 Giampiero I. Baroncelli1†
Giampiero I. Baroncelli1† Pasquale Comberiati1,2†
Pasquale Comberiati1,2† Tommaso Aversa3,4
Tommaso Aversa3,4 Federico Baronio5
Federico Baronio5 Alessandra Cassio5,6Mariangela Chiarito7Mirna Cosci o di Coscio1
Alessandra Cassio5,6Mariangela Chiarito7Mirna Cosci o di Coscio1 Luisa De Sanctis8Natascia Di Iorgi9,10
Luisa De Sanctis8Natascia Di Iorgi9,10 Maria Felicia Faienza7
Maria Felicia Faienza7 Danilo Fintini11
Danilo Fintini11 Roberto Franceschi12Mila Kalapurackal13Silvia Longhi13
Roberto Franceschi12Mila Kalapurackal13Silvia Longhi13 Michela Mariani11Marco Pitea14
Michela Mariani11Marco Pitea14 Andrea Secco15Daniele Tessaris8
Andrea Secco15Daniele Tessaris8 Francesco Vierucci16
Francesco Vierucci16 Malgorzata Wasniewska3Giovanna Weber14
Malgorzata Wasniewska3Giovanna Weber14 Stefano Mora17*
Stefano Mora17*- 1Pediatric and Adolescent Endocrinology, Division of Pediatrics, Department of Obstetrics, Gynecology and Pediatrics, University Hospital, Pisa, Italy
- 2Department of Clinical and Experimental Medicine, Section of Paediatrics, University of Pisa, Pisa, Italy
- 3Department of Human Pathology of Adulthood and Childhood, University of Messina, Messina, Italy
- 4Pediatric Unit, University Hospital “G. Martino”, Messina, Italy
- 5Pediatric Unit, IRCCS Azienda Ospedaliero-Universitaria di Bologna, Bologna, Italy
- 6Department of Medical and Surgical Sciences, University of Bologna, Bologna, Italy
- 7Pediatric Unit, Department of Precision and Regenerative Medicine and Ionian Area, University “A. Moro” of Bari, Bari, Italy
- 8Division of Pediatric Endocrinology, Department of Public Health and Pediatrics, University of Turin, Regina Margherita Children’s Hospital, Turin, Italy
- 9Department of Pediatrics, IRCCS Istituto Giannina Gaslini, Genova, Italy
- 10Department of Neuroscience, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health, University of Genova, Genova, Italy
- 11Endocrinology and Diabetology Unit, Bambino Gesù Children Hospital, IRCCS, Rome, Italy
- 12Department of Pediatrics, Santa Chiara Hospital of Trento, APSS, Trento, Italy
- 13Department of Pediatrics, Hospital of Bolzano (SABES-ASDAA), Teaching Hospital of Paracelsus Medical University (PMU), Bolzano, Italy
- 14Pediatric Endocrinology Unit, Department of Pediatrics, IRCCS Ospedale San Raffaele, Milan, Italy
- 15Pediatric and Pediatric Emergency Unit, Children Hospital, Azienda Ospedaliera SS Antonio e Biagio e C. Arrigo, Alessandria, Italy
- 16Department of Pediatrics, San Luca Hospital, Lucca, Italy
- 17Laboratory of Pediatric Endocrinology, Department of Pediatrics, IRCCS Ospedale San Raffaele, Milan, Italy
Rickets results from impaired mineralization of growing bone due to alterations in calcium and phosphate homeostasis. Clinical signs of rickets are related to the age of the patient, the duration of the disease, and the underlying disorder. The most common signs of rickets are swelling of the wrists, knees or ankles, bowing of the legs (knock-knees, outward bowing, or both) and inability to walk. However, clinical features alone cannot differentiate between the various forms of rickets. Rickets includes a heterogeneous group of acquired and inherited diseases. Nutritional rickets is due to a deficiency of vitamin D, dietary calcium or phosphate. Mutations in genes responsible for vitamin D metabolism or function, the production or breakdown of fibroblast growth factor 23, renal phosphate regulation, or bone mineralization can lead to the hereditary form of rickets. This position paper reviews the relevant literature and presents the expertise of the Bone and Mineral Metabolism Group of the Italian Society of Pediatric Endocrinology and Diabetology (SIEDP). The aim of this document is to provide practical guidance to specialists and healthcare professionals on the main criteria for diagnosis, treatment, and management of patients with rickets. The various forms of rickets are discussed, and detailed references for the discussion of each form are provided. Algorithms to guide the diagnostic approach and recommendations to manage patients with rare forms of hereditary rickets are proposed.
1 Introduction
Rickets is a skeletal disease characterized by deficient mineralization of growth plates and bone matrix (osteomalacia) associated with low concentrations of calcium and/or phosphate in the blood. Rickets includes a heterogeneous group of acquired and inherited diseases. Nutritional rickets is the most common form of rickets globally. On the other hand, the development of molecular genetic techniques increased identification of the inherited forms of rickets. The primary method for diagnosing rickets involves examining the patient’s medical history, conducting biochemical tests, and performing radiologic examinations. Genetic analyses are particularly important in cases where patients are suspected to have a genetic form of rickets despite the absence of a family history of the disease. Treatment and management of rickets should be targeted on the pathogenesis and both are strictly connected with the diagnosis.
The aim of this document is to provide practical guidance to specialists and healthcare professionals about the main criteria for diagnosis, treatment, and management of patients with rickets.
2 Methods
This position paper focuses on the main criteria for the diagnosis, treatment, and management of patients with rickets. Relevant studies were identified independently by three authors (G.B., S.M., and P.C.) from the Medline/PubMed database. This document is the result of a thorough discussion by the members of the Bone and Mineral Metabolism Group of the Italian Society of Pediatric Endocrinology and Diabetology (SIEDP) to standardize the procedures based on the relevant literature and on the expertise of the panel members. We did not grade the strength of our recommendations, but much information reported in this paper was obtained from studies that used this method.
The aim of the document was not to provide a literature review on rickets or to discuss in detail the various form of rickets. The intention was to provide stakeholders with selected data and suggest a correct approach for an early diagnosis of rickets. Some algorithms to facilitate the diagnosis are also presented. Moreover, some important aspects of the management of patients with different forms of rickets are proposed. This position paper presents a therapeutic approach to the various forms of rickets in light of recent guidelines, and a specific treatment for patients with X-linked hypophosphatemic rickets (XLH).
3 Classification of rickets
Rickets may be classified in various forms based on the primary biochemical alteration, such as hypocalcemia or hypophosphatemia, or subdivided into two main groups, such as nutritional and hereditary forms. Moreover, some rare forms of rickets may be associated with direct bone damage due to acquired or inherited disorders.
In this position paper, the various forms of rickets are subdivided into four major categories based on the main pathogenesis, such as nutritional, hereditary, non-hereditary hypophosphatemic, and other forms (Figure 1).
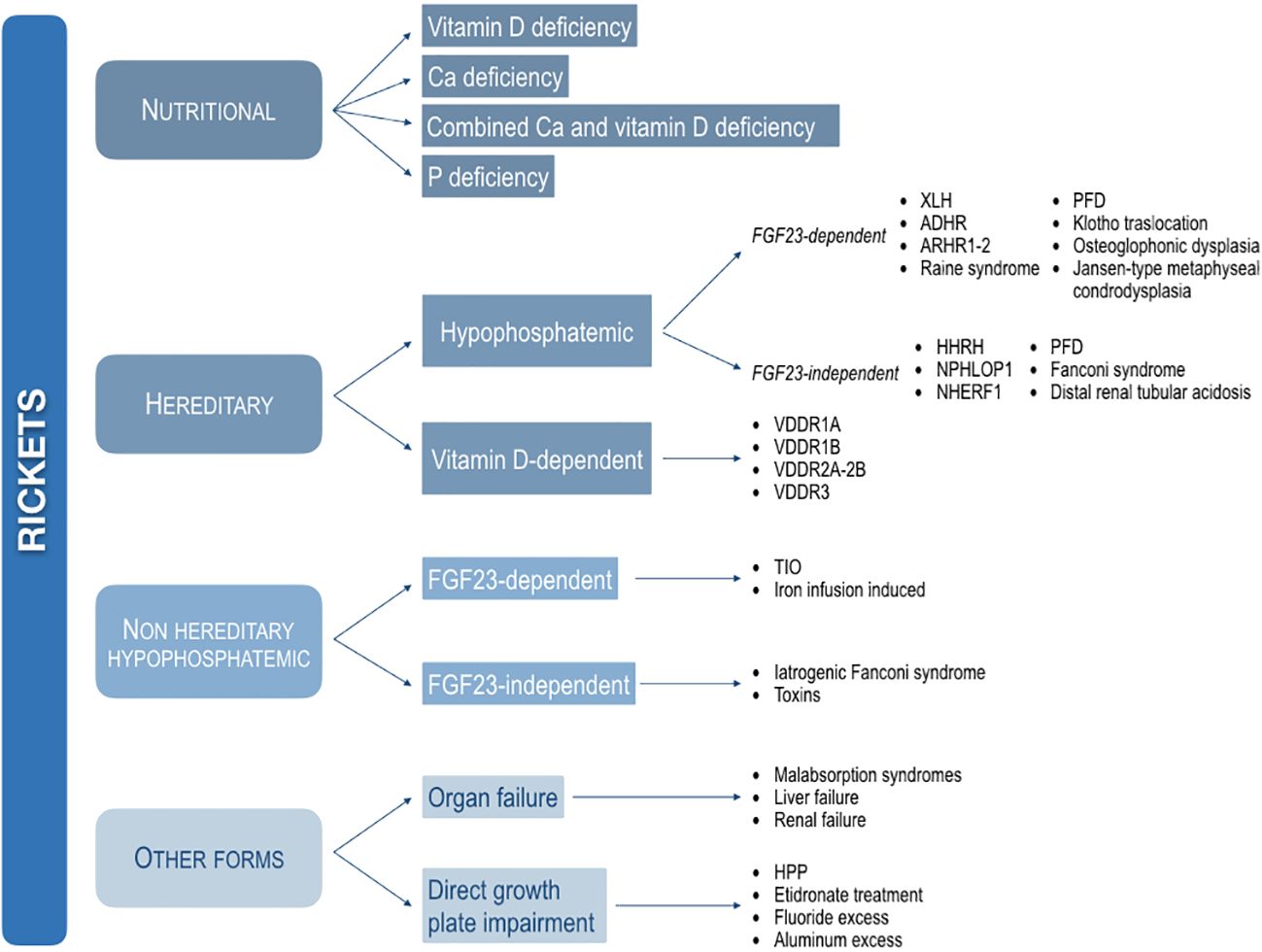
Figure 1 Classification of the main forms of rickets based on the pathogenesis. XLH, X-linked hypophosphatemic rickets; ADHR, autosomal dominant hypophosphatemic rickets; ARHR1, autosomal recessive hypophosphatemic rickets type 1; ARHR2, autosomal recessive hypophosphatemic rickets type 2; PFD, polyostotic fibrous dysplasia; HHRH, hypophosphatemic rickets with hypercalciuria; NPHLPO1, nephrolithiasis/osteoporosis, hypophosphatemic, 1. NHERF1, nephrolithiasis/osteoporosis, hypophosphatemic, 2; VDDR1A, vitamin D-dependent rickets type 1A; VDDR1B, vitamin D-dependent rickets type 1B; VDDR2A, vitamin D-dependent rickets type 2A; VDDR2B, vitamin D-dependent rickets type 2B; VDDR3, vitamin D-dependent rickets type 3; TIO, Tumor-induced osteomalacia; HPP, hypophosphatasia.
3.1 Nutritional rickets
Nutritional rickets is the most common form of rickets in developing countries, and it is frequently diagnosed in western countries among immigrant children, mainly from Africa and eastern Europe. Although it can rarely be caused by a phosphate-deficient diet, nutritional rickets is commonly the result of vitamin D deficiency due to insufficient UV exposure and/or insufficient dietary calcium intake, or a combination of both. This is particularly worrisome as nutritional rickets can profoundly affect the well-being of infants, children, and adolescents, with consequences that can persist into adulthood (1, 2).
In regions where calcium consumption is typically scant, with limited or absent dairy intake, dietary calcium deficiency stands as the primary culprit behind nutritional rickets among children beyond infancy (1, 3).
Dietary phosphate deficiency is a rare cause of nutritional rickets, but it is infrequent, because phosphate is abundant in natural foods. However, extreme starvation, a diet of elemental or hypoallergenic formula or parenteral nutrition, gastrointestinal surgery or short bowel syndrome, overuse of phosphate binders, or overly restrictive phosphate intake in managing chronic kidney disease may result in low phosphate intake (2, 4). Moreover, very low birthweight preterm infants are at high risk of developing nutritional rickets due to phosphate deficiency especially if they are breast-fed without phosphate supplements (2, 5).
3.2 Hereditary rickets
Hereditary rickets, caused by genetic mutations, may be subdivided into two main groups of disorders based on the association with impaired phosphate metabolism (hypophosphatemic rickets) or impaired vitamin D metabolism (vitamin D-dependent rickets) (Figure 1).
3.2.1 Hypophosphatemic rickets
Hypophosphatemic rickets include some disorders that may be associated with overproduction of fibroblast growth factor 23 (FGF23), namely FGF23-dependent forms, and disorders associated with a primary renal defect with normal concentrations of circulating FGF23, namely FGF23-independent forms (Figure 1).
3.2.1.1 FGF23-dependent hypophosphatemic rickets
X-linked hypophosphatemic rickets (XLH) is the commonest inherited form of rickets (6). Its prevalence is 1:20,000-60,000 (7, 8). XLH is due to mutation of PHEX gene encoding for phosphate regulating endopeptidase homolog X-linked, that regulates the expression of FGF23. The excessive production of FGF23 reduces tubular phosphate reabsorption and 1α-hydroxylase activity and stimulates renal 24-hydroxylase activity. Hypophosphatemia and a low or inappropriately normal serum 1,25-dihydroxyvitamin D (1,25(OH)2D) concentration in the setting of hypophosphatemia are the main biochemical findings of the patients with XLH (2, 9). Several other genes, such as FGF23, DMP1, and ENPP1, have a role in the synthesis, signaling, and regulation of FGF23. Mutations in these genes give rise to other rare variants of hypophosphatemic rickets, including autosomal dominant hypophosphatemic rickets (ADHR), autosomal recessive hypophosphatemic rickets type 1 (ARHR1) and autosomal recessive hypophosphatemic rickets type 2 (ARHR2), collectively constituting less than 20% of the cases (10).
Other rare forms of FGF23-dependent hypophosphatemic rickets include Raine syndrome due to mutation in the FAM20C gene (11), polyostotic fibrous dysplasia (PFD) due to gain-of-function mutation of the GNAS gene (12), hypophosphatemic rickets and hyperparathyroidism due to translocation of the KLOTHO promoter (13), osteoglophonic dysplasia caused by dominant activation variants in FGFR1 (14), opsismodysplasia (15), and Jansen-type metaphyseal chondrodysplasia due to mutations in the parathyroid hormone 1 receptor gene (16, 17). Further details are reported by Ackah et al. (14) and Shore (18).
3.2.1.2 FGF23-independent hypophosphatemic rickets
These disorders include some forms of hypophosphatemic rickets due to mutations in SLC34A3 encoding for NaPi2C (hypophosphatemic rickets with hypercalciuria, HHRH), SLC34A1 gene encoding for NaPi2a (nephrolithiasis/osteoporosis, hypophosphatemic 1, NPHLOP1), or mutation in NHERF1 gene (nephrolithiasis/osteoporosis, hypophosphatemic, 2) (19, 20). Patients affected by Fanconi syndrome or distal renal tubular acidosis may develop hypophosphatemic rickets (2).
3.2.2 Vitamin D-dependent rickets
Vitamin D-dependent rickets are caused by mutations affecting the enzymes involved in the activation or degradation of vitamin D or the action/expression of the vitamin D receptor (VDR). Vitamin D-dependent rickets type 1A (VDDR1A) is due to impaired synthesis of 1,25(OH)2D caused by a mutation in the enzyme (CYP27B1) encoding for renal 1α-hydroxylase. Vitamin D-dependent rickets type 1B (VDDR1B) is due to an impaired synthesis of 25-hydroxyvitamin D (25(OH)D) caused by a mutation in the enzyme (CYP2R1) encoding for hepatic 25-hydroxylase. Vitamin D-dependent rickets type 2 (VDDR2) is the result of impaired signaling of the VDR due to mutations in the VDR gene (VDDR2A) or the presence of a nuclear ribonucleoprotein that interferes with the VDR-DNA interaction (VDDR2B) (21). Vitamin D-dependent rickets type 3 (VDDR3) is due to increased inactivation of 1,25(OH)2D caused by a gain-of-function mutation in a gene encoding a vitamin D-degrading enzyme (CYP3A4) (21, 22). The most striking biochemical sign of these forms of rickets is severe hypocalcemia with secondary hyperparathyroidism.
3.3 Non-hereditary hypophosphatemic rickets
This group of hypophosphatemic rickets includes some heterogeneous acquired disorders causing phosphate wasting. They may be associated with increased production of FGF23 or iatrogenic causes.
3.3.1 Acquired FGF23-dependent hypophosphatemic rickets
Tumor-induced osteomalacia (TIO) is due to the overproduction of FGF23 and other phosphaturic factors by benign slow-growing mesenchymal tumors (23, 24). TIO has rarely been described in children (25–30).
Iron infusion-induced reversible hypophosphatemic osteomalacia may occur in patients receiving intravenous iron preparations, especially with iron carboxymaltose or iron polymaltose, for treatment of anemia when oral iron is either ineffective or contraindicated (31). Iron infusion acutely impairs FGF23 cleavage, triggering transient increase in intact FGF23 and hypophosphatemia in association with an inappropriately low concentration of 1,25(OH)2D, similar to genetic diseases of primary FGF23 excess (14, 31).
3.3.2 Acquired FGF23-independent hypophosphatemic rickets
Iatrogenic renal Fanconi syndromes with hypophosphatemia may be caused by some drugs, including valproate, cisplatin, ifosfamide, gentamycin, and excessive use of phosphate binders (32, 33) and by some toxins, such as heavy metals antibiotics, or antiretroviral and anticancer medications (14).
3.4 Other causes of rickets
Rickets may be a consequence of congenital or acquired chronic disorders affecting the organs involved in the activation of vitamin D. Liver failure and renal failure are the main causes of impaired vitamin D metabolism. Moreover, malabsorption syndromes, i.e., cystic fibrosis, inflammatory bowel diseases, coeliac disease, or extensive surgical intestinal resection, may affect the intestinal absorption of vitamin D. These heterogeneous forms of rickets are also associated with various degrees of reduced bone mineral density that may be assessed by dual energy X-ray absorptiometry (DXA) (2, 34, 35).
Some rare forms of rickets are caused by direct impairment of the process of mineralization at the growth plate. Protracted etidronate disodium (ethane 1-hydroxy-1, 1-diphosphonate) administration in patients with generalized arterial calcification has been associated with radiologic signs of rickets likely due to impaired growth plate mineralization (36, 37). Patients with the infantile form of hereditary hypophosphatasia (HPP) show radiological lesions that may resemble rickets. A poorly mineralized ribcage may be associated with respiratory insufficiency (38, 39).
A rare form of hypophosphatemic rickets with hypocalciuria has been reported after long-term treatment with aluminum-containing antacids (40, 41).
Furthermore, a particular form of rickets was observed in Indian children exposed to a high intake of endemic fluoride in the drinking water since their birth. The association with calcium deficiency was a trigger for the development of fluoride toxicity in bone. In calcium-deficient children, the toxic effects of fluoride manifested even at marginally high (> 2.5 mg/day) exposures to fluoride (42).
4 Clinical approach to rickets
Medical history, physical examination, and the biochemical results are the main findings to diagnose a form of rickets. The diagnosis of rickets should be confirmed by radiologic examination. The administration of vitamin D supplements, the evidence of relatives affected by rickets, the estimation of dietary calcium intake, and data on gestation and birth should be accurately investigated. Some clinical signs of rickets are more evident based on the age of the patient. Craniotabes (softening or thinning of the skull bones) may be the first sign of rickets in infants; it is detected by an inward collapse when applying pressure to the skull, typically followed by a snapping back after removing the pressure (43). In infants, other early signs of rickets are delayed fontanelle closure and frontal bossing (43, 44). In older children, swelling of the wrists, knees, or ankles and deformation of the legs such as knock-knees (genu valgum), or outward bowing (genu varum) with inability to walk are the main features (2, 44). Other skeletal signs of rickets are rachitic rosary, caused by swelling of the costochondral joints of the ribs, deformity of the soft rib cage, and bone pain (2, 44). Several non-osseous features may be associated with rickets: failure to thrive, delayed motor development, convulsions (due to hypocalcemia), muscular hypotonia, dilated cardiomyopathy, anemia, lethargy, irritability, delayed tooth eruption with altered enamel, and predisposition to respiratory infections (1, 43–48). HPP is characterized by diverse phenotypes that contribute to the diagnostic difficulties. In particular, neurological and dental involvements advocate for a multidisciplinary approach in the diagnosis of HPP (49).
Physical examination cannot differentiate children with active rickets from children recovering from rickets, and between the various forms of rickets (43). In addition, if abnormal bone features are detected clinically, the disease is already well established. Relying only on clinical signs may lead to overestimation of the prevalence of active rickets and lead to unnecessary treatment. The main skeletal signs of rickets are summarized in Table 1.
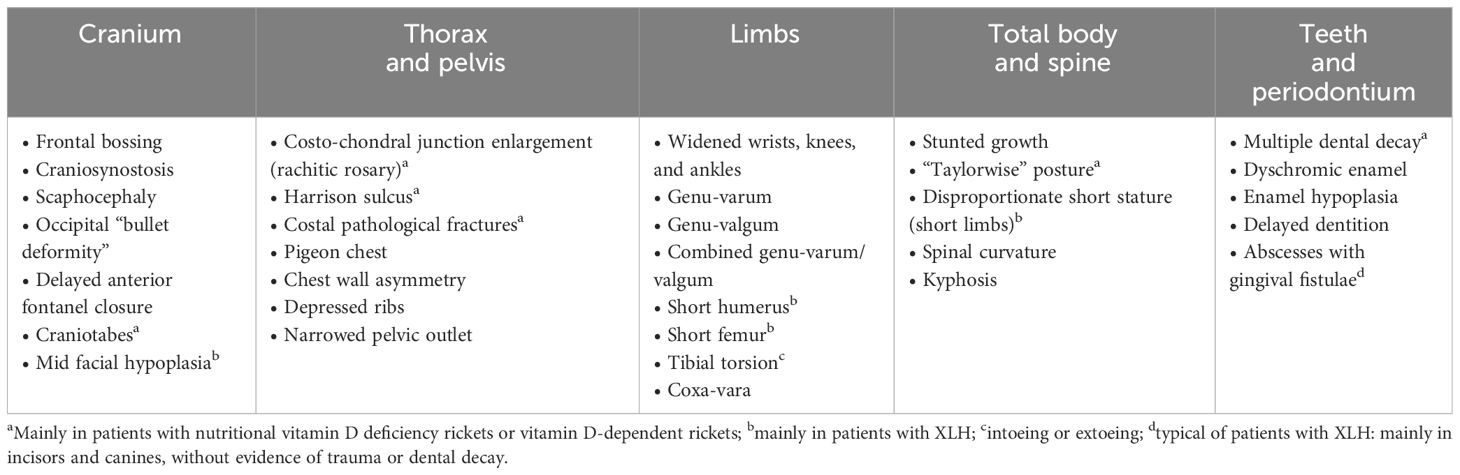
Table 1 Main skeletal and dental-periodontal signs in patients with rickets.
5 Biochemical features of the various forms of rickets
Serum calcium, phosphate, alkaline phosphatase, parathyroid hormone (PTH), and vitamin D metabolites are crucial biochemical parameters for the diagnosis of the various forms of rickets. Increased alkaline phosphatase activity is evident in all forms of rickets, except HPP (2, 4). Serum concentrations of phosphate and alkaline phosphatase activity should be interpreted according to appropriate age-specific reference values (49, 50).
Hypocalcemia is a main biochemical finding in patients with severe nutritional vitamin D deficiency rickets and in patients with vitamin D-dependent rickets. Hypophosphatemia (with normal serum calcium concentrations) is a specific feature in patients with hypophosphatemic rickets. However, hypophosphatemia may be also evident (usually associated with hypocalcemia) in patients with severe nutritional vitamin D deficiency rickets as a consequence of secondary hyperparathyroidism (2, 4, 35). It has been demonstrated that hypophosphatemia is the common denominator of all rickets affecting the process of apoptosis in the hypertrophic cells in the growth plate. In the absence of apoptosis, the hypertrophic cells accumulate in the growth plate and form the rachitic bone (51).
Renal tubular reabsorption of phosphate can be assessed by calculating the ratio of the tubular maximum reabsorption of phosphate to the glomerular filtration rate (TmP/GFR) (52). A reduced TmP/GFR ratio in patients with nutritional vitamin D deficiency rickets or vitamin D-dependent rickets reflects secondary hyperparathyroidism (2). The reduced TmP/GFR ratio found in patients with hypophosphatemic rickets is due to an overproduction or reduced degradation of fibroblast growth factor 23 (FGF23) (4, 20, 53).
Hypercalciuria may be an important diagnostic criterion in patients with HHRH (2, 4, 20, 35).
Secondary hyperparathyroidism is a major biochemical feature in patients with hypocalcemic forms of rickets. Serum PTH concentrations are normal or only slightly increased in patients with hypophosphatemic rickets, except in rare instances, such as in patients with HHRH who showed low normal o reduced serum PTH concentrations (2, 4, 20, 53).
There is strong evidence that nutritional vitamin D deficiency rickets develops with serum 25(OH)D concentrations <30 nmol/L (<12 ng/mL) (1), but it may also be associated with serum 25(OH)D concentrations >30 nmol/L (>12 ng/mL) (1, 54, 55). Furthermore, most children with vitamin D deficiency are asymptomatic (2, 56–58), and a normal calcium intake could be able to maintain bone integrity (1). Some evidence suggested that a 25(OH)D concentration of 30 - 34 nmol/L (12 - 13.6 ng/mL) may be the critical cutoff value below which nutritional vitamin D deficiency rickets is more likely to occur (1, 59). A recent systematic review and multi-level meta-analyses by odds, sensitivities and specificities for nutritional rickets at different serum 25(OH)D thresholds suggested a minimal risk threshold of around 28 nmol/L (11 ng/ml) for children with adequate calcium intakes and 40 nmol/L (16 ng/ml) for children with low calcium intakes (60). Indeed, the total concentration of 25(OH)D and its circulating components may be influenced by factors such as the severity and duration of reduced 25(OH)D levels, habitual intake and bioavailability of dietary calcium, the rate of bone growth and mineralization, as well as genetic polymorphisms (2, 55, 61).
Reduced serum 25(OH)D concentrations are observed in patients with vitamin D hydroxylation-deficient rickets type 1B (62) and VDDR3 (22); conversely, patients with VDDR1A and VDDR2A or VDDR2B have normal or increased serum 25(OH)D concentrations (21).
Serum 1,25(OH)2D concentrations vary with the time of diagnosis in nutritional vitamin D deficiency rickets. They are generally increased at the early stages of the disease to compensate for hypocalcemia and secondary hyperparathyroidism, whereas they are decreased at the late stages of the disease due to substrate depletion (48, 51). Therefore, measurement of 1,25(OH)2D is not useful in patients with nutritional vitamin D deficiency rickets. In patients with hypophosphatemic rickets, serum 1,25(OH)2D concentrations may be reduced, but in most patients, they are inappropriately normal in the setting of hypophosphatemia (2, 4, 35, 53). Serum 1,25(OH)2D concentrations are increased in patients with VDDR2A, VDDR2B, nutritional calcium deficiency rickets, X-linked recessive hypophosphatemic rickets, HHRH or NPHLPO1 (2, 4, 20, 35).
FGF23 concentrations are increased in FGF23-dependent hypophosphatemic rickets (2, 4, 20, 35, 50); therefore, these conditions can be distinguished from FGF23-independent hypophosphatemic disorders by measuring the FGF23 concentration. Hypophosphatemia associated with intact FGF23 concentration greater than 40 pg/ml may be a crucial marker for the early diagnosis of XLH in pediatric patients (50). Assessment of FGF23 is also indicated in the diagnosis of patients with suspected TIO. Furthermore, FGF23 is the most critical biochemical marker for distinguishing nutritional vitamin D deficiency rickets from hypophosphatemic rickets, mainly in patients with an overlap of serum 25(OH)D concentrations (63). Moreover, increased FGF23 concentration is found in patients with calcium deficiency rickets (64).
Hypercalcemia and hyperphosphatemia, in addition to reduced alkaline phosphatase activity, are useful parameters for the differential diagnosis of the various forms of rickets (38, 39).
Biochemical features of the main forms of rickets are shown in Table 2.
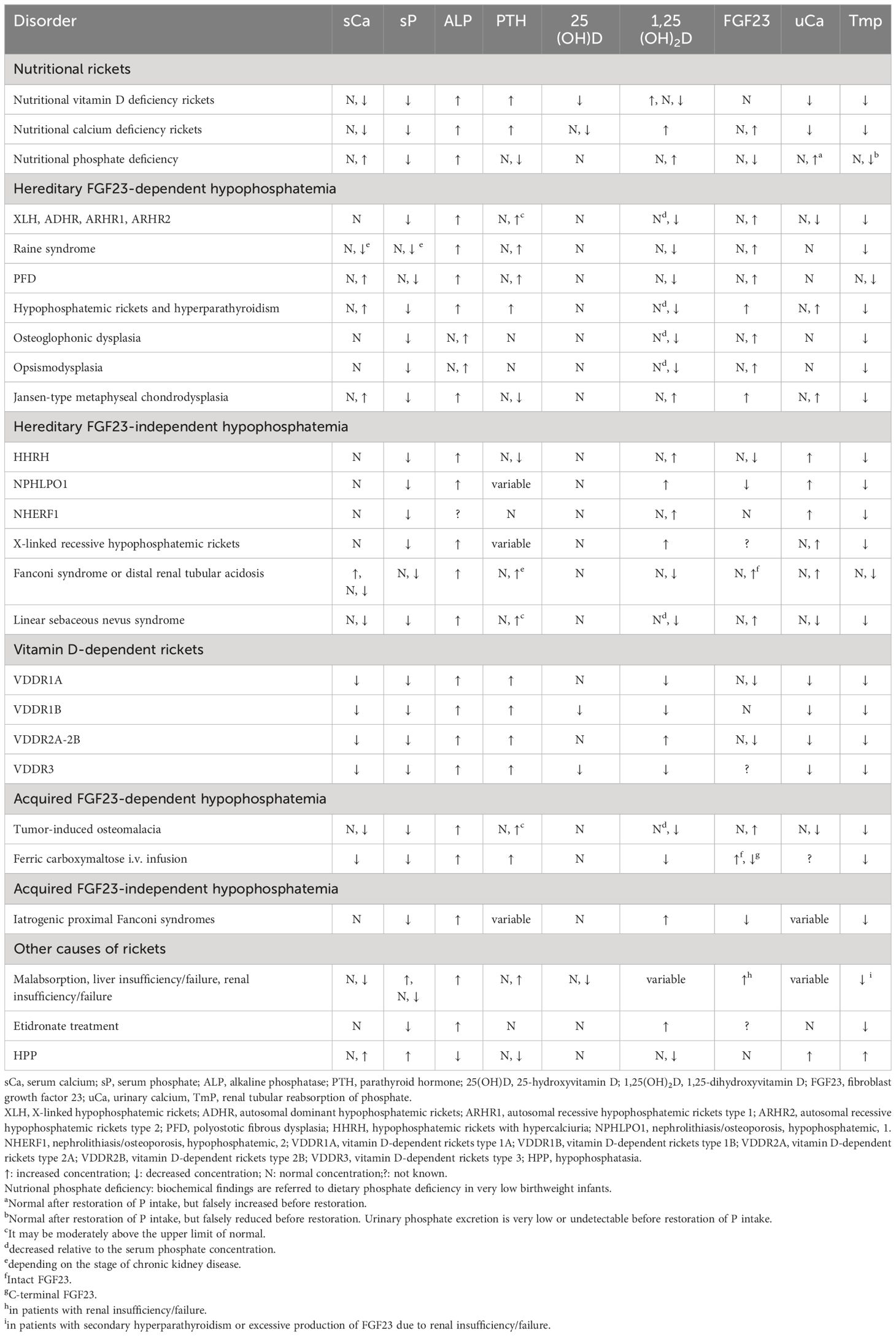
Table 2 Biochemical findings in patients with different types of rickets or rickets-like disorders.
6 Radiological assessment of rickets: diagnosis and follow-up
Plain radiography is the primary imaging tool for the assessment of rickets; the earliest signs are osteopenia and cortical thinning of long bones (48, 65). The initial assessment of a child suspected of having rickets should involve radiographs of skeletal sites where bones are undergoing the most rapid growth. These sites may differ depending on the child’s age.
A radiological examination of the chest may be useful to assess rickets in neonates, mainly in preterm infants, and in infants in the first year of life (65, 66). Insufficient mineralization of the metaphyseal-physeal region can be observed in the proximal humerus and anterior rib ends (rachitic rosary). In most children, radiological signs of rickets are evident at the wrists and knees, where rapid growth occurs. The distal ulna is the best site to demonstrate early changes of rickets, mostly in young children (67). The classical rachitic changes are the widening and irregularity of all the physes, with metaphyseal widening, splaying, cupping, and fraying. Moreover, a coarse trabecular pattern may be observed in the metaphysis (48, 65). Epiphyseal ossification centers become blurred or apparent (65). Cranial deformities and bowing of the legs are the typical signs of osteomalacia due to unmineralized osteoid (66).
Pathologic fractures (including rib fractures) are rare except in very preterm babies and are very uncommon beyond age 6 months corrected postnatal age (66). The occurrence of fractures is usually evident in children with a severe form of rickets (68). Fractures of long bones may occur occasionally in children affected by calciopenic and phosphopenic (excluding XLH) forms of rickets in association with radiographic signs of osteopenia and the evidence of typical metaphyseal rachitic lesions (2).
Moreover, by radiologic examination is possible to estimate the severity of rickets by using the Rickets Severity Score (RSS). This score is determined based on the degree of metaphyseal fraying, concavity, and the proportion of the growth plate affected at the wrists, knees, and ankles (69, 70). The RSS employs a 10-point scale, with 10 indicating the utmost severity of rickets, and 0 denoting the absence of any radiographic alterations. The radiographic response after treatment of nutritional vitamin D deficiency rickets (69) and XLH (70) can be estimated by the RSS, which correlates with serum alkaline phosphatase activity.
The Radiographic Global Impression of Change (RGI-C) represents a complementary assessment of RSS. A change score is assigned based on differences in the appearance of rickets on pairs of radiographs compared side by side. The RGI-C has been validated to evaluate skeletal changes in patients with HPP (71). Moreover, Lim et al. (72) demonstrated that RGI-C is a reliable, valid, and sensitive tool in patients with XLH, and complementary to the RSS.
Radiologic examination is also useful to confirm the diagnosis of rickets suspected by clinical signs and biochemical data, but it does not indicate the pathogenesis, because the radiological signs of rickets are similar in patients with nutritional rickets, hypophosphatemic rickets, and vitamin D-dependent rickets (Figure 2).
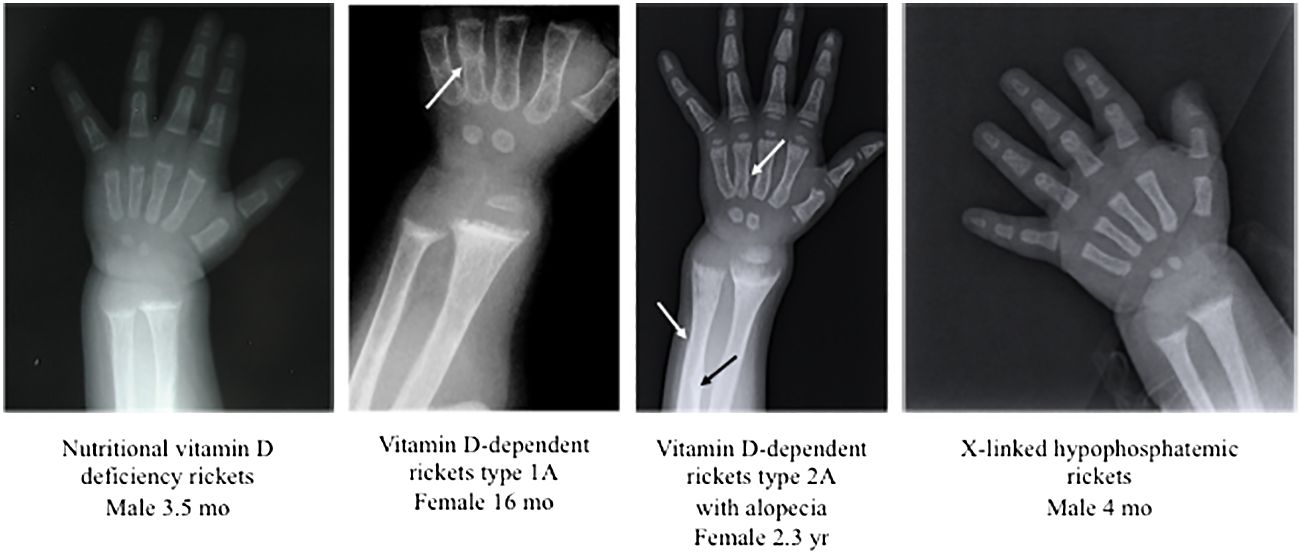
Figure 2 Radiographic features at the wrist and at the hand in patients with nutritional vitamin D deficiency rickets and some forms of hereditary rickets. In patients with hereditary rickets, the diagnosis was confirmed by genetic analyses. All patients showed widening and fraying of the epiphyseal plate and metaphyseal concavity of the ulna. The white and black arrows showed fractures.
Finally, radiological examination is useful to examine the effect of treatment with vitamin D in patients with nutritional vitamin D deficiency rickets showing the appearance of the zone of provisional calcification at the ends of the metaphyses that is usually seen within 3-4 weeks of treatment (67).
7 Algorithms for the diagnosis of rickets
Figure 3 shows a management algorithm based on the biochemical parameters for the evaluation of patients with rickets confirmed by the evidence of clinical signs and the typical radiologic lesions of rickets and associated with increased serum alkaline phosphatase activity. The algorithm is primarily based on the pathogenesis of hypocalcemia and hypophosphatemia; serum concentrations of calcium, phosphate, and PTH represent the key biochemical parameters to differentiate hypocalcemic from hypophosphatemic form of rickets. Hypocalcemic forms of rickets are usually characterized by hypocalcemia, a low or normal serum phosphate concentration, and increased PTH values; whereas, hypophosphatemia with normocalcemia and normal PTH concentrations are the main biochemical signs of most of the hypophosphatemic forms of rickets.
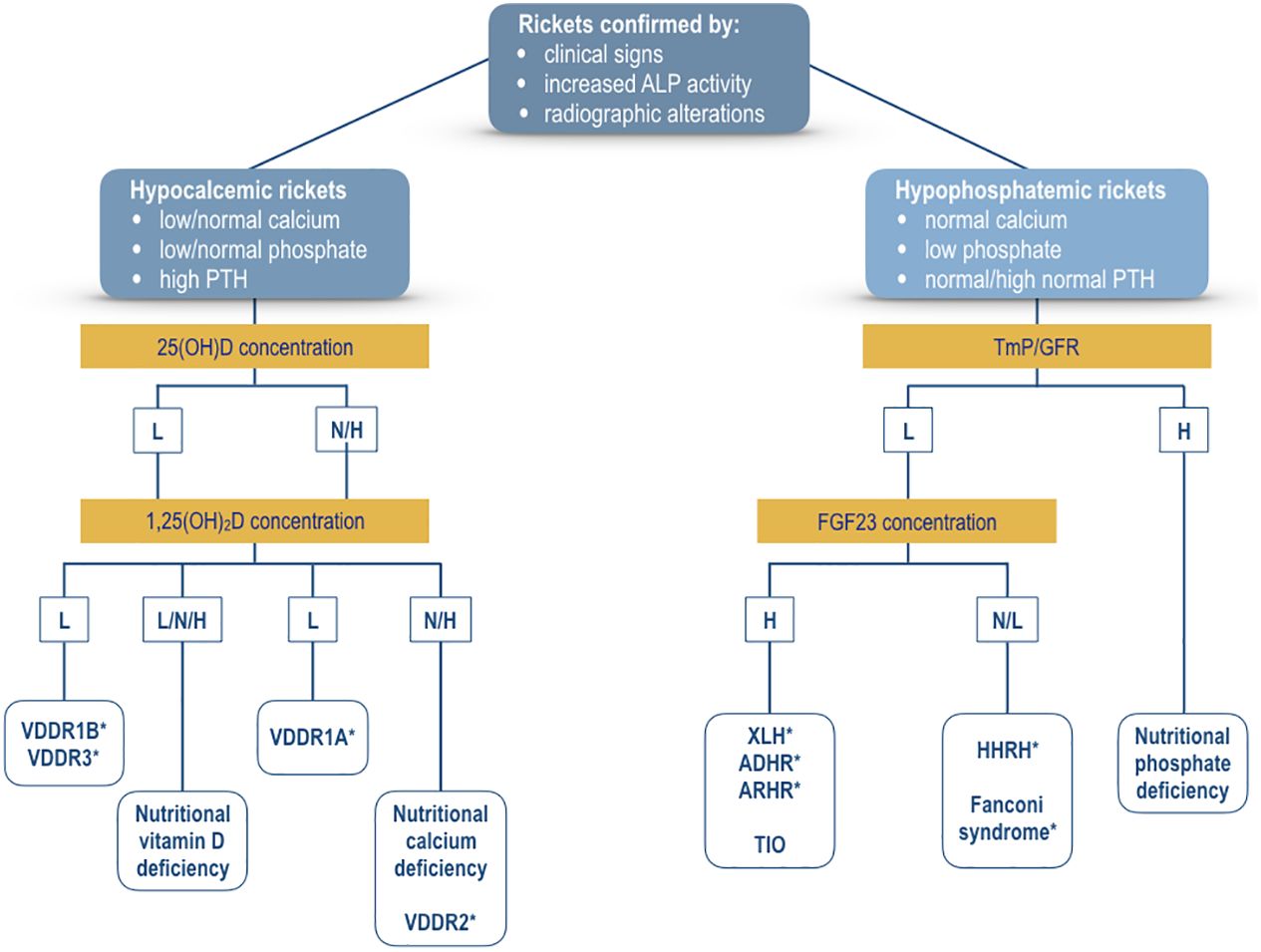
Figure 3 Algorithm for the evaluation of a patient with confirmed rickets by clinical signs, increased alkaline phosphatase activity, and radiographic lesions of rickets. The primary differential diagnosis is based on the presence of hypocalcemia (hypocalcemic rickets) or hypophosphatemia (hypophosphatemic rickets). Measurement of serum concentrations of 25(OH)D and 1,25(OH)2D may indicate the biochemical diagnosis of the various forms of hypocalcemic rickets. Measurement of TmP/GFR values represents the first step for the selection of the patients with hypophosphatemic rickets. Thereafter, measurement of FGF23 concentration may suggest the biochemical diagnosis of the various forms of hypophosphatemic rickets. For additional detais see text. XLH, X-linked hypophosphatemic rickets; ADHR, autosomal dominant hypophosphatemic rickets; ARHR1, autosomal recessive hypophosphatemic rickets type 1; ARHR2, autosomal recessive hypophosphatemic rickets type 2; HHRH, hypophosphatemic rickets with hypercalciuria; VDDR1A, vitamin D-dependent rickets type 1A; VDDR1B, vitamin D-dependent rickets type 1B; VDDR2A, vitamin D-dependent rickets type 2A; VDDR2B, vitamin D-dependent rickets type 2B; VDDR3, vitamin D-dependent rickets type 3; TIO, Tumor-induced osteomalacia. *Genetic testing is important to confirm the suspected diagnosis. L, low values; N, normal values; H, high values.
The measurement of serum concentrations of vitamin D metabolites may indicate the pathogenesis of rickets in patients with hypocalcemic forms. Reduced serum values of 25(OH)D and 1,25(OH)2D suggest the diagnosis of VDDR1B or VDDR3, whereas normal or increased serum 25(OH)D values associated with low 1,25(OH)2D may be diagnostic for VDDR1A. A normal or increased serum 25(OH)D concentration combined with a normal or increased serum 1,25(OH)2D concentration may characterize two conditions, nutritional calcium deficiency or VDDR2.
The measurement of FGF23 may be useful to identify the FGF23-mediated from the non-FGF23-mediated hypophosphatemic disorders. High FGF23 concentrations are evident in both hereditary and acquired forms of hypophosphatemic rickets such as TIO. Hypophosphatemia associated with normal or low FGF23 concentrations suggests the diagnosis of a form of Fanconi syndrome or HHRH. Patients with HHRH usually show hypercalciuria. Hypophosphatemia with high TmP/GFR values may indicate the diagnosis of nutritional phosphate deficiency rickets.
8 Genetics
Whenever the clinical, biochemical, and radiologic evidence indicates that the cause of rickets might be of genetic origin, specific molecular testing is necessary. Table 3 summarizes the genes involved in the genesis of genetically determined rickets or hypophosphatemia.
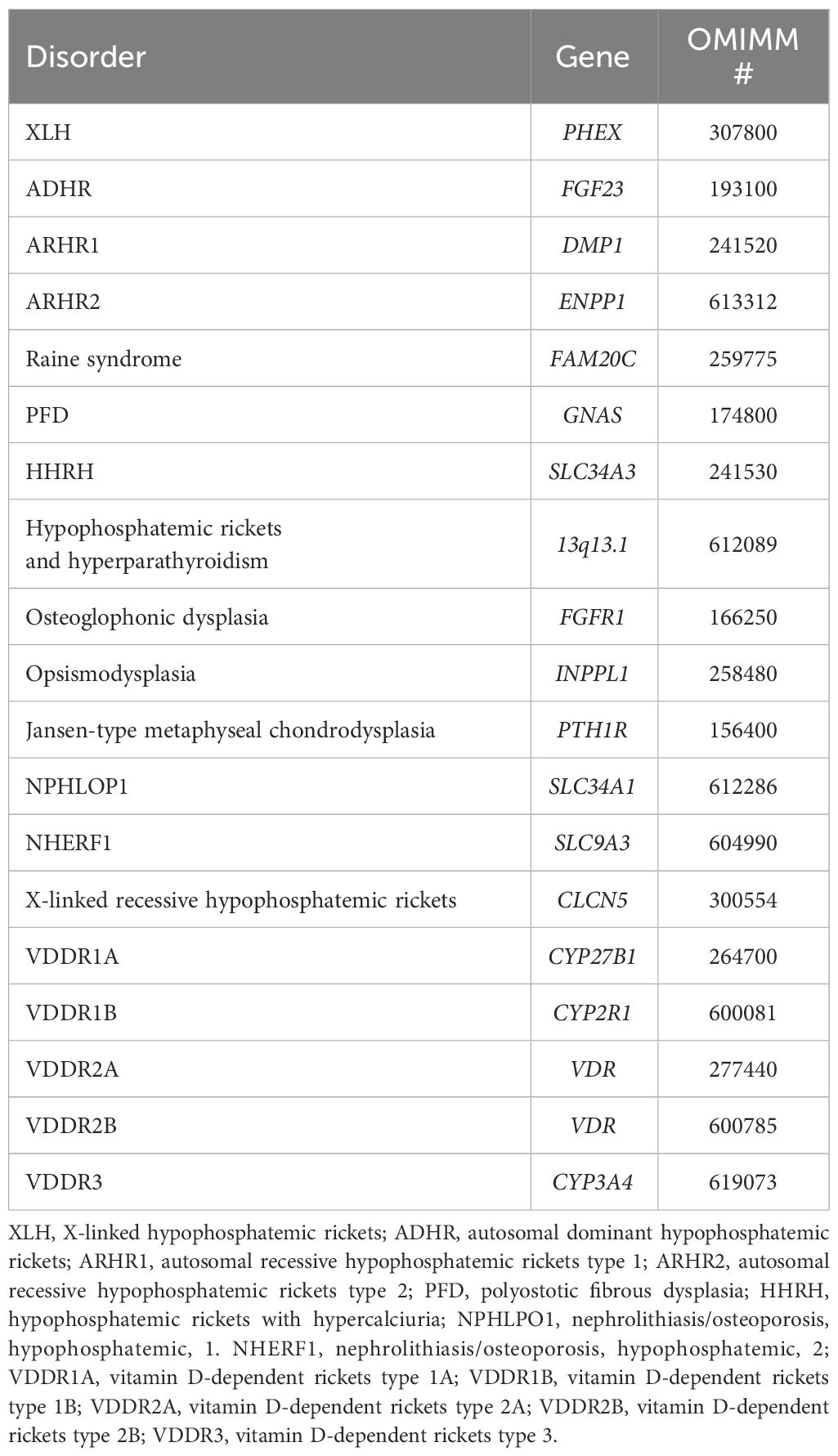
Table 3 Genes involved in the genesis of genetically determined rickets or hypophosphatemia.
The analysis of the PHEX gene in the suspect of XLH should be performed, bearing in mind the extensive heterogeneity of the pathogenic variants. These have been found in all exons and in the non-coding regions. Numerous are the insertions and deletions that have also been reported (73, 74).
9 Treatment and management of nutritional rickets
9.1 Nutritional vitamin D deficiency rickets
Vitamin D supplements may be administered as daily therapy or as a single bolus; the suggested doses are reported in Table 4. Alternatively, a bolus of 100,000 IU every 20 days for 3-4 times may be effective in patients with poor compliance with the treatment (75). Administration of a single-day high-dose of vitamin D therapy (stoss therapy), with doses reaching up to 600,000 IU, has been proposed. However, hypercalcemia may occur (76, 77). Therefore, the use of high and repeated doses of vitamin D is not recommended. Daily treatment with vitamin D should be continued for 3-4 months (1, 2, 35, 48, 75, 78), but the duration of treatment should be individualized. Normalization of skeletal lesions is usually reached during this time. If radiographic recovery remains incomplete or serum biochemistry hasn’t normalized, vitamin D treatment should be extended (79). Inadequate adherence to the treatment regimen may lead to failure or delayed healing of rickets. In this occurrence, vitamin D stoss therapy by a single intramuscular injection ranging from 50,000 IU for children aged 3 months and older, up to 300,000 IU for individuals over 12 years may be recommended (1).
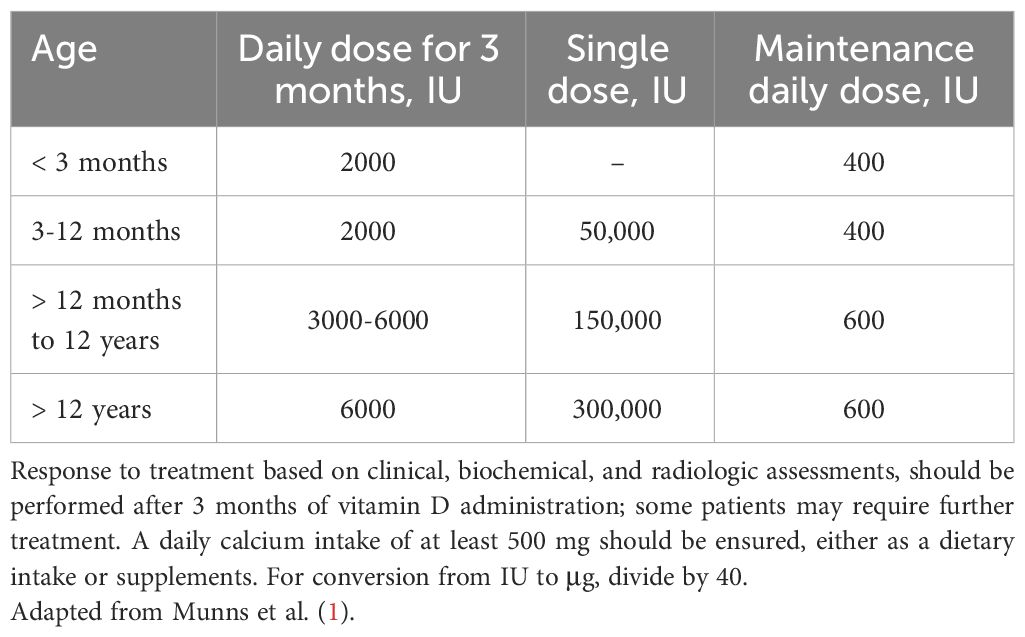
Table 4 Vitamin D treatment in patients with nutritional vitamin D deficiency rickets.
Some studies showed that in increasing serum 25(OH)D concentration the daily administration of vitamin D3 was similar to vitamin D2, whereas, vitamin D3 was more effective than vitamin D2 when administered as bolus (80). Moreover, there is no indication for the use of vitamin D metabolites for the treatment of patients with nutritional vitamin D deficiency rickets, because they do not restore vitamin D status and hypercalcemia may occur (66, 67, 81).
Patients experiencing symptomatic hypocalcemia, such as tetany, convulsions, or dilated cardiomyopathy, should receive intravenous calcium gluconate until serum calcium levels are normalized (35, 75, 78). Once normocalcemia is achieved, transitioning to oral calcium supplementation is appropriate. A routine oral calcium supplementation of 500 mg/day, obtained through dietary intake or supplements, should be implemented concurrently with vitamin D, irrespective of age or weight. Duration of calcium supplementation is variable and is related to the timing of normalization of serum calcium concentration (1). Alternatively, 30-75 mg/kg/day elemental calcium in 3 divided doses may be administered over 2-4 weeks. Calcium supplements are important to avoid hypocalcemia as the concentration of PTH normalizes (“hungry-bone” syndrome), particularly in patients treated with stoss therapy (48). Doses of intravenous and oral calcium supplements are reported in Table 5.
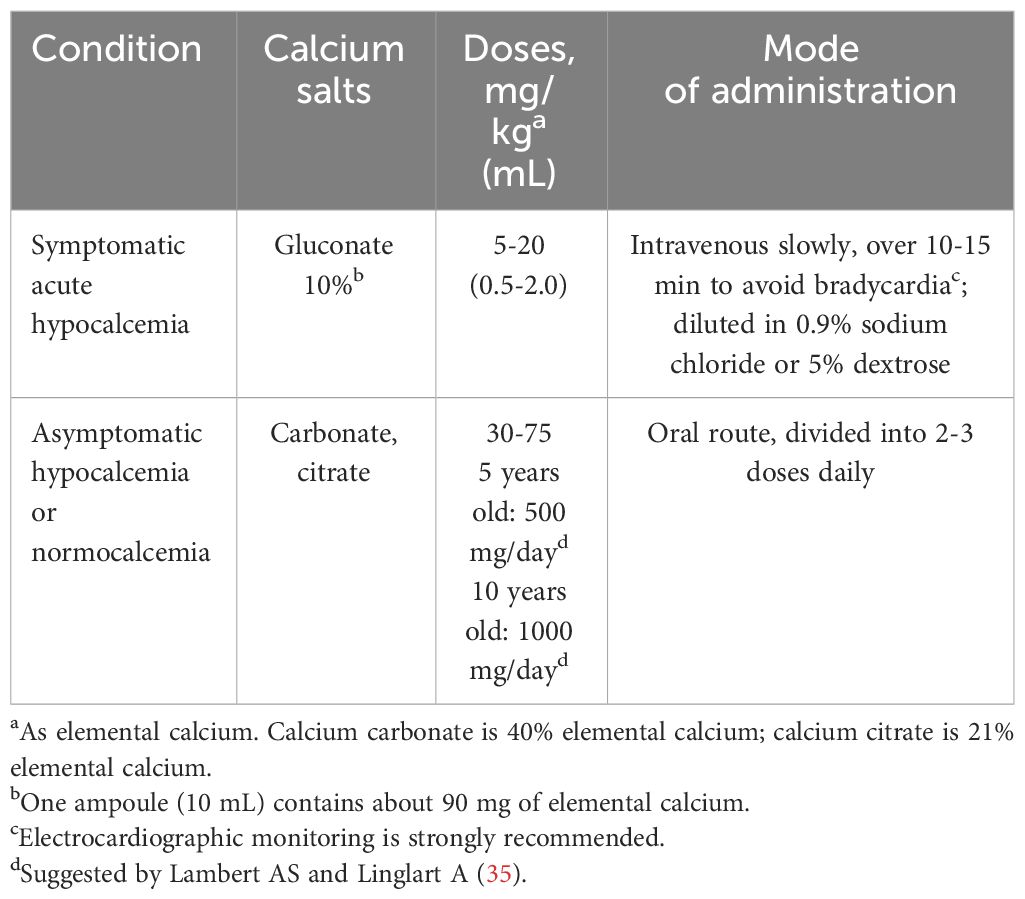
Table 5 Calcium treatment in patients with symptomatic or asymptomatic hypocalcemia associated with rickets.
Monitoring of treatment with vitamin D and calcium supplements varies with the severity of rickets and response to therapy. Normalization of serum calcium and phosphate concentrations usually occurs within 3 weeks (67, 75), but it may also be evident after only 6-10 days of treatment (48). Serum PTH concentrations usually fall within the normal range as normocalcemia is restored (75, 81). Serum 25(OH)D concentrations increase rapidly and normal values may be reached after 4-6 weeks (75). Serum 1,25(OH)2D concentrations increase rapidly with treatment and remain elevated for up to 10 weeks (81). Alkaline phosphatase activity declines progressively but it may remain increased for several months (3-6 months) depending on the severity of the vitamin D deficiency (48, 82). Moreover, alkaline phosphatase activity is associated with the recovery of skeletal rachitic lesions, suggesting that it is a reliable and economic biochemical marker for monitoring the effectiveness of treatment in the clinical setting (82).
In order to prevent the resurgence of nutritional vitamin D deficiency rickets vitamin D supplementation should be continued. Following the resolution of rickets, at least 400 IU/day before the age of 12 months and 600 IU/day of vitamin D are recommended (1, 67, 78).
9.2 Nutritional calcium deficiency rickets
A randomized controlled study in children with nutritional calcium-deficiency rickets showed that radiographic healing of the rachitic lesions was obtained by daily administration of with 500 mg, 1000 mg, or 2000 mg of elemental calcium salts. However, daily supplementation with 1000 mg and 2000 mg was more effective than 500 mg. The treatment with 2000 mg was similar to 1000 mg of supplemental calcium (83). Therefore, it is recommended that 1000 mg of elemental calcium daily (subdivided into 2-3 doses) should be used in children with nutritional calcium deficiency rickets, until healing is complete; in some children, it may take more than 24 weeks (84). The rate of healing of rickets may be improved by vitamin D supplements (50,000 IU every 4 weeks) (84). Radiologic healing of the growth plate usually occurred after 3-6 months of calcium administration, even though the clinical signs of rickets required a longer time to resolve than biochemical and radiologic alterations (3).
9.3 Nutritional phosphate deficiency rickets
Management of dietary phosphate deficiency is based primarily on adequate dietary supplements. Oral or parenteral phosphate salts are usually administered in patients with impaired phosphate absorption after extensive gastrointestinal surgery, short bowel syndrome, or severe gastrointestinal disorders (78).
Biochemical parameters, including relative hypocalcemia or hypophosphatemia, are the main factors for the management of metabolic bone disease of prematurity. Measurement of PTH has a pivotal role for the treatment, because increased PTH occurs in calcium deficiency, while it is normal or reduced in phosphate deficiency (85, 86). Human milk fortifiers and special preterm formulas are usually indicated to improve the needs for growth in very low birthweight infants. Phosphate supplementation should be considered for serum phosphate concentrations <4 mg/dL (1 mmol/L), but it can be also indicated if values fall below 5.5 mg/dL (1.3 mmol/L), especially if associated with increased alkaline phosphatase activity, to improve bone mineralization and to prevent hypercalciuria (87). The recommended daily oral intake of calcium and phosphate varies between 150-220 mg/kg/day and 75-140 mg/kg/day through enteral feeds, respectively (87–91); an enteral calcium to phosphate intake ratio at 1.5:1 to 1.7:1 on mg-to-mg basis is proposed (86, 87). For parental nutrition, calcium 75-100 mg/kg/day and phosphate 50-80 mg/kg/day are recommended (85, 86, 90, 91).
In infants unable to tolerate human milk fortifier or preterm formula, elemental minerals may be added by oral route. Supplementation usually starts at 20 mg/kg/day of elemental calcium and 10-20 mg/kg/day of elemental phosphorus. It is increased, as tolerated, to a maximum of 70-80 mg/kg/day of elemental calcium and 40-50 mg/kg/day of elemental phosphorus (87, 91). Serum PTH concentrations and alkaline phosphatase activity should be measured every 1-2 weekly for adjusting phosphate and calcium to phosphate ratios (86).
Vitamin D supplements in preterm infants differ between United States and Europe recommendations. The European Society for Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) recommends a dose of 800-1000 IU vitamin D daily, whereas 200 IU/day is recommended in the United States for infants weighing less than 1500 g and 400 IU/day for infants weighing more than 1500 g (87, 88, 91, 92). A lower calcium intake may benefit from a higher vitamin D intake as suggested by ESPGHAN recommendations, whereas a higher calcium intake may be preferred in association with a lower vitamin D intake as indicated in the United States (91, 92).
10 Treatment and management of vitamin D-dependent rickets
10.1 VDDR1A
Conventional treatment for patients with VDDR1A is based on the association of a vitamin D active metabolite and calcium supplements. Patients must be treated lifelong with physiologic doses of 1,25(OH)2D given twice daily due to its short half-life (21). Alternatively, alfacalcidol may be administered once a day due to its longer half-life. Alfacalcidol treatment by drop formulation may be useful in affected infants. The dosage of both calcitriol and alfacalcidol should be titrated according to serum calcium concentrations. The high calcium demands of the unmineralized skeleton occurring in the first months of treatment may require 2-5 times higher dosages of active vitamin D metabolites than during maintenance treatment (21, 93). The maintenance doses of active vitamin D metabolites and calcium supplements after normalization of serum calcium and PTH concentration are reported in Table 6.
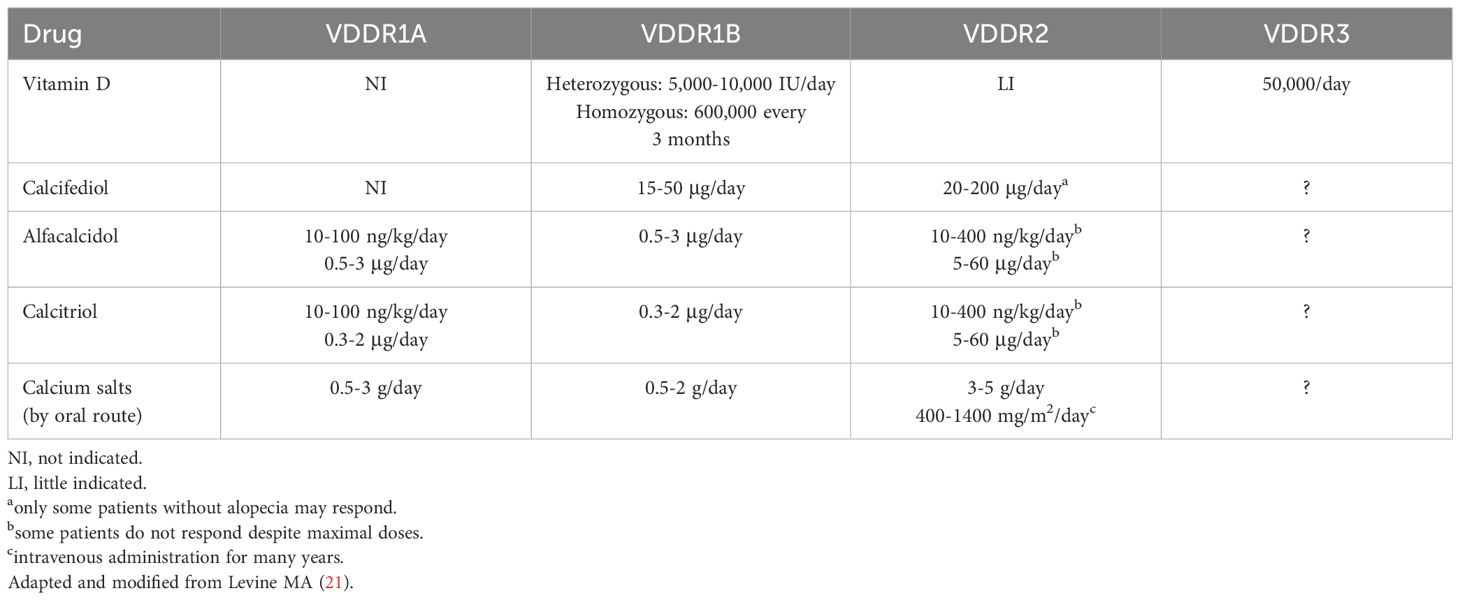
Table 6 Maintenance treatment in patients with vitamin D-dependent rickets.
Response to calcitriol or alfacalcidol is usually rapid, with healing of rickets and normalization of the biochemical parameters within 7-9 weeks (94). Long-term compliance with the treatment is important to maintain normal calcium and PTH concentrations. Serum concentrations of calcium, phosphate, and PTH, and urinary calcium excretion should be measured regularly during the maintenance treatment (at least every 4-6 months). However, time intervals may be reduced if hypocalcemia persists during the first weeks of treatment. Serum calcium concentration should be maintained in the low-normal range to keep PTH secretion below the upper limit of normal (95). Calcium excretion should be maintained below the normal limit for age and weight. Renal ultrasound examination should be performed every 1-2 years to monitor for renal calcification, and more frequently if there is evidence of hypercalciuria (21). However, based on personal expertise renal ultrasonography every 6-12 months may be useful for the early diagnosis of a mild form of nephrolithiasis.
10.2 VDDR1B
The best treatment for the patients with this form of rickets requires the association of calcifediol, as vitamin D has not/poor effect due to the altered hepatic conversion, and calcium supplements (Table 6). Serum 25(OH)D concentrations increased in response to a vitamin D bolus in heterozygous patients, although the achieved concentrations were lower than those found in controls. Homozygous patients with mutations in CYP2R1 showed a very limited or no increase in serum 25(OH)D concentrations in response to the same vitamin D bolus (62, 96). Alternatively, other vitamin D active metabolites, such as calcitriol or alfacalcidol, may be effective in improving serum calcium and PTH concentrations.
10.3 VDDR2A and VDDR2B
The success of the treatment of patients with VDDR2A varies according to the VDR mutation (97). It has been found that mutations in the ligand-binding domain are associated with variable resistance to 1,25(OH)2D; whereas, mutations in the DNA binding domain cause almost total 1,25(OH)2D resistance and alopecia (2). Patients with alopecia are usually less responsive to treatment in comparison with patients without alopecia (98). However, the response to treatment is highly variable and the doses of vitamin D metabolites and calcium salts should be individualized. About half of patients with alopecia are resistant even to the highest vitamin D metabolites doses. The other half require ten times higher vitamin D metabolites doses than patients without alopecia (21, 78).
Levine (21) suggested that maintenance therapy is based upon several factors: (a) some patients will respond to calciferols (vitamin D2, vitamin D3, or calcifediol), which are substrates for the generation of 1,25(OH)2D; (b) other patients may respond only to high doses of vitamin D metabolites (calcitriol or alfacalcidol) that possess 1α-hydroxylation; (c) a minority of patients will not respond to calciferols Some patients affected by severe forms of the disease required long-term intravenous calcium administration (given over 12 h, often during the night) for many months in order to achieve normocalcemia (21, 78, 99).
During and after puberty, the patients may develop an increased intestinal calcium absorption to concentrations that are even greater than those of normal individuals. Indeed, some patients maintain normocalcemia with modest oral calcium supplementation, and a small number of patients may have normocalcemia even without calcium supplements (21).
Management of patients with VDDR2B is similar to that indicated in patients with VDDR2A (21, 99).
10.4 VDDR3
Few data on the treatment of patients with VDDR3 are available. These patients may respond to vitamin D or vitamin D metabolites, but greater doses are required in comparison within patients with nutritional vitamin D deficiency rickets. Lifelong treatment with a high dose of vitamin D are needed to ensure clinical and biochemical remission (21, 22). Some suggestions for the treatment of these patients are reported in Table 6.
11 Treatment and management of hypophosphatemic rickets
11.1 FGF23-dependent hypophosphatemic rickets
Conventional treatment of patients with FGF23-dependent hypophosphatemic rickets, including XLH, ADHR, ARHR1, and ARHR2, consists of inorganic oral phosphate salts combined with vitamin D active metabolites, such as calcitriol or alfacalcidol. Most of the data on the use of conventional treatment in hypophosphatemic rickets comes from the management of patients with XLH. The recommended starting and maintenance doses of inorganic phosphate salts and vitamin D active metabolites in patients with hypophosphatemic rickets (2, 100–103) are summarized in Table 7. Conventional treatment may be associated with a transient increase in serum phosphate concentration only in some patients, without significant changes in TmP/GFR. The treatment started in early infancy improves outcomes even if it did not completely normalize skeletal abnormalities (2, 104–106). It is generally associated with slow improvement in the healing of rickets and with residual skeletal deformities in most patients. Moreover, it has been shown that patients with XLH treated with conventional treatment had decreased height gain by 1 year of age and remained below population norms thereafter (107). This suggests that XLH affects skeletal growth and bone mineralization from the earliest years of life and that conventional treatment may not improve the evolution of the disease.
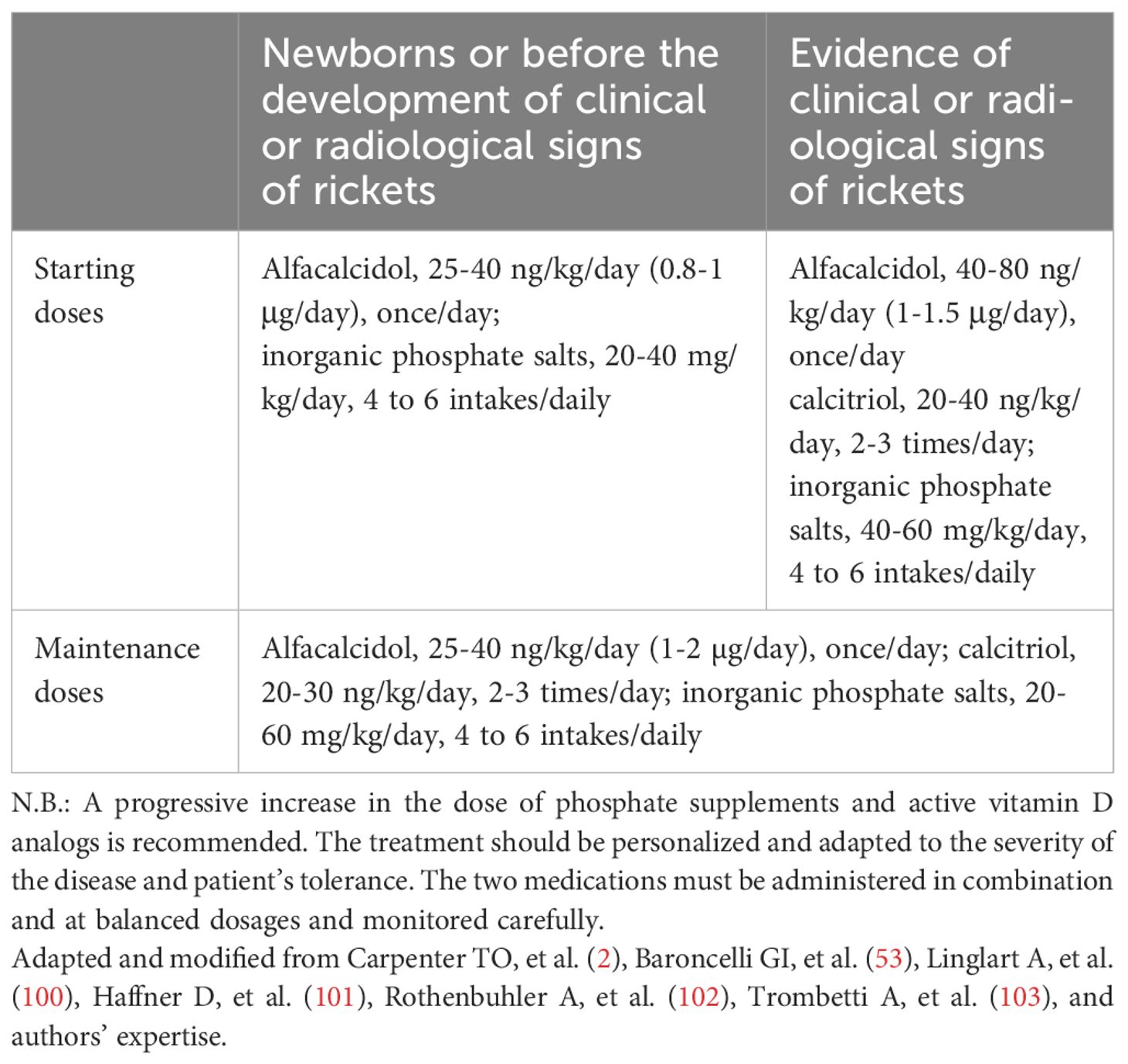
Table 7 Starting and maintenance doses of inorganic phosphate salts and active vitamin D analogs in patients with hypophosphatemic rickets.
Conventional treatment may be effective in improving dental and parodontal lesions (100, 108, 109) depending on the onset, compliance, and duration of therapy, even though some studies demonstrated a poor effect of the conventional treatment on oral phenotype (110, 111).
In patients with ADHR and ARHR1 the dosages of phosphate supplements and vitamin D active metabolites may vary according to the severity of the disease and the response to treatment. In patients with ADHR, it has been shown that iron deficiency affected the severity of the phenotype, and that iron repletion normalized previously increased FGF23 concentrations and improved serum phosphate concentration (112). Therefore, reduction or withdrawal of conventional treatment should be attempted only after the optimization of iron status by oral iron administration (3 to 6 mg/kg elemental Fe, max 200 mg/day for 3 months) (113, 114). Measurement of ferritin is suggested to assess the need for iron supplementation in patients with ADHR (114).
Inorganic oral phosphate salts and vitamin D active metabolites constitute the conventional treatment also for patients with ARHR2 (115–117). However, the fact that ENPP1 deficiency may also be associated with arterial, cardiac, and articular calcification, or may present as generalized arterial calcification (115, 118, 119), should be taken into consideration because these phenotypes preclude the administration of the conventional treatment; therefore, the assessment of carotid intima media thickness and cardiac ultrasonography are strongly recommended in patients with ARHR2 (78). However, recent studies in patients with ARHR2 indicated that conventional treatment did not result in increased calcification in patients with ENPP1 deficiency (120, 121). Furthermore, patients with biallelic ENPP1 mutations may develop normocalcemic primary hyperparathyroidism, which may require partial parathyroidectomy (116).
In patients with a form of FGF23-dependent hypophosphatemic rickets, the benefit of the conventional treatment should be balanced with the potential risks of overtreatment (53). Compliance with the treatment is poor in many patients, mainly in infants, due to gastrointestinal symptoms, including diarrhea, bloody stools, and abdominal pain; compliance may improve by reducing the dose of oral phosphate salts (53). Some complications have been reported in patients with XLH during conventional treatment, including hypercalcemia, secondary/tertiary hyperparathyroidism, hypercalciuria, nephrocalcinosis, and nephrolithiasis (53, 100–103). Some recommendations for follow-up in patients with XLH receiving conventional treatment have been suggested (2, 53, 100–103, 122) (Table 8).
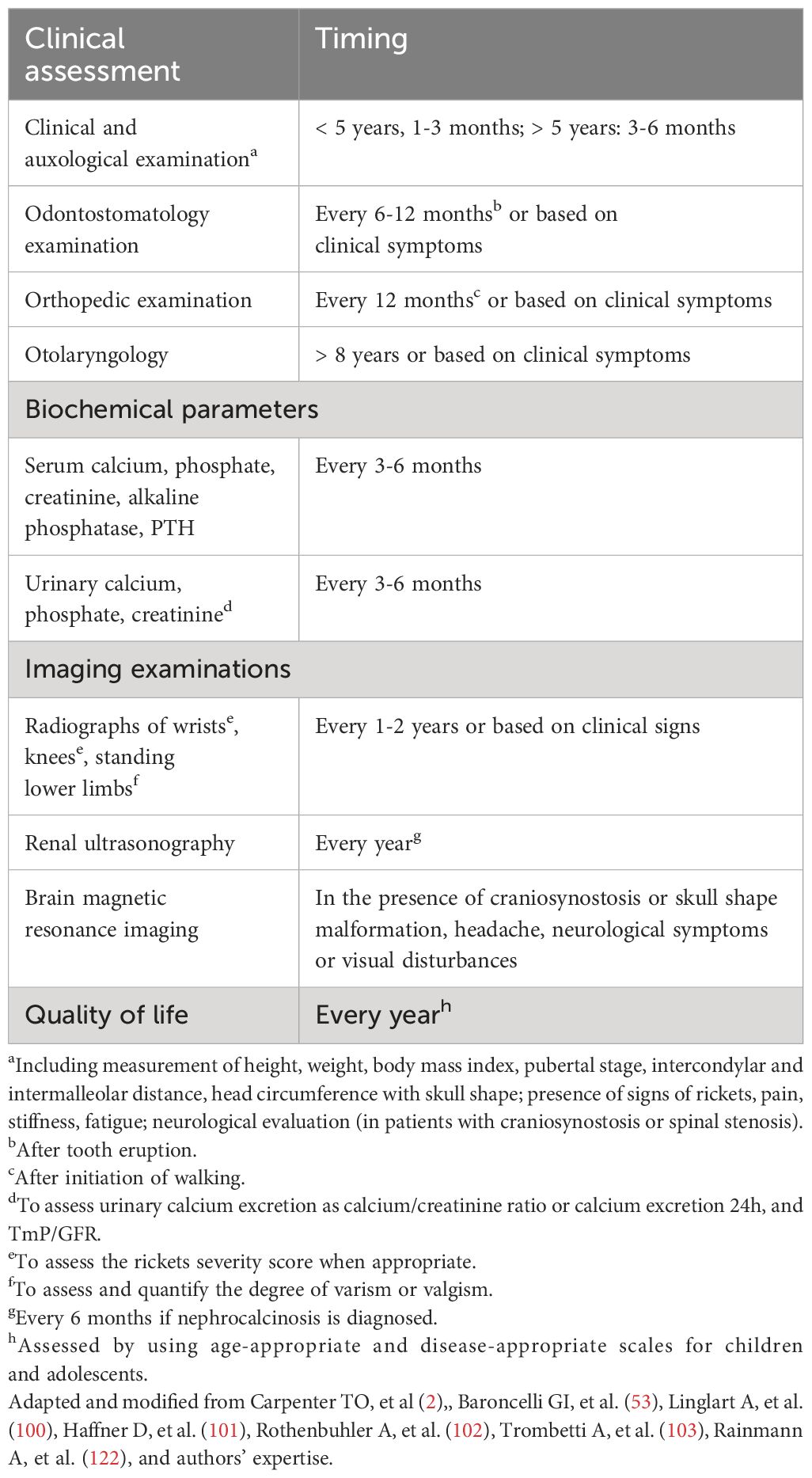
Table 8 Some general recommendations for the follow-up of patients with XLH receiving conventional treatment.
11.1.1 Burosumab treatment in patients with XLH
Burosumab, a recombinant human IgG1 monoclonal antibody that targets FGF23, has been approved by the US Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the Japan’s Health Authority (PMDA) for the treatment of patients with XLH. In Europe, burosumab is indicated in patients in children 1 year of age and older and adolescents with growing skeletons with radiographic evidence of rickets. The criteria for receiving burosumab vary between European countries. They are based according to eligibility rules for early access programs or qualification for reimbursement from health insurance or public health systems (123).
Some studies showed that burosumab treatment improved serum phosphate concentration and TmP/GFR values and were associated with rapid healing of radiographic sings of rickets, improved osteoarticular and muscular pain, and physical function (124, 125). Furthermore, a greater clinical improvement in rickets severity, longitudinal growth, and biochemical findings has been demonstrated in patients with XLH and an RSS of at least 2 treated with burosumab compared with patients continuing on conventional treatment (126). Beneficial effects of burosumab treatment on phosphate metabolism, skeletal lesions of rickets, and linear growth are reported in other recent studies (127–133). The main recommendations for treatment with burosumab for patients with XLH are summarized in Table 9 (134–136). Serum phosphate concentration represents a main biochemical parameter to titrate the dose of burosumab. Dose adjustments should be continued until the target low-normal serum phosphate concentration is reached or no further increase is observed after dose escalation (101, 135). Likely, targeting fasting serum phosphate at the lower end of the normal reference range for age is the safest approach to reduce the risk of ectopic mineralization (101). Serial measurement of alkaline phosphatase activity may also have a pivotal role to assess the progression of disease (135). A progressive decline in serum alkaline phosphatase activity, despite still being above the age- and sex-specific upper limit of normal, may indicate a sufficient clinical response (135). Serum phosphate concentrations below the age- and sex-specific low limit of normal may be considered as a sufficient treatment response if is associated with a sustained decrease in serum alkaline phosphatase activity and improved signs of rickets (135). Periodic measurements of serum 25(OH)D concentration are suggested, because a decline in vitamin D status may be the consequence of reduced sunlight exposure but also to the effect of burosumab treatment (14, 134, 135). We recommend maintaining serum 25(OH)D >50 nmol/L (20 ng/mL) to prevent secondary hyperparathyroidism and to facilitate burosumab-mediated 1,25(OH)2D synthesis (135). Measurement of serum 1,25(OH)2D concentration during burosumab treatment is debated (14). A Consensus Statement recommended monitoring 1,25(OH)2D during burosumab treatment to assess the increment of this vitamin D metabolite (101), but its role in adjusting burosumab dose has not been documented. In the opinion of other authors, measurement of serum 1,25(OH)2D concentrations during burosumab adds cost without clear benefit, so it is not recommended (14).
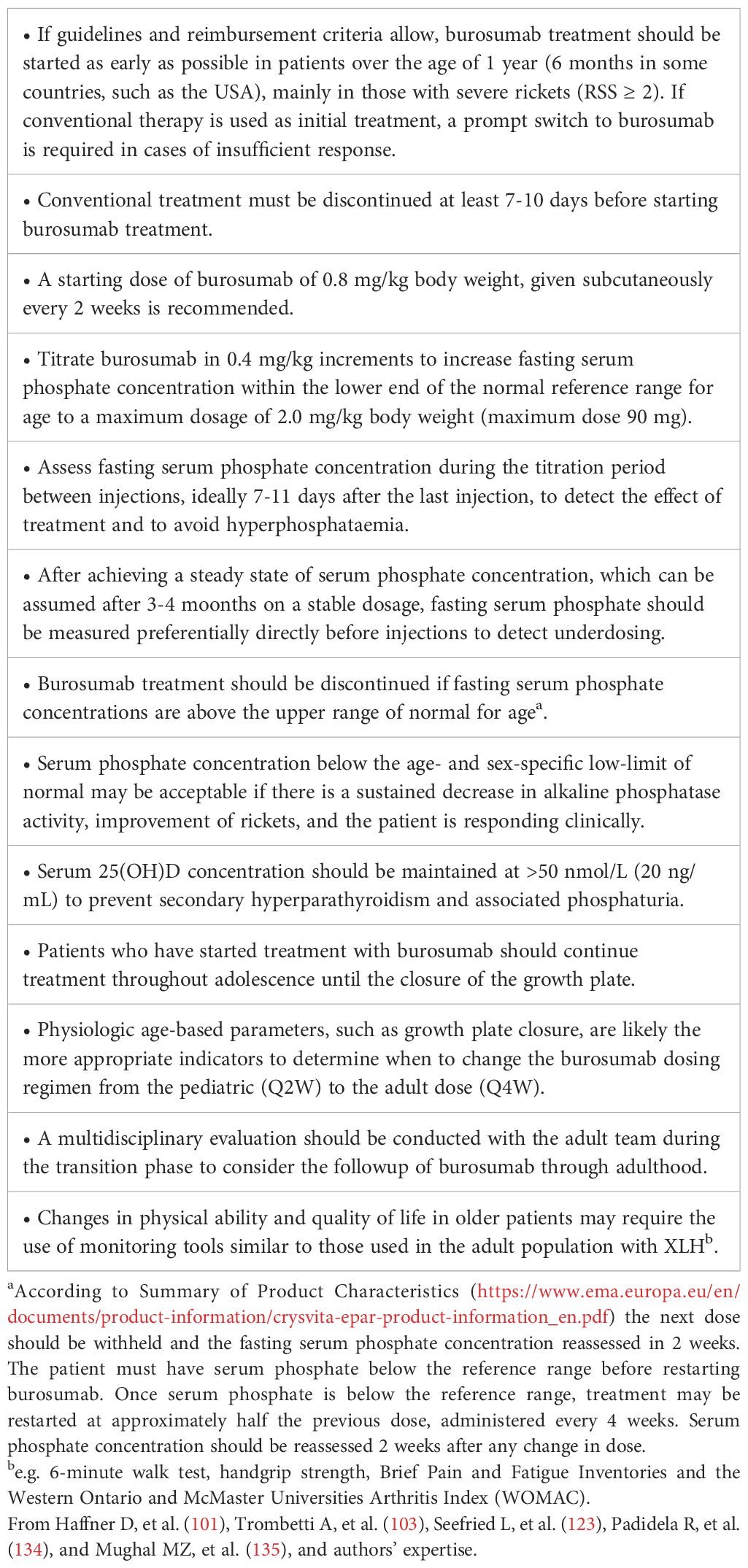
Table 9 Main recommendations for the treatment with burosumab in patients with XLH.
Some recommendations for the follow-up of patients with XLH treated with burosumab are reported in Table 10. The timing of clinical evaluation and biochemical measurements may vary according to the expertise of the authors and should be tailored for each patient. Burosumab treatment is well tolerated and safe. The most common adverse drug reactions reported in pediatric patients were transient injection site reaction, headache, and pain in the extremities (101, 124, 132, 134–137).
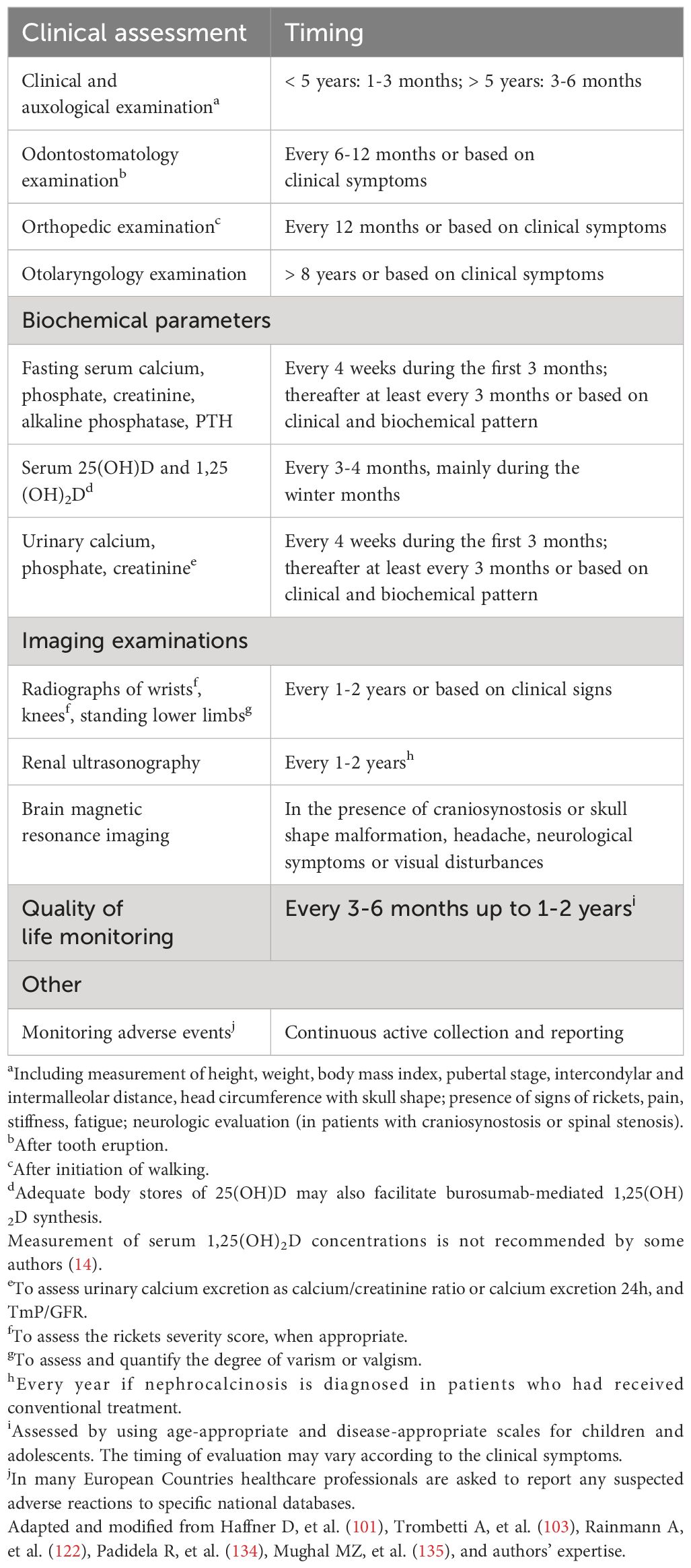
Table 10 Some general recommendations for the follow-up of patients with XLH receiving burosumab treatment.
11.1.2 Growth hormone treatment in patients with XLH
Some studies showed that growth hormone (GH) in association with conventional treatment may increase short-term linear growth in patients with XLH (53). However, the effects on final height are not conclusive (138–140). A recent large study suggested that continuing GH treatment in patients with XLH after switching from conventional therapy to burosumab, if the height prognosis was compromised, might be beneficial for the final height (141). Nevertheless, GH treatment in association with conventional treatment (101) or burosumab is not recommended routinely, and further longer studies are required to examine the role of GH in addition to burosumab.
11.2 Other forms of FGF23-dependent hypophosphatemic bone disorders
Hypophosphatemic rickets may be a rare complication in some patients with non-lethal Raine syndrome (ARHR3) (78, 142–144) and in patients with fibrous dysplasia (145, 146). Hypophosphatemia has been reported in approximately 50% of patients with fibrous dysplasia/McCune Albright syndrome and is associated with rickets or osteomalacia. Serum FGF23 concentrations are increased due to a mass of FGF23-producing cells in fibrous bone lesions (145). Hypophosphatemic rickets may be treated with the conventional treatment by oral inorganic phosphate salts associated with vitamin D active metabolites.
TIO may be solved definitively by removing the tumor causing excessive production of FGF23 (23, 78, 147). Measurement of circulating FGF23 concentration may be useful to detect the progressive reduction after tumor resection or tumor recurrence (78). When tumor resection is not possible or the tumor is not found, and in patients awaiting surgery, conventional treatment similar to that used in patients with XLH should be administered (23, 24, 147).
11.2.1 Burosumab treatment in patients with other FGF23-dependent hypophosphatemic bone disorders
Burosumab has been administered in patients with other forms of rickets, including two adult patients with ARHR1 who showed normalization of serum phosphate concentrations, healing of pseudofracture, reduced fatigue and bone pain (148), and in two children with fibrous dysplasia (149, 150) whose serum phosphate concentrations normalized (149) and alkaline phosphatase activity progressively reduced (150). Furthermore, some case reports in pediatric patients with cutaneous skeletal hypophosphatemia syndrome (27–30) showed that burosumab improved phosphate metabolism, bone turnover, skeletal abnormalities, and quality of life, suggesting that it could be a promising treatment for patients with the FGF23-mediated hypophosphatemia associated with this rare disorder.
11.3 FGF23-independent hypophosphatemic rickets
Monotherapy with oral inorganic phosphate supplements is the usual treatment for patients with HHRH, whereas active vitamin D metabolites are contraindicated due to the increased 1,25(OH)2D concentration (2, 151, 152). Treatment with phosphate should be targeted to decrease the 1,25(OH)2D concentration, reduce intestinal calcium absorption and hypercalciuria, and improve bone mineralization (2). Potassium citrate, salt restriction, and hyperhydration may be useful tools to reduce hypercalciuria. Moreover, treatment with fluconazole (100 mg once daily) has been proposed to inhibit the 1α-hydroxylase and reduce 1,25(OH)2D concentration and hypercalciuria to almost normal values (153). Furthermore, GH treatment associated with oral phosphate supplements improved the renal phosphate leakage and resulted in accelerated linear growth (154). The addition of GH, fluconazole, and salt restriction improved the effect of the conventional therapy with phosphate supplementation and potassium citrate (155). However, additional studies are required to provide the long-term efficacy and safety of fluconazole and GH alone or in combination.
Phosphate supplements and vitamin D active metabolites are suggested for the treatment of patients with hypophosphatemic rickets secondary to tubulopathies, Fanconi syndrome, Dent disease, or other systemic diseases, in association with the recommended treatments for each disease (2).
12 Conclusions
Rickets should be considered a health priority in infants and children. A correct clinical and biochemical approach is fundamental to identify the subjects with suspected rickets. Early diagnosis and adequate treatment are primary targets to avoid severe consequences later in life. The differentiation between the nutritional forms from the genetic forms of rickets is an important step for the correct treatment. Each form of rickets requires a specific diagnostic trail, distinct treatment and an interdisciplinary approach to management (53, 103, 123). Some algorithms to facilitate the diagnostic approach are suggested. Furthermore, some recommendations for the most appropriate treatments for patients with rare forms of hereditary rickets are proposed.
Author contributions
GB: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. PC: Conceptualization, Data curation, Formal analysis, Investigation, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. TA: Writing – original draft, Writing – review & editing. FB: Writing – original draft, Writing – review & editing. AC: Writing – original draft, Writing – review & editing. MC: Writing – original draft, Writing – review & editing. MC: Writing – original draft, Writing – review & editing. LD: Writing – original draft, Writing – review & editing. ND: Writing – original draft, Writing – review & editing. MF: Writing – original draft, Writing – review & editing. DF: Writing – original draft, Writing – review & editing. RF: Writing – original draft, Writing – review & editing. MK: Writing – original draft, Writing – review & editing. SL: Writing – original draft, Writing – review & editing. MM: Writing – original draft, Writing – review & editing. MP: Writing – original draft, Writing – review & editing. AS: Writing – original draft, Writing – review & editing. DT: Writing – original draft, Writing – review & editing. FV: Writing – original draft, Writing – review & editing. MW: Writing – original draft, Writing – review & editing. GW: Writing – original draft, Writing – review & editing. SM: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Kyowa Kirin srl supported EDRA srl for editorial assistance in preparing the manuscript and publication, without interfering with authorship and intellectual property of the contents.
Acknowledgments
Linguistic revision was performed by Dr Lorna O’Brien, Edra. This work is generated within the European Reference Networks for Rare Bone Diseases (BOND-ERN), for Rare Endocrine Conditions (ENDO-ERN), for Hereditary Metabolic Disorders (METAB-ERN), and the European Registries for Rare Endocrine Conditions (EuRRECa). GB is a representative of the BOND-ERN, and AC and GW are representatives of the ENDO-ERN. PC, ND, DF, and DT are part of the Main Thematic Groups in the Reference Centres of the BOND-ERN. LD, ND, SM, and MW are part of the Main Thematic Groups in the Reference Centres of the ENDO-ERN. AC and DT are part of the Main Thematic Groups in the Reference Centres of the EuRRECa.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Munns CF, Shaw N, Kiely M, Specker BL, Thacher TD, Ozono K, et al. Global consensus recommendations on prevention and management of nutritional rickets. J Clin Endocrinol Metab. (2016) 101:394–415. doi: 10.1210/jc.2015-2175
2. Carpenter TO, Shaw NJ, Portale AA, Ward LM, Abrams SA, Pettifor JM. Rickets. Nat Rev Dis Primers. (2017) 3:17101. doi: 10.1038/nrdp.2017.101
3. Thacher TD. Calcium-deficiency rickets. In: Hochberg Z, editor. Vitamin D and rickets. Karger AG, P.O. Box, CH-4009 Basel, Switzerland (2003). p. 105–25.
4. Haffner D, Leifheit-Nestler M, Grund A, Schnabel D. Rickets guidance: part I-diagnostic workup. Pediatr Nephrol. (2022) 37:2013–36. doi: 10.1007/s00467-021-05328-w
5. Chacham S, Pasi R, Chegondi M, Ahmad N, Mohanty SB. Metabolic bone disease in premature neonates: An unmet challenge. J Clin Res Pediatr Endocrinol. (20202020) 12:332–9. doi: 10.4274/jcrpe.galenos.2019.2019.0091
6. Imel EA, Econs MJ. Fibroblast growth factor 23: roles in health and disease. J Am Soc Nephrol. (2005) 16:2565–75. doi: 10.1681/ASN.2005050573
7. Beck-Nielsen SS, Brock-Jacobsen B, Gram J, Brixen K, Jensen TK. Incidence and prevalence of nutritional and hereditary rickets in Southern Denmark. Eur J Endocrinol. (2009) 160:491–7. doi: 10.1530/EJE-08-0818
8. Rafaelsen S, Johansson S, Raeder H, Bjerknes R. Hereditary hypophosphatemia in Norway: a retrospective population-based study of genotypes, phenotypes, and treatment complications. Eur J Endocrinol. (2016) 174:125–36. doi: 10.1530/EJE-15-0515
9. Beck-Nielsen SS, Mughal Z, Haffner D, Nilsson O, Levtchenko E, Ariceta G, et al. FGF23 and its role in X-linked hypophosphatemia-related morbidity. Orphanet J Rare Dis. (2019) 14:58. doi: 10.1186/s13023-019-1014-8
10. Pavone V, Testa G, Gioitta Iachino S, Evola FR, Avondo S, Sessa G. Hypophosphatemic rickets: etiology, clinical features and treatment. Eur J Orthop Surg Traumatol. (2015) 25:221–6. doi: 10.1007/s00590-014-1496-y
11. Eltan M, Alavanda C, Yavas Abali Z, Ergenekon P, Yalındag Ozturk N, Saka M, et al. A rare cause of hypophosphatemia: raine syndrome changing clinical features with age. Calcif Tissue Int. (2020) 107:96–103. doi: 10.1007/s00223-020-00694-3
12. Boyce AM, Collins MT. Fibrous dysplasia/McCune-Albright syndrome: a rare, mosaic disease of Gαs activation. End Rev. (2020) 41:345–70. doi: 10.1210/endrev/bnz011
13. Brownstein CA, Adler F, Nelson-Williams C, Iijima J, Li P, Imura A, et al. A translocation causing increased α-Klotho level results in hypophosphatemic rickets and hyperparathyroidism. Proc Natl Acad Sci USA. (2008) 105:3455–60. doi: 10.1073/pnas.0712361105
14. Ackah SA, Imel EA. Approach to hypophosphatemic rickets. J Clin Endocrinol Metab. (2023) 108:209–20. doi: 10.1210/clinem/dgac488
15. Cormier-Daire V, Delezoide AL, Philip N, Marcorelles P, Casas K, Hillion Y, et al. Clinical, radiological, and chondro-osseous findings in opsismodysplasia: survey of a series of 12 unreported cases. J Med Genet. (2003) 40:195–200. doi: 10.1136/jmg.40.3.195
16. Brown WW, Juppner H, Langman CB, Price H, Farrow EG, White KE, et al. Hypophosphatemia with elevations in serum fibroblast growth factor 23 in a child with Jansen’s metaphyseal chondrodysplasia. J Clin Endocrinol Metab. (2009) 94:17–20. doi: 10.1210/jc.2008-0220
17. Fukumoto S. Targeting fibroblast growth factor 23 signaling with antibodies and inhibitors, is there a rationale? Front Endocrinol. (2018) 9:48. doi: 10.3389/fendo.2018.00048
18. Shore RM. Disorders of phosphate homeostasis in children, part 2: hypophosphatemic and hyperphosphatemic disorders. Pediatr Radiol. (2022) 52:2290–305. doi: 10.1007/s00247-022-05373-z
19. Amatschek S, Haller M, Oberbauer R. Renal phosphate handling in human: what can we learn from hereditary hypophosphataemias? Eur J Clin Invest. (2010) 40:552–60. doi: 10.1111/j.1365-2362.2010.02286.x
20. Carpenter TO. The expanding family of hypophosphatemic syndromes. J Bone Miner Metab. (2012) 30:1–9. doi: 10.1007/s00774-011-0340-2
21. Levine MA. Diagnosis and management of vitamin D dependent rickets. Front Pediatr. (2020) 8:315. doi: 10.3389/fped.2020.00315
22. Roizen JD, Li D, O’Lear L, Javaid MK, Shaw NJ, Ebeling PR, et al. CYP3A4 mutation causes vitamin D-dependent rickets type 3. J Clin Invest. (2018) 128:1913–18. doi: 10.1172/JCI98680
23. Florenzano P, Hartley IR, Jimenez M, Roszko K, Gafni RI, Collins MT. Tumor-induced osteomalacia. Calcif Tissue Int. (2021) 108:128–42. doi: 10.1007/s00223-020-00691-6
24. Bosman A, Palermo A, Vanderhulst J, De Beur SMJ, Fukumoto S, Minisola S, et al. Tumor-induced osteomalacia: a systematic clinical review of 895 cases. Calcif Tissue Int. (2022) 111:367–79. doi: 10.1007/s00223-022-01005-8
25. Haeusler G, Freilinger M, Dominkus M, Egerbacher M, Amann G, Kolb A, et al. Tumor- tumor-induced hypophosphatemic rickets in an adolescent boy: clinical presentation, diagnosis, and histological findings in growth plate and muscle tissue. J Clin Endocrinol Metab. (2010) 95:4511–7. doi: 10.1210/jc.2010-0543
26. Crossen SS, Zambrano E, Newman B, Bernstein JA, Messner AH, Bachrach L, et al. Tumor-induced osteomalacia in a 3-year-old with unresectable central giant cell lesions. J Pediatr Hematol Oncol. (2017) 39:e21–4. doi: 10.1097/MPH.0000000000000686
27. Khadora M, Mughal MZ. Burosumab treatment in a child with cutaneous skeletal hypophosphatemia syndrome: A case report. Bone Rep. (2021) 15:101138. doi: 10.1016/j.bonr.2021.101138
28. Huynh C, Gillis A, Fazendin J, Abdullatif H. A case report to assess the safety and efficacy of burosumab, an investigational antibody to FGF23, in a single pediatric patient with epidermal nevus syndrome and associated hypophosphatemic rickets. Bone Rep. (2022) 17:101605. doi: 10.1016/j.bonr.2022.101605
29. Merz LM, Buerger F, Ziegelasch N, Zenker M, Wieland I, Tobias Lipek T, et al. A case report: first long-term treatment with burosumab in a patient with cutaneous-skeletal hypophosphatemia syndrome. Front Endocrinol. (2022) 13:866831. doi: 10.3389/fendo.2022.866831
30. Sugarman J, Maruri A, Hamilton DJ, Tabatabai L, Luca D, Cimms T, et al. The efficacy and safety of burosumab in two patients with cutaneous skeletal hypophosphatemia syndrome. Bone. (2023) 166:116598. doi: 10.1016/j.bone.2022.116598
31. Wolf M, Rubin J, Achebe M, Econs MJ, Peacock M, Imel EA, et al. Effects of iron isomaltoside vs ferric carboxymaltose on hypophosphatemia in iron-deficiency anemia: two randomized clinical trials. JAMA. (2020) 323:432–43. doi: 10.1001/jama.2019.22450
32. Hall AM, Bass P, Unwin RJ. Drug-induced renal Fanconi syndrome. QJM. (20142014) 107:261–9. doi: 10.1093/qjmed/hct258
33. Foreman JW. Fanconi syndrome. Pediatr Clin North Am. (20192019) 66:159–67. doi: 10.1016/j.pcl.2018.09.002
34. Bikle DD. Vitamin D insufficiency/deficiency in gastrointestinal disorders. J Bone Miner Res. (2007) 22:V50–4. doi: 10.1359/JBMR.07S208
35. Lambert AS, Linglart A. Hypocalcemic and hypophosphatemic rickets. Best Pract Res Clin Endocrinol Metab. (2018) 32:455–76. doi: 10.1016/j.beem.2018.05.009
36. Otero JE, Gottesman GS, McAlister WH, Mumm S, Madson KL, Kiffer-Moreira T, et al. Severe skeletal toxicity from protracted etidronatetherapy for generalized arterial calcification of infancy. J Bone Miner Res. (2013) 28:419–30. doi: 10.1002/jbmr.1752
37. Miyai K, Ariyasu D, Numakura C, Yoneda K, Nakazato H, Hasegawa Y. Hypophosphatemic rickets developed after treatment with etidronate disodium in a patient with generalized arterial calcification in infancy. Bone Rep. (2015) 3:57–60. doi: 10.1016/j.bonr.2015.09.001
38. Linglart A, Biosse-Duplan M. Hypophosphatasia. Curr Osteoporos Rep. (2016) 3:95–105. doi: 10.1007/s11914-016-0309-0
39. Whyte MP, Wenkert D, Zhang F. Hypophosphatasia: natural history study of 101 affected children investigated at one research center. Bone. (2016) 93:125–38. doi: 10.1016/j.bone.2016.08.019
40. Foldes J, Balena R, Ho A, Parfitt AM, Kleerekoper M. Hypophosphatemic rickets with hypocalciuria following long-term treatment with aluminum-containing antacid. Bone. (1991) 12:67–71. doi: 10.1016/8756-3282(91)90002-Z
41. Vukicevic S. First case of rickets following prolonged treatment with aluminum containing antacids that bind phosphate. Bone. (1992) 13:119. doi: 10.1016/8756-3282(92)90449-7
42. Teotia M, Teotia SP, Singh KP. Endemic chronic fluoride toxicity and dietary calcium deficiency interaction syndromes of metabolic bone disease and deformities in India: year 2000. Indian J Pediatr. (1998) 65:371–81. doi: 10.1007/BF02761130
43. Pettifor JM. Screening for nutritional rickets in a community. J Steroid Biochem Mol Biol. (2016) 164:139–44. doi: 10.1016/j.jsbmb.2015.09.008
44. Heimburger DC. Clinical manifestations of nutrient deficiencies and toxicities. In: Ross AC, Cabellero B, Cousins RJ, Tucker KL, Ziegler TR, editors. Modern nutrition in health and disease, 11th ed. Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia (2014). p. 757–70.
45. Holick MF. Resurrection of vitamin D deficiency and rickets. J Clin Invest. (2006) 116:2062–72. doi: 10.1172/JCI29449
46. Lerch C, Meissner T. Interventions for the prevention of nutritional rickets in term born children. Cochrane Database Syst Rev. (2007) 4:CD006164. doi: 10.1002/14651858.CD006164.pub2
47. Wagner CL, Greer FR. American Academy of Pediatrics Section on Breastfeeding, American Academy of Pediatrics Committee on Nutrition. Prevention of rickets and vitamin D deficiency in infants, children, and adolescents. Pediatrics. (2008) 122:1142–52. doi: 10.1542/peds.2008-1862
48. Misra M, Pacaud D, Petryk A, Collett-Solberg PF, Kappy M. Drug and Therapeutics Committee of the Lawson Wilkins Pediatric Endocrine Society. Vitamin D deficiency in children and its management: review of current knowledge and recommendations. Pediatrics. (2008) 122:398–417. doi: 10.1542/peds.2007-1894
49. Baroncelli GI, Carlucci G, Freri E, Giuca MR, Guarnieri V, Navarra G, et al. The diagnosis of hypophosphatasia in children as a multidisciplinary effort: an expert opinion. J Endocrinol Invest. (2024) 47:739–47. doi: 10.1007/s40618-023-02199-w
50. Baroncelli GI, Sessa MR, Pelosini C, Bertelloni S, Michelucci A, Toschi B, et al. Intact FGF23 concentration in healthy infants, children, and adolescents, and diagnostic usefulness in patients with X-linked hypophosphatemic rickets. J Endocrinol Invest. (2024) 47:873–82. doi: 10.1007/s40618-023-02202-4. in press.
51. Tiosano D, Hochberg Z. Hypophosphatemia: the common denominator of all rickets. J Bone Miner Metab. (2009) 27:392–401. doi: 10.1007/s00774-009-0079-1
52. Stark H, Eisenstein B, Tieder M, Rachmel A, Alpert G. Direct measurement of TP/GFR: a simple and reliable parameter of renal phosphate handling. Nephron. (1986) 44:125–8. doi: 10.1159/000184216
53. Baroncelli GI, Mora S. (2021) X-linked hypophosphatemic rickets: multisystemic disorder in children requiring multidisciplinary management Front Endocrinol. 12:688309. doi: 10.3389/fendo.2021.688309
54. Thacher TD, Fischer PR, Pettifor JM, Lawson JO, Isichei CO, Reading JC, et al. A comparison of calcium, vitamin D, or both for nutritional rickets in Nigerian children. N Engl J Med. (1999) 341:563–8. doi: 10.1056/NEJM199908193410803
55. Baroncelli GI, Bereket A, El Kholy M, Audì L, Cesur Y, Ozkan B, et al. Rickets in the Middle East: role of environment and genetic predisposition. J Clin Endocrinol Metab. (2008) 93:1743–50. doi: 10.1210/jc.2007-1413
56. Dahifar H, Faraji A, Ghorbani A, Yassobi S. Impact of dietary and lifestyle on vitamin D in healthy student girls aged 11–15 years. J Med Investig. (2006) 53:204–8. doi: 10.2152/jmi.53.204
57. Agarwal A, Gulati D. Early adolescent nutritional rickets. J Orthop Surg. (2009) 17:340–5. doi: 10.1177/230949900901700320
58. Hazzazi M, Alzeer I, Tamimi W, Al Atawi M, Al Alwan I. Clinical presentation and etiology of osteomalacia/rickets in adolescents. Saudi J Kidney Dis Transplant. (2013) 24:938–4. doi: 10.4103/1319-2442.118087
59. Atapattu N, Shaw N, Hogler W. Relationship between serum 25-hydroxyvitamin D and parathyroid hormone in the search for a biochemical definition of vitamin D deficiency in children. Pediatr Res. (2013) 74:552–6. doi: 10.1038/pr.2013.139
60. Rios-Leyvraz M, Thacher TD, Dabas A, Hassan Elsedfy H, Baroncelli GI, Cashman KD. Serum 25-hydroxyvitamin D threshold and risk of rickets in young children: a systematic review and individual participant data meta-analysis to inform the development of dietary requirements for vitamin D. Eur J Nutr. (2024) 63:673–95. doi: 10.1007/s00394-023-03299-2
61. El Kholy M, Elsedfy H, Fernandez-Cancio M, Hamza RT, Amr NH, Ahmed AY, et al. Nutritional rickets: vitamin D, calcium, and the genetic make-up. Pediatr Res. (2017) 81:356–63. doi: 10.1038/pr.2016.222
62. Molin A, Wiedemann A, Demers N, Kaufmann M, Do Cao J, Mainard L, et al. Vitamin D-dependent rickets type 1B (25-hydroxylase deficiency): a rare condition or a misdiagnosed condition? J Bone Miner Res. (2017) 32:1893–9. doi: 10.1002/jbmr.3181
63. Kubota T, Kitaokaa T, Miura K, Fujiwara M, Ohata Y, Miyoshi Y, et al. Serum fibroblast growth factor 23 is a useful marker to distinguish vitamin D-deficient rickets from hypophosphatemic rickets. Horm Res Paediatr. (2014) 81:251–7. doi: 10.1159/000357142
64. Prentice A, Ceesay M, Nigdikar S, Allen SJ, Pettifor JM. FGF23 is elevatewd in Gambian children with rickets. Bone. (2008) 42:788–97. doi: 10.1016/j.bone.2007.11.014
65. States LJ. Imaging of rachitic bone. In: Hochberg Z, editor. Vitamin D and rickets, vol. . p . Karger AG, P.O. Box, CH-4009 Basel, Switzerland (2003). p. 80–92.
67. Shaw NJ. Vitamin D deficiency rickets. In: Hochberg Z, editor. Vitamin D and rickets, vol. . p . Karger AG, P.O. Box, CH-4009 Basel, Switzerland (2003). p. 93–104.
68. Chapman T, Sugar N, Done S, Marasigan J, Wambold N, Feldman K. Fractures in infants and toddlers with rickets. Pediatr Radiol. (2010) 40:1184–9. doi: 10.1007/s00247-009-1470-8
69. Thacher TD, Fischer PR, Pettifor JM, Lawson JO, Manaster BJ, Reading JC. Radiographic scoring method for the assessment of the severity of nutritional rickets. J Trop Pediatr. (2000) 46:132–9. doi: 10.1093/tropej/46.3.132
70. Thacher TD, Pettifor JM, Tebben PJ, Creo AL, Skrinar A, Mao M, et al. Rickets severity predicts clinical outcomes in children with X-linked hypophosphatemia: utility of the radiographic rickets severity score. Bone. (2019) 122:76–81. doi: 10.1016/j.bone.2019.02.010
71. Whyte MO, Fujita KP, Moseley S, Thompson DD, McAlister WH. Validation of a novel scoring system for changes in skeletal manifestations of hypophosphatasia in newborns, infants, and children: the radiographic global impression of change scale. J Bone Miner Res. (2018) 33:868–74. doi: 10.1002/jbmr.3377
72. Lim R, Shailam R, Hulett R, Skrinar A, Nixon A, Williams A, et al. Validation of the radiographic global impression of change (RGI-C) score to assess healing of rickets in pediatric X-linked hypophosphatemia (XLH). Bone. (2021) 148:115964. doi: 10.1016/j.bone.2021.115964
73. Capelli S, Donghi V, Maruca K, Vezzoli G, Corbetta S, Brandi ML, et al. Clinical and molecular heterogeneity in a large series of patients with hypophosphatemic rickets. Bone. (2015) 79:143–9. doi: 10.1016/j.bone.2015.05.040
74. Yasuhisa Ohata Y, Ishihara Y. Pathogenic variants of the PHEX gene. Endocrines. (2022) 3:498–511. doi: 10.3390/endocrines3030040
75. Baroncelli GI, Vierucci F, Bertelloni S. Rachitismo da deficit di vitamina D: indicazioni per la diagnosi e la terapia. In: Baroncelli GI, editor. Ipovitaminosi D. Prevenzione e trattamento nel neonato, nel bambino e nell'adolescente, vol. p . Edizioni Mattioli, Fidenza, Italy (2011). p. 99–121.
76. Cesur Y, Çaksen H, Gündem A, Kirimi E, Odabas D. Comparison of low and high dose of vitamin D treatment in nutritional vitamin D deficiency rickets. J Pediatr Endocrinol Metab. (2003) 16:1105–9. doi: 10.1515/jpem.2003.16.8.1105
77. Mittal H, Rai S, Shah D, Madhu SV, Mehrotra G, Malhotra RK, et al. 300,000 IU or 600,000 IU of oral vitamin D3 for treatment of nutritional rickets: a randomized controlled trial. Indian Pediatr. (2014) 51:265–72. doi: 10.1007/s13312-014-0399-7
78. Haffner D, Leifheit-Nestler M, Grund A, Schnabel D. Rickets guidance: part II-management. Pediatr Nephrol. (2022) 37:2289–302. doi: 10.1007/s00467-022-05505-5
79. Pearce SH, Cheetham TD. Diagnosis and management of vitamin D deficiency. BMJ. (2010) 340:b5664. doi: 10.1136/bmj.b5664
80. Tripkovic L, Lambert H, Hart K, Smith CP, Bucca G, Penson S, et al. Comparison of vitamin D2 and vitamin D3 supplementation in raising serum 25-hydroxyvitamin D status: a systematic review and meta-analysis. Am J Clin Nutr. (2012) 95:1357–64. doi: 10.3945/ajcn.111.031070
81. Allgrove J, Shaw NJ. A practical approach to vitamin D deficiency and rickets. In: Allgrove J, Shaw NJ, editors. Calcium and bone disorders in children and adolescents. Karger AG, P.O. Box, CH-4009 Basel, Switzerland (2015). p. 119–33.
82. Baroncelli GI, Bertelloni S, Ceccarelli C, Amato V, Saggese G. Bone turnover in children with vitamin D deficiency rickets before and during treatment. Acta Paediatr. (2000) 89:513–8. doi: 10.1080/080352500750027763
83. Thacher TD, Smith L, Fischer PR, Isichei CO, Cha SS, Pettifor JM. Optimal dose of calcium for treatment of nutritional rickets: a randomized controlled trial. J Bone Miner Res. (2016) 31:2024–31. doi: 10.1002/jbmr.2886
84. Thacher TD, Fischer PR, Pettifor JM. Vitamin D treatment in calcium- deficiency rickets: a randomised controlled trial. Arch Dis Child. (2014) 99:807–11. doi: 10.1136/archdischild-2013-305275
85. Faienza MF, D'Amato E, Natale MP, Grano M, Chiarito M, Brunetti G, et al. Metabolic bone disease of prematurity: diagnosis and management. Front Pediatr. (2019) 7:143. doi: 10.3389/fped.2019.00143
86. Chinoy A, Mughal MZ, Raja Padidela R. Metabolic bone disease of prematurity: causes, recognition, prevention, treatment and longterm consequences. Arch Dis Child Fetal Neonatal Ed. (2019) 104:F560–6. doi: 10.1136/archdischild-2018-316330
87. Abrams SA, Committee on Nutrition. Calcium and vitamin D requirements of enterally fed preterm infants. Pediatrics. (2013) 131:e1676–83. doi: 10.1542/peds.2013-0420
88. Agostoni C, Buonocore G, Carnielli VP, De Curtis M, Darmaun D, Decsi T, et al. Enteral nutrient supply for preterm infants: commentary from the European Society of Paediatric Gastroenterology, Hepatology and Nutrition Committee on Nutrition. J Pediatr Gastroenterol Nutr. (2010) 50:85–91. doi: 10.1097/MPG.0b013e3181adaee0
89. Koletzko B, Poindexter B, Uauy R. Nutritional care of preterm infants: scientific basis and practical guidelines. Switzerland: Karger AG P.O. Box, CH-4009 Basel (2014). doi: 10.1159/isbn.978-3-318-02641-2
90. Nehra D, Carlson SJ, Fallon EM, Kalish B, Potemkin AK, Gura KM, et al. A.S.P.E.N. clinical guidelines: nutrition support of neonatal patients at risk for metabolic bone disease. J Parenter Enteral Nutr. (2013) 37:570–98. doi: 10.1177/0148607113487216
91. Lee B, De Beritto T. Metabolic bone disease of prematurity. Neoreviews. (2022) 23:e311–8. doi: 10.1542/neo.23-5-e311
92. Abrams SA. Vitamin D in preterm and full-term infants. Ann Nutr Metab. (2020) 76:6–14. doi: 10.1159/000508421
93. Reade TM, Scriver CR, Glorieux FH, Nogrady B, Delvin E, Poirier R, et al. Response to crystalline 1alpha-hydroxyvitamin D3 in vitamin D dependency. Pediatr Res. (1975) 9:593–9. doi: 10.1203/00006450-197507000-00008
94. Delvin EE, Glorieux FH, Marie PJ. Pettifor JM Vitamin D dependency: replacement therapy with calcitriol. J Pediatr. (1981) 99:26–34. doi: 10.1016/s0022-3476(81)80952-3
95. Miller WL, Portale AA. Vitamin D biosynthesis and vitamin D 1 α-hydroxylase deficiency. In: Hochberg Z, editor. Vitamin D and rickets. Karger AG, P.O. Box, CH-4009 Basel, Switzerland (2003). p. 156–74.
96. Thacher TD, Fischer PR, Singh RJ, Roizen J, Levine MA. CYP2R1 mutations impair generation of 25-hydroxyvitamin D and cause an atypical form of vitamin D deficiency. J Clin Endocrinol Metab. (2015) 100:E1005–13. doi: 10.1210/jc.2015-1746
97. Chaturvedi D, Garabedian M, Carel J-C, Léger J. Different mechanisms of intestinal calcium absorption at different life stages: therapeutic implications and long-term responses to treatment in patients with hereditary vitamin D-resistant rickets. Horm Res Paediatr. (2012) 78:326–31. doi: 10.1159/000341405
98. Marx SJ, Bliziotes MM, Nanes M. Analysis of the relation between alopecia and resistance to 1,25-dihydroxyvitamin D. Clin Endocrinol (Oxf.). (1986) 25:373–81. doi: 10.1111/j.1365-2265.1986.tb01703.x
99. Malloy PJ, Feldman D. Hereditary 1,25-dihydroxyvitamin D-resistant rickets. In: Hochberg Z, editor. Vitamin D and rickets, vol. . p . Karger AG, P.O. Box, CH-4009 Basel, Switzerland (2003). p. 175–99.
100. Linglart A, Biosse-Duplan M, Briot K, Chaussain C, Esterle L, Guillaume-Czitrom S, et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect. (2014) 3:R13–30. doi: 10.1530/EC-13-0103
101. Haffner D, Emma F, Eastwood DM, Biosse Duplan M, Bachetta J, Schnabel D, et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat Rev Nephrol. (2019) 15:435–55. doi: 10.1038/s41581-019-0152-5
102. Rothenbuhler A, Schnabel D, Högler W, Linglart A. Diagnosis, treatment-monitoring and follow-up of children and adolescents with X-linked hypophosphatemia (XLH). Metabolism. (2020) 103S:153892. doi: 10.1016/j.metabol.2019.03.009
103. Trombetti A, Al-Daghri N, Brandi ML, Cannata-Andia JB, Cavalier E, Chandran M, et al. Interdisciplinary management of FGF23-related phosphate wasting syndromes: a Consensus Statement on the evaluation, diagnosis and care of patients with X-linked hypophosphataemia. Nat Rev Endocrinol. (2022) 18:366–84. doi: 10.1038/s41574-022-00662-x
104. Makitie O, Doria A, Kooh SW, Cole WG, Daneman A, Sochett E. Early treatment improves growth and biochemical and radiographic outcome in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab. (2003) 88:3591–7. doi: 10.1210/jc.2003-030036
105. Zivicňjak M, Schnabel D, Staude H, Even G, Marx M, Beetz R, et al. Three-year growth hormone treatment in short children with X-linked hypophosphatemic rickets: effects on linear growth and body disproportion. J Clin Endocrinol Metab. (2011) 96:E2097–105. doi: 10.1210/jc.2011-0399
106. Fuente R, Gil-Peña H, Claramunt-Taberner D, Hernández O, Fernández- Iglesias A, Alonso-Durán L, et al. X-linked hypophosphatemia and growth. Rev Endocr Metab Disord. (2017) 18:107–15. doi: 10.1007/s11154-017-9408-1
107. Mao M, Carpenter TO, Whyte MP, Skrinar A, Chen CY, San Martin J, et al. Growth curves for children with X-linked hypophosphatemia. J Clin Endocrinol Metab. (2020) 105:1–7. doi: 10.1210/clinem/dgaa495
108. Chaussain-Miller C, Sinding C, Septier D, Wolikow M, Goldberg M, Garabedian M. Dentin structure in familial hypophosphatemic rickets: benefits of vitamin D and phosphate treatment. Oral Dis. (2007) 13:482–9. doi: 10.1016/j.metabol.2019.03.009
109. Connor J, Olear EA, Insogna KL, Katz L, Baker S, Kaur R, et al. Conventional therapy in adults with X-linked hypophosphatemia: effects on enthesopathy and dental disease. J Clin Endocrinol Metab. (2015) 100:3625–32. doi: 10.1210/JC.2015-2199
110. Baroncelli GI, Angiolini M, Ninni E, Galli V, Saggese R, Giuca MR. Prevalence and pathogenesis of dental and periodontal lesions in children with X-linked hypophosphatemic rickets. Eur J Paediatr Dent. (2006) 7:61–6.
111. Souza MA, Soares LA, Santos MA, Vaisbich MH. Dental abnormalities and oral health in patients with hypophosphatemic rickets. Clinics. (2010) 65:1023–6. doi: 10.1590/s1807-59322010001000017
112. Imel EA, Biggin A, Schindeler A, Munns CF. FGF23, hypophosphatemia, and emerging treatments. J Bone Miner Res Plus. (2019) 3:e10190. doi: 10.1002/jbm4.10190
113. Kapelari K, Köhle J, Kotzot D, Högler W. Iron supplementation associated with loss of phenotype in autosomal dominant hypophosphatemic rickets. J Clin Endocrinol Metab. (2015) 100:3388 –92. doi: 10.1210/jc.2015-2391
114. Hogler W, Kapelari K. Oral iron for prevention and treatment of rickets and osteomalacia in autosomal dominant hypophosphatemia. J Bone Miner Res. (2020) 35:226–30. doi: 10.1002/jbmr.3941
115. Saito T, Shimizu Y, Hori M, Taguchi M, Igarashi T, Fukumoto S, et al. A patient with hypophosphatemic rickets and ossification of posterior longitudinal ligament caused by a novel homozygous mutation in ENPP1 gene. Bone. (2011) 49:913–6. doi: 10.1016/j.bone.2011.06.029
116. Kotwal A, Ferrer A, Kumar R, Singh RJ, Murthy V, Schultz-Rogers L, et al. Clinical and biochemical phenotypes in a family with ENPP1 mutations. J Bone Min Res. (2020) 35:662–70. doi: 10.1002/jbmr.3938
117. Hoppner J, Kornak U, Sinningen K, Rutsch F, Oheim R, Grasemanna C. Autosomal recessive hypophosphatemic rickets type 2 (ARHR2) due to ENPP1-deficiency. Bone. (2021) 153:116111. doi: 10.1016/j.bone.2021.116111
118. Lorenz-Depiereux B, Schnabel D, Tiosano D, Hausler G, Strom TM. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet. (2010) 86:267–72. doi: 10.1016/j.ajhg.2010.01.006
119. Levy-Litan V, Hershkovitz E, Avizov L, Leventhal N, Bercovich D, Chalifa-Caspi V, et al. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet. (2010) 86:273–8. doi: 10.1016/j.ajhg.2010.01.010
120. Ferreira CR, Ziegler SG, Gupta C, Groden C, Hsu KS, Gahl WA. Treatment of hypophosphatemic rickets in generalized arterial calcification of infancy (GACI) without worsening of vascular calcification. Am J Med Genet. (2016) 170A:1308–11. doi: 10.1002/ajmg.a.37574
121. Ferreira CR, Hackbarth ME, Ziegler SG, Pan KS, Roberts MS, Rosing DR, et al. Prospective phenotyping of long-term survivors of generalized arterial calcification of infancy (GACI). Genet Med. (2021) 23:396–407. doi: 10.1038/s41436-020-00983-0
122. Raimann A, Mindler GT, Kocijan R, Bekes K, Zwerina J, Haeusler G, et al. Multidisciplinary patient care in X-linked hypophosphatemic rickets: one challenge, many perspectives. Wien Med Wochenschr. (2020) 170:116–23. doi: 10.1007/s10354-019-00732-2
123. Seefried L, Alzahrani A, Sancho PA, Bacchetta J, Crowley R, Emma F, et al. XLH Matters 2022: Insights and recommendations to improve outcomes for people living with X-linked hypophosphataemia (XLH). Orphanet J Rare Dis. (2023) 18:333. doi: 10.1186/s13023-023-02883-3
124. Carpenter TO, Whyte MP, Imel EA, Boot AM, Hogler W, Linglart A, et al. Burosumab therapy in children with X-linked hypophosphatemia. N Engl J Med. (2018) 378:1987–98. doi: 10.1056/NEJMoa1714641
125. Whyte MP, Carpenter TO, Gottesman GS, Mao M, Skrinar A, San Martin J, et al. Efficacy and safety of burosumab in children aged 1-4 years with X-linked hypophosphataemia: a multicentre, open-label, phase 2 trial. Lancet. (2019) 7:189–99. doi: 10.1016/S2213-8587(18)30338-3
126. Imel EA, Glorieux FH, Whyte MP, Munns CF, Ward LM, Nilsson O, et al. Burosumab versus conventional therapy in children with X-linked hypophosphataemia: a randomised, active-controlled, open-label, phase 3 trial. Lancet. (2019) 393:2416–27. doi: 10.1016/S2213-8587(18)30338-3
127. Martín Ramos S, Gil-Calvo M, Roldán V, Castellano Martínez A, Santos F. Positive response to one-year treatment with burosumab in pediatric patients with X-linked hypophosphatemia. Front Pediatr. (2020) 8:48. doi: 10.3389/fped.2020.00048
128. Linglart A, Imel EA, Whyte MP, Portale AA, Högler W, Boot AM. Sustained efficacy and safety of burosumab, a monoclonal antibody to FGF23, in children with X-linked hypophosphatemia. J Clin Endocrinol Metab. (2022) 107:813–24. doi: 10.1210/clinem/dgab729
129. Namba N, Kubota T, Muroya K, Tanaka H, Kanematsu M, Kojima M, et al. Safety and efficacy of burosumab in pediatric patients with X-linked hypophosphatemia: a phase 3/4 open-label trial. J Endocr Soc. (2022) 6:bvac021. doi: 10.1210/jendso/bvac021
130. Paloian NJ, Blaise Nemeth B, Sharafinski M, Modaff P, Steiner RD. Real-world effectiveness of burosumab in children with X-linked hypophosphatemic rickets. Pediatr Nephrol. (2022) 37:2667–77. doi: 10.1007/s00467-022-05484-7
131. Ward LM, Glorieux FH, Whyte MP, Munns CF, Portale AA, Högler W, et al. Effect of burosumab compared with conventional therapy on younger vs older children with X-linked hypophosphatemia. J Clin Endocrinol Metab. (2022) 107:e3241–53. doi: 10.1210/clinem/dgac296
132. Walker EYX, Lindsay TAJ, Allgrove J, Marlais M, Bockenhauer D, Hayes W. Burosumab in management of X-linked hypophosphataemia: a retrospective cohort study of growth and serum phosphate levels. Arch Dis Child. (2023) 108:379–84. doi: 10.1136/archdischild-2022-324962
133. Filler G, Tremblay O, Chen E, Huang SSH, Stein R. Sex differences of burosumab in children with X-linked hypophosphataemic rickets. Pediatr Nephrol. (2023) 38:3183–7. doi: 10.1007/s00467-022-05822-9
134. Padidela R, Cheung MS, Saraff V, Dharmaraj P. Clinical guidelines for burosumab in the treatment of XLH in children and adolescents: British paediatric and adolescent bone group recommendations. Endocr Connect. (2020) 9:1051–6. doi: 10.1530/EC-20-0291
135. Mughal MZ, Baroncelli GI, de Lucas-Collantes C, Linglart A, Magnolato A, Raimann A, et al. Burosumab for X-linked hypophosphatemia in children and adolescents: opinion based on early experience in seven European countries. Front Endocrinol. (2023) 13:1034580. doi: 10.3389/fendo.2022.1034580
136. Bacchetta J, Rothenbuhler A, Gueorguieva I, Kamenicky P, Salles J-P, Briot K, et al. X-linked hypophosphatemia and burosumab: practical clinical points from the French experience. Joint Bone Spine. (2021) 88:105208. doi: 10.1016/j.jbspin.2021.105208
137. Imel EA. Burosumab for pediatric X-linked hypophosphatemia. Curr Osteoporos Rep. (2021) 19:271–7. doi: 10.1007/s11914-021-00669-9
138. Baroncelli GI, Bertelloni S, Ceccarelli C, Saggese G. Effect of growth hormone treatment on final height, phosphate metabolism, and bone mineral density in children with X-linked hypophosphatemic rickets. J Pediatr. (2001) 138:236–43. doi: 10.1067/mpd.2001.108955
139. Huiming Y, Meng M, Chaomin W, Fan Y. Recombinant growth hormone therapy for X-linked hypophosphatemia in children (Review). Cochrane Database Syst Rev. (2005) 1:CD004447. doi: 10.1002/14651858.CD004447.pub2
140. Meyerhoff N, Haffner D, Staude H, Wühl E, Marx M, Beetz R, et al. Effects of growth hormone treatment on adult height in severely short children with X-linked hypophosphatemic rickets. Pediatr Nephrol. (2018) 33:447–56. doi: 10.1007/s00467-017-3820-3
141. Ertl DA, Le Lorier J, Gleiss A, Trabado S, Bensignor C, Audrain C, et al. Growth pattern in children with X-linked hypophosphatemia treated with burosumab and growth hormone. Orphanet J Rare Dis. (2022) 17:412. doi: 10.1186/s13023-022-02562-9
142. Rafaelsen SH, Ræder H, Fagerheim AK, Knappskog P, Carpenter TO, Johansson S, et al. Exome sequencing reveals FAM20C mutations associated with fibroblast growth factor 23– related hypophosphatemia, dental anomalies, and ectopic calcification. J Bone Miner Res. (2013) 28:1378–85. doi: 10.1002/jbmr.1850
143. Mameli C, Zichichi G, Mahmood N, Elalaoui SC, Mirza A, Dharmaraj P, et al. Natural history of non-lethal Raine syndrome during childhood. Orphanet J Rare Dis. (2020) 15:93. doi: 10.1186/s13023-020-01373-0
144. Hirst L, Abou-Ameira G, Critchlow S. Hypophosphataemic rickets secondary to Raine syndrome: a review of the literature and case reports of three paediatric patients’ dental management. Case Rep Pediatr. (2021) 6637180. doi: 10.1155/2021/6637180
145. Riminucci M, Collins MT, Fedarko NS, Cherman N, Corsi A, Kenneth E, et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting. J Clin Invest. (2003) 112:683–92. doi: 10.1172/JCI200318399
146. de Castro LF, Ovejero D, Alison M Boyce AM. Mosaic disorders of FGF23 excess: fibrous dysplasia/McCune-Albright syndrome and cutaneous skeletal hypophosphatemia syndrome. Eur J Endocrinol. (2020) 182:R83–99. doi: 10.1530/EJE-19-0969
147. Dahir K, Roberts MS, Krolczyk S, Simmons JH. X-linked hypophosphatemia: a new era in management. J Endocr Soc. (2020) 4:bvaa151. doi: 10.1210/jendso/bvaa151
148. Bai X, Levental M, Karaplis AC. Burosumab treatment for autosomal recessive hypophosphatemic rickets type 1 (ARHR1). J Clin Endocrinol Metab. (2022) 107:2777–83. doi: 10.1210/clinem/dgac433
149. Gladding A, Vivian Szymczuk V, Auble BA, Boyce AM. Burosumab treatment for fibrous dysplasia. Bone. (2021) 150:116004. doi: 10.1016/j.bone.2021.116004
150. Sato T, Azuma Y, Tauchi R, Matsushita M, Ozone C. Burosumab treatment for fibrous dysplasia/McCune-Albright syndrome with severe spine deformity. J Endocrine Soc. (2022) 6:A160. Abstract. doi: 10.1210/jendso/bvac150.327
151. Tieder M, Modai D, Samuel R, Arie R, Halabe A, Bab I, et al. Hereditary hypophosphatemic rickets with hypercalciuria. N Engl J Med. (1985) 312:611–7. doi: 10.1056/NEJM198503073121003
152. Bergwitz C, Miyamoto KI. Hereditary hypophosphatemic rickets with hypercalciuria: pathophysiology, clinical presentation, diagnosis and therapy. Pflugers Arch. (2019) 471:149–63. doi: 10.1007/s00424-018-2184-2
153. Bertholet-Thomas A, Tram N, Dubourg L, Lemoine S, Molin A, Bacchetta J. Fluconazole as a new therapeutic tool to manage patients with NPTIIc (SLC34A3) mutation: a case report. Am J Kidney Dis. (2019) 73:886–9. doi: 10.1053/j.ajkd.2018.12.026
154. Dreimane D, Chen A, Bergwitz C. Description of a novel SLC34A3.c.671delT mutation causing hereditary hypophosphatemic rickets with hypercalciuria in two adolescent boys and response to recombinant human growth hormone. Ther Adv Musculoskelet Dis. (2020) 12. doi: 10.1177/1759720X20912862
Keywords: hereditary rickets, management, non-hereditary rickets, nutritional rickets, treatment
Citation: Baroncelli GI, Comberiati P, Aversa T, Baronio F, Cassio A, Chiarito M, Cosci o di Coscio M, De Sanctis L, Di Iorgi N, Faienza MF, Fintini D, Franceschi R, Kalapurackal M, Longhi S, Mariani M, Pitea M, Secco A, Tessaris D, Vierucci F, Wasniewska M, Weber G and Mora S (2024) Diagnosis, treatment, and management of rickets: a position statement from the Bone and Mineral Metabolism Group of the Italian Society of Pediatric Endocrinology and Diabetology. Front. Endocrinol. 15:1383681. doi: 10.3389/fendo.2024.1383681
Received: 07 February 2024; Accepted: 03 April 2024;
Published: 19 April 2024.
Edited by:
Daniela Merlotti, University of Siena, ItalyReviewed by:
Luciano Colangelo, Sapienza University of Rome, ItalyHelen McDevitt, Royal Hospital for Children, United Kingdom
Copyright © 2024 Baroncelli, Comberiati, Aversa, Baronio, Cassio, Chiarito, Cosci o di Coscio, De Sanctis, Di Iorgi, Faienza, Fintini, Franceschi, Kalapurackal, Longhi, Mariani, Pitea, Secco, Tessaris, Vierucci, Wasniewska, Weber and Mora. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefano Mora, bW9yYS5zdGVmYW5vQGhzci5pdA==
†These authors have contributed equally to this work and share first authorship