 Francesca Orsolini1*
Francesca Orsolini1* Luisa Pignata1
Luisa Pignata1 Fulvia Baldinotti2Silvia Romano3Massimo Tonacchera1Domenico Canale1
Fulvia Baldinotti2Silvia Romano3Massimo Tonacchera1Domenico Canale1- 1Department of Clinical and Experimental Medicine, Endocrine Unit, University of Pisa, Pisa, Italy
- 2Department of Laboratory Medicine, Section of Molecular Genetics, Pisa University Hospital, Pisa, Italy
- 3Departmental Section of Medical Genetics, Pisa University Hospital, Pisa, Italy
Noonan syndrome (NS) is a genetic disorder characterized by multiple congenital defects caused by mutations in the RAS/mitogen-activated protein kinase pathway. Male fertility has been reported to be impaired in NS, but only a few studies have focused on fertility status in NS patients and underlying mechanisms are still incompletely understood. We describe the case of a 35-year-old man who underwent an andrological evaluation due to erectile dysfunction and severe oligospermia. A syndromic facial appearance and reduced testis size were present on clinical examination. Hormonal evaluation showed normal total testosterone level, high FSH level, and low–normal AMH and inhibin B, compatible with primary Sertoli cell dysfunction. Genetic analysis demonstrated the pathogenetic heterozygous variant c.742G>A, p.(Gly248Arg) of the LZTR1 gene (NM_006767.3). This case report provides increased knowledge on primary gonadal dysfunction in men with NS and enriches the clinical spectrum of NS from a rare variant in the novel gene LZTR1.
Introduction
Noonan syndrome (NS) is a relatively common genetic disorder characterized by multiple congenital defects with an estimated incidence of one in 1,000 to 2,500 live births (1). It has been classically considered an autosomal dominant disorder with variable expressivity, but most cases are sporadic (de novo), and recently, cases with autosomal recessive inheritance have been described (2, 3). It is caused by variants in the RAS/mitogen-activated protein kinase (MAPK) pathway involved in cell proliferation, differentiation, and survival (4). Approximately 50% of individuals with NS have a pathogenic missense variant in the PTPN11 gene, which encodes the non-receptor protein tyrosine phosphatase SHP-2. In the remaining cases (25%–30% of the cases), other RAS/MAPK genes, such as KRAS, SOS1, RAF1, KRAS, NRAS, BRAF, and MAP2K1, are implicated, and during the last years, novel genes have been identified in association with NS, including RIT1, RRAS, RASA2, A2ML1, SOS2, and LZTR1 (4, 5).
Characteristic dysmorphic facial appearance is evident in children and evolves with age, becoming quite undetectable in adulthood (6, 7), such that mildly affected cases may be misdiagnosed. More than 80% of affected individuals have congenital heart defects, particularly pulmonary valve stenosis, often of mild entity, followed by atrial septal defects and hypertrophic cardiomyopathy (8). Birth weight and length are normal, although more than half of individuals with NS experienced short stature during childhood because of apparent aberrations in growth hormone secretion/responsiveness (7). Puberty can be delayed with the mean age of pubertal onset of 14 years in boys and 13.5 years in girls, but it generally starts spontaneously (9–12). Despite the lack of evidence, female gonadal function seems not to be affected in NS. In male individuals, cryptorchidism is common, occurring in up to 80% of the cases (13), and male fertility has been reported to be decreased with oligospermia or non-obstructive azoospermia (14–16). Several previous studies recognized cryptorchidism as the main factor contributing to infertility (13, 15, 17, 18). However, recent evidence suggests that it could be the result of primary gonadal dysgenesis occurring during fetal development with intrinsic Sertoli cell defects with/without Leydig cell impairment (9, 11, 19). However, only a few studies have focused on gonadal function and fertility in NS patients.
In this article, we described a case of male infertility in a patient with NS due to a pathogenic variant of the LZTR1 gene.
Case description
A 35-year-old man was admitted to the Section of Andrology of Pisa University Hospital in September 2022 because of erectile dysfunction. He had a stable relationship with a female partner for 5 years. Sexual desire was present and spontaneous nocturnal/morning erections were reported. He declared that he has not used medications or smoked and did not engage in physical activity. The patient had no children.
He was born from a spontaneous pregnancy. His birth weight and length are normal, but he had a short stature in childhood. Thus, he underwent treatment with recombinant human growth hormone from 5 to 16 years old. The patient had a spontaneous onset of puberty at 14 years of age.
He suffered from congenital pulmonary valve stenosis, atrial septal defects, and mitral valve prolapse of mild entity, requiring periodic follow-up. Electrocardiography showed left-axis deviation and incomplete right bundle branch block.
Medical history also revealed incisor agenesis, chronic bilateral deep venous insufficiency, surgical correction of the right inguinal hernia at the age of 32 years, and removal of three nevi.
His elder sister was stillborn, and his parents died at the age of 52 and 64 years because of breast cancer and bowel infarction, respectively.
On clinical evaluation (Figure 1), the following data were recorded: height 1.73 m, weight 60 kg, and BMI 20 kg/m2. Multiple nevi were present. His facial features were characterized as follows: triangle-shaped head with a tall forehead and high anterior hairline, bilateral palpebral ptosis, low-set and posteriorly rotated ears, and short neck with low posterior hairline. He also had chest deformity (superior pectus carinatum and inferior pectus excavatum) with widely spaced and low-set nipples. Genital examinations showed reduced testis size with a volume of 10 ml (Prader orchidometer).
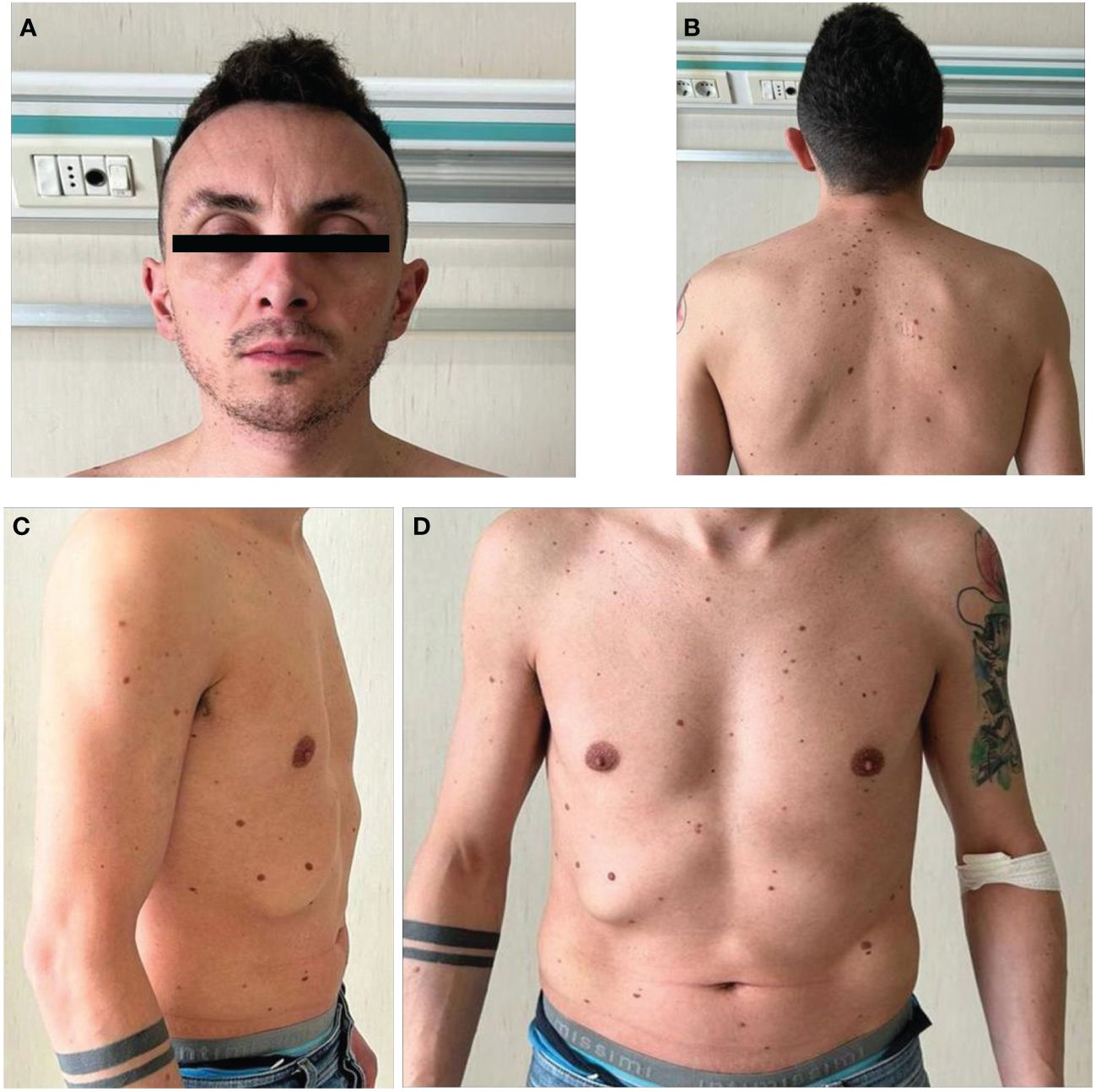
Figure 1 Facial and body appearance of the patient. Physical examination showed multiple nevi (B–D), a triangle-shaped head (A) with a tall forehead and high anterior hairline, bilateral palpebral ptosis, low-set and posteriorly rotated ears, short neck with low posterior hairline (B). The patient also showed chest deformity (superior pectus carinatum and inferior pectus excavatum) with widely spaced and low-set nipples (C, D).
Recent hormonal evaluation showed a normal total testosterone level [22.2 nmol/L, normal range (n.r.) 8.7–29.1] with high serum FSH (19.3 IU/L, n.r. 1.5–12.4). Semen analysis (Table 1) documented severe oligospermia (sperm concentration 1.5 × 106 per ml; total sperm number 6.2 × 106 per ejaculate).
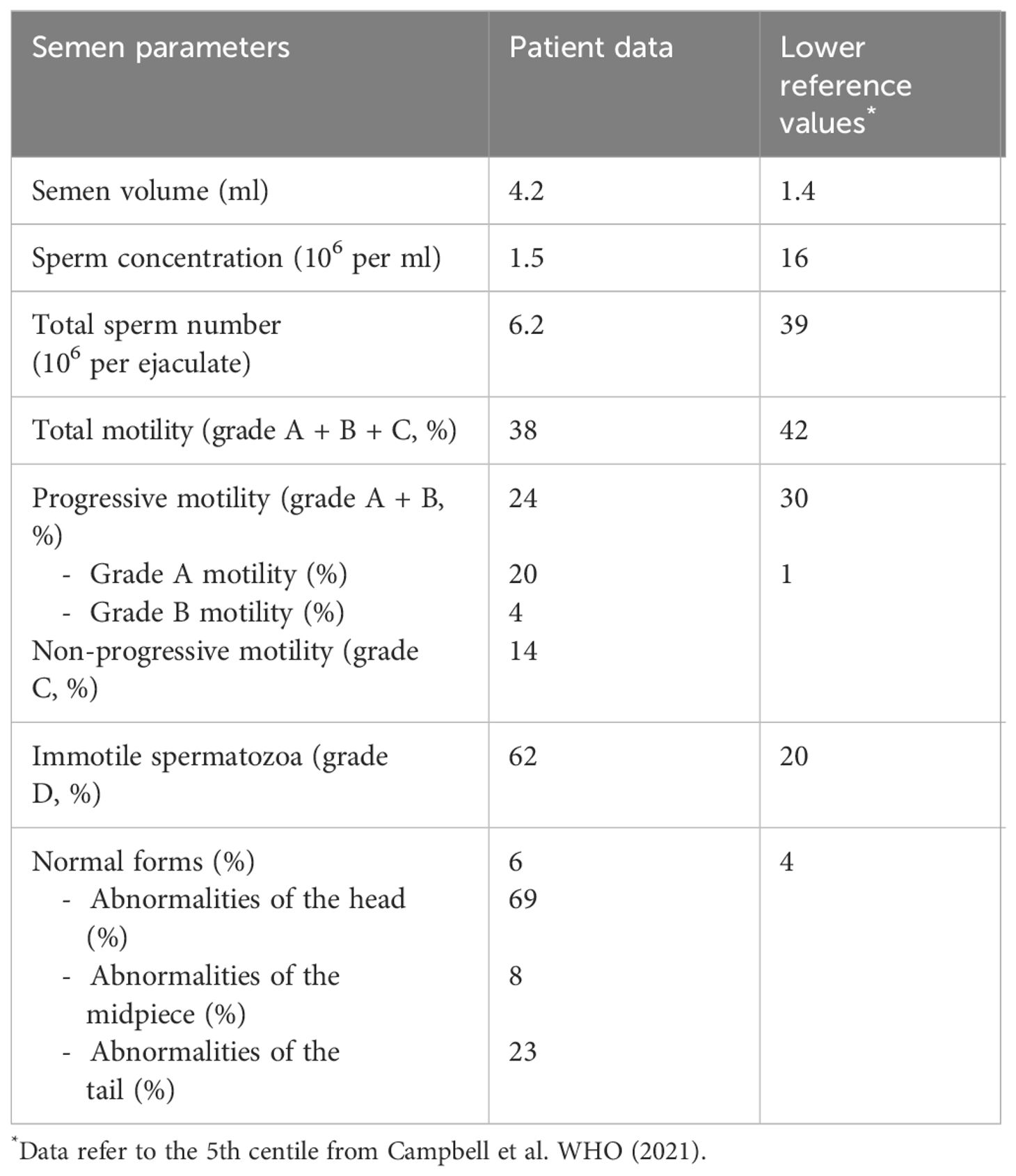
Table 1 Semen analysis (performed after 3 days of sexual abstinence).
We repeated hormonal analysis that confirmed normal total testosterone level (19 nmol/L, n.r. 6.2–27), high FSH level (20.7 IU/L, n.r. 1.3–19.5), and normal LH level (4.2 IU/L, n.r. 1.4–12.7). AMH and inhibin B were low–normal (AMH 1.4 µg/L, n.r. 0.73–16; inhibin B 31 pg/ml, n.r. 25–325). Thyroid function and prolactin were normal. Insulin-like growth factor 1 was in the normal range (138.5 µg/L, n.r. 85–256). Laboratory results are reported in Table 2. Baseline coagulation screening was normal.
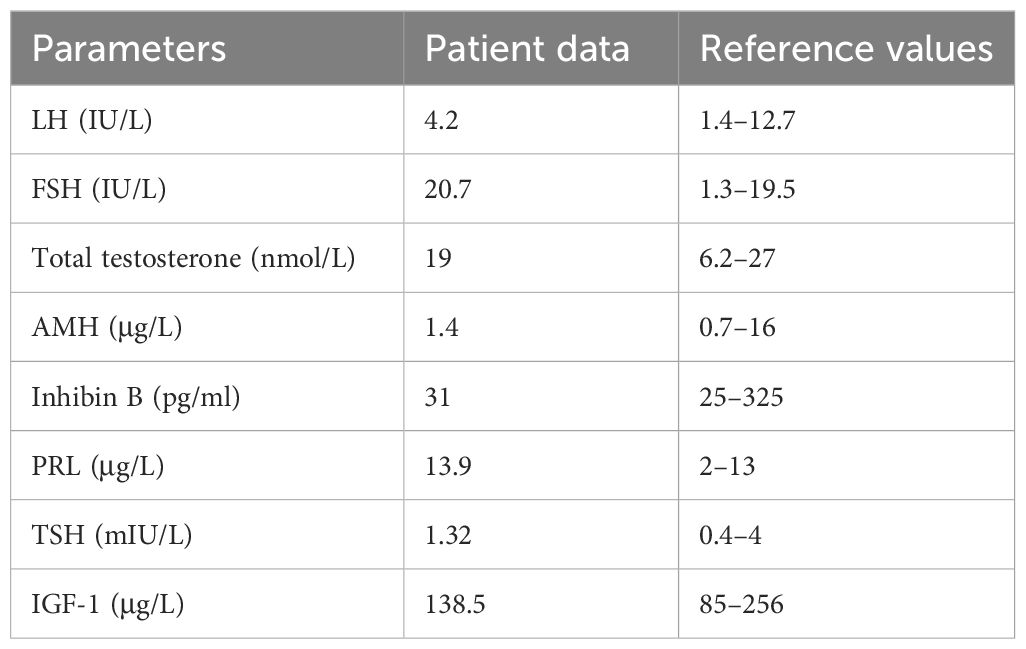
Table 2 Laboratory results.
Testicular ultrasound confirmed reduced testes volume (right testis volume 9 cc and left testis volume 6 cc) without focal lesions. Abdomen ultrasonography excluded hepatosplenomegaly or renal anomalies.
We performed next-generation sequencing (NGS) of the genes involved in the RAS/MAPK pathway (the custom panel having 19 genes including the coding sequence and the donor and acceptor splice sites: PTPN11, SOS1, SOS2, RAF1, BRAF, CBL, KRAS, NRAS, HRAS, RIT1, LZTR1, SPRED1, SHOC2, MAP2K1, MAP2K2, MRAS, PPP1CB, RASA2, RRAS2). NGS was performed through Nextera Flex IDT-custom panel (Illumina-IDT) in paired-end mode 2 × 150 using the MiSeq (Illumina) platform. Sequencing analysis demonstrated the presence of the pathogenic heterozygous variant c.742G>A, p.(Gly248Arg) of the LZTR1 gene (NM_006767.3). The variant was validated by Sanger sequencing (Figure 2). The karyotype was normal (46,XY), and no Y chromosome microdeletions in azoospermia factor (AZF) regions were found.
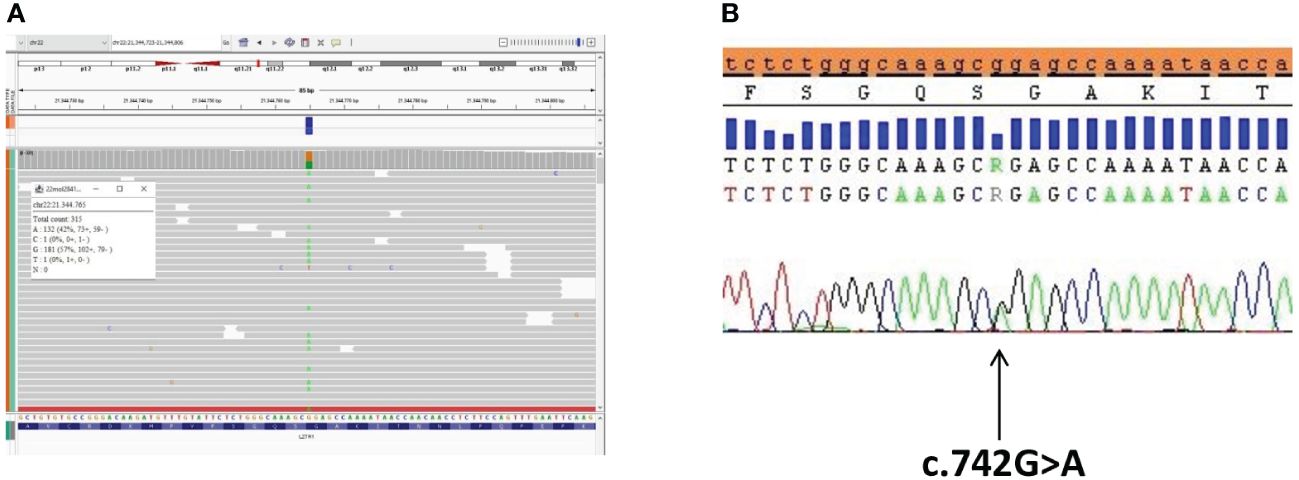
Figure 2 The LZTR1 variant identified by exome sequencing analysis and confirmation via Sanger sequencing. (A) The LZTR1 variant visualized by the Integrative Genomics Viewer (IGV) (reference base G in orange, variant base A in green) in the proband. (B) Sanger sequencing analysis of the LZTR1 variant visualized by SeqScape in the proband.
Patient consent
Written informed consent for the publication of the article and accompanying clinical images was obtained from the patient.
Discussion
NS is a complex and relatively common disorder that affects multiple organ systems with variable severity, caused by variants in the RAS/MAPK pathway. The phenotype is well characterized in childhood, with distinctive facial features and organ abnormalities; however, it is more subtle in adulthood which is often misdiagnosed or not detected by clinicians.
In this article, we reported the case of a 35-year-old man with no previous diagnosis of NS, who was referred to us because of andrological concern (erectile dysfunction and severe oligospermia). The other typical abnormalities associated (mainly congenital pulmonary valve stenosis and other cardiac anomalies and a history of short stature) allowed us to suppose that it was an NS, which was subsequently confirmed by genetic analysis. Clinical features were very mild but still visible. Typical ECG pattern (20), dental anomalies (incisor agenesis), multiple lentigines and nevi, and chronic bilateral deep venous insufficiency supported the diagnosis. Andrological history/examination revealed delayed puberty from 14 years with low testes volume and previous surgical correction of inguinal hernia. The patient’s biochemical profile showed high serum FSH levels associated with low–normal AMH and inhibin B values, compatible with primary Sertoli cell defects. No cryptorchidism was reported at birth, and the levels of testosterone were normal, reflecting normal Leydig cell function.
Gonadal dysfunction and subfertility are rarely investigated in these subjects, and the underlying mechanisms are still incompletely understood. Several articles report that gonadal dysfunction in NS is secondary to cryptorchidism (which is present at birth in the majority of cases) and consequent testes damage (13, 15, 17, 18, 21). However, recent evidence suggests that gonadal dysfunction is a feature of the syndrome and is rather related to abnormal testes development (9, 11, 19, 22). Gonadal impairment can also affect NS individuals with normal testicular descent (19, 22, 23). Moreover, genital tract defects other than cryptorchidism, such as hypoplastic external genitalia, penile hypospadias, and inguinal hernia, have been reported in NS individuals (24–26). Primary gonadal dysfunction is supported by the demonstration that the PTPN11 gene (which variant is the most implicated in the syndrome) is expressed in germ cells and Sertoli cells and has a pivotal role in the regulation of spermatogenesis (27–29). As for other clinical manifestations of NS, as well as for Sertoli cell dysfunction, a genotype/phenotype correlation has been observed (9). Aside from patients with the PTPN11 variant, significantly low levels of Sertoli cell markers (AMH and inhibin B) were found by Moniez et al. in two patients with RAF1 variants (23), while Marcus et al. showed Sertoli cell dysfunction (raised FSH level in combination with low inhibin B concentration) in one BRAF patient with normal testicular descent (22). SHOC2 and KRAS variants were associated with delayed puberty and/or low testicular volume in the study of Patti et al., but no information on fertility was available in these patients (11). In the other cases of male gonadal dysfunction, genetic characterization was lacking or only the PTPN11 gene was tested.
In our case report, we identified the pathogenetic heterozygous variant c.742G>A, p.(Gly248Arg) of the leucine-zipper-like transcriptional regulator 1 (LZTR1) gene [a rare variant that accounts for approximately 2%–3% of NS cases (30)].
The LZTR1 gene is a tumor suppressor that encodes a protein member of the BTB-kelch superfamily, located within the 3-Mb-long region. Its function is lacking, although Nacak et al. (31) characterized the LZTR1 protein on the cellular level as a BTB-kelch protein that exclusively localizes to the Golgi complex (maybe to stabilize it). The LZTR1 protein has been only recently associated with the RAS–MAPK signaling pathway; Motta et al. (32) proposed a model in which LZTR1 negatively modulates this pathway; consequently, pathogenetic variants that inactivate LZTR1 result in a RAS–MAPK signaling upregulation. LZTR1 variants can be associated with either the dominant or recessive form of NS; the variants associated with dominant NS are located in the highly conserved kelch domain, while the variants associated with recessive forms have been found throughout the protein (2, 3, 33).
The detected variant c.742G>A, p.(Gly248Arg) of the LZTR1 gene results in a non-conservative amino acid change located in the Kelch repeat type 1 of the encoded protein sequence and has been reported in the literature in individuals affected with NS (including familial and de-novo cases). Yamamoto et al. (34) identified the variant in a cohort of NS probands with typical facial features (downslanting palpebral fissures, hypertelorism, ptosis, short/webbed neck) and cardiac abnormalities (pulmonary stenosis), but not short stature and cognitive disabilities; furthermore, one individual of the cohort developed multiple schwannomas. Chinton et al. (33) detected the same variant in five NS patients with heart defects, neurodevelopmental delay, and short stature and described acute lymphoblastic leukemia in one subject. We had no notice about gonadal function and fertility status in these cohorts of NS patients. Umeki et al. (25) reported the variant in a child with NS who showed pulmonary stenosis, anomalous origin of the coronary artery, and a concealed penis but no cryptorchidism. Finally, Güemes (35) identified the variant, which appeared de novo, in a boy with typical dysmorphic features, cardiac abnormalities, and bilateral cryptorchidism. Table 3 summarizes the clinical manifestations of the reported cases of NS from the heterozygous LZTR1 variant c.742G>A, p.(Gly248Arg).
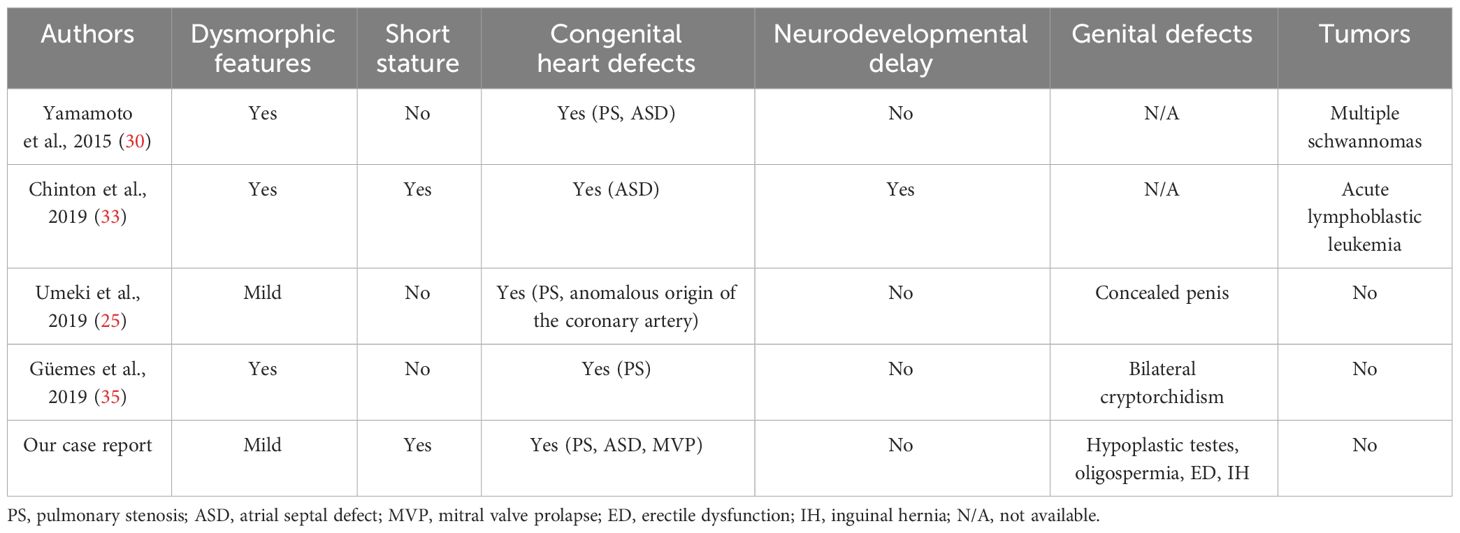
Table 3 List of case reports of male individuals with the heterozygous LZTR1 variant c.742G>A, p.(Gly248Arg): clinical findings.
Hence, although gonadal function was not available in most of the cases, we could suppose that, similar to PTPN11, also the LZTR1 gene has a role in testicular development and spermatogenesis. More studies are necessary to define the exact role of the LZTR1 gene in germ cell development. However, fertility status assessment should be performed in NS individuals with LZTR1 variants, and andrological evaluation should be offered to the affected individuals. Furthermore, acting like a tumor-suppressor gene, NS individuals with LZTR1 variants need a more careful follow-up because of the potential higher risk of malignant tumors.
In conclusion, NS is a relatively common disorder, and the characteristic features, although subtle in adulthood, should induce clinicians to suspect the syndrome and to propose a genetic test. Variants in the LZTR1 gene are rare, and their manifestation is still not clearly delineated because of the few reported cases. This case report enriches the clinical spectrum of LZTR1 variants that seem to be associated with the complete phenotype of the syndrome (facial dysmorphology, cardiac defects, short stature), including male semen abnormalities. Finally, it provides increased knowledge on reproductive disorders affecting male NS individuals, focusing on primary gonadal dysfunction and subfertility as part of the syndrome.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any identifiable images or data included in this article.
Author contributions
FO: Writing – original draft, Methodology, Investigation, Data curation, Conceptualization. LP: Writing – original draft. FB: Writing – review & editing, Visualization, Data curation, Conceptualization. SR: Writing – review & editing, Visualization. MT: Writing – review & editing, Visualization, Supervision. DC: Writing – review & editing, Visualization, Validation, Supervision.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Mendez HMM, Opitz JM. Noonan syndrome: A review. Am J Med Genet. (1985) 21:493–506. doi: 10.1002/ajmg.1320210312
2. Johnston JJ, van der Smagt JJ, Rosenfeld JA, Pagnamenta AT, Alswaid A, Baker EH, et al. Autosomal recessive Noonan syndrome associated with biallelic LZTR1 variants. Genet Med. (2018) 20:1175–85. doi: 10.1038/gim.2017.249
3. Pagnamenta AT, Kaisaki PJ, Bennett F, Burkitt-Wright E, Martin HC, Ferla MP, et al. Delineation of dominant and recessive forms of LZTR1-associated Noonan syndrome. Clin Genet. (2019) 95:693–703. doi: 10.1111/cge.13533
4. Aoki Y, Niihori T, Inoue SI, Matsubara Y. Recent advances in RASopathies. J Hum Genet. (2016) 61:33–9. doi: 10.1038/jhg.2015.114
5. Grant AR, Cushman BJ, Cavé H, Dillon MW, Gelb BD, Gripp KW, et al. Assessing the gene–disease association of 19 genes with the RASopathies using the ClinGen gene curation framework. Hum Mutat. (2018) 39:1485–93. doi: 10.1101/323303
7. Romano AA, Allanson JE, Dahlgren J, Gelb BD, Hall B, Pierpont ME, et al. Noonan syndrome: Clinical features, diagnosis, and management guidelines. Pediatrics. (2010) 126:746–59. doi: 10.1542/peds.2009-3207
8. Marino B, Digilio MC, Toscano A, Giannotti A, Dallapiccola B. Congenital heart diseases in children with Noonan syndrome: An expanded cardiac spectrum with high prevalence of atrioventricular canal. J Pediatr. (1999) 135:703–6. doi: 10.1016/S0022-3476(99)70088-0
9. Edouard T, Cartault A. Gonadal function in Noonan syndrome. Ann Endocrinol (Paris). (2022) 83:203–6. doi: 10.1016/j.ando.2022.04.008
10. Theintz G, Savage MO. Growth and pubertal development in five boys with Noonan’s syndrome. Arch Dis Child. (1982) 57:13–7.
11. Patti G, Scaglione M, Maiorano NG, Rosti G, Divizia MT, Camia T, et al. Abnormalities of pubertal development and gonadal function in Noonan syndrome. Front Endocrinol. (2023) 14. doi: 10.3389/fendo.2023.1213098
12. Kelnar CJH. Noonan Syndrome: The hypothalamo-adrenal and hypothalamo-gonadal axes. Hormone Res. (2009) 72(Suppl 2):24–30. doi: 10.1159/000243775
13. Sharland M, Burch M, McKenna WM, Paton MA. A clinical study of Noonan syndrome. Arch Dis Child. (1992) 67:178–83. doi: 10.1136/adc.67.2.178
14. Mikec Š, Kolenc Ž, Peterlin B, Horvat S, Pogorevc N, Kunej T. Syndromic male subfertility: A network view of genome–phenome associations. Andrology. (2022) 10:720–32. doi: 10.1111/andr.13167
15. Elsawi MM, Pryor JP, Klufio G, Barnes C, Patton MA. Genital tract function in men with Noonan syndrome. J Med Genet. (1994) 31:468–70. doi: 10.1136/jmg.31.6.468
16. Shaw AC, Kalidas K, Crosby AH, Jeffery S, Patton MA. The natural history of Noonan syndrome: A long-term follow-up study. Arch Dis Child. (2007) 92:128–32. doi: 10.1136/adc.2006.104547
17. Cortes D, Thorup J, Lindenberg S, Visfeldt J. Infertility despite surgery for cryptorchidism in childhood can be classified by patients with normal or elevated follicle-stimulating hormone and identified at orchidopexy. BJU Int. (2003) 91:670–4. doi: 10.1046/j.1464-410X.2003.04177.x
18. Gracia J, Sánchez Zalabardo J, Sánchez García J, García C, Ferrández A. Clinical, physical, sperm and hormonal data in 251 adults operated on for cryptorchidism in childhood. BJU Int. (2000) 85:1100–3. doi: 10.1046/j.1464-410x.2000.00662.x
19. Ankarberg-Lindgren C, Westphal O, Dahlgren J. Testicular size development and reproductive hormones in boys and adult males with Noonan syndrome: A longitudinal study. Eur J Endocrinol. (2011) 165:137–44. doi: 10.1530/EJE-11-0092
20. Raaijmakers R, Noordam C, Noonan JA, Croonen EA, van der Burgt CJAM, Draaisma JMT. Are ECG abnormalities in Noonan syndrome characteristic for the syndrome? Eur J Pediatr. (2008) 167:1363–7. doi: 10.1007/s00431-008-0670-9
21. Trsinar B, Muravec UR. Fertility potential after unilateral and bilateral orchidopexy for cryptorchidism. World J Urol. (2009) 27:513–9. doi: 10.1007/s00345-009-0406-0
22. Marcus KA, Sweep CGJ, van der Burgt I, Noordam C. Impaired sertoli cell function in males diagnosed with Noonan syndrome. J Pediatr Endocrinol Metab. (2008) 21:1079–84. doi: 10.1515/JPEM.2008.21.11.1079
23. Moniez S, Pienkowski C, Lepage B, Hamdi S, Daudin M, Oliver I, et al. Noonan syndrome males display Sertoli cell-specific primary testicular insufficiency. Eur J Endocrinol. (2018) 179:409–18. doi: 10.1530/EJE-18-0582
24. Kosaki K, Suzuki T, Muroya K, Hasegawa T, Sato S, Matsuo N, et al. PTPN11 (protein-tyrosine phosphatase, nonreceptor-type 11) mutations in seven Japanese patients with Noonan syndrome. J Clin Endocrinol Metab. (2002) 87:3529–33. doi: 10.1210/jcem.87.8.8694
25. Umeki I, Niihori T, Abe T, Kanno SI, Okamoto N, Mizuno S, et al. Delineation of LZTR1 mutation-positive patients with Noonan syndrome and identification of LZTR1 binding to RAF1–PPP1CB complexes. Hum Genet. (2019) 138:21–35. doi: 10.1007/s00439-018-1951-7
26. Nistal M, Paniagua R, Pallardo LF. Testicular biopsy and hormonal study in a male with Noonan’s syndrome. Andrologia. (1983) 15:415–25. doi: 10.1111/and.1983.15.issue-5
27. Puri P, Walker WH. The tyrosine phosphatase shp2 regulates sertoli cell junction complexes. Biol Reprod. (2013) 88:59. doi: 10.1095/biolreprod.112.104414
28. Puri P, Phillips BT, Suzuki H, Orwig KE, Rajkovic A, Lapinski PE, et al. The transition from stem cell to progenitor spermatogonia and male fertility requires the SHP2 protein tyrosine phosphatase. Stem Cells. (2014) 32:741–53. doi: 10.1002/stem.1572
29. Hu X, Tang Z, Li Y, Liu W, Zhang S, Wang B, et al. Deletion of the tyrosine phosphatase Shp2 in Sertoli cells causes infertility in mice. Sci Rep. (2015) 5:12982. doi: 10.1038/srep12982
30. Tajan M, Paccoud R, Branka S, Edouard T, Yart A. The RASopathy family: Consequences of germline activation of the RAS/MAPK pathway. Endocr Rev. (2018) 39:676–700. doi: 10.1210/er.2017-00232
31. Nacak TG, Leptien K, Fellner D, Augustin HG, Kroll J. The BTB-kelch protein LZTR-1 is a novel Golgi protein that is degraded upon induction of apoptosis. J Biol Chem. (2006) 281:5065–71. doi: 10.1074/jbc.M509073200
32. Motta M, Fidan M, Bellacchio E, Pantaleoni F, Schneider-Heieck K, Coppola S, et al. Dominant Noonan syndrome-causing LZTR1 mutations specifically affect the Kelch domain substrate-recognition surface and enhance RAS-MAPK signaling. Hum Mol Genet. (2019) 28:1007–22. doi: 10.1093/hmg/ddy412
33. Chinton J, Huckstadt V, Mucciolo M, Lepri F, Novelli A, Gravina LP, et al. Providing more evidence on LZTR1 variants in Noonan syndrome patients. Am J Med Genet A. (2020) 182:409–14. doi: 10.1002/ajmg.a.61445
34. Yamamoto GL, Aguena M, Gos M, Hung C, Pilch J, Fahiminiya S, et al. Rare variants in SOS2 and LZTR1 are associated with Noonan syndrome. J Med Genet. (2015) 52:413–21. doi: 10.1136/jmedgenet-2015-103018
Keywords: Noonan syndrome, gonadal function, fertility, LZTR1 variant, case report
Citation: Orsolini F, Pignata L, Baldinotti F, Romano S, Tonacchera M and Canale D (2024) Gonadal dysfunction in a man with Noonan syndrome from the LZTR1 variant: case report and review of literature. Front. Endocrinol. 15:1354699. doi: 10.3389/fendo.2024.1354699
Received: 12 December 2023; Accepted: 22 March 2024;
Published: 16 April 2024.
Edited by:
Gedis Grudzinskas, Consultant, London, United KingdomReviewed by:
Monika Obara-Moszynska, Poznan University of Medical Sciences, PolandJayant Mehta, Barking, Havering and Redbridge University Hospitals NHS Trust, United Kingdom
Copyright © 2024 Orsolini, Pignata, Baldinotti, Romano, Tonacchera and Canale. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Orsolini, b3Jzb2xpbmkuZnJhbmNlc2NhQGxpYmVyby5pdA==