 Martin O. Savage
Martin O. Savage Rosario Ferrigno
Rosario Ferrigno
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Endocrinol., 22 January 2024
Sec. Pediatric Endocrinology
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1345174
This article is part of the Research TopicRare Forms of Pediatric Adrenal Disorders: Beyond Congenital Adrenal Hyperplasia due to 21-Hydroxylase DeficiencyView all 10 articles
Paediatric Cushing’s disease (CD) is characterized by excess ACTH secretion from a pituitary adenoma, leading to hypercortisolism. It has approximately 5% of the incidence of adult CD and is a rare disorder in the paediatric age range. The four most specific presenting features of hypercortisolism are: change in facial appearance, weight gain, decreased linear growth and virilisation shown by advanced pubic hair for the stage of breast development or testicular volume. The main diagnostic priority is the demonstration of hypercortisolism followed by distinction between its ACTH-dependent and ACTH-independent origin, thus leading to identification of aetiology. All treatment options aim to resolve or control hypercortisolism. Consensus favours transsphenoidal (TSS) pituitary surgery with selective removal of the corticotroph adenoma. TSS in children with CD is now well established and induces remission in 70-100% of cases. External pituitary radiotherapy and bilateral adrenalectomy are second-line therapeutic approaches in subjects not responding to TSS. Long-term medical treatment is less frequently adopted. Recurrence in paediatric CD cases is low with factors predicting relapse being higher post-TSS cortisol and ACTH levels and rapid recovery of the hypothalamic-pituitary-adrenal axis after TSS. In summary, complete excision of the microadenoma with histological and biochemical evidence for this, predicts a low rate of recurrence of CD. Due to the need for rapid diagnosis and management to avoid the burden of prolonged exposure to hypercortisolism, tertiary university centres comprising both paediatric and adult endocrinology specialists together with experienced pituitary surgery and, eventually, radiotherapy units are recommended for referral of these patients.
Cushing’s disease (CD), characterised by hypercortisolism due to excess ACTH secretion by a pituitary adenoma, is essentially an adult disorder which occasionally presents in children (1, 2). Its incidence in the paediatric age range is considered to be approximately 5% of that seen in adults (3). Consequently, a paediatric endocrinologist is likely to see only a few cases during their career, which strengthens the case for specialist university centres, staffed by both paediatric and adult endocrinologists, neurosurgeons specialising in transsphenoidal pituitary surgery, and specialised pituitary radiotherapists, to be the optimal institutions for the care of these patients. There is no doubt that collaboration and cooperation between paediatric and adult endocrinologists in the components of clinical management is beneficial to patient care (4).
Because the morbidity of hypercortisolism, also named Cushing’s syndrome (CS), in children is serious, early and rapid investigation is indicated in suspected cases. Two main goals dominate the investigational process. First, it is essential to confirm or exclude the presence of endogenous CS. Secondly, the etiologic cause of CS needs to be defined. Cushing’s syndrome can be divided into two main aetiological groups, namely ACTH-dependent and ACTH-independent CS: the first group comprises CD and ectopic ACTH syndrome, whereas the second one comprises adrenal cortical neoplasms, both benign and malignant, and adrenocortical hyperplasia. Exogenous glucocorticoid administration should be ruled out in the early phases of the diagnostic work-up to avoid needless investigations.
There are four key presenting features which are currently recognised to have a relevant diagnostic value (Table 1), whereas other features tend to be non-specific and are therefore less reliable in suspecting CD in children. These four key diagnostic features are; change in facial appearance with rounding of the face; weight gain, which is almost universal; retardation of linear growth, which may or may not lead to clinical short stature (i.e. height <-2 SDS); and increased virilisation (4). Although being present in 100% of cases in an English series of 41 patients (7), the change in facial appearance usually occurs slowly over several months or years, thus it may not be recognised as being pathological by parents or family doctors. In the same English series, weight gain was present in 98% of cases, and it usually leads to a marked increase in few months, thus being more easily recognized as a pathological sign by patients’ relatives. Growth retardation was present in all cases where low height velocity was documented, but actual short stature was reported only in 42% (10) and 56% of cases (6) in two large paediatric CD series, so short stature should not be considered as a CS sign per se (11). Virilisation presents as a disharmony of secondary sexual characteristics with Tanner stage pubic hair growth inappropriately advanced compared to breast development or testicular volume (12), although it may not be easy to identify in patients which are already in pubertal age. Other features which are less specific, but nevertheless important, include osteopaenia, hirsutism, mood changes, headache, striae, hypertension, acne, and pubertal delay (4). Bone age is usually within the normal range and not significantly decreased despite the short stature. Increased adrenal androgen secretion may contribute to this (12). Hypertension was common at diagnosis, being present in 36 -71% of cases (Table 1). Growth hormone responses to stimulation are usually normal at presentation, however gonadotrophin deficiency is a complication of long-term hypercorticism and the combination of decreased testicular volume or decreased breast development and advanced pubic hair growth is suggestive of Cushing’s syndrome (12).

Table 1 Frequency of clinical findings at diagnosis of paediatric Cushing’s disease.
Clinical skills remain important, and the history must exclude administration of exogenous glucocorticoids. Height and weight measurements, pubertal development using Tanner’s criteria and bone age assessment should also be performed. Confirmation of hypercortisolism can be assessed using three tests (4); 24-h urinary-free cortisol, late-night sleeping salivary/serum cortisol, and dexamethasone testing. None of these tests has 100% diagnostic accuracy, so multiple tests are usually required to confirm hypercortisolism. Among them, late night serum cortisol had the highest sensitivity and specificity in children (13, 14).
ACTH-dependent CS needs to be differentiated from ACTH-independent CS. First, hormonal investigations should be performed. In CD, morning plasma ACTH is typically detectable (>5 pg/ml) compared with suppressed values in primary adrenal disorders (13). The CRH stimulation test may also be useful but is not widely available. Patients with excess ACTH-secreting pituitary adenomas typically give an exaggerated response to CRH, resulting in anelevated cortisol response (15). Second, imaging techniques are required. Pituitary MRI is the optimal method for pituitary visualisation and should be performed after hormonal confirmation of ACTH-dependent CS in patients with suspect CD. However, MRI was only able to positively identify an adenoma in 16-71% of CD cases and in an English series of 41 patients appearances were consistent with a microadenoma in 55% (21/38) of paediatric patients compared with 76% (50/66) of adult patients (P=0.045) (7).
Although a positive MRI is beneficial for adenoma identification and supports the diagnosis of CD, the relatively low prediction rate requires a more precise localisation technique, the bilateral inferior petrosal sinus sampling (BIPSS). BIPSS with CRH stimulation (1 µg/kg, max 100 µg/kg) has been suggested in paediatric patients with a negative MRI and confirmed hypercortisolism (16). The best practical organisation is for the BIPSS procedure to be performed by a radiologist who has extensive experience of this investigation in adults with CS. The aim of this investigation is to demonstrate excess central ACTH secretion, thereby excluding ectopic ACTH syndrome, which remains very rare in children.
The overall opinion of expert adult endocrinologists treating Cushing’s disease is that the treatment of choice is removal of the microadenoma by transsphenoidal pituitary resection (TSS) (17). This neurosurgical approach was developed in the 1980s and has now become the standard therapy for Cushing’s disease. TSS can be very challenging in children because the microadenomas are often extremely small and difficult to locate and remove, as well as due to the specific features of skull base in paediatric patients, including anatomy of the sellar region varying with age, variable pneumatisation of the sphenoid bone according to age, reduced inter-carotid distance in younger children, and high frequency of anatomic variants, namely shorter nare-sellar and vomer-clivus distances and smaller transsphenoidal angles (18). Total excision of a corticotroph microadenoma results in immediate post-operative ACTH and cortisol deficiency (19). The histological appearance of normal corticotroph cells surrounding the adenoma are morphologically abnormal, the appearance being known as Crooke’s change.
There is no international consensus on the definition of success of TSS. The term ‘cure’ has generally been replaced by ‘remission’, which is usually defined as <5 µg/dL (<138 nmol/L) or urinary free cortisol <28-56 nmol/day (<10-20 µg/day) within 7 days of selective tumour resection (20). According to these criteria, remission rates of 70-98% have been reported (8, 14, 21, 22), but also remission rates of 100% and 69% were reported in two large paediatric CD series which used stricter definitions of remission, namely post-TSS serum cortisol of <1 µg/dL (28 nmol/L) and <1.8 µg/dL (50 nmol/L), respectively (7, 23). In summary, remission rates of ~70% or greater are now expected from specialist centres with experience of paediatric CD. The most common complication of TSS was post-operative diabetes insipidus, present in 5% of patients at discharge from neurosurgical care (8). Overall, TSS is effective and safe first-line treatment for paediatric CD. An endo-nasal approach with endoscopy may be also used for access to the pituitary and this is now gaining popularity, particularly in adult patients. A small paediatric series of patients from an English reference centre showed biochemical remission in 5 out of 6 patients, with no recurrence after a mean follow-up of 55 months (24). Second-line therapy in the form of reoperation, pituitary radiotherapy, bilateral adrenalectomy or long-term medical therapy to control cortisol synthesis may be required in approximately 30% of cases.
Adrenal insufficiency follows successful pituitary adenoma resection and may persists for many months. A median time of recovery of the HPA axis in 106 paediatric patients successfully treated by TSS at the NIH was 12.3 months with a range of 3-35 months (25). The level of pre-operative urinary free cortisol related to recovery time with high values being associated with longer intervals of recovery of normal adrenal function.
There are relatively few studies of recurrence of CD following treatment during the paediatric age range. Yordanova et al. studied long-term follow-up of 21 paediatric CD patients in remission following definitive TSS or pituitary radiotherapy. During an interval of 2 to 7.6 years, the recurrence rate was 14.3% (26). The most comprehensive data comes from the NIH. Interesting results were reported showing a relationship (p=0.0342) between a shorter HPA axis recovery time and the likelihood of recurrence of CD (25). All patients with recurrence of hypercortisolism had recovery of the HPA axis by 15 months post-TSS. It is helpful to look at risk of recurrence in the context of prediction of short-term remission following TSS – because the two phenomena are linked. Essentially the factors significantly predicting remission (P=<0.05) after TSS are: lack of prior surgery, younger age, identification of adenoma during surgery, the presence of a positive ACTH-producing immunohistochemical adenoma and a non-invasive and smaller adenoma (8) (Figure 1). All these factors favour complete removal of the adenoma. In a series of 78 paediatric CD patients from the NIH, 94% had sustained remission for 5.8 – 18.3 years with 6 patients showing recurrence of CD. Children who remained in remission had; lower morning ACTH and cortisol levels during the post-operative period after TSS compared to those who relapsed (P <0.001). Relapse was associated with; higher cortisol response to CRH pre-TSS, lack of histological confirmation of adenoma at surgery, normal serum cortisol and ACTH (as opposed to subnormal values) post-TSS and the need for glucocorticoid replacement for less than 6 months after surgery, ie a rapid recovery of the HPA axis (10). The confirmation of these factors, in an expanded report of 179 NIH patients (8) reinforced the message that a positive predictive value for lasting remission (96%), was associated with a minimum morning cortisol of <1 µg/dL (28 nmol/L) during the immediate post-operative period. Paediatric patients harbouring somatic USP8 mutations were reported to have a higher likelihood of recurrence of CD following TSS compared with patients without mutations (46.2% vs 10.3%, P=0.009) (27).
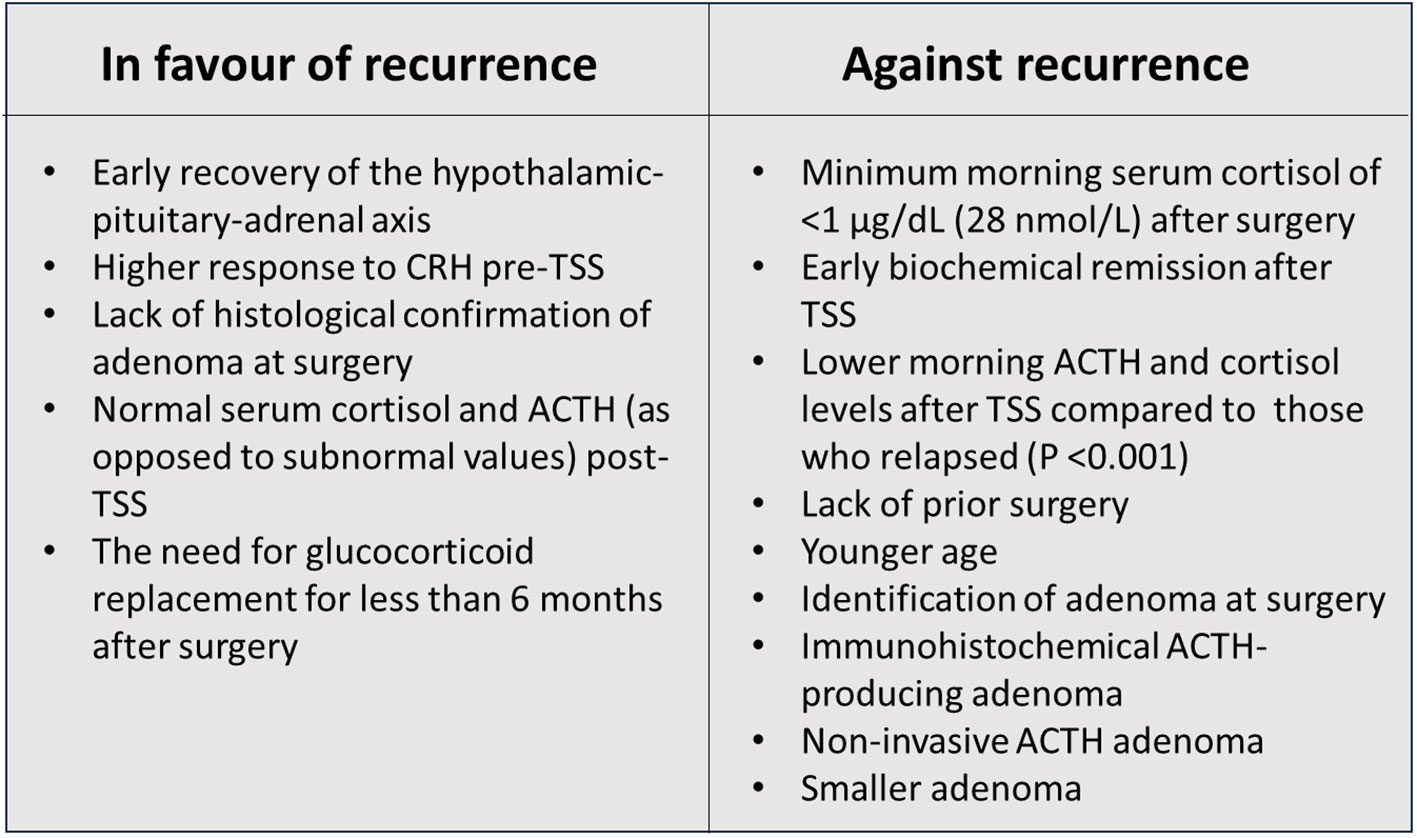
Figure 1 Factors in favour and against recurrence of paediatric Cushing’s disease.
External pituitary radiotherapy (RT) is an optional second-line therapy for patients not in remission after TSS. However, this treatment in children is controversial because of concern related to cognitive effects and possible post-RT hypopituitarism. Corticotroph microadenomas respond well to conventional fractionated external RT, as first documented in 1977 (28). An English series of 7 paediatric patients reported treatment by a 6-MV linear accelerator delivering a dose of 45 Gy in 25 fractions over 35 days, which induced remission in all subjects at a mean interval of 0.94 years (range 0.25-2.86) (29). These data were later confirmed in a multi-centre study focusing on the gamma knife radiotherapy technique on 24 children with recurring CD, which induced remission in 87.5% of subjects after a mean interval of 12 months (30). The effects of RT take several months during which control of hypercortisolism is required using medical therapy.
Bilateral adrenalectomy may represent a life-saving procedure in children with very severe hypercortisolism and life-threatening clinical morbidities per se or which may prevent safely approaching more definitive treatments, as TSS. Virtually, bilateral adrenalectomy has a remission rate of 100%, as it removes the source of cortisol production, and only patients with subtotal or incomplete surgical removal may experience recurrence after bilateral adrenalectomy (4). However, bilateral adrenalectomy requires life-long replacement treatment with glucocorticoids and mineralocorticoids and may be associated with Nelson’s syndrome. The current definition of Nelson’s syndrome is ‘radiological progression or new detection of a pituitary tumour on thin-section MRI’ (31). Clinically this is associated with increasing levels of ACTH causing hyperpigmentation, that may even occur several years after bilateral adrenalectomy (31). Although few paediatric series have been reported, a mean interval of 8.4 years post-adrenalectomy was reported in one series (32). The mean cumulative incidence of Nelson’s syndrome was considerably higher (45%, 25–67%) compared to results in adult patients (31). This emphasizes the importance of life-long follow-up of all paediatric CD patients irrespective of the therapy they have received.
Whereas extensively used in adult CD patients (33), few reports are available for the medical treatment of children with CD. Ketoconazole, a multi-enzyme steroidogenesis inhibitor, is the most frequently reported drug in children with CD, mainly in patients needing fast symptomatic relief from CS comorbidities and in patients waiting for pituitary radiotherapy to be effective (4). A French study reported the effects of “low-dose” mitotane, an adrenolytic drug used in adrenocortical cancer and in patients with very severe CS. In 9 CD children, there was significant improvement in weight and growth rate after 12 months of treatment, as well as a general improvement of clinical features, although the high rate of reported adverse events suggests caution in the widespread use of this treatment in children (34). Therapeutic trials are currently in progress in children using the 11-beta hydroxylase blocking agent osilodrostat, which is now approved for treatment of adults with CD (35).
Growth hormone deficiency (GHD) is common after TSS in children (5) and may also be a complication of pituitary RT (9, 20). In an English series, GHD occurred in 86% of paediatric patients treated with RT, but after 10 years of follow-up 3/4 boys showed recovery of GH secretion to a peak GH value of >10 ng/ml (9). Gonadotrophin deficiency was also present in 9/20 subjects causing delayed puberty and several required sex steroid replacement (26). A major goal in the management of paediatric CD is the restoration of normal linear growth during remission after successful TSS. As CD often presents shortly before or during puberty, the time for catch-up growth is frequently limited and decreased adult height in paediatric CD has been well documented (5, 11, 36) (Figure 2). We advocate testing for GH deficiency 3 months after TSS and a low threshold for hGH replacement, if necessary in combination with a GnRH analogue (4).
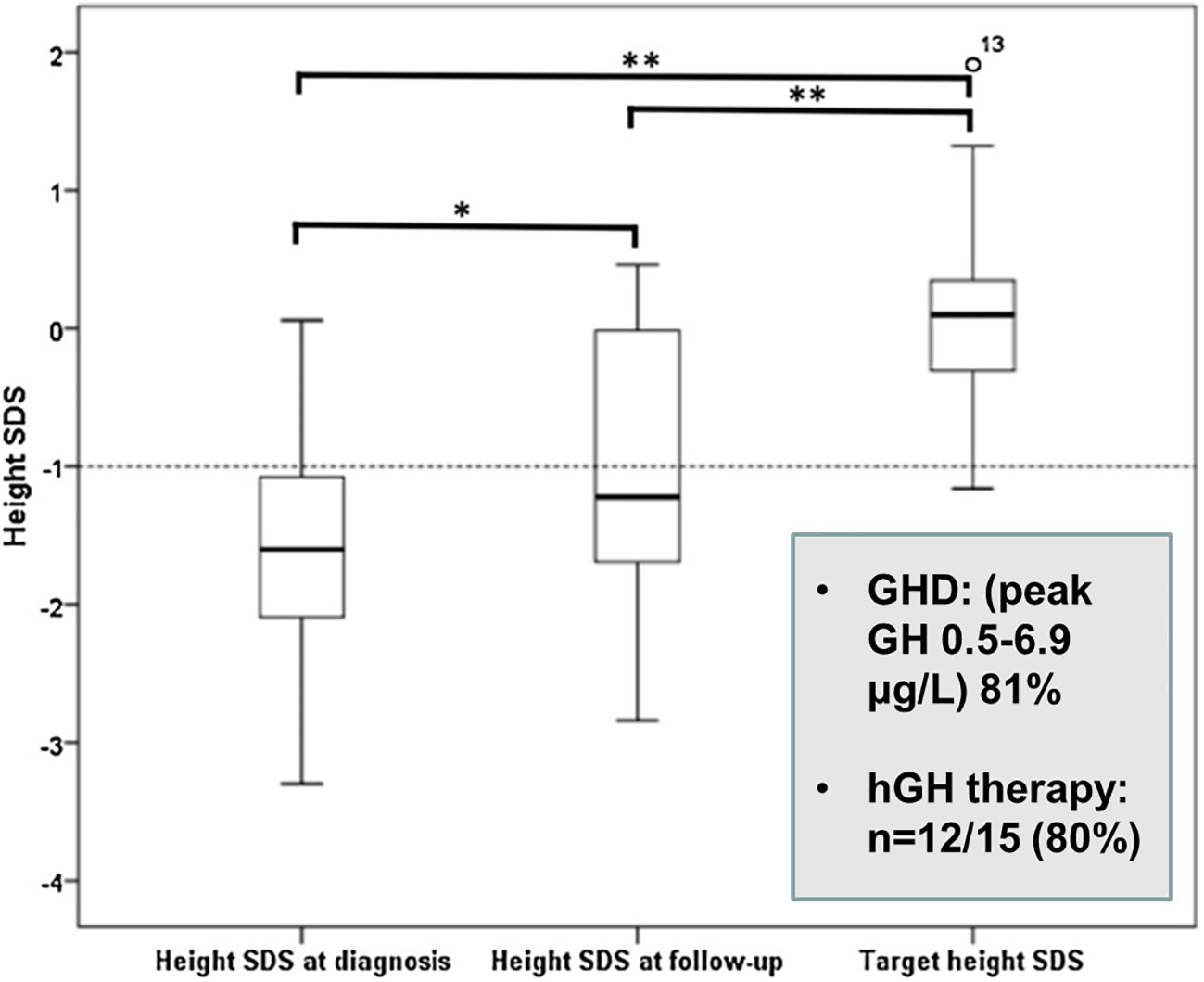
Figure 2 Height SDS values at diagnosis, latest assessment and target height in paediatric Cushing’s disease (26). *p = 0.033, **p = 0.000.
There are follow-up data on two other variables, namely body composition and bone mineral density. In 14 patients treated by TSS alone (n=8) or TSS followed by external pituitary radiotherapy (n=6), body mass index (BMI) SDS was elevated at +2.7 (0.8 - 5.1) at diagnosis. At a mean interval of 4.1 (1.1 -10.7) years after remission of hypercortisolaemia (postoperative serum cortisol <50 nmol/L), BMI SDS remained elevated above the mean at 1.7 SDS, being lower than at diagnosis (P < 0.05), but elevated compared to the normal population (P < 0.01) (37). It is often difficult for children to normalise their BMI after remission. Careful dieting is required to minimise the risk of continuing insulin resistance.
Bone mineral density (BMD) is frequently reduced at diagnosis of paediatric CD (4). Two groups of patients were studied using DEXA, the first comprising 8 patients, mean age 12.4 yr (8.2 – 16.8 yr) had a mean L2-L4 volumetric BMD at diagnosis of -1.04 (-3.2 – 0.11) with values corelating negatively with midnight serum cortisol (P = < 0.05) (38). The second group comprised 11 subjects with hypercortisolaemia in remission following TSS or external RT, studied at a mean of 4.5 yr after remission, during which 8/11 had received hGH replacement (37). Mean L2-L4vBMD SDS was -0.38 (-1.0 – 0.13) with mean femoral neck areal BMD SDS of 0.14 (-1.62 – 2.46). These data show variable BMD SDS at diagnosis and near normal BMD SDS after induction of remission in paediatric CD (38).
Paediatric CD can be regarded as a niche disorder, which ideally requires joint input from paediatric and adult endocrinologists in terms of diagnosis and therapeutic strategy. Ideally a small number of tertiary centres should be reserved for the management of these patients. Following diagnosis, it is of crucial importance that a pituitary surgeon with prior experience of transsphenoidal surgery in children is identified for referral and becomes part of the management team. Complete selective excision of the corticotroph adenoma is difficult due to its very small size, but is directly related to long-term post-operative remission and quality of life. In experienced hands, the prognosis is good and the rate of recurrence is low. Post-operative challenges relate to catch-up linear growth and resumption of normal body composition. Life-long follow-up is required.
MS: Conceptualization, Writing – original draft, Writing – review & editing. RF: Conceptualization, Formal analysis, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Newell-Price J, Bertagna X, Grossman AB, Nieman LK. Cushing’s syndrome. Lancet (2006) 367(9522):1605–17. doi: 10.1016/s0140-6736(06)68699-6
2. Lacroix A, Feelders RA, Stratakis CA, Nieman LK. Cushing’s syndrome. Lancet (2015) 386(9996):913–27. doi: 10.1016/S0140-6736(14)61375-1
3. Ragnarsson O, Olsson DS, Chantzichristos D, Papakokkinou E, Dahlqvist P, Segerstedt E, et al. The incidence of Cushing’s disease: a nationwide Swedish study. Pituitary (2019) 22(2):179–86. doi: 10.1007/s11102-019-00951-1
4. Ferrigno R, Hasenmajer V, Caiulo S, Minnetti M, Mazzotta P, Storr HL, et al. Paediatric Cushing’s disease: epidemiology, pathogenesis, clinical management and outcome. Rev Endocr Metab Disord (2021) 22:817–35. doi: 10.1007/s11154-021-09626-4
5. Magiakou MA, Mastorakos G, Gomez MT, Rose SR, Chrousos GP. Suppressed spontaneous and stimulated growth hormone secretion in patients with Cushing’s disease before and after surgical cure. J Clin Endocrinol Metab (1994) 78(1):131–7. doi: 10.1210/jcem.78.1.7507118
6. Shah NS, George J, Acharya SV, Lila AR, Sarathi V, Bandgar TR, et al. Cushing disease in children and adolescents: twenty years’ experience in a tertiary care center in India. Endocr Pract (2011) 17(3):369–76. doi: 10.4158/EP10143.OR
7. Storr HL, Alexandraki KI, Martin L, Isidori AM, Kaltsas GA, Monson JP, et al. Comparisons in the epidemiology, diagnostic features and cure rate by transsphenoidal surgery between paediatric and adult-onset Cushing’s disease. Eur J Endocrinol (2011) 164(5):667–74. doi: 10.1530/EJE-10-1120
8. Lonser RR, Wind JJ, Nieman LK, Weil RJ, DeVroom HL, Oldfield EH. Outcome of surgical treatment of 200 children with Cushing’s disease. J Clin Endocrinol Metab (2013) 98(3):892–901. doi: 10.1210/jc.2012-3604
9. Chan LF, Storr HL, Plowman PN, Perry LA, Besser GM, Grossman AB, et al. Long-term anterior pituitary function in patients with paediatric Cushing’s disease treated with pituitary radiotherapy. Eur J Endocrinol (2007) 156(4):477–82. doi: 10.1530/EJE-06-0588
10. Batista DL, Oldfield EH, Keil MF, Stratakis CA. Postoperative testing to predict recurrent Cushing disease in children. J Clin Endocrinol Metab (2009) 94(8):2757–65. doi: 10.1210/jc.2009-0302
11. Minnetti M, Caiulo S, Ferrigno R, Baldini-Ferroli B, Bottaro G, Gianfrilli D, et al. Abnormal linear growth in paediatric adrenal diseases: Pathogenesis, prevalence and management. Clin Endocrinol (Oxf) (2020) 92(2):98–108. doi: 10.1111/cen.14131
12. Dupuis CC, Storr HL, Perry LA, Ho JTF, Ahmed L, Ong KK, et al. Abnormal puberty in paediatric Cushing’s disease; relationships with adrenal androgens, sex hormone binding globulin and gonadotrophin concentrations. Clin Endocrinol (Oxf) (2007) 66:838–43. doi: 10.1111/j.1365-2265.2007.02822.x
13. Batista DL, Riar J, Keil M, Stratakis CA. Diagnostic tests for children who are referred for the investigation of Cushing syndrome. Paediatrics (2007) 120(3):e575–e86. doi: 10.1542/peds.2006-2402
14. Tarçın G, Çatlı G, Çetinkaya S, Eren E, Kardelen AD, Akıncı A, et al. Clinical features, diagnosis and treatment outcomes of Cushing’s disease in children: A multicentre study. Clin Endocrinol (Oxf) (2023) 100, 19–28. doi: 10.1111/cen.14980
15. Nieman LK, Biller BM, Findling JW, Newell-Price J, Savage MO, Stewart PM, et al. The diagnosis of Cushing’s syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab (2008) 93(5):1526–40. doi: 10.1210/jc.2008-0125
16. Batista D, Gennari M, Riar J, Chang R, Keil MF, Oldfield EH, et al. An assessment of petrosal sinus sampling for localization of pituitary microadenomas in children with Cushing disease. J Clin Endocrinol Metab (2006) 91(1):221–4. doi: 10.1210/jc.2005-1096
17. Fleseriu M, Auchus R, Bancos I, Ben-Shlomo A, Bertherat J, Biermasz NR, et al. Consensus on diagnosis and management of Cushing’s disease: a guideline update. Lancet Diabetes Endocrinol (2021) 9(12):847–75. doi: 10.1016/S2213-8587(21)00235-7
18. Marino AC, Taylor DG, Desai B, Jane JA Jr. Surgery for paediatric pituitary adenomas. Neurosurg Clin N Am (2019) 30(4):465–71. doi: 10.1016/j.nec.2019.05.008
19. Crock PA, Ludecke DK, Knappe UJ, Saeger W. A personal series of 100 children operated for Cushing’s disease (CD): optimizing minimally invasive diagnosis and transnasal surgery to achieve nearly 100% remission including reoperations. J Pediatr Endocrinol Metab (2018) 31(9):1023–31. doi: 10.1515/jpem-2018-0262
20. Nieman LK, Biller BM, Findling JW, Murad MH, Newell-Price J, Savage MO, et al. Treatment of Cushing’s syndrome: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab (2015) 100(8):2807–31. doi: 10.1210/jc.2015-1818
21. Dyer EH, Civit T, Visot A, Delalande O, Derome P. Transsphenoidal surgery for pituitary adenomas in children. Neurosurgery (1994) 34(2):207–12. doi: 10.1227/00006123-199402000-00001
22. Devoe DJ, Miller WL, Conte FA, Kaplan SL, Grumbach MM, Rosenthal SM, et al. Long-term outcome in children and adolescents after transsphenoidal surgery for Cushing’s disease. J Clin Endocrinol Metab (1997) 82(10):3196–202. doi: 10.1210/jcem.82.10.4290
23. Magiakou MA, Mastorakos G, Oldfield EH, Gomez MT, Doppman JL, Cutler GB, et al. Cushing’s syndrome in children and adolescents. Presentation, diagnosis, and therapy. N Engl J Med (1994) 331(10):629–36. doi: 10.1056/NEJM199409083311002
24. Storr HL, Drake WM, Evanson J, Matson M, Berney DM, Grossman AB, et al. Endonasal endoscopic transsphenoidal pituitary surgery: early experience and outcome in paediatric Cushing’s disease. Clin Endocrinol (Oxf) (2014) 80(2):270–6. doi: 10.1111/cen.12275
25. Tatsi C, Neely M, Flippo C, Bompou ME, Keil M, Stratakis CA. Recovery of hypothalamic-pituitary-adrenal Axis in paediatric Cushing disease. Clin Endocrinol (Oxf) (2020) 94:40–7. doi: 10.1111/cen.14300
26. Yordanova G, Martin L, Afshar F, Sabin I, Alusi G, Plowman NP, et al. Long-term outcomes of children treated for Cushing’s disease: a single center experience. Pituitary (2016) 19(6):612–24. doi: 10.1007/s11102-016-0756-8
27. Faucz FR, Tirosh A, Tatsi C, Berthon A, Hernandez-Ramirez LC, Settas N, et al. Somatic USP8 gene mutations are a common cause of paediatric Cushing disease. J Clin Endocrinol Metab (2017) 102(8):2836–43. doi: 10.1210/jc.2017-00161
28. Jennings AS, Liddle GW, Orth DN. Results of treating childhood Cushing’s disease with pituitary irradiation. N Engl J Med (1977) 297(18):957–62. doi: 10.1056/NEJM197711032971801
29. Storr HL, Plowman PN, Carroll PV, Francois I, Krassas GE, Afshar F, et al. Clinical and endocrine responses to pituitary radiotherapy in paediatric Cushing’s disease: an effective second-line treatment. J Clin Endocrinol Metab (2003) 88(1):34–7. doi: 10.1210/jc.2002-021032
30. Shrivastava A, Mohammed N, Xu Z, Liščák R, Kosak M, Krsek M, et al. Outcomes after gamma knife stereotactic radiosurgery in paediatric patients with Cushing disease or acromegaly: A multi-institutional study. World Neurosurg (2019) 125:e1104–13. doi: 10.1016/j.wneu.2019.01.252
31. Reincke M, Albani A, Assie G, Bancos I, Brue T, Buchfelder M, et al. Corticotroph tumour progression after bilateral adrenalectomy (Nelson’s syndrome): systematic review and expert consensus recommendations. Eur J Endocrinol (2021) 184(3):P1–P16. doi: 10.1530/EJE-20-1088
32. Thomas CG Jr, Smith AT, Benson M, Griffith J. Nelson’s syndrome after Cushing’s disease in childhood: a continuing problem. Surgery (1984) 96(6):1067–77.
33. Pivonello R, De Leo M, Cozzolino A, Colao A. The treatment of Cushing’s disease. Endocr Rev (2015) 36(4):385–486. doi: 10.1210/er.2013-1048
34. Motte E, Rothenbuhler A, Gaillard S, Lahlou N, Teinturier C, Coutant R, et al. Mitotane (op’DDD) restores growth and puberty in nine children with Cushing’s disease. Endocr Connect (2018) 7(12):1280–7. doi: 10.1530/EC-18-0215
35. Pivonello R, Fleseriu M, Newell-Price J, Bertagna X, Findling J, Shimatsu A, et al. Efficacy and safety of osilodrostat in patients with Cushing’s disease (LINC 3): a multicentre phase III study with a double-blind, randomised withdrawal phase. Lancet Diabetes Endocrinol (2020) 8(9):748–61. doi: 10.1016/S2213-8587(20)30240-0
36. Guemes M, Murray PG, Brain CE, Spoudeas HA, Peters CJ, Hindmarsh PC, et al. Management of Cushing syndrome in children and adolescents: experience of a single tertiary Centre. Eur J Pediatr (2016) 175(7):967–76. doi: 10.1007/s00431-016-2727-5
37. Davies JH, Storr HL, Davies K, Monson JP, Besser GM, Afshar F, et al. Final height and body mass index after cure of paediatric Cushing’s disease. Clin Endocrinol (2005) 62:466–72. doi: 10.1111/j.1365-2265.2005.02244.x
Keywords: Cushing’s, pituitary adenoma, transsphenoidal surgery, pituitary radiotherapy, recurrence
Citation: Savage MO and Ferrigno R (2024) Paediatric Cushing’s disease: long-term outcome and predictors of recurrence. Front. Endocrinol. 15:1345174. doi: 10.3389/fendo.2024.1345174
Received: 27 November 2023; Accepted: 02 January 2024;
Published: 22 January 2024.
Edited by:
Federico Baronio, Dpt Hospital of Woman and Child, ItalyReviewed by:
Elena Varlamov, Oregon Health and Science University, United StatesCopyright © 2024 Savage and Ferrigno. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin O. Savage, bS5vLnNhdmFnZUBxbXVsLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.