 Qiaohui Shen
Qiaohui Shen Ming Yang
Ming Yang Song Wang2
Song Wang2 Yan Leng
Yan Leng- 1College of Traditional Chinese Medicine, Changchun University of Chinese Medicine, Changchun, China
- 2Department of Liver, Spleen and Gastroenterology, First Affiliated Hospital to Changchun University of Chinese Medicine, Changchun, China
Non-alcoholic fatty liver disease (NAFLD) is a clinicopathologic syndrome characterized by excessive fat deposition in hepatocytes and a major cause of end-stage liver disease. Autophagy is a metabolic pathway responsible for degrading cytoplasmic products and damaged organelles, playing a pivotal role in maintaining the homeostasis and functionality of hepatocytes. Recent studies have shown that pharmacological intervention to activate or restore autophagy provides benefits for liver function recovery by promoting the clearance of lipid droplets (LDs) in hepatocytes, decreasing the production of pro-inflammatory factors, and inhibiting activated hepatic stellate cells (HSCs), thus improving liver fibrosis and slowing down the progression of NAFLD. This article summarizes the physiological process of autophagy, elucidates the close relationship between NAFLD and autophagy, and discusses the effects of drugs on autophagy and signaling pathways from the perspectives of hepatocytes, kupffer cells (KCs), and HSCs to provide assistance in the clinical management of NAFLD.
1 Introduction
NAFLD is a common chronic liver disease characterized by excessive accumulation of hepatic fat in the absence of chronic viral infection and alcohol abuse (1, 2). NAFLD is a metabolic disease involving abnormal liver fat metabolism, which has recently been renamed as metabolism-associated fatty liver disease (MAFLD) (3), as represented by NAFLD in the following article. Non-alcoholic fatty liver (NAFL) continues to progress, leading to hepatocyte swelling, liver damage, inflammation, and varying degrees of fibrosis. This promotes the occurrence of non-alcoholic steatohepatitis (NASH), which can eventually progress to cirrhosis over time (4). The incidence and prevalence of NAFLD have been reported to be rapidly increasing worldwide, reaching approximately 30%. It is projected that the prevalence of NASH will experience a significant rise of 56% by the year 2030 (5). The traditional treatment of NAFLD focuses on addressing pathological changes caused by excessive fatty accumulation through exercise, dietary adjustments, lifestyle improvements, and reduction of liver fat accumulation (6, 7). While these methods alleviate NAFLD to some extent, they are not sufficient to prevent the progression of NAFLD. Therefore, there is an urgent need for new treatment methods for NAFLD in clinical practice.
The pathogenesis of NAFLD is mainly characterized by the accumulation of triglycerides (TGs) in hepatocytes, and the long-term accumulation of liver lipids promotes the occurrence of steatosis and lipotoxicity (8, 9). However, lipotoxicity induces mitochondrial dysfunction, triggering endoplasmic reticulum stress (ERS) and lipid peroxidation (10). This process further promotes the activation of KCs, leading to inflammation and the release of inflammatory cytokines and transforming growth factors, and activates HSCs, thus facilitating the progression of NAFLD (11, 12).
Autophagy is an important process for maintaining cellular homeostasis, and forms autophagosomes by degrading unnecessary organelles, proteins and other components in cells, and forms autolysosomes by combining with lysosomes to eliminate phagocytized goods and maintain intracellular homeostasis (13, 14). The term “autophagy” was initially coined by Christian de Duve in 1967, coined from the Greek terms “auto” meaning self, and “phagy” meaning to consume (15). In recent years, researchers have conducted in-depth studies on autophagy and believe that autophagy is a widely existing necessary physiological regulatory process in mammalian cells (16). Significantly, autophagosomes and autolysosomes are two essential components in the formation of autophagy (17). It is generally believed that the fusion between autophagosomes and lysosomes is necessary for the formation of autolysosomes. However, distinct mechanisms regulate the formation of autophagosomes as well as their subsequent fusion with lysosomes to form autolysosomes.
An increasing number of studies have shown a close relationship between the development of NAFLD and impaired autophagy function (18). Restoring autophagy can promote the breakdown of fat, improve hepatic steatosis, and reduce liver cell damage (19). In addition, autophagy can maintain the homeostasis and function of non-parenchymal cell types, thereby inhibiting liver inflammation and fibrosis and slowing down the progression of NAFLD. This makes it one of the important targets for treating NAFLD (20, 21). However, the current literature on the relationship between drugs and autophagy in NAFLD needs to be better organized. Therefore, this review will discuss the important role of autophagy in NAFLD specifically focusing on the two processes of autophagosome formation and autolysosome formation, to provide theoretical support for the clinical treatment of NAFLD.
2 Autophagy
In mammals, autophagy mainly includes macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) (22). Microautophagy is selective organelle degradation that occurs through the interaction of proteins with lysosomes, vacuolar membranes, or late endosomal surface proteins (23). CMA is a highly selective form of autophagy that often involves binding to the molecular chaperone heat-shock cognate protein in order to transport substrate proteins to the lysosomal surface for internalization and rapid degradation within the lysosomal lumen (24). This review concentrates on macroautophagy (hereinafter referred to as autophagy) and its role in NAFLD.
Autophagy is governed by a complex series of regulatory mechanisms, including initiation, formation of autophagosomes, formation of autolysosomes, degradation, and termination of recycling (25). Meanwhile, autophagy is primarily initiated by autophagy-related (ATGs) proteins, which are encoded and modified by highly conserved ATG genes. These ATG proteins, in conjunction with autophagy regulatory factors, play a crucial role in regulating the autophagic process (26, 27). Moreover, the AMP-activated protein kinase (AMPK) and mammalian target of rapamycin (mTOR) are two pivotal components involved in the regulation of autophagy (28).
When autophagy is initiated, AMPK is regulated by upstream kinases such as liver kinase B1 (LKB1) and calcium/calmodulin-dependent protein kinase kinase 2 or β (CaMKK2/β), while simultaneously inhibiting the expression of mammalian target of rapamycin 1 (mTOR1) (29, 30). Subsequently, AMPK phosphorylates the downstream UNC-51-like kinase 1 (ULK1) complex, which serves as the initiation kinase complex of autophagy and primarily consists of ATG13, FIP200, ATG101, ULK1 or ULK2 in mammals (31, 32). Activation of ULK1 promotes phosphorylation of the downstream III phosphoinositide 3-kinase (PI3K) complex I, which is composed of autophagy and Beclin 1 regulator 1 (AMBRA1), Beclin-1, class III PI3K vacuolar protein sorting 34 (Vps34), p115 and ATG14 (17). Subsequently, downstream proteins of the class III PI3K complex, including double-FYVE-containing protein 1 (DFCP1) and WD repeat domain phosphoinositide-interacting protein (WIPI), are recruited to the cell wall for the production phosphatidylinositol 3-phosphate (PI3P), which contributes to phagosome extension (33, 34). Simultaneously, PI3P recruits ATG2 and transmembrane protein ATG9 as lipid transporters within the autophagosome intermembrane and interfollicular regions to facilitate phagocytosis (35) (Figure 1).
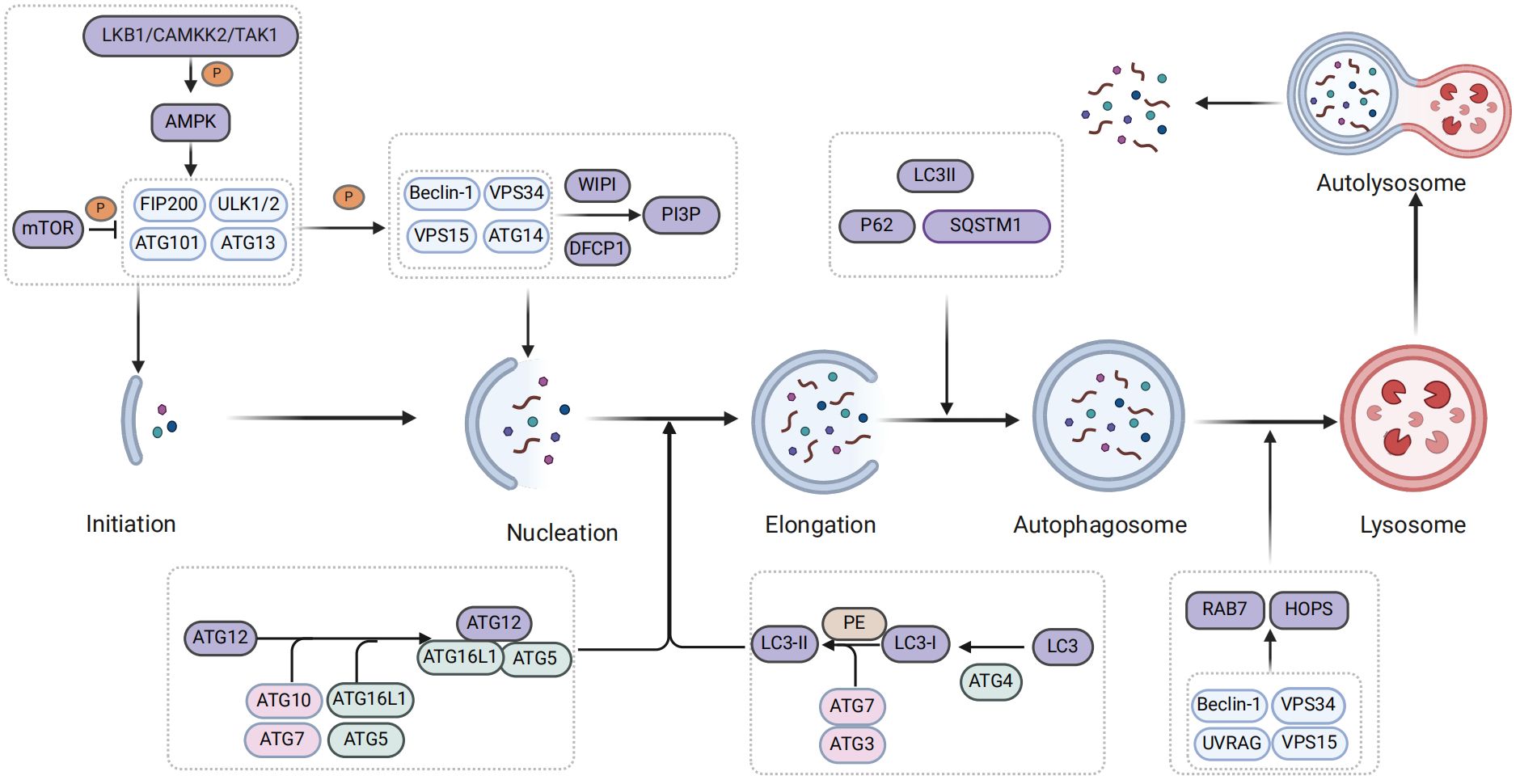
Figure 1 The biological process of autophagy.
Subsequently, the engagement system of two ubiquitin-like proteins promotes phagosome extension and formation of autophagosomes (36). Ubiquitin-like protein ATG12 is bound to ATG5 by the combination of E1 activase ATG7 and E2 binding enzyme ATG10 and further bound to ATG16L1 to form the ATG12-ATG5-ATG16L1 ubiquitin-like complex (37, 38). Furthermore, the homolog of ATG8, microtubule-associated protein 1A/1B-light chain 3B (LC3), undergoes hydrolysis by ATG4 protein to generate LC3I. Subsequently, the ubiquitin-like binding of E1 activase enzyme ATG7, the E2 binding enzyme ATG3, and the E3 ligase ATG12-ATG5-ATG16L1 activates LC3I and facilitates its interaction with phosphatidylethanolamine (PE) to form lipidized ATG8/LC3-II (39–42). Subsequently, P62/sequestosome1 (SQSTM1) functions as an autophagy adaptor protein that mediates the interaction between ubiquitin-like proteins and LC3II, facilitating expansion of phagocytic cells and promoting closure of the isolation membrane to facilitate formation of autophagosomes (43) (Figure 1).
Finally, autophagosomes form autolysosomes by transporting their cargo into lysosomes and fusing with them (44). In the process of autophagosome-lysosome fusion, the ADP-ribosylation factor-like GTPase AR18 and Ras-associated protein 7 (Rab7) play a pivotal role in localizing autophagosomes and lysosomes, as well as mediating fusion through recruitment of specific adapter proteins and motor proteins (45, 46). Additionally, tethering factors such as homotypic fusion and vacuole protein sorting (HOPS) are crucial for autolysosomal formation (47). They are recruited through interaction with pleckstrin homology and RUN domain containing M1 (PLEKHM1) and soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complexes to facilitate lipid and content mixing, promoting fusion between autophagosomes and lysosomes (47). The pacer autophagy enhancer is targeted by STX17 and phosphatidylinositol 3-phosphate (PtdIns3P) within the SNARE complex localized in autophagy vacuoles. This targeting facilitates the recruitment of the VP34-Beclin 1-UVRAG complex to the autophagy vacuoles (48). Meanwhile, the PtdIns3P protein on autophagosomes facilitates fusion with lysosomes by recruiting the HOPS complex tethering factor (48, 49). Notably, transcription factor EB (TFEB) and transcription factor enhancer 3 (TFE3) are also involved in the formation of autolysosomes (50). Ultimately, lysosomal proteases release degraded products into extracellular environment to promote recycling of these materials (51) (Figure 1).
Autophagy degradation can be classified into two categories: non-selective and selective. Non-selective autophagy refers to the random phagocytosis and degradation of cytoplasmic substances such as proteins and organelles. However, selective autophagy is a cellular response mechanism that specifically targets cargoes for degradation in lysosomes upon exposure of cells to various stresses, such as DNA damage (52, 53). Furthermore, studies have found that the progression of NAFLD is not only related to non-selective autophagy (hereinafter referred to as autophagy) but also closely associated with selective forms of autophagy, including mitophagy, lipophagy, and endoplasmic reticulum (ER) autophagy (54).
The liver utilizes non-selective autophagy to eliminate, degrade, and recycle senescent or dysfunctional cellular components, organelles, and proteins in response to nutrient deficiency or stressful conditions (55). This ensures the maintenance of cellular energy balance. In a state of starvation, autophagy degrades LDs in hepatocytes to release fatty acids to the mitochondria for β-oxidation, thereby regulating lipid storage and energy balance in hepatocytes (55). On the contrary, dysregulation of autophagy weakens lipolysis and fat phagocytosis in hepatocytes, leading to mitochondrial dysfunction and increased production of reactive oxygen species (ROS) in the liver (56). This activation of KCs induces the proliferation and activation of HSCs, thus promoting the development of NAFLD. However, improving specific selective autophagy can alleviate the progression of NAFLD (57).
For instance, lipophagy is the process through which cells selectively recognize lipids and, activated by autophagy-related molecules, contribute to the degradation of LDs in hepatocytes (58). Under normal conditions, the liver mediates the decomposition of triacylglycerols (TAGs) stored in LDs and induces mitochondrial β-oxidation through two major pathways: lipid catabolism and lipophagy (59). Thus, adipose triglyceride lipase (ATGL) is a cytosolic lipase involved in lipolysis, which releases free fatty acids (FFAs) that can serve as substrates for mitochondrial β-oxidation or as potent signaling molecules for various cellular processes (60). The ATGL and patatin-like phospholipase domain-containing protein 8 (PNPLA8) act as selective autophagy receptors for lipid engulfment, facilitating lipolysis and β-oxidation of FFAs (61). During the process of lipophagy, lipid droplet-associated proteins perilipin 2 (PLIN2) and perilipin 3 (PLIN3) are degraded through CMA by a synergistic effect of Hsc70 and lysosome-associated membrane protein 2A (LAMP-2A) receptors (62). Similarly, the interactions of ATG14 with ULK1 and LC3 also induce lipophagy, leading to the release of FFAs and mitochondrial β-oxidation (63). The regulation of TFEB can enhance lysosome biosynthesis, autophagosome formation, and their fusion with lysosomes and promote autophagy flux, a crucial regulatory factor in lipolysis (64). The studies have shown that overexpression of TFE3 can regulate coordinated lysosomal expression and regulation (CLEAR) elements, leading to a significant reduction in hepatocyte steatosis (65). Another study found that important proteins and pathways involved in maintaining lipid phagocytosis and lipid droplet homeostasis include the Rab GTPase in LDs, Rab10, and Rab7 (primarily activated during autophagy), as well as the mTORC1-perilipin-3 pathway (66).
In addition, mitophagy can degrade functionally impaired mitochondria and misfolded proteins in the liver, regulating cell death to maintain a stable state of liver lipid metabolism and inhibit the production of ROS. This process is important in the treatment of NAFLD (67, 68). Simultaneously, mitophagy also triggers the activation of inflammatory vesicles (56). Normally, PINK1 facilitates the translocation of Parkin from cytoplasmic lysates to depolarized mitochondria. Parkin ubiquitinates outer membrane proteins and recruits P62 to damaged mitochondria, resulting in changes in autophagosome membrane structure and initiating selective mitophagy (69, 70). During hypoxia, HIF1-α promotes the expression of BNIP3 (BCL2/adenovirus E1B interacting protein 3) and its relocation to the mitochondrial envelope. Subsequently, BNIP3 interacts with Bcl-2 to facilitate mitochondrial remodeling, while NIX directly interacts with LC3 through the LIR domain, recruiting autophagosomes to mitochondria and inducing mitophagy (71–73). Studies have demonstrated that macrophage-stimulating protein 1 (Mst1) mediates mitochondrial autophagy by regulating Parkin expression through the AMPK pathway, which further promotes NAFLD (74). However, the early pathogenesis of NAFLD involves the loss of mitophagy due to the deletion of a key regulator, Parkin, which accelerates the onset of crucial disease features in NAFLD (68). Another study demonstrated that increased expression and mitochondrial localization of PINK1/Parkin promoted PINK1-mediated mitophagy for clearing damaged mitochondria (75). This reduction in oxidative stress inhibited NOD-like receptor thermal protein domain-associated protein 3 (NLRP3) inflammasome activation and slowed hepatic steatosis progression in NAFLD patients (75).
Interestingly, ERS activates the unfolded protein response (UPR) and induces ER autophagy, which is an essential factor in restoring the degradation of substances in the ER to normal levels (76). In the normal liver, ERS facilitates autophagy-mediated lipid degradation in hepatocytes to maintain the protective mechanism of physiological hepatocyte function (77). The ER autophagy receptor is a crucial factor that affects the level of ER autophagy. Notably, current investigations have identified several ER-phagy receptors, namely FAM134, SEC62, RTN3L, CCPG1, ATL, TEX264, CALCOCO1, C53, and p63 (78). Under ERS induction, the disruption of the ER structure is recognized by the ER receptor and leads to binding with LC3/GABARAP/ATG8, thereby promoting ER autophagy (79). However, during the process of ERS-induced autophagy, UPR mainly regulates autophagy through inositol requiring enzyme 1 (IRE1α), double-stranded RNA-activated protein kinase (PKR)–like ER kinase (PERK), activating transcription factor 6 (ATF6), and Ca2+ regulation. By upregulating ER autophagy receptors, UPR promotes autophagy, and C/EBP homologous protein (CHOP) plays a critical regulatory role in this process (80, 81). Specifically, IRE1α promotes autophagy by modulating Beclin1 through the tumor necrosis factor receptor-associated factor 2 (TRAF2) and c-Jun N-terminal kinase (JNK) pathways. Additionally, the PERK pathway induces autophagy by suppressing mTORC1 activity (82, 83). ATF6 activates death-associated protein kinase 1 downstream through Beclin1, thereby triggering autophagy (84). Additionally, Ca2+ in the ER can also participate in autophagy via protein kinase C (PKC) and calcium Ca2+/calcium calmodulin-dependent kinase II (CaMKII). Animal studies have shown that autophagy induces autophagy by downregulating mTOR signaling, ERS-related genes (PERK, IRE1, ATF6, BIP, and CHOP), and autophagy-related genes. This reduces hepatic steatosis and lipid accumulation (85). Another study demonstrated that exposure of human liver cells to low concentrations of per- and polyfluoroalkyl substances induces ERS through activation of the UPR signaling pathway, and modulates NAFLD (86).
Many clinical drugs for treating NAFLD have been shown to activate autophagy and affect the progression of NAFLD through different signaling pathways, including the classic hypoglycemic drug metformin (87), the well-established lipid-lowering agent simvastatin (88), and the mTOR target inhibitor rapamycin (89). Emerging therapeutic drugs, such as glucagon-like peptide-1 (GLP-1), sodium glucose co-transporter 2 (SGLT2) inhibitors, and immune-related GTPase M (IRGM), have been studied extensively (90). Additionally, studies have demonstrated that herbal extracts containing active ingredients such as resveratrol, ginsenoside, and curcumin exhibit hepatoprotective effects by inducing autophagy in the liver (91). More detailed information will be provided in the subsequent sections.
3 Hepatic parenchymal and non-parenchymal cells in NAFLD
In the pathological progression of NAFLD, various cell types including parenchymal and non-parenchymal hepatocytes, KCs, HSCs, liver sinusoidal endothelial cells (LSECs), and cholangiocytes are implicated (92) (Figure 2).
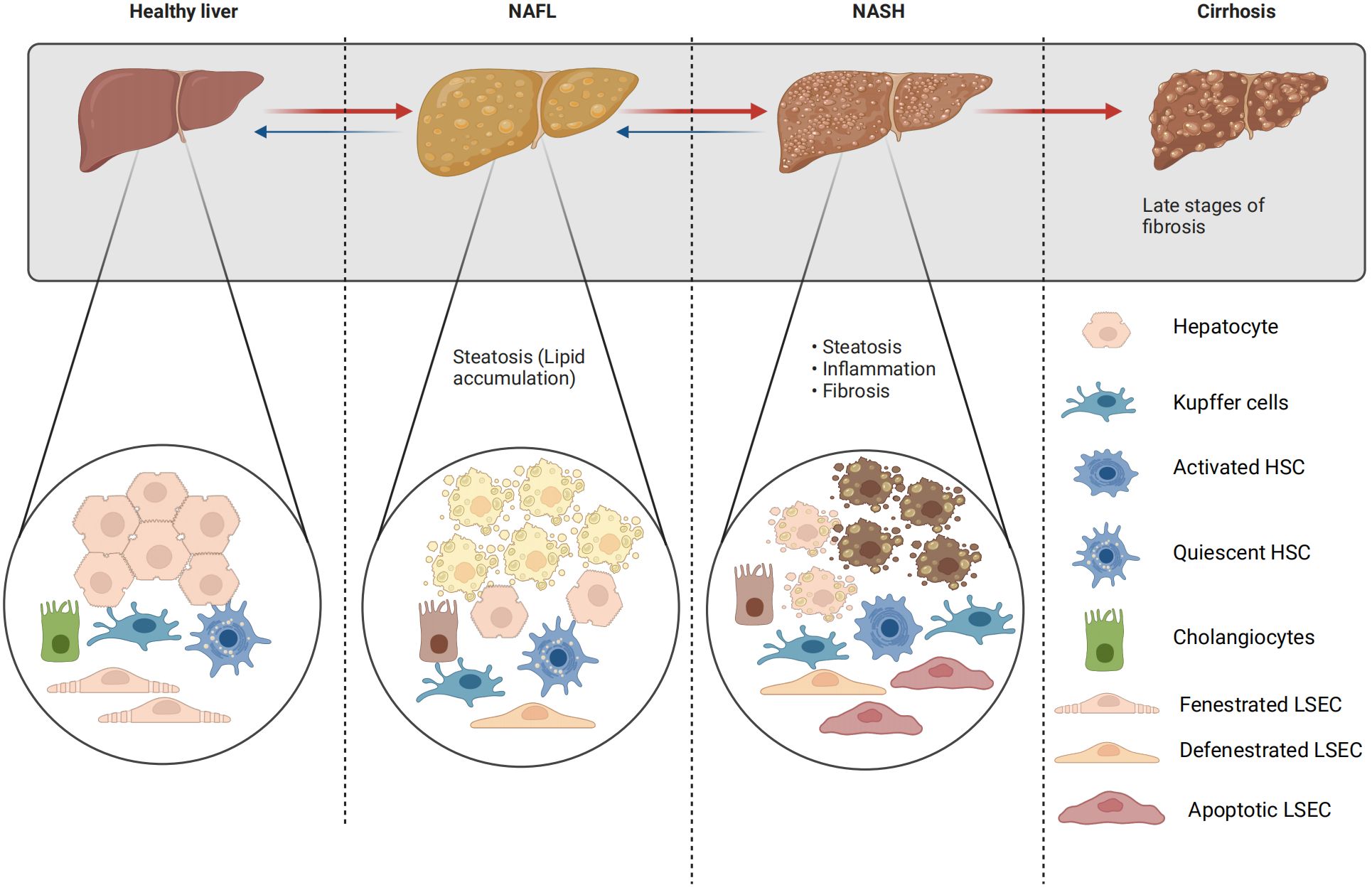
Figure 2 Changes of liver parenchyma and non-parenchymal cells in NAFLD. The transition between fenestrated and defenestrated LSECs: under physiological conditions, LSECs are perforated by fenestrations and lack a basement membrane; however, under pathological conditions, LSECs lose their fenestrations and form a continuous basement membrane. This phenomenon is called “capillarization”. Capillarization, i.e. loss of LSECs fenestrae, and LSECs dysfunction, i.e. the loss of the ability of LSECs to generate vasodilator agents in response to increased shear stress, are two events occurring early in NAFLD. The transition from quiescent to active HSCs: a distinct feature of quiescent hepatic stellate cells is the storage of retinoids (vitamin A and its metabolites) within their cytoplasmic lipid droplets. However, activated hepatic stellate cells exhibit a contractile, proliferative, and fibrogenic phenotype, which can be further distinguished from quiescent hepatic stellate cells by the loss of their retinol-containing lipid droplets.
3.1 Hepatocytes in NAFLD
Under physiological conditions, hepatic lipid metabolism is typically mediated by fatty acid synthase (FAS), which facilitates the esterification of TG and very low-density lipoprotein (VLDL) for secretion into the circulation (93). Meanwhile, it can be oxidized in mitochondria through β-oxidation or stored as LDs, these LDs can then undergo lipolysis and fat phagocytosis to provide fatty acids for β-oxidation (94). In NAFLD, the increased lipolysis in the liver leads to the excessive release of FFA, which stimulates lipid accumulation and degeneration in hepatocytes, triggers mitochondrial dysfunction, activates ERS, and results in lipid peroxidation in the liver (8, 9). This process promotes excessive production of reactive ROS and damages liver autophagy. Furthermore, the generation of reactive ROS activates inflammatory signaling pathways, such as the nuclear factor κB (NF-κB) and JNK pathways, leading to an increase in inflammatory cytokines. As a result, this activation triggers KCs and accelerates the progression of NAFLD (94). Overexpression of miR-1297 in exosomes from lipotoxic hepatocytes can activate the PTEN/PI3K/AKT signaling pathway, promoting HSC activation and proliferation, inducing fibrosis, and accelerating the progression of NAFLD (95). Hepatocyte autophagy can enhance lipophagy, selectively degrade LDs, and thus improve lipid metabolism in NAFLD (96, 97). This topic will be further discussed in the subsequent section of the article.
3.2 Kupffer cells in NAFLD
KCs, the liver-resident macrophages, originate from yolk sac progenitor cells and are integral to liver homeostasis, inflammation, and fibrosis regulation (98, 99). The phagocytosis of apoptotic steatosis hepatocytes and uptake of free cholesterol are critical features of KCs in NAFLD. However, in NAFLD-damaged hepatocytes, resident KCs sense the disturbance of homeostasis and are activated. This activation stimulates the production of inflammation and the release of pro-inflammatory factors (IL-1β, IL-6, TNF-α) and pro-fibrotic factors, promoting the progression of NAFLD (11, 100). In addition, hepatic macrophages function by undergoing classical M1 activation or M2 activation (101). The regulation of NASH involves a delicate balance between pro-inflammatory M1 KCs and anti-inflammatory M2 KCs, with the activation of NLRP3 as a classical inflammasome in KCs (102). There is an increasing body of evidence suggesting that hepatic macrophages play a crucial role in the development of NAFLD (103–105). Studies have found that autophagy is an upstream regulator of KCs inflammasome polarization and activation. Increasing autophagy flux and inhibiting inflammasome activation are the main approaches to improving the inflammatory state of NAFLD by inhibiting M1 polarization and NLRP3 inflammasome activation (106).
3.3 Hepatic stellate cells in NAFLD
HSCs, situated in the Disse space between the lateral basal region of hepatocytes and the surface of sinusoidal endothelial cells, serve as the principal producers of collagen within the liver (107). In healthy livers, quiescent HSCs facilitate the intercellular transport of cytokines and soluble mediators (108). However, activation of HSCs enhances extracellular matrix (ECM) production, which is a key initiating event in hepatic fibrosis and plays a crucial role in NAFLD progression (109, 110). Specifically, factors such as inflammation, lipotoxicity, lipid mediators, and growth factors trigger hepatocyte death. A distinctive feature of HSCs is the storage of retinoids (vitamin A and its metabolites) within their cytoplasmic lipid droplets (111). Lipophagy occurs in HSCs, mediating the extensive β-oxidation of FFA, providing the necessary energy for HSC activation during the fibrosis process (66). Meanwhile, lipophagy promotes the decomposition of LDs and accelerates the activation of HSCs, which eventually leads to the progression of liver fibrosis (66). They also stimulate the recruitment of KCs, induce the production of transforming growth factor beta (TGF-β) and expression of alpha-smooth muscle actin (α-SMA), along with ECM synthesis. This leads to proliferation and activation of HSCs that transform into “myofibroblasts” (112). Activated HSCs contribute to inflammation and fibrosis in NAFLD by secreting proinflammatory cytokines and expressing adhesion molecules that promote the recruitment and infiltration of immune cells into the liver (113). The inhibition of the TGF-β pathway in a mouse model of NAFLD has been demonstrated to result in reduced activation of HSCs and alleviated fibrosis, with the most pronounced effect observed when IL13 is concurrently suppressed (114). However, the regulation of autophagy in HSCs is intricate, encompassing the activation of quiescent HSCs and the inhibition of activated HSCs. Recent studies have shown that activated HSCs are essential in the deposition of fibrotic tissue. However, autophagy inhibits the release of fibrogenic extracellular vesicles (EVs), thereby alleviating liver fibrosis (115). Furthermore, in mouse models of NAFLD, the activation of the PGE2/PGE2 receptor 4 (EP4) axis stimulates extracellular signal-regulated kinase pathways, enhances autophagy, and induces HSC activation and fibrosis (116).
3.4 Liver sinusoidal endothelial cells in NAFLD
LSECs are the predominant non-parenchymal cells in the liver and play a pivotal role in mediating the transport of nutrients, lipids, and lipoproteins (117). Under physiological conditions, LSECs are perforated by fenestrations and lack a basement membrane; however, under pathological conditions, LSECs lose their fenestrations and form a continuous basement membrane, this phenomenon is called ‘capillarization’, also known as defenestration, refers to the loss of fenestrae (118). LSECs facilitate bidirectional lipid exchange between the bloodstream and hepatic tissue (119). Additionally, they serve as a crucial defense barrier within the liver microenvironment by actively maintaining quiescence in HSCs and KCs (119). Conversely, the dysfunction of LSECs are an early event in the progression of NAFLD, leading to impaired hepatic lipid uptake and metabolism, inflammation, and hepatocyte damage (120–122). Specifically, impaired LSEC function reduces their permeability, resulting in formation of capillarization and hindering the release of VLDL from hepatocytes into the hepatic sinusoidal cavity (123). Consequently, cholesterol and TG accumulate in the liver, promoting the development of NAFLD. As this condition progresses, autophagy impairment, LSEC capillarization, inability to maintain KC dormancy, activation of the NF-κB pathway, reduction in endothelial nitric oxide synthase (eNOS), and increase in inducible nitric oxide synthase (iNOS) occur. Also, there is an upregulation of adhesion molecules and chemokines, all of which contribute to inflammation in patients with NAFLD (119, 124). Interestingly, capillarization LSECs also secrete fibrosis factors, such as TGF-β1 and extracellular matrix proteins, thereby stimulating the activation of adjacent HSCs and promoting the progression of NAFLD to liver fibrosis (125, 126). Studies have shown that nitric oxide in LSECs can temporarily activate KC while down-regulating proinflammatory chemokines involved in monocyte and macrophage recruitment through MAPK-dependent pathways (127). Hammoutene et al. (128) revealed LSEC apoptosis and inflammatory responses following a high-fat diet in LSEC-specific Atg5 knockout mice.
3.5 Cholangiocytes in NAFLD
The epithelium of intra- and extra-hepatic bile ducts is lined with cholangiocytes, which include two distinct types, small and large (129, 130). In the physiological state, cholangiocytes have a low proliferative capacity and are in a mitotic quiescent phase (131). In contrast, during the development of NAFLD, hepatocyte apoptosis leads to cholangiocyte injury (132). However, cholangiocytes participate in the progressive portal and bridging fibrosis of NAFLD through stimulation by fatty acids, injury caused by bile stasis, and influences on repair and proliferation. These factors promote the progression of NAFLD to liver fibrosis (133, 134). Specifically, damaged cholangiocytes rapidly activate into proliferating progenitor cells by releasing various cytokines, growth factors, neuropeptides, and hormones. This leads to manifestations such as proliferation promotion, inflammation promotion, fibrosis promotion, or aging (135, 136). Additionally, the expression and localization of bile acid receptors TGR5 and S1PR2 (sphingosine 1-phosphate receptor 2) in cholangiocytes are diminished, accompanied by a decrease in FXR activation (137, 138). This impairment in promoting downstream pathways of bile acids disrupts cholangiocyte bile acid homeostasis, elevating total bile acids and contributing to the NAFLD phenotype (137, 138). Research has shown that cholangiocytes promote hepatic steatosis by upregulating lipid biosynthesis genes by regulating the SCT/SCTR/miR-125b axis, which is crucial for improving the phenotype of NAFLD/NASH in humans (139).
4 NAFLD and autophagy
As previously described, we discussed the biological processes and degradation of autophagy, as well as its function in cellular metabolism. Through a literature review, we demonstrated the close association between autophagy and the progression of NAFLD in both liver parenchyma and non-parenchymal cells (Figure 3). However, there is a lack of research on the treatment of NAFLD by regulating autophagy through drug-targeting signaling pathways for LSECs and cholangiocytes. Therefore, we will start by focusing on the formation of autophagosomes and autolysosomes in three specific cell types, namely hepatocytes, KCs, and HSCs. This will allow us to explore how drugs targeting autophagy through signaling pathways can be used to treat the progression of NAFLD, thereby deepening our understanding of the relationship between autophagy and NAFLD.
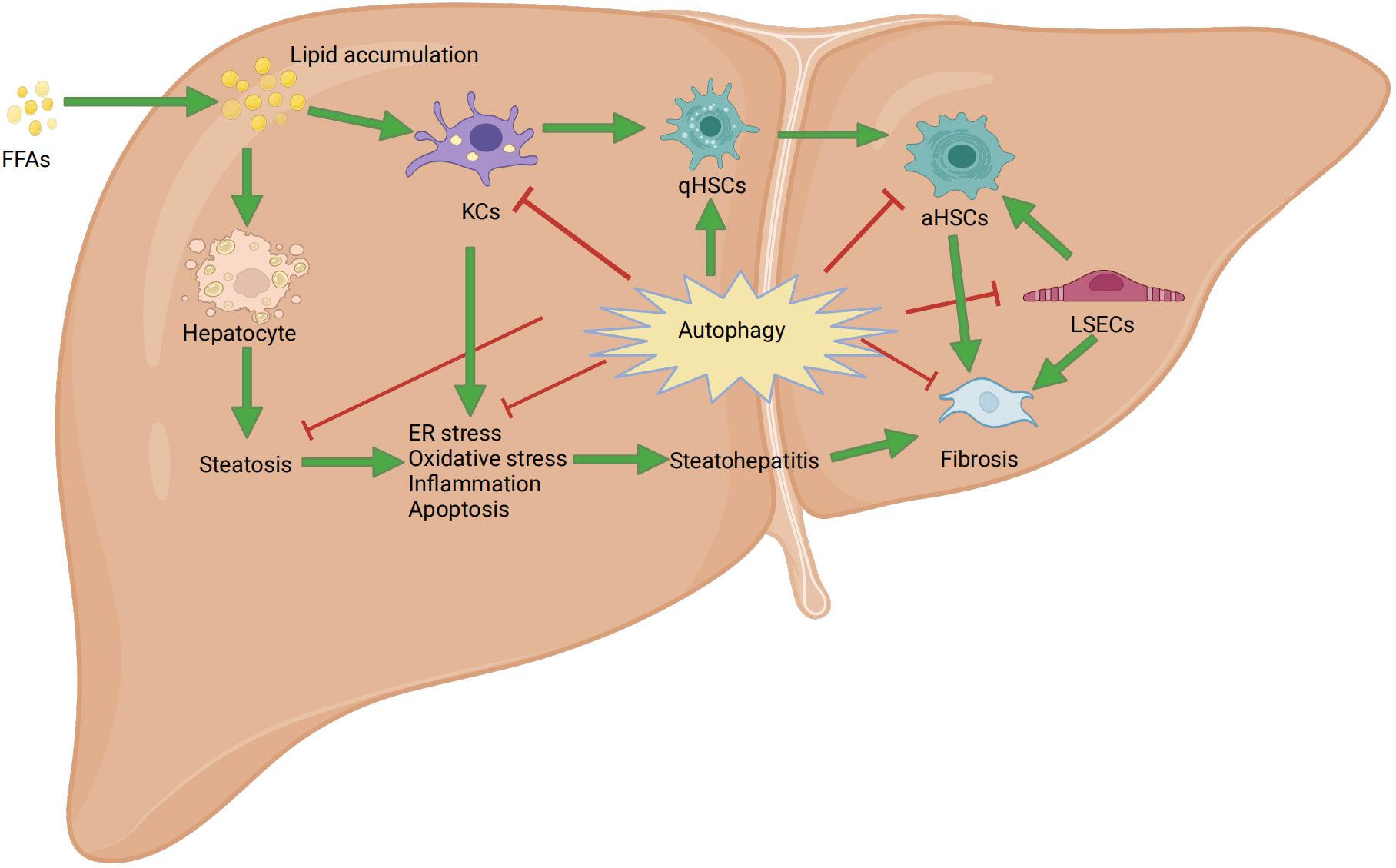
Figure 3 Mechanism of autophagy in nonalcoholic fatty liver disease. FFAs: free fatty acids; ER: endoplasmic reticulum stress; aHSCs: activated hepatic stellate cells; qHSCs: Quiescent hepatic stellate cells; KCs: Kupffer cells; LSECs: liver sinusoidal endothelial cells.
4.1 The role of autophagosome formation in NAFLD
4.1.1 The role of autophagy in hepatocytes
In mouse models of NAFLD, pharmacological agents have been shown to modulate autophagy via key metabolic pathways, thus influencing the disease’s progression. Compounds such as schisandrin B and acetylshikonin (AS) activate the AMPK/mTOR pathway, stimulating hepatocyte autophagy and attenuating NAFLD progression (140, 141). Similarly, empagliflozin activates the AMPK/mTOR pathway, upregulates LC3B expression, induces autophagy, and improves NAFLD in apolipoprotein E (ApoE) knockout mouse models (142). In the ob/ob mouse model of NAFLD, metformin enhances the activity of PRKA by activating SIRT1-FOXO signaling pathways, thereby inducing autophagy and facilitating lipid metabolism in NAFLD (143). Berbamine (BBM) similarly invokes the SIRT1/LKB1/AMPK pathway to enhance autophagy markers and induce autophagy, slowing down NAFLD progression (144). However, in a HepG2 model induced by palmitic acid (PA), dapagliflozin, an SGLT2 inhibitor, induces fatty acid oxidation and autophagy through the AMPK-mTOR-ULK1 pathway (145). Pueraria radix flavonoids stimulate autophagy by inhibiting the PI3K/Akt/mTOR signaling pathway, reducing intracellular lipid accumulation and inflammation, while ginsenoside Rg1 modulates the PTEN-AKT pathway to mitigate NAFLD progress (146, 147). Dual inhibitions of soluble epoxide hydrolase (sEH) and cyclooxygenase-2 (COX-2), along with the GLP-1 analog liraglutide (LRG), activate the Sirt1/PI3K/AKT/mTOR and AMPK/mTOR/Beclin1 pathways to promote autophagy, alleviating NAFLD (148–150). Ursodeoxycholic acid (UDCA) modulates the Bcl-2/Beclin-1 and Bcl-2/Bax complexes via an AMPK pathway, inducing autophagy and impeding the progression of NAFLD (151). Conversely, acetaminophen (APAP) overdose reduces LC3-II, Beclin1, and AMPK levels while increasing mTOR and SREBP-1c levels, this inhibits autophagy and exacerbates NAFLD lipid accumulation (152). Chemerin/CMKLR1 enhances autophagy and reduces hepatic oxidative stress via the JAK2-STAT3 pathway (153). Interestingly, fasting-induced fibroblast growth factor-21 (FGF21) signaling activates PKA, which phosphorylates JMJD3, enhancing its nuclear localization and interaction with the nuclear receptor PPARα, this promotes hepatic autophagy and lipid degradation in hepatocytes (154). In another experimental study, aescinate activated the Keap1-Nrf2 pathway and autophagy, thereby ameliorating the progression of NAFLD (155). Compounds like magnolol (MG), in HepG2 and Wistar rat models, demonstrate therapeutic potential by inhibiting mTOR and activating the Nrf2-ARE pathway, enhancing autophagic flux and reversing hepatic steatosis (156). Additional studies with curcumin, 1,3-dichloro-2-propanol, icariin, baicalein, and verapamil have elucidated its effects on autophagic processes in hepatocyte models of NAFLD (157–161). Thus, autophagy regulates hepatocyte metabolism through mTOR, AMPK, PI3K, AKT, and PPAR pathways. These pathways can mitigate steatosis, ameliorate oxidative stress and liver injury, and slow the progression of NAFLD (Figure 4, Tables 1–3).
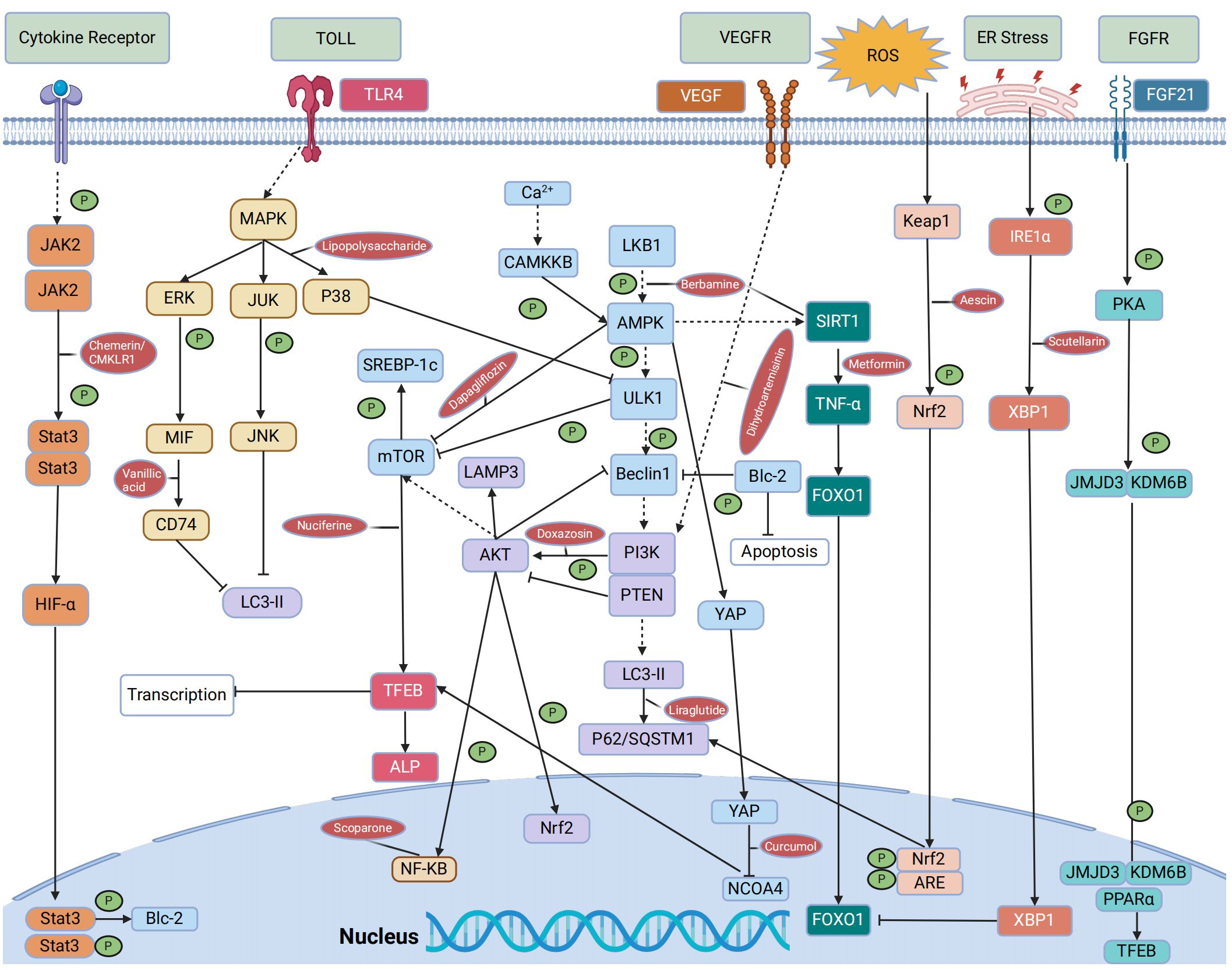
Figure 4 Signaling pathways that regulate autophagy or are regulated by autophagy in NAFLD.
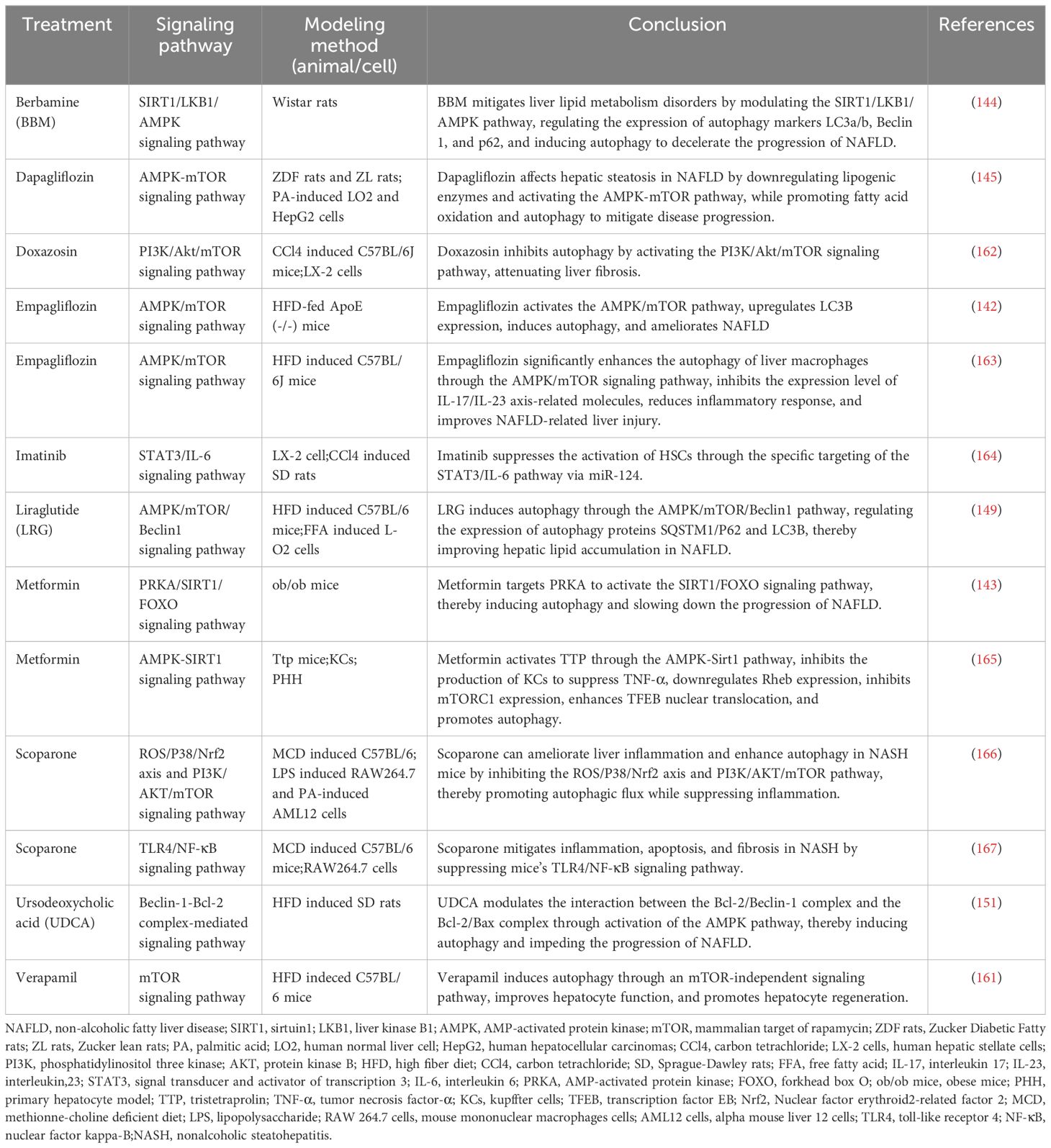
Table 1 The role of autophagosome formation in NAFLD (drugs with clinical trials).
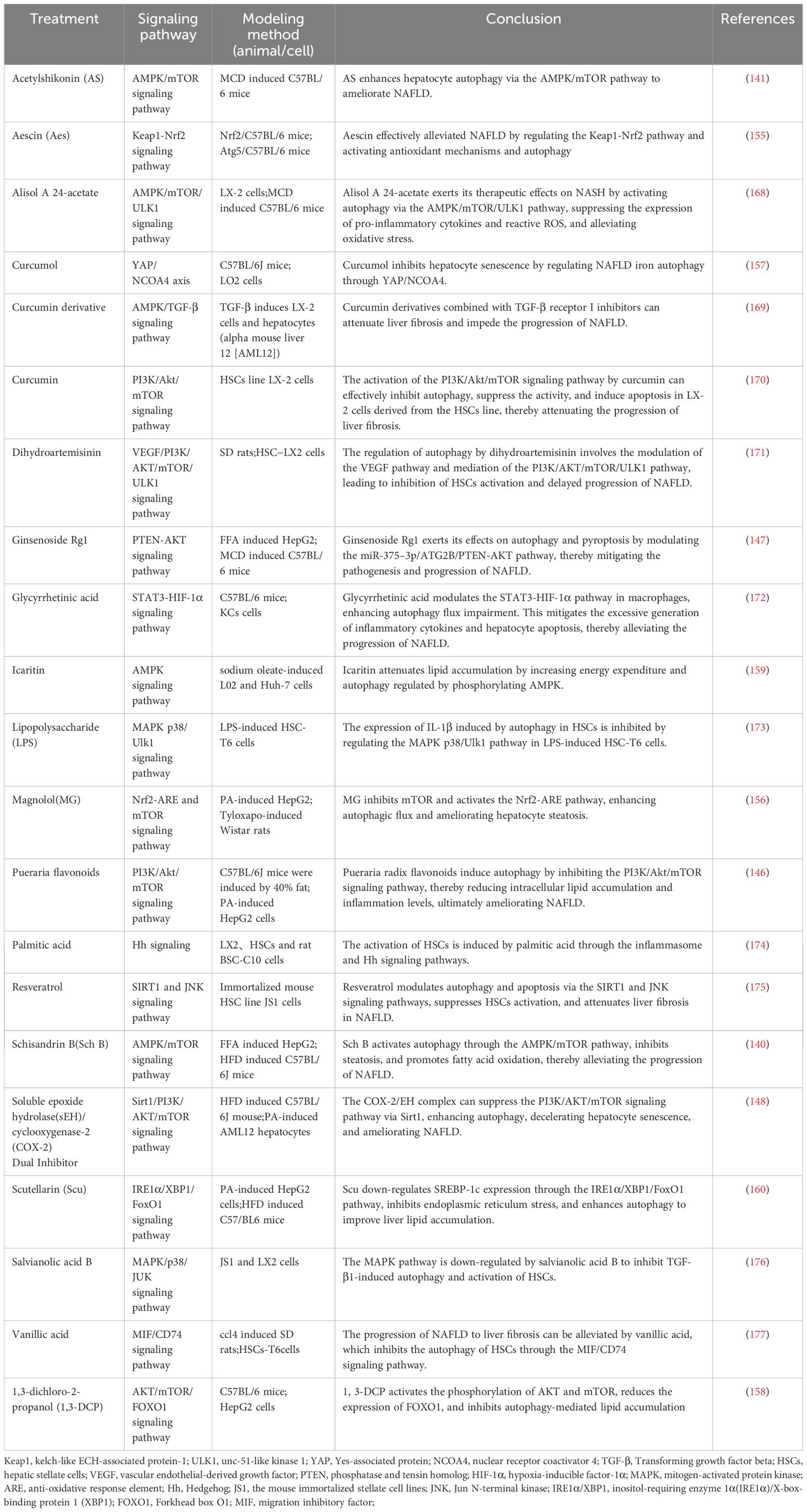
Table 2 The role of autophagosome formation in NAFLD (drugs without clinical trials).
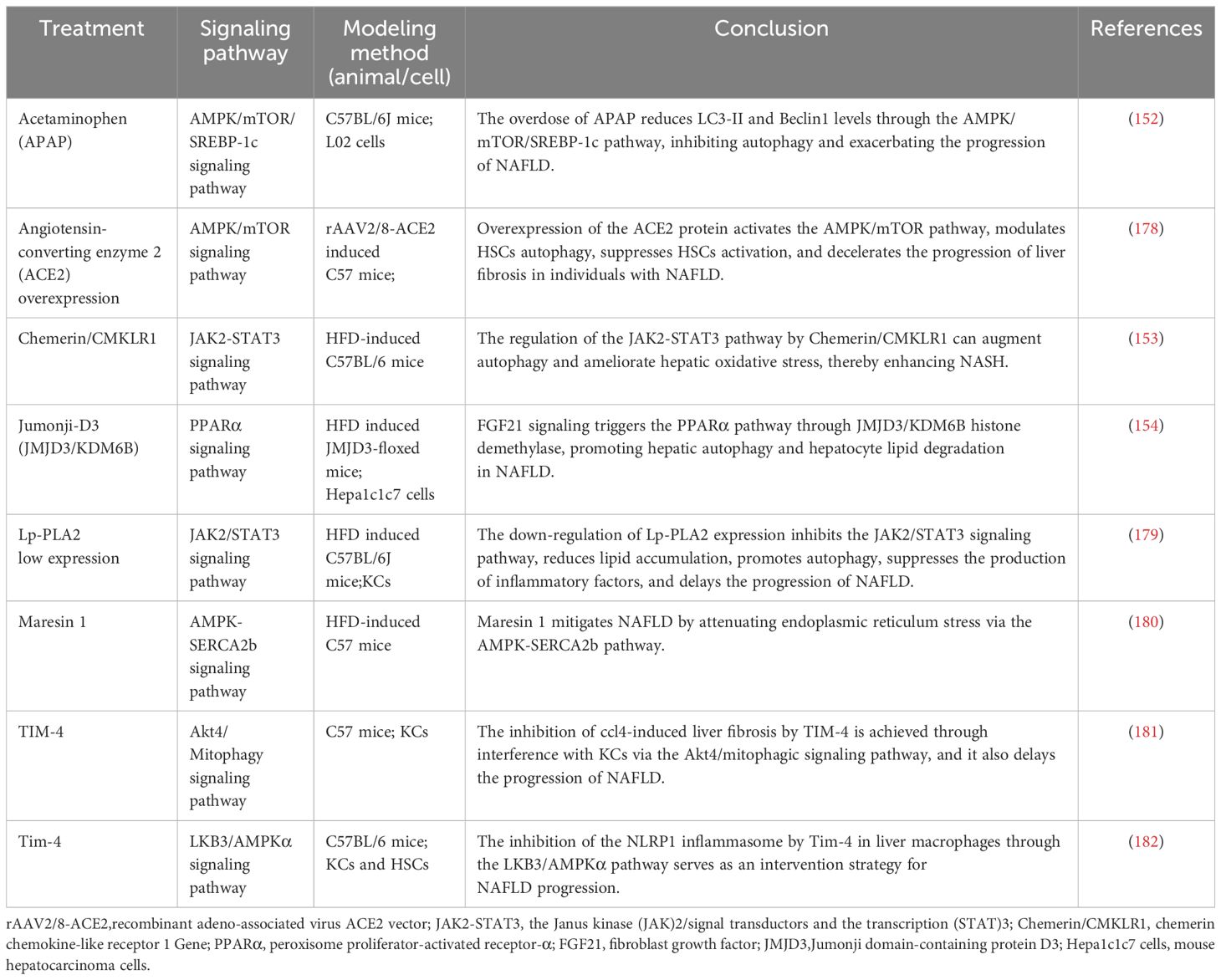
Table 3 The role of autophagosome formation in NAFLD (low or high expression of a particular target).
4.1.2 The role of autophagy in Kupffer cells
Scoparone, for instance, has been shown to enhance autophagic flux and suppress inflammation by inhibiting the ROS/P38/Nrf2 axis and the PI3K/AKT/mTOR pathway in an LPS-induced liver macrophage model (166). It also attenuates inflammation, apoptosis, and fibrosis progression in NAFLD through the downregulation of the TLR4/NF-κB signaling pathway (167). Furthermore, silencing lipoprotein-associated phospholipase A2 (Lp-PLA2) in KCs and NAFLD model mice inhibited the activation of the JAK2/STAT3 signaling pathway, mitigated lipid accumulation, promoted autophagy, reduced production of inflammatory factors, and inhibited the progression of NAFLD (179). In Ttp mice models of KCs and primary hepatocytes, metformin activates the mRNA-binding protein tristetraproline (TTP) through the AMPK-Sirt1 pathway in both hepatocytes and KCs, this activation curtails TNF-α production in KCs, downregulates Rheb expression, suppresses mTORC1 expression, and enhances TFEB nuclear translocation (165). Collectively, these effects promote autophagy and alleviate NAFLD progression. In addition, the presence of TIM-4 on KCs acts as a negative regulator, reinstating autophagy through the Akt4/mitophagy signaling pathway and modulating the progression of NAFLD by exerting inhibitory effects on the NLRP1 inflammasome via the LKB3/AMPKα pathway (181, 182). Glycyrrhizic acid has been demonstrated to enhance impaired autophagy flux, mitigate excessive production of inflammatory cytokines, and ameliorate hepatocyte apoptosis through the regulation of the STAT3-HIF-1α pathway in macrophages (172). Empagliflozin has also been reported to significantly enhance autophagy in hepatic macrophages through the AMPK/mTOR pathway, suppressing the IL-17/IL-23 axis and improving liver injury related to NAFLD (163). Moreover, maresin 1, a twenty-two carbon polyunsaturated fatty acid synthesized by macrophages, reinstates autophagy and suppresses ER via the AMPK-SERCA2b pathway (180). In summary, autophagy plays a crucial role in maintaining the homeostasis and functionality of KCs. Autophagy in hepatic macrophages can modulate the polarization and activation of inflammasomes, thereby mitigating the progression of NAFLD through its antioxidant and anti-inflammatory effects. Conversely, impaired autophagy can worsen inflammatory responses in KCs, induce ERS, and expedite the onset and progression of diseases. This detrimental cascade can be mitigated by pharmacological agents such as metformin, scoparone, and empagliflozin, which modulate autophagy-related pathways (Figure 4; Tables 1–3).
4.1.3 The role of autophagy in hepatic stellate cells
Resveratrol has been shown to induce autophagy in immortalized mouse HSCs, dampening HSC activation through the SIRT1 and JNK signaling pathways, thus attenuating liver fibrosis (175). Salvianolic acid B, by influencing the MAPK pathway, has been effective in suppressing TGF-β1-induced autophagy and HSC activation, as well as modulating ERK, p38, and JNK signaling cascades (176). In both CCl4-induced SD rat and HSC models, vanillic acid treatment was found to inhibit the MIF/CD74 signaling pathway, subsequently suppressing autophagy (177). When curcumin derivatives are combined with a TGF-β receptor I inhibitor in LX-2 liver fibrosis models, they activate the AMPK/TGF-β signaling pathway, regulate autophagy, and alleviate liver fibrosis (169). Curcumin itself activates the PI3K/Akt/mTOR signaling pathway, inhibiting autophagy and promoting apoptosis in LX-2 cells (170). Alisol A 24-acetate induces autophagy via the AMPK/mTOR/ULK1 pathway in MCD mice and LX-2 models (168). However, the activation of the VEGF pathway by dihydroartemisinin, along with its regulation of the PI3K/AKT/mTOR/ULK1 pathway, facilitates autophagy and retards the progression of NAFLD (171). Doxazosin has been observed to inhibit autophagy by stimulating the PI3K/Akt/mTOR pathway, attenuating liver fibrosis (162). Overexpression of angiotensin-converting enzyme 2 (ACE2) modulates autophagy via the AMPK/mTOR pathway in HSCs, suppressing HSC activation and promoting apoptosis (178). In the mouse model of HSCs, imatinib specifically targeted the STAT3/IL-6 pathway via miR-124, leading to the induction of autophagy and suppression of HSC activation (164). Conversely, PA induces HSC activation through the NLRP3 inflammasome and Hedgehog (Hh) signaling pathways, diminishing autophagic flux, inhibiting autophagy, and promoting the progression of NASH toward fibrosis (174). Additionally, in LPS-induced HSC-T6 cells, activation of the MAPK p38/Ulk1 pathway inhibits autophagy in HSCs and induces IL-1β expression (173). Based on the literature above, it has been observed that autophagy plays a dual role in HSCs. Dysfunction of autophagy facilitates energy supply for HSC activation during the development of fibrosis and contributes to the progression of NAFLD. On the other hand, autophagy also plays a role as an anti-fibrotic factor. It may promote HSC death, mediate the degradation of profibrotic mediators such as collagens and metalloproteinases, or diminish the release of exosomes that carry pro-fibrotic signals. This decelerates the progression of NAFLD to liver fibrosis. However, modulation of autophagy-related pathways through drug administration, such as resveratrol, dihydroartemisinin, and doxazosin, can enhance autophagic processes (Figure 4; Tables 1–3).
4.2 The role of autolysosome in NAFLD
In a NAFLD mouse model and a PA-induced HepG2 cell model, liraglutide has been observed to alleviate hepatic steatosis by modulating the expression of autophagy substrates LC3-II and SQSTM1/P62, thereby activating the autophagy-lysosomal pathway (ALP) regulated through the TFEB pathway (183). The PPARα agonist fenofibrate activates lysosomal Ca2+ via mucolipin 1, leading to stimulates the calcineurin phosphatase and CaMKKβ-AMPK-ULK1 signaling pathways (184). These pathways trigger TFEB activation and enhance lipophagy, which ameliorates fat accumulation in NAFLD. Nuciferine has been shown to effectively regulate the mTORC1-TFEB-ALP axis, suppressing the lysosomal localization and activity of mTORC1, activating TFEB-mediated ALP, and restoring autophagy (185). Simultaneously, quercetin enhances the co-localization of lysosomes and LDs through the IRE1a/XBP1s pathway while reducing their accumulation. It also stimulates autophagy to ameliorate HFD-induced NAFLD (186). Phillygenin reduces hepatocyte lipid deposition by regulating the Ca2+ calcineurin-TFEB axis, thereby enhancing lysosomal biogenesis and autophagic flux (187). The upregulation of lysosome-associated membrane protein 3 (LAMP3) in patients with NAFLD and in mouse models has been correlated with the activation of the PI3K/Akt pathway, which facilitates autolysosome fusion (188). Additionally, in HepG2 cells and HFD-induced C57BL/6J mice, the administration of lipid-lowering granules and activation of the PI3K-AKT-mTOR signaling pathway resulted in the induction of autophagosomes or their co-localization with lysosomes, this led to autophagosomal degradation, activation of the autophagy process, reduction in oxidative stress levels, and improvement in lipid accumulation and inflammatory response (189). Baicalein has been found to inhibit the mTOR signaling pathway, enhance lysosomal membrane permeability, and restore autophagy, offering a therapeutic strategy for NAFLD (190). Ajugol, an active alkaloid derived from the root of rehmanniobium, effectively inhibits mTOR and induces the nuclear translocation of TFEB, promoting the restoration of the autophagy-lysosomal pathway and lipophagy (191). In conclusion, the formation of autolysosomes is another key pathway during autophagy related to the degradation of intracellular metabolites (192). The dysregulation of the vital autophagy factor TFEB impaired lysosomal membrane activity and lysosomal biogenesis, leading to impaired lysosomal function and disruption of the autophagic degradation pathway (193). However, treatments such as liraglutide and quercetin have shown potential in restoring lysosomal function by promoting autophagy and decelerating the progression of NAFLD (Figure 4; Table 4).
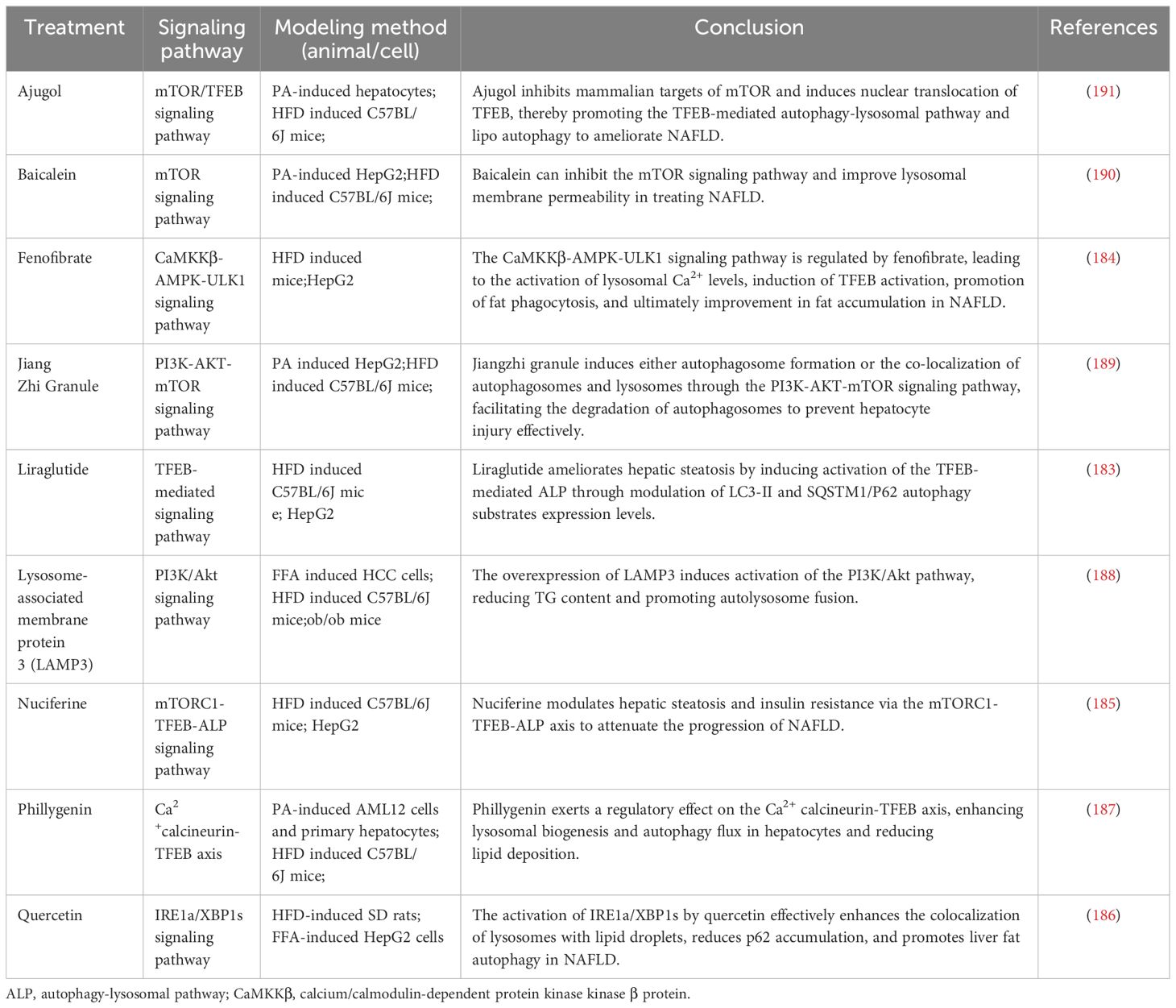
Table 4 The role of autolysosome formation in NAFLD.
5 Discussion
Defects in autophagy are implicated in the pathogenesis of numerous diseases, including tumors, neurodegenerative disorders, and metabolic conditions (16). Moreover, dysfunctional autophagy is intricately associated with the pathological progression of NAFLD (194).
The upstream proteins activate AMPK in autophagy to facilitate phosphorylation of the ULK-1 complex and suppress activation of mTOR (29, 30). Subsequently, phosphorylation of downstream PI3KC3 complex I induces recruitment of ATG9, a downstream autophagy factor, onto the cell membrane and initiates autophagosome nucleation. Through interaction between DFCP1 and WIPI proteins, PI3P is generated, which facilitates cell nucleation and autophagosome formation (33, 34). Concurrently, the induction of WIPI and ATG16L1 triggers the formation of two downstream ubiquitin complexes: ATG12-ATG5-ATG16L1 and LC3-II (33, 35, 37, 38). These complexes are crucial in facilitating autophagosome elongation. Subsequently, the ubiquitination of LC3II through its association with p62/SQSTM1 as an autophagosomal substrate facilitates autophagosome maturation (43). In conjunction with the HOPS complex, VPS34 complex II and Rab7 further regulate this process, enabling fusion between cargo-containing autophagosome sand lysosomes (45, 47, 48). Ultimately, lysosomes digest this fused product, leading to degradation, reactivating mTORC1, inhibiting autophagy, and terminating the autophagic process (51). The degradation modes within autophagy encompass both non-selective and selective processes. Non-selective autophagy is a cellular process that eliminates senescent or dysfunctional components like organelles and proteins due to nutrient deficiency or stress conditions while degrading them for recycling. This process helps maintain cellular energy balance (54). On the other hand, selective autophagy is a cellular response mechanism that targets explicit cargoes for degradation within lysosomes when cells are exposed to various stresses like DNA damage (52, 53). However, during the development of NAFLD pathology, selective autophagy primarily plays a role in degrading LDs through mitophagy and ER-phagy processes (57).
The pathogenesis of NAFLD is intricate and primarily associated with the interplay of steatosis-induced inflammation, lipotoxicity, oxidative stress, and the ER (8). During disease progression, excessive accumulation of fat in hepatocytes, increased levels of FFAs and TG stimulate hepatocyte steatosis, induce mitochondrial dysfunction, and ERS. This promotes excessive production of ROS, impairs autophagy, promotes the capillarization of LSECs, and stimulates the activation of inflammasomes in KCs. Simultaneously, it also enhances the secretion of fibrotic factors that stimulate the activation of adjacent HSCs and facilitate the progression of NAFLD to liver fibrosis (126). However, autophagy has a dual role in HSCs, promoting the growth of quiescent HSCs, while inhibiting the growth of activated HSCs. This prevents fibrosis changes in NAFLD and its involvement in all stages of NAFLD progression (115). In recent years, there have been reports discussing drugs that target autophagy for the treatment of NAFLD, including herbal extracts, emerging GLP-1 activators, and SGLT2 inhibitors (91). These drugs have demonstrated their ability to induce or restore the process of autophagy, ameliorate hepatocyte steatosis, reduce inflammation caused by KCs, inhibit HSC activation, decrease the occurrence of liver fibrosis, and delay the progression of NAFLD (115).
Three types of autophagy have been identified, namely microautophagy, macroautophagy, and CMA. However, numerous unresolved questions remain regarding the intricate interplay among these processes and the specific triggering mechanisms involved in autophagy. Unresolved inquiries persist concerning identification of novel receptors involved in selective autophagy for autophagy-mediated degradation. Despite extensive knowledge of liver autophagy, the specific mechanism of action of drugs targeting autophagy in the pathogenesis of NAFLD remains to be determined. Certain drugs have not transitioned from animal experiments to clinical practice due to a lack of theoretical support from human clinical research. Similarly, the current research stage in NAFLD pathology lacks sufficient investigation into drug targeting of autophagy through signaling pathways, specifically in hepatic sinusoidal endothelial cells and cholangiocytes. In the future, by regulating the role of autophagy through signaling pathways, we will be able to identify better therapeutic targets to treat NAFLD more effectively.
6 Conclusions
The activation of autophagy can inhibit hepatocyte steatosis, suppress the production of inflammatory factors in KCs, prevent the activation of HSCs, and maintain LSECs. In contrast, impaired autophagy is involved in different stages of NAFLD. However, despite ongoing clinical studies and the use of some drugs to improve autophagy in NAFLD patients, the incidence of this disease continues to rise. The maintenance of autophagy in innate liver cells is an urgent problem that needs to be addressed in the medical field. This study aims to provide novel perspectives and insights for the clinical management of NAFLD by summarizing the physiological process of autophagy, elucidating the association between autophagy and NAFLD, and exploring the role and relevant pathways of intracellular therapeutic agents.
Author contributions
QS: Writing – original draft, Writing – review & editing, Conceptualization, Project administration, Visualization. MY: Writing – review & editing, Project administration, Visualization. SW: Investigation, Writing – review & editing. XC: Writing – original draft, Investigation, Project administration. SC: Writing – original draft, Project administration. RZ: Investigation, Writing – original draft. ZX: Writing – review & editing, Visualization. YL: Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by the Natural Science Foundation of Jilin Province (20210101208JC) and the Natural Science Foundation of Jilin Province (YDZJ202201ZYTS263).
Acknowledgments
Author thanks the Jilin Provincial Natural Science Foundation for the financial support.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fendo.2024.1374644/full#supplementary-material
References
1. Nassir F. NAFLD: mechanisms, treatments, and biomarkers. Biomolecules. (2022) 12(6):824. doi: 10.3390/biom12060824
2. Yin X, Guo X, Liu Z, Wang J. Advances in the diagnosis and treatment of non-alcoholic fatty liver disease. Int J Mol Sci. (2023) 24(3):2844. doi: 10.3390/ijms24032844
3. Eslam M, Sanyal AJ, George J. MAFLD: A consensus-driven proposed nomenclature for metabolic associated fatty liver disease. Gastroenterology. (2020) 158:1999–2014.e1. doi: 10.1053/j.gastro.2019.11.312
4. Huang TD, Behary J, Zekry A. Non-alcoholic fatty liver disease: a review of epidemiology, risk factors, diagnosis and management. Intern Med J. (2020) 50:1038–47. doi: 10.1111/imj.14709
5. Han SK, Baik SK, Kim MY. Non-alcoholic fatty liver disease: Definition and subtypes. Clin Mol Hepatol. (2023) 29:S5–s16. doi: 10.3350/cmh.2022.0424
6. Raza S, Rajak S, Upadhyay A, Tewari A, Anthony Sinha R. Current treatment paradigms and emerging therapies for NAFLD/NASH. Front Biosci (Landmark Ed). (2021) 26:206–37. doi: 10.2741/4892
7. Grander C, Grabherr F, Tilg H. Non-alcoholic fatty liver disease: pathophysiological concepts and treatment options. Cardiovasc Res. (2023) 119:1787–98. doi: 10.1093/cvr/cvad095
8. Badmus OO, Hillhouse SA, Anderson CD, Hinds TD, Stec DE. Molecular mechanisms of metabolic associated fatty liver disease (MAFLD): functional analysis of lipid metabolism pathways. Clin Sci (Lond). (2022) 136:1347–66. doi: 10.1042/CS20220572
9. Clare K, Dillon JF, Brennan PN. Reactive oxygen species and oxidative stress in the pathogenesis of MAFLD. J Clin Transl Hepatol. (2022) 10:939–46. doi: 10.14218/JCTH.2022.00067
10. Marra F, Svegliati-Baroni G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J Hepatol. (2018) 68:280–95. doi: 10.1016/j.jhep.2017.11.014
11. Chen J, Deng X, Liu Y, Tan Q, Huang G, Che Q, et al. Kupffer cells in non-alcoholic fatty liver disease: friend or foe? Int J Biol Sci. (2020) 16:2367–78. doi: 10.7150/ijbs.47143
12. Wang S, Li K, Pickholz E, Dobie R, Matchett KP, Henderson NC, et al. An autocrine signaling circuit in hepatic stellate cells underlies advanced fibrosis in nonalcoholic steatohepatitis. Sci Transl Med. (2023) 15:eadd3949. doi: 10.1126/scitranslmed.add3949
13. Vargas JNS, Hamasaki M, Kawabata T, Youle RJ, Yoshimori T. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol. (2023) 24:167–85. doi: 10.1038/s41580-022-00542-2
14. Kitada M, Koya D. Autophagy in metabolic disease and ageing. Nat Rev Endocrinol. (2021) 17:647–61. doi: 10.1038/s41574-021-00551-9
15. Deter RL, De Duve C. Influence of glucagon, an inducer of cellular autophagy, on some physical properties of rat liver lysosomes. J Cell Biol. (1967) 33:437–49. doi: 10.1083/jcb.33.2.437
16. Klionsky DJ, Petroni G, Amaravadi RK, Baehrecke EH, Ballabio A, Boya P, et al. Autophagy in major human diseases. EMBO J. (2021) 40:e108863. doi: 10.15252/embj.2021108863
17. Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. (2018) 19:349–64. doi: 10.1038/s41580-018-0003-4
18. Ren Q, Sun Q, Fu J. Dysfunction of autophagy in high-fat diet-induced non-alcoholic fatty liver disease. Autophagy. (2024) 20:221–41. doi: 10.1080/15548627.2023.2254191
19. Martinez-Lopez N, Singh R. Autophagy and lipid droplets in the liver. Annu Rev Nutr. (2015) 35:215–37. doi: 10.1146/annurev-nutr-071813-105336
20. Weiskirchen R, Tacke F. Relevance of autophagy in parenchymal and non-parenchymal liver cells for health and disease. Cells. (2019) 8(1):16. doi: 10.3390/cells8010016
21. Qian H, Chao X, Williams J, Fulte S, Li T, Yang L, et al. Autophagy in liver diseases: A review. Mol Aspects Med. (2021) 82:100973. doi: 10.1016/j.mam.2021.100973
22. Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxid Redox Signal. (2014) 20:460–73. doi: 10.1089/ars.2013.5371
23. Wang L, Klionsky DJ, Shen HM. The emerging mechanisms and functions of microautophagy. Nat Rev Mol Cell Biol. (2023) 24:186–203. doi: 10.1038/s41580-022-00529-z
24. Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. (2018) 19:365–81. doi: 10.1038/s41580-018-0001-6
25. Kim KH, Lee MS. Autophagy–a key player in cellular and body metabolism. Nat Rev Endocrinol. (2014) 10:322–37. doi: 10.1038/nrendo.2014.35
26. Shu F, Xiao H, Li QN, Ren XS, Liu ZG, Hu BW, et al. Epigenetic and post-translational modifications in autophagy: biological functions and therapeutic targets. Signal Transduct Target Ther. (2023) 8:32. doi: 10.1038/s41392-022-01300-8
27. Levine B, Kroemer G. Biological functions of autophagy genes: A disease perspective. Cell. (2019) 176:11–42. doi: 10.1016/j.cell.2018.09.048
28. Inoki K, Kim J, Guan KL. AMPK and mTOR in cellular energy homeostasis and drug targets. Annu Rev Pharmacol Toxicol. (2012) 52:381–400. doi: 10.1146/annurev-pharmtox-010611-134537
29. Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, et al. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. (2004) 101:3329–35. doi: 10.1073/pnas.0308061100
30. Saikia R, Joseph J. AMPK: a key regulator of energy stress and calcium-induced autophagy. J Mol Med (Berl). (2021) 99:1539–51. doi: 10.1007/s00109-021-02125-8
31. Alers S, Löffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. (2012) 32:2–11. doi: 10.1128/MCB.06159-11
32. Hao X, Sun J, Zhong L, Baudry M, Bi X. UBE3A deficiency-induced autophagy is associated with activation of AMPK-ULK1 and p53 pathways. Exp Neurol. (2023) 363:114358. doi: 10.1016/j.expneurol.2023.114358
33. Almannai M, Marafi D, El-Hattab AW. WIPI proteins: Biological functions and related syndromes. Front Mol Neurosci. (2022) 15:1011918. doi: 10.3389/fnmol.2022.1011918
34. Boukhalfa A, Nascimbeni AC, Ramel D, Dupont N, Hirsch E, Gayral S, et al. PI3KC2α-dependent and VPS34-independent generation of PI3P controls primary cilium-mediated autophagy in response to shear stress. Nat Commun. (2020) 11:294. doi: 10.1038/s41467-019-14086-1
35. Noda NN. Atg2 and Atg9: Intermembrane and interleaflet lipid transporters driving autophagy. Biochim Biophys Acta Mol Cell Biol Lipids. (2021) 1866:158956. doi: 10.1016/j.bbalip.2021.158956
36. Nakatogawa H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. (2013) 55:39–50. doi: 10.1042/bse0550039
37. Popelka H, Reinhart EF, Metur SP, Leary KA, Ragusa MJ, Klionsky DJ. Membrane binding and homodimerization of atg16 via two distinct protein regions is essential for autophagy in yeast. J Mol Biol. (2021) 433:166809. doi: 10.1016/j.jmb.2021.166809
38. Lystad AH, Carlsson SR, Simonsen A. Toward the function of mammalian ATG12-ATG5-ATG16L1 complex in autophagy and related processes. Autophagy. (2019) 15:1485–6. doi: 10.1080/15548627.2019.1618100
39. Martens S, Fracchiolla D. Activation and targeting of ATG8 protein lipidation. Cell Discovery. (2020) 6:23. doi: 10.1038/s41421-020-0155-1
40. Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. (2007) 130:165–78. doi: 10.1016/j.cell.2007.05.021
41. Abreu S, Kriegenburg F, Gómez-Sánchez R, Mari M, Sánchez-Wandelmer J, Skytte Rasmussen M, et al. Conserved Atg8 recognition sites mediate Atg4 association with autophagosomal membranes and Atg8 deconjugation. EMBO Rep. (2017) 18:765–80. doi: 10.15252/embr.201643146
42. Walczak M, Martens S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy. (2013) 9:424–5. doi: 10.4161/auto.22931
43. Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. (2015) 282:4672–8. doi: 10.1111/febs.13540
44. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. (2011) 147:728–41. doi: 10.1016/j.cell.2011.10.026
45. Jaber N, Mohd-Naim N, Wang Z, DeLeon JL, Kim S, Zhong H, et al. Vps34 regulates Rab7 and late endocytic trafficking through recruitment of the GTPase-activating protein Armus. J Cell Sci. (2016) 129:4424–35. doi: 10.1242/jcs.192260
46. Carroll B, Mohd-Naim N, Maximiano F, Frasa MA, McCormack J, Finelli M, et al. The TBC/RabGAP Armus coordinates Rac1 and Rab7 functions during autophagy. Dev Cell. (2013) 25:15–28. doi: 10.1016/j.devcel.2013.03.005
47. Zhang S, Li L, Liu X, Zhong Q. The hookup model of the HOPS complex in autophagosome-lysosome fusion. Autophagy. (2024) 20:714–5. doi: 10.1080/15548627.2023.2291938
48. Cheng X, Ma X, Ding X, Li L, Jiang X, Shen Z, et al. Pacer mediates the function of class III PI3K and HOPS complexes in autophagosome maturation by engaging Stx17. Mol Cell. (2017) 65:1029–43.e5. doi: 10.1016/j.molcel.2017.02.010
49. Zhao YG, Codogno P, Zhang H. Machinery, regulation and pathophysiological implications of autophagosome maturation. Nat Rev Mol Cell Biol. (2021) 22:733–50. doi: 10.1038/s41580-021-00392-4
50. Raben N, Puertollano R. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu Rev Cell Dev Biol. (2016) 32:255–78. doi: 10.1146/annurev-cellbio-111315-125407
51. Yang C, Wang X. Lysosome biogenesis: Regulation and functions. J Cell Biol. (2021) 220(6):eaal3694. doi: 10.1083/jcb.202102001
52. Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. (2018) 20:233–42. doi: 10.1038/s41556-018-0037-z
53. Kirkin V, Rogov VV. A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol Cell. (2019) 76:268–85. doi: 10.1016/j.molcel.2019.09.005
54. Li W, He P, Huang Y, Li YF, Lu J, Li M, et al. Selective autophagy of intracellular organelles: recent research advances. Theranostics. (2021) 11:222–56. doi: 10.7150/thno.49860
55. Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab. (2011) 13:495–504. doi: 10.1016/j.cmet.2011.04.004
56. Zheng Y, Wang S, Wu J, Wang Y. Mitochondrial metabolic dysfunction and non-alcoholic fatty liver disease: new insights from pathogenic mechanisms to clinically targeted therapy. J Transl Med. (2023) 21:510. doi: 10.1186/s12967-023-04367-1
57. Alim Al-Bari A, Ito Y, Thomes PG, Menon MB, García-Macia M, Fadel R, et al. Emerging mechanistic insights of selective autophagy in hepatic diseases. Front Pharmacol. (2023) 14:1149809. doi: 10.3389/fphar.2023.1149809
58. An L, Wirth U, Koch D, Schirren M, Drefs M, Koliogiannis D, et al. Metabolic role of autophagy in the pathogenesis and development of NAFLD. Metabolites. (2023) 13(1):101. doi: 10.3390/metabo13010101
59. Kloska A, Węsierska M, Malinowska M, Gabig-Cimińska M, Jakóbkiewicz-Banecka J. Lipophagy and lipolysis status in lipid storage and lipid metabolism diseases. Int J Mol Sci. (2020) 21(17):6113. doi: 10.3390/ijms21176113
60. Schott MB, Weller SG, Schulze RJ, Krueger EW, Drizyte-Miller K, Casey CA, et al. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J Cell Biol. (2019) 218:3320–35. doi: 10.1083/jcb.201803153
61. Zechner R, Madeo F, Kratky D. Cytosolic lipolysis and lipophagy: two sides of the same coin. Nat Rev Mol Cell Biol. (2017) 18:671–84. doi: 10.1038/nrm.2017.76
62. Kaushik S, Cuervo AM. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol. (2015) 17:759–70. doi: 10.1038/ncb3166
63. Mukhopadhyay S, Schlaepfer IR, Bergman BC, Panda PK, Praharaj PP, Naik PP, et al. ATG14 facilitated lipophagy in cancer cells induce ER stress mediated mitoptosis through a ROS dependent pathway. Free Radic Biol Med. (2017) 104:199–213. doi: 10.1016/j.freeradbiomed.2017.01.007
64. Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. (2013) 15:647–58. doi: 10.1038/ncb2718
65. Xiong J, Wang K, He J, Zhang G, Zhang D, Chen F. TFE3 alleviates hepatic steatosis through autophagy-induced lipophagy and PGC1α-mediated fatty acid β-oxidation. Int J Mol Sci. (2016) 17:387. doi: 10.3390/ijms17030387
66. Zhang S, Peng X, Yang S, Li X, Huang M, Wei S, et al. The regulation, function, and role of lipophagy, a form of selective autophagy, in metabolic disorders. Cell Death Dis. (2022) 13:132. doi: 10.1038/s41419-022-04593-3
67. Niture S, Lin M, Rios-Colon L, Qi Q, Moore JT, Kumar D. Emerging roles of impaired autophagy in fatty liver disease and hepatocellular carcinoma. Int J Hepatol. (2021) 2021:6675762. doi: 10.1155/2021/6675762
68. Undamatla R, Fagunloye OG, Chen J, Edmunds LR, Murali A, Mills A, et al. Reduced mitophagy is an early feature of NAFLD and liver-specific PARKIN knockout hastens the onset of steatosis, inflammation and fibrosis. Sci Rep. (2023) 13:7575. doi: 10.1038/s41598-023-34710-x
69. Lu Y, Li Z, Zhang S, Zhang T, Liu Y, Zhang L. Cellular mitophagy: Mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics. (2023) 13:736–66. doi: 10.7150/thno.79876
70. Yamada T, Dawson TM, Yanagawa T, Iijima M, Sesaki H. SQSTM1/p62 promotes mitochondrial ubiquitination independently of PINK1 and PRKN/parkin in mitophagy. Autophagy. (2019) 15:2012–8. doi: 10.1080/15548627.2019.1643185
71. Flores-Toro JA, Go KL, Leeuwenburgh C, Kim JS. Autophagy in the liver: cell's cannibalism and beyond. Arch Pharm Res. (2016) 39:1050–61. doi: 10.1007/s12272-016-0807-8
72. Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. (2010) 11:45–51. doi: 10.1038/embor.2009.256
73. Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson ÅB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. (2012) 287:19094–104. doi: 10.1074/jbc.M111.322933
74. Zhou T, Chang L, Luo Y, Zhou Y, Zhang J. Corrigendum to "Mst1 inhibition attenuates non-alcoholic fatty liver disease via reversing Parkin-related mitophagy. Redox Biol. (2020) 28:101299. doi: 10.1016/j.redox.2019.101299
75. Li X, Shi Z, Zhu Y, Shen T, Wang H, Shui G, et al. Cyanidin-3-O-glucoside improves non-alcoholic fatty liver disease by promoting PINK1-mediated mitophagy in mice. Br J Pharmacol. (2020) 177:3591–607. doi: 10.1111/bph.15083
76. Bernales S, Schuck S, Walter P. ER-phagy: selective autophagy of the endoplasmic reticulum. Autophagy. (2007) 3:285–7. doi: 10.4161/auto.3930
77. Lebeaupin C, Vallée D, Hazari Y, Hetz C, Chevet E, Bailly-Maitre B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol. (2018) 69:927–47. doi: 10.1016/j.jhep.2018.06.008
78. Mochida K, Nakatogawa H. ER-phagy: selective autophagy of the endoplasmic reticulum. EMBO Rep. (2022) 23:e55192. doi: 10.15252/embr.202255192
79. Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature. (2015) 522:354–8. doi: 10.1038/nature14498
80. Bhardwaj M, Leli NM, Koumenis C, Amaravadi RK. Regulation of autophagy by canonical and non-canonical ER stress responses. Semin Cancer Biol. (2020) 66:116–28. doi: 10.1016/j.semcancer.2019.11.007
81. Zhang J, Guo J, Yang N, Huang Y, Hu T, Rao C. Endoplasmic reticulum stress-mediated cell death in liver injury. Cell Death Dis. (2022) 13:1051. doi: 10.1038/s41419-022-05444-x
82. Fernández A, Ordóñez R, Reiter RJ, González-Gallego J, Mauriz JL. Melatonin and endoplasmic reticulum stress: relation to autophagy and apoptosis. J Pineal Res. (2015) 59:292–307. doi: 10.1111/jpi.12264
83. Cybulsky AV. Endoplasmic reticulum stress, the unfolded protein response and autophagy in kidney diseases. Nat Rev Nephrol. (2017) 13:681–96. doi: 10.1038/nrneph.2017.129
84. Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. (2008) 454:455–62. doi: 10.1038/nature07203
85. Zheng X, Dai W, Chen X, Wang K, Zhang W, Liu L, et al. Caffeine reduces hepatic lipid accumulation through regulation of lipogenesis and ER stress in zebrafish larvae. J BioMed Sci. (2015) 22:105. doi: 10.1186/s12929-015-0206-3
86. Qi Q, Niture S, Gadi S, Arthur E, Moore J, Levine KE, et al. Per- and polyfluoroalkyl substances activate UPR pathway, induce steatosis and fibrosis in liver cells. Environ Toxicol. (2023) 38:225–42. doi: 10.1002/tox.23680
87. Li YL, Li XQ, Wang YD, Shen C, Zhao CY. Metformin alleviates inflammatory response in non-alcoholic steatohepatitis by restraining signal transducer and activator of transcription 3-mediated autophagy inhibition in vitro and in vivo. Biochem Biophys Res Commun. (2019) 513:64–72. doi: 10.1016/j.bbrc.2019.03.077
88. Ciric D, Kravic-Stevovic T, Bumbasirevic V, Petricevic S, Jovanovic S, Trajkovic V, et al. Effects of metformin and simvastatin treatment on ultrastructural features of liver macrophages in HFD mice. Ultrastruct Pathol. (2023) 47:1–11. doi: 10.1080/01913123.2022.2156639
89. Garcia-Macia M, Santos-Ledo A, Leslie J, Paish HL, Collins AL, Scott RS, et al. A mammalian target of rapamycin-perilipin 3 (mTORC1-plin3) pathway is essential to activate lipophagy and protects against hepatosteatosis. Hepatology. (2021) 74:3441–59. doi: 10.1002/hep.32048
90. Chen CL, Lin YC. Autophagy dysregulation in metabolic associated fatty liver disease: A new therapeutic target. Int J Mol Sci. (2022) 23(17):10055. doi: 10.3390/ijms231710055
91. Zhou H, Ma C, Wang C, Gong L, Zhang Y, Li Y. Research progress in use of traditional Chinese medicine monomer for treatment of non-alcoholic fatty liver disease. Eur J Pharmacol. (2021) 898:173976. doi: 10.1016/j.ejphar.2021.173976
92. Kumar S, Duan Q, Wu R, Harris EN, Su Q. Pathophysiological communication between hepatocytes and non-parenchymal cells in liver injury from NAFLD to liver fibrosis. Adv Drug Delivery Rev. (2021) 176:113869. doi: 10.1016/j.addr.2021.113869
93. Fujita K, Nozaki Y, Wada K, Yoneda M, Fujimoto Y, Fujitake M, et al. Dysfunctional very-low-density lipoprotein synthesis and release is a key factor in nonalcoholic steatohepatitis pathogenesis. Hepatology. (2009) 50:772–80. doi: 10.1002/hep.23094
94. Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. (2015) 21:739–46. doi: 10.1016/j.cmet.2015.04.004
95. Luo X, Luo SZ, Xu ZX, Zhou C, Li ZH, Zhou XY, et al. Lipotoxic hepatocyte-derived exosomal miR-1297 promotes hepatic stellate cell activation through the PTEN signaling pathway in metabolic-associated fatty liver disease. World J Gastroenterol. (2021) 27:1419–34. doi: 10.3748/wjg.v27.i14.1419
96. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, et al. Autophagy regulates lipid metabolism. Nature. (2009) 458:1131–5. doi: 10.1038/nature07976
97. Filali-Mouncef Y, Hunter C, Roccio F, Zagkou S, Dupont N, Primard C, et al. The ménage à trois of autophagy, lipid droplets and liver disease. Autophagy. (2022) 18:50–72. doi: 10.1080/15548627.2021.1895658
98. Yang T, Wang X, Jiang LF, Li J. [Macrophage heterogeneity role in NAFLD and NASH disease progression]. Zhonghua Gan Zang Bing Za Zhi. (2023) 31:770–5. doi: 10.3760/cma.j.cn501113-20220428-00223
99. Wynn TA, Barron L. Macrophages: master regulators of inflammation and fibrosis. Semin Liver Dis. (2010) 30:245–57. doi: 10.1055/s-0030-1255354
100. Wen Y, Lambrecht J, Ju C, Tacke F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell Mol Immunol. (2021) 18:45–56. doi: 10.1038/s41423-020-00558-8
101. Luo W, Xu Q, Wang Q, Wu H, Hua J. Effect of modulation of PPAR-γ activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci Rep. (2017) 7:44612. doi: 10.1038/srep44612
102. Pan J, Ou Z, Cai C, Li P, Gong J, Ruan XZ, et al. Fatty acid activates NLRP3 inflammasomes in mouse Kupffer cells through mitochondrial DNA release. Cell Immunol. (2018) 332:111–20. doi: 10.1016/j.cellimm.2018.08.006
103. He K, Zhu X, Liu Y, Miao C, Wang T, Li P, et al. Inhibition of NLRP3 inflammasome by thioredoxin-interacting protein in mouse Kupffer cells as a regulatory mechanism for non-alcoholic fatty liver disease development. Oncotarget. (2017) 8:37657–72. doi: 10.18632/oncotarget.v8i23
104. Yu Z, Xie X, Su X, Lv H, Song S, Liu C, et al. ATRA-mediated-crosstalk between stellate cells and Kupffer cells inhibits autophagy and promotes NLRP3 activation in acute liver injury. Cell Signal. (2022) 93:110304. doi: 10.1016/j.cellsig.2022.110304
105. Park SJ, Garcia Diaz J, Um E, Hahn YS. Major roles of kupffer cells and macrophages in NAFLD development. Front Endocrinol (Lausanne). (2023) 14:1150118. doi: 10.3389/fendo.2023.1150118
106. Ge J, Li H, Yang JQ, Yue Y, Lu SY, Nie HY, et al. Autophagy in hepatic macrophages can be regulator and potential therapeutic target of liver diseases: A review. Med (Baltimore). (2023) 102:e33698. doi: 10.1097/MD.0000000000033698
107. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. (2008) 88:125–72. doi: 10.1152/physrev.00013.2007
108. Friedman SL, Roll FJ, Boyles J, Bissell DM. Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci U S A. (1985) 82:8681–5. doi: 10.1073/pnas.82.24.8681
109. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. (2017) 14:397–411. doi: 10.1038/nrgastro.2017.38
110. Wiering L, Subramanian P, Hammerich L. Hepatic stellate cells: dictating outcome in nonalcoholic fatty liver disease. Cell Mol Gastroenterol Hepatol. (2023) 15:1277–92. doi: 10.1016/j.jcmgh.2023.02.010
111. Sufleţel RT, Melincovici CS, Gheban BA, Toader Z, Mihu CM. Hepatic stellate cells - from past till present: morphology, human markers, human cell lines, behavior in normal and liver pathology. Rom J Morphol Embryol. (2020) 61:615–42. doi: 10.47162/rjme.61.3.01
112. Schwabe RF, Tabas I, Pajvani UB. Mechanisms of fibrosis development in nonalcoholic steatohepatitis. Gastroenterology. (2020) 158:1913–28. doi: 10.1053/j.gastro.2019.11.311
113. Carter JK, Friedman SL. Hepatic stellate cell-immune interactions in NASH. Front Endocrinol (Lausanne). (2022) 13:867940. doi: 10.3389/fendo.2022.867940
114. Hart KM, Fabre T, Sciurba JC, Gieseck RL, Borthwick LA, Vannella KM, et al. Type 2 immunity is protective in metabolic disease but exacerbates NAFLD collaboratively with TGF-β. Sci Transl Med. (2017) 9(396):eaal3694. doi: 10.1126/scitranslmed.aal3694
115. Gao J, Wei B, de Assuncao TM, Liu Z, Hu X, Ibrahim S, et al. Hepatic stellate cell autophagy inhibits extracellular vesicle release to attenuate liver fibrosis. J Hepatol. (2020) 73:1144–54. doi: 10.1016/j.jhep.2020.04.044
116. Cao Y, Mai W, Li R, Deng S, Li L, Zhou Y, et al. Macrophages evoke autophagy of hepatic stellate cells to promote liver fibrosis in NAFLD mice via the PGE2/EP4 pathway. Cell Mol Life Sci. (2022) 79:303. doi: 10.1007/s00018-022-04319-w
117. Wisse E, De Zanger RB, Charels K, van der Smissen P, McCuskey RS. The liver sieve: considerations concerning the structure and function of endothelial fenestrae, the sinusoidal wall and the space of Disse. Hepatology. (1985) 5:683–92. doi: 10.1002/(ISSN)1527-3350
118. Sun X, Harris EN. New aspects of hepatic endothelial cells in physiology and nonalcoholic fatty liver disease. Am J Physiol Cell Physiol. (2020) 318:C1200–c13. doi: 10.1152/ajpcell.00062.2020
119. Nasiri-Ansari N, Androutsakos T, Flessa CM, Kyrou I, Siasos G, Randeva HS, et al. Endothelial cell dysfunction and nonalcoholic fatty liver disease (NAFLD): A concise review. Cells. (2022) 11(16):2511. doi: 10.3390/cells11162511
120. Wang XK, Peng ZG. Targeting liver sinusoidal endothelial cells: an attractive therapeutic strategy to control inflammation in nonalcoholic fatty liver disease. Front Pharmacol. (2021) 12:655557. doi: 10.3389/fphar.2021.655557
121. Lafoz E, Ruart M, Anton A, Oncins A, Hernández-Gea V. The endothelium as a driver of liver fibrosis and regeneration. Cells. (2020) 9(4):929. doi: 10.3390/cells9040929
122. Pasarín M, La Mura V, Gracia-Sancho J, García-Calderó H, Rodríguez-Vilarrupla A, García-Pagán JC, et al. Sinusoidal endothelial dysfunction precedes inflammation and fibrosis in a model of NAFLD. PloS One. (2012) 7:e32785. doi: 10.1371/journal.pone.0032785
123. He Q, He W, Dong H, Guo Y, Yuan G, Shi X, et al. Role of liver sinusoidal endothelial cell in metabolic dysfunction-associated fatty liver disease. Cell Commun Signal. (2024) 22:346. doi: 10.1186/s12964-024-01720-9
124. Miyao M, Kotani H, Ishida T, Kawai C, Manabe S, Abiru H, et al. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab Invest. (2015) 95:1130–44. doi: 10.1038/labinvest.2015.95
125. Wang R, Ding Q, Yaqoob U, de Assuncao TM, Verma VK, Hirsova P, et al. Exosome adherence and internalization by hepatic stellate cells triggers sphingosine 1-phosphate-dependent migration. J Biol Chem. (2015) 290:30684–96. doi: 10.1074/jbc.M115.671735
126. Wells RG. Cellular sources of extracellular matrix in hepatic fibrosis. Clin Liver Dis. (2008) 12:759–68. doi: 10.1016/j.cld.2008.07.008
127. McMahan RH, Porsche CE, Edwards MG, Rosen HR. Free fatty acids differentially downregulate chemokines in liver sinusoidal endothelial cells: insights into non-alcoholic fatty liver disease. PloS One. (2016) 11:e0159217. doi: 10.1371/journal.pone.0159217
128. Hammoutene A, Rautou PE. Role of liver sinusoidal endothelial cells in non-alcoholic fatty liver disease. J Hepatol. (2019) 70:1278–91. doi: 10.1016/j.jhep.2019.02.012
129. Maroni L, Haibo B, Ray D, Zhou T, Wan Y, Meng F, et al. Functional and structural features of cholangiocytes in health and disease. Cell Mol Gastroenterol Hepatol. (2015) 1:368–80. doi: 10.1016/j.jcmgh.2015.05.005
130. Cheung AC, Lorenzo Pisarello MJ, LaRusso NF. Pathobiology of biliary epithelia. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:1220–31. doi: 10.1016/j.bbadis.2017.06.024
131. Sato K, Meng F, Giang T, Glaser S, Alpini G. Mechanisms of cholangiocyte responses to injury. Biochim Biophys Acta Mol Basis Dis. (2018) 1864:1262–9. doi: 10.1016/j.bbadis.2017.06.017
132. Martínez AK, Glaser SS. Cholangiocyte lipoapoptosis: implications for biliary damage during nonalcoholic fatty liver disease. Hepatology. (2014) 60:1809–11. doi: 10.1002/hep.27341
133. Zhou T, Kundu D, Robles-Linares J, Meadows V, Sato K, Baiocchi L, et al. Feedback signaling between cholangiopathies, ductular reaction, and non-alcoholic fatty liver disease. Cells. (2021) 10(8):2072. doi: 10.3390/cells10082072
134. Chiba M, Sasaki M, Kitamura S, Ikeda H, Sato Y, Nakanuma Y. Participation of bile ductular cells in the pathological progression of non-alcoholic fatty liver disease. J Clin Pathol. (2011) 64:564–70. doi: 10.1136/jcp.2011.090175
135. Zhou T, Wu N, Meng F, Venter J, Giang TK, Francis H, et al. Knockout of secretin receptor reduces biliary damage and liver fibrosis in Mdr2(-/-) mice by diminishing senescence of cholangiocytes. Lab Invest. (2018) 98:1449–64. doi: 10.1038/s41374-018-0093-9
136. Alvaro D, Mancino MG, Glaser S, Gaudio E, Marzioni M, Francis H, et al. Proliferating cholangiocytes: a neuroendocrine compartment in the diseased liver. Gastroenterology. (2007) 132:415–31. doi: 10.1053/j.gastro.2006.07.023
137. Nagahashi M, Takabe K, Liu R, Peng K, Wang X, Wang Y, et al. Conjugated bile acid-activated S1P receptor 2 is a key regulator of sphingosine kinase 2 and hepatic gene expression. Hepatology. (2015) 61:1216–26. doi: 10.1002/hep.27592
138. Pathak P, Liu H, Boehme S, Xie C, Krausz KW, Gonzalez F, et al. Farnesoid X receptor induces Takeda G-protein receptor 5 cross-talk to regulate bile acid synthesis and hepatic metabolism. J Biol Chem. (2017) 292:11055–69. doi: 10.1074/jbc.M117.784322
139. Chen L, Wu N, Kennedy L, Francis H, Ceci L, Zhou T, et al. Inhibition of secretin/secretin receptor axis ameliorates NAFLD phenotypes. Hepatology. (2021) 74:1845–63. doi: 10.1002/hep.31871
140. Yan LS, Zhang SF, Luo G, Cheng BC, Zhang C, Wang YW, et al. Schisandrin B mitigates hepatic steatosis and promotes fatty acid oxidation by inducing autophagy through AMPK/mTOR signaling pathway. Metabolism. (2022) 131:155200. doi: 10.1016/j.metabol.2022.155200
141. Zeng J, Zhu B, Su M. Autophagy is involved in acetylshikonin ameliorating non-alcoholic steatohepatitis through AMPK/mTOR pathway. Biochem Biophys Res Commun. (2018) 503:1645–50. doi: 10.1016/j.bbrc.2018.07.094
142. Nasiri-Ansari N, Nikolopoulou C, Papoutsi K, Kyrou I, Mantzoros CS, Kyriakopoulos G, et al. Empagliflozin attenuates non-alcoholic fatty liver disease (NAFLD) in high fat diet fed ApoE((-/-)) mice by activating autophagy and reducing ER stress and apoptosis. Int J Mol Sci. (2021) 22(2):818. doi: 10.3390/ijms22020818
143. Song YM, Lee YH, Kim JW, Ham DS, Kang ES, Cha BS, et al. Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy. (2015) 11:46–59. doi: 10.4161/15548627.2014.984271
144. Sharma A, Anand SK, Singh N, Dwarkanath A, Dwivedi UN, Kakkar P. Berbamine induced activation of the SIRT1/LKB1/AMPK signaling axis attenuates the development of hepatic steatosis in high-fat diet-induced NAFLD rats. Food Funct. (2021) 12:892–909. doi: 10.1039/D0FO02501A
145. Li L, Li Q, Huang W, Han Y, Tan H, An M, et al. Dapagliflozin alleviates hepatic steatosis by restoring autophagy via the AMPK-mTOR pathway. Front Pharmacol. (2021) 12:589273. doi: 10.3389/fphar.2021.589273
146. Sun C, Zhang J, Hou J, Hui M, Qi H, Lei T, et al. Induction of autophagy via the PI3K/Akt/mTOR signaling pathway by Pueraria flavonoids improves non-alcoholic fatty liver disease in obese mice. BioMed Pharmacother. (2023) 157:114005. doi: 10.1016/j.biopha.2022.114005
147. Chen X, Xue W, Zhang J, Peng J, Huang W. Ginsenoside Rg1 attenuates the NASH phenotype by regulating the miR-375-3p/ATG2B/PTEN-AKT axis to mediate autophagy and pyroptosis. Lipids Health Dis. (2023) 22:22. doi: 10.1186/s12944-023-01787-2
148. Zhang CY, Tan XH, Yang HH, Jin L, Hong JR, Zhou Y, et al. COX-2/sEH dual inhibitor alleviates hepatocyte senescence in NAFLD mice by restoring autophagy through Sirt1/PI3K/AKT/mTOR. Int J Mol Sci. (2022) 23(15):8267. doi: 10.3390/ijms23158267
149. He Q, Sha S, Sun L, Zhang J, Dong M. GLP-1 analogue improves hepatic lipid accumulation by inducing autophagy via AMPK/mTOR pathway. Biochem Biophys Res Commun. (2016) 476:196–203. doi: 10.1016/j.bbrc.2016.05.086
150. Hao T, Chen H, Wu S, Tian H. LRG ameliorates steatohepatitis by activating the AMPK/mTOR/SREBP1 signaling pathway in C57BL/6J mice fed a high−fat diet. Mol Med Rep. (2019) 20:701–8. doi: 10.3892/mmr.2019.10304
151. Wu P, Zhao J, Guo Y, Yu Y, Wu X, Xiao H. Ursodeoxycholic acid alleviates nonalcoholic fatty liver disease by inhibiting apoptosis and improving autophagy via activating AMPK. Biochem Biophys Res Commun. (2020) 529:834–8. doi: 10.1016/j.bbrc.2020.05.128
152. Shi C, Xue W, Han B, Yang F, Yin Y, Hu C. Acetaminophen aggravates fat accumulation in NAFLD by inhibiting autophagy via the AMPK/mTOR pathway. Eur J Pharmacol. (2019) 850:15–22. doi: 10.1016/j.ejphar.2019.02.005
153. An X, Liu J, Li Y, Dou Z, Li N, Suo Y, et al. Chemerin/CMKLR1 ameliorates nonalcoholic steatohepatitis by promoting autophagy and alleviating oxidative stress through the JAK2-STAT3 pathway. Peptides. (2021) 135:170422. doi: 10.1016/j.peptides.2020.170422
154. Byun S, Seok S, Kim YC, Zhang Y, Yau P, Iwamori N, et al. Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat Commun. (2020) 11:807. doi: 10.1038/s41467-020-14384-z
155. Yu H, Yan S, Jin M, Wei Y, Zhao L, Cheng J, et al. Aescin can alleviate NAFLD through Keap1-Nrf2 by activating antioxidant and autophagy. Phytomedicine. (2023) 113:154746. doi: 10.1016/j.phymed.2023.154746
156. Kuo NC, Huang SY, Yang CY, Shen HH, Lee YM. Involvement of HO-1 and autophagy in the protective effect of magnolol in hepatic steatosis-induced NLRP3 inflammasome activation in vivo and in vitro. Antioxidants (Basel). (2020) 9(10):924. doi: 10.3390/antiox9100924
157. Qi X, Song A, Ma M, Wang P, Zhang X, Lu C, et al. Curcumol inhibits ferritinophagy to restrain hepatocyte senescence through YAP/NCOA4 in non-alcoholic fatty liver disease. Cell Prolif. (2021) 54:e13107. doi: 10.1111/cpr.13107
158. Fan Y, Lu J, Liu J, Zhang R, Yu Z, Guan S. 1,3-dichloro-2-propanol induced hepatic lipid accumulation by inhibiting autophagy via AKT/mTOR/FOXO1 pathway in mice. Food Chem Toxicol. (2021) 157:112578. doi: 10.1016/j.fct.2021.112578
159. Wu Y, Yang Y, Li F, Zou J, Wang YH, Xu MX, et al. Icaritin attenuates lipid accumulation by increasing energy expenditure and autophagy regulated by phosphorylating AMPK. J Clin Transl Hepatol. (2021) 9:373–83. doi: 10.14218/JCTH.2021.00050
160. Zhang X, Huo Z, Luan H, Huang Y, Shen Y, Sheng L, et al. Scutellarin ameliorates hepatic lipid accumulation by enhancing autophagy and suppressing IRE1α/XBP1 pathway. Phytother Res. (2022) 36:433–47. doi: 10.1002/ptr.7344
161. Lai JL, Lian YE, Wu JY, Wang YD, Bai YN. Verapamil induces autophagy to improve liver regeneration in non-alcoholic fatty liver mice. Adipocyte. (2021) 10:532–45. doi: 10.1080/21623945.2021.1983241
162. Xiu AY, Ding Q, Li Z, Zhang CQ. Doxazosin attenuates liver fibrosis by inhibiting autophagy in hepatic stellate cells via activation of the PI3K/Akt/mTOR signaling pathway. Drug Des Devel Ther. (2021) 15:3643–59. doi: 10.2147/DDDT.S317701
163. Meng Z, Liu X, Li T, Fang T, Cheng Y, Han L, et al. The SGLT2 inhibitor empagliflozin negatively regulates IL-17/IL-23 axis-mediated inflammatory responses in T2DM with NAFLD via the AMPK/mTOR/autophagy pathway. Int Immunopharmacol. (2021) 94:107492. doi: 10.1016/j.intimp.2021.107492
164. Alavifard H, Mazhari S, Meyfour A, Tokhanbigli S, Ghavami S, Zali MR, et al. Imatinib suppresses activation of hepatic stellate cells by targeting STAT3/IL-6 pathway through miR-124. Cell Biol Int. (2023) 47:969–80. doi: 10.1002/cbin.11992
165. Park J, Rah SY, An HS, Lee JY, Roh GS, Ryter SW, et al. Metformin-induced TTP mediates communication between Kupffer cells and hepatocytes to alleviate hepatic steatosis by regulating lipophagy and necroptosis. Metabolism. (2023) 141:155516. doi: 10.1016/j.metabol.2023.155516
166. Liu B, Deng X, Jiang Q, Li G, Zhang J, Zhang N, et al. Scoparone improves hepatic inflammation and autophagy in mice with nonalcoholic steatohepatitis by regulating the ROS/P38/Nrf2 axis and PI3K/AKT/mTOR pathway in macrophages. BioMed Pharmacother. (2020) 125:109895. doi: 10.1016/j.biopha.2020.109895
167. Liu B, Deng X, Jiang Q, Li G, Zhang J, Zhang N, et al. Scoparone alleviates inflammation, apoptosis and fibrosis of non-alcoholic steatohepatitis by suppressing the TLR4/NF-κB signaling pathway in mice. Int Immunopharmacol. (2019) 75:105797. doi: 10.1016/j.intimp.2019.105797
168. Wu C, Jing M, Yang L, Jin L, Ding Y, Lu J, et al. Alisol A 24-acetate ameliorates nonalcoholic steatohepatitis by inhibiting oxidative stress and stimulating autophagy through the AMPK/mTOR pathway. Chem Biol Interact. (2018) 291:111–9. doi: 10.1016/j.cbi.2018.06.005
169. Ha KB, Lee ES, Park NW, Jo SH, Shim S, Kim DK, et al. Beneficial effects of a curcumin derivative and transforming growth factor-β Receptor I inhibitor combination on nonalcoholic steatohepatitis. Diabetes Metab J. (2023) 47:500–13. doi: 10.4093/dmj.2022.0110
170. Shu Y, He Y, Ye G, Liu X, Huang J, Zhang Q, et al. Curcumin inhibits the activity and induces apoptosis of activated hepatic stellate cell by suppressing autophagy. J Cell Biochem. (2023) 124:1764–78. doi: 10.1002/jcb.30487
171. Huan S, Sun S, Song S, Dai J, Zhu G, Zhong Y, et al. Dihydroartemisinin inhibits the activation and proliferation of hepatic stellate cells by regulating miR−29b−3p. Int J Mol Med. (2023) 51(5):40. doi: 10.3892/ijmm.2023.5243
172. Fan Y, Dong W, Wang Y, Zhu S, Chai R, Xu Z, et al. Glycyrrhetinic acid regulates impaired macrophage autophagic flux in the treatment of non-alcoholic fatty liver disease. Front Immunol. (2022) 13:959495. doi: 10.3389/fimmu.2022.959495
173. Jin L, Li J, Yang S, Zhang R, Hu C, Chen Y, et al. MAPK p38/Ulk1 pathway inhibits autophagy and induces IL-1β expression in hepatic stellate cells. Am J Physiol Gastrointest Liver Physiol. (2022) 322:G360–g7. doi: 10.1152/ajpgi.00230.2021
174. Duan NN, Liu XJ, Wu J. Palmitic acid elicits hepatic stellate cell activation through inflammasomes and hedgehog signaling. Life Sci. (2017) 176:42–53. doi: 10.1016/j.lfs.2017.03.012
175. Zhang J, Ping J, Jiang N, Xu L. Resveratrol inhibits hepatic stellate cell activation by regulating autophagy and apoptosis through the SIRT1 and JNK signaling pathways. J Food Biochem. (2022) 46:e14463. doi: 10.1111/jfbc.14463
176. Jiang N, Zhang J, Ping J, Xu L. Salvianolic acid B inhibits autophagy and activation of hepatic stellate cells induced by TGF-β1 by downregulating the MAPK pathway. Front Pharmacol. (2022) 13:938856. doi: 10.3389/fphar.2022.938856
177. Qin L, Tan J, Lv X, Zhang J. Vanillic acid alleviates liver fibrosis through inhibiting autophagy in hepatic stellate cells via the MIF/CD74 signaling pathway. BioMed Pharmacother. (2023) 168:115673. doi: 10.1016/j.biopha.2023.115673
178. Wu Y, Yin AH, Sun JT, Xu WH, Zhang CQ. Angiotensin-converting enzyme 2 improves liver fibrosis in mice by regulating autophagy of hepatic stellate cells. World J Gastroenterol. (2023) 29:4975–90. doi: 10.3748/wjg.v29.i33.4975
179. Yao J, Zhao Y. Lp-PLA2 silencing ameliorates inflammation and autophagy in nonalcoholic steatohepatitis through inhibiting the JAK2/STAT3 pathway. PeerJ. (2023) 11:e15639. doi: 10.7717/peerj.15639
180. Jung TW, Kim HC, Abd El-Aty AM, Jeong JH. Maresin 1 attenuates NAFLD by suppression of endoplasmic reticulum stress via AMPK-SERCA2b pathway. J Biol Chem. (2018) 293:3981–8. doi: 10.1074/jbc.RA117.000885
181. Wu H, Chen G, Wang J, Deng M, Yuan F, Gong J. TIM-4 interference in Kupffer cells against CCL4-induced liver fibrosis by mediating Akt1/Mitophagy signalling pathway. Cell Prolif. (2020) 53:e12731. doi: 10.1111/cpr.12731
182. Liu W, Bai F, Wang H, Liang Y, Du X, Liu C, et al. Tim-4 inhibits NLRP3 inflammasome via the LKB1/AMPKα Pathway in macrophages. J Immunol. (2019) 203:990–1000. doi: 10.4049/jimmunol.1900117
183. Fang Y, Ji L, Zhu C, Xiao Y, Zhang J, Lu J, et al. Liraglutide alleviates hepatic steatosis by activating the TFEB-regulated autophagy-lysosomal pathway. Front Cell Dev Biol. (2020) 8:602574. doi: 10.3389/fcell.2020.602574
184. Yoo J, Jeong IK, Ahn KJ, Chung HY, Hwang YC. Fenofibrate, a PPARα agonist, reduces hepatic fat accumulation through the upregulation of TFEB-mediated lipophagy. Metabolism. (2021) 120:154798. doi: 10.1016/j.metabol.2021.154798
185. Du X, Di Malta C, Fang Z, Shen T, Niu X, Chen M, et al. Nuciferine protects against high-fat diet-induced hepatic steatosis and insulin resistance via activating TFEB-mediated autophagy-lysosomal pathway. Acta Pharm Sin B. (2022) 12:2869–86. doi: 10.1016/j.apsb.2021.12.012
186. Zhu X, Xiong T, Liu P, Guo X, Xiao L, Zhou F, et al. Quercetin ameliorates HFD-induced NAFLD by promoting hepatic VLDL assembly and lipophagy via the IRE1a/XBP1s pathway. Food Chem Toxicol. (2018) 114:52–60. doi: 10.1016/j.fct.2018.02.019
187. Zhou W, Yan X, Zhai Y, Liu H, Guan L, Qiao Y, et al. Phillygenin ameliorates nonalcoholic fatty liver disease via TFEB-mediated lysosome biogenesis and lipophagy. Phytomedicine. (2022) 103:154235. doi: 10.1016/j.phymed.2022.154235
188. Liao X, Song L, Zhang L, Wang H, Tong Q, Xu J, et al. LAMP3 regulates hepatic lipid metabolism through activating PI3K/Akt pathway. Mol Cell Endocrinol. (2018) 470:160–7. doi: 10.1016/j.mce.2017.10.010
189. Zheng YY, Wang M, Shu XB, Zheng PY, Ji G. Autophagy activation by Jiang Zhi Granule protects against metabolic stress-induced hepatocyte injury. World J Gastroenterol. (2018) 24:992–1003. doi: 10.3748/wjg.v24.i9.992
190. Zhu X, Yao P, Liu J, Guo X, Jiang C, Tang Y. Baicalein attenuates impairment of hepatic lysosomal acidification induced by high fat diet via maintaining V-ATPase assembly. Food Chem Toxicol. (2020) 136:110990. doi: 10.1016/j.fct.2019.110990
191. Zhang H, Lu J, Liu H, Guan L, Xu S, Wang Z, et al. Ajugol enhances TFEB-mediated lysosome biogenesis and lipophagy to alleviate non-alcoholic fatty liver disease. Pharmacol Res. (2021) 174:105964. doi: 10.1016/j.phrs.2021.105964
192. Byrnes K, Blessinger S, Bailey NT, Scaife R, Liu G, Khambu B. Therapeutic regulation of autophagy in hepatic metabolism. Acta Pharm Sin B. (2022) 12:33–49. doi: 10.1016/j.apsb.2021.07.021
193. Zeng J, Acin-Perez R, Assali EA, Martin A, Brownstein AJ, Petcherski A, et al. Restoration of lysosomal acidification rescues autophagy and metabolic dysfunction in non-alcoholic fatty liver disease. Nat Commun. (2023) 14:2573. doi: 10.1038/s41467-023-38165-6
Keywords: non-alcoholic fatty liver disease, autophagy, autophagosome, autolysosome, hepatocytes, kupffer cells, hepatic stellate cells
Citation: Shen Q, Yang M, Wang S, Chen X, Chen S, Zhang R, Xiong Z and Leng Y (2024) The pivotal role of dysregulated autophagy in the progression of non-alcoholic fatty liver disease. Front. Endocrinol. 15:1374644. doi: 10.3389/fendo.2024.1374644
Received: 23 January 2024; Accepted: 23 July 2024;
Published: 08 August 2024.
Edited by:
Toru Hosoi, Sanyo-Onoda City University, JapanReviewed by:
Sangam Rajak, Sanjay Gandhi Post Graduate Institute of Medical Sciences (SGPGI), IndiaAaron Balasingam Koenig, INOVA Health System, United States
Copyright © 2024 Shen, Yang, Wang, Chen, Chen, Zhang, Xiong and Leng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Leng, bGVuZ3lhbjIyOUAxNjMuY29t