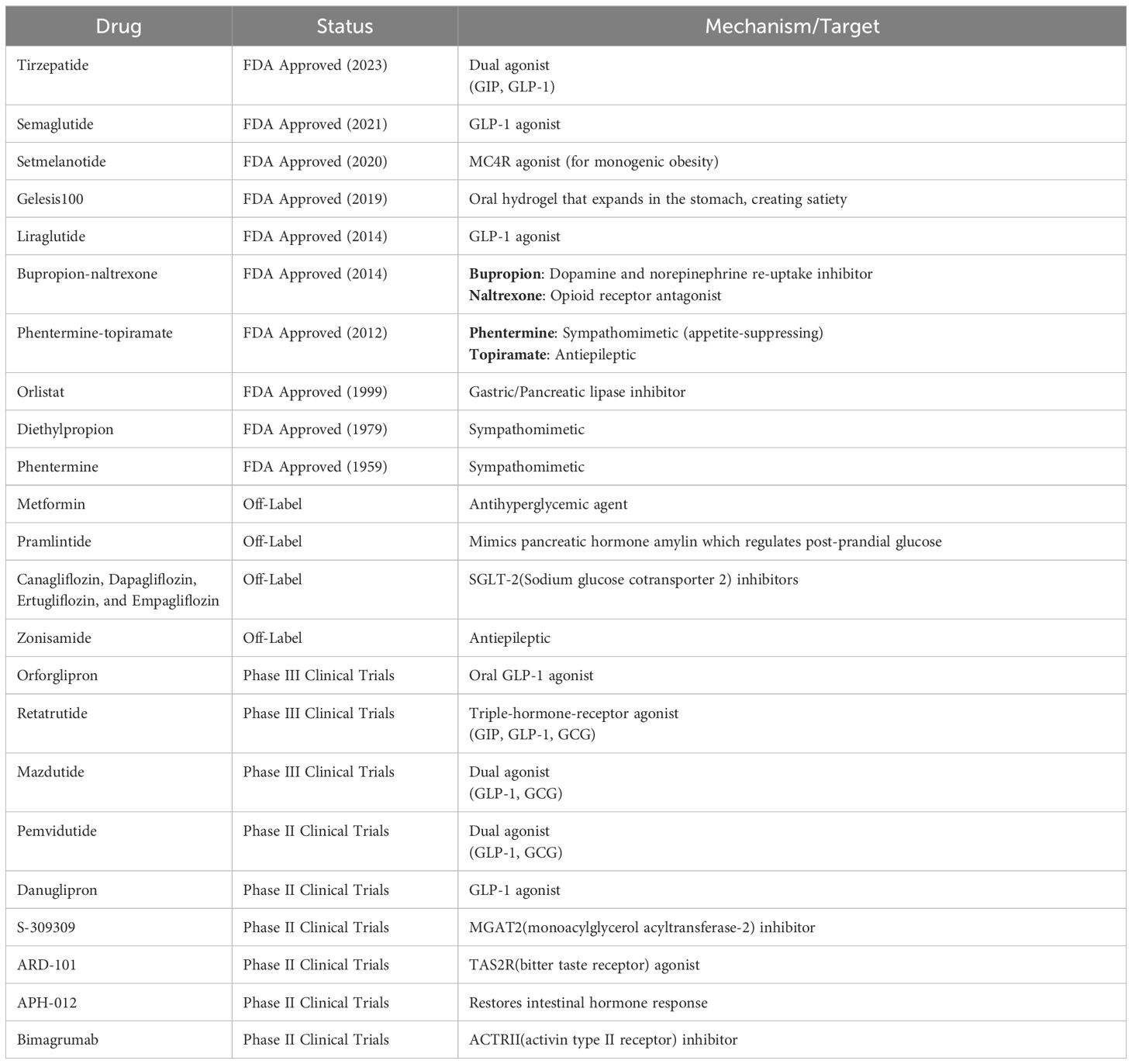
Table 2 List of drugs approved or currently in trial for obesity treatment and their targets for treatment (185).
Current research unequivocally illustrates the transgenerational transmission of obesity (Figure 3). Recent studies in both mice and humans have revealed that both undernutrition and overnutrition contribute to metabolic disorders in offspring, shedding light on some of the underlying mechanisms. However, numerous critical areas warrant future investigation to comprehensively grasp the central mechanisms that drive the perpetuation of obesity across generations. With emerging evidence indicating sexual dimorphism in the transmission of obesity, elucidating its extent will be invaluable for tailoring future pharmacological interventions. Additionally, further studies are imperative to thoroughly dissect the impact of maternal obesity on the metabolic organs of offspring and to ascertain whether the pathophysiological mechanisms promoting cardiometabolic complications in adulthood are equivalent across generations of obesity. For instance, because obesity occurs earlier and manifests more severely in offspring, future research should adopt a holistic approach to analyze inherited epigenetic alterations in the metabolic organs of offspring. The placental–fetal system represents another focal point for offspring developmental programming, given its significant role during the critical windows of prenatal development. Identifying the mediating factors and signaling pathways is essential for human translation, particularly given the escalating global obesity epidemic, which renders these issues increasingly pertinent for the future.
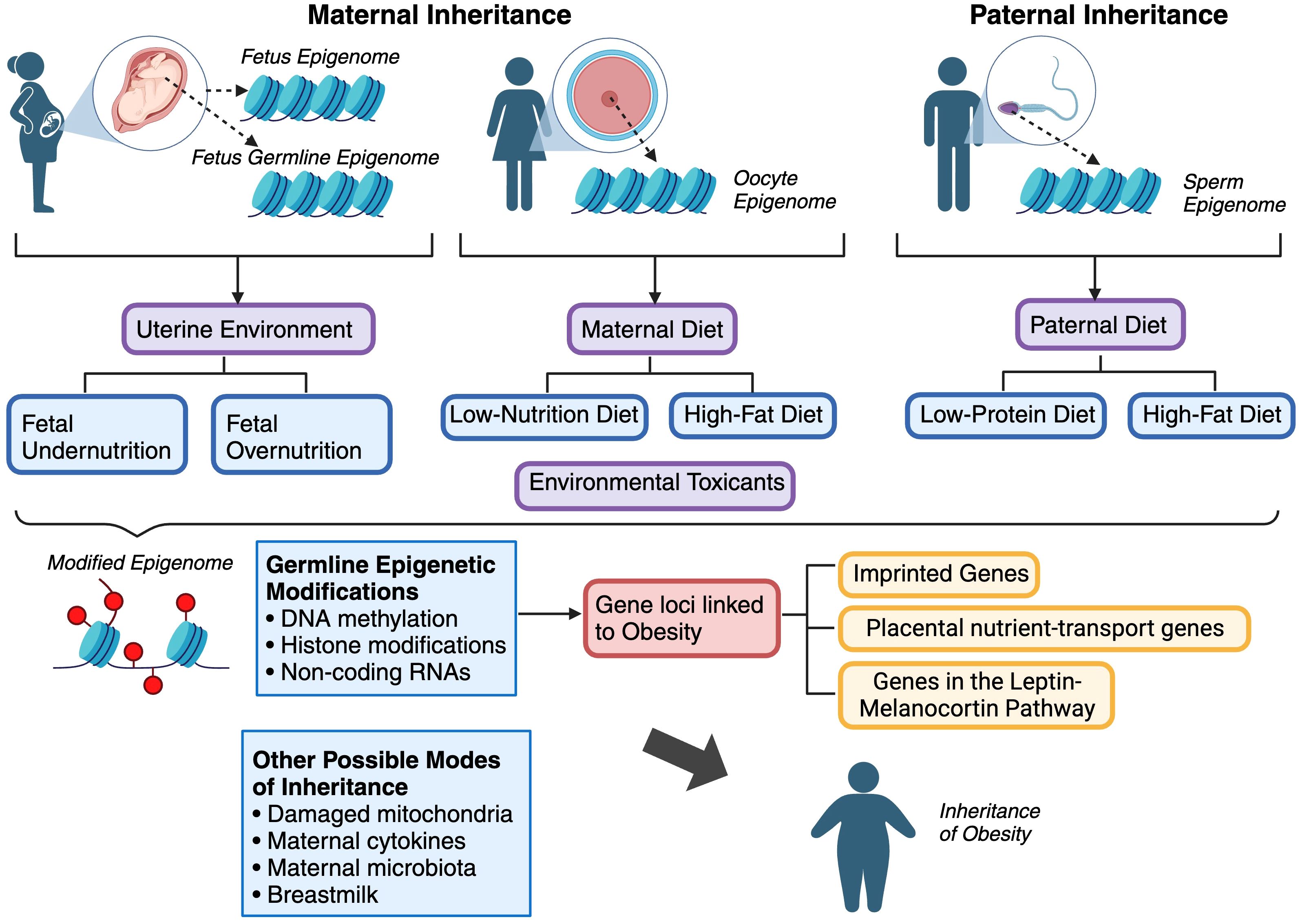
Figure 3 Summary of the various modes of inheritance of obesity.
SS: Writing – original draft. DL: Writing – original draft. NR: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The authors were supported by grant# U24DK097771 from the National Institute of Diabetes, Digestive, and kidney diseases via the NIDDK Information Network’s (dkNET) New Investigator Pilot Program in Bioinformatics (NR) & the Department of Biochemistry & Cell Biology at Early Career Award (NR).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Lim Y, Boster J. Obesity and Comorbid Conditions. In: StatPearls. StatPearls Publishing, Treasure Island (FL (2024). Available at: http://www.ncbi.nlm.nih.gov/books/NBK574535/.
2. Reilly JJ, Kelly J. Long-term impact of overweight and obesity in childhood and adolescence on morbidity and premature mortality in adulthood: systematic review. Int J Obes. (2011) 35:891–8. doi: 10.1038/ijo.2010.222
3. Simmonds M, Llewellyn A, Owen CG, Woolacott N. Predicting adult obesity from childhood obesity: a systematic review and meta-analysis. Obes Rev Off J Int Assoc Study Obes. (2016) 17:95–107. doi: 10.1111/obr.12334
4. Park MH, Falconer C, Viner RM, Kinra S. The impact of childhood obesity on morbidity and mortality in adulthood: a systematic review. Obes Rev Off J Int Assoc Study Obes. (2012) 13:985–1000. doi: 10.1111/j.1467-789X.2012.01015.x
5. Quek YH, Tam WWS, Zhang MWB, Ho RCM. Exploring the association between childhood and adolescent obesity and depression: a meta-analysis. Obes Rev Off J Int Assoc Study Obes. (2017) 18:742–54. doi: 10.1111/obr.12535
6. NCD Risk Factor Collaboration (NCD-RisC). Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128·9 million children, adolescents, and adults. Lancet Lond Engl. (2017) 390:2627–42. doi: 10.1016/S0140-6736(17)32129-3
7. GBD 2015 Obesity Collaborators, Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, et al. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med. (2017) 377:13–27. doi: 10.1056/NEJMoa1614362
8. Lartey ST, Jayawardene WP, Dickinson SL, Chen X, Gletsu-Miller N, Lohrmann DK. Evaluation of unintended consequences of COVID-19 pandemic restrictions and obesity prevalence among youths. JAMA Netw Open. (2023) 6:e2323596. doi: 10.1001/jamanetworkopen.2023.23596
9. Leibel RL, Rosenbaum M, Hirsch J. Changes in energy expenditure resulting from altered body weight. N Engl J Med. (1995) 332:621–8. doi: 10.1056/NEJM199503093321001
10. Romieu I, Dossus L, Barquera S, Blottière HM, Franks PW, Gunter M, et al. Energy balance and obesity: what are the main drivers? Cancer Causes Control CCC. (2017) 28:247–58. doi: 10.1007/s10552-017-0869-z
11. McAllister EJ, Dhurandhar NV, Keith SW, Aronne LJ, Barger J, Baskin M, et al. Ten putative contributors to the obesity epidemic. Crit Rev Food Sci Nutr. (2009) 49:868–913. doi: 10.1080/10408390903372599
12. Brown RE, Sharma AM, Ardern CI, Mirdamadi P, Mirdamadi P, Kuk JL. Secular differences in the association between caloric intake, macronutrient intake, and physical activity with obesity. Obes Res Clin Pract. (2016) 10:243–55. doi: 10.1016/j.orcp.2015.08.007
13. Chaput JP, McHill AW, Cox RC, Broussard JL, Dutil C, da Costa BGG, et al. The role of insufficient sleep and circadian misalignment in obesity. Nat Rev Endocrinol. (2023) 19:82–97. doi: 10.1038/s41574-022-00747-7
14. Takeuchi T, Kameyama K, Miyauchi E, Nakanishi Y, Kanaya T, Fujii T, et al. Fatty acid overproduction by gut commensal microbiota exacerbates obesity. Cell Metab. (2023) 35:361–75. doi: 10.1016/j.cmet.2022.12.013
15. Keith SW, Redden DT, Katzmarzyk PT, Boggiano MM, Hanlon EC, Benca RM, et al. Putative contributors to the secular increase in obesity: exploring the roads less traveled. Int J Obes. (2006) 30:1585–94. doi: 10.1038/sj.ijo.0803326
16. Grarup N, Sandholt CH, Hansen T, Pedersen O. Genetic susceptibility to type 2 diabetes and obesity: from genome-wide association studies to rare variants and beyond. Diabetologia. (2014) 57:1528–41. doi: 10.1007/s00125-014-3270-4
17. Albuquerque D, Nóbrega C, Manco L, Padez C. The contribution of genetics and environment to obesity. Br Med Bull. (2017) 123:159–73. doi: 10.1093/bmb/ldx022
18. Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, et al. Fetal and infant growth and impaired glucose tolerance at age 64. BMJ. (1991) 303:1019–22. doi: 10.1136/bmj.303.6809.1019
19. Ardissino M, Morley AP, Slob EAW, Schuermans A, Rayes B, Raisi-Estabragh Z, et al. Birth weight influences cardiac structure, function, and disease risk: evidence of a causal association. Eur Heart J. (2024) 45:443–54. doi: 10.1093/eurheartj/ehad631
20. Agius R, Savona-Ventura C, Vassallo J. Transgenerational metabolic determinants of fetal birth weight. Exp Clin Endocrinol Diabetes Off J Ger Soc Endocrinol Ger Diabetes Assoc. (2013) 121:431–5. doi: 10.1055/s-00000017
21. Lahti-Pulkkinen M, Bhattacharya S, Wild SH, Lindsay RS, Räikkönen K, Norman JE, et al. Consequences of being overweight or obese during pregnancy on diabetes in the offspring: a record linkage study in Aberdeen, Scotland. Diabetologia. (2019) 62:1412–9. doi: 10.1007/s00125-019-4891-4
22. Lobo E, Ana Y, Deepa R, Shriyan P, Sindhu ND, Karthik M, et al. Cohort profile: maternal antecedents of adiposity and studying the transgenerational role of hyperglycaemia and insulin (MAASTHI). BMJ Open. (2022) 12:e063794. doi: 10.1136/bmjopen-2022-063794
23. Shokry E, Marchioro L, Uhl O, Bermúdez MG, García-Santos JA, Segura MT, et al. Impact of maternal BMI and gestational diabetes mellitus on maternal and cord blood metabolome: results from the PREOBE cohort study. Acta Diabetol. (2019) 56:421–30. doi: 10.1007/s00592-019-01291-z
24. Lee JS, Jin MH, Lee HJ. Global relationship between parent and child obesity: a systematic review and meta-analysis. Clin Exp Pediatr. (2022) 65:35–46. doi: 10.3345/cep.2020.01620
25. Whitaker KL, Jarvis MJ, Beeken RJ, Boniface D, Wardle J. Comparing maternal and paternal intergenerational transmission of obesity risk in a large population-based sample. Am J Clin Nutr. (2010) 91:1560–7. doi: 10.3945/ajcn.2009.28838
26. Morandi A, Meyre D, Lobbens S, Kleinman K, Kaakinen M, Rifas-Shiman SL, et al. Estimation of newborn risk for child or adolescent obesity: lessons from longitudinal birth cohorts. PloS One. (2012) 7:e49919. doi: 10.1371/journal.pone.0049919
27. Moschonis G, Siopis G, Anastasiou C, Iotova V, Stefanova T, Dimova R, et al. Prevalence of childhood obesity by country, family socio-demographics, and parental obesity in Europe: the feel4Diabetes study. Nutrients. (2022) 14:1830. doi: 10.3390/nu14091830
28. Linabery AM, Nahhas RW, Johnson W, Choh AC, Towne B, Odegaard AO, et al. Stronger influence of maternal than paternal obesity on infant and early childhood body mass index: the Fels Longitudinal Study. Pediatr Obes. (2013) 8:159–69. doi: 10.1111/j.2047-6310.2012.00100.x
29. Næss M, Holmen TL, Langaas M, Bjørngaard JH, Kvaløy K. Intergenerational transmission of overweight and obesity from parents to their adolescent offspring – the HUNT study. PloS One. (2016) 11:e0166585. doi: 10.1111/j.2047-6310.2012.00100.x
30. Parikka S, Mäki P, Levälahti E, Lehtinen-Jacks S, Martelin T, Laatikainen T. Associations between parental BMI, socioeconomic factors, family structure and overweight in Finnish children: a path model approach. BMC Public Health. (2015) 15:271. doi: 10.1186/s12889-015-1548-1
31. Jääskeläinen A, Pussinen J, Nuutinen O, Schwab U, Pirkola J, Kolehmainen M, et al. Intergenerational transmission of overweight among Finnish adolescents and their parents: a 16-year follow-up study. Int J Obes 2005. (2011) 35:1289–94. doi: 10.1038/ijo.2011.150
32. Skinner MK. What is an epigenetic transgenerational phenotype? F3 or F2. Reprod Toxicol Elmsford N. (2008) 25:2–6. doi: 10.1016/j.reprotox.2007.09.001
33. Heard E, Martienssen RA. Transgenerational epigenetic inheritance: myths and mechanisms. Cell. (2014) 157:95–109. doi: 10.1016/j.cell.2014.02.045
34. Perez MF, Lehner B. Intergenerational and transgenerational epigenetic inheritance in animals. Nat Cell Biol. (2019) 21:143–51. doi: 10.1038/s41556-018-0242-9
35. Wardle J, Carnell S, Haworth CM, Plomin R. Evidence for a strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am J Clin Nutr. (2008) 87:398–404. doi: 10.1093/ajcn/87.2.398
36. Cummings DE, Schwartz MW. Genetics and pathophysiology of human obesity. Annu Rev Med. (2003) 54:453–71. doi: 10.1146/annurev.med.54.101601.152403
37. Farooqi S, O’Rahilly S. Genetics of obesity in humans. Endocr Rev. (2006) 27:710–8. doi: 10.1210/er.2006-0040
39. Kaur Y, de Souza RJ, Gibson WT, Meyre D. A systematic review of genetic syndromes with obesity. Obes Rev Off J Int Assoc Study Obes. (2017) 18:603–34. doi: 10.1111/obr.12531
40. Choquet H, Meyre D. Genomic insights into early-onset obesity. Genome Med. (2010) 2:36. doi: 10.1186/gm157
41. Farooqi IS, O’Rahilly S. Mutations in ligands and receptors of the leptin-melanocortin pathway that lead to obesity. Nat Clin Pract Endocrinol Metab. (2008) 4:569–77. doi: 10.1038/ncpendmet0966
42. Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. (2015) 518:197–206. doi: 10.1038/nature14177
43. Dina C, Meyre D, Gallina S, Durand E, Körner A, Jacobson P, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. (2007) 39:724–6. doi: 10.1038/ng2048
44. Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. (2007) 316:889–94. doi: 10.1126/science.1141634
45. Loos RJF, Yeo GSH. The genetics of obesity: from discovery to biology. Nat Rev Genet. (2022) 23:120–33. doi: 10.1038/s41576-021-00414-z
46. Chami N, Preuss M, Walker RW, Moscati A, Loos RJF. The role of polygenic susceptibility to obesity among carriers of pathogenic mutations in MC4R in the UK Biobank population. PloS Med. (2020) 17:e1003196. doi: 10.1371/journal.pmed.1003196
47. Montague CT, Farooqi IS, Whitehead JP, Soos MA, Rau H, Wareham NJ, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. (1997) 387:903–8. doi: 10.1038/43185
48. Wabitsch M, Funcke JB, von Schnurbein J, Denzer F, Lahr G, Mazen I, et al. Severe early-onset obesity due to bioinactive leptin caused by a p.N103K mutation in the leptin gene. J Clin Endocrinol Metab. (2015) 100:3227–30. doi: 10.1210/jc.2015-2263
49. Clément K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature. (1998) 392:398–401. doi: 10.1038/32911
50. Farooqi IS, Wangensteen T, Collins S, Kimber W, Matarese G, Keogh JM, et al. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N Engl J Med. (2007) 356:237–47. doi: 10.1056/NEJMoa063988
51. Yeo GS, Farooqi IS, Aminian S, Halsall DJ, Stanhope RG, O’Rahilly S. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. (1998) 20:111–2. doi: 10.1038/2404
52. Vaisse C, Clement K, Guy-Grand B, Froguel P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet. (1998) 20:113–4. doi: 10.1038/2407
53. Krude H, Biebermann H, Luck W, Horn R, Brabant G, Grüters A. Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat Genet. (1998) 19:155–7. doi: 10.1038/509
54. Jackson RS, Creemers JW, Ohagi S, Raffin-Sanson ML, Sanders L, Montague CT, et al. Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene. Nat Genet. (1997) 16:303–6. doi: 10.1038/ng0797-303
55. Michaud JL, Boucher F, Melnyk A, Gauthier F, Goshu E, Lévy E, et al. Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus. Hum Mol Genet. (2001) 10:1465–73. doi: 10.1093/hmg/10.14.1465
56. Ramachandrappa S, Raimondo A, Cali AMG, Keogh JM, Henning E, Saeed S, et al. Rare variants in single-minded 1 (SIM1) are associated with severe obesity. J Clin Invest. (2013) 123:3042–50. doi: 10.1172/JCI68016
57. Holder JL, Butte NF, Zinn AR. Profound obesity associated with a balanced translocation that disrupts the SIM1 gene. Hum Mol Genet. (2000) 9:101–8. doi: 10.1093/hmg/9.1.101
58. van der Klaauw AA, Croizier S, Mendes de Oliveira E, Stadler LKJ, Park S, Kong Y, et al. Human semaphorin 3 variants link melanocortin circuit development and energy balance. Cell. (2019) 176:729–742.e18. doi: 10.1016/j.cell.2018.12.009
59. Rios M, Fan G, Fekete C, Kelly J, Bates B, Kuehn R, et al. Conditional deletion of brain-derived neurotrophic factor in the postnatal brain leads to obesity and hyperactivity. Mol Endocrinol Baltim Md. (2001) 15:1748–57. doi: 10.1210/mend.15.10.0706
60. Gray J, Yeo GSH, Cox JJ, Morton J, Adlam ALR, Keogh JM, et al. Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene. Diabetes. (2006) 55:3366–71. doi: 10.2337/db06-0550
61. Gray J, Yeo G, Hung C, Keogh J, Clayton P, Banerjee K, et al. Functional characterization of human NTRK2 mutations identified in patients with severe early-onset obesity. Int J Obes 2005. (2007) 31:359–64. doi: 10.1038/sj.ijo.0803390
62. Xu B, Goulding EH, Zang K, Cepoi D, Cone RD, Jones KR, et al. Brain-derived neurotrophic factor regulates energy balance downstream of melanocortin-4 receptor. Nat Neurosci. (2003) 6:736–42. doi: 10.1038/nn1073
63. Yeo GSH, Connie Hung CC, Rochford J, Keogh J, Gray J, Sivaramakrishnan S, et al. A de novo mutation affecting human TrkB associated with severe obesity and developmental delay. Nat Neurosci. (2004) 7:1187–9. doi: 10.1038/nn1336
64. Lopez-Tello J, Yong HEJ, Sandovici I, Dowsett GKC, Christoforou ER, Salazar-Petres E, et al. Fetal manipulation of maternal metabolism is a critical function of the imprinted Igf2 gene. Cell Metab. (2023) 35:1195–1208.e6. doi: 10.1016/j.cmet.2023.06.007
65. Moon YS, Smas CM, Lee K, Villena JA, Kim KH, Yun EJ, et al. Mice lacking paternally expressed Pref-1/Dlk1 display growth retardation and accelerated adiposity. Mol Cell Biol. (2002) 22:5585–92. doi: 10.1128/MCB.22.15.5585-5592.2002
66. Nikonova L, Koza RA, Mendoza T, Chao PM, Curley JP, Kozak LP. Mesoderm-specific transcript is associated with fat mass expansion in response to a positive energy balance. FASEB J Off Publ Fed Am Soc Exp Biol. (2008) 22:3925–37. doi: 10.1096/fj.08-108266
67. Shi W, Lefebvre L, Yu Y, Otto S, Krella A, Orth A, et al. Loss-of-imprinting of Peg1 in mouse interspecies hybrids is correlated with altered growth. Genes N Y N 2000. (2004) 39:65–72. doi: 10.1002/gene.20027
68. Dalgaard K, Landgraf K, Heyne S, Lempradl A, Longinotto J, Gossens K, et al. Trim28 haploinsufficiency triggers bi-stable epigenetic obesity. Cell. (2016) 164:353–64. doi: 10.1016/j.cell.2015.12.025
69. Flaherty SE, Bezy O, Paulhus BL, Song L, Piper M, Pang J, et al. SPAG7 deletion causes intrauterine growth restriction, resulting in adulthood obesity and metabolic dysfunction. bioRxiv. (2023), 2023.09.05.555087. doi: 10.1101/2023.09.05.555087v1
70. Alegría-Torres JA, Baccarelli A, Bollati V. Epigenetics and lifestyle. Epigenomics. (2011) 3:267–77. doi: 10.2217/epi.11.22
71. Sharma M, Li Y, Stoll ML, Tollefsbol TO. The epigenetic connection between the gut microbiome in obesity and diabetes. Front Genet. (2019) 10:1329. doi: 10.3389/fgene.2019.01329
72. King SE, Skinner MK. Epigenetic transgenerational inheritance of obesity susceptibility. Trends Endocrinol Metab TEM. (2020) 31:478–94. doi: 10.1016/j.tem.2020.02.009
73. Moore LD, Le T, Fan G. DNA methylation and its basic function. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. (2013) 38:23–38. doi: 10.1038/npp.2012.112
74. Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. (2011) 21:381–95. doi: 10.1038/cr.2011.22
75. Wei JW, Huang K, Yang C, Kang CS. Non-coding RNAs as regulators in epigenetics (Review). Oncol Rep. (2017) 37:3–9. doi: 10.3892/or.2016.5236
76. Bartolomei MS, Ferguson-Smith AC. Mammalian genomic imprinting. Cold Spring Harb Perspect Biol. (2011) 3:a002592. doi: 10.1101/cshperspect.a002592
77. Palmer NO, Bakos HW, Owens JA, Setchell BP, Lane M. Diet and exercise in an obese mouse fed a high-fat diet improve metabolic health and reverse perturbed sperm function. Am J Physiol Endocrinol Metab. (2012) 302:E768–780. doi: 10.1152/ajpendo.00401.2011
78. Arnheim N, Calabrese P. Understanding what determines the frequency and pattern of human germline mutations. Nat Rev Genet. (2009) 10:478–88. doi: 10.1038/nrg2529
79. Sharp GC, Lawlor DA. Paternal impact on the life course development of obesity and type 2 diabetes in the offspring. Diabetologia. (2019) 62:1802–10. doi: 10.1007/s00125-019-4919-9
80. Rando OJ. Daddy issues: paternal effects on phenotype. Cell. (2012) 151:702–8. doi: 10.1016/j.cell.2012.10.020
81. Bakos HW, Mitchell M, Setchell BP, Lane M. The effect of paternal diet-induced obesity on sperm function and fertilization in a mouse model. Int J Androl. (2011) 34:402–10. doi: 10.1111/j.1365-2605.2010.01092.x
82. Chavarro JE, Toth TL, Wright DL, Meeker JD, Hauser R. Body mass index in relation to semen quality, sperm DNA integrity, and serum reproductive hormone levels among men attending an infertility clinic. Fertil Steril. (2010) 93:2222–31. doi: 10.1016/j.fertnstert.2009.01.100
83. Koloszár S, Fejes I, Závaczki Z, Daru J, Szöllosi J, Pál A. Effect of body weight on sperm concentration in normozoospermic males. Arch Androl. (2005) 51:299–304. doi: 10.1080/01485010590919701
84. Kort HI, Massey JB, Elsner CW, Mitchell-Leef D, Shapiro DB, Witt MA, et al. Impact of body mass index values on sperm quantity and quality. J Androl. (2006) 27:450–2. doi: 10.2164/jandrol.05124
85. Shayeb AG, Harrild K, Mathers E, Bhattacharya S. An exploration of the association between male body mass index and semen quality. Reprod BioMed Online. (2011) 23:717–23. doi: 10.1016/j.rbmo.2011.07.018
86. Crean AJ, Bonduriansky R. What is a paternal effect? Trends Ecol Evol. (2014) 29:554–9. doi: 10.1016/j.tree.2014.07.009
87. Cooper R, Hyppönen E, Berry D, Power C. Associations between parental and offspring adiposity up to midlife: the contribution of adult lifestyle factors in the 1958 British Birth Cohort Study. Am J Clin Nutr. (2010) 92:946–53. doi: 10.3945/ajcn.2010.29477
88. Li L, Law C, Lo Conte R, Power C. Intergenerational influences on childhood body mass index: the effect of parental body mass index trajectories. Am J Clin Nutr. (2009) 89:551–7. doi: 10.3945/ajcn.2008.26759
89. Figueroa-Colon R, Arani RB, Goran MI, Weinsier RL. Paternal body fat is a longitudinal predictor of changes in body fat in premenarcheal girls. Am J Clin Nutr. (2000) 71:829–34. doi: 10.1093/ajcn/71.3.829
90. Fullston T, Ohlsson Teague EMC, Palmer NO, DeBlasio MJ, Mitchell M, Corbett M, et al. Paternal obesity initiates metabolic disturbances in two generations of mice with incomplete penetrance to the F2 generation and alters the transcriptional profile of testis and sperm microRNA content. FASEB J Off Publ Fed Am Soc Exp Biol. (2013) 27:4226–43. doi: 10.1096/fj.12-224048
91. Ng SF, Lin RCY, Laybutt DR, Barres R, Owens JA, Morris MJ. Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nature. (2010) 467:963–6. doi: 10.1038/nature09491
92. Aizawa S, Tochihara A, Yamamuro Y. Paternal high-fat diet alters triglyceride metabolism-related gene expression in liver and white adipose tissue of male mouse offspring. Biochem Biophys Rep. (2022) 31:101330. doi: 10.1016/j.bbrep.2022.101330
93. Wei S, Luo S, Zhang H, Li Y, Zhao J. Paternal high-fat diet altered SETD2 gene methylation in sperm of F0 and F1 mice. Genes Nutr. (2023) 18:12. doi: 10.1186/s12263-023-00731-4
94. Chen Q, Yan M, Cao Z, Li X, Zhang Y, Shi J, et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science. (2016) 351:397–400. doi: 10.1126/science.aad7977
95. Wang B, Xia L, Zhu D, Zeng H, Wei B, Lu L, et al. Paternal high-fat diet altered sperm 5’tsRNA-gly-GCC is associated with enhanced gluconeogenesis in the offspring. Front Mol Biosci. (2022) 9:857875. doi: 10.3389/fmolb.2022.857875
96. Pentinat T, Ramon-Krauel M, Cebria J, Diaz R, Jimenez-Chillaron JC. Transgenerational inheritance of glucose intolerance in a mouse model of neonatal overnutrition. Endocrinology. (2010) 151:5617–23. doi: 10.1210/en.2010-0684
97. Masuyama H, Mitsui T, Eguchi T, Tamada S, Hiramatsu Y. The effects of paternal high-fat diet exposure on offspring metabolism with epigenetic changes in the mouse adiponectin and leptin gene promoters. Am J Physiol Endocrinol Metab. (2016) 311:E236–245. doi: 10.1152/ajpendo.00095.2016
98. Deshpande SSS, Bera P, Khambata K, Balasinor NH. Paternal obesity induces epigenetic aberrations and gene expression changes in placenta and fetus. Mol Reprod Dev. (2023) 90:109–26. doi: 10.1002/mrd.23660
99. Masuyama H, Mitsui T, Nobumoto E, Hiramatsu Y. The effects of high-fat diet exposure in utero on the obesogenic and diabetogenic traits through epigenetic changes in adiponectin and leptin gene expression for multiple generations in female mice. Endocrinology. (2015) 156:2482–91. doi: 10.1210/en.2014-2020
100. Soubry A, Murphy SK, Vansant G, He Y, Price TM, Hoyo C. Opposing epigenetic signatures in human sperm by intake of fast food versus healthy food. Front Endocrinol. (2021) 12:625204. doi: 10.3389/fendo.2021.625204
101. Soubry A, Guo L, Huang Z, Hoyo C, Romanus S, Price T, et al. Obesity-related DNA methylation at imprinted genes in human sperm: Results from the TIEGER study. Clin Epigenetics. (2016) 8:51. doi: 10.1186/s13148-016-0217-2
102. Potabattula R, Dittrich M, Schorsch M, Hahn T, Haaf T, El Hajj N. Male obesity effects on sperm and next-generation cord blood DNA methylation. PloS One. (2019) 14:e0218615. doi: 10.1371/journal.pone.0218615
103. Watkins AJ, Dias I, Tsuro H, Allen D, Emes RD, Moreton J, et al. Paternal diet programs offspring health through sperm- and seminal plasma-specific pathways in mice. Proc Natl Acad Sci U S A. (2018) 115:10064–9. doi: 10.1073/pnas.1806333115
104. Masuyama H, Hiramatsu Y. Effects of a high-fat diet exposure in utero on the metabolic syndrome-like phenomenon in mouse offspring through epigenetic changes in adipocytokine gene expression. Endocrinology. (2012) 153:2823–30. doi: 10.1210/en.2011-2161
105. Masuyama H, Hiramatsu Y. Treatment with constitutive androstane receptor ligand during pregnancy prevents insulin resistance in offspring from high-fat diet-induced obese pregnant mice. Am J Physiol Endocrinol Metab. (2012) 303:E293–300. doi: 10.1152/ajpendo.00167.2012
106. Soubry A, Schildkraut JM, Murtha A, Wang F, Huang Z, Bernal A, et al. Paternal obesity is associated with IGF2 hypomethylation in newborns: results from a Newborn Epigenetics Study (NEST) cohort. BMC Med. (2013) 11:29. doi: 10.1186/1741-7015-11-29
107. Pepin AS, Lafleur C, Lambrot R, Dumeaux V, Kimmins S. Sperm histone H3 lysine 4 tri-methylation serves as a metabolic sensor of paternal obesity and is associated with the inheritance of metabolic dysfunction. Mol Metab. (2022) 59:101463. doi: 10.1016/j.molmet.2022.101463
108. Portha B, Grandjean V, Movassat J. Mother or father: who is in the front line? Mechanisms underlying the non-genomic transmission of obesity/diabetes via the maternal or the paternal line. Nutrients. (2019) 11:233.
109. Hillier TA, Pedula KL, Schmidt MM, Mullen JA, Charles MA, Pettitt DJ. Childhood obesity and metabolic imprinting: the ongoing effects of maternal hyperglycemia. Diabetes Care. (2007) 30:2287–92. doi: 10.2337/dc06-2361
110. Morris MJ. Early life influences on obesity risk: maternal overnutrition and programming of obesity. Expert Rev Endocrinol Metab. (2009) 4:625–37. doi: 10.1586/eem.09.45
111. Vickers MH. Developmental programming and transgenerational transmission of obesity. Ann Nutr Metab. (2014) 64 Suppl 1:26–34. doi: 10.1159/000360506
112. Mameli C, Mazzantini S, Zuccotti GV. Nutrition in the first 1000 days: the origin of childhood obesity. Int J Environ Res Public Health. (2016) 13:838. doi: 10.3390/ijerph13090838
113. Szostak-Wegierek D. Intrauterine nutrition: long-term consequences for vascular health. Int J Womens Health. (2014) 6:647–56. doi: 10.2147/IJWH
114. Aiken CE, Ozanne SE. Transgenerational developmental programming. Hum Reprod Update. (2014) 20:63–75. doi: 10.1093/humupd/dmt043
115. Neel JV. Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet. (1962) 14:353–62.
116. Ramirez D, Haas SA. Windows of vulnerability: consequences of exposure timing during the dutch hunger winter. Popul Dev Rev. (2022) 48:959–89. doi: 10.1111/padr.12513
117. Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A. (2008) 105:17046–9. doi: 10.1073/pnas.0806560105
118. Argentato PP, Marchesi JAP, Dejani NN, Nakandakare PY, Teles L de F da S, Batista LPR, et al. The relationship between obesity-related H19DMR methylation and H19 and IGF2 gene expression on offspring growth and body composition. Front Nutr. (2023) 10:1170411. doi: 10.3389/fnut.2023.1170411
119. Perkins E, Murphy SK, Murtha AP, Schildkraut J, Jirtle RL, Demark-Wahnefried W, et al. Insulin-like growth factor 2/H19 methylation at birth and risk of overweight and obesity in children. J Pediatr. (2012) 161:31–9. doi: 10.1016/j.jpeds.2012.01.015
120. Hollstein T, Vinales K, Chen KY, Cypess AM, Basolo A, Schlögl M, et al. Reduced brown adipose tissue activity during cold exposure is a metabolic feature of the human thrifty phenotype. Metabolism. (2021) 117:154709. doi: 10.1016/j.metabol.2021.154709
121. Gaillard R, Durmuş B, Hofman A, Mackenbach JP, Steegers EAP, Jaddoe VWV. Risk factors and outcomes of maternal obesity and excessive weight gain during pregnancy. Obes Silver Spring Md. (2013) 21:1046–55. doi: 10.1002/oby.20088
122. Gaillard R, Welten M, Oddy WH, Beilin LJ, Mori TA, Jaddoe VWV, et al. Associations of maternal prepregnancy body mass index and gestational weight gain with cardio-metabolic risk factors in adolescent offspring: a prospective cohort study. BJOG Int J Obstet Gynaecol. (2016) 123:207–16. doi: 10.1111/1471-0528.13700
123. Yu Z, Han S, Zhu J, Sun X, Ji C, Guo X. Pre-pregnancy body mass index in relation to infant birth weight and offspring overweight/obesity: a systematic review and meta-analysis. PloS One. (2013) 8:e61627. doi: 10.1371/journal.pone.0061627
124. Godfrey KM, Barker DJ. Fetal programming and adult health. Public Health Nutr. (2001) 4:611–24. doi: 10.1079/PHN2001145
125. Gluckman PD, Hanson MA, Cooper C, Thornburg KL. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. (2008) 359:61–73. doi: 10.1056/NEJMra0708473
126. Tyrrell J, Richmond RC, Palmer TM, Feenstra B, Rangarajan J, Metrustry S, et al. Genetic evidence for causal relationships between maternal obesity-related traits and birth weight. JAMA. (2016) 315:1129–40. doi: 10.1001/jama.2016.1975
127. Chen J, Bacelis J, Sole-Navais P, Srivastava A, Juodakis J, Rouse A, et al. Dissecting maternal and fetal genetic effects underlying the associations between maternal phenotypes, birth outcomes, and adult phenotypes: A mendelian-randomization and haplotype-based genetic score analysis in 10,734 mother-infant pairs. PloS Med. (2020) 17:e1003305. doi: 10.1371/journal.pmed.1003305
128. BIRTH-GENE (BIG) Study Working Group, Huang T, Wang T, Zheng Y, Ellervik C, Li X, et al. Association of birth weight with type 2 diabetes and glycemic traits: A mendelian randomization study. JAMA Netw Open. (2019) 2:e1910915. doi: 10.1001/jamanetworkopen.2019.10915
129. Dabelea D, Mayer-Davis EJ, Lamichhane AP, D’Agostino RB, Liese AD, Vehik KS, et al. Association of intrauterine exposure to maternal diabetes and obesity with type 2 diabetes in youth: the SEARCH Case-Control Study. Diabetes Care. (2008) 31:1422–6. doi: 10.2337/dc07-2417
130. Ramsay JE, Ferrell WR, Crawford L, Wallace AM, Greer IA, Sattar N. Maternal obesity is associated with dysregulation of metabolic, vascular, and inflammatory pathways. J Clin Endocrinol Metab. (2002) 87:4231–7. doi: 10.1210/jc.2002-020311
131. Smith J, Cianflone K, Biron S, Hould FS, Lebel S, Marceau S, et al. Effects of maternal surgical weight loss in mothers on intergenerational transmission of obesity. J Clin Endocrinol Metab. (2009) 94:4275–83. doi: 10.1210/jc.2009-0709
132. Guénard F, Deshaies Y, Cianflone K, Kral JG, Marceau P, Vohl MC. Differential methylation in glucoregulatory genes of offspring born before vs. after maternal gastrointestinal bypass surgery. Proc Natl Acad Sci U S A. (2013) 110:11439–44. doi: 10.1073/pnas.1216959110
133. Jansson N, Nilsfelt A, Gellerstedt M, Wennergren M, Rossander-Hulthén L, Powell TL, et al. Maternal hormones linking maternal body mass index and dietary intake to birth weight. Am J Clin Nutr. (2008) 87:1743–9. doi: 10.1093/ajcn/87.6.1743
134. Kimyon Comert G, Esin S, Caglar GS, Yirci B, Ozdemir S, Demirtas S, et al. The correlation between birth weight and insulin-like growth factor-binding protein-1 (IGFBP-1), kisspeptin-1 (KISS-1), and three-dimensional fetal volume. J Matern-Fetal Neonatal Med Off J Eur Assoc Perinat Med Fed Asia Ocean Perinat Soc Int Soc Perinat Obstet. (2019) 32:2152–8. doi: 10.1080/14767058.2018.1427720
135. Valsamakis G, Papatheodorou DC, Margeli A, Bakoulas V, Kapantais E, Papassotiriou I, et al. First trimester maternal BMI is a positive predictor of cord blood c-peptide levels while maternal visfatin levels is a negative predictor of birth weight. Horm Athens Greece. (2014) 13:87–94. doi: 10.1007/BF03401324
136. Leus X, Watson F, Forte G. Humanitarian assistance: technical assessment and public health support for coordinated relief in the former Yugoslavia. World Health Stat Q Rapp Trimest Stat Sanit Mond. (1993) 46:199–203.
137. Godfrey KM, Sheppard A, Gluckman PD, Lillycrop KA, Burdge GC, McLean C, et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes. (2011) 60:1528–34. doi: 10.2337/db10-0979
138. Ma Z, Wang Y, Quan Y, Wang Z, Liu Y, Ding Z. Maternal obesity alters methylation level of cytosine in CpG island for epigenetic inheritance in fetal umbilical cord blood. Hum Genomics. (2022) 16:34. doi: 10.1186/s40246-022-00410-2
139. Berglind D, Müller P, Willmer M, Sinha I, Tynelius P, Näslund E, et al. Differential methylation in inflammation and type 2 diabetes genes in siblings born before and after maternal bariatric surgery. Obes Silver Spring Md. (2016) 24:250–61. doi: 10.1002/oby.21340
140. Li J, Huang J, Li JS, Chen H, Huang K, Zheng L. Accumulation of endoplasmic reticulum stress and lipogenesis in the liver through generational effects of high fat diets. J Hepatol. (2012) 56:900–7. doi: 10.1016/j.jhep.2011.10.018
141. Huang J, Ru G, Sun J, Sun L, Li Z. Elevated RIF1 participates in the epigenetic abnormalities of zygotes by regulating histone modifications on MuERV-L in obese mice. Mol Med Camb Mass. (2022) 28:17. doi: 10.1186/s10020-022-00446-z
142. Chen YT, Yang QY, Hu Y, Liu XD, de Avila JM, Zhu MJ, et al. Imprinted lncRNA Dio3os preprograms intergenerational brown fat development and obesity resistance. Nat Commun. (2021) 12:6845. doi: 10.1038/s41467-021-27171-1
143. Wang H, Chen Y, Mao X, Du M. Maternal obesity impairs fetal mitochondriogenesis and brown adipose tissue development partially via upregulation of miR-204-5p. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:2706–15. doi: 10.1016/j.bbadis.2019.07.012
144. Boudoures AL, Saben J, Drury A, Scheaffer S, Modi Z, Zhang W, et al. Obesity-exposed oocytes accumulate and transmit damaged mitochondria due to an inability to activate mitophagy. Dev Biol. (2017) 426:126–38. doi: 10.1016/j.ydbio.2017.04.005
145. Saben JL, Boudoures AL, Asghar Z, Thompson A, Drury A, Zhang W, et al. Maternal Metabolic Syndrome Programs Mitochondrial Dysfunction via Germline Changes across Three Generations. Cell Rep. (2016) 16:1–8. doi: 10.1016/j.celrep.2016.05.065
146. Chao S, Lu J, Li LJ, Guo HY, Xu KP, Wang N, et al. Maternal obesity may disrupt offspring metabolism by inducing oocyte genome hyper-methylation via increased DNMTs [Internet]. bioRxiv. (2024), 2024.04.24.590938. doi: 10.1101/2024.04.24.590938v1
147. Goldstein JA, Gallagher K, Beck C, Kumar R, Gernand AD. Maternal-fetal inflammation in the placenta and the developmental origins of health and disease. Front Immunol. (2020) 11:531543. doi: 10.3389/fimmu.2020.531543
148. Jones HN, Woollett LA, Barbour N, Prasad PD, Powell TL, Jansson T. High-fat diet before and during pregnancy causes marked up-regulation of placental nutrient transport and fetal overgrowth in C57/BL6 mice. FASEB J Off Publ Fed Am Soc Exp Biol. (2009) 23:271–8. doi: 10.1096/fj.08-116889
149. Rosario FJ, Kanai Y, Powell TL, Jansson T. Increased placental nutrient transport in a novel mouse model of maternal obesity with fetal overgrowth. Obes Silver Spring Md. (2015) 23:1663–70. doi: 10.1002/oby.21165
150. Garcia-Santillan JA, Lazo-de-la-Vega-Monroy ML, Rodriguez-Saldaña GC, Solis-Barbosa MA, Corona-Figueroa MA, Gonzalez-Dominguez MI, et al. Placental nutrient transporters and maternal fatty acids in SGA, AGA, and LGA newborns from mothers with and without obesity. Front Cell Dev Biol. (2022) 10:822527. doi: 10.3389/fcell.2022.822527
151. Jansson N, Rosario FJ, Gaccioli F, Lager S, Jones HN, Roos S, et al. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J Clin Endocrinol Metab. (2013) 98:105–13. doi: 10.1210/jc.2012-2667
152. Merech F, Hauk V, Paparini D, Fernandez L, Naguila Z, Ramhorst R, et al. Growth impairment, increased placental glucose uptake and altered transplacental transport in VIP deficient pregnancies: Maternal vs. placental contributions. Biochim Biophys Acta Mol Basis Dis. (2021) 1867:166207. doi: 10.1016/j.bbadis.2021.166207
153. Lim MA, Stack CM, Cuasay K, Stone MM, McFarlane HG, Waschek JA, et al. Regardless of genotype, offspring of VIP-deficient female mice exhibit developmental delays and deficits in social behavior. Int J Dev Neurosci Off J Int Soc Dev Neurosci. (2008) 26:423–34. doi: 10.1016/j.ijdevneu.2008.03.002
154. Page L, Younge N, Freemark M. Hormonal determinants of growth and weight gain in the human fetus and preterm infant. Nutrients. (2023) 15:4041. doi: 10.3390/nu15184041
155. Chavira-Suárez E, Reyes-Castro LA, López-Tenorio II, Vargas-Hernández L, Rodríguez-González GL, Chavira R, et al. Sex-differential RXRα gene methylation effects on mRNA and protein expression in umbilical cord of the offspring rat exposed to maternal obesity. Front Cell Dev Biol. (2022) 10:892315. doi: 10.3389/fcell.2022.892315
156. Rasool A, Mahmoud T, Mathyk B, Kaneko-Tarui T, Roncari D, White KO, et al. Obesity downregulates lipid metabolism genes in first trimester placenta. Sci Rep. (2022) 12:19368. doi: 10.1038/s41598-022-24040-9
157. Rabhi N, Hannou SA, Froguel P, Annicotte JS. Cofactors as metabolic sensors driving cell adaptation in physiology and disease. Front Endocrinol. (2017) 8:304. doi: 10.3389/fendo.2017.00304
158. Manikkam M, Tracey R, Guerrero-Bosagna C, Skinner MK. Plastics derived endocrine disruptors (BPA, DEHP and DBP) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. PloS One. (2013) 8:e55387. doi: 10.1371/journal.pone.0055387
159. Brulport A, Le Corre L, Maquart G, Barbet V, Dastugue A, Severin I, et al. Multigenerational study of the obesogen effects of bisphenol S after a perinatal exposure in C57BL6/J mice fed a high fat diet. Environ pollut Barking Essex 1987. (2021) 270:116243. doi: 10.1016/j.envpol.2020.116243
160. Kubsad D, Nilsson EE, King SE, Sadler-Riggleman I, Beck D, Skinner MK. Assessment of glyphosate induced epigenetic transgenerational inheritance of pathologies and sperm epimutations: generational toxicology. Sci Rep. (2019) 9:6372. doi: 10.1038/s41598-019-42860-0
161. Skinner MK, Manikkam M, Tracey R, Guerrero-Bosagna C, Haque M, Nilsson EE. Ancestral dichlorodiphenyltrichloroethane (DDT) exposure promotes epigenetic transgenerational inheritance of obesity. BMC Med. (2013) 11:228. doi: 10.1186/1741-7015-11-228
162. Manikkam M, Haque MM, Guerrero-Bosagna C, Nilsson EE, Skinner MK. Pesticide methoxychlor promotes the epigenetic transgenerational inheritance of adult-onset disease through the female germline. PloS One. (2014) 9:e102091. doi: 10.1371/journal.pone.0102091
163. Chamorro-Garcia R, Diaz-Castillo C, Shoucri BM, Käch H, Leavitt R, Shioda T, et al. Ancestral perinatal obesogen exposure results in a transgenerational thrifty phenotype in mice. Nat Commun. (2017) 8:2012. doi: 10.1038/s41467-017-01944-z
164. Knecht AL, Truong L, Marvel SW, Reif DM, Garcia A, Lu C, et al. Transgenerational inheritance of neurobehavioral and physiological deficits from developmental exposure to benzo[a]pyrene in zebrafish. Toxicol Appl Pharmacol. (2017) 329:148–57. doi: 10.1016/j.taap.2017.05.033
165. Tracey R, Manikkam M, Guerrero-Bosagna C, Skinner MK. Hydrocarbons (jet fuel JP-8) induce epigenetic transgenerational inheritance of obesity, reproductive disease and sperm epimutations. Reprod Toxicol Elmsford N. (2013) 36:104–16. doi: 10.1016/j.reprotox.2012.11.011
166. Kim JH, Sartor MA, Rozek LS, Faulk C, Anderson OS, Jones TR, et al. Perinatal bisphenol A exposure promotes dose-dependent alterations of the mouse methylome. BMC Genomics. (2014) 15:30. doi: 10.1186/1471-2164-15-30
167. Kochmanski JJ, Marchlewicz EH, Cavalcante RG, Perera BPU, Sartor MA, Dolinoy DC. Longitudinal effects of developmental bisphenol A exposure on epigenome-wide DNA hydroxymethylation at imprinted loci in mouse blood. Environ Health Perspect. (2018) 126:077006. doi: 10.1289/EHP3441
168. Junge KM, Leppert B, Jahreis S, Wissenbach DK, Feltens R, Grützmann K, et al. MEST mediates the impact of prenatal bisphenol A exposure on long-term body weight development. Clin Epigenetics. (2018) 10:58. doi: 10.1186/s13148-018-0478-z
169. Li Y, Duan F, Zhou X, Pan H, Li R. Differential responses of GC−1 spermatogonia cells to high and low doses of bisphenol A. Mol Med Rep. (2018) 18:3034–40. doi: 10.3892/mmr
170. Ding ZM, Jiao XF, Wu D, Zhang JY, Chen F, Wang YS, et al. Bisphenol AF negatively affects oocyte maturation of mouse in vitro through increasing oxidative stress and DNA damage. Chem Biol Interact. (2017) 278:222–9. doi: 10.1016/j.cbi.2017.10.030
171. Chamorro-García R, Sahu M, Abbey RJ, Laude J, Pham N, Blumberg B. Transgenerational inheritance of increased fat depot size, stem cell reprogramming, and hepatic steatosis elicited by prenatal exposure to the obesogen tributyltin in mice. Environ Health Perspect. (2013) 121:359–66. doi: 10.1289/ehp.1205701
172. Sun W, Dong H, Becker AS, Dapito DH, Modica S, Grandl G, et al. Cold-induced epigenetic programming of the sperm enhances brown adipose tissue activity in the offspring. Nat Med. (2018) 24:1372–83. doi: 10.1038/s41591-018-0102-y
173. Thum C, Cookson AL, Otter DE, McNabb WC, Hodgkinson AJ, Dyer J, et al. Can nutritional modulation of maternal intestinal microbiota influence the development of the infant gastrointestinal tract? J Nutr. (2012) 142:1921–8. doi: 10.3945/jn.112.166231
174. Browne HP, Shao Y, Lawley TD. Mother-infant transmission of human microbiota. Curr Opin Microbiol. (2022) 69:102173. doi: 10.1016/j.mib.2022.102173
175. Li H t, Zhou Y b, Liu J m. The impact of cesarean section on offspring overweight and obesity: a systematic review and meta-analysis. Int J Obes. (2013) 37:893–9. doi: 10.1038/ijo.2012.195
176. Abraham MJ, El Sherbini A, El-Diasty M, Askari S, Szewczuk MR. Restoring epigenetic reprogramming with diet and exercise to improve health-related metabolic diseases. Biomolecules. (2023) 13:318. doi: 10.3390/biom13020318
177. Petridou A, Siopi A, Mougios V. Exercise in the management of obesity. Metabolism. (2019) 92:163–9. doi: 10.1016/j.metabol.2018.10.009
178. Son JS, Liu X, Tian Q, Zhao L, Chen Y, Hu Y, et al. Exercise prevents the adverse effects of maternal obesity on placental vascularization and fetal growth. J Physiol. (2019) 597:3333–47. doi: 10.1113/JP277698
179. Mangwiro YTM, Cuffe JSM, Mahizir D, Anevska K, Gravina S, Romano T, et al. Exercise initiated during pregnancy in rats born growth restricted alters placental mTOR and nutrient transporter expression. J Physiol. (2019) 597:1905–18. doi: 10.1113/JP277227
180. Kusuyama J, Alves-Wagner AB, Conlin RH, Makarewicz NS, Albertson BG, Prince NB, et al. Placental superoxide dismutase 3 mediates benefits of maternal exercise on offspring health. Cell Metab. (2021) 33:939–956.e8. doi: 10.1016/j.cmet.2021.03.004
181. Kusuyama J, Alves-Wagner AB, Makarewicz NS, Goodyear LJ. Effects of maternal and paternal exercise on offspring metabolism. Nat Metab. (2020) 2:858–72. doi: 10.1038/s42255-020-00274-7
182. Moholdt T, Stanford KI. Exercised breastmilk: a kick-start to prevent childhood obesity? Trends Endocrinol Metab TEM. (2024) 35:23–30. doi: 10.1016/j.tem.2023.08.019
183. Cilar Budler L, Budler M. Physical activity during pregnancy: a systematic review for the assessment of current evidence with future recommendations. BMC Sports Sci Med Rehabil. (2022) 14:133. doi: 10.1186/s13102-022-00524-z
184. Ding L, Liu J, Zhou L, Xiao X. Maternal exercise impacts offspring metabolic health in adulthood: A systematic review and meta-analysis of animal studies. Nutrients. (2023) 15:2793. doi: 10.3390/nu15122793
185. Tchang BG, Aras M, Kumar RB, Aronne LJ. Pharmacologic Treatment of Overweight and Obesity in Adults. In: Feingold KR, Anawalt B, Blackman MR, Boyce A, Chrousos G, Corpas E, et al, editors. Endotext. (2019) MDText.com, Inc, South Dartmouth (MA (2000). Available at: http://www.ncbi.nlm.nih.gov/books/NBK279038/.
186. Garvey WT, Batterham RL, Bhatta M, Buscemi S, Christensen LN, Frias JP, et al. Two-year effects of semaglutide in adults with overweight or obesity: the STEP 5 trial. Nat Med. (2022) 28:2083–91. doi: 10.1038/s41591-022-02026-4
Keywords: obesity, maternal obesity, paternal obesity, transgenerational, intergenerational, epigenetics
Citation: Sivakumar S, Lama D and Rabhi N (2024) Childhood obesity from the genes to the epigenome. Front. Endocrinol. 15:1393250. doi: 10.3389/fendo.2024.1393250
Received: 28 February 2024; Accepted: 25 June 2024;
Published: 09 July 2024.
Edited by:
Deanne Helena Hryciw, Griffith University, AustraliaReviewed by:
Erika Chavira-Suárez, National Autonomous University of Mexico, MexicoCopyright © 2024 Sivakumar, Lama and Rabhi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nabil Rabhi, cmFiaGlAYnUuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.