 Dorota Leszczyńska1*
Dorota Leszczyńska1* Alicja Szatko1,2*
Alicja Szatko1,2* Julia Latocha3Magdalena Kochman1Maria Duchnowska1Anna Wójcicka4,5Waldemar Misiorowski1Wojciech Zgliczyníski1
Julia Latocha3Magdalena Kochman1Maria Duchnowska1Anna Wójcicka4,5Waldemar Misiorowski1Wojciech Zgliczyníski1 Piotr Glinicki1,2
Piotr Glinicki1,2- 1Department of Endocrinology, Centre of Postgraduate Medical Education, Warsaw, Poland
- 2EndoLab Laboratory, Centre of Postgraduate Medical Education, Warsaw, Poland
- 3Students’ Scientific Group Affiliated with the Department of Endocrinology, Centre of Postgraduate Medical Education, Warsaw, Poland
- 4Warsaw Genomics, Warsaw, Poland
- 5Fundacja Wiedzieć Więcej, Warsaw, Poland
Introduction: 24-Hydroxylase, encoded by the CYP24A1 gene, is a crucial enzyme involved in the catabolism of vitamin D. Loss-of-function mutations in CYP24A1 result in PTH-independent hypercalcaemia with high levels of 1,25(OH)2D3. The variety of clinical manifestations depends on age, and underlying genetic predisposition mutations can lead to fatal infantile hypercalcaemia among neonates, whereas adult symptoms are usually mild.
Aim of the study: We report a rare case of an adult with primary hyperparathyroidism and loss-of-function mutations in the CYP24A1 gene and a review of similar cases.
Case presentation: We report the case of a 58-year-old woman diagnosed initially with primary hyperparathyroidism. Preoperatively, the suspected mass adjoining the upper pole of the left lobe of the thyroid gland was found via ultrasonography and confirmed by 99mTc scintigraphy and biopsy as the parathyroid gland. The patient underwent parathyroidectomy (a histopathology report revealed parathyroid adenoma), which led to normocalcaemia. After 10 months, vitamin D supplementation was introduced due to deficiency, and the calcium level remained within the reference range. Two years later, biochemical tests showed recurrence of hypercalcaemia with suppressed parathyroid hormone levels and elevated 1,25(OH)2D3 concentrations. Further investigation excluded the most common causes of PTH-independent hypercalcaemia, such as granulomatous disease, malignancy, and vitamin D intoxication. Subsequently, vitamin D metabolites were measured using LC–MS/MS, which revealed high levels of 25(OH)D3, low levels of 24,25(OH)2D3 and elevated 25(OH)2D3/24,25(OH)2D3 ratios, suggesting a defect in vitamin D catabolism. Molecular analysis of the CYP24A1 gene using the NGS technique revealed two pathogenic variants: p.(Arg396Trp) and p.(Glu143del) (rs114368325 and rs777676129, respectively).
Conclusions: The diagnostic process for hypercalcaemia becomes complicated when multiple causes of hypercalcaemia coexist. The measurement of vitamin D metabolites using LC–MS/MS may help to identify carriers of CYP24A1 mutations. Subsequent molecular testing may contribute to establishing the exact frequency of pathogenic variants of the CYP24A1 gene and introducing personalized treatment.
Introduction
The active form of vitamin D (1,25(OH)2D3 or calcitriol), in addition to parathyroid hormone (PTH) and fibroblast growth factor 23 (FGF23), is critical for maintaining calcium haemostasis (1). Primary hyperparathyroidism (PHPT) and malignancy are the most common causes of hypercalcaemia. Among the less frequent aetiologies, vitamin D-mediated hypercalcaemia should be considered.
The main source of vitamin D is skin synthesis from 7-dehydrocholestrol (7-DHC); however, it can also be obtained from the diet. For full hormonal activity, vitamin D3 requires two-stage hydroxylation. First, 25 hydroxylase generates 25(OH)D3 in the liver. The second hydroxylation by 1-alpha-hydroxylase (CYP27B1, Cytochrome P450 Family 27 Subfamily B Member 1) leads to the conversion of 25(OH)D3 to the biologically active metabolite of vitamin D (1,25(OH)2D3) (2). This process occurs mainly in the kidneys, but CYP27B1 is also expressed at several external sites (3). 1,25(OH)2D3 and 25(OH)D3 are inactivated by 24-hydroxylase (24HD) (cytochrome P450 family 24 subfamily a member 1 (CYP24A1)), the key enzymes involved in vitamin D catabolism. The direct products of the CYP24A1 reaction are 24,25(OH)2D3 and 1,24,25(OH)2D3, which are further converted to calcitroic acid destined for biliary excretion (4) (Figure 1). The activity of CYP24A1 is regulated by numerous biochemical factors. Hypocalcaemia and PTH downregulate CYP24A1 expression and enzymatic activity (5). The opposite stimulating effect is caused by 1,25(OH)2D3 and FGF23, together with its coreceptor α-Klotho protein (5).
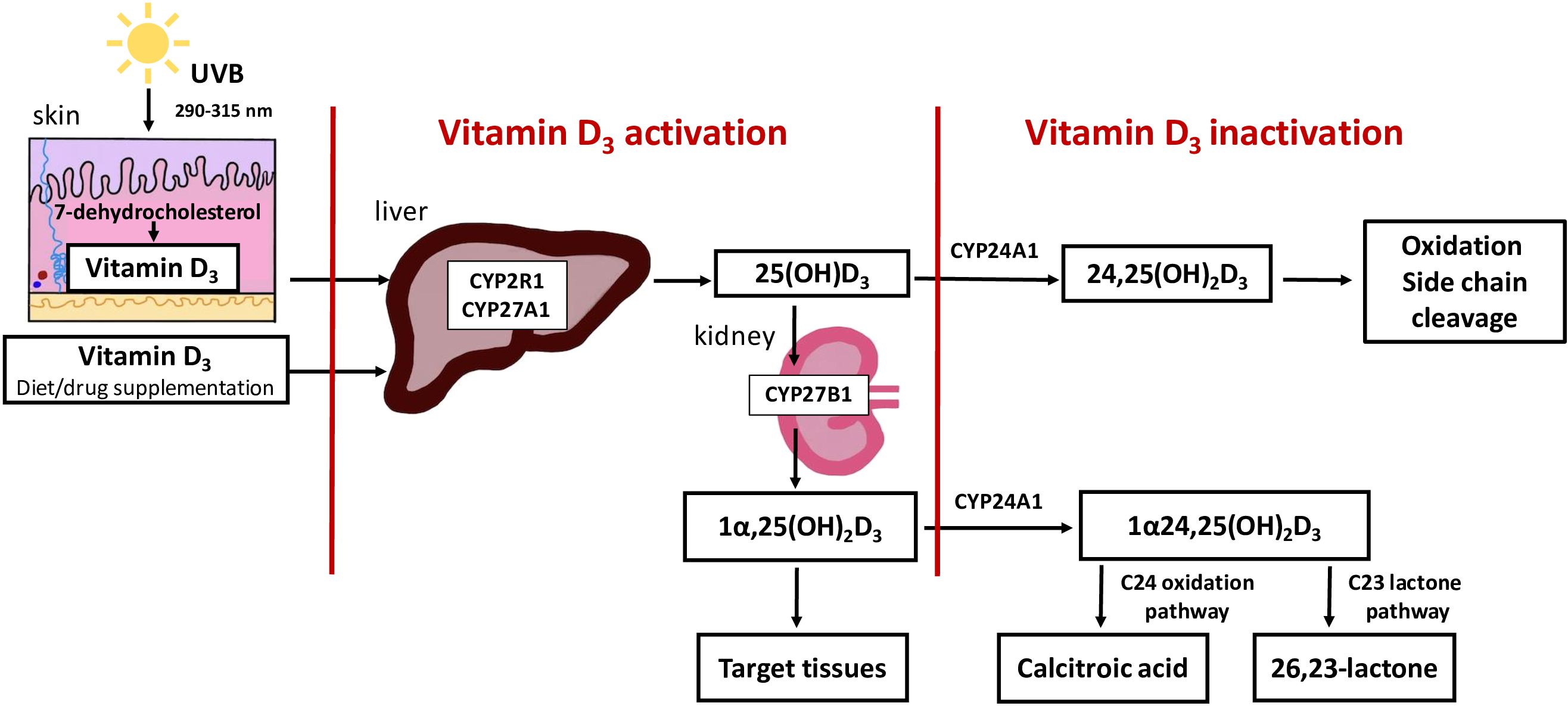
Figure 1 Overview of vitamin D synthesis, activation and inactivation. The main source of vitamin D is skin synthesis from 7-dehydrocholestrol (7-DHC), however, it can also be obtained from the diet. For full hormonal activity, vitamin D3, requires two-stage hydroxylation. First, 25 hydroxylase generates 25(OH)D3 in the liver. The second hydroxylation by 1-alpha-hydroxylase (CYP2781, Cytochrome P450 Family 27 Subfamily B Member 1) leads to the conversion of 25(OH)D3 to the biologically active metabolite of vitamin D (1,25(OH)2D3). This process occurs mainly in the kidneys. 1,25(OH)2 D3 and 25(OH)D3 are inactivated by 24-hydroxylase (CYP24A1, cytochrome P450 family 24 subfamily a member 1) the key enzymes involved in vitamin D catabolism. The direct products of the CYP24A1 reaction are 24,25(OH)2D3 and 1,24,25(OH)2D3, which are further converted to calcitroic acid or a 26,23- lactone derivative.
The concentration of 25(OH)D, the best indicator of vitamin D status, can be measured by numerous diagnostic methods. The following immunoassays were used: chemiluminescence immunoassay (CLIA), electrochemiluminescence immunoassay (ECLIA), vitamin D binding protein (DBP-based assays), high-performance liquid chromatography (HPLC) and liquid chromatography with tandem mass spectrometry (LC−MS/MS). However, the ‘gold standard’ and reference technique that allows for the most reliable assessment of many vitamin D metabolites simultaneously is LC–MS/MS (6).
Excessive vitamin D or calcitriol ingestion, ectopic production of 1,25(OH)2D3 in granulomatous or lymphoproliferative disease and genetically determined dysregulation of vitamin D metabolism may lead to PTH-independent hypercalcaemia (7). There are two known underlying genetic mechanisms resulting from either mutations in CYP24A1encoding 24HD leading to an inability to deactivate 25(OH)D3 and 1,25(OH)2D3 or in the sodium phosphate cotransporter IIa gene (SLC34A1, Solute Carrier Family 27 Subfamily B Member 1), resulting in excessive 1,25(OH)2D3 biosynthesis secondary to chronic phosphate wasting (8). Both genetic disorders cause idiopathic infantile hypercalcaemia (HCINF), which is associated with hypersensitivity to vitamin D, especially in patients receiving vitamin D supplementation. In patients with CYP24A1 mutations, laboratory tests revealed an increased 25(OH)D3/24,25(OH)2D3 ratio. This investigation has been recommended as a screening tool whenever genetic causes of hypercalcaemia are considered (9).
Aim of the study
The aim of this study was to determine the importance of determining vitamin D metabolites in the differential diagnosis of hypercalcaemia and the possibility of co-occurring various causes of hypercalcaemia. In this study, we present a rare case of an adult patient with PHPT and hypercalcaemia caused by two heterozygous pathogenic CYP24A1 variants (confirmed by next-generation sequencing [NGS]) with a review of reported cases of patients with hypercalcaemia associated with pathogenic variants of CYP24A1 and concomitant PHPT.
Case presentation
Clinical data
A 58-year-old woman was referred to the Endocrinology Unit in 2019 due to incidental findings of hypercalcaemia. Her medical history included hypertension, stage 3a chronic kidney disease (CKD), and carpal tunnel syndrome treated surgically. She had no family history of endocrine diseases or nephrolithiasis.
Biochemical tests
A laboratory test revealed hypercalcaemia, hypercalciuria, and unsuppressed PTH levels (27 pg/mL, reference range, 15-65 pg/mL) (Table 1; Figure 2).
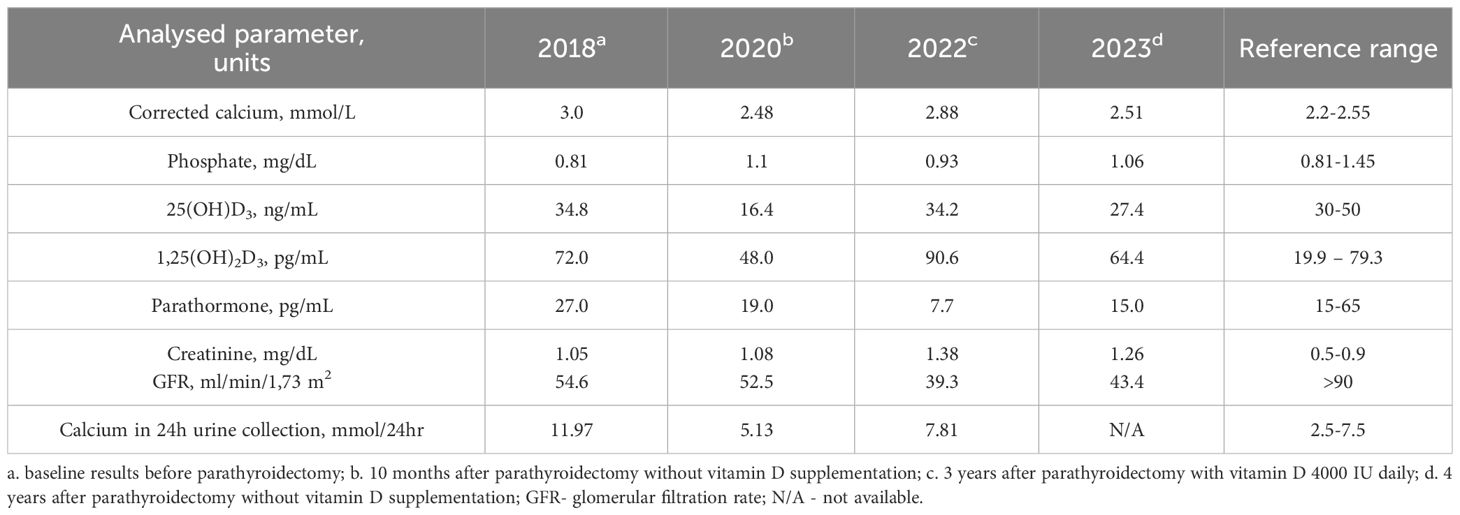
Table 1 Laboratory results presented in chronological order.
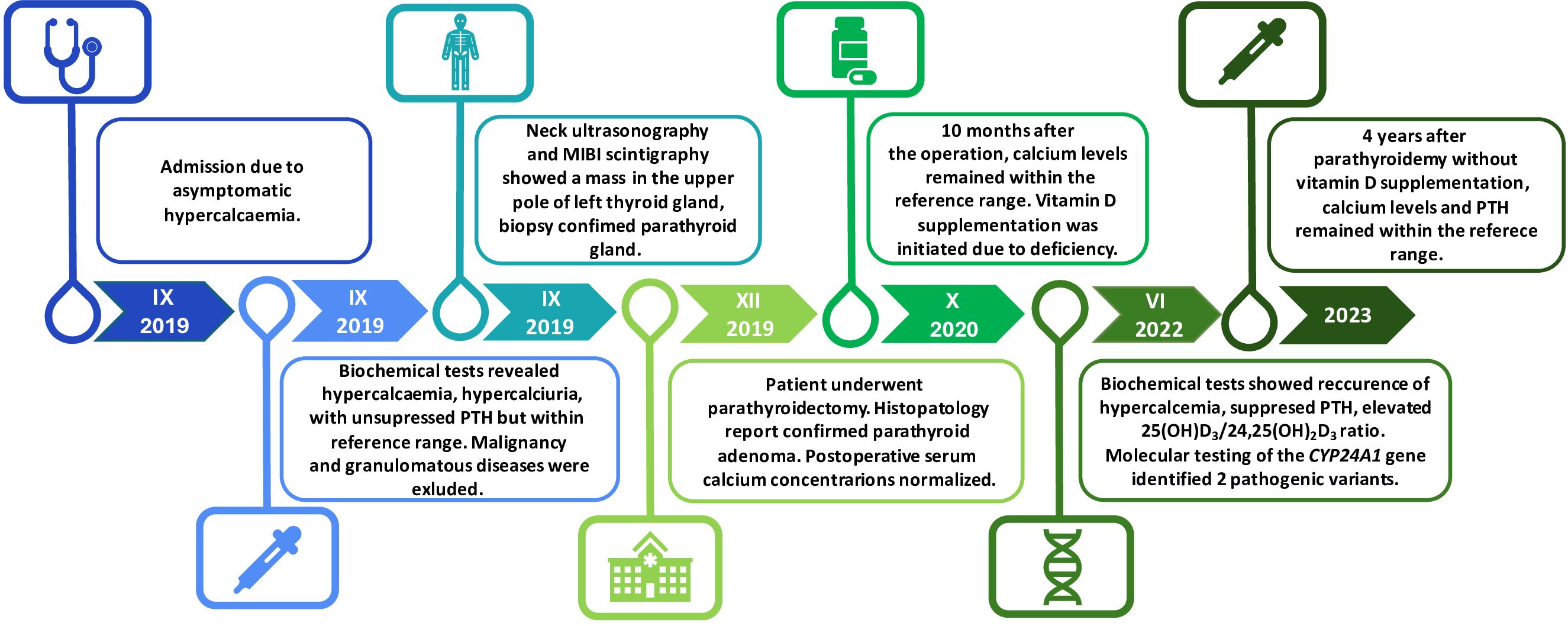
Figure 2 Timeline summarization of the patient’s clinical course. Asymptomatic hypercalcaemia was incidentally found in 2019. The same year, primary hyperparathyroidism (PHPT) was diagnosed, and the presence of suspected lesion was confirmed in the imaging studies. The patient underwent parathyroidectomy which led to normocaicaemia. After 10 months, vitamin D supplementation was initiated due to deficiency. In 2022, biochemical tests revealed recurrence of hypercalcaemia with suppressed PTH concentration. After the exclusion of the common causes of PTH-independent hypercalceamia, the presence of two heterozygous loss-of-function mutations in the CYP24A1 gene was confirmed in Next-Generation Sequencing (NGS). After discontinuation of vitamin D supplementation, calcium and PTH concentrations normalized. PTH-parathormone; MIBI -technetium-99m-sestamibi scintigraphy; CYP24A1- cytochrome P450 family 24 subfamily a member 1.
Imaging
Computed tomography (CT) scans of the chest, abdomen and pelvis were conducted and showed no signs of malignancy or granulomatous disease.
Neck ultrasonography and technetium 99m sestamibi (MIBI) scintigraphy revealed a mass in the upper pole of the left thyroid gland lobe, which was confirmed to be the parathyroid gland after biopsy via immunohistochemical staining (Figure 2).
There were no abnormalities in the densitometry results (T score of the lumbar spine 1.1; T score of the proximal femur -0.2; T score of the radius 0.5). However, an X-ray revealed advanced subperiosteal bone resorption in the fingers and bone loss in the thoracic spine and clavicles. There were no renal stones or fractures.
Treatment and outcomes
The patient underwent effective parathyroidectomy, after which the calcium level was normalized (2.45 mmol/L, reference range, 2.2-2.55 mmol/L). Histopathology confirmed parathyroid adenoma.
Ten months after the operation, postoperative assessment revealed a normal calcium concentration (2.48 mmol/L), but vitamin D deficiency (16.4 ng/mL, reference range, 30-50 ng/mL); thus, supplementation with cholecalciferol was administered (4000 IU/daily) (Table 1; Figure 2). The patient did not attend her follow-up appointments for two years. In 2022, a laboratory test showed hypercalcaemia and hypercalciuria recurrence, which was initially identified as recurrent primary hyperparathyroidism. However, blood tests revealed PTH suppression with elevated 1,25(OH)2D3 concentrations (Table 1; Figure 2).
The patient was screened for granulomatous disease and hypercalcaemia of malignancy again, and the results were negative.
Vitamin D metabolites were measured using LC–MS/MS, which revealed high 25(OH)D3 (72.62 ng/mL) and low 24,25(OH)2D3 (0.09 ng/mL) concentrations and an elevated 25(OH)D3/24,25(OH)2D3 ratio 806,9 (reference range, 7.0-23.6), suggesting a defect in vitamin D catabolism.
Molecular testing
The genetic testing of the CYP24A1 gene was conducted using the NGS technique, and two pathogenic variants were identified NM_000782.5:c; 1186C>T(p.Arg396Trp, rs114368325) and NM_000782.5:c; 428_430del (p.Glu143del, rs777676129). Both variants are classified as pathogenic/likely pathogenic, and associated with hypercalcemia in the available databases, including ClinVar NIH (ClinVar archives, National Institutes of Health). Due to the unavailability of family members, it was impossible to assess whether the variants were located in the same of different alleles of CYP24A1, what would be a direct proof of their dominant or recessive involvement in development of the observed symptoms.
The recommendation was to discontinue vitamin D supplementation, maintain adequate hydration and avoid excessive sunlight exposure. Follow-up evaluation showed normalization of calcium and PTH concentrations (Table 1).
Discussion
Hypercalcaemia is a common clinical abnormality. PTH measurement is the key initial test for differentiating between PTH-dependent and PTH-independent hypercalcaemia.
A high or inappropriately normal PTH level in relation to a high calcium concentration is typical for PTH-dependent causes. Among them, the most common is PHPT, whose reported incidence varies between 0.2% and 1.3% across the population (10). In PTH-independent hypercalcaemia, the PTH level is appropriately suppressed for hypercalcaemia status. Malignancy is the most common disorder in this population. Among other causes of PTH-independent hypercalcaemia, vitamin D-mediated hypercalcaemia should be considered.
Currently, in many countries where the evaluation of serum calcium levels has become routine, PHPT appears to be the most common cause of hypercalcaemia and is identified most frequently in the initial asymptomatic stage (11). A growing prevalence of PHPT results in the identification of unusual cases of overlapping PTH-dependent and PTH-independent causes of hypercalcaemia. We described a woman with asymptomatic mild hypercalcaemia and ambiguous low-normal PTH, which made the initial classification of hypercalcaemia difficult. For this reason, in the differentiation, we considered both PTH-dependent and PTH-independent causes of hypercalcaemia. After excluding malignancy and granulomatous diseases, PHTP was diagnosed based on imaging studies and parathyroid biopsy. The patient underwent successful parathyroidectomy. Only the recurrence of PTH-independent hypercalcaemia after vitamin D supplementation revealed the other cause of hypercalcaemia, which turned out to be a genetic disorder of vitamin D catabolism.
Pathogenic mutations in the CYP24A1 gene clinically manifest as infantile hypercalcaemia-1 (HCINF1, OMIM 143880), a rare disorder linked with a disturbance in vitamin D degradation (12). HCINF1 is classically characterized by severe symptoms such as vomiting, polysomnia, hypotonia, constipation, failure to thrive, and renal stone disease (13). The first case reports of HCINF1 were published in the 1950s, when approximately 200 cases of PTH-independent hypercalcaemia in infants were noted in the United Kingdom because of the wide use of formula milk enriched with increased doses of vitamin D (up to 4000 International Units (IU)) (14, 15). Although the majority of affected children developed mild hypercalcaemia not associated with other syndromic features (HCINF1), the percentage of fatal cases was remarkable, and some affected children had multisystem disorders, which were later described as Willams–Beuren syndrome (OMIM 194050) (12, 16). At that time, vitamin D dietary intake was identified as a precipitating factor of HCINF1, yet the exact pathogenesis has remained unknown (12, 14).
Only recently, the structure of the CYP24A1 gene and the pathways involved in vitamin D metabolism leading to HCINF1 were revealed. In 1991, Ohyama et al. described the isolation of complementary deoxyribonucleic acid (cDNA) from a rat kidney cDNA library utilizing antibodies specific for the enzyme (17). Two years later, Chen et al. published a paper on the isolation, sequencing and expression of cDNA encoding the human 24HD (18). In the same year, Hahn et al. independently isolated human kidney cDNA encoding vitamin D 24HD (19). In 2011, Schlingmann et al. confirmed that HCINF1 is mutated in CYP24A1 based on 8 cases of infants receiving vitamin D supplementation (either daily or bolus doses) (12). The affected children were homozygotes or compound heterozygotes for nonsense or missense mutations of CYP24A1 (inherited as autosomal recessive trait), leading to complete loss of function of 24HD confirmed in the transfected eukaryotic cell line (12). Since then, multiple pathogenic variants (PVs) in the CYP24A1 gene have been identified, revealing that the spectrum of biallelic variants (either homozygous or compound heterozygous) ranges from severe hypercalcaemia in infants to mild hypercalcaemia in adults (20–23). Heterozygotes usually present with milder or asymptomatic phenotypes (24). However, in the case of a 44-year-old patient with intermittent hypercalcaemia and two intron−exon splice junction mutations (IVS5 + 1G>A and IVS6–2A>G) of CYP24A1, an analysis of family members suggested autosomal dominant inheritance with partial penetrance (25). Furthermore, the clinical phenotype is dependent not only on the PV but also on vitamin D intake, sunlight exposure or pregnancy (12, 26, 27). It is known that 1,25(OH)2D3 is elevated during normal pregnancy, which additionally promotes hypercalcaemia in women with disturbed calcitriol catabolism associated with CYP24A1 mutation (5, 28).
Hypercalcaemia related to the presence of PVs in the CYP24A1 gene is often overlooked. Although epidemiological data concerning the prevalence of PVs of the CYP24A1 gene are scarce, the estimated frequency of deleterious minor alleles is 0.140 (29). Thus, the calculated frequency (using the Hardy−Weinberg equilibrium) of recessive disorders is 1960 per 100.000 people (29). By adding patients harbouring monoallelic mutations who may present with a clinical phenotype, the number of affected individuals is significant, and this should be considered more often in the differential diagnosis of hypercalcaemia (30, 31).
In 2016, an autosomal recessive loss-of-function mutation in the SLC34A1 gene was identified as the second cause of infantile hypercalcaemia-2 (HCINF2, OMIM 616963) (32). Like in patients with CYP24A1 mutations, hypercalcaemia, suppressed PTH and inappropriately high 1,25(OH)2D3 are observed. The distinguishing feature is hypophosphatemia due to renal phosphate wasting. (32)
Low PTH and high 1,25(OH)2D3 levels are specific to the whole group of endogenous causes of vitamin D related to hypercalcaemia (including HCINF, granulomatous disease and some lymphomas); therefore, 24,25(OH)2D3 assessment is crucial for differentiation. The distinguishing feature of HCINF1 is the very low concentration of 24,25(OH)2D3. Therefore, the measurement of this metabolite is the first screening tool for HCINF1, allowing to select subjects for further genetic tests. However, in healthy individuals, there is a positive linear correlation between the concentration of 25(OH)D3 and 24,25(OH)2D3, which results in physiological inhibition of the production of 24,25(OH)2D3 when the serum 25OHD3 concentration falls into the vitamin D deficiency range (33). For this reason, a more accurate parameter for expressing the absence of 24,25(OH)2D3 in HCINF1 patients is the ratio of 25(OH)D3 to 24,25(OH)2D3, especially because some of these patients have a low vitamin D status (34). The technique allowing a reliable assessment of 24,25(OH)2D3 is LC−MS/MS. Chromatographic separation is the reference method that allows for the precise resolution of 24,25(OH)2D3 from other vitamin D metabolites. Using the LC−MS/MS method involving derivatization with DMEQ-TAD {4-[2-(6,7-dimethoxy-4-methyl-3,4-dihydroquinoxalinyl)ethyl]-1,2,4-triazoline-3,5-dione}, a ratio greater than 80 (normal ratio, 5 to 25) allows for the identification of HCINF1 due to the CYP24A1 mutation (34). Using extended chromatography, which resolves 24,25(OH)2D3, 25,26-(OH)2D3 and 1,25(OH)2D3, a ratio > 140 was used as the cut-off value (35).
To date, 6 cases of primary hyperparathyroidism coexisting with a CYP24A1 mutation have been reported in 5 publications (Table 2) (36–40). The diagnosis of PHPT was made due to elevated serum calcium and inadequate nonsuppressed PTH. In most of these patients, parathyroidectomy was initially performed following the diagnosis of primary hyperparathyroidism, and after surgery, due to the persistence of hypercalcaemia, a diagnosis of 24-hydroxylase deficiency was established. Pathological examination revealed hyperplasia in the parathyroid glands in 2 patients after total or partial parathyroidectomy (37, 38). For the other subjects, adenomas were found; in one patient, a complete histopathology report was unavailable (36, 37, 39, 40). In all patients, removal of autonomous parathyroid tissue resulted in a significant decrease in the serum calcium concentration. PTH reduction may have additional value for individuals with a CYP24A1 mutation because it eliminates the continuous stimulation of 1,25(OH)2D3 synthesis, whose catabolism is disrupted. In the present case, the relatively low concentration of PTH (27 pg/mL) before parathyroidectomy was noteworthy. Primary hyperparathyroidism is characterized by elevated calcium concentrations and a lack of portal feedback between calcium concentration and PTH secretion. Usually, the PTH level is significantly elevated but can also be within the normal range. In both situations, PTH levels are clearly inadequate for elevating serum calcium levels. However, well-documented PHPT has been reported with PTH levels as low as 20 pg/mL to 25 pg/mL, given a reference range of 10 pg/mL to 65 pg/mL (11).
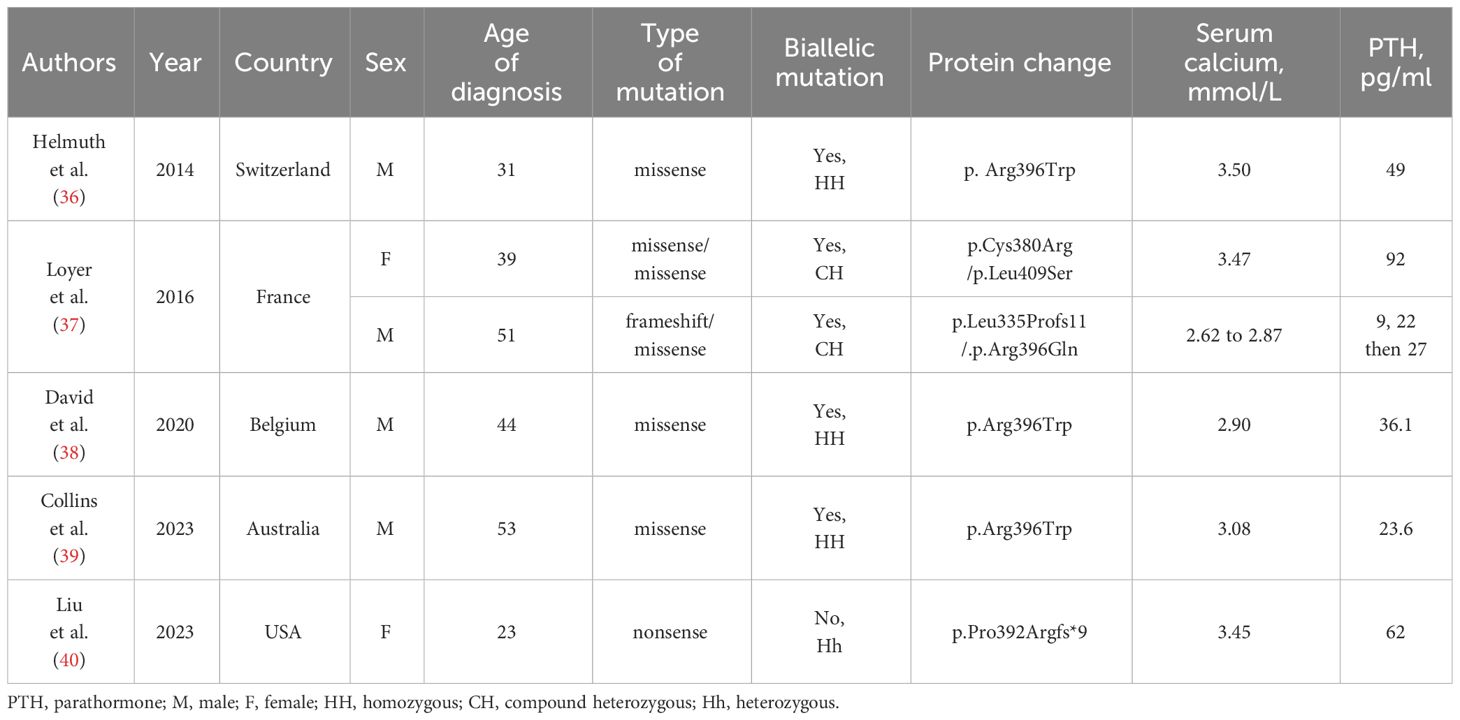
Table 2 The summary of reported cases of concomitant primary hyperparathyroidism and mutations in the CYP24A1 (human cytochrome P450 24 subfamily A member 1) gene leading to impaired function of 24-hydroxylas.
Vitamin D and PTH are conjugated: 1,25(OH)2D3 reduces PTH directly by inhibiting its transcription and indirectly by increasing calcium absorption from the gastrointestinal tract (41). Furthermore, calcitriol induces the expression of FGF23 in bones, and FGF23 decreases PTH secretion and PTH mRNA levels (42). (42). On the other hand, PTH is the main stimulator of 1,25(OH)2D3 synthesis in the kidneys (43).
As shown in a systematic review and meta-analysis by Song et al., vitamin D supplementation in patients with PHPT and vitamin D deficiency significantly reduces PTH without causing hypercalcaemia or hypercalciuria (44). The coexistence of 24-hydroxylase insufficiency and the consequent extraordinarily high 1,25(OH)2D3 level may intensify this process. The observed concentrations of PTH in earlier described patients with HPT coexisting with CYP24A1 mutations differed greatly from each other. Loyer et al. described 2 patients with inappropriate/high PTH levels (22 pg/mL to 92 pg/mL) (37). In the case described by Helmuth et al., the preoperative PTH concentration was 58 pg/mL (36). Another patient had PTH ranging between 36.6 pg/mL and 80.3 pg/mL during the year preceding parathyroid surgery (38). In a recent case, low-normal PTH concentrations (23.6 pg/mL) before and after the first partial parathyroidectomy were observed, and after the second parathyroid surgery, PTH became undetectable (39). The only known patient with a heterozygous pathogenic variant in CYP24A1 had a preoperative PTH level of 63 pg/mL (40).
It is unclear whether the co-occurrence of PHPT with a CYP24A1 mutation in these patients is a unique coincidence or is a new phenotype of CYP24A1 mutation combined with hyperparathyroidism. The biochemical profile of patients with 24-hydroxylase deficiency-hypercalcaemia and especially high 1,25(OH)2D3 decreases not only PTH secretion but also parathyroid cell proliferation. Calcitriol reduces parathyroid volume through the suppression of the cell cycle regulator c-Myc, the suppression of transforming growth factor-α (TGF-α), and the induction of p21 (p21 protein), which is an inhibitor of the cell cycle (45). The opposite condition in which parathyroid hyperplasia occurs as a result of hyperphosphatemia, calcitriol deficiency, or hypocalcaemia is renal failure. In our patient, as well as in the other 5 described patients with the CYP24A1 mutation and PHPT, mild renal failure was noted; however, the phosphate concentration remained low (36–39).
In most of the discussed cases, the 25(OH)D3:24.25(OH)2D3 ratio was determined before genetic testing (37, 38, 40). Except for one patient with a heterozygous pathogenic variant in CYP24A1 (25(OH)D3:24.25(OH)2D3 ratio of 25.18), the ratios were above 100 (37, 38, 40).
In the first reported case, a homozygous loss-of-function mutation (p.Arg396Trp) in the gene encoding vitamin D 24-hydroxylase was identified; second, the most prevalent mutation in the CYP24A1 gene was found in patients with HCINF1 (5, 30, 36). In patients with the p.Arg396Trp mutation, the arginine-to-tryptophane substitution leads to complete loss of catabolic activity of 24-hydroxylase due to destruction of the hydrogen bonds between arginine and the haem propionate group, thus blocking transient but crucial 24-hydroxylase substrate binding to haem (46, 47). Interestingly, the homozygous p.Arg396Trp mutation was also found in the cases described by David et al. and Collins et al. (38, 39). In most of the reported cases, the genetic panel was extended by the analysis of the genes associated with hyperparathyroidism, i.e., MEN1 (encoding the tumour suppressor protein menin; MEN1, Multiple Endocrine Neoplasia type 1), CaSR (encoding the G protein-coupled extracellular calcium-sensing receptor), HRPT2 (encoding the tumour suppressor parafibromin) and CDK (encoding the tumour suppressor cyclin-dependent kinase) (37, 48, 49). In the case reported by David et al., CaSR and MEN1 mutations, which are underlying causes of hyperparathyroidism, were excluded (38). In the case described by Collins et al., genetic testing confirmed that the patient was homozygous for the pathogenic variant c.1186C>T, p.Arg396Trp in the CYP24A1 gene (39). (39) Documented cases of primary hyperparathyroidism and missense p.Arg396Trp mutation raise the question of whether the described genetic alteration predisposes patients to parathyroid autonomy. The link between loss-of-function CYP24A1 mutations and the development of primary hyperparathyroidism has not been confirmed (50). In the present case, a heterozygous missense variant (rs114368325; p.Arg396Trp) was also identified, together with the second pathogenic variant. In this study, we were unable to confirm the heterozygosity or compound heterozygosity of both variants; therefore, we cannot discuss the recessive or dominant inheritance of the disease.
The coexistence of parathyroid autonomy and pathogenic variants in the CYP24A1 gene is not associated only with p.Arg396Trp mutations. Loyer et al. reported two cases of concomitant PTH-dependent hypercalcaemia and deleterious compound heterozygous mutations in the gene encoding vitamin D 24-hydroxylase (37). In the first case, a 39-year-old female with persistent hypercalcaemia after the removal of hyperplastic parathyroid fluid, p.Cys380Arg/p. Leu409Ser compound heterozygous mutations were identified (37). Interestingly, in the same patient, an intronic HRPT2 gene polymorphism (c.1418-17C>G) was found (37). In the second case, genetic testing confirmed compound heterozygous (p.Leu335Profs11/.p.Arg396Gln) mutations in the CYP24A1 gene, intronic polymorphisms (c.237 + 28 T>C) in the HRPT2 gene and the p.Gln1011Glu variant in the CaSR gene (37). Hypercalcaemia due to a novel heterozygous pathogenic variant (p.Pro392Argfs*9) in the CYP24A1 gene and concomitant primary hyperparathyroidism were documented in the case of a 23-year-old female by Liu et al. (40).
In PHPT, the only curative treatment is parathyroidectomy. In patients with proven CYP24A1 mutations, management should concentrate on eliminating or reducing hypercalcaemia and hypercalciuria. Thus, the basis of long-term strategies is a low vitamin D and calcium diet, in which vitamin D supplementation is omitted and sunlight exposure is avoided. In patients for whom this approach is not sufficient, various pharmacologic therapies have been described. Glucocorticoids that reduce enteral calcium absorption and inhibit the conversion of serum 25(OH)D3 to active 1,25(OH)2D3 were found to be ineffective in patients with CYP24A1 defects (7, 51). However, effective therapeutic outcomes have been described in patients treated with imidazoles, such as ketoconazole or less toxic fluconazole, which reduce 1,25(OH)2D3 synthesis by inhibiting CYP enzymes (cytochrome enzymes) (25, 52).
The limitation of this study was the unavailability of patient family members, which allowed us to clearly determine whether the variants were located cis or trans within the CYP24A1 gene and therefore how their concurrence affected the clinical outcome of the patient.
Conclusions
The present case underlines the importance of accurate clinical evaluation of hypercalcaemia. Rarely may multiple causes of hypercalcaemia coexist, which complicates the diagnostic process. Identification of such conditions often requires a wider range of diagnostic techniques. In patients with PTH-independent hypercalcaemia, the measurement of vitamin D metabolites using LC–MS/MS analytical technique, followed by genetic testing (e.g., NGS technique), may help to identify carriers of CYP24A1 mutations.
Patient perspective
Currently, the patient reports an improvement in her wellbeing. The patient is pleased that the present treatment was limited by adequate hydration, the avoidance of sun exposure and the lack of vitamin D supplementation.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding authors.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
DL: Project administration, Methodology, Writing – review & editing, Writing – original draft, Investigation, Data curation, Conceptualization. AS: Writing – review & editing, Writing – original draft, Investigation, Data curation, Conceptualization. JL: Writing – review & editing, Writing – original draft, Investigation, Data curation. MK: Writing – review & editing, Writing – original draft, Investigation, Data curation. MD: Writing – review & editing, Writing – original draft, Investigation, Data curation. AW: Writing – review & editing, Writing – original draft, Methodology, Investigation, Data curation. WM: Writing – review & editing, Writing – original draft, Supervision. WZ: Validation, Writing – review & editing, Writing – original draft, Supervision, Conceptualization. PG: Validation, Project administration, Writing – review & editing, Writing – original draft, Supervision, Methodology, Investigation, Conceptualization.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Goltzman D. Pathophysiology of hypercalcemia. Endocrinol Metab Clinics North America. (2021) 50:591–607. doi: 10.1016/j.ecl.2021.07.008
2. Christakos S, Ajibade DV, Dhawan P, Fechner AJ, Mady LJ. Vitamin D: metabolism. Endocrinol Metab Clinics North America. (2010) 39:243–53. doi: 10.1016/j.ecl.2010.02.002
3. Bikle D. Nonclassic actions of vitamin D. J Clin Endocrinol Metab. (2009) 94:26–34. doi: 10.1210/jc.2008-1454
4. Prosser DE, Jones G. Enzymes involved in the activation and inactivation of vitamin D. Trends Biochem Sci. (2004) 29:664–73. doi: 10.1016/j.tibs.2004.10.005
5. De Paolis E, Scaglione GL, De Bonis M, Minucci A, Capoluongo E. CYP24A1 and SLC34A1 genetic defects associated with idiopathic infantile hypercalcemia: From genotype to phenotype. Clin Chem Lab Med (CCLM). (2019) 57:1650–67. doi: 10.1515/cclm-2018-1208
6. Saponaro F, Saba A, Frascarelli S, Prontera C, Clerico A, Scalese M, et al. Vitamin D measurement and effect on outcome in a cohort of patients with heart failure. Endocrine Connections. (2018) 7:957–64. doi: 10.1530/ec-18-0207
7. Schlingmann KP. Vitamin D–dependent hypercalcemia. Endocrinol Metab Clinics North America. (2021) 50:729–42. doi: 10.1016/j.ecl.2021.08.005
8. Kaufmann M, Schlingmann KP, Berezin L, Molin A, Sheftel J, Vig M, et al. Differential diagnosis of vitamin D related hypercalcemia using serum vitamin D metabolite profiling. J Bone Mineral Res. (2021) 36(7):1340–50. doi: 10.1002/jbmr.4306
9. Dirks NF, Ackermans MT, de Jonge R, Heijboer AC. Reference values for 24,25-dihydroxyvitamin D and the 25-hydroxyvitamin D/24,25-dihydroxyvitamin D ratio. Clin Chem Lab Med. (2019) 57:e259–61. doi: 10.1515/cclm-2018-1096
10. Soto-Pedre E, Newey PJ, Leese GP. Stable incidence and increasing prevalence of primary hyperparathyroidism in a population-based study in scotland. J Clin Endocrinol Metab. (2023) 108(10):e1117–e1124. doi: 10.1210/clinem/dgad201
11. Bilezikian JP. Primary hyperparathyroidism. J Clin Endocrinol Metab. (2018) 103:3993–4004. doi: 10.1210/jc.2018-01225
12. Schlingmann KP, Kaufmann M, Weber S, Irwin A, Goos C, John U. Mutations in CYP24A1 and idiopathic infantile hypercalcemia. New Engl J Med. (2011) 365:410–21. doi: 10.1056/NEJMoa1103864
13. Marks BE, Doyle DA. Idiopathic infantile hypercalcemia: Case report and review of the literature. J Pediatr Endocrinol Metab. (2016) 29. doi: 10.1515/jpem-2015-0133
14. Group BMJP. Hypercalcaemia in infants and vitamin D. Br Med J. (1956) 2:149–9. doi: 10.1136/bmj.2.4985.149
15. Lightwood R, Stapleton T. Idiopathic hypercalcæmia in infants. Lancet. (1953) 262:255–6. doi: 10.1016/s0140-6736(53)90187-1
16. Pober BR. Williams–beuren syndrome. New Engl J Med. (2010) 362:239–52. doi: 10.1056/nejmra0903074
17. Ohyama Y, Noshiro M, Okuda K. Cloning and expression of cDNA encoding 25-hydroxyvitamin D3 24-hydroxylase. FEBS Lett. (1991) 278:195–8. doi: 10.1016/0014-5793(91)80115-J
18. Chen K-S, Prahl JM, DeLuca HF. Isolation and expression of human 1,25-dihydroxyvitamin D3 24-hydroxylase cDNA. Proc Natl Acad Sci United States America. (1993) 90:4543–7. doi: 10.1073/pnas.90.10.4543
19. Hahn C, Baker E, Laslo P, May BK, Omdahl J, Sutherland GR. Localization of the human vitamin D 24-hydroxylase gene (CYP24) to chromosome 20q13.2→q13.3. Cytogenetic Genome Res. (1993) 62:192–3. doi: 10.1159/000133473
20. Jones G, Kottler ML, Schlingmann KP. Genetic diseases of vitamin D metabolizing enzymes. Endocrinol Metab Clinics North America. (2017) 46:1095–117. doi: 10.1016/j.ecl.2017.07.011
21. Carpenter TO. CYP24A1 loss of function: Clinical phenotype of monoallelic and biallelic mutations. J Steroid Biochem Mol Biol. (2017) 173:337–40. doi: 10.1016/j.jsbmb.2017.01.006
22. Cappellani D, Brancatella A, Kaufmann M, Minucci A, Vignali E, Canale D, et al. Hereditary hypercalcemia caused by a homozygous pathogenic variant in the CYP24A1 gene: a case report and review of the literature. Case Rep Endocrinol. (2019) 2019:4982621. doi: 10.1155/2019/4982621
23. Tebben PJ, Singh RJ, Kumar R. Vitamin D-mediated hypercalcemia: Mechanisms, diagnosis, and treatment. Endocrine Rev. (2016) 37:521–47. doi: 10.1210/er.2016-1070
24. Cools M, Goemaere S, Baetens D, Raes A, Desloovere A, Kaufman JM. Calcium and Bone Homeostasis in Heterozygous Carriers of CYP24A1 mutations: a cross-sectional Study. Bone. (2015) 81:89–96. doi: 10.1016/j.bone.2015.06.018
25. Tebben PJ, Milliner DS, Horst RL, Harris PC, Singh RJ, Wu Y. Hypercalcemia, hypercalciuria, and elevated calcitriol concentrations with autosomal dominant transmission due toCYP24A1Mutations: Effects of ketoconazole therapy. J Clin Endocrinol Metab. (2012) 97:E423–7. doi: 10.1210/jc.2011-1935
26. Figueres M-L, Linglart A, Bienaime F, Allain-Launay E, Roussey-Kessler G, Ryckewaert A. Kidney function and influence of sunlight exposure in patients with impaired 24-hydroxylation of vitamin D due to CYP24A1 mutations. Am J Kidney Diseases: Off J Natl Kidney Foundation. (2015) 65:122–6. doi: 10.1053/j.ajkd.2014.06.037
27. Shah A, Hsiao EC, O’Donnell B, Salmeen K, Nussbaum RL, Krebs M. Maternal hypercalcemia due to failure of 1,25-dihydroxyvitamin-D3Catabolism in a patient withCYP24A1Mutations. J Clin Endocrinol Metab. (2015) 100:2832–6. doi: 10.1210/jc.2015-1973
28. Kumar R, Cohen WR, Lisbôa C, Epstein FH. Elevated 1,25-dihydroxyvitamin D plasma levels in normal human pregnancy and lactation. J Clin Invest. (1979) 63:342–4. doi: 10.1172/jci109308
29. Nesterova G, Malicdan MC, Yasuda K, Sakaki T, Vilboux T, Ciccone C. 1,25-(OH)2D-24 hydroxylase (CYP24A1) deficiency as a cause of nephrolithiasis. Clin J Am Soc Nephrol CJASN. (2013) 8:649–57. doi: 10.2215/CJN.05360512
30. Cappellani D, Brancatella A, Morganti R, Borsari S, Baldinotti F, Caligo MA. Hypercalcemia due to CYP24A1 mutations: A systematic descriptive review. Eur J Endocrinol. (2021) 186:137–49. doi: 10.1530/eje-21-0713
31. Brancatella A, Cappellani D, Kaufmann M, Borsari S, Piaggi P, Baldinotti F. Do the heterozygous carriers of a CYP24A1 mutation display a different biochemical phenotype than wild types? J Clin Endocrinol Metab. (2020) 106:708–17. doi: 10.1210/clinem/dgaa876
32. Schlingmann KP, Ruminska J, Kaufmann M, Dursun I, Patti M, Kranz B. Autosomal-recessive mutations in SLC34A1 encoding sodium-phosphate cotransporter 2A cause idiopathic infantile hypercalcemia. J Am Soc Nephrol. (2016) 27:604–14. doi: 10.1681/asn.2014101025
33. Leszczyńska D, Szatko A, Raczkiewicz D, Ostrowska M, Misiorowski W, Zgliczyński W. The effect of a single high dose of vitamin D on serum levels of its metabolites in the elderly. Front Bioscience. (2022) 27:289–9. doi: 10.31083/j.fbl2710289
34. Kaufmann M, Gallagher JC, Peacock M, Schlingmann K-P, Konrad M, DeLuca HF. Clinical utility of simultaneous quantitation of 25-hydroxyvitamin D and 24,25-dihydroxyvitamin D by LC-MS/MS involving derivatization with DMEQ-TAD. J Clin Endocrinol Metab. (2014) 99:2567–74. doi: 10.1210/jc.2013-4388
35. Kaufmann M, Morse N, Molloy BJ, Cooper DP, Schlingmann KP, Molin A. Improved screening test for idiopathic infantile hypercalcemia confirms residual levels of serum 24,25-(OH) 2 D 3 in affected patients. J Bone Mineral Res. (2017) 32:1589–96. doi: 10.1002/jbmr.3135
36. Helmuth A, Konrad M, Schlingmann KP, Pasch A. The case | Hypercalcemia in a 60-year-old male. Kidney Int. (2014) 85:219–21. doi: 10.1038/ki.2013.184
37. Loyer C, Leroy C, Molin A, Odou MF, Huglo D, Lion G. Hyperparathyroidism complicating CYP 24A1 mutations. Annales d’Endocrinologie. (2016) 77:615–9. doi: 10.1016/j.ando.2016.03.002
38. David K, Khalil R, Hannon H, Evenepoel P, Decallonne B. Therapy-resistant hypercalcemia in a patient with inactivating CYP24A1 mutation and recurrent nephrolithiasis: Beware of concomitant hyperparathyroidism. Calcified Tissue Int. (2020) 107:524–8. doi: 10.1007/s00223-020-00738-8
39. Collins L, Boehm E, Luxford C, Clifton-Bligh R, Grill V. Hypervitaminosis D secondary to a CYP24A1 loss-of-function mutation: An unusual cause of hypercalcemia in two siblings. JBMR plus. (2023) 7(9):e10788. doi: 10.1002/jbm4.10788
40. Liu J, Angelos P, Barhoum M, Jain R. Severe hypercalcemia due to primary hyperparathyroidism and heterozygous pathogenic variant of CYP24A1. JCEM Case Rep. (2023) 1(4):luad071. doi: 10.1210/jcemcr/luad071
41. Bienaimé F, Prié D, Friedlander G, Souberbielle JC. Vitamin D metabolism and activity in the parathyroid gland. Mol Cell Endocrinol. (2011) 347:30–41. doi: 10.1016/j.mce.2011.05.031
42. Martin A, David V, Quarles LD. Regulation and function of the FGF23/klotho endocrine pathways. Physiol Rev. (2012) 92:131–55. doi: 10.1152/physrev.00002.2011
43. Saliba W, Barnett O, Rennert HS, Lavi I, Rennert G. The relationship between serum 25(OH)D and parathyroid hormone levels. Am J Med. (2011) 124:1165–70. doi: 10.1016/j.amjmed.2011.07.009
44. Song A, Zhao H, Yang Y, Liu S, Nie M, Wang O, et al. Safety and efficacy of common vitamin D supplementation in primary hyperparathyroidism and coexistent vitamin D deficiency and insufficiency: a systematic review and meta-analysis. J Endocrinological Invest. (2021) 44(8):1667–77. doi: 10.1007/s40618-020-01473-5
45. Mizobuchi M, Ogata H, Koiwa F. Secondary hyperparathyroidism: pathogenesis and latest treatment. Ther Apheresis Dialysis. (2018) 23:309–18. doi: 10.1111/1744-9987.12772
46. Hong-Fang J, Shen L. CYP24A1Mutations in idiopathic infantile hypercalcemia. New Engl J Med. (2011) 365:1741–3. doi: 10.1056/nejmc1110226
47. Jones G, Prosser DE, Kaufmann M. Cytochrome P450-mediated metabolism of vitamin D. J Lipid Res. (2013) 55:13–31. doi: 10.1194/jlr.r031534
48. Falchetti A, Marini F, Luzi E, Giusti F, Cavalli L, Cavalli T. Multiple endocrine neoplasia type 1 (MEN1): Not only inherited endocrine tumors. Genet Med. (2009) 11:825–35. doi: 10.1097/gim.0b013e3181be5c97
49. Hannan FM, Thakker RV. Calcium-sensing receptor (CaSR) mutations and disorders of calcium, electrolyte and water metabolism. Best Pract Res Clin Endocrinol Metab. (2013) 27:359–71. doi: 10.1016/j.beem.2013.04.007
50. Chorti A, Cheva A, Chatzikyriakidou A, Achilla C, Boulogeorgou K, Despoina K. Sporadic parathyroid adenoma: an updated review of molecular genetics. Front Endocrinol. (2023) 14:2023.1180211. doi: 10.3389/fendo.2023.1180211
51. Colussi G, Ganon L, Penco S, De Ferrari ME, Ravera F, Querques M. Chronic hypercalcaemia from inactivating mutations of vitamin D 24-hydroxylase (CYP24A1): implications for mineral metabolism changes in chronic renal failure. Nephrology Dialysis Transplantation: Off Publ Eur Dialysis Transplant Assoc - Eur Renal Assoc. (2014) 29:636–43. doi: 10.1093/ndt/gft460
Keywords: hypercalcaemia, CYP24A1 gene, vitamin D, primary hyperparathyroidism, 25(OH)D3, 1,25(OH)2D3, 24,25(OH)2D3, 24-hydroxylase deficiency
Citation: Leszczyńska D, Szatko A, Latocha J, Kochman M, Duchnowska M, Wójcicka A, Misiorowski W, Zgliczyníski W and Glinicki P (2024) Persistent hypercalcaemia associated with two pathogenic variants in the CYP24A1 gene and a parathyroid adenoma—a case report and review. Front. Endocrinol. 15:1355916. doi: 10.3389/fendo.2024.1355916
Received: 03 January 2024; Accepted: 19 March 2024;
Published: 11 April 2024.
Edited by:
Giacomina Brunetti, University of Bari Aldo Moro, ItalyReviewed by:
Khashayar Sakhaee, University of Texas Southwestern Medical Centre, United StatesEjigayehu Gigi Abate, Mayo Clinic, United States
Copyright © 2024 Leszczyńska, Szatko, Latocha, Kochman, Duchnowska, Wójcicka, Misiorowski, Zgliczyníski and Glinicki. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dorota Leszczyńska, ZC5sZXN6Y3p5bnNrYUB0bGVuLnBs; Alicja Szatko, YWxpY2phLnN6YXRrb0BnbWFpbC5jb20=