 Xiaohui Fu
Xiaohui Fu Shuli Chen1
Shuli Chen1 Qin Yang
Qin Yang- 1Department of Inherited Metabolic Disorders, Shenzhen Children’s Hospital, Shenzhen, China
- 2Department of Respiratory Diseases, Shenzhen Children’s Hospital, Shenzhen, China
Background: Mandibuloacral dysplasia (MAD) syndrome is a rare genetic disease. Several progeroid syndromes including mandibuloacral dysplasia type A (MADA), mandibuloacral dysplasia type B(MADB), Hutchinson-Gilford progeria (HGPS) and mandibular hypoplasia, deafness, and lipodystrophy syndrome (MDPL) have been reported previously. A novel MAD progeroid syndrome (MADaM) has recently been reported. So far, 7 cases of MADaM diagnosed with molecular diagnostics have been reported in worldwide. In the Chinese population, cases of MAD associated with the MTX2 variant have never been reported.
Methods: The clinical symptoms and the genetic analysis were identified and investigated in patients presented with the disease. In addition, we analyzed and compared 7 MADaM cases reported worldwide and summarized the progeroid syndromes reported in the Chinese population to date.
Results: The present study reports a case of a novel homozygous mutation c.378 + 1G > A in the MTX2 gene, which has not been previously reported in the literature. Patients present with early onset and severe symptoms and soon after birth are found to have growth retardation. In addition to the progeroid features, skeletal deformities, generalized lipodystrophy reported previously, and other multisystem involvement, e.g. hepatosplenic, renal, and cardiovascular system, this case was also reported to have combined hypogammaglobulinemia. She has since been admitted to the hospital several times for infections. Among 22 previously reported progeroid syndromes, 16/22 were MADA or HGPS caused by LMNA gene mutations, and the homozygous c.1579C > T (p.R527C) mutation may be a hot spot mutation for MAD in the Chinese population. MAD and HGPS mostly present in infancy with skin abnormalities or alopecia, MDPL mostly presents in school age with growth retardation as the first manifestation, and is often combined with an endocrine metabolism disorder after several decades.
Conclusion: This is the first case of MAD syndrome caused by mutations in MTX2 gene reported in the Chinese population. MTX2 gene c.378 + 1G > A homozygous mutation has not been previously reported and the report of this patient expands the spectrum of MTX2 mutations. In addition, we summarized the genotypes and clinical characteristics of patients with progeroid syndromes in China.
1 Introduction
Mandibuloacral dysplasia progeriod syndromes is a rare disabling autosomal recessive genetic disease first reported by Young et al. in 1971 (1). It is mainly characterized by severe skeletal deformities, abnormalities of skin, and lipodysplasia (2). It is estimated that there are about 400 children with progeria worldwide, it affects 1 in 4-8 million people approximately, irrespective of sex and race(Progeria 101 FAQ | The Progeria Research Foundation).Several types of MAD have been reported, of which the MAD associated with mutations in the LMNA gene is the first MAD syndrome with an identified gene (3). LMNA mainly causes Hutchinson-Gilford Progeria (HGPS) and type A MAD (MADA) (3, 4). HGPS is one of the progeria syndromes, Jonathan Hutchinson discovered it in 1886. The main features are progeroid face, short stature, skin abnormalities, lipodystrophy, skeletal abnormalities, etc. The differences between HGPS and MADA mainly lie in the molecular genetic pattern and the severity of the clinical phenotype, HGPS is usually characterized by earlier onset and more severe clinical symptoms than MADA, and in terms of molecular genetics, HGPS is predominantly autosomal dominant and is almost entirely caused by codon 608 mutation on chromosome 1 exon 11 of the LMNA gene, whereas MADA shows autosomal recessive inheritance (2). The other type of MAD, type B MAD (MADB), is caused by mutations in the gene encoding zinc metalloprotease ZMPSTE24 (5). The clinical manifestations of MADB are similar to those of MADA. The main difference is that MADA is partial lipodystrophy, while MADB is generalized lipodystrophy and MADB presents with a more severe metabolic syndrome (6). Nestor-Guillermo Progeria Syndrome (NGPS) was first described and diagnosed in 2011 by Cabanillas R et al., with biallelic pathogenic variants in the BANF1 gene (7). The product encoded by the BANF1 gene is required for the reaggregation of lamin A during the remodeling of the nuclear envelope at the end of mitosis (7). Although the causative genes for several of the above syndromes are different, they are all thought to be associated with toxic accumulation of either prelamin A or lamin A/C.
In 2013, Weedon et al. identified a mandibular hypoplasia, deafness, and lipodystrophy syndrome (MDPL) caused by mutations in POLD1 gene, which encodes the catalytic subunit (p125) of DNA polymerase δ(POLδ) (8). The catalytic subunit is responsible for synthesizing the lagging strand DNA during DNA replication with both 5′- to 3′-polymerase activity and 3′- to 5′-exonuclease activity (9). Polδ is involved in DNA replication and maintains genomic stability (9). The p125 mutation leads to reduced genomic stability, cellular senescence, and apoptosis, which may be the pathogenic mechanism of MDPL (9, 10). In contrast to other progeria syndromes, MDPL is characterized by early-onset hearing loss.
The novel MAD progeroid syndrome (MADaM: Mandibuloacral dysplasia associated to MTX2) reported in this study was first described in 2020 by Elouej et al., due to recessive mutations in MTX2 encoding Metaxin-2 (MTX2), an outer mitochondrial membrane (OMM) protein (11). MADaM is clinically characterized by mandibuloacral dysplasia, generalized lipodystrophy, hypotonia, acro osteolysis, skin abnormalities, renal impairment, and cardiovascular system damage such as hypertension and left ventricular hypertrophy. To date, only 2 studies have reported 7 genetically definite diagnosed MADaM (11, 12). This study is the third, and the first MADaM from China.
2 Materials and methods
2.1 Patients and clinical investigations
A 2-year-4-month-old girl was admitted to Shenzhen Children’s Hospital in 2023, due to progeroid facial features, severe generalized lipodystrophy, hypotonia, skeletal malformations, recurrent liver enzyme elevations, eyelid edema and tachypnea. Multiple previous hospitalizations due to recurrent infections. Whole-exome sequencing was performed. A signed informed consent form was obtained from the proband’s parents.
2.2 Whole-exome sequencing
Whole blood (3 ml) was collected from the affected proband and her parents. DNA was isolated from peripheral blood with CWE9600 Automated Nucleic Acid Extraction System using CWE2100 Blood DNA Kit V2 (CWBiotech, China, CW2553). 750ng genomic DNA was fragmented into 200-300bp length using Scientz08-III Ultrasonic Homogenizer (SCIENTZ, China). The DNA fragments were then processed by end-repairing, A-tailing and adaptor ligation using KAPA Library Preparation Kit (Illumina, KR0453, v3.13), followed by an 8-cycle pre-capture polymerase chain reaction (PCR) amplification. Then, the amplified DNA sample was captured in the Agilent SureSelect XT2 Target Enrichment System (Agilent Technologies, Inc., USA). Captured DNA fragments were purified by Dynabeads MyOne Streptavidin T1 (Invitrogen, Thermo Fisher Scientific, USA) and amplified by 13 cycle post-capture PCR. The final products were purified by Agencourt AMPure XP (Beckman Coulter, Inc., USA) and quantitated with Life Invitrogen Qubit 3.0 by Qubit dsDNA HS Assay Kit (Invitrogen, Thermo Fisher Scientific, USA). Eventually, quantified DNA was sequenced in 150-bp paired-end modes on Illumina Novaseq 6000 platform (Illumina, Inc., USA) according to the standard manual.
The raw data produced on Novaseq platform were filtered and aligned against the human reference genome (hg19) using the BWA Aligner (http://bio-bwa.sourceforge.net/) after evaluated according to Illumina Sequence Control Software (SCS). The single-nucleotide variants (SNVs) were called using GATK software (Genome Analysis ToolKit) (www.broadinstitute.org/gatk). Variants were annotated using ANNOVAR (annovar.openbioinformatics.org/en/latest/). Effects of SNVs were predicted by SIFT, Polyphen-2, and MutationTaster programs. All variants were interpreted according to ACMG standards and categorized to be pathogenic, likely pathogenic, variants of unknown clinical significance (VUS), likely benign and benign.
3 Results
3.1 Case presentation
The proband, a 2-year-4-month-old girl, G3P2, was born to fourth-degree consanguineous parents at 38 weeks gestation after an uneventful pregnancy. The birth weight was 3350g and the height was in the normal range. Intellectual and motor development was within normal limits at the age of 1 year. After 1 year of age, language and motor development began to lag behind.
She had been suffering from lipodysplasia and recurrent diarrhea since about 5 months after birth. Elevated liver enzymes were noted at 8 months of age, at which point oral hepatoprotective drugs were started for more than a year. She was prone to recurrent infections and had eyelid edema that began when she was approximately 1 year old. A urine examination revealed massive proteinuria and microhematuria, and she had hypertension. Captopril and amlodipine were added to control her blood pressure, but the results were not satisfactory.
On physical examination, her weight was 6 kg (<3 p), height was 66.5 cm (<3 p), head circumference was 41.5 cm (<3 p) and blood pressure was 122/70mmHg. She had tachypnea at 42 breaths per minute, generalized lipodystrophy, poor skin elasticity, wrinkled skin, wide forehead, sparse hair and eyebrows, small jaw, full cheeks, patent fontanel, wet rales in both lungs, distend abdomen, generalized joint hypermobility, muscle hypotonia, and dystrophic nails.
X-ray examination suggested pneumonia, mandibuloacral dysplasia, thoracolumbar kyphosis, developmental hip dislocation, gracile long bones of ribs, clavicles, and extremities, osteoporosis, osteolysis of the proximal radius and distal parts of both toes (Figure 1B). Previously elevated transaminases, ALT up to 471 IU/L, oral administration of liver protective drugs, this admission examination showed that the patient’s ALT was in the normal range, immune tests suggested IgG2.2g/l (3.2-11.5), cholesterol was 6.62mmol/L(0-5.18), urine examination suggested protein 3 +, occult blood 3 +, red blood cells 254 cells/UL (0-10).
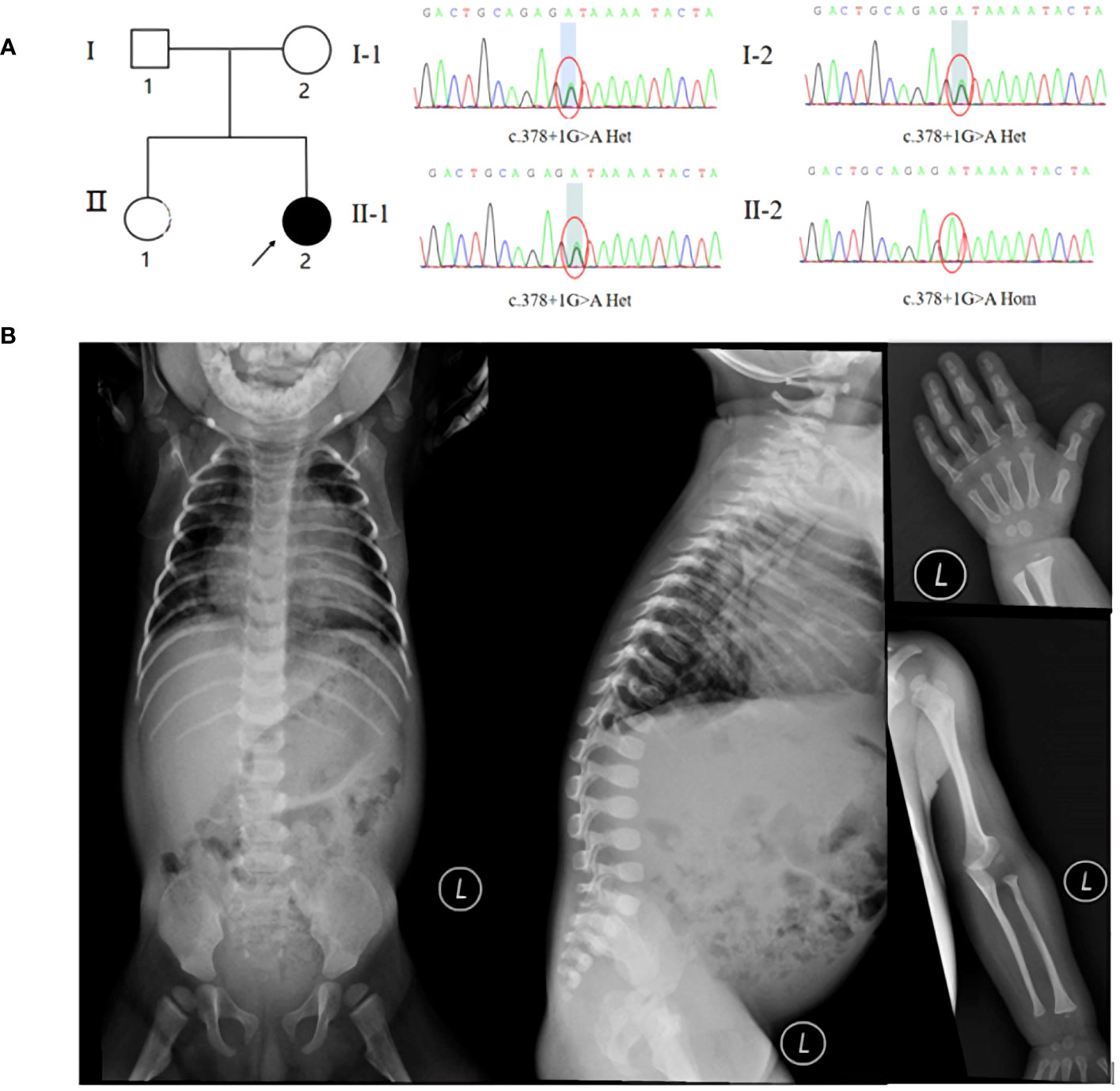
Figure 1 (A) Pedigree and Sanger sequencing of the MADaM family. (B) X-ray examination suggested pneumonia, mandibuloacral dysplasia, thoracolumbar kyphosis, developmental hip dislocation, gracile long bones of ribs, clavicles, and extremities, osteoporosis, osteolysis of the proximal radius and distal part of finger.
3.2 Molecular diagnosis
Coverage was 99.80%, the exon region and flanking splicing or intronic junctions of the MTX2 gene were well covered, and the sequencing depth was >100×. Whole-exome sequencing revealed a homozygous MTX2 gene mutation, NM_006554.5: c.378 + 1G > A, which had not been reported previously. The variant was inherited from his parents, and the mutation prediction retained the reading frame (Figure 1A). AlphaFold2 software was used to develop a suitable model to simulate the effect of the mutation region, the 3D protein modeling predicted that compared with wild type, the mutation would result in a truncated protein with an absence of the translated portion of exon 6 protein (Figure 2A). The mutation had not been reported in the normal population variant database (PM2), which was homozygous variation (PM3), but the deletion length accounted for more than 10% of the total protein length (PVS1). This variant can be rated as “likely pathogenic” (PVS1+PM3+PM2) according to ACMG guidelines.
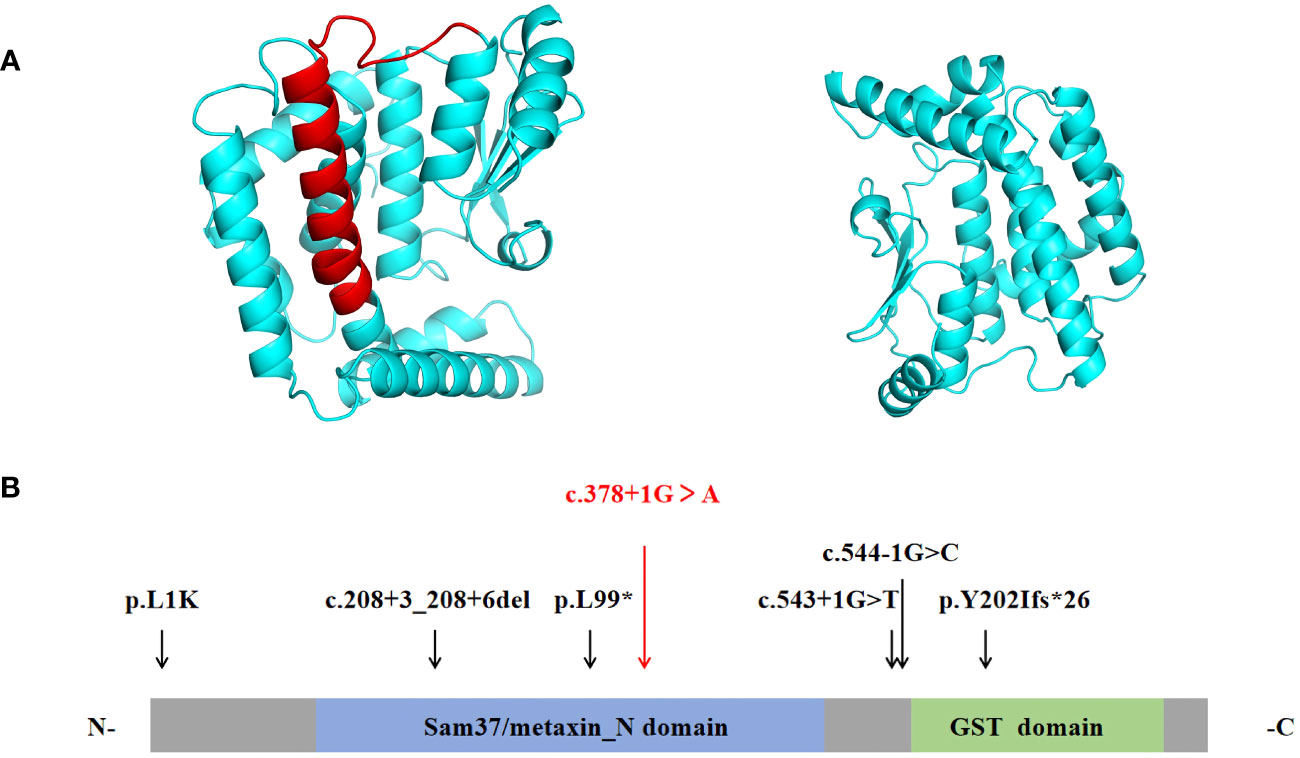
Figure 2 (A) The ribbon protein models of wild-type (left) and c.378 + 1G > A mutant forms (right) are displayed. The red section indicates the area where the mutant protein is lost. (B) Protein structure of MTX2. Previously reported mutations in the MTX2 gene are indicated by black vertical arrows, and the mutation reported in this patient is shown in red. (N- N terminus; C-C terminus).
4 Discussion
4.1 Mechanism
MADaM is caused by a mutation in the MTX2 gene, which encodes a mitochondrial outer membrane protein known as Metaxin-2 (11, 13). Metaxin-2 (MTX2) interacts directly with Metaxin-1(MTX1), being part of the mitochondrial sorting and assembly machinery (SAM) responsible for the correct integration of β-barrel proteins into the outer mitochondrial membrane (14). Previous functional studies have shown that loss of MTX2 leads to decreased MTX1 protein levels, and causes mitochondrial network fragmentation, decreased oxidative phosphorylation, resistance to apoptosis triggering by TNF-α, increased senescence and autophagy, reduced proliferation, and abnormal nuclear morphology (11). These ultimately lead to severe features of progeroid in the patient.
4.2 Molecular mapping
MTX2 located on chromosome 2q31.1, consists of 263 amino acids and contained two functional domains: Sam37/metaxin_N domain (41-162AA) and glutathione S-transferase (GST) domain (187-249AA) (https://www.ebi.ac.uk/interpro/protein/UniProt/O75431). The six previously reported MTX2 variants in MADaM patients are summarized in Figure 2B, all variants were homozygous and from six consanguineous families (11, 12). There were three splicing mutations, two frameshift mutations, and one missense mutation. The c.378 + 1G > A mutation reported in this study is a splicing mutation, which is predicted to cause exon 6 skipping. Exon 6 of MTX2 encodes 96 to 126 amino acids, and skipping of exon 6 is predicted to lose a portion of the Sam37/metaxin_N domain.
4.3 Clinical features
MADaM, like other types of MAD, also shown as autosomal recessive, and the patients reported so far are all homozygous mutations in the MTX2 gene, which may be related to their consanguineous family history (11, 12). The clinical symptoms of MADA typically appeared at the age of 4-5 years, with most cases being partial lipodystrophy in the torso and limbs. The symptoms of MADB were more severe than those of MADA (6). All MADaM patients had a typical progeroid facial appearance, severe growth retardation, generalized lipodystrophy, clavicular hypoplasia, and distal osteolysis, indicating a severe progeroid form of MAD. In addition, 5/7 of the MADaM patients had hypotonia, which had not been reported in MAD patients previously. At present, MAD is considered to be a segmental form of progeria, mainly affecting bone, skin and adipose tissue, with the brain barely involved. So far, the reported cognitive development of MADA, MADB and MADaM is normal (Table 1) (11, 12, 15, 16).
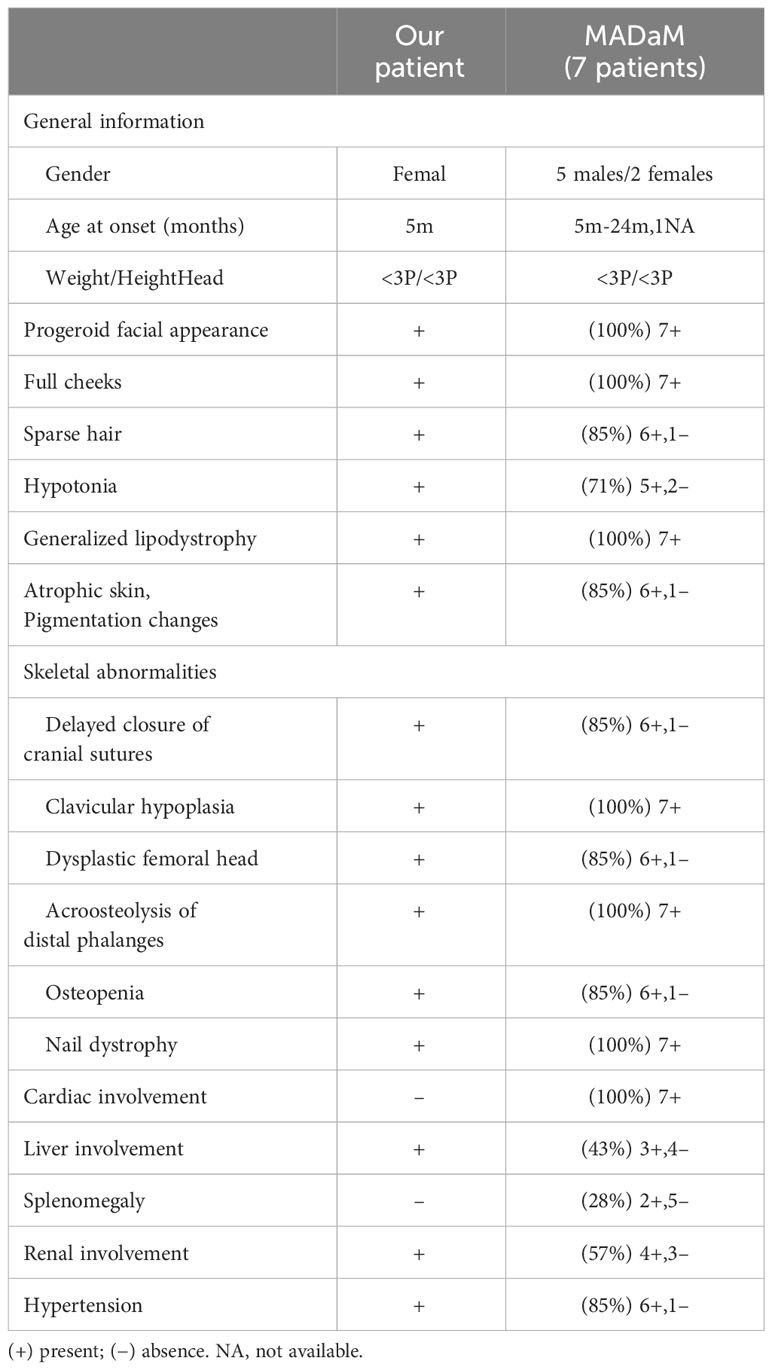
Table 1 Clinical features of our patients and those 7 MADaM individuals reported in the literature.
Delayed closure of the cranial suture, clavicle hypoplasia and thin ribs due to osteolysis, acral osteolysis, joint contracture and nail dysplasia are common in almost all MAD cases (2). Dysplasia of the femoral head is rarely reported and has been reported in 6/7 of MADaM cases (11, 12). Occipital ossification defect is thought to be a unique feature of MADB (17), showing fragmented ossification on cranial imaging, which has not been found in MADaM to date. The ‘Beaten silver’ skull observed on the skull radiograph of the MADaM patient reported by Yeter Doğan et al. (12), may be related to increased intracranial pressure caused by cranial suture atresia, which was not found in our patient.
MAD patients tend to have thin wrinkled skin with pigmentary changes, which are also found in 6/7 of MADaM patients (11, 12). In addition, subcutaneous calcification has been found in patients with MADB (15), but not in patients with MADaM.
In contrast to other types of MAD, MADaM appears to be more likely to involve other organs at an early stage. Cardiac involvement was present in all 7 previously reported MADaM cases, including 4 cases with left ventricular hypertrophy and 3 others with dilated cardiomyopathy, mitral valve prolapse and patent foramen ovale, respectively (11, 12). Cardiac involvement has been identified in only 4 MAD patients so far (15, 18–20). With the exception of 1 case, a 4-year-old girl, who developed right heart failure due to recurrent infections, all but one occurred after the age of 20 years, and 2 after the age of 40 years. 6/7 of cardiac involvement in MADaM was left ventricular hypertrophy, probably related to severe hypertension in the patient.
Currently, there is no case report of MADA patients with renal involvement, and there are 6 cases of MADB (5, 6, 17, 18, 21), 2 of them eventually developed chronic renal failure in their second or third decade (5, 18). Most of MADaM had renal involvement (4/7) and hypertension (6/7) in the early stage, including 3 cases of focal segmental glomerulosclerosis and 1 case of IgM nephropathy (11, 12). The main manifestations of MADaM were proteinuria and microscopic hematuria. The patient reported in this study had massive proteinuria, hematuria and severe hypertension from the age of one year, and he gradually developed hyperlipidemia, despite treatment with enalapril with poor efficacy, similar to previous reports of MADB with chronic kidney disease.
Our patient presented with recurrent transaminase elevations before one year of age without hepatic dysfunction manifestations such as decreased albumin or increased bilirubin, and we excluded viral infections or other factors for hereditary liver disease. Hepatosplenic involvement is uncommon in MAD, but 3/7 of MADaM from our statistics had liver involvement (hepatomegaly or elevated transaminases) and 2/7 had splenomegaly (11, 12). Whether hepatosplenic involvement is a unique clinical feature of MADaM to distinguish it from other MAD requires more data in the future.
In this case, the patient also had a significant decrease in plasma IgG levels, which led to multiple hospitalizations due to infection. Low IgG levels have not been reported in previous patients with MADA, MADB, and MADaM. Clinical data have shown that the vast majority of children with nephropathy have obvious hypogammaglobulinemia, and the specific mechanism is not clear, and some studies suggest that it may be related to B-cell immunoglobulin class conversion dysfunction (22). Therefore, hypogammaglobulinemia in this patient may be a secondary manifestation of her nephropathy.
Few cases of MADaM have been reported so far, and its long-term prognosis is unknown. The oldest reported case of MADA is 56 years old, a Japanese woman who was paraplegic due to vertebral destruction at age 56 (23). A total of 5 patient deaths occurred, including 1 case of MADA and 4 cases of MADB (5, 15, 18, 24, 25). Two of the five patients died of respiratory failure in early stage (24, 25), and the rest died of kidney or heart failure in the second or third decade (5, 15, 18). In this case, the patient has been hospitalized for infection many times, and even developed respiratory failure, accompanied by complications such as elevated aminotransferases, nephropathy and hypertension. In addition to enalapril, dipyridamole and gamma globulin supplementation, we also tried to give the patient cocktail therapy, and the specific effect still needs long-term follow-up.
4.4 Characteristics of progeria syndrome in Chinese patients
So far, a total of 22 cases of progeria syndrome in the Chinese population have been reported, and their clinical features and genotypes are shown in Table 2. Among them, MADA or HGPS caused by LMNA gene mutation was the most common (72.7%), followed by MDPL caused by POLD1 gene mutation (22.7%), and MADB was only 1 case, brain imaging examination showed typical failure of occipital ossification (35). The end-product of the LMNA gene lamin A is formed from the prelamin A (precursor protein of lamin A) through a series of shearing processing, which undergoes a series of processes such as farnesylation and hydrolysis by ZMPSTE24 enzyme to form mature lamin A (2). Lamins are part of the nuclear structural scaffold and play an important role in the structure and function of the nucleus. When LMNA or ZMPSTE24 genes are mutated, they can lead to the toxic accumulation of prelamin A, leading to multiple types of premature aging laminopathies. The severity of the disease is mainly related to the degree of prelamin A accumulation (2).
Both MADA and HGPS are caused by mutations in the LMNA gene. The inheritance pattern of MADA is autosomal recessive, which is usually caused by homozygous or compound heterozygous mutations of LMNA (2). Previous reports have suggested that p.R527H is the most common mutation (20, 41). However, genetic analysis of Chinese progeria patients revealed that the LMNA gene p. R527C homozygous mutation is the most common mutation, and it is presumed that R527C is a hot spot mutation in Chinese MADA patients (Table 2). HGPS is thought to be autosomal dominant inherited disease and mostly caused by the p.G608G heterozygous mutation, which is known as classical HGPS (31). Individuals who exhibit the characteristic clinical features of HGPS, accompanied by other LMNA mutations, are often referred to as atypical HGPS(AHGPS) (33). However, since MADA has more overlap with HGPS clinical manifestations and is caused by mutations in the same gene, the clinical features cannot be easily distinguished (24). Plasilova et al. described the LMNA homozygous mutation p.K542N in four patients with an overlapping phenotype of MAD and early-onset progeria/HGPS, concluding that MAD and HGPS are a clinical spectrum disorder rather than separate disorders (24). HGPS clinical symptoms are generally characterized by an earlier onset and more severe clinical manifestations than MAD, with a mean life expectancy of 14.6 years, and the leading cause of death being stroke or myocardial obstruction (42). At present, three cases of HGPS caused by p.G608G heterozygous mutation have been reported in China, two of whom had cerebral infarction in childhood (31), which is a serious cardiovascular disease, suggesting the early involvement of cardiovascular system in HGPS.
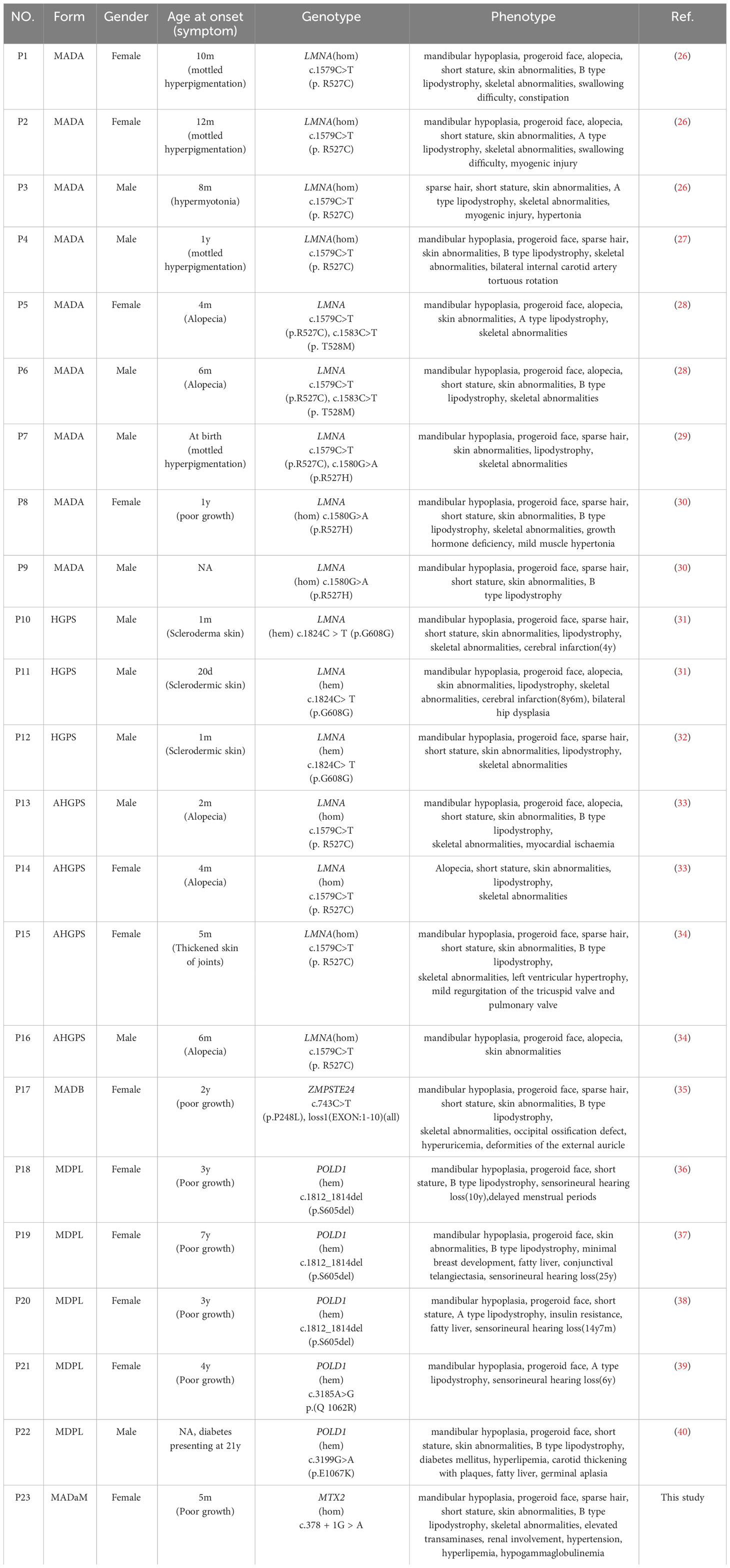
Table 2 Clinical data review of Chinese progeria patients.
Although previous reports suggested that clinical symptoms of MADA usually appear in early childhood (4-5 years) and MADB signs and symptoms can appear around the age of 2 years, our analysis of 22 progeria patients reported in China revealed that, except for five cases of MDPL mostly in preschool or school age where growth retardation was the predominant manifestation, most other progeria patients had clinical manifestations of progeria from infancy, mainly skin changes (8/17) and alopecia (6/17) (Table 2), and progressive skeletal and lipodystrophy with growth retardation, the main skeletal manifestation being progressive osteolysis of the extremities, which leads to hypoplasia of the mandible, terminal phalanges, clavicles, and ribs. Xu et al. performed a growth hormone provocation test in a Chinese MADA patient with the p.R527H homozygous mutation in the LMNA gene, which suggested partial growth hormone deficiency (30), and Sakka et al. reported that three MADA patients with the p.R527H homozygous mutation also had growth hormone deficiency, and they treated three patients with growth hormone for 18 months, but none of them had any effect (20), failure of response to trials of GH therapy in this patients could be a result of GH-IGF1 signaling disruption by pro-inflammatory cytokines, indeed IL-6 inappropriate secretion.
The early stage of MDPL is mainly characterized by growth retardation. PODL1 gene c.1812_1814del(p.S605del) mutation has been reported as a hot spot mutation, which is also the most common mutation in China (3/5). One Japanese patient was examined with a growth hormone provocation test, which showed results suggestive of a normal range, sensorineural deafness was noted at ages 6-25 years, and effective treatment with hearing aids (43). In addition to this progeria, growth retardation and deafness, 4/5 patients had endocrine abnormalities, such as menstrual delay, gonadal dysgenesis, fatty liver, insulin resistance, diabetes and hyperlipidemia, which is similar to what has been reported abroad, and most MDPL patients had combined endocrine and metabolic abnormalities (Table 2).
5 Conclusion
We report the first case of MADaM syndrome caused by mutations in the MTX2 gene in China, with some of the clinical features have not been previously reported. The clinical phenotypes of patients with progeroid syndromes often overlap, and early genetic testing is helpful for typing to provide clinical guidance for their treatment and prognosis follow-up.
Data availability statement
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Ethics Committee of Shenzhen Children’s Hospital. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s), and minor(s)’ legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
Author contributions
XHF: Software, Resources, Investigation, Funding acquisition, Data curation, Conceptualization, Writing – review & editing, Writing – original draft. SC: Writing – original draft, Methodology, Investigation. XH: Writing – original draft, Formal analysis, Data curation. QHL: Writing – original draft, Formal analysis, Data curation. YFC: Writing – original draft, Formal analysis, Data curation. WNL: Writing – original draft, Visualization. QY: Writing – review & editing, Supervision, Resources.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Shenzhen Key Medical Discipline Construction Fund(No.SZXK033).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Young LW, Radebaugh JF, Rubin P, Sensenbrenner JA, Fiorelli G, McKusick VA. New syndrome manifested by mandibular hypoplasia, acroosteolysis, stiff joints and cutaneous atrophy (mandibuloacral dysplasia) in two unrelated boys. Birth Defects Orig Artic Ser. (1971) 7:291–7.
2. Cenni V, D’Apice MR, Garagnani P, Columbaro M, Novelli G, Franceschi C, et al. Mandibuloacral dysplasia: A premature ageing disease with aspects of physiological ageing. Ageing Res Rev. (2018) 42:1–13. doi: 10.1016/j.arr.2017.12.001
3. Novelli G, Muchir A, Sangiuolo F, Helbling-Leclerc A, D’Apice MR, Massart C, et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-encoding lamin A/C. Am J Hum Genet. (2002) 71:426–31. doi: 10.1086/341908
4. Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. (2003) 423:293–8. doi: 10.1038/nature01629
5. Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum Mol Genet. (2003) 12:1995–2001. doi: 10.1093/hmg/ddg213
6. Cunningham VJ, D’Apice MR, Licata N, Novelli G, Cundy T. Skeletal phenotype of mandibuloacral dysplasia associated with mutations in ZMPSTE24. Bone. (2010) 47:591–7. doi: 10.1016/j.bone.2010.06.004
7. Puente XS, Quesada V, Osorio FG, Cabanillas R, Cadiñanos J, Fraile JM, et al. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am J Hum Genet. (2011) 88:650–6. doi: 10.1016/j.ajhg.2011.04.010
8. Weedon MN, Ellard S, Prindle MJ, Caswell R, Lango Allen H, Oram R, et al. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet. (2013) 45:947–50. doi: 10.1038/ng.2670
9. Murdocca M, Spitalieri P, De Masi C, Udroiu I, Marinaccio J, Sanchez M, et al. Functional analysis of POLD1 p.ser605del variant: the aging phenotype of MDPL syndrome is associated with an impaired DNA repair capacity. Aging (Albany NY). (2021) 13:4926–45. doi: 10.18632/aging.202680
10. Elouej S, Beleza-Meireles A, Caswell R, Colclough K, Ellard S, Desvignes JP, et al. Exome sequencing reveals a de novo POLD1 mutation causing phenotypic variability in mandibular hypoplasia, deafness, progeroid features, and lipodystrophy syndrome (MDPL). Metabolism. (2017) 71:213–25. doi: 10.1016/j.metabol.2017.03.011
11. Elouej S, Harhouri K, Le Mao M, Baujat G, Nampoothiri S, Kayserili H, et al. Loss of MTX2 causes mandibuloacral dysplasia and links mitochondrial dysfunction to altered nuclear morphology. Nat Commun. (2020) 11:4589. doi: 10.1038/s41467-020-18146-9
12. Yeter Doğan B, Günay N, Ada Y, Doğan ME. A novel MTX2 gene splice site variant resulting in exon skipping, causing the recently described mandibuloacral dysplasia progeroid syndrome. Am J Med Genet A. (2023) 191:173–82. doi: 10.1002/ajmg.a.63010
13. Armstrong LC, Saenz AJ, Bornstein P. Metaxin 1 interacts with metaxin 2, a novel related protein associated with the mammalian mitochondrial outer membrane. J Cell Biochem. (1999) 74:11–22. doi: 10.1002/(ISSN)1097-4644
14. Paschen SA, Waizenegger T, Stan T, Preuss M, Cyrklaff M, Hell K, et al. Evolutionary conservation of biogenesis of beta-barrel membrane proteins. Nature. (2003) 426:862–6. doi: 10.1038/nature02208
15. Hitzert MM, van der Crabben SN, Baldewsingh G, van Amstel HKP, van den Wijngaard A, van Ravenswaaij-Arts CMA, et al. Mandibuloacral dysplasia type B (MADB): a cohort of eight patients from Suriname with a homozygous founder mutation in ZMPSTE24 (FACE1), clinical diagnostic criteria and management guidelines. Orphanet J Rare Dis. (2019) 14:294. doi: 10.1186/s13023-019-1269-0
16. Jéru I, Nabil A, El-Makkawy G, Lascols O, Vigouroux C, Abdalla E. Two decades after mandibuloacral dysplasia discovery: additional cases and comprehensive view of disease characteristics. Genes (Basel). (2021) 12:1508. doi: 10.3390/genes12101508
17. Haye D, Dridi H, Levy J, Lambert V, Lambert M, Agha M, et al. Failure of ossification of the occipital bone in mandibuloacral dysplasia type B. Am J Med Genet A. (2016) 170:2750–5. doi: 10.1002/ajmg.a.37825
18. Ben Yaou R, Navarro C, Quijano-Roy S, Bertrand AT, Massart C, De Sandre-Giovannoli A, et al. Type B mandibuloacral dysplasia with congenital myopathy due to homozygous ZMPSTE24 missense mutation. Eur J Hum Genet. (2011) 19:647–54. doi: 10.1038/ejhg.2010.256
19. Al-Haggar M, Madej-Pilarczyk A, Kozlowski L, Bujnicki JM, Yahia S, Abdel-Hadi D, et al. A novel homozygous p.Arg527Leu LMNA mutation in two unrelated Egyptian families causes overlapping mandibuloacral dysplasia and progeria syndrome. Eur J Hum Genet. (2012) 20:1134–40. doi: 10.1038/ejhg.2012.77
20. Sakka R, Marmouch H, Trabelsi M, Achour A, Golli M, Hannachi I, et al. Mandibuloacral dysplasia type a in five Tunisian patients. Eur J Med Genet. (2021) 64:104138. doi: 10.1016/j.ejmg.2021.104138
21. Miyoshi Y, Akagi M, Agarwal AK, Namba N, Kato-Nishimura K, Mohri I, et al. Severe mandibuloacral dysplasia caused by novel compound heterozygous ZMPSTE24 mutations in two Japanese siblings. Clin Genet. (2008) 73:535–44. doi: 10.1111/j.1399-0004.2008.00992.x
22. Yang X, Tang X, Li T, Man C, Yang X, Wang M, et al. Circulating follicular T helper cells are possibly associated with low levels of serum immunoglobulin G due to impaired immunoglobulin class-switch recombination of B cells in children with primary nephrotic syndrome. Mol Immunol. (2019) 114:162–70. doi: 10.1016/j.molimm.2019.07.001
23. Kosho T, Takahashi J, Momose T, Nakamura A, Sakurai A, Wada T, et al. Mandibuloacral dysplasia and a novel LMNA mutation in a woman with severe progressive skeletal changes. Am J Med Genet A. (2007) 143A:2598–603. doi: 10.1002/ajmg.a.31983
24. Plasilova M, Chattopadhyay C, Pal P, Schaub NA, Buechner SA, Mueller H, et al. Homozygous missense mutation in the lamin A/C gene causes autosomal recessive Hutchinson-Gilford progeria syndrome. J Med Genet. (2004) 41:609–14. doi: 10.1136/jmg.2004.019661
25. Shackleton S, Smallwood DT, Clayton P, Wilson LC, Agarwal AK, Garg A, et al. Compound heterozygous ZMPSTE24 mutations reduce prelamin A processing and result in a severe progeroid phenotype. J Med Genet. (2005) 42:e36. doi: 10.1136/jmg.2004.029751
26. Luo DQ, Wang XZ, Meng Y, He DY, Chen YM, Ke ZY, et al. Mandibuloacral dysplasia type A-associated progeria caused by homozygous LMNA mutation in a family from Southern China. BMC Pediatr. (2014) 14:256. doi: 10.1186/1471-2431-14-256
27. Yang F, Li Q, Liu BH, Huang JM, Jiang BX, Mei ZX, et al. Mutation analysis of the LMNA gene in a child with mandibuloacral dysplasia and progeria. J Clin Dermatol. (2023) 52:7–10. doi: 10.16761/j.cnki.1000-4963.2023.01.003
28. Xiang S, Zhang X, Li XY, Bi Y, Xiao N. A family study of mandibuloacral dysplasia with type A lipodystrophy. J Clin Pediatr. (2014) 32:1084–8. doi: 10.3969/j.issn.1000-3606.2014.11.021
29. Li X, Jiang Y, Wang O, Li M, Xing XP, Xia WB. Clinical features of mandibuloacral dysplasia caused by LMNA mutation. Chin J OSTEOPOR OSIS &BONE MINER Res. (2019) 12:347–55.
30. Xu XM, Chen Y, Li X, Li Q, Wang YR, Shen YN, et al. Mandibuloacral dysplasia type A: a familial report and literature review. J Clin Pediatr. (2019) 37:677–81. doi: 10.3969/j.issn.1000-3606.2019.09.010
31. Wang J, Yu Q, Ma X, Yuan Z, Mao J. Hutchinson-Gilford progeria syndrome complicated with stroke: A report of 2 cases and literature review. Front Pediatr. (2022) 10:1056225. doi: 10.3389/fped.2022.1056225
32. Chu Y, Xu ZG, Xu Z, Ma L. Hutchinson-Gilford progeria syndrome caused by an LMNA mutation: a case report. Pediatr Dermatol. (2015) 32:271–5. doi: 10.1111/pde.12406
33. Liang L, Zhang H, Gu X. Homozygous LMNA mutation R527C in atypical Hutchinson-Gilford progeria syndrome: evidence for autosomal recessive inheritance. Acta Paediatr. (2009) 98:1365–8. doi: 10.1111/j.1651-2227.2009.01324.x
34. Xiong Z, Lu Y, Xue J, Luo S, Xu X, Zhang L, et al. Hutchinson-Gilford progeria syndrome accompanied by severe skeletal abnormalities in two Chinese siblings: two case reports. J Med Case Rep. (2013) 7:63. doi: 10.1186/1752-1947-7-63
35. Wu DD, Li R, Li XN, Liu QQ, Dou LH. Diagnosis and genetic analysis of a case with mandibuloacral dysplasia type B due to compound heterozygous mutations of the ZMPSTE24 gene. Yi Chuan. (2022) 44:1167–74. doi: 10.16288/j.yczz.22-117
36. Liu ZQ, Chen XB. Madibular hypoplasia, deafness, progeroid features, and lipodystrophy syndrome: One case report. Chin J Endocrinol Metab. (2018) 34:246–8. doi: 10.3760/cma.j.issn.1000-6699.2018.03.011
37. Yu PT, Luk HM, Mok MT, Lo FI. Evolving clinical manifestations of mandibular hypoplasia, deafness, progeroid features, and lipodystrophy syndrome: From infancy to adulthood in a 31-year-old woman. Am J Med Genet A. (2021) 185:995–8. doi: 10.1002/ajmg.a.62035
38. Wang XW, Lu LY, Xie Y, Yu XJ. A Chinese girl with mandibular hypoplasia, deafness, progeroid features, and lipodystrophy (MDPL) diagnosed via POLD1 mutation detection. Chin Med J (Engl). (2020) 133:2009–11. doi: 10.1097/CM9.0000000000000986
39. Zuo B, Xu H, Pan Z, Mao L, Feng H, Zeng B, et al. A likely pathogenic POLD1 variant associated with mandibular hypoplasia, deafness, progeroid features, and lipodystrophy syndrome in a Chinese patient. BMC Med Genomics. (2022) 15:220. doi: 10.1186/s12920-022-01374-x
40. He YF, Li H, Lu J. Mandibular hypoplasia, deafness, progeroid features and lipodystrophy syndrome caused by POLD1 mutation: a case report. Acad J Naval Med University. (2022) 43:848–50. doi: 10.16781/j.CN31-2187/R.20200934
41. Garavelli L, D’Apice MR, Rivieri F, Bertoli M, Wischmeijer A, Gelmini C, et al. Mandibuloacral dysplasia type A in childhood. Am J Med Genet A. (2009) 149A:2258–64. doi: 10.1002/ajmg.a.33005
42. Gordon LB, Massaro J, D’Agostino RB Sr., Campbell SE, Brazier J, Brown WT, et al. Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation. (2014) 130:27–34. doi: 10.1161/CIRCULATIONAHA.113.008285
Keywords: MTX2 gene, mandibular hypoplasia (MAD) syndrome, progeria syndrome, splice-site mutation, exon skipping
Citation: Fu XH, Chen SL, Huang X, Lu QH, Cui YF, Lin WN and Yang Q (2024) Case report: A novel splice-site mutation of MTX2 gene caused mandibuloacral dysplasia progeroid syndrome: the first report from China and literature review. Front. Endocrinol. 15:1345067. doi: 10.3389/fendo.2024.1345067
Received: 27 November 2023; Accepted: 23 February 2024;
Published: 13 March 2024.
Edited by:
Ahmed Rebai, Centre of Biotechnology of Sfax, TunisiaReviewed by:
Hisaya Kato, Chiba University, JapanEmilia Severin, Carol Davila University of Medicine and Pharmacy, Romania
Copyright © 2024 Fu, Chen, Huang, Lu, Cui, Lin and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qin Yang, eWFuZ3FpbjA1MzlAc2luYS5jb20=