
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol., 23 May 2024
Sec. Reproduction
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1393111
This article is part of the Research TopicNon-Invasive Biomarkers for Sperm Retrieval in Non-Obstructive PatientsView all 6 articles
 Haohui Xu1,2†
Haohui Xu1,2† Yixin Zhang1,2†
Yixin Zhang1,2† Caiqin Wang1,2†Zhuoyan Fu1,3Jing Lv1,3Yufang Yang1,4Zihan Zhang1,2Yuanmin Qi1,3
Caiqin Wang1,2†Zhuoyan Fu1,3Jing Lv1,3Yufang Yang1,4Zihan Zhang1,2Yuanmin Qi1,3 Kai Meng1*
Kai Meng1* Jinxiang Yuan1*
Jinxiang Yuan1* Xiaomei Wang5*
Xiaomei Wang5*Non-obstructive azoospermia (NOA) is a disease characterized by spermatogenesis failure and comprises phenotypes such as hypospermatogenesis, mature arrest, and Sertoli cell-only syndrome. Studies have shown that FA cross-linked anemia (FA) pathway is closely related to the occurrence of NOA. There are FA gene mutations in male NOA patients, which cause significant damage to male germ cells. The FA pathway is activated in the presence of DNA interstrand cross-links; the key step in activating this pathway is the mono-ubiquitination of the FANCD2-FANCI complex, and the activation of the FA pathway can repair DNA damage such as DNA double-strand breaks. Therefore, we believe that the FA pathway affects germ cells during DNA damage repair, resulting in minimal or even disappearance of mature sperm in males. This review summarizes the regulatory mechanisms of FA-related genes in male azoospermia, with the aim of providing a theoretical reference for clinical research and exploration of related genes.
Male infertility is a common reproductive disease that affects multiple families eager to create a new life. Existing research statistics have found that approximately seven out of every 100 males suffer from infertility symptoms (1). Of these, 13-17% of infertile men are due to azoospermia, which accounts for 55-65% of non-obstructive azoospermia (2). It is generally believed that the main cause of male infertility may be abnormal reduction in sperm count or even azoospermia caused by factors such as congenital inheritance or acquired living environment (3). Azoospermia is an extreme form of decreased sperm count during ejaculation, divided into two types: obstructive azoospermia (OA) and non-obstructive azoospermia (NOA) (4). The viability of sperm is closely related to the health status of the testes, and NOA is the result of abnormal gene expression in the testes. The entire ejaculatory duct of NOA is open and functioning normally, but the number of healthy sperm is very low, which may be related to genetic mutations (5). Interstrand cross-linking (ICL) has a direct effect on the proliferation of male germ cells and sperm maturation, and the error repair of ICL will lead to the production of NOA (6). Fanconi anemia (FA) is a chromosomally inherited disease caused by mutations in the FA genes. It affects the same signaling pathway through multiple sets of genes and is highly sensitive to damage of intracellular genetic material (7). Studies have found that the deletion of FA gene is closely related to reproductive diseases, especially interference with primordial germ cells (PGCs), including ovarian and breast cancer in women and azoospermia in men (8–10). Some of the genes in the FA pathway are important for the repair process of ICL of DNA (11). In this review, we focus on exploring the relationship between FA genes and male azoospermia. For example, Krausz et al. (12)reported a high correlation between NOA and the pathogenic variation of the Fanconi gene in the FA pathway; Tsui et al. (13)also suspected that homozygous mutations in FANCM can lead to the occurrence of NOA. The association between the Fanconi gene and male NOA deserves further research, such as whether abnormal DNA damage and calcium signaling abnormalities in FA have an impact on sperm formation and maturation (14). Based on the relevant research results in recent years, this review summarizes the molecular-level interaction mechanism and relationship between the specific genes of FA and NOA, aiming to provide helpful insights for the treatment of azoospermia and research on FA from a genetic perspective.
The FA pathway is associated with genetic disorders that primarily play a role in DNA replication (11). DNA replication includes processes such as unwinding, catalysis by DNA polymerase, and DNA ligase connecting Okazaki fragments, and replication is carried out in a semiconservative replication manner (15). This process requires the collaboration of multiple enzymes and is prone to errors. Damaged DNA needs to be repaired or cleared in a timely manner, and FA signals are involved in this process (15, 16). There are currently 22 known genes related to the FA pathway (16): FANCA (17), FANCB (18), FANCC (19), FANCD1 (BRCA2) (20), FANCD2 (21), FANCE (22), FANCF (21), FANCG (XRCC9) (17, 23), FANCI (21), FANCJ (BRIP1/BACH1) (24), FANCL (25), FANCM (26), FANCN (PALB2) (27), FANCO (RAD51C) (28), FANCP (SLX4) (29), FANCQ (ERCC4) (30, 31), FANCR (RAD51) (31), FANCS (BRCA1) (32), FANCT (UBE2T) (25), FANCU (XRCC2) (33), FANCV (REV7) (34) and FANCW (35, 36) (Table 1). Research has shown that the FA pathway can be classified upstream, middle, and downstream of FANCD2 ubiquitination: Upstream is mediated by a complex enzyme composed of FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCI, FANCL and FANCM (110). The ubiquitination of FANCD2 by these complex results from the activation of FANCT by FANCL, and FANCD2 often binds to FANCI, resulting in the ubiquitination of FANCD2-FANCI (ID2) complex (111–113). The ubiquitination of the ID2 complex induces processes such as cleavage and homologous recombination of endonucleases, leading to de-ubiquitination and ultimately completing DNA damage repair (114, 115) (Figure 1). In the FA pathway, signaling networks formed by protein complex enzymes such as FANCA can dynamically monitor the process of DNA changes (116). Damage or termination of DNA replication forks can activate upstream FA proteins, thereby regulating downstream genes such as FANCP to correct DNA damage and restore normal replication process (116). One of the most important links connecting the upstream and downstream FA signaling pathways is the ubiquitination of FANCI-FANCD2 (116). The FRT-flanked neomycin cassette can target the FANCG gene in mouse embryonic stem cells, causing the defect of this gene and further blocking the protein signal network that binds FANCG to the SH3 domain of RIISp, and the DNA repair of embryonic stem cells cannot be completed (117, 118). Embryonic stem cells can differentiate into germ cells, so blocking the FNACG signaling pathway may reduce the number of sperm in mice (117–119). In addition, exogenous stimuli such as cisplatin and mitomycin C lead to the production of cellular ICL, where the FA protein recognizes and repairs damaged DNA or excises faulty coding (120). When the FA gene is missing, the damaged DNA accumulates in the cell, The resulting self-nucleic acids activate the cyclic GMP-AMP synthase (cGAS) in the cytoplasm to produce secondary messenger cGAMP, which in turn activates type I interferons (IFN-I), which is involved in the emergence of Fanconi related disease phenotypes via the cGAS-STING-IFN-I axis (120, 121). It can be seen that FA, as a signal pathway of target gene, is closely related to DNA damage repair. In addition, studies have shown that abnormal DNA repair can lead to the occurrence of NOA (6). Taking FA signaling pathway as the starting point, we will elaborate on the effects and possible roles of various FA genes in male NOA according to the upstream, middle, and downstream processes of FANCD2 ubiquitination (Figure 2).
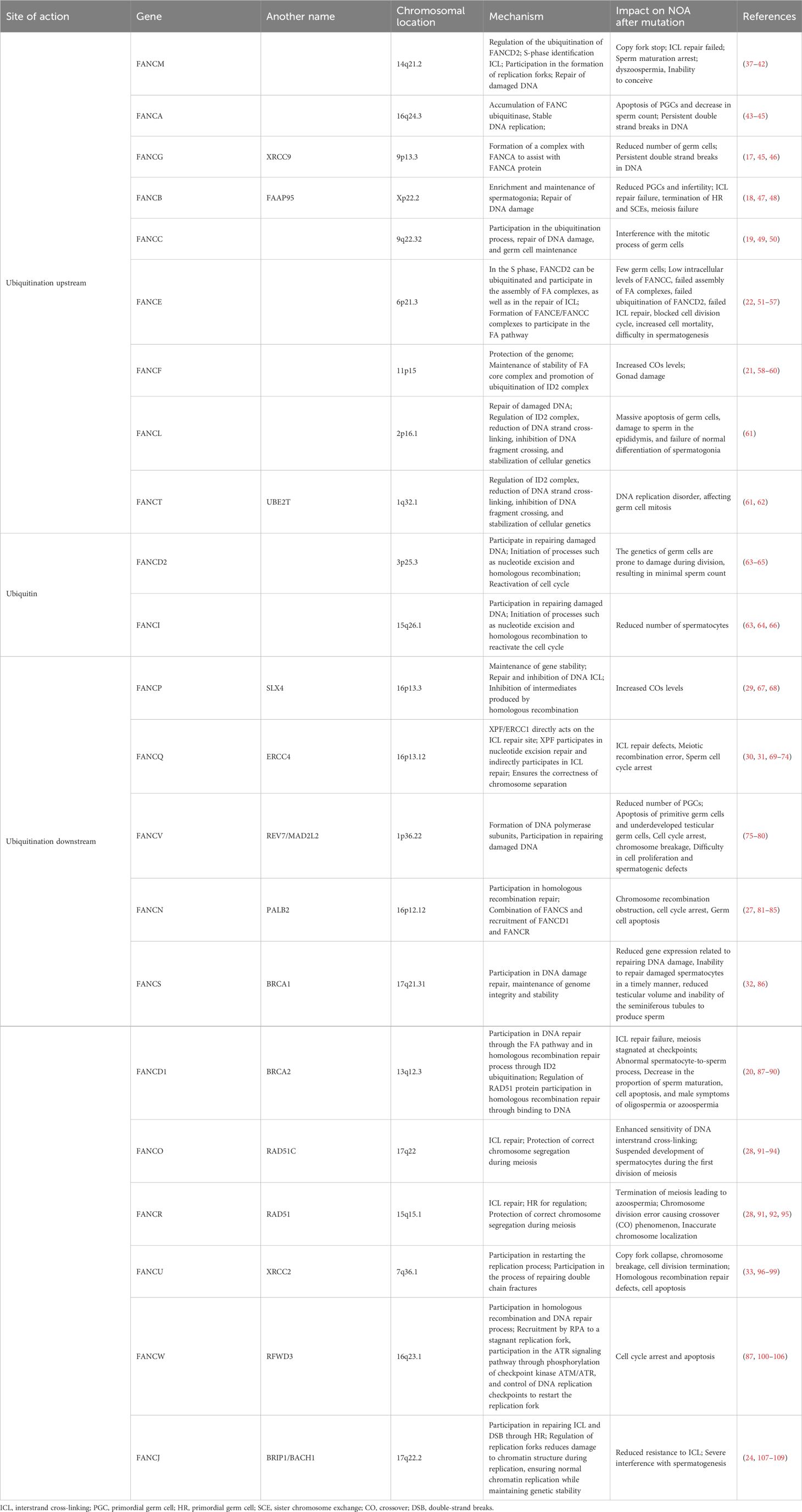
Table 1 The position of human FA gene and its mechanism and influence on NOA.
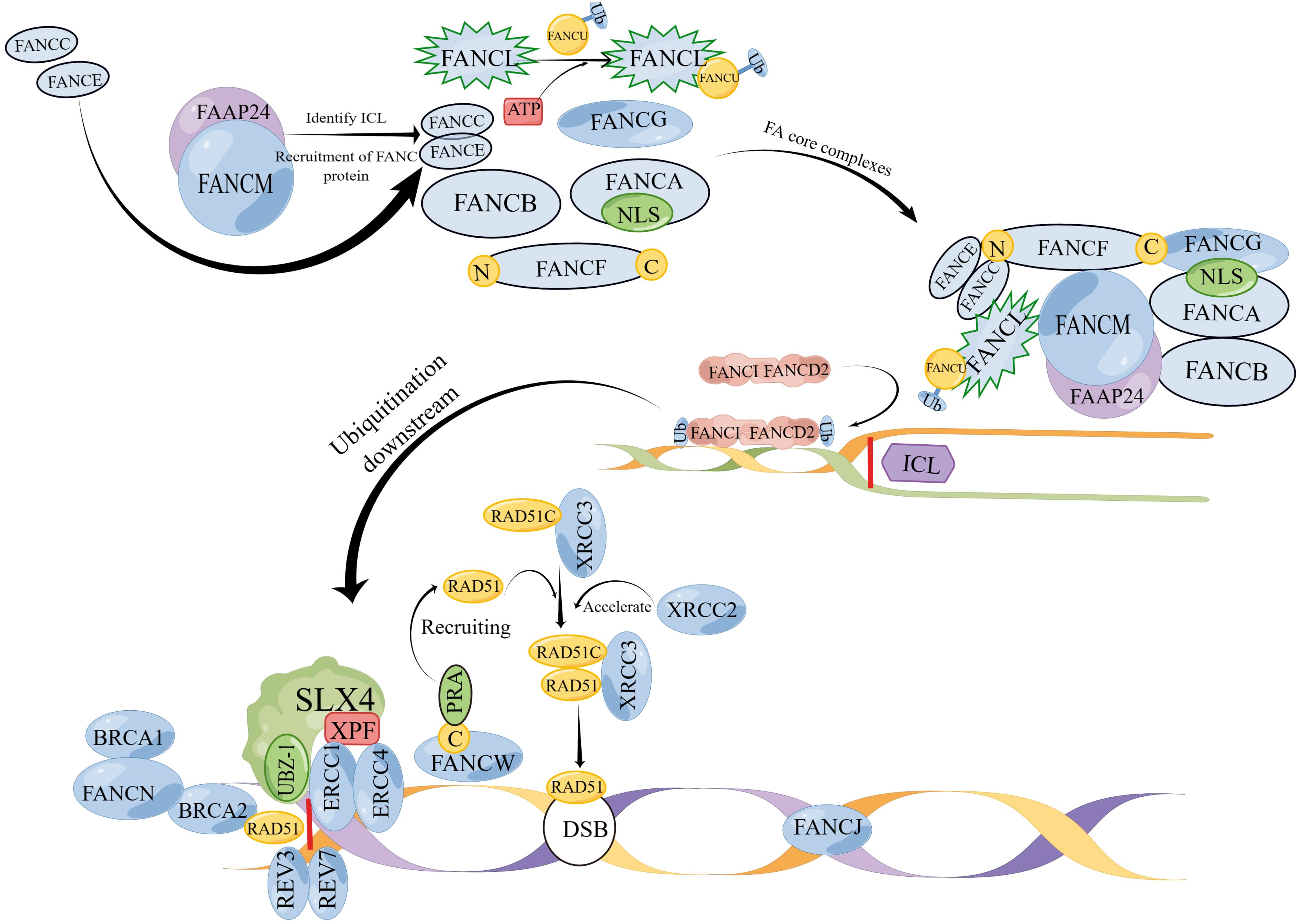
Figure 1 (By Figdraw) A model of the mechanism of action of FA genes. The FA gene action sites are divided into three parts: upper, middle, and lower. Proteins such as FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCI, FANCL and FANCM that exist upstream combine to form FA core complexes; ubiquitination of the ID2 complex located in the middle reaches is the main step in repairing DNA damage such as ICL, and the ID2 complex is the main pathway for activating downstream; the downstream protein SLX4 (FANCP) catalyzes ICL decoupling and binding to ERCC4 (FANCQ) through the UBZ-1 domain. REV7 (FANCV) and REV3 form a DNA polymerase complex for ICL repair. Under the action of FANCN, BRCA1 (FANCS) binds to BRCA2 (FANCD1) and acts together with RAD51 (FANCR) to repair DSB. RAD51C binds to XRCC3 to form a complex and is dependent on RAD51 to maintain normal division.However, XRCC2 (FANCU) forms a complex with RAD51 and has replication restart effect, which can repair homologous recombination. The C end of FANCW binds to PRA and is recruited by it to restart the replication fork. RAD51 (FANCR) can be used to repair DNA double-strand break. FANCJ repairs DNA interstrand cross-linking through homologous recombination (HR). (Note: The diagram is not the actual spatial configuration of the FA core complex, it is only a schematic diagram).
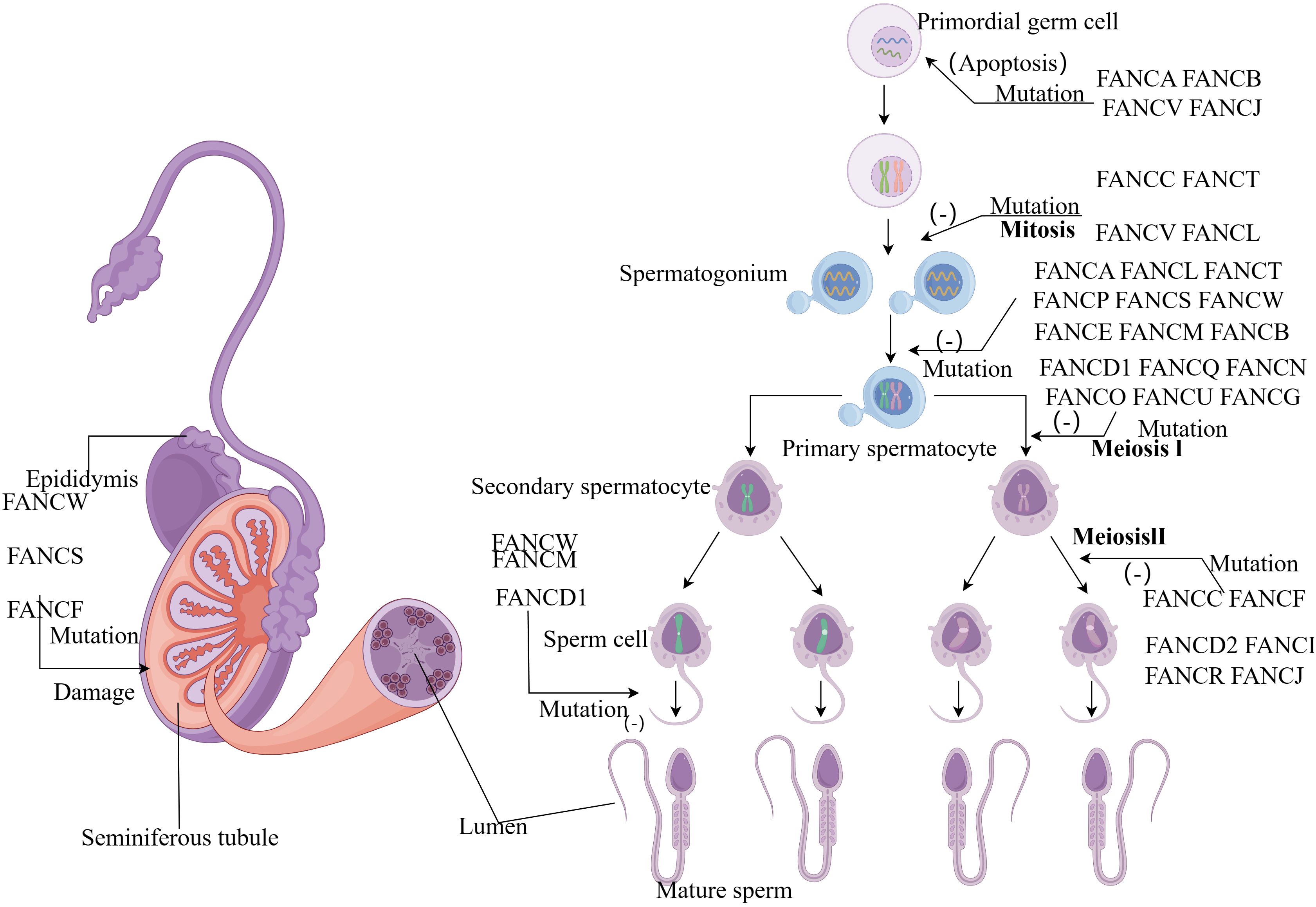
Figure 2 (By Figdraw) Schematic diagram of the influence of FA gene on sperm development process. FA genes affect almost every stage from primitive germ cells to sperm development. Mutations in the FANCA, FANCB, FANCV and FANCJ gene can cause apoptosis in primordial germ cells. During the mitotic stage, FANCC, FANCT, FANCV and FANCL mutations prevent the formation of spermatogonia, and a group of genes also play an important role in the formation of primary spermatocytes. Most genes affect the meiotic stage, while FANCD1, FANCM and FANCW mutant spermatocytes cannot form mature sperm.
In physiological conditions, DNA replication is more common in poorly differentiated embryonic stem cells with the ability to divide (122). ICL is a type of DNA damage that poses great harm during DNA replication, known as the obstruction of replication forks, which has a significant effect on the development of germ cells (123). ICL can terminate normal DNA unwinding, causing replication and transcription abnormalities and leading to meiosis arrest, chromosome breakage, and abnormal chromosome binding, significantly damaging the cell’s proliferative ability and causing abnormal germ cell development (124, 125). Zhang et al. found that the Sertoli cell-only syndrome phenotype of NOA may be related to ICL repair defects and is associated with DNA replication in germ cells (126). In the upstream of the FA signaling pathway, mutations in the FANCM/A/G/B/C gene have been shown to be associated with human NOA (12, 46, 47, 49, 127). Among them, FANCM defect will cause the FA core complex enzyme in the cell to be unable to recognize ICL in time, and the deletion of FANCA/G/B/C gene will cause the FA core complex enzyme to be unable to associate with FANCD2 ubiquitination, resulting in abnormal repair of ICL (17, 18, 37, 50). ICL persists and interferes with the normal replication of DNA, eventually leading to the proliferation of male germ cells and a decrease in male sperm count (124, 125). The ubiquitination of FANCD2 is closely related to the repair of ICL, and FANCD2 mutations occur at the K561 site in patients with FA. Due to genetic mutations, FANCD2 is unable to complete ubiquitination (51). Failure of the FANCD2 ubiquitination process causes cells to stall in the S phase (51), unable to produce mature sperm. At the same time, in the downstream of the FA pathway, homologous recombination mediated by FANCN/FANCU and other proteins is involved in the subsequent ICL repair process, and the defects of related genes lead to the reduction of sperm quantity and quality (27, 51, 81, 95, 96). The essence of NOA is the reduction of sperm count, which is often caused by the failure of migration of PGCs (128) and abnormal mitosis and meiosis of PGCs (8). For men, a defect in the repair of DNA cross-linking between strands during the formation of sperm can cause problems in the gene expression of PGCs, leading to defects in the sperm and even the inability to form mature sperm, leading to NOA or oligospermia (6). Therefore, the FA signaling pathway is related to the occurrence of NOA, and the defects of related genes will hinder the repair process of ICL and eventually lead to the occurrence of NOA.
The expression product of FANCM is a protein comprising the FA core complex. The FANCM protein has seven domains: aa 5-12, aa 77-590, aa 661-800, aa 826-967, aa 1218-1251, aa 1818-1956, and aa 1971-2030. Each domain is involved in different stages of FA and has genetic significance for the formation of NOA (26, 129). According to reports, FANCM mutants are present in patients with NOA (129). Kherraf et al. analyzed 151 genes using whole-exome sequencing and found a homozygous mutation in the FANCM gene of patients with NOA (NM_020937.4: c.5791C>T; p.Arg1931Ter) (127). Previous studies have shown that after truncated FANCM mutations, the major histone fold binding domain, MM1 (aa 826-967) domain, MM2 (aa 1218-1251) domain, and ERCC4 (aa 1818-1956) domain cannot express the corresponding proteins, leading to spermatogenesis disorders and infertility (38, 39). Furthermore, FANCM can regulate the ubiquitination process of FANCD2 and play an important role in DNA repair. Studies have shown that mutations in the DEAH (aa 77-590), MM1 (aa 826-967), and FANCM-PIPbox (aa 5-12) domains of FANCM can lead to the inability to recruit FA complexes and FANCD2 to undergo ubiquitination, leading to the disruption of DNA repair processes (38, 40, 41), thereby blocking the proliferation of germ cells. Yin et al. found that adult mouse sperm inside the testicles with FANCM mutations were lost and sperm maturation was delayed (42), which may be related to DNA damage, such as FANCM binding to FAAP24 protein at the termination of a replication fork and recognition of ICL in the S phase (37). Therefore, during cell division, FANCM mutations may lead to the formation of FA complexes and the inability of FANCD2 to complete ubiquitination, resulting in the inability to repair ICL. Ultimately, the number of sperm in seminiferous tubules may decrease because of the inability of spermatogonia cells to proliferate and mature, leading to the occurrence of NOA.
The FANCA gene is currently one of the genes generating the most interest (43), along with FANCG, also known as XRCC9 (23). FANCA is an important gene for repairing DNA breakage and is mainly expressed in the testes (130). The complex composed of two gene-encoded proteins is one of the main elements involved in the formation of ubiquitination complex enzymes (17). FANCA uses the amino terminal nuclear localization signal to bind to the FANCG protein to increase the content of the FANCA protein, leading to the accumulation of FANC ubiquitinase in the nucleus (44), which is conducive to stable DNA replication. In contrast, the FANCG protein can assist the FANCA protein in DNA repair (17). Krausz et al. found that pathogenic mutations in FANCA lead to NOA (12). They sequenced two types of patients and found that the first type had c.2639G>A mutations, while the second type had two types of mutations, [NM_000135.2: c.3788-3790delTCT, p.Phe1263del] and [NM_000135.2: c.3913C>T, p.Leu1305Phe], both of which had significant pathogenicity (12). Simultaneously, studies have found that FANCA mutated and showed high expression (12) after biopsy of tissues from patients with NOA (131). Furthermore, Wong et al. found that under the action of FANCAtm1.1Hsc homozygotes, the number PGCs may decrease due to their inability to survive or divide (43). Simultaneously, the number of errors during meiosis in spermatocytes and apoptosis in spermatocytes increase, ultimately resulting in almost zero sperm count in the vas deferens (43). After mutation of the FANCG gene, PGCs are blocked during the S phase, resulting in a decrease in the number of PGCs and can interfere with the migration process of PGCs to the testes through Rac1 (46). After a FANCA or FANCG defect mutation, double stranded breaks in DNA become difficult to repair (45). The FANCA-FANCG complex can interact with each other, and the phenotype of the double mutation is consistent with that of FANCA or FANCG single mutation, but with a higher degree of harm (132). From this, it can be seen that the FANCA-FANCG complex has a regulatory effect on DNA interstrand cross-linking repair and can lead to NOA by affecting the cell proliferation cycle, especially the meiosis process of sperm.
The FANCB gene is currently the only known FA gene located on the X chromosome. However most current research focuses on the relationship between this gene and female ovaries, breasts, and other aspects and the research on male azoospermia is scarce. The effects of FANCB on male reproduction can be attributed to two pathways. The first pathway is for FANCB can accumulate in spermatogonia and participate in the maintenance of undifferentiated spermatogonia (47, 48). Kato et al. found that the number of PGCs in male Fancb-mutant mice was significantly reduced compared with that in the control group, and infertility occurred (47). As PGCs decreased, the number of spermatogonial cells that divided into spermatogonia also decreased, leading to a decrease in sperm count and the occurrence of NOA. The second pathway is that FANCB can participate in DNA damage repair, ensuring normal meiosis. Studies have shown that the FA core complex cannot ubiquitinate FANCD2 because of the mutant FANCB, which leads to the failure of ICL repair. This prevents normal homologous recombination (HR) and sister chromosome exchange (SCEs), leading to failure of division (18). Therefore, the FANCB mutation decreases the number of spermatogonia in male testes and may reduce the success rate of meiosis by hindering the repair of ICL and meiotic recombination process, inducing NOA.
The expression product of the FANCC gene exists in the cytoplasm and nuclear compartments (133). As a core complex of FA, the function of FANCC to repair DNA damage can maintain the activity of sperms and ensure their survival (19). FANCC does not change during the cell cycle, but there is a certain degree of dependent regulation (133). Nadler et al. found that mutations in the FANCC gene can interfere with the mitotic process of PGCs, and in males, these exhibit infertility (49). FANCC participates in the ubiquitination process of the FA core complex (50). A mutation in this gene will result in the inability of the FA core complex to ubiquitinate, inactivating the FA pathway and preventing it from participating in replication fork stability and cytoplasmic division, which may further lead to the inability of primordial germ cells to undergo mitosis and meiosis. The division of primordial germ cells is impaired, preventing the formation of mature sperm. In experiments in mice, mice with Fancc gene mutations experienced a significant decrease in the number of sperms within more than ten days (49). PGCs are precursors of sperm, and a decrease in PGCs indicates a corresponding decrease in the number of subsequently produced sperm (134). Low sperm count leads to a series of male infertility diseases such as NOA.
The product of the FANCE gene is a member of the FA core complex (50), which is mainly involved in ICL repair. Fu et al. (52) reported very few spermatocytes in the Fance mutant mouse model, and that spermatocytes are directly related to spermatogenesis. Therefore, similar to NOA, Fance deficiency significantly reduces the sperm count, leading to impaired reproductive function and infertility symptoms in male mice (52, 135). Research has shown that FANCE participates in ICL repair through both direct and indirect pathways. In the direct pathway, FANCE can ubiquitinate FANCD2 in the S phase and participate in the assembly of FA complexes (51, 53). Then, FANCD2 locates the DNA damage site and recruits FA protein for ICL repair (51). In the indirect pathway, FANCE and FANCC participate in each other’s nuclear accumulation and form the FANCE/FANCC complex regulated by FANCF (54)to participate in the FA pathway (55, 56). After FANCE mutation, FANCC cannot accumulate in the nucleus, FA complex cannot assemble (22), and FANCD2 cannot complete ubiquitination, ultimately leading to ICL repair failure. Data shows that FANCE mutations can arrest the cell division cycle (57). Loss of FANCE leads to increase cell mortality rate, and cause difficulty in spermatogenesis, often resulting in NOA (52, 57). Simultaneously, cell cycle arrest can affect the development of spermatogonial stem cells (136). As the earliest sperm cells, spermatogonial stem cells are of great significance in male reproduction. Therefore, FANCE deficiency can lead to cell cycle arrest, affect spermatocyte and sperm development, and lead to NOA.
FANCF mainly exists in the nucleus and is an important component of the core complex that constitutes the FA pathway, playing a protective role on the genome (21). FANCF is an adapter protein and has two relatively stable regions located at the N-terminus and C-terminus. The C-terminal binding of FANCG to FANCF seems more stable, while the FANCC/FANCE complex can bind to the N-terminal of FANCF (54). In this way, FANCF protein tightly binds to other core complex components to maintain the stability of the FA core complex and promote the ubiquitination of the ID2 complex (58). Compared with other Fanconi genes, many mechanisms of FANCF are not yet clear. Previous studies have shown that FANCF mutations can cause instability in the FA genome, damaging the gonads (59). According to relevant data (137), FANCF defects can cause testicular lesions and testicular cancer. Spermatogonia develop into sperm in the seminiferous tubules within the testicles (138) (139),. Therefore, mutations in FANCF can further damage the seminiferous tubules by causing testicular damage and making them unable to provide a place for sperm production, resulting in immature sperm development and ultimately NOA. Furthermore, Mutations in FANCF protect the crossover phenomenon of DNA, making it difficult to repair its function and adversely affecting fertility (140). Therefore, the deletion of the FANCF gene not only causes testicular lesions, causing difficulty in spermatogenesis, but also affects seminiferous tubules, leading to NOA.
FANCL encodes the FA core complex protein E3 ubiquitin ligase (141), and its encoded product plays an important role in DNA damage repair. FANCT, also known as UBE2T, encodes E2 ubiquitin-conjugating enzyme (142). The complex formed by FANCL/FANCT (E2-E3) (25) is the only known complex that directly regulates FANCD2-FANCI (ID2) and positively affects the ubiquitination of ID2 complexes. This complex can reduce the DNA interstrand cross-links to a certain extent, inhibit DNA segment crossovers that occur in some cells during DNA replication, and ensure the stability of cellular genetics (61). FANCD2, as a key factor in activating the FA pathway, appears around the nucleus of S-phase PGCs in normal development, indicating a correlation between the FA pathway and fertility (62). Although there are not many reports on these two genes in NOA diseases, we can speculate based on the above research results that UBE2T mutation or knockout will result in disorder in the PGCs in DNA replication and also affect normal mitosis (62), and FANCL mutations may cause apoptosis in a large number of sperms in the seminiferous tubules, and resulting in a sudden decrease in sperm. Therefore, both FANCL and FANCT defects can damage sperm formation and lead to NOA in men (143).
FANCD2 is one of the most conserved members of the FA family, and its mono-ubiquitination is key to activating the FA pathway. FANCD2 can interact with other proteins to repair DNA damage and play an important role in many stages of cell development (144, 145). FANCI is mainly located in the nucleolus and can regulate the transcription of ribosomes, maintaining the relative stability of DNA (146, 147). FANCD2 and FANCI combine to form an ID2 complex, both of which are mono-ubiquitinated substrates (21). ICL cannot be repaired without the activation of the FA pathway, and this depends on the mono-ubiquitination of FANCD2 and FANCI, which activates the downstream FA pathway (148, 149). At the molecular level, the ubiquitinated ID2 complex initiates downstream processes such as nucleotide cleavage and homologous recombination, while at the cellular level, it activates the cell cycle to continue dividing (63). Ubiquitination binds the ID2 complex more firmly in space, thus better protecting damaged DNA from external interference and ensuring normal DNA damage response and repair (64). Moreover, by phosphorylating FANCI with ATR kinase, the ID2 complex can maintain its ubiquitination (150). After participating in the FA pathway, the ID2 complex needs to be immediately de-ubiquitinated, which is mediated by the ubiquitin-specific peptidase USP1. Blocking the de-ubiquitination process will lead to the failure of chromosomal homologous recombination and the inability to produce spermatogenic cells (151). Hypospermatogenesis is the testicular phenotype of patients with NOA (152). Furthermore, in mice with Fancd2 deficiency, double strand break (DSB) and crossovers cannot be repaired in a timely manner, resulting in genetic information damage to PGCs during division and minimal sperm count (65). Simultaneously, mutated FANCI will induce FANCD2 lesions and lead to a decrease in the number of undifferentiated spermatocytes, which may develop into NOA (143). In summary, the ubiquitination, ATM and Rad3-related phosphorylation, and de-ubiquitination processes of the ID2 complex are indispensable. A mistake in any link can seriously affect downstream homologous recombination processes (69), leading to ICL repair failure and affecting the normal development of male testes (66), thereby inducing NOA.
FANCP, also known as SLX4, encodes a large scaffold protein that assists structure-specific endonucleases. This protein is important in maintaining gene stability in repairing various types of DNA damage, such as DNA interstrand cross-links (29). This gene is precisely located in pachytene sex chromatin and is closely related to the genetic stability of sex chromosomes (153). The more stable the inheritance of XY chromatin, the stronger the SLX4 intensity and the more complete the genetic material possessed by the sperm (153, 154). We believe that the probability of NOA occurring in a stable genetic environment will also be minimal. In the process of repairing interchain crosslinks, FANCP decouple the cross-linked portion (67). FANCP (SLX4) acts as a scaffold protein and acts on DNA strand cross-linking through the UBZ-1 domain to release the cross-linked portion (68). Simultaneously, this protein can effectively inhibit the intermediate produced by homologous recombination, and SLX4IP regulates the SLX4-XPF-ERCC1 complex, which can, to some extent, inhibit ICL (68). FANCP regulates the correct segregation of chromosomes within PGCs. Furthermore, research has found that the absence of SLX4 can lead to the abnormal development of spermatogonia, which cannot develop into mature sperm in the seminiferous tubules (139), and the number of sperm decreases. A low number of mature sperm can lead to a low number of sperm in the seminal vesicles, resulting in NOA (155). We speculate that if SLX4 undergoes mutations or deletions, the meiotic process may be halted owing to genetic instability, resulting in the inability to produce sperm and NOA or oligospermia.
FANCQ, also known as ERCC4 (156), encodes a protein mainly involved in the repair of ICL. FANCQ plays a role downstream of the FA pathway. XPF expressed through ERCC4 can form a complex with ERCC1, which is recruited by SLX4 with the help of FANCD2 mono-ubiquitination and directly acts on the ICL repair site (30, 31, 69). In contrast, ERCC4 expresses a specific nuclease XPF that participates in the nucleotide excision repair necessary for ICL repair, thereby indirectly participating in ICL repair (30, 70). FANCQ mutations exhibit cell cycle arrest induced by mitomycin C, which may be due to chromosome breakage caused by ICL repair defects (71, 72). Therefore, FANCQ mutations may disrupt the process of sperm production or reduce the number of sperm, and hypospermatogenesis and cell maturity arrest are the histological phenotypes of the testicles in NOA (157). Furthermore, FANCQ also plays an important role in the meiotic recombination process. The SHOC1 protein is an ERCC4-like protein, and plays a role in the meiotic process of spermatocytes, promoting the formation of crossovers and ensuring correct chromosome separation to produce haploid gametes, and the ERCC4 protein also has this function (73, 74). Therefore, FANCQ mutations may result in chromosome recombination errors during meiosis and arrest the spermatocyte cycle (73, 74). We summarized the possible effects of FANCQ on NOA pathogenesis. FANCQ plays important roles in ICL repair and meiotic recombination. FANCQ mutation may prevent ICL repair, cause meiotic recombination errors, and hinder the spermatocyte division cycle, leading to apoptosis, and then lead to hypospermatogenesis often leads to NOA (72).
FANCV, also known as REV7/MAD2L2 (34, 75), encodes a protein that can form a DNA polymerase complex with REV3 (76), enabling FANCV to greatly promote REV3’s ability to repair DNA damage and play an expanding role in ICL repair in the form of enzymes (77, 78). ICL repair is closely related to spermatogenesis, indicating the importance of FANCV in regulating cell mitosis and cell development. Experimental reports show the extreme abundance of REV7 in adult male testes (158). After the deletion of this gene, the number of PGCs in mice decreases and they exhibit infertility (79). Furthermore, studies have shown that after FANCV mutation, PGCs undergo apoptosis during the embryonic stage, resulting in spermatogenesis defects and testicular sperms are underdeveloped (75). The structural domain mutations in the interaction between FANCV and REV3 manifest as cell cycle arrest, chromosome breakage, and difficulty in cell proliferation, leading to difficulty in spermatogenesis (80). Therefore, REV7 can maintain the normal morphology and function of sperms, and FANCV mutations can lead to sperm maturity arrest, leading to NOA (157).
Homologous recombination is important in chromosomal inheritance and DNA repair during mitosis and meiosis. Homologous recombination maintains genome stability by regulating damaged replication forks or filling DNA gaps, and can be used to repair DNA double-strand break (159, 160). Studies have shown that the FA pathway can also repair DNA damage (161), and many genes in the FA gene family are involved in homologous recombination processes, such as FANCR (RAD51), FANCS (BRCA1), FANCD1 (BRCA2), and other genes that encode crucial homologous recombination proteins (162). Homologous recombination repair affects spermatogonial stem cells and may be associated with NOA (163).
FANCN, also known as PALB2, encodes a gene product that can participate in homologous recombination repair, which is related to the repair of ICL and the smooth progression of the cell cycle (27). Cell cycle arrest and cell apoptosis often cause NOA. FANCN recruits DNA damage sites by connecting one end of the coil domain to FANCS (BRCA1), while the other end recruits FANCD1 (BRCA2) and FANCR (RAD51) to DNA damage sites to initiate homologous recombination repair (82). Furthermore, phosphorylation of FANCN by the cell cycle checkpoint regulatory factor ataxia telangiectasia mutated can promote the repair process of homologous recombination by RAD51 (164, 165). The n-terminal mutation of FANCN prevents homologous recombination repair involving FANCN (81, 83). According to Simhadri et al., removing the Palb2 gene in mice can lead to a significant reduction or apoptosis of male sperms, affecting fertility (81). Defects in homologous recombination repair of cells can hinder chromosomal recombination during decelerative division, and spermatocytes exhibit cell cycle arrest (81). Male spermatogonial stem cells will gradually develop into sperm (166), and in mice lacking Palb2, the number of spermatogonial stem cells significantly decreases. We speculate that the number of male sperms lacking this gene will also show a downward trend, resulting in a sudden decrease in the number of sperm formed in the future. Therefore, FANCN mutations may be potential risk factors for NOA.
FANCS, also known as BRCA1 (167), encodes a protein that is important in DNA damage repair and transcriptional regulation (168, 169), and can also maintain genomic integrity and stability by participating in cellular DNA damage repair (32). Research has shown that BRCA1 can interact with Werner syndrome protein to jointly repair ICL (170). According to Durocher et al., the BRCA1 gene is present in interstitial cells of the testis, and BRCA1 deficiency can lead to impaired expression in interstitial cells of the testis (171). Interstitial cells of the testis play a controlling role in male fertility, and if damaged, they can lead to a decrease in sperm quality and quantity (172). Therefore, mutations in BRCA1 damage interstitial cells of the testis, further affecting sperm count and indirectly leading to NOA. Furthermore, research has found that when FANCS mutates, the testicular volume significantly decreases, the seminiferous tubules cannot produce sperm, and the expression of genes related to the repair of DNA damage is reduced, making it difficult to repair damaged spermatocytes in a timely manner (86). Sciurano et al. found that spermatocyte division was stagnant and spermatocyte death occurred in a patient with azoospermia (173). Therefore, patients with FANCS mutations are likely to further develop NOA due to irreversible damage to spermatocytes.
BRCA2, also known as FANCD1, encodes a protein that regulates the RAD51 protein by participating in ID2 ubiquitination and binding to DNA, thereby participating in homologous recombination repair processes (87, 88). BRCA2 can control the stability of the genome while maintaining normal meiotic processes, playing an important role in the maturation and development of sperms (174). Studies have shown that BRCA2 is associated with male azoospermia and plays a controlling role in meiosis (89). BRCA2 deficiency can lead to a meiotic disorder in spermatocytes, leading to infertility and directly affecting spermatogenesis, leading to male azoospermia or oligospermia (89). Shukron et al. compared the structure of male fruit flies with Brca2 homozygous and heterozygous deletions, as well as wild-type fruit flies, and found that male fruit flies with homozygous mutations experienced sperm arrest, testicular development abnormalities, and infertility (175). Shive et al. found oligospermia in male zebrafish in a homozygous mutation model of Brca2 (176). Simultaneously, a report showed that homozygous deletion of FANCD1 resulted lower sperm maturation (90). Therefore, mutated FANCD1 may lead to the failure of ICL repair, meiosis stagnation at the S-phase checkpoint, failure of the spermatocyte-to-sperm process to proceed normally, gradual apoptosis, and oligospermia or aspermia (20, 87, 89).
FANCO is also known as RAD51C, and FANCR is also known as RAD51 (177). FANCO, a newly isolated member of the RAD51 (FANCR) family, shares genetic similarities with other members. Therefore, RAD51C is a collateral homolog of RAD51 (178). Both play important roles in repairing DNA interstrand cross-links. In DNA damage, overexpression of the RAD51 paraprotein XRCC3 increases resistance to ICL inducers, while RAD51C binds to XRCC3 to form a complex dependent on RAD51 to ensure proper chromosome separation during meiosis (28, 91, 92). Mutation or deletion of RAD51C can enhance the sensitivity of cross-linking between DNA strands (93), and RAD51C can transmit DNA damage signals to induce DNA damage repair (179). Some studies have found that RAD51 mutants develop azoospermia due to meiosis termination (95). Simultaneously, in a mouse model, Qin et al. found (180) that the knockout of Rad51 led to abnormal development of spermatogonocytes, a reduced number of pachytene spermatocytes, or apoptosis. If defective RAD51 results in a reduced sperm count, NOA is further increased. Similarly, mutations in RAD51C can induce the suspension of spermatocyte development, which mainly occurs in the first phase of meiotic division and is related to abnormal chromosome breakage or premature sister chromosome separation (94). The further development of meiotic arrest in spermatocytes may result in NOA (173) Therefore, FANCO and FANCR are interdependent, ensuring normal progression of meiosis. If both mutations occur, they can affect spermatogonia and spermatocytes, causing NOA.
The alias of FANCU is XRCC2, and its expression product can form a complex BCDX2 with FANCR (RAD51) homologous compounds (33, 178, 181). This complex plays a role in replication restart in homologous recombination repair (96). FANCU mutation reduces the stability of BCDX2 (96), resulting in the collapse of the replication fork due to the inability to restart, resulting in chromosome breakage and forced termination of cell division (97, 98). Sanger sequencing of Fancu mutant mice revealed a recessive mutation in Fancu (C. 41T>C/p.Leu14Pro), which stopped the meiosis of cells, thus leading to azoospermia (95). BCDX2 complex participates in the formation of RAD51 nuclear protein filament by binding with single-stranded DNA, and plays a role in double-strand break repair in the assembly of RAD51 lesions (33). FANCU mutant cells showed defects in homologous recombination damage repair (99). Simultaneously, Zhang et al. used whole-exome sequencing to sequence two patients with infertility and found that male patients with NOA had C. 41T>C (p.Leu14Pro) XRCC2 mutation, and believed that the occurrence of NOA in the family was caused by XRCC2 mutation (182). Therefore, we concluded that the mutation of FANCU led to the defective assembly of RAD51 lesions, which resulted in the failure of homologous recombination repair and apoptosis (99). The consequence of incomplete spermatocyte division may be sperm apoptosis, resulting in NOA.
FANCW, also known as RFWD3, encodes a protein that plays a role in the synthesis of homologous recombination repair DNA (87). After binding to RPA, the C-terminal of FANCW is recruited by RPA to the stalled replication fork, and the protein is phosphorylated by checkpoint kinase Ataxia-telangiectasia mutated (ATM) and Rad3-related (ATR) to participate in the ATM/ATR signaling pathway and control the DNA replication checkpoint to restart the replication fork (100–104). FANCW accelerates the separation of RPA from single-stranded DNA by regulating its ubiquitination (105), while also promoting the recruitment of repair proteins such as RAD51 and RAD52 by RPA, enabling them to jointly locate DNA damage sites and initiate RAD51-mediated homologous recombination repair (35, 36, 183). This may be due to the FANCW mutation, which prevents isolation of RPA from the RAD51 lesion, prevents the formation of RAD51 nuclear filaments, and leads to cell cycle arrest (105, 106). Cell cycle arrest greatly affects the development of spermatogonial stem cells, causing them to stagnate, unable to produce spermatogonial cells or sperm, resulting in a decreased sperm count (136). Simultaneously, the interaction between spermatogonial stem cells and Sertoli cells is regulated, and Sertoli cells promote the maturation and development of testicular seminiferous tubules (166). Although there is currently no relevant literature on FANCW mutations directly leading to NOA, we speculate that FANCW defects can cause low sperm levels and immature spermatogenic tubules, affecting the maturation and development of spermatogonial stem cells and supporting cells, thereby inducing NOA.
The FANCJ gene is called BRIP1 or BACH1 (184). The protein encoded by this gene acts DNA helicase and plays an important role in repairing ICL and DNA DSB through HR (24). Studies have found that mutated FANCJ seriously affects spermatogenesis (107). Compared with normal cells, cells with FANCJ deficiency or lack of FANCJ catalytic activity have an increased sensitivity to DNA ICL (185), while cells with FANCJ mutations significantly reduce their resistance to ICL (108), resulting in a significant increase in the number of ICL within cells. Furthermore, FANCJ can control the normal division of chromosomes by regulating the replication fork, reducing the destruction of chromatin structure during the replication process, ensuring normal chromatin replication, and maintaining genetic stability (109). According to experimental studies in mice, FANCJ causes genetic defects or mutations, with only some males exhibiting fertility. However, there is a significant reduction in testicular volume and a sudden decrease in the number of spermatogonial stem cells (107). NOA is characterized by a low sperm level, whereas spermatogonial stem cells are the most primitive spermatogonial cells, which gradually develop into sperm (186). Although no direct evidence currently links FANCJ mutations to the occurrence of NOA, based on our research, we speculate that FANCJ mutations may cause genetic and DNA disorders in primordial germ cells, preventing the formation of normal sperms and leading to NOA.
The treatment methods for infertility, including for NOA, are not mature internationally and can be roughly divided into auxiliary technologies, such as spermatogonial stem cell transplantation and intracytoplasmic sperm injection (187, 188). The diagnosis and treatment of NOA through the FA pathway has broad research prospects. If activation of the FA pathway fails, it can lead to unstable DNA replication in PGCs, exacerbating their damage and significantly reducing their number during division, leading to NOA or oligospermia in males. Using the FA pathway to repair the ICL and restore the number of mature sperm in the testes of patients with NOA may cure the disease. Mutations in any FA gene may cause irreparable damage to male reproductive function through the FA pathway and may even be fatal. A key step in the FA pathway is successful ubiquitination of the ID2 complex to repair the damage caused by ICL. Activation of the FA pathway controls the rapidly proliferating PGCs to stabilize the replication and transcription of their DNA (8). At present, relatively novel studies mainly focus on gene level changes such as mRNA therapy and lentiviral vector therapy (189, 190). At the same time, adeno-associated virus integration site 1 has also been found to guide FANCA to target the binding of DNA missing this gene fragment to achieve changes in cell phenotype (191). In the future, perhaps we can actively encourage genetic testing and premarital physical examination to reduce the birth of NOA children to some extent from the perspective of prevention. For patients who have been clinically diagnosed with NOA, we can use special mRNA or other special carriers to bind the DNA of FA gene defect to make the expression of FA gene complete again, so as to maintain the number of viable sperm in NOA patients. At the same time, the development of FA protein replacement drugs may be of great significance for maintaining the normal function of FA pathway and improving the reproductive defects in NOA patients, and it is expected to become a new therapeutic strategy. This article reviews the correlation between NOA and FA, categorizes them according to the different gene properties, and attempts to elucidate the relevant mechanisms and significance. We hope that this review will be helpful in solving the problems of DNA damage and FA pathway activation failure caused by FA gene mutations and provide new development ideas and concepts for NOA researchers.
HX: Writing – original draft, Writing – review & editing. YZ: Writing – original draft, Writing – review & editing. CW: Writing – original draft, Writing – review & editing. ZF: Writing – original draft. JL: Writing – original draft. YY: Writing – original draft. ZZ: Writing – review & editing. YQ: Writing – review & editing. KM: Writing – review & editing. JY: Writing – review & editing. XW: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by grants from Research Fund for Lin He’s Academician Workstation of New Medicine and Clinical Translation in Jining Medical University (JYHL2021MS13,JYHL2021MS10), the Research Start-up Fund of Jining Medical University (600791001) and College Students’ Innovation Training Program of Jining Medical University (cx2023136z).
We thank to Figdraw for providing scientific researchers with exquisite materials and convenient drawing platform. All the figures in this article are made by Figdraw (www.figdraw.com).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Krausz C. Male infertility: pathogenesis and clinical diagnosis. Best Pract Res Clin Endocrinol Metab. (2011) 25:271–85. doi: 10.1016/j.beem.2010.08.006
2. Wosnitzer M, Goldstein M, Hardy MP. Review of azoospermia. Spermatogenesis. (2014) 4:e28218. doi: 10.4161/spmg.28218
3. Agarwal A, Baskaran S, Parekh N, Cho C-L, Henkel R, Vij S, et al. Male infertility. Lancet. (2021) 397:319–33. doi: 10.1016/S0140-6736(20)32667-2
4. Arshad MA, Majzoub A, Esteves SC. Predictors of surgical sperm retrieval in non-obstructive azoospermia: summary of current literature. Int Urol Nephrol. (2020) 52:2015–38. doi: 10.1007/s11255-020-02529-4
5. Klami R, Mankonen H, Perheentupa A. Successful microdissection testicular sperm extraction for men with non-obstructive azoospermia. Reprod Biol. (2018) 18:137–42. doi: 10.1016/j.repbio.2018.03.003
6. Hill RJ, Crossan GP. DNA cross-link repair safeguards genomic stability during premeiotic germ cell development. Nat Genet. (2019) 51:1283–94. doi: 10.1038/s41588-019-0471-2
7. Pickering A, Zhang J, Panneerselvam J, Fei P. Advances in the understanding of the Fanconi anemia tumor suppressor pathway. Cancer Biol Ther. (2013) 14:1089–91. doi: 10.4161/cbt.26380
8. Yang Y, Xu W, Gao F, Wen C, Zhao S, Yu Y, et al. Transcription–replication conflicts in primordial germ cells necessitate the Fanconi anemia pathway to safeguard genome stability. Proc Natl Acad Sci. (2022) 119:e2203208119. doi: 10.1073/pnas.2203208119
9. Kwong A, Ho CY, Shin VY, Au CH, Chan TL, Ma ES. A case report of germline compound heterozygous mutations in the BRCA1 gene of an ovarian and breast cancer patient. Int J Mol Sci. (2021) 22:889. doi: 10.3390/ijms22020889
10. Daum H, Zlotogora J. Fanconi anemia gene variants in patients with gonadal dysfunction. Reprod Sci. (2022) 29:1408–13. doi: 10.1007/s43032-021-00582-7
11. Auerbach AD. Fanconi anemia and its diagnosis. Mutat Res/Fundamental Mol Mech Mutagenesis. (2009) 668:4–10. doi: 10.1016/j.mrfmmm.2009.01.013
12. Krausz C, Riera-Escamilla A, Chianese C, Moreno-Mendoza D, Ars E, Rajmil O, et al. From exome analysis in idiopathic azoospermia to the identification of a high-risk subgroup for occult Fanconi anemia. Genet Med. (2019) 21:189–94. doi: 10.1038/s41436-018-0037-1
13. Tsui V, Crismani W. The Fanconi anemia pathway and fertility. Trends Genet. (2019) 35:199–214. doi: 10.1016/j.tig.2018.12.007
14. Degan P, Cappelli E, Regis S, Ravera S. New insights and perspectives in Fanconi anemia research. Trends Mol Med. (2019) 25:167–70. doi: 10.1016/j.molmed.2019.01.003
15. Ekundayo B, Bleichert F. Origins of DNA replication. PloS Genet. (2019) 15:e1008320. doi: 10.1371/journal.pgen.1008320
16. Milletti G, Strocchio L, Pagliara D, Girardi K, Carta R, Mastronuzzi A, et al. Canonical and noncanonical roles of fanconi anemia proteins: Implications in cancer predisposition. Cancers. (2020) 12:2684. doi: 10.3390/cancers12092684
17. Jeong E, Lee S-G, Kim H-S, Yang J, Shin J, Kim Y, et al. Structural basis of the fanconi anemia-associated mutations within the FANCA and FANCG complex. Nucleic Acids Res. (2020) 48:3328–42. doi: 10.1093/nar/gkaa062
18. Kim TM, Ko JH, Choi YJ, Hu L, Hasty P. The phenotype of FancB-mutant mouse embryonic stem cells. Mutat Res/Fundamental Mol Mech Mutagenesis. (2011) 712:20–7. doi: 10.1016/j.mrfmmm.2011.03.010
19. Noll M, Battaile KP, Bateman R, Lax TP, Rathbun K, Reifsteck C, et al. Fanconi anemia group A and C double-mutant mice: functional evidence for a multi-protein Fanconi anemia complex. Exp Hematol. (2002) 30:679–88. doi: 10.1016/S0301-472X(02)00838-X
20. Godthelp BC, van Buul PP, Jaspers NG, Elghalbzouri-Maghrani E, van Duijn-Goedhart A, Arwert F, et al. Cellular characterization of cells from the Fanconi anemia complementation group, FA-D1/BRCA2. Mutat Res/Fundamental Mol Mech Mutagenesis. (2006) 601:191–201. doi: 10.1016/j.mrfmmm.2006.07.003
21. Walden H, Deans AJ. The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu Rev Biophys. (2014) 43:257–78. doi: 10.1146/annurev-biophys-051013-022737
22. Gordon SM, Alon N, Buchwald M, FANCC FANCE. and FANCD2 form a ternary complex essential to the integrity of the Fanconi anemia DNA damage response pathway. J Biol Chem. (2005) 280:36118–25. doi: 10.1074/jbc.M507758200
23. Auerbach AD, Greenbaum J, Pujara K, Batish SD, Bitencourt MA, Kokemohr I, et al. Spectrum of sequence variation in the FANCG gene: an International Fanconi Anemia Registry (IFAR) study. Hum Mutat. (2003) 21:158–68. doi: 10.1002/(ISSN)1098-1004
24. Nath S, Somyajit K, Mishra A, Scully R, Nagaraju G. FANCJ helicase controls the balance between short-and long-tract gene conversions between sister chromatids. Nucleic Acids Res. (2017) 45:8886–900. doi: 10.1093/nar/gkx586
25. Frost MG, Mazloumi Aboukheili AM, Toth R, Walden H. Characterization of FANCL variants observed in patient cancer cells. Biosci Rep. (2020) 40:BSR20191304. doi: 10.1042/BSR20191304
26. Domingues-Silva B, Silva B, Azzalin CM. ALTernative functions for human FANCM at telomeres. Front Mol Biosci. (2019) 6:84. doi: 10.3389/fmolb.2019.00084
27. Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. (2010) 11:196–207. doi: 10.1038/nrm2851
28. Xu Z-Y, Loignon M, Han F-Y, Panasci L, Aloyz R. Xrcc3 induces cisplatin resistance by stimulation of Rad51-related recombinational repair, S-phase checkpoint activation, and reduced apoptosis. J Pharmacol Exp Ther. (2005) 314:495–505. doi: 10.1124/jpet.105.084053
29. Lachaud C, Castor D, Hain K, Munoz I, Wilson J, MacArtney TJ, et al. Distinct functional roles for the two SLX4 ubiquitin-binding UBZ domains mutated in Fanconi anemia. J Cell Sci. (2014) 127:2811–7. doi: 10.1242/jcs.146167
30. De Silva IU, McHugh PJ, Clingen PH, Hartley JA. Defining the roles of nucleotide excision repair and recombination in the repair of DNA interstrand cross-links in mammalian cells. Mol Cell Biol. (2000) 20:7980–90. doi: 10.1128/.20.21.7980-7990.2000
31. Dehé P-M, Gaillard P-HL. Control of structure-specific endonucleases to maintain genome stability. Nat Rev Mol Cell Biol. (2017) 18:315–30. doi: 10.1038/nrm.2016.177
32. Bunting SF, Callén E, Kozak ML, Kim JM, Wong N, López-Contreras AJ, et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol Cell. (2012) 46:125–35. doi: 10.1016/j.molcel.2012.02.015
33. Greenhough LA, Liang C-C, Belan O, Kunzelmann S, Maslen S, Rodrigo-Brenni MC, et al. Structure and function of the RAD51B–RAD51C–RAD51D–XRCC2 tumour suppressor. Nature. (2023) 619:1–8. doi: 10.1038/s41586-023-06179-1
34. Mamrak NE, Shimamura A, Howlett NG. Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev. (2017) 31:93–9. doi: 10.1016/j.blre.2016.10.002
35. Elia AE, Wang DC, Willis NA, Boardman AP, Hajdu I, Adeyemi RO, et al. RFWD3-dependent ubiquitination of RPA regulates repair at stalled replication forks. Mol Cell. (2015) 60:280–93. doi: 10.1016/j.molcel.2015.09.011
36. Wu X, Yang Z, Liu Y, Zou Y. Preferential localization of hyperphosphorylated replication protein A to double-strand break repair and checkpoint complexes upon DNA damage. Biochem J. (2005) 391:473–80. doi: 10.1042/BJ20050379
37. Deans AJ, West SC. FANCM connects the genome instability disorders Bloom's Syndrome and Fanconi Anemia. Mol Cell. (2009) 36:943–53. doi: 10.1016/j.molcel.2009.12.006
38. Schwab RA, Blackford AN, Niedzwiedz W. ATR activation and replication fork restart are defective in FANCM-deficient cells. EMBO J. (2010) 29:806–18. doi: 10.1038/emboj.2009.385
39. Rohleder F, Huang J, Xue Y, Kuper J, Round A, Seidman M, et al. FANCM interacts with PCNA to promote replication traverse of DNA interstrand crosslinks. Nucleic Acids Res. (2016) 44:3219–32. doi: 10.1093/nar/gkw037
40. Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, Meetei AR, et al. Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol Cell. (2007) 25:331–43. doi: 10.1016/j.molcel.2007.01.003
41. Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, et al. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol. (2003) 23:3417–26. doi: 10.1128/MCB.23.10.3417-3426.2003
42. Yin H, Ma H, Hussain S, Zhang H, Xie X, Jiang L, et al. A homozygous FANCM frameshift pathogenic variant causes male infertility. Genet Med. (2019) 21:62–70. doi: 10.1038/s41436-018-0015-7
43. Wong JC, Alon N, Mckerlie C, Huang JR, Meyn MS, Buchwald M. Targeted disruption of exons 1 to 6 of the Fanconi Anemia group A gene leads to growth retardation, strain-specific microphthalmia, meiotic defects and primordial germ cell hypoplasia. Hum Mol Genet. (2003) 12:2063–76. doi: 10.1093/hmg/ddg219
44. Garcia-Higuera I, Kuang Y, Denham J, D'Andrea AD. The Fanconi anemia proteins FANCA and FANCG stabilize each other and promote the nuclear accumulation of the Fanconi anemia complex. Blood J Am Soc Hematol. (2000) 96:3224–30. doi: 10.1182/blood.V96.9.3224.h8003224_3224_3230
45. Benitez A, Liu W, Palovcak A, Wang G, Moon J, An K, et al. FANCA promotes DNA double-strand break repair by catalyzing single-strand annealing and strand exchange. Mol Cell. (2018) 71:621–628. e4. doi: 10.1016/j.molcel.2018.06.030
46. Jarysta A, Riou L, Firlej V, Lapoujade C, Kortulewski T, Barroca V, et al. Abnormal migration behavior linked to Rac1 signaling contributes to primordial germ cell exhaustion in Fanconi anemia pathway-deficient Fancg–/– embryos. Hum Mol Genet. (2022) 31:97–110. doi: 10.1093/hmg/ddab222
47. Kato Y, Alavattam KG, Sin H-S, Meetei AR, Pang Q, Andreassen PR, et al. FANCB is essential in the male germline and regulates H3K9 methylation on the sex chromosomes during meiosis. Hum Mol Genet. (2015) 24:5234–49. doi: 10.1093/hmg/ddv244
48. Jamsai D, O’Connor AE, O’Donnell L, Lo JCY, O’Bryan MK. Uncoupling of transcription and translation of Fanconi anemia (FANC) complex proteins during spermatogenesis. Spermatogenesis. (2015) 5:e979061. doi: 10.4161/21565562.2014.979061
49. Nadler JJ, Braun RE. Fanconi anemia complementation group C is required for proliferation of murine primordial germ cells. genesis. (2000) 27:117–23. doi: 10.1002/(ISSN)1526-968X
50. Taniguchi T, D'Andrea AD. The Fanconi anemia protein, FANCE, promotes the nuclear accumulation of FANCC. Blood J Am Soc Hematol. (2002) 100:2457–62. doi: 10.1182/blood-2002-03-0860
51. Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D'Andrea AD. S-phase–specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood J Am Soc Hematol. (2002) 100:2414–20. doi: 10.1182/blood-2002-01-0278
52. Fu C, Begum K, Jordan PW, He Y, Overbeek PA. Dearth and delayed maturation of testicular germ cells in Fanconi anemia E mutant male mice. PloS One. (2016) 11:e0159800. doi: 10.1371/journal.pone.0159800
53. Pace P, Johnson M, Tan WM, Mosedale G, Sng C, Hoatlin M, et al. FANCE: the link between Fanconi anaemia complex assembly and activity. EMBO J. (2002) 21:3414–23. doi: 10.1093/emboj/cdf355
54. Léveillé F, Blom E, Medhurst AL, Bier P, Laghmani EH, Johnson M, et al. The Fanconi anemia gene product FANCF is a flexible adaptor protein. J Biol Chem. (2004) 279:39421–30. doi: 10.1074/jbc.M407034200
55. Youssoufian H. Localization of Fanconi anemia C protein to the cytoplasm of mammalian cells. Proc Natl Acad Sci. (1994) 91:7975–9. doi: 10.1073/pnas.91.17.7975
56. Léveillé F, Ferrer M, Medhurst AL, Laghmani EH, Rooimans MA, Bier P, et al. The nuclear accumulation of the Fanconi anemia protein FANCE depends on FANCC. DNA Repair. (2006) 5:556–65. doi: 10.1016/j.dnarep.2006.01.005
57. Bouffard F, Plourde K, Bélanger S, Ouellette G, Labrie Y, Durocher F. Analysis of a FANCE splice isoform in regard to DNA repair. J Mol Biol. (2015) 427:3056–73. doi: 10.1016/j.jmb.2015.08.004
58. De Winter JP, van der Weel L, De Groot J, Stone S, Waisfisz Q, Arwert F, et al. The Fanconi anemia protein FANCF forms a nuclear complex with FANCA, FANCC and FANCG. Hum Mol Genet. (2000) 9:2665–74. doi: 10.1093/hmg/9.18.2665
59. Bakker ST, van de Vrugt HJ, Visser JA, Delzenne-Goette E, Wal Avd, Berns MA, et al. Fancf-deficient mice are prone to develop ovarian tumours. J Pathol. (2012) 226:28–39. doi: 10.1002/path.2992
60. Singh DK, Gamboa RS, Singh AK, Walkemeier B, Van Leene J, De Jaeger G, et al. The FANCC–FANCE–FANCF complex is evolutionarily conserved and regulates meiotic recombination. Nucleic Acids Res. (2023) 51:2516–28. doi: 10.1093/nar/gkac1244
61. Cornwell MJ, Thomson GJ, Coates J, Belotserkovskaya R, Waddell ID, Jackson SP, et al. Small-molecule inhibition of UBE2T/FANCL-mediated ubiquitylation in the Fanconi anemia pathway. ACS Chem Biol. (2019) 14:2148–54. doi: 10.1021/acschembio.9b00570
62. Yu Y, Xu W, Wen C, Zhao S, Li G, Liu R, et al. UBE2T resolves transcription-replication conflicts and protects common fragile sites in primordial germ cells. Cell Mol Life Sci. (2023) 80:92. doi: 10.1007/s00018-023-04733-8
63. Dubois EL, Guitton-Sert L, Béliveau M, Parmar K, Chagraoui J, Vignard J, et al. and distinct functions for FANCI and FANCD2. Nucleic Acids Res. (2019) 47:7532–47. doi: 10.1093/nar/gkz514
64. Tan W, van Twest S, Leis A, Bythell-Douglas R, Murphy VJ, Sharp M, et al. Monoubiquitination by the human Fanconi anemia core complex clamps FANCI: FANCD2 on DNA in filamentous arrays. elife. (2020) 9:e54128. doi: 10.7554/eLife.54128
65. Zhao S, Huang C, Yang Y, Xu W, Yu Y, Wen C, et al. DNA repair protein FANCD2 has both ubiquitination-dependent and ubiquitination-independent functions during germ cell development. J Biol Chem. (2023) 299:102905. doi: 10.1016/j.jbc.2023.102905
66. Al Jabri A, Al Naim N, Al Dossari A. Homozygous mutation in the FANCD2 gene observed in a Saudi male infant with severe ambiguous genitalia. Case Rep Endocrinol. (2021) 2021:6686312. doi: 10.1155/2021/6686312
67. Hoogenboom WS, Boonen RA, Knipscheer P. The role of SLX4 and its associated nucleases in DNA interstrand crosslink repair. Nucleic Acids Res. (2019) 47:2377–88. doi: 10.1093/nar/gky1276
68. Zhang H, Chen Z, Ye Y, Ye Z, Cao D, Xiong Y, et al. SLX4IP acts with SLX4 and XPF–ERCC1 to promote interstrand crosslink repair. Nucleic Acids Res. (2019) 47:10181–201. doi: 10.1093/nar/gkz769
69. Douwel DK, Boonen RA, Long DT, Szypowska AA, Räschle M, Walter JC, et al. XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell. (2014) 54:460–71. doi: 10.1016/j.molcel.2014.03.015
70. Crossan GP, Patel KJ. The Fanconi anaemia pathway orchestrates incisions at sites of crosslinked DNA. J Pathol. (2012) 226:326–37. doi: 10.1002/path.3002
71. Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, Raams A, et al. Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet. (2013) 92:800–6. doi: 10.1016/j.ajhg.2013.04.002
72. Marín M, Ramírez MJ, Aza Carmona M, Jia N, Ogi T, Bogliolo M, et al. Functional comparison of XPF missense mutations associated to multiple DNA repair disorders. Genes. (2019) 10:60. doi: 10.3390/genes10010060
73. Guiraldelli MF, Felberg A, Almeida LP, Parikh A, de Castro RO, Pezza RJ. SHOC1 is a ERCC4-(HhH) 2-like protein, integral to the formation of crossover recombination intermediates during mammalian meiosis. PloS Genet. (2018) 14:e1007381. doi: 10.1371/journal.pgen.1007381
74. Wang W, Meng L, He J, Su L, Li Y, Tan C, et al. Bi-allelic variants in SHOC1 cause non-obstructive azoospermia with meiosis arrest in humans and mice. Mol Hum Reprod. (2022) 28:gaac015. doi: 10.1093/molehr/gaac015
75. Bluteau D, Masliah-Planchon J, Clairmont C, Rousseau A, Ceccaldi R, d’Enghien CD, et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. J Clin Invest. (2016) 126:3580–4. doi: 10.1172/JCI88010
76. Sharma S, Helchowski CM, Canman CE. The roles of DNA polymerase ζ and the Y family DNA polymerases in promoting or preventing genome instability. Mutat Res/Fundamental Mol Mech Mutagenesis. (2013) 743:97–110. doi: 10.1016/j.mrfmmm.2012.11.002
77. Räschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, et al. Mechanism of replication-coupled DNA interstrand crosslink repair. Cell. (2008) 134:969–80. doi: 10.1016/j.cell.2008.08.030
78. Hara K, Shimizu T, Unzai S, Akashi S, Sato M, Hashimoto H. Purification, crystallization and initial X-ray diffraction study of human REV7 in complex with a REV3 fragment. Acta Crystallographica Section F: Struct Biol Crystallization Commun. (2009) 65:1302–5. doi: 10.1107/S1744309109046181
79. Watanabe N, Mii S, Asai N, Asai M, Niimi K, Ushida K, et al. The REV7 subunit of DNA polymerase ζ is essential for primordial germ cell maintenance in the mouse. J Biol Chem. (2013) 288:10459–71. doi: 10.1074/jbc.M112.421966
80. Tomida J, K.-i. Takata SS, Schibler AC, Yousefzadeh MJ, Bhetawal S, Dent SY, et al. REV7 is essential for DNA damage tolerance via two REV3L binding sites in mammalian DNA polymerase ζ. Nucleic Acids Res. (2015) 43:1000–11. doi: 10.1093/nar/gku1385
81. Simhadri S, Peterson S, Patel DS, Huo Y, Cai H, Bowman-Colin C, et al. Male fertility defect associated with disrupted BRCA1-PALB2 interaction in mice. J Biol Chem. (2014) 289:24617–29. doi: 10.1074/jbc.M114.566141
82. Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res. (2009) 7:1110–8. doi: 10.1158/1541-7786.MCR-09-0123
83. Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc Natl Acad Sci. (2009) 106:7155–60. doi: 10.1073/pnas.0811159106
84. Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, et al. PALB2 links BRCA1 and BRCA2 in the DNA-damage response. Curr Biol. (2009) 19:524–9. doi: 10.1016/j.cub.2009.02.018
85. Sy SM-H, Huen MS, Zhu Y, Chen J. PALB2 regulates recombinational repair through chromatin association and oligomerization. J Biol Chem. (2009) 284:18302–10. doi: 10.1074/jbc.M109.016717
86. Xu X, Aprelikova O, Moens P, Deng C-X, Furth PA. Impaired meiotic DNA-damage repair and lack of crossing-over during spermatogenesis in BRCA1 full-length isoform deficient mice. Development. (2003) 130:2001–12. doi: 10.1242/dev.00410
87. Gallina I, Hendriks IA, Hoffmann S, Larsen NB, Johansen J, Colding-Christensen CS, et al. The ubiquitin ligase RFWD3 is required for translesion DNA synthesis. Mol Cell. (2021) 81:442–58. doi: 10.1016/j.molcel.2020.11.029
88. Rodríguez-Marí A, Wilson C, Titus TA, Canestro C, BreMiller RA, Yan Y-L, et al. Roles of brca2 (fancd1) in oocyte nuclear architecture, gametogenesis, gonad tumors, and genome stability in zebrafish. PloS Genet. (2011) 7:e1001357. doi: 10.1371/journal.pgen.1001357
89. Zhoucun A, Zhang S, Yang Y, Ma Y, Zhang W, Lin L. The common variant N372H in BRCA2 gene may be associated with idiopathic male infertility with azoospermia or severe oligozoospermia. Eur J Obstet Gynecol Reprod Biol. (2006) 124:61–4. doi: 10.1016/j.ejogrb.2005.09.001
90. Ramanagoudr-Bhojappa R, Carrington B, Ramaswami M, Bishop K, Robbins GM, Jones M, et al. Multiplexed CRISPR/Cas9-mediated knockout of 19 Fanconi anemia pathway genes in zebrafish revealed their roles in growth, sexual development and fertility. PloS Genet. (2018) 14:e1007821. doi: 10.1371/journal.pgen.1007821
91. Liu N, Schild D, Thelen MP, Thompson LH. Involvement of Rad51C in two distinct protein complexes of Rad51 paralogs in human cells. Nucleic Acids Res. (2002) 30:1009–15. doi: 10.1093/nar/30.4.1009
92. Su H, Cheng Z, Huang J, Lin J, Copenhaver GP, Ma H, et al. RAD51C and XRCC3 proteins form a complex and facilitate RAD51 localization on chromosomes for meiotic recombination. PloS Genet. (2017) 13:e1006827. doi: 10.1371/journal.pgen.1006827
93. Somyajit K, Subramanya S, Nagaraju G. Distinct roles of FANCO/RAD51C protein in DNA damage signaling and repair. J Biol Chem. (2012) 287:3366–80. doi: 10.1074/jbc.M111.311241
94. Kuznetsov S, Pellegrini M, Shuda K, Fernandez-Capetillo O, Liu Y, Martin BK, et al. RAD51C deficiency in mice results in early prophase I arrest in males and sister chromatid separation at metaphase II in females. J Cell Biol. (2007) 176:581–92. doi: 10.1083/jcb.200608130
95. Yang Y, Guo J, Dai L, Zhu Y, Hu H, Tan L, et al. XRCC2 mutation causes meiotic arrest, azoospermia and infertility. J Med Genet. (2018) 55:628–36. doi: 10.1136/jmedgenet-2017-105145
96. Park J-Y, Virts EL, Jankowska A, Wiek C, Othman M, Chakraborty SC, et al. Complementation of hypersensitivity to DNA interstrand crosslinking agents demonstrates that XRCC2 is a Fanconi anaemia gene. J Med Genet. (2016) 53:672–80. doi: 10.1136/jmedgenet-2016-103847
97. Somyajit K, Saxena S, Babu S, Mishra A, Nagaraju G. Mammalian RAD51 paralogs protect nascent DNA at stalled forks and mediate replication restart. Nucleic Acids Res. (2015) 43:9835–55. doi: 10.1093/nar/gkv880
98. Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. (2010) 11:208–19. doi: 10.1038/nrm2852
99. Takata M, Sasaki MS, Tachiiri S, Fukushima T, Sonoda E, Schild D, et al. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol Cell Biol. (2001) 21:2858–66. doi: 10.1128/MCB.21.8.2858-2866.2001
100. Knies K, Inano S, Ramírez MJ, Ishiai M, Surrallés J, Takata M, et al. Biallelic mutations in the ubiquitin ligase RFWD3 cause Fanconi anemia. J Clin Invest. (2017) 127:3013–27. doi: 10.1172/JCI92069
101. Moore CE, Yalcindag SE, Czeladko H, Ravindranathan R, Wijesekara Hanthi Y, Levy JC, et al. RFWD3 promotes ZRANB3 recruitment to regulate the remodeling of stalled replication forks. J Cell Biol. (2023) 222:e202106022. doi: 10.1083/jcb.202106022
102. Liu S, Chu J, Yucer N, Leng M, Wang S-Y, Chen BP, et al. RING finger and WD repeat domain 3 (RFWD3) associates with replication protein A (RPA) and facilitates RPA-mediated DNA damage response. J Biol Chem. (2011) 286:22314–22. doi: 10.1074/jbc.M111.222802
103. Gong Z, Chen J. E3 ligase RFWD3 participates in replication checkpoint control. J Biol Chem. (2011) 286:22308–13. doi: 10.1074/jbc.M111.222869
104. Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. (2008) 9:616–27. doi: 10.1038/nrm2450
105. Feeney L, Munoz IM, Lachaud C, Toth R, Appleton PL, Schindler D, et al. RPA-mediated recruitment of the E3 ligase RFWD3 is vital for interstrand crosslink repair and human health. Mol Cell. (2017) 66:610–21. doi: 10.1016/j.molcel.2017.04.021
106. Inano S, Sato K, Katsuki Y, Kobayashi W, Tanaka H, Nakajima K, et al. RFWD3-mediated ubiquitination promotes timely removal of both RPA and RAD51 from DNA damage sites to facilitate homologous recombination. Mol Cell. (2017) 66:622–634. e8. doi: 10.1016/j.molcel.2017.04.022
107. Sun X, Brieño-Enríquez MA, Cornelius A, Modzelewski AJ, Maley TT, Campbell-Peterson KM, et al. FancJ (Brip1) loss-of-function allele results in spermatogonial cell depletion during embryogenesis and altered processing of crossover sites during meiotic prophase I in mice. Chromosoma. (2016) 125:237–52. doi: 10.1007/s00412-015-0549-2
108. Calvo JA, Fritchman B, Hernandez D, Persky NS, Johannessen CM, Piccioni F, et al. Comprehensive mutational analysis of the BRCA1-associated DNA helicase and tumor-suppressor FANCJ/BACH1/BRIP1. Mol Cancer Res. (2021) 19:1015–25. doi: 10.1158/1541-7786.MCR-20-0828
109. Brosh RM Jr., Cantor SB. Molecular and cellular functions of the FANCJ DNA helicase defective in cancer and in Fanconi anemia. Front Genet. (2014) 5:372. doi: 10.3389/fgene.2014.00372
110. Patel KJ, Joenje H. Fanconi anemia and DNA replication repair. DNA Repair. (2007) 6:885–90. doi: 10.1016/j.dnarep.2007.02.002
111. Joo W, Xu G, Persky NS, Smogorzewska A, Rudge DG, Buzovetsky O, et al. Structure of the FANCI-FANCD2 complex: insights into the Fanconi anemia DNA repair pathway. Science. (2011) 333:312–6. doi: 10.1126/science.1205805
112. Chaugule VK, Arkinson C, Rennie ML, Kämäräinen O, Toth R, Walden H. Allosteric mechanism for site-specific ubiquitination of FANCD2. Nat Chem Biol. (2020) 16:291–301. doi: 10.1038/s41589-019-0426-z
113. Alpi AF, Chaugule V, Walden H. Mechanism and disease association of E2-conjugating enzymes: lessons from UBE2T and UBE2L3. Biochem J. (2016) 473:3401–19. doi: 10.1042/BCJ20160028
114. Lachaud C, Moreno A, Marchesi F, Toth R, Blow JJ, Rouse J. Ubiquitinated Fancd2 recruits Fan1 to stalled replication forks to prevent genome instability. Science. (2016) 351:846–9. doi: 10.1126/science.aad5634
115. Murina O, von Aesch C, Karakus U, Ferretti LP, Bolck HA, Hänggi K, et al. FANCD2 and CtIP cooperate to repair DNA interstrand crosslinks. Cell Rep. (2014) 7:1030–8. doi: 10.1016/j.celrep.2014.03.069
116. Zhan S, Siu J, Wang Z, Yu H, Bezabeh T, Deng Y, et al. Focal point of Fanconi anemia signaling. Int J Mol Sci. (2021) 22:12976. doi: 10.3390/ijms222312976
117. Lefferts JA, Wang C, Sridharan D, Baralt M, Lambert MW. The SH3 domain of αII spectrin is a target for the Fanconi anemia protein, FANCG. Biochemistry. (2009) 48:254–63. doi: 10.1021/bi801483u
118. Yang Y, Kuang Y, De Oca RM, Hays T, Moreau L, Lu N, et al. Targeted disruption of the murine Fanconi anemia gene, Fancg/Xrcc9. Blood J Am Soc Hematol. (2001) 98:3435–40. doi: 10.1182/blood.V98.12.3435
119. Hassani Moghaddam M, Eskandari N, Nikzad H, Miryounesi M, Karimian M, Amini Mahabadi J, et al. Primordial germ cells can be differentiated by retinoic acid and progesterone induction from embryonic stem cells. J Biosci. (2021) 46:1–10. doi: 10.1007/s12038-021-00210-1
120. Landelouci K, Sinha S, Pépin G. Type-I interferon signaling in fanconi anemia. Front Cell Infect Microbiol. (2022) 12:820273. doi: 10.3389/fcimb.2022.820273
121. Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, et al. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. (2013) 498:380–4. doi: 10.1038/nature12306
122. Kermi C, Lo Furno E, Maiorano D. Regulation of DNA replication in early embryonic cleavages. Genes. (2017) 8:42. doi: 10.3390/genes8010042
123. Mutreja K, Krietsch J, Hess J, Ursich S, Berti M, Roessler FK, et al. ATR-mediated global fork slowing and reversal assist fork traverse and prevent chromosomal breakage at DNA interstrand cross-links. Cell Rep. (2018) 24:2629–2642. e5. doi: 10.1016/j.celrep.2018.08.019
124. Lopez-Martinez D, Liang C-C, Cohn MA. Cellular response to DNA interstrand crosslinks: the Fanconi anemia pathway. Cell Mol Life Sci. (2016) 73:3097–114. doi: 10.1007/s00018-016-2218-x
125. Chesner LN, Degner A, Sangaraju D, Yomtoubian S, Wickramaratne S, Malayappan B, et al. Cellular repair of DNA–DNA cross-links induced by 1, 2, 3, 4-diepoxybutane. Int J Mol Sci. (2017) 18:1086. doi: 10.3390/ijms18051086
126. Zhang Y, Li P, Liu N, Jing T, Ji Z, Yang C, et al. Novel bi-allelic variants of FANCM cause sertoli cell-only syndrome and non-obstructive azoospermia. Front Genet. (2021) 12:799886. doi: 10.3389/fgene.2021.799886
127. Kherraf Z-E, Cazin C, Bouker A, Mustapha SFB, Hennebicq S, Septier A, et al. Whole-exome sequencing improves the diagnosis and care of men with non-obstructive azoospermia. Am J Hum Genet. (2022) 109:508–17. doi: 10.1016/j.ajhg.2022.01.011
128. Cantú AV, Laird DJ. A pilgrim's progress: Seeking meaning in primordial germ cell migration. Stem Cell Res. (2017) 24:181–7. doi: 10.1016/j.scr.2017.07.017
129. Kasak L, Punab M, Nagirnaja L, Grigorova M, Minajeva A, Lopes AM, et al. Bi-allelic recessive loss-of-function variants in FANCM cause non-obstructive azoospermia. Am J Hum Genet. (2018) 103:200–12. doi: 10.1016/j.ajhg.2018.07.005
130. Van De Vrugt HJ, Cheng NC, De Vries Y, Rooimans MA, De Groot J, Scheper RJ, et al. Cloning and characterization of murine fanconi anemia group A gene: Fanca protein is expressed in lymphoid tissues, testis, and ovary. Mamm Genome. (2000) 11:326–31. doi: 10.1007/s003350010060
131. Tang D, Li K, Geng H, Xu C, Lv M, Gao Y, et al. Identification of deleterious variants in patients with male infertility due to idiopathic non-obstructive azoospermia. Reprod Biol Endocrinol. (2022) 20:63. doi: 10.1186/s12958-022-00936-z
132. Van de Vrugt HJ, Koomen M, Bakker S, Berns MA, Cheng NC, van der Valk MA, et al. Evidence for complete epistasis of null mutations in murine Fanconi anemia genes Fanca and Fancg. DNA Repair. (2011) 10:1252–61. doi: 10.1016/j.dnarep.2011.09.015
133. Heinrich MC, Silvey KV, Stone S, Zigler AJ, Griffith DJ, Montalto M, et al. Posttranscriptional cell cycle–dependent regulation of human FANCC expression. Blood J Am Soc Hematol. (2000) 95:3970–7. doi: 10.1182/blood.V95.12.3970
134. Odroniec A, Olszewska M, Kurpisz M. Epigenetic markers in the embryonal germ cell development and spermatogenesis. Basic Clin Androl. (2023) 33:6. doi: 10.1186/s12610-022-00179-3
135. Tournaye H, Krausz C, Oates RD. Novel concepts in the aetiology of male reproductive impairment. Lancet Diabetes Endocrinol. (2017) 5:544–53. doi: 10.1016/S2213-8587(16)30040-7
136. Lee SJ, Kim KH, Lee DJ, Kim P, Park J, Kim SJ, et al. MAST4 controls cell cycle in spermatogonial stem cells. Cell Prolif. (2023) 56:e13390. doi: 10.1111/cpr.13390
137. Koul S, McKiernan JM, Narayan G, Houldsworth J, Bacik J, Dobrzynski DL, et al. Role of promoter hypermethylation in Cisplatin treatment response of male germ cell tumors. Mol Cancer. (2004) 3:1–12. doi: 10.1186/1476-4598-3-16
138. Su J, Yang Y, Zhao F, Zhang Y, Su H, Wang D, et al. Study of spermatogenic and Sertoli cells in the Hu sheep testes at different developmental stages. FASEB J. (2023) 37:e23084. doi: 10.1096/fj.202300373R
139. Wu X, Yun D, Sang M, Liu J, Zhou L, Shi J, et al. Defects of microtubule cytoskeletal organization in NOA human testes. Reprod Biol Endocrinol. (2022) 20:1–17. doi: 10.1186/s12958-022-01026-w
140. Singh DK, Gamboa RS, Singh AK, Walkemeier B, Van Leene J, De Jaeger G, et al. NAR Breakthrough Article The FANCC–FANCE–FANCF complex is evolutionarily. Nucleic Acids Res. (2023) 51:2517. doi: 10.1093/nar/gkac1244
141. D'Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nat Rev Cancer. (2003) 3:23–34. doi: 10.1038/nrc970
142. Machida YJ, Machida Y, Chen Y, Gurtan AM, Kupfer GM, D'Andrea AD, et al. UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol Cell. (2006) 23:589–96. doi: 10.1016/j.molcel.2006.06.024
143. Xu L, Xu W, Li D, Yu X, Gao F, Qin Y, et al. FANCI plays an essential role in spermatogenesis and regulates meiotic histone methylation. Cell Death Dis. (2021) 12:780. doi: 10.1038/s41419-021-04096-7
144. Nepal M, Che R, Ma C, Zhang J, Fei P. FANCD2 and DNA damage. Int J Mol Sci. (2017) 18:1804. doi: 10.3390/ijms18081804
145. Chen X, Bosques L, Sung P, Kupfer GM. A novel role for non-ubiquitinated FANCD2 in response to hydroxyurea-induced DNA damage. Oncogene. (2016) 35:22–34. doi: 10.1038/onc.2015.68
146. Sondalle SB, Longerich S, Ogawa LM, Sung P, Baserga SJ. Fanconi anemia protein FANCI functions in ribosome biogenesis. Proc Natl Acad Sci. (2019) 116:2561–70. doi: 10.1073/pnas.1811557116
147. Zhang J, Wang J, Wu J, Huang J, Lin Z, Lin X. UBE2T regulates FANCI monoubiquitination to promote NSCLC progression by activating EMT. Oncol Rep. (2022) 48:1–13. doi: 10.3892/or
148. Lemonidis K, Arkinson C, Rennie ML, Walden H. Mechanism, specificity, and function of FANCD2-FANCI ubiquitination and deubiquitination. FEBS J. (2022) 289:4811–29. doi: 10.1111/febs.16077
149. Rickman KA, Lach FP, Abhyankar A, Donovan FX, Sanborn EM, Kennedy JA, et al. Deficiency of UBE2T, the E2 ubiquitin ligase necessary for FANCD2 and FANCI ubiquitination, causes FA-T subtype of Fanconi anemia. Cell Rep. (2015) 12:35–41. doi: 10.1016/j.celrep.2015.06.014
150. Tan W, Van Twest S, Murphy VJ, Deans AJ. ATR-mediated FANCI phosphorylation regulates both ubiquitination and deubiquitination of FANCD2. Front Cell Dev Biol. (2020) 8:2. doi: 10.3389/fcell.2020.00002
151. Kim JM, Parmar K, Huang M, Weinstock DM, Ruit CA, Kutok JL, et al. Inactivation of murine Usp1 results in genomic instability and a Fanconi anemia phenotype. Dev Cell. (2009) 16:314–20. doi: 10.1016/j.devcel.2009.01.001
152. Arafat M, Har-Vardi I, Harlev A, Levitas E, Zeadna A, Abofoul-Azab M, et al. Mutation in TDRD9 causes non-obstructive azoospermia in infertile men. J Med Genet. (2017) 54:633–9. doi: 10.1136/jmedgenet-2017-104514
153. Alavattam KG, Kato Y, Sin H-S, Maezawa S, Kowalski IJ, Zhang F, et al. Elucidation of the Fanconi anemia protein network in meiosis and its function in the regulation of histone modifications. Cell Rep. (2016) 17:1141–57. doi: 10.1016/j.celrep.2016.09.073
154. Lu C, Wen Y, Hu W, Lu F, Qin Y, Wang Y, et al. Y chromosome haplogroups based genome-wide association study pinpoints revelation for interactions on non-obstructive azoospermia. Sci Rep. (2016) 6:33363. doi: 10.1038/srep33363
155. Zhang W, Zhang Y, Zhao M, Ding N, Yan L, Chen J, et al. MicroRNA expression profiles in the seminal plasma of nonobstructive azoospermia patients with different histopathologic patterns. Fertil Steril. (2021) 115:1197–211. doi: 10.1016/j.fertnstert.2020.11.020
156. Osorio A, Bogliolo M, Fernández V, Barroso A, de la Hoya M, Caldés T, et al. Evaluation of rare variants in the new F anconi anemia gene ERCC 4 (FANCQ) as familial breast/ovarian cancer susceptibility alleles. Hum Mutat. (2013) 34:1615–8. doi: 10.1002/humu.22438
157. Ghieh F, Barbotin A, Swierkowski-Blanchard N, Leroy C, Fortemps J, Gerault C, et al. Whole-exome sequencing in patients with maturation arrest: a potential additional diagnostic tool for prevention of recurrent negative testicular sperm extraction outcomes. Hum Reprod. (2022) 37:1334–50. doi: 10.1093/humrep/deac057
158. Sakurai Y, Ichinoe M, Yoshida K, Nakazato Y, Saito S, Satoh M, et al. Inactivation of REV7 enhances chemosensitivity and overcomes acquired chemoresistance in testicular germ cell tumors. Cancer Lett. (2020) 489:100–10. doi: 10.1016/j.canlet.2020.06.001
159. Huselid E, Bunting SF. The regulation of homologous recombination by helicases. Genes. (2020) 11:498. doi: 10.3390/genes11050498
160. Dhingra N, Zhao X. Intricate SUMO-based control of the homologous recombination machinery. Genes Dev. (2019) 33:1346–54. doi: 10.1101/gad.328534.119
161. Ali AM, Pradhan A, Singh TR, Du C, Li J, Wahengbam K, et al. FAAP20: a novel ubiquitin-binding FA nuclear core-complex protein required for functional integrity of the FA-BRCA DNA repair pathway. Blood J Am Soc Hematol. (2012) 119:3285–94. doi: 10.1182/blood-2011-10-385963
162. Michl J, Zimmer J, Tarsounas M. Interplay between Fanconi anemia and homologous recombination pathways in genome integrity. EMBO J. (2016) 35:909–23. doi: 10.15252/embj.201693860
163. Le W, Qi L, Xu C, Xiang Z, Mao Z, Zhang J, et al. Preliminary study of the homologous recombination repair pathway in mouse spermatogonial stem cells. Andrology. (2018) 6:488–97. doi: 10.1111/andr.12481
164. Ahlskog JK, Larsen BD, Achanta K, Sørensen CS. ATM/ATR-mediated phosphorylation of PALB 2 promotes RAD 51 function. EMBO Rep. (2016) 17:671–81. doi: 10.15252/embr.201541455
165. Guo Y, Feng W, Sy SM, Huen MS. ATM-dependent phosphorylation of the Fanconi anemia protein PALB2 promotes the DNA damage response. J Biol Chem. (2015) 290:27545–56. doi: 10.1074/jbc.M115.672626
166. Jensen CFS, Wang D, Mamsen LS, Giwercman A, Jørgensen N, Fode M, et al. Sertoli and germ cells within atrophic seminiferous tubules of men with non-obstructive azoospermia. Front Endocrinol. (2022) 13:825904. doi: 10.3389/fendo.2022.825904
167. Sawyer SL, Tian L, Kähkönen M, Schwartzentruber J, Kircher M, U.o.W.C.f.M. Genomics, F.C. Consortium, et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discovery. (2015) 5:135–42. doi: 10.1158/2159-8290.CD-14-1156
168. Yang J, Qi L, Chiang H-C, Yuan B, Li R, Hu Y. BRCA1 antibodies matter. Int J Biol Sci. (2021) 17:3239. doi: 10.7150/ijbs.63115
169. Li M, Yu X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer Cell. (2013) 23:693–704. doi: 10.1016/j.ccr.2013.03.025
170. Cheng W-H, Kusumoto R, Opresko PL, Sui X, Huang S, Nicolette ML, et al. Collaboration of Werner syndrome protein and BRCA1 in cellular responses to DNA interstrand cross-links. Nucleic Acids Res. (2006) 34:2751–60. doi: 10.1093/nar/gkl362
171. Durocher F, Simard J, Ouellette J, Richard V, Labrie F, Pelletier G. Localization of BRCA1 gene expression in adult cynomolgus monkey tissues. J Histochem Cytochem. (1997) 45:1173–88. doi: 10.1177/002215549704500901
172. Adamczewska D, Słowikowska-Hilczer J, Walczak-Jędrzejowska R. The fate of leydig cells in men with spermatogenic failure. Life. (2022) 12:570. doi: 10.3390/life12040570
173. Sciurano RB, Rahn M, Pigozzi MI, Olmedo SB, Solari AJ. An azoospermic man with a double-strand DNA break-processing deficiency in the spermatocyte nuclei: case report. Hum Reprod. (2006) 21:1194–203. doi: 10.1093/humrep/dei479
174. Sharan SK, Pyle A, Coppola V, Babus J, Swaminathan S, Benedict J, et al. BRCA2 deficiency in mice leads to meiotic impairment and infertility. Development. (2004) 131:131–42. doi: 10.1242/dev.00888
175. Weinberg-Shukron A, Rachmiel M, Renbaum P, Gulsuner S, Walsh T, Lobel O, et al. Essential role of BRCA2 in ovarian development and function. New Engl J Med. (2018) 379:1042–9. doi: 10.1056/NEJMoa1800024
176. Shive HR, West RR, Embree LJ, Azuma M, Sood R, Liu P, et al. brca2 in zebrafish ovarian development, spermatogenesis, and tumorigenesis. Proc Natl Acad Sci. (2010) 107:19350–5. doi: 10.1073/pnas.1011630107
177. St Martin EC, Ferrer A, Wudhikarn K, Mangaonkar A, Hogan W, Tefferi A, et al. Clinical features and survival outcomes in patients with chronic myelomonocytic leukemia arising in the context of germline predisposition syndromes. Am J Hematol. (2021) 96:E327–30. doi: 10.1002/ajh.26250
178. Dosanjh MK, Collins DW, Schild D, Fan W, Lennon GG, Albala JS, et al. Isolation and characterization of RAD51C, a new human member of the RAD51 family of related genes. Nucleic Acids Res. (1998) 26:1179–84. doi: 10.1093/nar/26.5.1179
179. Badie S, Liao C, Thanasoula M, Barber P, Hill MA, Tarsounas M. RAD51C facilitates checkpoint signaling by promoting CHK2 phosphorylation. J Cell Biol. (2009) 185:587–600. doi: 10.1083/jcb.200811079
180. Qin J, Huang T, Wang J, Xu L, Dang Q, Xu X, et al. RAD51 is essential for spermatogenesis and male fertility in mice. Cell Death Discovery. (2022) 8:118. doi: 10.1038/s41420-022-00921-w
181. Braybrooke JP, Spink KG, Thacker J, Hickson ID. The RAD51 family member, RAD51L3, is a DNA-stimulated ATPase that forms a complex with XRCC2. J Biol Chem. (2000) 275:29100–6. doi: 10.1074/jbc.M002075200
182. Zhang Y-X, Li H-Y, He W-B, Tu C, Du J, Li W, et al. XRCC2 mutation causes premature ovarian insufficiency as well as non-obstructive azoospermia in humans. Clin Genet. (2019) 95:442–3. doi: 10.1111/cge.13475
183. Lee D-H, Pan Y, Kanner S, Sung P, Borowiec JA, Chowdhury D. A PP4 phosphatase complex dephosphorylates RPA2 to facilitate DNA repair via homologous recombination. Nat Struct Mol Biol. (2010) 17:365–72. doi: 10.1038/nsmb.1769
184. Voutsadakis IA. Landscape of BRIP1 molecular lesions in gastrointestinal cancers from published genomic studies. World J Gastroenterol. (2020) 26:1197. doi: 10.3748/wjg.v26.i11.1197
185. Peng M, Litman R, Xie J, Sharma S, Brosh RM Jr., Cantor SB. The FANCJ/MutLα interaction is required for correction of the cross-link response in FA-J cells. EMBO J. (2007) 26:3238–49. doi: 10.1038/sj.emboj.7601754
186. Kubota H, Brinster RL. Spermatogonial stem cells. Biol Reprod. (2018) 99:52–74. doi: 10.1093/biolre/ioy077
187. Vij SC, Sabanegh E Jr., Agarwal A. Biological therapy for non-obstructive azoospermia. Expert Opin Biol Ther. (2018) 18:19–23. doi: 10.1080/14712598.2018.1380622
188. Ishikawa T, Mizuta S, Yamaguchi K, Ohara Y, Doshida M, Takeuchi T, et al. P-030 Clinical outcomes of microdissection testicular sperm extraction (micro TESE) and intracytoplasmic sperm injection (ICSI) in non-obstructive azoospermia (NOA) with the history of cryptorchidism. Hum Reprod. (2023) 38:dead093. 397. doi: 10.1093/humrep/dead093.397
189. Elena M-B, Jean-Hugues G, Belen P-OA, Christophe L. Beyond current treatment of Fanconi Anemia: What do advances in cell and gene-based approaches offer? Blood Rev. (2023) 60:101094. doi: 10.1016/j.blre.2023.101094
190. Verhoeyen E, Jose Roman-Rodriguez F, Cosset F-L, Levy C, Rio P. Gene therapy in Fanconi anemia: a matter of time, safety and gene transfer tool efficiency. Curr Gene Ther. (2016) 16:297–308. doi: 10.2174/1566523217666170109114309
Keywords: non-obstructive azoospermia, Fanconi anemia pathway, Fanconi anemia gene, interstrand crosslinks, homologous recombination
Citation: Xu H, Zhang Y, Wang C, Fu Z, Lv J, Yang Y, Zhang Z, Qi Y, Meng K, Yuan J and Wang X (2024) Research progress on the fanconi anemia signaling pathway in non-obstructive azoospermia. Front. Endocrinol. 15:1393111. doi: 10.3389/fendo.2024.1393111
Received: 28 February 2024; Accepted: 13 May 2024;
Published: 23 May 2024.
Edited by:
Marjan Sabbaghian, Royan Institute, IranReviewed by:
Panneerselvam Jayabal, The University of Texas Health Science Center at San Antonio, United StatesCopyright © 2024 Xu, Zhang, Wang, Fu, Lv, Yang, Zhang, Qi, Meng, Yuan and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kai Meng, bWVuZ2thaTUyMTg4OEAxMjYuY29t; Jinxiang Yuan, eXVhbmppbnhpYW5nMThAMTYzLmNvbQ==; Xiaomei Wang, eGljaGViaW5ncWluZ0AxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.