 Min Wang1
Min Wang1 Bo-Qi Zhang2
Bo-Qi Zhang2 Shuai Ma1
Shuai Ma1 Ying Xu1Dong-Hai Zhao3Jing-Shun Zhang1
Ying Xu1Dong-Hai Zhao3Jing-Shun Zhang1 Chun-Jin Li2*Xu Zhou2*
Chun-Jin Li2*Xu Zhou2* Lian-Wen Zheng1*
Lian-Wen Zheng1*- 1Department of Obstetrics and Gynecology, The Second Hospital of Jilin University, Changchun, China
- 2College of Animal Sciences, Jilin University, Changchun, China
- 3Department of Pathology, Jilin Medical College, Jilin, China
Polycystic ovarian syndrome (PCOS) is a common heterogeneous reproductive endocrine metabolic disorder in women of reproductive age characterized by clinical and biochemical hyperandrogenemia, ovulation disorders, and polycystic ovarian morphology. Ferroptosis is a novel type of cell death driven by iron accumulation and lipid peroxidation. Ferroptosis plays a role in maintaining redox balance, iron metabolism, lipid metabolism, amino acid metabolism, mitochondrial activity, and many other signaling pathways linked to diseases. Iron overload is closely related to insulin resistance, decreased glucose tolerance, and the occurrence of diabetes mellitus. There is limited research on the role of ferroptosis in PCOS. Patients with PCOS have elevated levels of ferritin and increased reactive oxygen species in ovarian GCs. Studying ferroptosis in PCOS patients is highly important for achieving personalized treatment. This article reviews the progress of research on ferroptosis in PCOS, introduces the potential connections between iron metabolism abnormalities and oxidative stress-mediated PCOS, and provides a theoretical basis for diagnosing and treating PCOS.
1 Introduction
PCOS is a complex endocrine and metabolic disorder (1). The primary characteristics of PCOS patients are long-term anovulation or infrequent ovulation, insulin resistance (IR), hyperandrogenemia, polycystic ovarian morphology, and decreased female fertility (2–4). In addition to having abnormal reproductive function, many PCOS patients suffer from metabolic syndrome, IR, impaired glucose tolerance, obesity, atherosclerosis, and other metabolic abnormalities (5). At present, it is believed that the occurrence of PCOS is mainly related to genetic factors, environmental factors, and metabolic factors (6, 7). The specific causes of PCOS have not been fully elucidated (8, 9). Ferroptosis is caused by iron buildup and lipid peroxidation. It differs from cell apoptosis and necrosis (10). Ferroptosis is involved in regulating the occurrence and progression of diseases such as cancer, respiratory system diseases, cardiovascular system diseases, diabetes mellitus, and urinary system diseases and plays a key role in disease treatment (11) (Figure 1). There is limited and incomplete research on ferroptosis in the reproductive system. The role of ferroptosis in the reproductive system deserves further investigation (12, 13). Ferroptosis in PCOS is accompanied by the dysregulation of mitochondrial dynamics, the promotion of an inflammatory response, and the intensification of oxidative stress (14–16). This article reviews the correlation between ferroptosis and PCOS, providing ideas for exploring the underlying mechanisms of PCOS.
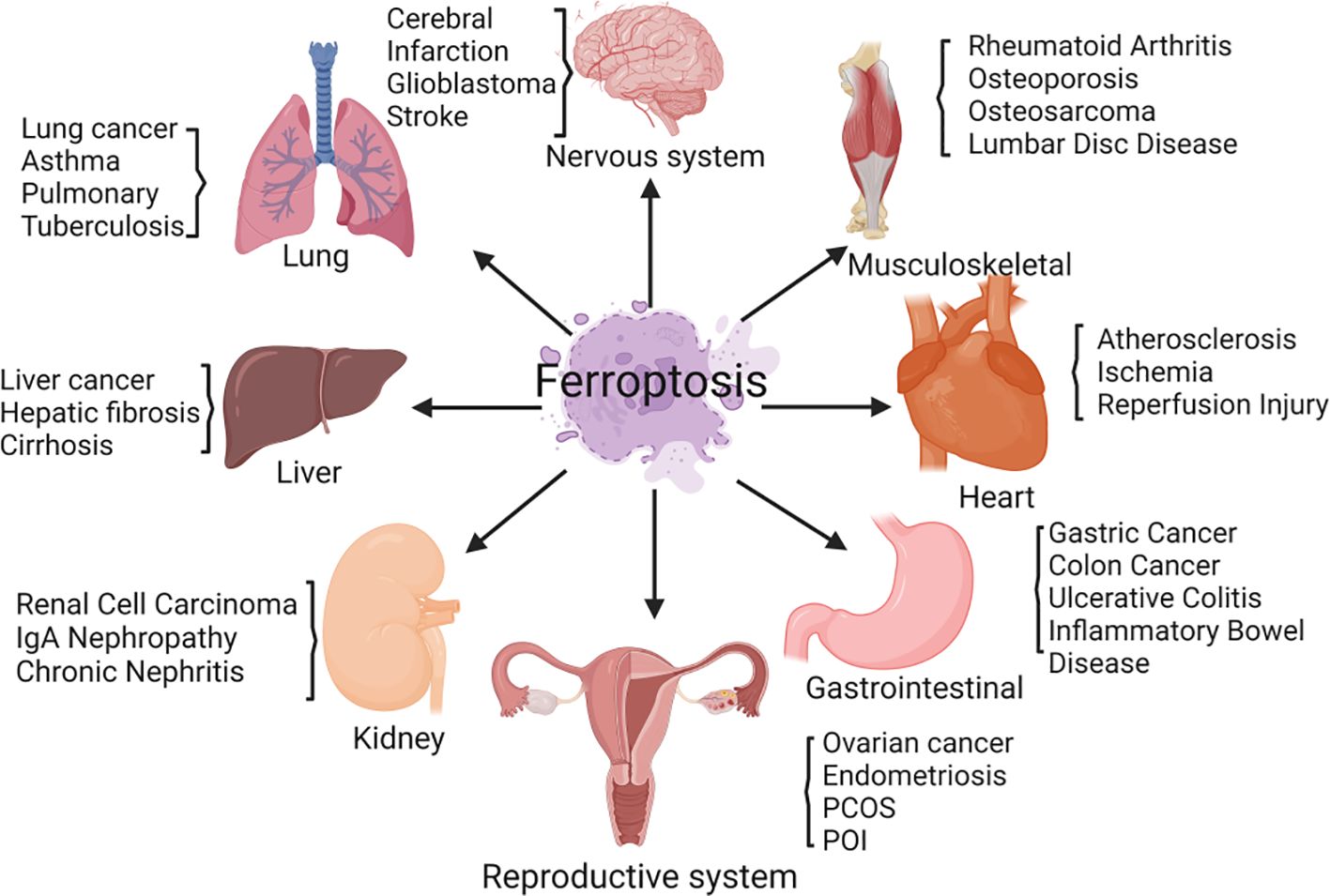
Figure 1 Ferroptosis is involved in regulating the occurrence and progression of various diseases, including those of the nervous system, respiratory system, cardiovascular system, diabetes mellitus, urinary system, and female reproductive system.
2 Ferroptosis
In 2012, Dixon et al. proposed ferroptosis as a novel iron-dependent programmed cell death mechanism that is distinct from apoptosis, necrosis, and autophagy (17) (Table 1). Its main characteristics are as follows (1): During the process of cell death, a large amount of iron accumulates, and lipid peroxidation occurs, activating signaling pathways such as oxidative stress (18) (2); In the ultrastructure, cell atrophy, membrane rupture, and mitochondrial membrane wrinkling, and no significant nuclear morphological changes are observed (19). Ferroptosis is a nonenzymatic and enzymatic reaction that occurs under iron catalysis, leading to lipid peroxidation of cell membranes (20). Polyunsaturated fatty acids (PUFAs) are the main targets of lipid peroxidation of cell membranes (21). Glutathione peroxidase 4 (GPX4) regulates ferroptosis (22). Under antioxidant conditions, GPX4 can reduce the accumulation of intracellular reactive oxygen species (ROS) and reduce the sensitivity of cells to ferroptosis, thus affecting the occurrence of ferroptosis (23).
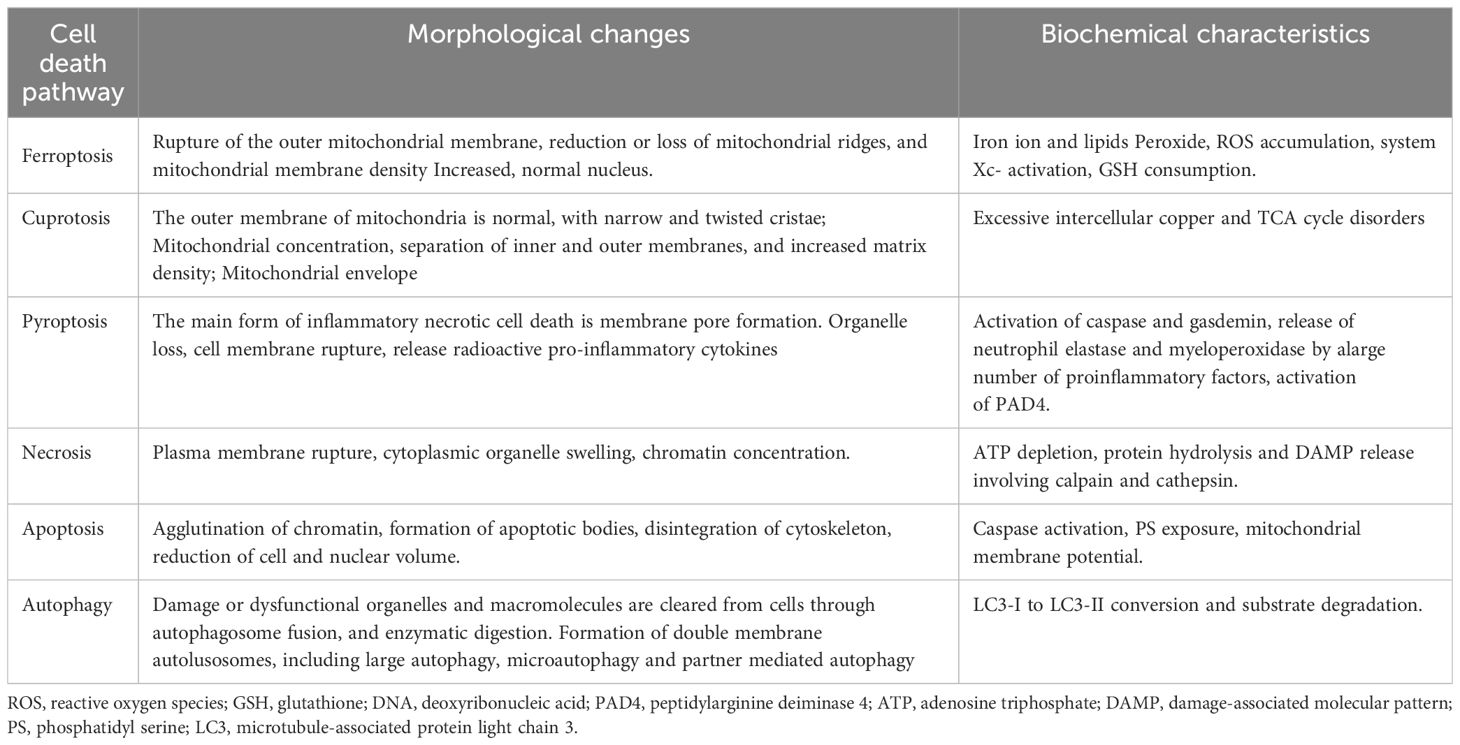
Table 1 Comparison of cell death pathways.
The Fenton reaction first removes a hydrogen atom from poly-unsaturated fatty acid-phosphatidyl ethanolamine (24). This leads to the formation of carbon-centered phospholipids, which then react with molecular oxygen to generate PLOO·, which can remove hydrogen from another PUFA and form phospholipid hydroperoxides (25). If antioxidants such as GPX4 are unable to promptly convert phospholipid hydroperoxides to the appropriate alcohols, phospholipid hydroperoxides and lipid free radicals will react with polyunsaturated fatty acid-phosphatidyl ethanolamine to further promote the production of phospholipid hydroperoxides (26). Ultimately, this leads to the production of many secondary products, such as lipid peroxidation products, leading to ferroptosis (27). Ferroptosis mainly causes oxidative metabolic disorders of cell membrane phospholipids through iron metabolism, lipid metabolism, and amino acid metabolism (28) (Figure 2).
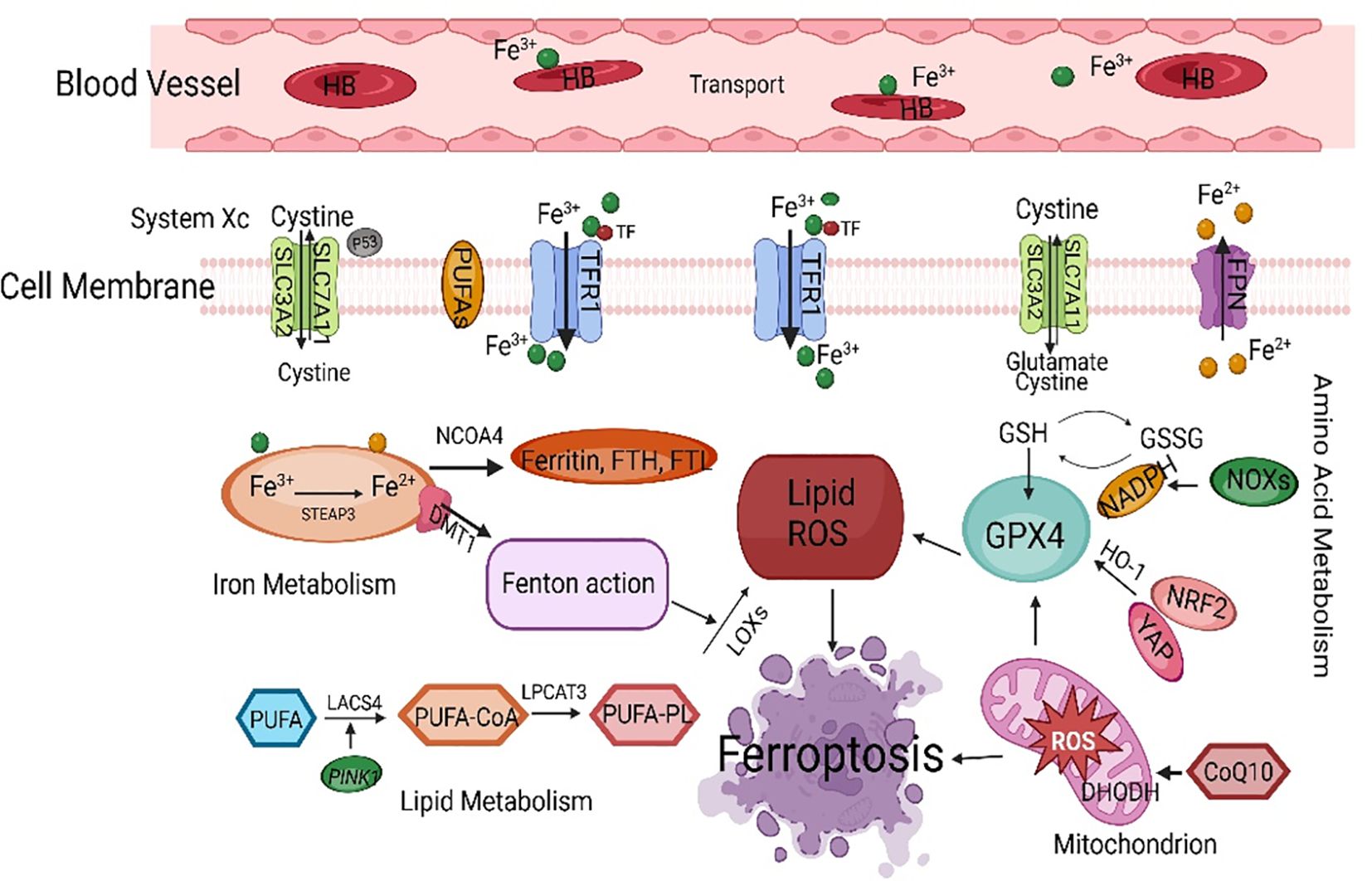
Figure 2 The main regulatory mechanisms of ferroptosis. SLC7A11, solute carrier family 7 member 11; PUFAs, polyunsaturated fatty acids; TFCR1, transferrin receptor 1; FPN, ferroportin; NCOA4, nuclear receptor coactivator 4; FTH, ferritin heavy chain; FTL, ferritin light chain; GSH, glutathione; GPX4, glutathione peroxidase 4; HO-1, heme oxygenase-1; NRF2, nuclear factor-erythroid 2-related factor 2; ROS, reactive oxygen species; LACS4, acyl-CoA synthetase long-chain family member 4.
2.1 Ferroptosis and iron metabolism
2.1.1 Iron uptake, storage, and release
Ferroptosis requires the involvement of iron (29). The sensitivity of cells to ferroptosis depends on the input, output, storage, and transport of iron. There are two forms of iron in cells: Fe2+ and Fe3+ (30). A large amount of ROS is generated during the mutual conversion of Fe3+ and Fe2+ through redox reactions, leading to tissue damage and ferroptosis (31). Fe3+ has strong stability and is responsible for the storage and transportation of iron in the body; Fe2+ has an electron transport ability, and proteins containing Fe2+ can participate in various redox reactions (32). Transferrin (TF) carries Fe3+ in the bloodstream and transfers iron in cells through transferrin receptor 1 (TFCR1). Fe3+ is reduced to Fe2+ by the six-transmembrane epithelial antigen of prostate 3, which is stored in the ferritin heavy chain (FTH) and ferritin light chain (FTL) (33). Fe2+ is stored in ferritin (Fn) and becomes a part of the labile iron pool, which plays a dominant role in ferroptosis. TFCR1 is considered a marker protein for ferroptosis, and knocking out TFCR1 can block ferroptosis. In the absence of enough iron in cells, an “iron starvation response” begins, increasing iron availability by increasing TFCR1 and decreasing the levels of FTH and ferroportin (FPN) (34).
2.1.2 Iron homeostasis
Under normal circumstances, intracellular iron maintains a subtle balance through the iron transport system (35). Some iron is used for the biosynthesis of hemoglobin and iron sulfur clusters and for DNA synthesis. Cellular iron homeostasis also depends on iron regulatory proteins and iron-responsive elements (36). Iron regulatory proteins can bind to iron-responsive element mRNAs, regulating their translation process. Aberrant expression or dysfunction of the divalent metal transporter 1, TFRC1, and Fn genes and FPN leads to intracellular iron imbalance (37). Iron regulatory protein 2 can enhance the sensitivity of cells to erastin-induced ferroptosis by inhibiting the expression of FTH and FTL (38). When the plasma iron level meets the systemic iron demand, the liver will increase the secretion of hepcidin into the blood, reducing the plasma iron level and maintaining iron homeostasis in the body (39). Although every cell requires iron to generate energy, high iron levels can induce inflammation, oxidative stress, and lipid peroxidation in the cell membrane, leading to ferroptosis (40).
Iron homeostasis dysregulation is involved in ferroptosis, and multiple iron metabolism regulatory factors work together to maintain iron homeostasis (41). As mentioned, Fn autophagy is crucial for regulating ferroptosis (42). Nuclear receptor coactivator 4 (NCOA4) helps lysosomes breakdown intracellular Fn through autophagy, which releases free iron and causes oxidative damage (43). This selective autophagy process is called iron autophagy. Knockout of NCOA4 can inhibit ferroptosis caused by decreased amino acid or cysteine levels (44). Research has shown that knocking out autophagy-related genes 5 (ATG5) and ATG7 can reduce intracellular Fe2+ levels and lipid peroxidation, inhibiting ferroptosis (45). In addition to selective autophagy, activating the ubiquitin−proteasome system can promote Fn degradation. As an antioxidant, ubiquitin captures lipid peroxidation free radicals, prevents lipid peroxidation generation, and inhibits ferroptosis. Tang et al. reported that in treated rat hearts, ubiquitin-specific protease 7 increases iron uptake and promotes ferroptosis by activating the P53/TFRC1 pathway (46).
2.2 Ferroptosis and lipid metabolism
The conversion of lipids into membrane phospholipids and the occurrence of peroxidation are necessary conditions for ferroptosis (47). Lipid metabolism is closely related to ferroptosis. Most ROS related to ferroptosis originates from the Fenton and Haber-Weiss reactions, which then interact with PUFAs on the lipid membrane to form ROS (48). Phosphatidylethanolamine and arachidonic acid are essential membrane phospholipids that cause ferroptosis in the cell membrane. Lipoxygenases are nonheme iron-dependent dioxygenases that target polyunsaturated fatty acids (PUFAs). With the participation of iron in the cytoplasm, lipid free radicals are formed, which can directly oxidize PUFAs and PUFA-containing lipids in the biofilm, leading to cell damage (49). A decrease in lipoxygenase expression can also effectively improve ferroptosis induced by erastin (50). However, lipoxygenases cannot prevent ferroptosis induced by RSL3 (51). Although lipoxygenases do not have extensive regulatory effects on ferroptosis, they play essential roles in specific ferroptosis mechanisms.
Acyl-CoA synthetase long chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 are key enzymes involved in the process of lipid peroxidation (52). ACSL4 and lysophosphatidylcholine acyltransferase 3, which are involved in the synthesis of PUFAs, play essential roles in the ferroptosis pathway (53). ACSL4 binds PUFA to coenzyme A through acylation and further undergoes an esterification reaction with phosphatidylethanolamine under the action of LPCAT to generate PUFA-PE (54). Continuous oxidation reactions and consumption of PUFAs may alter the fluid structure of cell membranes, thereby increasing membrane permeability and ultimately leading to cell death. Inhibiting the expression of ACSL4 can increase the resistance of cells to ferroptosis and can be a target for inhibiting ferroptosis, which may provide new ideas for diagnosing and treating PCOS. Ferroptosis may play a role not only in pathological conditions but also in physiological processes. The normal development of the human fetal immune system depends on sufficient dietary intake of PUFAs (55). To summarize, studying the correlation between lipid metabolism and ferroptosis has essential research value (56).
2.3 Ferroptosis and amino acid metabolism
Ferroptosis is closely related to amino acid metabolism (57). The consumption of glutathione (GSH) can lead to the inactivation of GPX4 (58). Cysteine synthesizes GSH in the body through the cystine/glutamic acid reverse transporter (System Xc -) and the transsulfuration pathway (59). System Xc -, located on the cell membrane, is a heterodimer linked by disulfide bonds and contains the heavy chain subunits solute carrier family 3 member 2 and solute carrier family 7 member 11 (SLC7A11) (60). System Xc - is responsible for transporting extracellular cysteine into the cell. Glutamate exchanges cysteine at a 1:1 ratio, transporting intracellular glutamate to the outside of the cell (61). Cysteine provides the raw material for intracellular GSH synthesis (62). Another source of cysteine is cysteine sulfide, which is formed through the reverse sulfurization of methionine (63).
2.3.1 Negative regulatory factors of System Xc -
Inhibiting cysteine uptake by System Xc - can inhibit the synthesis of GSH, leading to the accumulation of peroxides in the body and the induction of ferroptosis (64). System Xc—inhibitors such as erastin, sulfasalazine, sorafenib, and glutamate—are considered class I ferroptosis inducers, and inhibition of System Xc - leads to compensatory transcriptional upregulation of SLC7A11 (65). SLC7A11, an essential component of System Xc -, is an upstream regulatory factor of ferroptosis (66). SLC7A11 can reduce lipid peroxidation accumulation and prevent cells from entering the ferroptosis program by introducing cysteine and promoting GSH synthesis (67). Song et al. reported that Beclin 1 directly stops System Xc - activity by attaching to the core region of SLC7A11 and encouraging ferroptosis (68). Nuclear factor erythroid 2-related factor 2 (NRF2) is a crucial antioxidant transcription factor. Silencing NRF2 can significantly reduce the expression of SLC7A11 and heme oxygenase-1 (HO-1) and inhibit ferroptosis (69).
2.3.2 GPX4: a core regulator of ferroptosis
Ferroptosis is related to the inactivation of GPX4 (70). As an important antioxidant in cells, GPX4 is a key ferroptosis regulator. GSH is used as a reducing substrate to promote the transformation of lipid peroxidation products into hydroxyl compounds, preventing ferroptosis in cells and protecting the structure and function of cell membranes from interference and damage (71). Under normal circumstances, fatty acid hydroperoxides can be converted into fatty acid alcohols through GPX4 mediation (72). Inhibiting GPX4 can interfere with intracellular iron homeostasis and reduce lipid peroxidation. The ferroptosis inducers RSL3 and erastin can both inactivate GPX4. RSL-3 blocks GPX4, which increases the levels of ROS and malondialdehyde (MDA) inside cells and promotes ferroptosis by blocking the SLC7A11/GSH/GPX4 pathway (73). Erastin indirectly inhibits GPX4 by inhibiting cysteine input, leading to cell membrane damage and death. Selenocysteine is an amino acid found in the active center of GPX4 (74). It can maintain GPX4 activity and help eliminate lipid peroxidation, which stops ferroptosis. Therefore, selenium deficiency can inhibit GPX4 activity and induce ferroptosis. GPX4 can also be turned off directly by squalene synthase, HMG CoA reductase, and other enzymes, which can change reduction reactions (75).
2.4 Ferroptosis and antioxidant pathways
Under physiological conditions, when cells undergo lipid peroxidation, multiple antioxidant pathways counteract this change. Research has shown that the intracellular ferroptosis antioxidant system is related to the SLC7A11/GSH/GPX4 signaling pathway (76). GPX4 reacts with GSH and lipid peroxidation products, efficiently clearing accumulated lipid peroxidation products and maintaining normal physiological functions. Ferroptosis suppressor protein 1 (FSP1) is a newly discovered inhibitory protein that inhibits GPX4 deficiency (77). The NAD(P)H/CoQ10/FSP1 signaling pathway is a crucial ferroptosis regulatory pathway (78). GTP cyclohydrolase 1 is involved in ferroptosis resistance, and its mechanism involves the generation of tetrahydrobipterin (BH4), which has redox activity, from GTP (79). BH4, a powerful antioxidant that captures free radicals, can promote CoQH2 regeneration and combat lipid peroxidation by activating downstream molecules such as GTP cyclohydrolase 1. These results indicate that the NADH-FSP1 CoQ10 and GTP cyclohydrolase 1/BH4 pathways work together with the SLC7A11/GPX4/GSH pathway to inhibit ferroptosis (80). Mao et al. reported that there is a ferroptosis system in mitochondria mainly composed of dihydrolactate dehydrogenase, which also induces lipid peroxidation in the mitochondrial membrane by promoting the reduction of CoQ10 to combat ferroptosis (81).
2.5 Ferroptosis and other regulatory mechanisms
2.5.1 p53-mediated ferroptosis
Research has shown that p53 has a complex dual mechanism for regulating cellular ferroptosis through transcription and translation (82). P53 can inhibit cysteine uptake by downregulating SLC7A11, reducing GPX activity and GSH synthesis and subsequently causing intracellular ROS accumulation and ferroptosis (83). On the other hand, p53 can promote the activity of glutaminase 2 and spermidine/spermine N1-acetyltransferase 1 (SAT1). Glutaminase 2 catalyzes the degradation of glutamine to glutamic acid, and the activation of SAT1 induces lipid peroxidation. The overexpression of glutaminase 2 and SAT1 promotes PUFA oxidation and lipid peroxidation, leading to ferroptosis. The transcription target gene cyclin-dependent kinase inhibitor 1A is used to reduce GSH and ROS and delay ferroptosis (84). The specific mechanism by which P53 regulates ferroptosis needs further study.
2.5.2 The Keap1-NRF2 pathway mediates ferroptosis
The NRF2 transcription factor stimulates the production of NADPH by blocking the expression of antioxidant genes and increasing the expression of enzymes in the pentose phosphate pathway, which decreases the sensitivity of cells to ferroptosis (85). Under normal physiological conditions, activation of the Keap1-NRF2 signaling pathway promotes the activation of System Xc - and the expression of GPX4 and accelerates cysteine glutamate transport, thereby clearing accumulated lipid peroxidation and inhibiting ferroptosis (86). Activation of NRF2 reduces iron absorption, limits ROS production, and enhances cellular antioxidant capacity (87). P62 strictly controls NRF2 and can inhibit ferroptosis. P62 is an autophagic receptor that directly inhibits Keap1 while promoting NRF2 activation (88). NRF2 regulates multiple targets, such as genes regulating GSH synthesis and encoding antioxidant proteins (89). The iron-chelating enzymes HO-1, FTH, and FTL are all strictly controlled by NRF2 (90). Glutamate cysteine ligase, GSH synthase, and SLC7A11 are also transcriptional targets of NRF2 (91).
2.6 The interaction between ferroptosis and other types of cell death
Ferroptosis may interact with other types of cell death (92). Anthraquinone modifications can significantly upregulate the expression of glucose-regulated protein 78, activate transcription factor 4, and downregulate the expression of the essential ferroptosis protein GPX4. Endoplasmic reticulum stress induces apoptosis and accompanies ferroptosis. The autophagy pathway can degrade ferritin, while ferroptosis regulatory proteins can regulate autophagy (93). Ferritin autophagy and lipid autophagy can promote ferroptosis by regulating iron metabolism and lipid peroxidation (94). Under oxidative stress, autophagy protects mitochondrial integrity by clearing ROS, preventing cell apoptosis, and exerting a protective effect (95). Excessive ROS-induced autophagy can also lead to cell death (96). Lipid peroxidation can attach to particular mitochondria and autophagy-related proteins, leading to autophagic cell death and cellular dysfunction (97). In summary, ferroptosis interacts with and promotes other types of cell death.
3 The role of ferroptosis in female reproductive disorders
3.1 The impact of ferroptosis on the process of follicular development
ROS and antioxidants in the ovaries play critical regulatory roles during oocyte maturation, fertilization, and embryonic development and implantation (98). Studies have shown that excessive iron in follicular fluid significantly increases ROS levels in mouse oocytes, leading to a decrease in the maturation rate of oocytes (99). Recurrent bleeding from ovarian lesions can lead to iron overload, increased ferroptosis, and decreased ovarian function, affecting follicular development and oocyte quality. ROS in follicles promotes apoptosis in most follicular cells (100). Studies have shown that blocking the production of GSH increases antral follicular atresia in rats (101). Research has shown that the expression of TF in early follicular atresia is significantly reduced, while the expression of the iron chaperone protein PCBP is increased considerably. During follicular development, Basonclin1 maintains lipid metabolism and redox homeostasis in oocytes. Research has confirmed abnormal levels and pathways of ferroptosis-related indicators in basonclin1-truncated mouse oocytes (102). Neurofibromin 2 expression was significantly reduced in basonclin1-deficient oocytes, while the expression levels of yes-associated protein, TFR, and ACSL4 increased considerably, triggering oocyte ferroptosis (103).
3.2 Ferroptosis of GCs leads to immature oocytes
Oogenesis is achieved through the interaction between the oocyte and the microenvironment of the follicle (104). The production of various cytokines and hormones in follicular fluid mainly relies on ovarian GCs, which play an essential role in oogenesis (105). Excessive iron in the internal environment can cause ferroptosis in ovarian GCs, which slows oocyte maturation and follicle development, increasing the risk of infertility (106, 107). Researchers have shown that a TFR-mediated increase in iron uptake in GCs induces the release of ROS, mitochondrial autophagy, and lipid peroxidation (108). The levels of FTH, TF, and TFRC in the ovarian GCs of infertile women are much lower than those in the ovarian GCs of healthy women (109). This finding suggested that FTH is a crucial regulator of ovarian follicle development and atresia (110). CircRHBG competes with SLC7A11 for binding to miR-515-5p, inhibiting GC ferroptosis. Therefore, the knockout of circRHBG can promote ferroptosis (111).
3.3 Oxidative stress impairs reproductive function
Mitochondrial dysfunction and excessive ROS production are characteristics of ferroptosis, and mitochondrial dysfunction further promotes ROS production, leading to oxidative stress, which induces the development and exacerbation of ferroptosis (112, 113). The other key indicator of ferroptosis is an increase in the levels of MDA, the final product of lipid oxidation (114). MDA can affect the activity of respiratory chain complexes and critical enzymes in mitochondria and aggravate membrane damage. A reduction in mitochondrial DNA is correlated with IR, hyperandrogenemia, and polycystic ovarian morphology in women with PCOS (115–117). Oxidative stress, ferroptosis, and PCOS interact with each other (118). An imbalance in total antioxidant levels in the serum of women with PCOS can exacerbate cell damage and reduce cellular defense ability (119). By comparing the oxidative stress indices of women with PCOS and healthy women, it was found that women with PCOS have significantly greater GPX and GSH reductase activities (120). The concentration of these substances directly affects the maturation and quality of oocytes, fertilization, and embryonic development (121). Metabolomic analysis of follicular fluid from clinical PCOS patients revealed mitochondrial dysfunction, oxidation−reduction potential imbalance, and increased oxidative stress in cumulus cells (122). The elevated levels of autoantibodies and ROS in the serum of PCOS patients suggest that oxidative stress may be one of the critical reasons for the abnormal endometrial environment in PCOS patients (123). Insulin is the primary regulator of oxidative phosphorylation, and its secretion can directly affect mitochondrial function (124, 125).
3.4 The impact of ferroptosis on pregnancy outcomes
Iron homeostasis plays an essential role in maintaining pregnancy. In vitro experimental studies have shown that appropriate iron in protein-free embryo culture medium can promote embryonic development, while iron overload is not conducive to embryonic development. Ferroptosis is associated with placental injury or miscarriage (126). During the abortion process, the large amount of ROS generated increases the generation of lipid peroxidation at the maternal-fetal interface, providing a primary condition for ferroptosis. Ferroptosis is triggered in the uterus and placenta of PCOS rats with hyperandrogenemia and IR. Compared with those in the control group, the expression levels of GPX4 and GSH in the uterus and placenta of PCOS rats were lower, and the expression levels of the ferroptosis-related genes ACSL4, TFCR1, SLC7A11, and glutamate cysteine ligase C were significantly increased (127). Ferroptosis is closely related to spontaneous preterm birth, and PLA2G6 can alleviate ferroptosis caused by GPX4 inhibition during pregnancy in mice (128).
4 Ferroptosis in PCOS
PCOS is a reproductive endocrine disorder that not only affects women’s reproductive and physiological health but also leads to complications such as diabetes mellitus type 2 (T2DM), obesity, familial cardiovascular disease, and cardiovascular disease (129). PCOS has transcended the field of obstetrics and gynecology and affects various significant systems throughout the body, seriously threatening women’s physical and mental health, affecting their quality of life, and causing lifelong endocrine and metabolic diseases (130, 131) (Figure 3). The etiology of PCOS has not yet been clarified, although much work has been done on this topic over the past few decades (132). The onset of PCOS involves multiple factors, such as hyperandrogenemia, IR, and abnormal follicular development (133–135). Ferroptosis affects the proliferation and apoptosis of granulosa cells (GCs), affecting endocrine and metabolic processes (Figure 4).
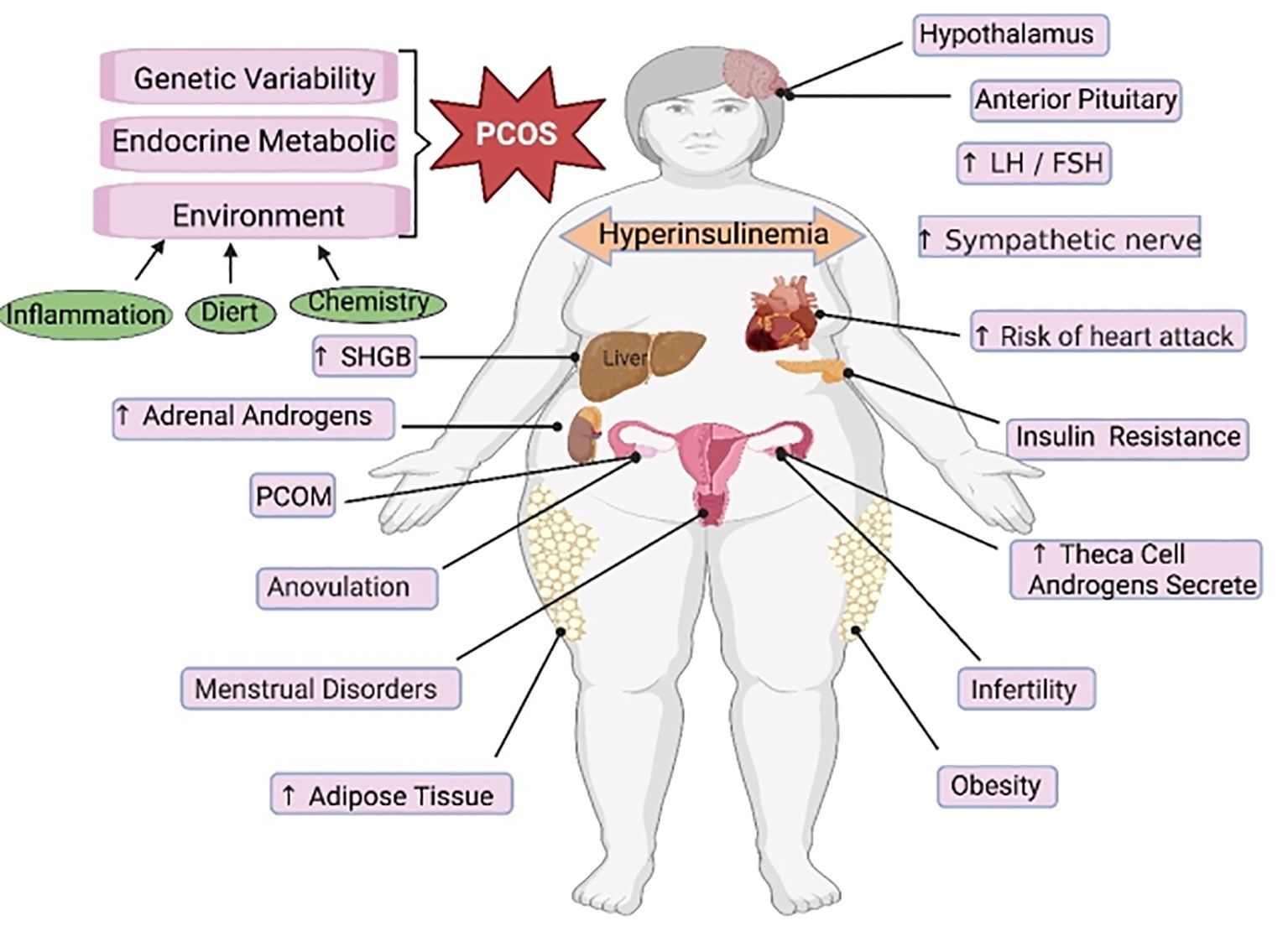
Figure 3 The main regulatory mechanisms of ferroptosis in PCOS. PCOS, polycystic ovarian syndrome; SHGB, sex hormone-binding globulin; LH, luteinizing hormone; FSH, follicle-stimulating hormone.
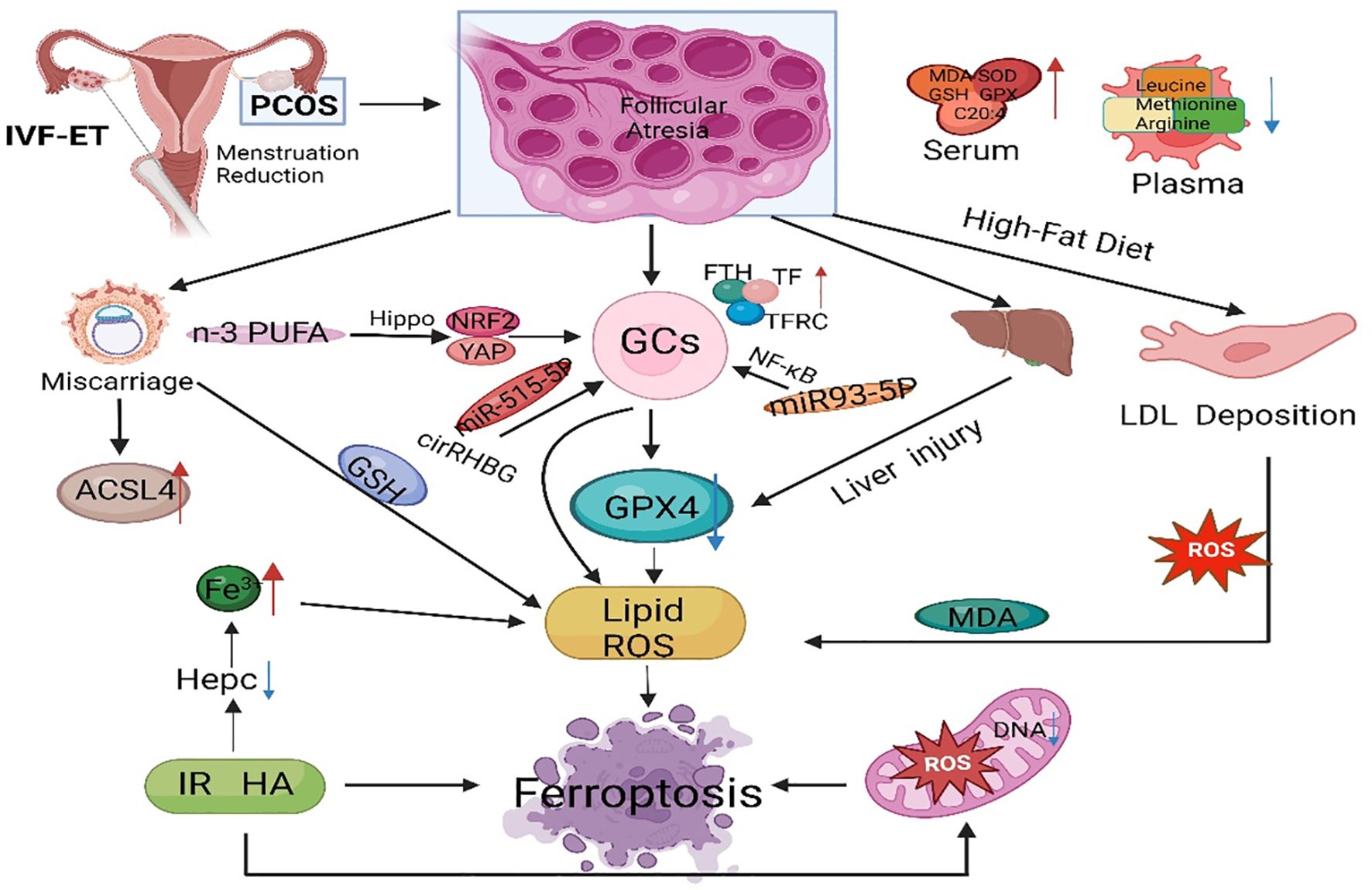
Figure 4 The main regulatory mechanisms of ferroptosis in PCOS. PCOS, polycystic ovarian syndrome; GCs, granulosa cells; GPX4, glutathione peroxidase 4; FTH, ferritin heavy chain; TF, transferrin; TFRC, transferrin receptor; IVF-ET, in vitro fertilization and embryo transfer; PUFA, polyunsaturated fatty acid; ACSL4, acyl-CoA synthetase long chain family member 4; IR, insulin resistance; ROS, reactive oxygen species; MDA, malondialdehyde; GSH, glutathione; NRF2, nuclear factor-erythroid 2-related factor 2.
Iron is an essential trace element for life and an important component of hemoglobin, myoglobin, and various enzymes (136). Iron participates in multiple critical physiological and biochemical processes in the body, including oxygen transport, DNA biosynthesis, and ATP synthesis (137). Research has shown that regardless of obesity, the serum Fn levels in PCOS patients are significantly greater than those in control individuals, indicating that abnormal iron metabolism may be involved in the occurrence and development of PCOS (138). Iron overload occurs in PCOS patients, potentially due to chronic oligomenorrhea and decreased hepcidin concentration (139). Research has shown that the serum Fn concentration is directly proportional to the severity of menstrual dysfunction, suggesting that the iron retention effect of chronic oligomenorrhea may be associated with increased iron storage in some PCOS patients (140). The compensatory hyperinsulinemia caused by PCOS may promote iron absorption (141, 142). Additionally, with the combined action of IR and excessive androgen in PCOS patients, TF inhibition is decreased, iron absorption in the intestine is increased, and iron release from macrophages is decreased, leading to iron overload (143).
4.1 Ferroptosis is involved in endocrine and metabolic disorders in PCOS
Iron overload can affect glucose metabolism, leading to IR and exacerbating metabolic abnormalities in PCOS patients (144). On the one hand, iron can affect the secretion and sensitivity of insulin and inhibit liver glycogen production, and insulin secretion decreases with increasing liver iron storage, leading to systemic hyperinsulinemia. On the other hand, insulin can promote iron absorption and Fn synthesis while also promoting glucose transport. Excessive androgen and insulin resistance can activate ferroptosis in the uterus and placenta of pregnant women with PCOS (145). The plasma levels of leucine, isoleucine, methionine, glutamine, and arginine were much lower in PCOS patients than in healthy controls (146). Compared to those in healthy women, serum bioactive lipid levels are much lower in women with PCOS (147). Arachidonic acid levels in the serum of PCOS rats were significantly greater than those in the serum of control rats (148). Phosphatidylethanolamine combined with arachidonic acid is an essential phospholipid that causes ferroptosis, which means that there are appropriate conditions for ferroptosis in PCOS patients (149).
4.2 Oxidative stress mediates metabolic abnormalities in PCOS
Oxidative stress is closely related to obesity, IR, and HA in PCOS, and the induced apoptosis of ovarian GCs leads to follicular atresia, which is one of the mechanisms of ovulation disorders in PCOS (150). SOD is an important antioxidant enzyme for scavenging oxygen-free radicals and catalyzes the dismutation reaction of ROS to eliminate oxygen-free radicals and reduce damage to ovarian cells (151). MDA levels are positively correlated with BMI, triglyceride levels, low-density lipoprotein levels, systolic blood pressure, diastolic blood pressure, and HOMA-IR (152). Research has shown that ROS produced by monocytes in the pancreas plays an essential role in the abnormal development of cells (153). ROS-induced NF-κB can enter the nucleus and bind to chromatin to promote tumor necrosis factor-α (TNF-α) (154). Transcription can activate the PI3K/Akt/mTOR pathway to inhibit insulin secretion (155). In PCOS animal models, free fatty acids in ovarian tissue increase, glucose oxidation is inhibited, free fatty acid oxidation is enhanced, and ROS increase through the TCA cycle, leading to IR (156).
4.3 Ferroptosis and a high-fat diet
Obesity is closely related to PCOS and plays a crucial role in the occurrence and development of PCOS (157). In obese PCOS patients, long-term high-fat diets can induce inflammatory reactions and oxidative damage and exacerbate iron deposition, which is an essential factor influencing ferroptosis (158). A long-term high-fat diet can cause low-density lipoprotein, which is deposited in the endothelium of tissues. The prolonged accumulation of low-density lipoprotein can induce an oxidative stress response (159). Oxidative stress is one of the pathogenic mechanisms of PCOS, and many studies have confirmed that oxidative stress indicators are positively correlated with obesity. Compared with those in nonobese patients, the serum levels of the antioxidant substances MDA, SOD, GSH, and GPX were significantly greater in the obese PCOS group (160). In the environment of mature follicles, oxidative damage can further trigger chronic inflammatory reactions in the ovaries. ROS plays a crucial role in follicular development disorders (161). In follicular fluid containing high levels of ROS, a large amount of highly toxic MDA is produced, which can exacerbate cell damage in the ovaries. Therefore, obesity, ferroptosis, and the development of PCOS are closely related.
5 Ferroptosis contributes to the diagnosis and treatment of PCOS
Follicular development is a complex biological process of continuous change involving multiple hormones and regulatory factors, and the loss of oocytes from the ovary is irreversible (162). Abnormal follicular development can cause a wide range of female disorders and lead to reduced female fertility (163). Early preventive measures could mitigate the development of PCOS (164). The substances that regulate ferroptosis can serve as targets for diagnosing and treating PCOS, exhibiting broad research prospects. Zheng et al. reported that unexplained liver injury in PCOS patients and animal models was accompanied by increased Fe deposition and downregulation of hepcidin and GPX4 expression in the liver, indicating the importance of iron metabolism in this type of unexplained liver injury (165). Tang et al. reported that NEDD4L promotes ferroptosis in GCs and promotes the occurrence of PCOS by promoting GPX4 ubiquitination and degradation. NEDD4L decreased the viability of KGN cells and increased the levels of MDA and ROS. Moreover, ferroptosis inhibitors can block NEDD4L-induced KGN cell death, suggesting that NEDD4L regulates ferroptosis in KGN cells (166). Jiang et al. reported that KGN cells treated with DHEA exhibited ferroptosis characterized by decreased viability, inhibited GPX4 and SLC7A11 expression, increased ACSL4 expression, increased MDA levels and ROS accumulation, and increased lipid peroxidation. These findings may provide new insights into the pathophysiology and treatment of PCOS (167).
Zhang et al. noted that differentially expressed ferroptosis-related genes are associated with reproductive outcomes in infertile POCS patients, and they constructed a FerSig risk prognosis model (168). Lin et al. identified five essential differentially expressed ferroptosis-related genes (NOX1, ACVR1B, PHF21A, FTL, and GALNT14) that may be related to the pathogenesis of PCOS, providing a new perspective for the clinical diagnosis and treatment of PCOS (169). Research has shown that miR-93-5p regulates the NF-κB signaling pathway and promotes apoptosis and ferroptosis in GCs (170). Silencing miR-93-5p can prevent GC dysfunction and provide new molecular targets for diagnosing and treating PCOS. N-3 PUFAs activate Hippo, promote yes-associated protein 1 exocytosis, weaken cross-talk between yes-associated protein 1 and Nrf2, and ultimately activate ferroptosis sensitivity in ovarian GCs. N-3 PUFAs also inhibit excessive proliferation of GCs in ovarian follicles, by which n-3 PUFAs weaken PCOS, and identifying yes-associated protein 1 (Nrf2) as a potential therapeutic target for regulating GCs in PCOS (171). Transferrin receptor-mediated ROS promote ferroptosis in KGN cells by regulating NADPH oxidase 1/PTEN-induced kinase 1/acyl-CoA synthetase long-chain family member 4 signaling, and the inhibitory effects of TFRC/NOX1/PINK1/ACSL4 signaling on folliculogenesis could be a potential target for PCOS treatment (172). Peng et al. reported that metformin regulates ferroptosis through the SIRT3/AMPK/mTOR pathway to improve weight, metabolic disorders, and ovarian dysfunction in PCOS mice (173). Therefore, exploring the role of ferroptosis in the occurrence and development of PCOS can provide more ideas for mechanistic research on PCOS and more potential targets for PCOS treatment.
6 Conclusions
In recent years, substantial progress has been made in exploring ferroptosis. The regulatory network of iron homeostasis, lipid metabolism, amino acid metabolism, and antioxidant pathways provides new ideas for diagnosing and treating human diseases. Ferroptosis is expected to become a new biomarker for the development, treatment efficacy, and prognostic evaluation of PCOS. Research on ferroptosis in PCOS is still lacking. Further research on PCOS animal models with larger sample sizes is needed to validate the potential effects of ferroptosis on ovarian GCs, follicles, ovaries, and even the entire female reproductive system. More clinical studies are urgently needed. Assessing whether ferroptosis and its related molecules play essential roles in infertility, metabolic abnormalities, and other aspects of clinical PCOS and whether the administration of antioxidants can prevent ferroptosis, lipid peroxidation, and adverse maternal and fetal outcomes caused by maternal hyperandrogenemia and IR will provide insights and directions for future clinical diagnosis and treatment. Based on existing prevention and treatment methods for PCOS, interventions targeting different nodes of the ferroptosis regulatory network combined with other treatment methods for various patient etiologies, treatments, and prognoses are expected to be effective and reasonable in the future, thus achieving personalized treatment of PCOS.
Author contributions
MW: Conceptualization, Formal analysis, Methodology, Project administration, Resources, Writing – original draft, Writing – review & editing. B-QZ: Conceptualization, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing. SM: Conceptualization, Data curation, Formal analysis, Funding acquisition, Methodology, Resources, Writing – review & editing. YX: Formal analysis, Funding acquisition, Investigation, Methodology, Software, Writing – review & editing. D-HZ: Conceptualization, Data curation, Funding acquisition, Investigation, Methodology, Resources, Writing – original draft. J-SZ: Conceptualization, Data curation, Formal analysis, Funding acquisition, Software, Writing – review & editing. C-JL: Conceptualization, Data curation, Formal analysis, Methodology, Project administration, Resources, Visualization, Writing – review & editing. XZ: Conceptualization, Data curation, Investigation, Methodology, Supervision, Validation, Writing – review & editing. L-WZ: Conceptualization, Investigation, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Natural Science Foundation of Jilin Province (NO YDZJ202301ZYTS434, NO YDZJ202201ZYTS007) and the Jilin Provincial Development and Reform Commission Project (2023C037-4).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
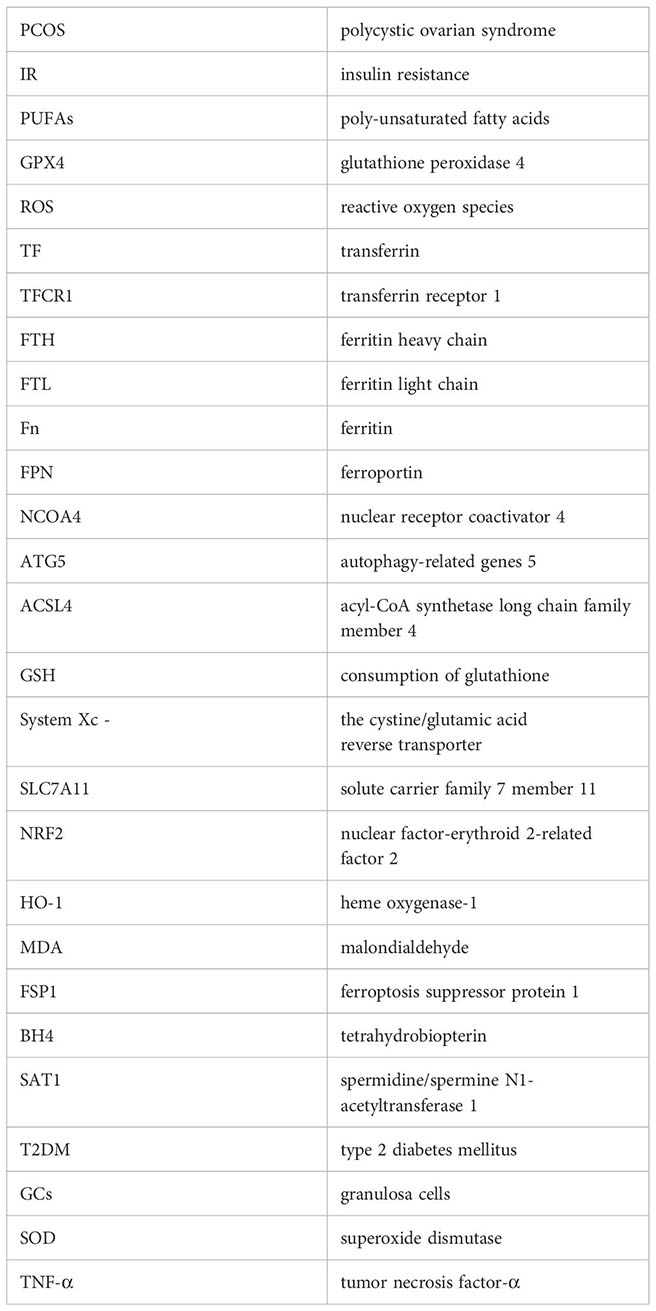
Glossary
References
1. Bell RJ, Islam RM, Skiba MA, Davis SR. Reply: A single cut-off value of anti-Mullerian hormone should not be used for the diagnosis of PCOS in all reproductive-aged women. Hum Reprod. (2022) 37:622. doi: 10.1093/humrep/deac013
2. Ehrmann DA. Polycystic ovary syndrome. N Engl J Med. (2005) 352:1223–36. doi: 10.1056/NEJMra041536
3. Essah PA, Nestler JE. The metabolic syndrome in polycystic ovary syndrome. J Endocrinol Invest. (2006) 29:270–80. doi: 10.1007/BF03345554
4. Dapas M, Dunaif A. Deconstructing a syndrome: genomic insights into PCOS causal mechanisms and classification. Endocr Rev. (2022) 43:927–65. doi: 10.1210/endrev/bnac001
5. Capelletti MM, Manceau H, Puy H, Peoc’h K. Ferroptosis in liver diseases: an overview. Int J Mol Sci. (2020) 21(14):4908. doi: 10.3390/ijms21144908
6. Fernandez-Real JM, Lopez-Bermejo A, Ricart W. Cross-talk between iron metabolism and diabetes. Diabetes. (2002) 51:2348–54. doi: 10.2337/diabetes.51.8.2348
7. Cooney LG, Lee I, Sammel MD, Dokras A. High prevalence of moderate and severe depressive and anxiety symptoms in polycystic ovary syndrome: a systematic review and meta-analysis. Hum Reprod. (2017) 32:1075–91. doi: 10.1093/humrep/dex044
8. Walters KA, Bertoldo MJ, Handelsman DJ. Evidence from animal models on the pathogenesis of PCOS. Best Pract Res Clin Endocrinol Metab. (2018) 32:271–81. doi: 10.1016/j.beem.2018.03.008
9. Tay CT, Teede HJ, Hill B, Loxton D, Joham AE. Increased prevalence of eating disorders, low self-esteem, and psychological distress in women with polycystic ovary syndrome: a community-based cohort study. Fertil Steril. (2019) 112:353–61. doi: 10.1016/j.fertnstert.2019.03.027
10. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. (2014) 16:1180–91. doi: 10.1038/ncb3064
11. Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. (2022) 22:381–96. doi: 10.1038/s41568-022-00459-0
12. Banuls C, Rovira-Llopis S, Martinez de Maranon A, Veses S, Jover A, Gomez M, et al. Metabolic syndrome enhances endoplasmic reticulum, oxidative stress, and leukocyte-endothelium interactions in PCOS. Metabolism. (2017) 71:153–62. doi: 10.1016/j.metabol.2017.02.012
13. Lai Q, Xiang W, Li Q, Zhang H, Li Y, Zhu G, et al. Oxidative stress in granulosa cells contributes to poor oocyte quality and IVF-ET outcomes in women with polycystic ovary syndrome. Front Med. (2018) 12:518–24. doi: 10.1007/s11684-017-0575-y
14. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell. (2019) 73:354–63 e3. doi: 10.1016/j.molcel.2018.10.042
15. Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J, et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol Cell. (2021) 81:355–69 e10. doi: 10.1016/j.molcel.2020.11.024
16. Nasri F, Zare M, Doroudchi M, Gharesi-Fard B. Proteome analysis of CD4(+) T cells reveals differentially expressed proteins in infertile polycystic ovary syndrome patients. Endocr Metab Immune Disord Drug Targets. (2021) 21:1998–2004. doi: 10.2174/1871530320666201119152323
17. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. (2012) 149:1060–72. doi: 10.1016/j.cell.2012.03.042
18. Liu Y, Zhou L, Xu Y, Li K, Zhao Y, Qiao H, et al. Heat shock proteins and ferroptosis. Front Cell Dev Biol. (2022) 10:864635.doi: 10.3389/fcell.2022.864635
19. Wen J, Chen H, Ren Z, Zhang P, Chen J, Jiang S. Ultrasmall iron oxide nanoparticles induced ferroptosis via Beclin1/ATG5-dependent autophagy pathway. Nano Converg. (2021) 8:10. doi: 10.1186/s40580-021-00260-z
20. D’Herde K, Krysko DV. Ferroptosis: Oxidized PEs trigger death. Nat Chem Biol. (2017) 13:4–5. doi: 10.1038/nchembio.2261
21. Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of poly-unsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. (2016) 113:E4966–75. doi: 10.1073/pnas.1603244113
22. Li C, Deng X, Zhang W, Xie X, Conrad M, Liu Y, et al. Novel allosteric activators for ferroptosis regulator glutathione peroxidase 4. J Med Chem. (2019) 62:266–75. doi: 10.1021/acs.jmedchem.8b00315
23. Forcina GC, Dixon SJ. GPX4 at the crossroads of lipid homeostasis and ferroptosis. Proteomics. (2019) 19:e1800311. doi: 10.1002/pmic.201800311
24. Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. (2020) 585:603–8. doi: 10.1038/s41586-020-2732-8
25. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. (2020) 16:302–9. doi: 10.1038/s41589-020-0472-6
26. Agmon E, Solon J, Bassereau P, Stockwell BR. Modelling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep. (2018) 8:5155. doi: 10.1038/s41598-018-23408-0
27. Hadian K, Stockwell BR. SnapShot: ferroptosis. Cell. (2020) 181:1188– e1. doi: 10.1016/j.cell.2020.04.039
28. Stockwell BR, Jiang X, Gu W. Emerging mechanisms and disease relevance of ferroptosis. Trends Cell Biol. (2020) 30:478–90. doi: 10.1016/j.tcb.2020.02.009
29. Salnikow K. Role of iron in cancer. Semin Cancer Biol. (2021) 76:189–94. doi: 10.1016/j.semcancer.2021.04.001
30. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell. (2017) 171:273–85. doi: 10.1016/j.cell.2017.09.021
31. Kuang F, Liu J, Tang D, Kang R. Oxidative damage and antioxidant defense in ferroptosis. Front Cell Dev Biol. (2020) 8:586578doi: 10.3389/fcell.2020.586578
32. Cepelak I, Dodig S, Dodig DC. Ferroptosis: regulated cell death. Arh Hig Rada Toksikol. (2020) 71:99–109. doi: 10.2478/aiht-2020-71-3366
33. Chen PH, Wu J, Ding CC, Lin CC, Pan S, Bossa N, et al. Kinome screen of ferroptosis reveals a novel role of ATM in regulating iron metabolism. Cell Death Differ. (2020) 27:1008–22. doi: 10.1038/s41418-019-0393-7
34. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. (2014) 156:317–31. doi: 10.1016/j.cell.2013.12.010
35. Galaris D, Barbouti A, Pantopoulos K. Iron homeostasis and oxidative stress: An intimate relationship. Biochim Biophys Acta Mol Cell Res. (2019) 1866:118535. doi: 10.1016/j.bbamcr.2019.118535
36. Turcu AL, Versini A, Khene N, Gaillet C, Caneque T, Muller S, et al. DMT1 inhibitors kill cancer stem cells by blocking lysosomal iron translocation. Chemistry. (2020) 26:7369–73. doi: 10.1002/chem.202000159
37. Rockfield S, Raffel J, Mehta R, Rehman N, Nanjundan M. Iron overload and altered iron metabolism in ovarian cancer. Biol Chem. (2017) 398:995–1007. doi: 10.1515/hsz-2016-0336
38. Pantopoulos K, Porwal SK, Tartakoff A, Devireddy L. Mechanisms of mammalian iron homeostasis. Biochemistry. (2012) 51:5705–24. doi: 10.1021/bi300752r
39. Angeli JPF, Shah R, Pratt DA, Conrad M. Ferroptosis inhibition: mechanisms and opportunities. Trends Pharmacol Sci. (2017) 38:489–98. doi: 10.1016/j.tips.2017.02.005
40. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. (2016) 23:369–79. doi: 10.1038/cdd.2015.158
41. Gao M, Monian P, Pan Q, Zhang W, Xiang J, Jiang X. Ferroptosis is an autophagic cell death process. Cell Res. (2016) 26:1021–32. doi: 10.1038/cr.2016.95
42. Park E, Chung SW. ROS-mediated autophagy increases intracellular iron levels and ferroptosis by ferritin and transferrin receptor regulation. Cell Death Dis. (2019) 10:822. doi: 10.1038/s41419-019-2064-5
43. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. (2014) 509:105–9. doi: 10.1038/nature13148
44. Gryzik M, Asperti M, Denardo A, Arosio P, Poli M. NCOA4-mediated ferritinophagy promotes ferroptosis induced by erastin, but not by RSL3 in HeLa cells. Biochim Biophys Acta Mol Cell Res. (2021) 1868:118913. doi: 10.1016/j.bbamcr.2020.118913
45. Kang B, Jiang D, Ma R, He H. Evidence for a role of ferritin heavy chain in mediating reproductive processes of geese. Reprod Biol. (2015) 15:205–9. doi: 10.1016/j.repbio.2015.10.001
46. Tang LJ, Zhou YJ, Xiong XM, Li NS, Zhang JJ, Luo XJ, et al. Ubiquitin-specific protease 7 promotes ferroptosis via activation of the p53/TFCR1 pathway in the rat hearts after ischemia/reperfusion. Free Radic Biol Med. (2021) 162:339–52. doi: 10.1016/j.freeradbiomed.2020.10.307
47. Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. (2014) 10:9–17. doi: 10.1038/nchembio.1416
48. Green DR. The coming decade of cell death research: five riddles. Cell. (2019) 177:1094–107. doi: 10.1016/j.cell.2019.04.024
49. Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. (2016) 12:497–503. doi: 10.1038/nchembio.2079
50. Wang L, Liu Y, Du T, Yang H, Lei L, Guo M, et al. ATF3 promotes erastin-induced ferroptosis by suppressing System Xc -(). Cell Death Differ. (2020) 27:662–75. doi: 10.1038/s41418-019-0380-z
51. Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. (2017) 13:81–90. doi: 10.1038/nchembio.2238
52. Yuan H, Li X, Zhang X, Kang R, Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem Biophys Res Commun. (2016) 478:1338–43. doi: 10.1016/j.bbrc.2016.08.124
53. Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. (2015) 10:1604–9. doi: 10.1021/acschembio.5b00245
54. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. (2017) 13:91–8. doi: 10.1038/nchembio.2239
55. Enke U, Seyfarth L, Schleussner E, Markert UR. Impact of PUFA on early immune and fetal development. Br J Nutr. (2008) 100:1158–68. doi: 10.1017/S000711450801413X
56. Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. (2016) 26:165–76. doi: 10.1016/j.tcb.2015.10.014
57. Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell. (2019) 35:830–49. doi: 10.1016/j.ccell.2019.04.002
58. Kinowaki Y, Kurata M, Ishibashi S, Ikeda M, Tatsuzawa A, Yamamoto M, et al. Glutathione peroxidase 4 overexpression inhibits ROS-induced cell death in diffuse large B-cell lymphoma. Lab Invest. (2018) 98:609–19. doi: 10.1038/s41374-017-0008-1
59. Sbodio JI, Snyder SH, Paul BD. Regulators of the transsulfuration pathway. Br J Pharmacol. (2019) 176:583–93. doi: 10.1111/bph.14446
60. Sun Y, Chen P, Zhai B, Zhang M, Xiang Y, Fang J, et al. The emerging role of ferroptosis in inflammation. BioMed Pharmacother. (2020) 127:110108. doi: 10.1016/j.biopha.2020.110108
61. Kobayashi S, Hamashima S, Homma T, Sato M, Kusumi R, Bannai S, et al. Cystine/glutamate transporter, system x(c)(-), is involved in nitric oxide production in mouse peritoneal macrophages. Nitric Oxide. (2018) 78:32–40. doi: 10.1016/j.niox.2018.05.005
62. Vaskova J, Kocan L, Vasko L, Perjesi P. Glutathione-related enzymes and proteins: A review. Molecules. (2023) 28(3):1447. doi: 10.3390/molecules28031447
63. McBean GJ. The transsulfuration pathway: a source of cysteine for glutathione in astrocytes. Amino Acids. (2012) 42:199–205. doi: 10.1007/s00726-011-0864-8
64. Hao S, Yu J, He W, Huang Q, Zhao Y, Liang B, et al. Cysteine dioxygenase 1 mediates erastin-induced ferroptosis in human gastric cancer cells. Neoplasia. (2017) 19:1022–32. doi: 10.1016/j.neo.2017.10.005
65. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. (2015) 59:298–308. doi: 10.1016/j.molcel.2015.06.011
66. Koppula P, Zhang Y, Zhuang L, Gan B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun (Lond). (2018) 38:12. doi: 10.1186/s40880-018-0288-x
67. Ye Z, Liu W, Zhuo Q, Hu Q, Liu M, Sun Q, et al. Ferroptosis: Final destination for cancer? Cell Prolif. (2020) 53:e12761. doi: 10.1111/cpr.12761
68. Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system X(c)(-) activity. Curr Biol. (2018) 28:2388–99 e5. doi: 10.1016/j.cub.2018.05.094
69. Dong H, Qiang Z, Chai D, Peng J, Xia Y, Hu R, et al. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging (Albany NY). (2020) 12:12943–59. doi: 10.18632/aging.v12i13
70. Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. (2021) 31:107–25. doi: 10.1038/s41422-020-00441-1
71. Latunde-Dada GO. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta Gen Subj. (2017) 1861:1893–900. doi: 10.1016/j.bbagen.2017.05.019
72. Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic Biol Med. (2019) 133:144–52. doi: 10.1016/j.freeradbiomed.2018.09.014
73. Goji T, Takahara K, Negishi M, Katoh H. Cystine uptake through the cystine/glutamate antiporter xCT triggers glioblastoma cell death under glucose deprivation. J Biol Chem. (2017) 292:19721–32. doi: 10.1074/jbc.M117.814392
74. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. (2018) 172:409–22.e21. doi: 10.1016/j.cell.2017.11.048
75. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. (2021) 22:266–82. doi: 10.1038/s41580-020-00324-8
76. Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): cystine supplier and beyond. Amino Acids. (2012) 42:231–46. doi: 10.1007/s00726-011-0867-5
77. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. (2019) 575:693–8. doi: 10.1038/s41586-019-1707-0
78. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. (2019) 575:688–92. doi: 10.1038/s41586-019-1705-2
79. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, Zandkarimi F, et al. GTP cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci. (2020) 6:41–53. doi: 10.1021/acscentsci.9b01063
80. Bridges R, Lutgen V, Lobner D, Baker DA. Thinking outside the cleft to understand synaptic activity: contribution of the cystine-glutamate antiporter (System Xc-) to normal and pathological glutamatergic signaling. Pharmacol Rev. (2012) 64:780–802. doi: 10.1124/pr.110.003889
81. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. (2021) 593:586–90. doi: 10.1038/s41586-021-03539-7
82. Liu J, Zhang C, Wang J, Hu W, Feng Z. The Regulation of Ferroptosis by Tumor Suppressor p53 and its pathway. Int J Mol Sci. (2020) 21(21):8387. doi: 10.3390/ijms21218387
83. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. (2015) 520:57–62. doi: 10.1038/nature14344
84. Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, et al. p53 suppresses metabolic stress-induced ferroptosis in cancer cells. Cell Rep. (2018) 22:569–75. doi: 10.1016/j.celrep.2017.12.077
85. Shimada K, Hayano M, Pagano NC, Stockwell BR. Cell-line selectivity improves the predictive power of pharmacogenomic analyses and helps identify NADPH as biomarker for ferroptosis sensitivity. Cell Chem Biol. (2016) 23:225–35. doi: 10.1016/j.chembiol.2015.11.016
86. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. (2016) 63:173–84. doi: 10.1002/hep.28251
87. Duarte TL, Talbot NP, Drakesmith H. NRF2 and hypoxia-inducible factors: key players in the redox control of systemic iron homeostasis. Antioxid Redox Signal. (2021) 35:433–52. doi: 10.1089/ars.2020.8148
88. Wang Y, Zhang L, Zhou X. Activation of Nrf2 signaling protects hypoxia-induced HTR-8/SVneo cells against ferroptosis. J Obstet Gynaecol Res. (2021) 47:3797–806. doi: 10.1111/jog.15009
89. Zhu H, Jia Z, Misra BR, Zhang L, Cao Z, Yamamoto M, et al. Nuclear factor E2-related factor 2-dependent myocardiac cytoprotection against oxidative and electrophilic stress. Cardiovasc Toxicol. (2008) 8:71–85. doi: 10.1007/s12012-008-9016-0
90. Kerins MJ, Ooi A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid Redox Signal. (2018) 29:1756–73. doi: 10.1089/ars.2017.7176
91. Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M, Buchfelder M, et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis. (2017) 6:e371. doi: 10.1038/oncsis.2017.65
92. Dai E, Han L, Liu J, Xie Y, Kroemer G, Klionsky DJ, et al. Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein. Autophagy. (2020) 16:2069–83. doi: 10.1080/15548627.2020.1714209
93. Kang R, Tang D. Autophagy and ferroptosis - what’s the connection? Curr Pathobiol Rep. (2017) 5:153–9. doi: 10.1007/s40139-017-0139-5
94. Liu J, Yang M, Kang R, Klionsky DJ, Tang D. Autophagic degradation of the circadian clock regulator promotes ferroptosis. Autophagy. (2019) 15:2033–5. doi: 10.1080/15548627.2019.1659623
95. Wang H, Liu C, Zhao Y, Gao G. Mitochondria regulation in ferroptosis. Eur J Cell Biol. (2020) 99:151058. doi: 10.1016/j.ejcb.2019.151058
96. Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. (2016) 12:1425–8. doi: 10.1080/15548627.2016.1187366
97. Bai Y, Meng L, Han L, Jia Y, Zhao Y, Gao H, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. (2019) 508:997–1003. doi: 10.1016/j.bbrc.2018.12.039
98. Babayev E, Seli E. Oocyte mitochondrial function and reproduction. Curr Opin Obstet Gynecol. (2015) 27:175–81. doi: 10.1097/GCO.0000000000000164
99. Fan H, He J, Bai Y, He Q, Zhang T, Zhang J, et al. Baicalin improves the functions of granulosa cells and the ovary in aged mice through the mTOR signaling pathway. J Ovarian Res. (2022) 15:34. doi: 10.1186/s13048-022-00965-7
100. Zhang J, Liu Y, Yao W, Li Q, Liu H, Pan Z. Initiation of follicular atresia: gene networks during early atresia in pig ovaries. Reproduction. (2018) 156:23–33. doi: 10.1530/REP-18-0058
101. Tsai-Turton M, Luong BT, Tan Y, Luderer U. Cyclophosphamide-induced apoptosis in COV434 human granulosa cells involves oxidative stress and glutathione depletion. Toxicol Sci. (2007) 98:216–30. doi: 10.1093/toxsci/kfm087
102. Zhang D, Liu Y, Zhang Z, Lv P, Liu Y, Li J, et al. Basonuclin 1 deficiency is a cause of primary ovarian insufficiency. Hum Mol Genet. (2018) 27:3787–800. doi: 10.1093/hmg/ddy261
103. Wang F, Liu Y, Ni F, Jin J, Wu Y, Huang Y, et al. BNC1 deficiency-triggered ferroptosis through the NF2-YAP pathway induces primary ovarian insufficiency. Nat Commun. (2022) 13:5871. doi: 10.1038/s41467-022-33323-8
104. Jakimiuk AJ, Weitsman SR, Navab A, Magoffin DA. Luteinizing hormone receptor, steroidogenesis acute regulatory protein, and steroidogenic enzyme messenger ribonucleic acids are overexpressed in thecal and granulosa cells from polycystic ovaries. J Clin Endocrinol Metab. (2001) 86:1318–23. doi: 10.1210/jcem.86.3.7318
105. Yang H, Xie Y, Yang D, Ren D. Oxidative stress-induced apoptosis in granulosa cells involves JNK, p53 and Puma. Oncotarget. (2017) 8:25310–22. doi: 10.18632/oncotarget.v8i15
106. Shen M, Cao Y, Jiang Y, Wei Y, Liu H. Melatonin protects mouse granulosa cells against oxidative damage by inhibiting FOXO1-mediated autophagy: Implication of an antioxidation-independent mechanism. Redox Biol. (2018) 18:138–57. doi: 10.1016/j.redox.2018.07.004
107. Ni Z, Li Y, Song D, Ding J, Mei S, Sun S, et al. Iron-overloaded follicular fluid increases the risk of endometriosis-related infertility by triggering granulosa cell ferroptosis and oocyte dysmaturity. Cell Death Dis. (2022) 13:579. doi: 10.1038/s41419-022-05037-8
108. Mancias JD, Pontano Vaites L, Nissim S, Biancur DE, Kim AJ, Wang X, et al. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife. (2015) 4:e10308. doi: 10.7554/eLife.10308
109. Wang S, Ji LY, Li L, Li JM. Oxidative stress, autophagy and pyroptosis in the neovascularization of oxygen−induced retinopathy in mice. Mol Med Rep. (2019) 19:927–34. doi: 10.3892/mmr.2018.9759
110. Moreno-Navarrete JM, Lopez-Navarro E, Candenas L, Pinto F, Ortega FJ, Sabater-Masdeu M, et al. Ferroportin mRNA is down-regulated in granulosa and cervical cells from infertile women. Fertil Steril. (2017) 107:236–42. doi: 10.1016/j.fertnstert.2016.10.008
111. Zhang D, Yi S, Cai B, Wang Z, Chen M, Zheng Z, et al. Involvement of ferroptosis in the granulosa cells proliferation of PCOS through the circRHBG/miR-515/SLC7A11 axis. Ann Transl Med. (2021) 9:1348. doi: 10.21037/atm
112. Nemati A, Alipanah-Moghadam R, Molazadeh L, Naghizadeh Baghi A. The effect of glutamine supplementation on oxidative stress and matrix metalloproteinase 2 and 9 after exhaustive exercise. Drug Des Devel Ther. (2019) 13:4215–23. doi: 10.2147/DDDT
113. Cacciottola L, Donnez J, Dolmans MM. Can endometriosis-related oxidative stress pave the way for new treatment targets? Int J Mol Sci. (2021) 22(13):7138. doi: 10.3390/ijms22137138
114. Saeed N, Hamzah IH, Al-Gharrawi SAR. Polycystic ovary syndrome dependency on mtDNA mutation; copy Number and its association with insulin resistance. BMC Res Notes. (2019) 12:455. doi: 10.1186/s13104-019-4453-3
115. Zhang M, Bener MB, Jiang Z, Wang T, Esencan E, Scott R, et al. Mitofusin 2 plays a role in oocyte and follicle development, and is required to maintain ovarian follicular reserve during reproductive aging. Aging (Albany NY). (2019) 11:3919–38. doi: 10.18632/aging.v11i12
116. Wang CH, Wei YH. Role of mitochondrial dysfunction and dysregulation of Ca(2+) homeostasis in the pathophysiology of insulin resistance and type 2 diabetes. J BioMed Sci. (2017) 24:70. doi: 10.1186/s12929-017-0375-3
117. Kim JA, Wei Y, Sowers JR. Role of mitochondrial dysfunction in insulin resistance. Circ Res. (2008) 102:401–14. doi: 10.1161/CIRCRESAHA.107.165472
118. Yang S, Lian G. ROS and diseases: role in metabolism and energy supply. Mol Cell Biochem. (2020) 467:1–12. doi: 10.1007/s11010-019-03667-9
119. Zhang J, Bao Y, Zhou X, Zheng L. Polycystic ovary syndrome and mitochondrial dysfunction. Reprod Biol Endocrinol. (2019) 17:67. doi: 10.1186/s12958-019-0509-4
120. Papalou O, Victor VM, Diamanti-Kandarakis E. Oxidative stress in polycystic ovary syndrome. Curr Pharm Des. (2016) 22:2709–22. doi: 10.2174/1381612822666160216151852
121. Ding Y, Xia BH, Zhang CJ, Zhuo GC. Mitochondrial tRNA(Leu(UUR)) C3275T, tRNA(Gln) T4363C and tRNA(Lys) A8343G mutations may be associated with PCOS and metabolic syndrome. Gene. (2018) 642:299–306. doi: 10.1016/j.gene.2017.11.049
122. Zhao H, Zhao Y, Li T, Li M, Li J, Li R, et al. Metabolism alteration in follicular niche: The nexus among intermediary metabolism, mitochondrial function, and classic polycystic ovary syndrome. Free Radic Biol Med. (2015) 86:295–307. doi: 10.1016/j.freeradbiomed.2015.05.013
123. Liu H, Xie J, Fan L, Xia Y, Peng X, Zhou J, et al. Cryptotanshinone Protects against PCOS-Induced Damage of Ovarian Tissue via Regulating Oxidative Stress, Mitochondrial Membrane Potential, Inflammation, and Apoptosis via Regulating Ferroptosis. Oxid Med Cell Longev. (2022) 2022:8011850. doi: 10.1155/2022/8011850
124. Boirie Y. Insulin regulation of mitochondrial proteins and oxidative phosphorylation in human muscle. Trends Endocrinol Metab. (2003) 14:393–4. doi: 10.1016/j.tem.2003.09.002
125. Evans JL, Maddux BA, Goldfine ID. The molecular basis for oxidative stress-induced insulin resistance. Antioxid Redox Signal. (2005) 7:1040–52. doi: 10.1089/ars.2005.7.1040
126. Babaei-Abraki S, Karamali F, Nasr-Esfahani MH. Monitoring the induction of ferroptosis following dissociation in human embryonic stem cells. J Biol Chem. (2022) 298:101855. doi: 10.1016/j.jbc.2022.101855
127. Zhang Y, Hu M, Jia W, Liu G, Zhang J, Wang B, et al. Hyperandrogenism and insulin resistance modulate gravid uterine and placental ferroptosis in PCOS-like rats. J Endocrinol. (2020) 246:247–63. doi: 10.1530/JOE-20-0155
128. Beharier O, Tyurin VA, Goff JP, Guerrero-Santoro J, Kajiwara K, Chu T, et al. PLA2G6 guards placental trophoblasts against ferroptotic injury. Proc Natl Acad Sci USA. (2020) 117:27319–28. doi: 10.1073/pnas.2009201117
129. Rodriguez Paris V, Wong XYD, Solon-Biet SM, Edwards MC, Aflatounian A, Gilchrist RB, et al. The interplay between PCOS pathology and diet on gut microbiota in a mouse model. Gut Microbes. (2022) 14:2085961. doi: 10.1080/19490976.2022.2085961
130. Song X, Long D. Nrf2 and ferroptosis: A new research direction for neurodegenerative diseases. Front Neurosci. (2020) 14:267doi: 10.3389/fnins.2020.00267
131. Zhu T, Cui J, Goodarzi MO. Polycystic ovary syndrome and risk of type 2 diabetes, coronary heart disease, and stroke. Diabetes. (2021) 70:627–37. doi: 10.2337/db20-0800
132. Greenwood EA, Huddleston HG. Insulin resistance in polycystic ovary syndrome: concept versus cutoff. Fertil Steril. (2019) 112:827–8. doi: 10.1016/j.fertnstert.2019.08.100
133. Mahalingaiah S, Diamanti-Kandarakis E. Targets to treat metabolic syndrome in polycystic ovary syndrome. Expert Opin Ther Targets. (2015) 19:1561–74. doi: 10.1517/14728222.2015.1101067
134. Yilmaz M, Bukan N, Ayvaz G, Karakoc A, Toruner F, Cakir N, et al. The effects of rosiglitazone and metformin on oxidative stress and homocysteine levels in lean patients with polycystic ovary syndrome. Hum Reprod. (2005) 20:3333–40. doi: 10.1093/humrep/dei258
135. Teede HJ, Misso ML, Costello MF, Dokras A, Laven J, Moran L, et al. Recommendations from the international evidence-based guideline for the assessment and management of polycystic ovary syndrome. Hum Reprod. (2018) 33:1602–18. doi: 10.1093/humrep/dey256
136. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N, et al. Ferroptosis: past, present and future. Cell Death Dis. (2020) 11:88. doi: 10.1038/s41419-020-2298-2
137. Liu S, Navarro G, Mauvais-Jarvis F. Androgen excess produces systemic oxidative stress and predisposes to beta-cell failure in female mice. PloS One. (2010) 5:e11302. doi: 10.1371/journal.pone.0011302
138. Chappel S. The role of mitochondria from mature oocyte to viable blastocyst. Obstet Gynecol Int. (2013) 2013:183024. doi: 10.1155/2013/183024
139. Escobar-Morreale HF, Luque-Ramirez M, Alvarez-Blasco F, Botella-Carretero JI, Sancho J, San Millan JL. Body iron stores are increased in overweight and obese women with polycystic ovary syndrome. Diabetes Care. (2005) 28:2042–4. doi: 10.2337/diacare.28.8.2042
140. Escobar-Morreale HF, Luque-Ramirez M. Role of androgen-mediated enhancement of erythropoiesis in the increased body iron stores of patients with polycystic ovary syndrome. Fertil Steril. (2011) 95:1730–5 e1. doi: 10.1016/j.fertnstert.2011.01.038
141. Escobar-Morreale HF, San Millan JL. Abdominal adiposity and the polycystic ovary syndrome. Trends Endocrinol Metab. (2007) 18:266–72. doi: 10.1016/j.tem.2007.07.003
142. Wang H, Wang X, Zhu Y, Chen F, Sun Y, Han X. Increased androgen levels in rats impair glucose-stimulated insulin secretion through disruption of pancreatic beta cell mitochondrial function. J Steroid Biochem Mol Biol. (2015) 154:254–66. doi: 10.1016/j.jsbmb.2015.09.003
143. Luque-Ramirez M, Alvarez-Blasco F, Alpanes M, Escobar-Morreale HF. Role of decreased circulating hepcidin concentrations in the iron excess of women with the polycystic ovary syndrome. J Clin Endocrinol Metab. (2011) 96:846–52. doi: 10.1210/jc.2010-2211
144. Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, et al. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. (2019) 12:34. doi: 10.1186/s13045-019-0720-y
145. Hu M, Zhang Y, Ma S, Li J, Wang X, Liang M, et al. Suppression of uterine and placental ferroptosis by N-acetylcysteine in a rat model of polycystic ovary syndrome. Mol Hum Reprod. (2021) 27(12):gaab067. doi: 10.1093/molehr/gaab067
146. Sun L, Hu W, Liu Q, Hao Q, Sun B, Zhang Q, et al. Metabonomics reveals plasma metabolic changes and inflammatory marker in polycystic ovary syndrome patients. J Proteome Res. (2012) 11:2937–46. doi: 10.1021/pr3000317
147. Li S, Chu Q, Ma J, Sun Y, Tao T, Huang R, et al. Discovery of novel lipid profiles in PCOS: do insulin and androgen oppositely regulate bioactive lipid production? J Clin Endocrinol Metab. (2017) 102:810–21. doi: 10.1210/jc.2016-2692
148. Huang R, Xue X, Li S, Wang Y, Sun Y, Liu W, et al. Alterations of poly-unsaturated fatty acid metabolism in ovarian tissues of polycystic ovary syndrome rats. J Cell Mol Med. (2018) 22:3388–96. doi: 10.1111/jcmm.13614
149. Sak S, Uyanikoglu H, Incebiyik A, Incebiyik H, Hilali NG, Sabuncu T, et al. Associations of serum fetuin-A and oxidative stress parameters with polycystic ovary syndrome. Clin Exp Reprod Med. (2018) 45:116–21. doi: 10.5653/cerm.2018.45.3.116
150. Raizel R, Leite JS, Hypolito TM, Coqueiro AY, Newsholme P, Cruzat VF, et al. Determination of the anti-inflammatory and cytoprotective effects of l-glutamine and l-alanine, or dipeptide, supplementation in rats submitted to resistance exercise. Br J Nutr. (2016) 116:470–9. doi: 10.1017/S0007114516001999
151. Ge MH, Tian H, Mao L, Li DY, Lin JQ, Hu HS, et al. Zinc attenuates ferroptosis and promotes functional recovery in contusion spinal cord injury by activating Nrf2/GPX4 defense pathway. CNS Neurosci Ther. (2021) 27:1023–40. doi: 10.1111/cns.13657
152. Uckan K, Demir H, Turan K, Sarikaya E, Demir C. Role of oxidative stress in obese and nonobese PCOS patients. Int J Clin Pract. (2022) 2022:4579831. doi: 10.1155/2022/4579831
153. Malin SK, Kirwan JP, Sia CL, Gonzalez F. Glucose-stimulated oxidative stress in mononuclear cells is related to pancreatic beta-cell dysfunction in polycystic ovary syndrome. J Clin Endocrinol Metab. (2014) 99:322–9. doi: 10.1210/jc.2013-3177
154. Palacio JR, Iborra A, Ulcova-Gallova Z, Badia R, Martinez P. The presence of antibodies to oxidative modified proteins in serum from polycystic ovary syndrome patients. Clin Exp Immunol. (2006) 144:217–22. doi: 10.1111/j.1365-2249.2006.03061.x
155. Chen PH, Tseng WH, Chi JT. The intersection of DNA damage response and ferroptosis-A rationale for combination therapeutics. Biol (Basel). (2020) 9(8):187. doi: 10.3390/biology9080187
156. Ryu Y, Kim SW, Kim YY, Ku SY. Animal models for human polycystic ovary syndrome (PCOS) focused on the use of indirect hormonal perturbations: A review of the literature. Int J Mol Sci. (2019) 20(11):2720. doi: 10.3390/ijms20112720
157. Choi HD, Kim JH, Chang MJ, Kyu-Youn Y, Shin WG. Effects of astaxanthin on oxidative stress in overweight and obese adults. Phytother Res. (2011) 25:1813–8. doi: 10.1002/ptr.3494
158. Azziz R, Carmina E, Chen Z, Dunaif A, Laven JS, Legro RS, et al. Polycystic ovary syndrome. Nat Rev Dis Primers. (2016) 2:16057. doi: 10.1038/nrdp.2016.57
159. Joham AE, Piltonen T, Lujan ME, Kiconco S, Tay CT. Challenges in diagnosis and understanding of natural history of polycystic ovary syndrome. Clin Endocrinol (Oxf). (2022) 97:165–73. doi: 10.1111/cen.14757
160. Jones DP. Redefining oxidative stress. Antioxid Redox Signal. (2006) 8:1865–79. doi: 10.1089/ars.2006.8.1865
161. Sulaiman MA, Al-Farsi YM, Al-Khaduri MM, Saleh J, Waly MI. Polycystic ovarian syndrome is linked to increased oxidative stress in Omani women. Int J Womens Health. (2018) 10:763–71. doi: 10.2147/IJWH
162. Hoeger KM, Dokras A, Piltonen T. Update on PCOS: consequences, challenges, and guiding treatment. J Clin Endocrinol Metab. (2021) 106:e1071–e83. doi: 10.1210/clinem/dgaa839
163. Deswal R, Narwal V, Dang A, Pundir CS. The prevalence of polycystic ovary syndrome: A brief systematic review. J Hum Reprod Sci. (2020) 13:261–71. doi: 10.4103/jhrs.JHRS_95_18
164. Kakoly NS, Earnest A, Teede HJ, Moran LJ, Joham AE. The impact of obesity on the incidence of type 2 diabetes among women with polycystic ovary syndrome. Diabetes Care. (2019) 42:560–7. doi: 10.2337/dc18-1738
165. Zheng R, Lin C, Mao Y, Jin F. miR-761-hepcidin/Gpx4 pathway contribute to unexplained liver dysfunction in polycystic ovary syndrome by regulating liver iron overload and ferroptosis. Gynecol Endocrinol. (2023) 39:2166483. doi: 10.1080/09513590.2023.2166483
166. Tang H, Jiang X, Hua Y, Li H, Zhu C, Hao X, et al. NEDD4L facilitates granulosa cell ferroptosis by promoting GPX4 ubiquitination and degradation. Endocr Connect. (2023) 12(4):e220459. doi: 10.1530/EC-22-0459
167. Jiang Y, Yang J, Du K, Luo K, Yuan X, Hua F. 1,25-Dihydroxyvitamin D3 alleviates hyperandrogen-induced ferroptosis in KGN cells. Hormones (Athens). (2023) 22:273–80. doi: 10.1007/s42000-023-00439-5
168. Zhang J, Ding N, Xin W, Yang X, Wang F. Quantitative proteomics reveals that a prognostic signature of the endometrium of the polycystic ovary syndrome women based on ferroptosis proteins. Front Endocrinol (Lausanne). (2022) 13:871945.doi: 10.3389/fendo.2022.871945
169. Lin S, Jin X, Gu H, Bi F. Relationships of ferroptosis-related genes with the pathogenesis in polycystic ovary syndrome. Front Med (Lausanne). (2023) 10:1120693.doi: 10.3389/fmed.2023.1120693
170. Tan W, Dai F, Yang D, Deng Z, Gu R, Zhao X, et al. MiR-93-5p promotes granulosa cell apoptosis and ferroptosis by the NF-kB signaling pathway in polycystic ovary syndrome. Front Immunol. (2022) 13:967151.doi: 10.3389/fimmu.2022.967151
171. Zhang P, Pan Y, Wu S, He Y, Wang J, Chen L, et al. n-3 PUFA promotes ferroptosis in PCOS GCs by inhibiting YAP1 through activation of the hippo pathway. Nutrients. (2023) 15(8):1927. doi: 10.3390/nu15081927
172. Zhang L, Wang F, Li D, Yan Y, Wang H. Transferrin receptor-mediated reactive oxygen species promotes ferroptosis of KGN cells via regulating NADPH oxidase 1/PTEN induced kinase 1/acyl-CoA synthetase long chain family member 4 signaling. Bioengineered. (2021) 12:4983–94. doi: 10.1080/21655979.2021.1956403
Keywords: ferroptosis, polycystic ovary syndrome, oxidative stress, metabolic disorders, expression, biomarkers
Citation: Wang M, Zhang B-Q, Ma S, Xu Y, Zhao D-H, Zhang J-S, Li C-J, Zhou X and Zheng L-W (2024) Broadening horizons: the role of ferroptosis in polycystic ovary syndrome. Front. Endocrinol. 15:1390013. doi: 10.3389/fendo.2024.1390013
Received: 22 February 2024; Accepted: 22 July 2024;
Published: 02 August 2024.
Edited by:
Katja Teerds, Wageningen University and Research, NetherlandsReviewed by:
Wen-Xiang Liu, Inner Mongolia University, ChinaAvi Lerner, Imperial College London, United Kingdom
Copyright © 2024 Wang, Zhang, Ma, Xu, Zhao, Zhang, Li, Zhou and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lian-Wen Zheng, ZGF2ZXpoZW5nQHNvaHUuY29t; Xu Zhou, enh1QGpsdS5lZHUuY24=; Chun-Jin Li, bGlfY2h1bmppbkBqbHUuZWR1LmNu