 Maria Eleni Chondrogianni1,2†
Maria Eleni Chondrogianni1,2† Ioannis Kyrou3,4,5,6,7,8,9†
Ioannis Kyrou3,4,5,6,7,8,9† Theodoros Androutsakos10
Theodoros Androutsakos10 Christina-Maria Flessa1Evangelos Menenakos11Kamaljit Kaur Chatha5,6,12Yekaterina Aranan4,5
Christina-Maria Flessa1Evangelos Menenakos11Kamaljit Kaur Chatha5,6,12Yekaterina Aranan4,5 Athanasios G. Papavassiliou1
Athanasios G. Papavassiliou1 Eva Kassi1,2*‡
Eva Kassi1,2*‡ Harpal S. Randeva4,5,6,7*‡
Harpal S. Randeva4,5,6,7*‡- 1Department of Biological Chemistry, Medical School, National and Kapodistrian University of Athens, Athens, Greece
- 2Endocrine Unit, 1st Department of Propaupedic Internal Medicine, Laiko Hospital, National and Kapodistrian University of Athens, Athens, Greece
- 3Laboratory of Dietetics and Quality of Life, Department of Food Science and Human Nutrition, School of Food and Nutritional Sciences, Agricultural University of Athens, Athens, Greece
- 4Warwickshire Institute for the Study of Diabetes, Endocrinology and Metabolism (WISDEM), University Hospitals Coventry and Warwickshire NHS Trust, Coventry, United Kingdom
- 5Institute for Cardiometabolic Medicine, University Hospitals Coventry and Warwickshire NHS Trust, Coventry, United Kingdom
- 6Warwick Medical School, University of Warwick, Coventry, United Kingdom
- 7Centre for Health & Life Sciences, Coventry University, Coventry, United Kingdom
- 8Aston Medical School, College of Health and Life Sciences, Aston University, Birmingham, United Kingdom
- 9College of Health, Psychology and Social Care, University of Derby, Derby, United Kingdom
- 10Department of Pathophysiology, Medical School, National and Kapodistrian University of Athens, Athens, Greece
- 115th Surgical Clinic, Department of Surgery, ‘Evgenidion Hospital’, National and Kapodistrian University of Athens Medical School, Athens, Greece
- 12Department of Biochemistry and Immunology, University Hospitals Coventry and Warwickshire NHS Trust, Coventry, United Kingdom
Over the last years non-alcoholic fatty liver disease (NAFLD) has grown into the most common chronic liver disease globally, affecting 17-38% of the general population and 50-75% of patients with obesity and/or type 2 diabetes mellitus (T2DM). NAFLD encompasses a spectrum of chronic liver diseases, ranging from simple steatosis (non-alcoholic fatty liver, NAFL) and non-alcoholic steatohepatitis (NASH; or metabolic dysfunction-associated steatohepatitis, MASH) to fibrosis and cirrhosis with liver failure or/and hepatocellular carcinoma. Due to its increasing prevalence and associated morbidity and mortality, the disease-related and broader socioeconomic burden of NAFLD is substantial. Of note, currently there is no globally approved pharmacotherapy for NAFLD. Similar to NAFLD, osteoporosis constitutes also a silent disease, until an osteoporotic fracture occurs, which poses a markedly significant disease and socioeconomic burden. Increasing emerging data have recently highlighted links between NAFLD and osteoporosis, linking the pathogenesis of NAFLD with the process of bone remodeling. However, clinical studies are still limited demonstrating this associative relationship, while more evidence is needed towards discovering potential causative links. Since these two chronic diseases frequently co-exist, there are data suggesting that anti-osteoporosis treatments may affect NAFLD progression by impacting on its pathogenetic mechanisms. In the present review, we present on overview of the current understanding of the liver-bone cross talk and summarize the experimental and clinical evidence correlating NAFLD and osteoporosis, focusing on the possible effects of anti-osteoporotic drugs on NAFLD.
1 Introduction
Non-alcoholic fatty liver disease (NAFLD; or metabolic dysfunction-associated fatty liver disease, MAFLD; or metabolic dysfunction-associated steatotic liver disease, MASLD) is one of the most common causes of chronic liver disease worldwide. Indeed, over the past couple of decades NAFLD has grown into the most common chronic liver disease, with prevalence of 17-38% in the general population (1), and 50-75% in patients with obesity and/or type 2 diabetes mellitus (T2DM) (2, 3). NAFLD is determined as steatosis affecting 5% of the liver volume or weight (accumulation of fat in more than 5% of hepatocytes) (4), and encompasses a spectrum of liver diseases, ranging from simple steatosis (non-alcoholic fatty liver, NAFL) and non-alcoholic steatohepatitis (NASH or metabolic dysfunction-associated steatohepatitis, MASH) to fibrosis and cirrhosis with liver failure or/and hepatocellular carcinoma. Notably, based on recent estimates, cirrhosis due to NAFLD is expected to be the leading cause of liver transplantation in the US by 2030 (5). Additionally, the economic burden of NAFLD/NASH on health systems is enormous, while that there is currently no globally approved treatment specifically for NAFLD/NASH (6).
Currently, NAFLD is diagnosed by detecting steatosis (either by imaging or histologically) and by excluding other causes of liver disease, including exclusion of alcoholic liver disease (ALD) which has similar pathologic spectra with NAFLD (in NAFLD the daily alcohol consumption should not exceed 20 g in women or 30 g in men) (7). For the diagnosis, as well as its staging, of NASH and cirrhosis, liver biopsy remains the ‘gold standard’, which, however, also has limitations since it is an invasive method with possible complications, sampling errors and high cost (8, 9). Of note the renaming of NAFLD as MAFLD or MASLD has recently been proposed, with new proposed diagnostic criteria, namely hepatic steatosis based on histological (biopsy), imaging or biochemical confirmation along with one of the following: (a) overweight/obesity; (b) T2DM; or (c) metabolic dysfunction as indicated by 2 of the following: increased waist circumference, hypertension, elevated triglycerides, low HDL, prediabetes (IGT, IFG), HOMA index>2.5, elevated CRP, which rather put a positive diagnosis and not an exclusion of other causes of liver disease (10).
Osteoporosis is also a chronic disease characterized by decreased bone density and a disruption of the bone’s architectural structure, resulting in bone fragility and increased fracture risk (11). Osteoporosis is particularly prevalent in postmenopausal women, where the noted estrogen decrease leads to an increase of the activity of the osteoclasts, increasing their responsiveness to RANKL which binds to RANK on the osteoclast membrane, and resulting in the differentiation of osteoclast precursors into mature osteoclasts (12). The drugs used for the treatment of osteoporosis are antiresorptives (e.g. bisphosphonates, denosumab and raloxifene), bone anabolic agents (e.g. teriparatide and romosozumab) and calcitonin (13). Similar to NAFLD, osteoporosis progresses as a silent disease until an osteoporotic fracture occurs, which also poses a very significant disease-related and socio-economic burden (6).
Recent data have been highlighting potential links between NAFLD and osteoporosis, linking the pathogenesis of NAFLD to the process of bone remodeling. In this context, it is considered that chronic low-grade inflammation plays a crucial role in the pathogenesis of both diseases (14, 15). However, this field is still open and more evidence is needed towards understanding the potential common pathogenetic mechanism(s) and the correlations/links between NAFLD and osteoporosis.
In the present review, we present an overview of the current understanding of the liver-bone cross talk and summarize the experimental and clinical evidence correlating NAFLD and osteoporosis. Since these two diseases frequently co-exist, medications for the treatment of osteoporosis may affect NAFLD progression by impacting on underlying pathogenetic mechanisms/links. As such, herein, we also focus on insights into the possible effects of anti-osteoporotic drugs on NAFLD.
2 Pathogenetic mechanisms linking NAFLD and osteoporosis
Potential common pathogenetic mechanisms linking NAFLD and osteoporosis have been recently described. The pathogenesis of NAFLD was initially described with the ‘two-hit’ hypothesis, with steatosis, i.e. the accumulation of triglycerides (TAGs) in the liver, representing the ‘first hit’ and triggering the expression of pro-inflammatory cytokines (e.g. NF-a, IL-6) which was described as the ‘second’ hit (16). The latter results to the activation of pro-inflammatory pathways and potential fibrogenesis in the liver. However, the spectrum of mechanisms implicated in NAFLD pathogenesis appears to be much more complex, and, thus, has been more recently described with the “multiple-hit hypothesis” which includes multiple genetic and environmental factors that may result to obesity, insulin resistance, gut microbiome alterations, adipose tissue dysfunction and liver fat accumulation with or without hepatic inflammation (17). For example, hepatic mitochondrial dysfunction leads to endoplasmic reticulum (ER) stress, oxidative stress and the production of reactive oxygen species (ROS), while autophagy and apoptosis also play crucial role in NAFLD (18–20). Of note, even certain gut microbiome modifications appear to trigger the expression of pro-inflammatory cytokines (e.g. IL-6, TNF-α), whilst all the aforementioned mechanisms combined with genetic factors and epigenetic alterations may lead to chronic liver inflammation (17).
In this context, focus has been placed not only on traditional cytokines (e.g. IL-6, TNF-α), but also on additional factors implicated in these pathways such as adipokines (21, 22). Among these, adiponectin is the most abundant and is secreted predominantly by the white adipose tissue. Notably, adiponectin has anti-inflammatory and anti-atherogenic effects, and plays an important role in lipid and glucose metabolism by increasing insulin sensitivity, promoting the oxidation of free fatty acids, decreasing de novo synthesis/accumulation of lipids and protecting hepatic cells from apoptosis (23, 24). The effects of adiponectin on the liver are mediated by its receptors (AdipoR1, AdipoR2), interacting with the adaptor protein phosphotyrosine interaction (APPL1). Indeed, AdipoR1 activates the AMP-activated protein kinase (AMPK) and AdipoR2 the peroxisome proliferator–activated receptor-alpha (PPAR-a) signaling, and, thus, through these pathways adiponectin acts against hepatic lipid accumulation and regulates glucose homeostasis. Moreover, through the blockage of nuclear factor kappa (NF-κβ), adiponectin reduces inflammation. Notably, a diet rich in saturated and trans-fats, which directly induces significant hepatic fatty infiltration, has been shown to also reduce the circulating levels of adiponectin (25, 26). Furthermore, data show that adiponectin impedes hepatic fibrosis, by inhibiting platelet-derived growth factor (PDGF) stimulation and downregulating the transforming growth factor beta 1 (TGF-β1) (26), whilst as NAFLD progresses, adiponectin levels appear to decline (27). Interestingly, expression of adiponectin receptors has also been found in bone cells, both osteoblasts and osteoclasts (28). In addition, data from in vitro experiments and animal studies support an osteogenic role for adiponectin, by promoting osteoblastogenesis and limiting osteoclastogenesis (29). Accordingly, since NAFLD is associated with decreased adiponectin levels, the adiponectin downstream signaling pathways may favor osteoclast function and bone loss in patients with NAFLD. However, it should be noted that human data do not consistently point towards a favorable effect of adiponectin on bone biology (28, 29). Bacchetta et al. showed a negative association between bone mineral density (BMD) and adiponectin in patients with chronic kidney disease (30), whilst Jürimäe et al. demonstrated a negative correlation between adiponectin and BMD in a group of middle-aged premenopausal women (31). Moreover, a recent case-control study, including 210 postmenopausal women, showed an inverse relationship between serum adiponectin levels and T-score in women with osteoporosis and osteopenia (32). Finally a prospective study by Barbour et al., showed that high adiponectin levels were correlated with a higher fracture risk in men, but not in women (33).
Recent research focus has also been placed on osteocalcin (OC) which is secreted by osteoblasts and constitutes the most abundant non-collagen protein in bone (34). In its uncarboxylated form, OC exerts effects on bone by binding calcium (35), whilst it also plays a role on the pancreas-liver crosstalk and metabolism by promoting directly insulin expression in the pancreas and by increasing GLP-1 and adiponectin expression in adipocytes (36). Conversely, OC expression in osteoblasts is promoted by insulin and adiponectin (37). Several studies have demonstrated an inverse association between NAFLD and serum OC levels, with Yilmaz et al. showing that patients with NAFLD and increased hepatocyte ballooning degree had lower OC levels (38). Furthermore, Yang et al. demonstrated that Korean men with NAFLD had lower BMD and OC levels compared to those without (39), whilst Luo et al. showed that among postmenopausal Chinese women with normal blood glucose levels those with NAFLD had lower OC levels (40). Finally, Fang et al. also revealed that lower serum OC levels was an independent risk factor for NAFLD and progression to NASH (41). Interestingly, the hepatic inflammation observed during the progression of NAFLD due to lipotoxicity and the production of pro-inflammatory (e.g., TNF-α, IL-1, IL-6, IL-17) and prothrombotic factors appears to affect both the pathogenesis of NAFLD and bone tissue metabolism (14). RANKL, RANK and osteoprotegerin (OPG) are osteokines which are expressed in bone cells and regulate bone remodeling (42). The OPG/RANKL balance is highly important for the maintenance of bone health, with denosumab, a RANKL-binding monoclonal antibody, being approved as an anti-osteoporotic treatment (42). Of note, upregulation of the RANK/RANKL pathway induces the expression of pro-inflammatory cytokines, such as IL-1, IL-6, and TNF-α, which, in turn, promote osteoclast activation and bone resorption (43). RANKL binds to RANK on the osteoclast membrane, resulting in the differentiation of osteoclast precursors into mature osteoclasts (12), whilst OPG diminishes osteoclastogenesis and, thus, bone loss by binding to RANKL, preventing the RANK-RANKL activation of osteoclasts. Potential associations between RANKL, OPG and NAFLD have been investigated, with experimental data showing that the hepatic expression of RANKL may be elevated in patients with NAFLD (44). Furthermore, a study by Amrousy et al. in children with obesity and NAFLD showed that these children had both higher TNF-a and IL-6 levels and lower OC, OPG and adiponectin levels compared to the study controls (45). Moreover, Mantovani et al., trying to access bone turnover markers in postmenopausal T2DM patients with and without NAFLD (10 patients with NAFLD and fibrosis, 52 with NAFLD and without fibrosis, and 15 without NAFLD), found that RANKL levels gradually diminished from the study patients without NAFLD, to those with steatosis and then to those with steatosis and fibrosis, while sclerostin levels were higher in patients with NAFLD (46). Another study by Niksersht et al. in a sample of 57 men with NAFLD and 25 controls demonstrated that patients with NAFLD had lower levels of RANKL and OPG compared to controls, whilst OPG and RANKL gene expression was also reduced, suggesting a potential role in NAFLD pathogenesis (47). Similarly, Hadinia et al. showed that patients with NAFLD exhibited lower plasma RANK levels compared to controls, with diminished mRNA RANK levels (48). A previous study by Yilmaz et al. had also demonstrated that serum OPG levels were lower in patients with NASH compared to controls, suggesting that OPG could be used as a biomarker for NASH (49). A case-control study Niu et al. including T2DM patients with NAFLD (N=367) and without (N=379) NAFLD showed that OPG levels were lower in those with NAFLD (50). Interestingly, Erol et al., trying to investigate whether there is a correlation between OPG and insulin resistance in children with obesity, found that OPG levels were lower in such children, but failed to detect a difference in OPG concentrations between children with both obesity and NAFLD compared to those with obesity without NAFLD (51). Finally, Ayaz et al. have shown that serum OPG levels and carotid intima media thickness (CIMT) were higher in patients with NAFLD, with a positive association between OPG and CIMT in these patients (52). It appears that most of the studies point towards a positive correlation of serum RANKL with NAFLD, while serum OPG decreases with disease severity. Though, high serum OPG and low serum RANKL levels have also been reported in patients with advanced NAFLD-related fibrosis (46). It is difficult to explain this discrepancy, however, it should be noted that the elevation of circulating OPG levels and the decreased RANKL levels could represent a compensatory mechanism to limit the liver damage during the progress of NAFLD to fibrosis.
Of note, OPG and other molecules involved in bone metabolism, such as osteopontin (OPN) have also been associated with the progression of hepatic fatty infiltration to fibrosis (14). Indeed, focus is now placed on OPG which is a member of the TNF-a receptor family and acts as a cytokine by preventing RANK from binding to RANKL (53). In this context, Yang et al., in an attempt to suggest OPG as a noninvasive biomarker for NASH diagnosis and NAFLD progression, found that serum OPG was lower in NASH patients compared to normal controls (54). Similarly, OPN is a glycoprotein that plays a role in bone remodeling, bone matrix mineralization, bone remodeling, cell chemotaxis and cell survival and apoptosis (55). Bertola et al. studied the hepatic expression of OPN and its surface receptor CD44, in patients with obesity, showing that hepatic OPN levels were higher in those with severe steatosis and insulin resistance, suggesting their local implication in the hepatic injury progression (56). Moreover, Gómez-Santos et al. showed that OPN levels are higher in older patients, whilst this finding did not apply to patients with NAFLD where higher OPN levels were noted in younger patients. By also studying OPN deficient mice during aging, this study also showed that in older mice decreased OPN levels resulted in augmented senescence, ER stress, hepatic steatosis, and inflammation (57).
Insulin-like growth factor-1 (IGF-1) is mainly secreted by the liver while is also locally produced in small amounts by bones, affecting positively bone remodeling (58). Yao et al. in their metanalysis demonstrated that IGF-1 levels were decreased in patients with NAFLD compared to healthy controls (59). Moreover, Dichtel et al. showed that decreased IGF-1 levels were correlated with higher histological severity of NAFLD (60). Decreased IGF-1 have been reported in both patients with osteoporosis and NAFLD, indicating the important role of IGF-1 in the liver-bone axis (58).
To this end, Wang et al. investigated the role of IGF-1 in the progression of both NAFLD and osteoporosis (61). Using 48 female mice divided into two groups, WT and fed with high fat diet, they showed that bone loss, deterioration of bone microarchitecture and NAFLD were progressing in parallel. They demonstrated that changes of the TNF‐α, IL‐6, as well as IGF‐ 1 and IGFBP‐1 levels appear to play crucial roles in the different stages of NAFLD in HFD-fed mice. In particular, they showed that in 24 weeks the levels of TNF-α and IL-6 were higher in mice fed with HFD compared to controls leading to changes in the OPG/RANK/RANKL pathway. They concluded that changes in bone microstructure and BMD regarding the ‘second hit’ were due to higher levels of TNF-a and IL-6. They also showed that, in 32 weeks, IGF-1 was lower in mice fed with HFD resulting to reduced osteoblast activity, justifying bone changes in the progressive stage of NAFLD (61).In addition to the aforementioned factors, vitamin D deficiency, which has known effects on bone metabolism, seems to also influence NAFLD progression by inducing pro-inflammatory processes and oxidative stress, as well as stimulating the proliferation of stellate cells and the production of pro-fibrotic factors (e.g. PDGF and TGFβ) (14). Indeed, The role of vitamin D deficiency in the progression of NAFLD has been demonstrated in animal models (14), whilst clinical data, such as those from Wang et al., also suggest that 25(OH)-vitamin D levels are lower in patients with NAFLD (62).
Advanced glycation end-products (AGEs), molecules deriving from the glycation of proteins or lipids, seem to play a role in both the pathogenesis of NAFLD and osteoporosis (63, 64). Asadipooya et al. in their review provided a thorough description of how AGEs, through their receptors (RAGE), provoke inflammation, cellular proliferation, and increased oxidative stress that lead to the progression of steatosis to NASH and fibrosis, while vice versa oxidative stress and inflammation trigger the AGEs production (63). Of note, AGEs are also involved in bone metabolism. At low concentrations, AGEs promote osteoblastic activity, but at higher concentrations impair mineralization, induce osteoclastogenesis by upregulating RANKL, restrain osteoblasts’ growth, inhibit their differentiation and promote their apoptosis (64). Thus, targeting AGE-RAGE signaling appear to be very promising in preventing the progression of both NAFLD and bone loss.
Finally, emerging data also suggest that the Wnt signaling pathway may contribute to the liver-bone crosstalk. The role of the Wnt signaling pathway in osteogenesis is well-described, with Wnt-derived proteins diminishing apoptosis in osteoblast precursor cells and promoting osteoblast differentiation (65). Similarly, the role of Wnt/beta catenin signaling - where the binding of a Wnt ligand with a surface receptor (Fzd) and a co-receptor (LRP5/6) is responsible for the stabilization of beta/catenin, its nuclear translocation and Wnt target gene expression - in NAFLD development has been described, as recently reviewed in detail by Harini et al. (66). In this context, the role of the canonical and non- canonical Wnt pathway is considered crucial in the development of NAFLD, with the latter being promoted or suppressed according to Wnt5a binding, whilst NAFLD can induced by the inhibition of the canonical pathway. As such, it is noteworthy that mutations in the Wnt co-receptor low density lipoprotein (LDL) receptor-related protein 6 (LRP6) can provoke NAFLD (66), with Liu et al. demonstrating that LRP6+/- mice were protected against insulin resistance and obesity (67).
3 Studies on NAFLD and osteoporosis links
A number of mostly cross-sectional studies have examined the interrelation between NAFLD and BMD, as presented in Table 1. Of these, several demonstrated that patients with NAFLD had higher risk of osteoporosis. For example, Chen et al. have shown that there is a decrease in the rate of bone production and an increase in the rate of bone resorption in elderly patients with NAFLD relative to individuals without NAFLD (78). Similarly, a recent study by Lee et al. in men older than 50 years showed a higher 10-year probability of a major osteoporotic fracture in those with NAFLD compared to those without, while this association was more pronounced in those with sarcopenia (82). Contrary, there are also studies demonstrating either no correlation or a positive correlation between NAFLD and BMD (Table 1). However, as summarized in Table 1, several limitations make the findings of these studies questionable, particularly since no liver biopsies were performed to conclusively diagnose/stage NAFLD and fractures were self-reported, whilst various confounding factors (e.g. vitamin D levels, metabolic bone markers, other medications) were not accessed/included in the analysis. Meta-analysis data correlating NAFLD and osteoporosis have been presented by Su et al. which showed that NAFLD is associated with decreased BMD and higher risk of osteoporosis or osteoporotic fractures, with male sex potentially being a risk factor for decreased BMD in adults with NAFLD, whilst ethnic disparities appear to be also present between non-Asian and Asian populations regarding both BMD and osteoporotic fractures (87). Moreover, the systematic review and meta-analysis by Pan et al. which included seven eligible studies showed a significant association between NAFLD and the prevalence and risk of osteoporosis or osteoporotic fractures in both men and women (88).
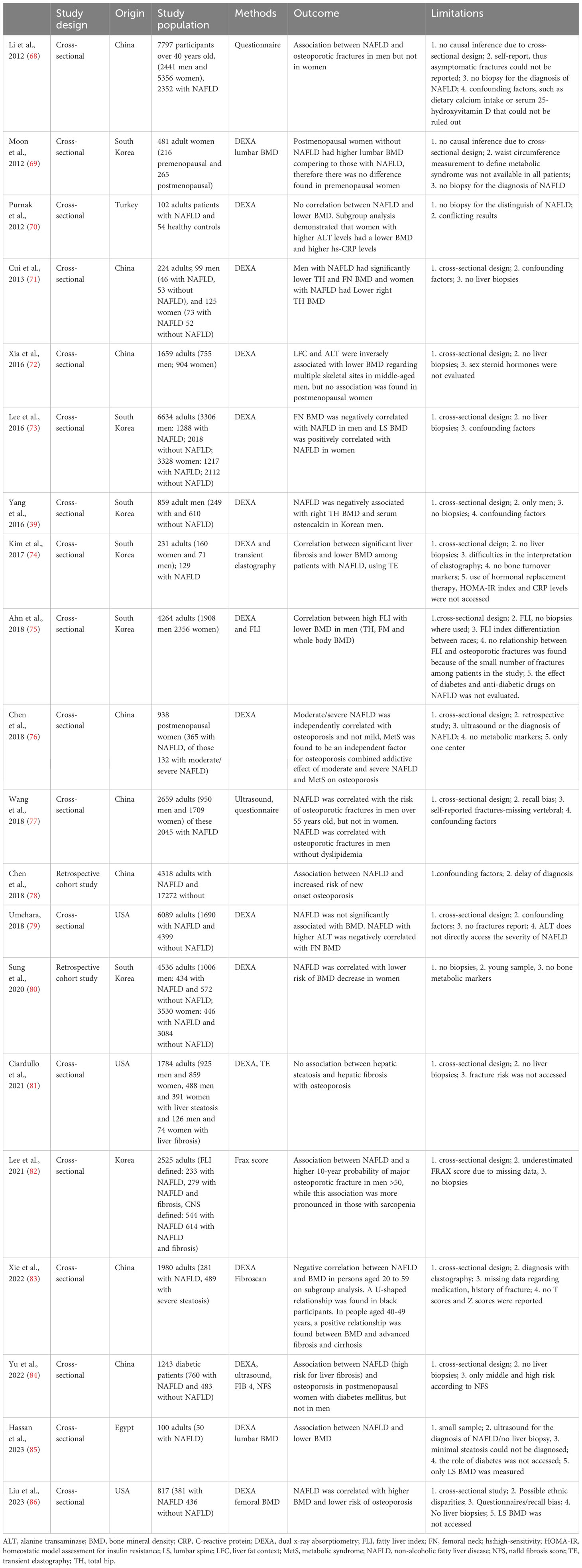
Table 1 Selected studies (in chronological order) on the association between non-alcoholic fatty liver disease (NAFLD) and osteoporosis/bone mineral density (BMD) in humans.
4 Anti-osteoporotic treatments and NAFLD
Several pharmacotherapies, including denosumab, bisphosphonates, teriparatide, raloxifene, calcitonin, and romosozumab, have well-established efficacy in the treatment of osteoporosis, reducing the risk of osteoporotic fractures (89). Given that osteoporosis and NAFLD frequently co-exist, particularly in older adults, such medications against osteoporosis may affect NAFLD progression by impacting on pathogenetic mechanisms/pathways shared by both these chronic diseases (Figure 1).
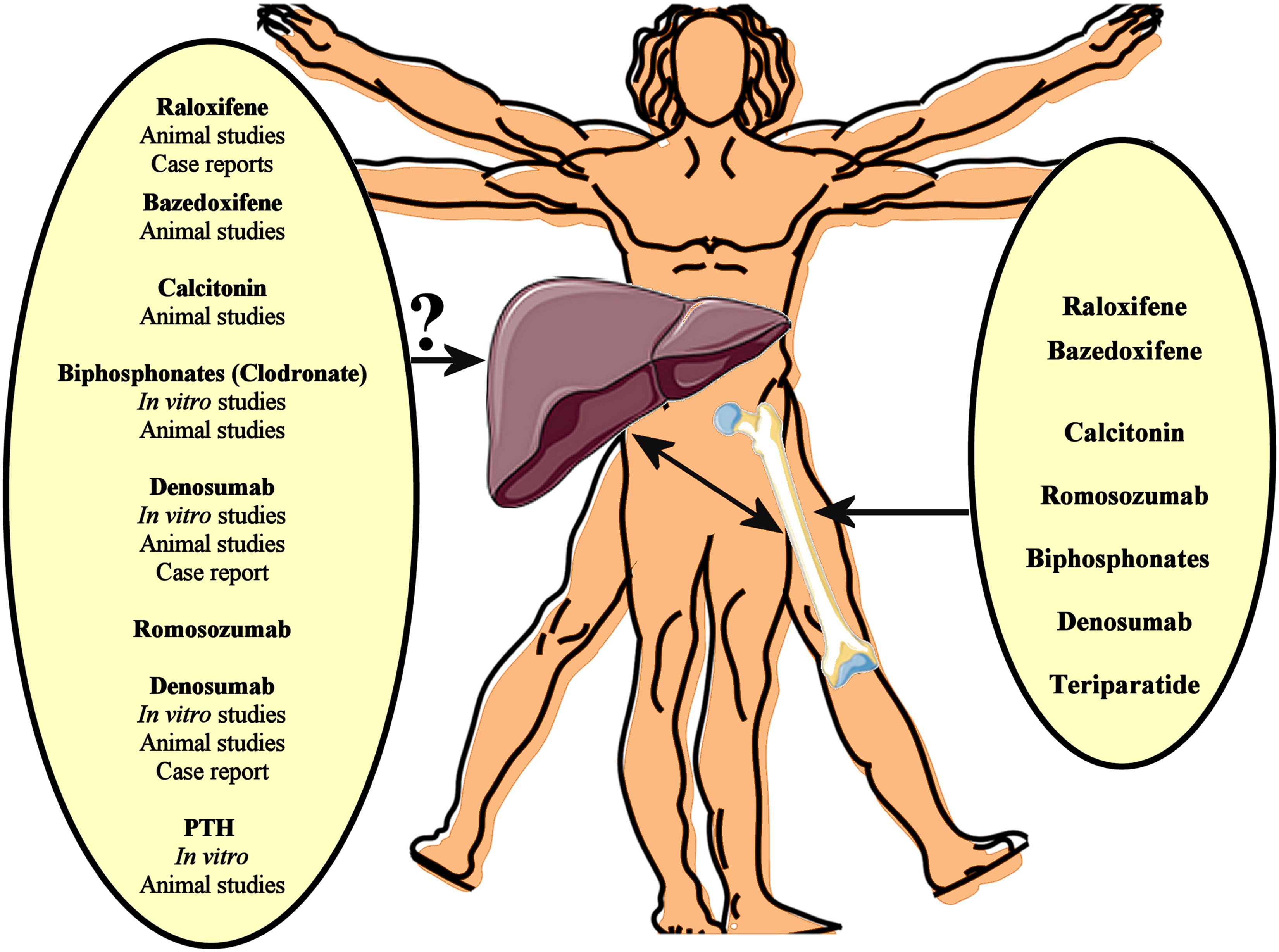
Figure 1 Several pharmacotherapies, including denosumab, bisphosphonates, teriparatide, raloxifene, calcitonin, and romosozumab, have well-established efficacy in the treatment of osteoporosis, reducing the risk of osteoporotic fractures. Given that osteoporosis and NAFLD frequently co-exist, such medications against osteoporosis may affect NAFLD progression by impacting on pathogenetic mechanisms/pathways shared by both these chronic diseases. In vitro data, animal studies and case reports support a beneficial effect of anti-osteoporotic drugs on the NAFLD/NASH progression. However, interventional studies could finally evaluate the potential impact of these anti-osteoporotic drugs on NAFLD.
4.1 Bisphosphonates and NAFLD
Bisphosphonates are a class of anti-osteoclastic drugs which are widely used as a pharmaceutical treatment for osteoporosis, constituting first-line therapeutic choices for osteoporosis. These have a structure like pyrophosphate and act by inhibiting bone resorption and remaining on the bone surface (90). Bisphosphonates are divided in nitrogen-containing and non-nitrogen-containing agents (91), and can be used continuously for 3-5 years. However, the long use of bisphosphonates may have side effects, such as atypical femoral fractures and jaw osteonecrosis (92). To date, no experimental or clinical study has showed that the nitrogen bisphosphonates may impact on NAFLD. However, there are experimental data from Hasuzawa et al. in a NASH mouse model induced by a methionine and choline deficient diet which show that clodronate (a non-nitrogen bisphosphonate which acts as a potent and selective inhibitor of the vesicular nucleotide transporter, VNUT) may improve NASH and diminish hepatic inflammation, steatosis, and fibrosis (93). In vitro experiments also showed that clodronate reduced hepatic neutrophil infiltration, hepatocyte apoptosis, and cytokine production, suggesting that VNUT-dependent vesicular ATP release plays a role in aggravating hepatic steatosis (93).
4.2 Selective estrogen receptor modulators and NAFLD
Selective estrogen receptor modulators (SERMS) act as estrogen agonists in the bone tissue, inhibiting the osteoclast activity, and as estrogen antagonists in breast and uterine tissues, thus exerting anti-osteoporotic effects without increasing the risk of breast cancer, as estrogen replacement therapy does; although, they increase the risk for thrombosis and pulmonary emboli (92). Raloxifene hydrochloride is the first SERM used for the treatment of osteoporosis (94), with data from the MORE study showing that it increases BMD in the spine and the femoral neck, whilst decreasing the vertebral fracture risk (95). Bazedoxifene is another SERM which has been approved for the treatment of osteoporosis in post-menopausal women (96); although, it is considered inferior compared to other anti-osteoporotic drugs since it has been shown to augment the lumbar spine BMD, but not the hip BMD. Furthermore, bazedoxifene is combined with estrogens forming a tissue selective estrogen complex (TSEC), which is used to moderate vasomotor symptoms (97). Interestingly, Takamura et al. presented a case report regarding a 53-year-old woman with liver impairment and histologically confirmed NASH after the initiation of raloxifene treatment (98). Matsumura et al. reported a similar case regarding a 70-year-old woman, whose NAFLD deteriorated within three months after starting raloxifene (99). Both these clinical cases suggested that liver function should be carefully surveilled following initiation of raloxifene treatment. Contrary, the findings from Luo et al. in a choline-deficient high-fat diet NASH mouse model showed an improvement in NASH after the administration of raloxifene (100). In line with this animal study, Barrera et al. examined a recent SERM, i.e. bazedoxifene acetate (BZA), in ovariectomized female mice fed a Western diet for 10-12 weeks. In this study, BZA administration, either alone or in combination with a conjugated estrogen (CE), resulted in attenuated liver steatosis along with increases in subcutaneous and visceral white adipose tissue induced by a high-fat diet (101). Moreover, Kim et al. studied the effects of BZA and TSEC on metabolic dysfunction in ovariectomized mice fed with a high-fat diet, demonstrating that BZA and TSEC promoted hepatic lipid oxidation and improved glucose homeostasis by raising the activity of Sirtuin1 (SIRT1), PPARα and hepatic AMPK of a different mechanism of action compared to E2 and CE (102). Interestingly, a natural SERM (genistein) given as monotherapy at high doses or in combination with CE in ovariectomized mice reduced fed with a high-fat diet significantly reduced the microvesicular fat infiltration in hepatocytes and hepatic TG accumulation induced by the high-fat diet (103). In an attempt to elucidate the underlying molecular mechanism, genistein was shown to decrease the expression of peroxisome proliferator-activated receptor-gamma (PPARγ) which is known to play a crucial role in the progression of hepatic steatosis (103).
4.3 Calcitonin and NAFLD
Calcitonin is a 32 amino acids hormone which acts by inhibiting the osteoclasts and stimulating the renal calcium excretion; hence, is regarded as an anti-osteoporotic treatment (not as effective as other anti-osteoporotic drugs) (104). Although, the nasal spray of calcitonin may be used in patients that cannot tolerate other therapies, there are concerns that calcitonin may provoke malignancies, thus, it was withdrawn from the market in Europe and Canada (92). Gydesen et al. investigated the effect of a dual amylin and calcitonin receptor agonist (DACRA) on rats fed with high-fat diet, showing that this treatment resulted in improved glucose homeostasis, higher weight loss, enhanced insulin action and decreased lipid accumulation in the liver and skeletal muscles, whilst improved food preferences was also noted in these rats (105, 106). Finally, Polymeris et al. demonstrated that serum calcitonin levels increased after a 75-g oral glucose tolerance test in healthy adults, suggesting that calcitonin may be stimulated by hyperinsulinemia (107).
4.4 Denosumab and NAFLD
Denosumab is a human monoclonal immunoglobulin G2 antibody used as a treatment of osteoporosis. which binds to RANKL, thus inhibiting RANK activation and the formation and survival of osteoclasts. As shown by the 10-year FREEDOM Extension study, denosumab can be safely used as an anti-osteoporotic treatment for 10 years with low rates of adverse events, whilst significantly increasing the lumbar spine BMD and decreasing the incidence of fractures (108, 109). However, multiple vertebral fractures have been reported after the discontinuation of this drug, and, thus, the transition to another anti-osteoporotic therapy is important after the discontinuation of denosumab (110).
As RANK/RANKL and OPG are also expressed in other tissues (e.g. in the liver and fibroblasts), it has been suggested that the RANK/RANKL/OPG system may play a physiologic role in organs/tissues other than bone tissue (44). Indeed, the inhibition of the RANKL/RANK signaling pathway has been reported as a potential target for the treatment of T2DM and insulin resistance in humans (111). Interestingly, Zhong et al. showed that RANKL levels were gradually higher when going from control mice to high-fat diet induced NAFLD and NASH, whilst RANKL appeared to also play a role in Runx2-prompted macrophage migration (112). In vitro, this study also showed that Runx2 regulated the production of RANKL in hepatic stellate cells (112). Furthermore, using a mouse model that expressed human RANKL, Rinotas et al. showed that RANKL overexpression increases insulin resistance and promotes the development of NAFLD, with these effects being exerted -at least partially- by acting at a post-receptor level, as well as by upregulating the secretion of inflammatory cytokines through NFκB activation (113). Of note, the administration of denosumab appeared to reverse the negative effect of RANKL on insulin resistance (113). In line with this finding, Kiechl et al. showed in a mouse model that RANKL blockage improved hepatic insulin resistance by preventing the activation of NFκB which is known to play a role in hepatic steatosis and NAFLD (114).
Moreover, Takeno et al. presented a case report, showing NASH improvement following denosumab treatment in a woman with growth hormone deficiency and NASH (115). Observational studies have also noted an association between serum RANKL levels and NAFLD, with. Lu et al. reporting a correlation between elevated RANKL levels and higher NAFLD risk in women with PCOS (116). In addition, RANKL levels have been associated with hyperglycemia and higher T2DM risk. Recently, taking into account that increased hepatic expression of RANKL may play a role in the progression of NAFLD, Polyzos et al. proposed the use of denosumab for the treatment of NAFLD (117). However, interventional studies are required to support this suggestion.
4.5 Romosozumab and NAFLD
Romosozumab is a monoclonal antibody which inhibits sclerostin, an inhibitor of the Wnt signaling pathway signaling, and increases bone formation whilst reducing bone resorption. Since there are studies that have documented a relation between romosozumab treatment and cardiovascular and cerebrovascular events, it is currently recommended not to use romosozumab in patients with myocardial infarction or stroke in the last year (118).
As aforementioned, the Wnt/beta-catenin pathway appears to have an important role in the development and the progression of NAFLD (66), hence, romosozumab has also been proposed as a potential treatment for NAFLD (119). In line with this, Kim et al. using two different mouse models, namely sclerostin-deficient mice and mice treated with a sclerostin-neutralizing antibody, showed significantly increased bone mass, as well as decreased hepatic lipid accumulation and liver inflammation (120). Furthermore, Zhou et al. also revealed that sclerostin levels were reduced in NAFLD mice compared to controls (121). Finally, Oh et al. reported higher sclerostin mRNA levels in both patients with obesity and mice fed with a high-fat diet., whilst further showed that sclerostin administration amplified lipid accumulation in hepatocytes (122). On the other hand, Polyzos et al. reported decreased sclerostin levels in patients with NAFLD and NASH (123), while Rhee et al. founded that patients with advanced liver cirrhosis had higher sclerostin levels compared to healthy controls and patients with early cirrhosis (124).
Overall, the role of sclerostin and Wnt/beta-catenin in the development and progression of NAFLD appears to be complex and further research on the potential clinical impact of romosozumab on NAFLD is required to elucidate the role of this anti-osteoporotic treatment in the context of NAFLD/NASH.
4.6 Teriparatide and NAFLD
Teriparatide [rhPTH(1-34), the bioactive portion (1-34) of endogenous human PTH] is an anti-osteoporotic/osteoanabolic treatment (89). Feng et al. in their recent study in animal models of NAFLD using intermittent PTH administration, showed an amelioration of hepatic steatosis. They demonstrated, using an in vitro model of hepatic steatosis, that PTH through its receptor, induces in hepatocytes the expression of genes involved in β-oxidation and reduces the expression of genes involved in lipid uptake and de novo lipogenesis (125).
A recent metanalysis of 10 studies by Jaroenlapnopparat et al. demonstrated that high PTH levels was correlated with NAFLD, and their relation was statistically important. They also showed an association between PTH level and NASH, which was not statistically important (126).
5 Perspectives and conclusion
NAFLD and osteoporosis are highly prevalent diseases which frequently co-exist with increasing incidence globally. Although common molecular pathogenetic mechanisms/pathways (e.g. the RANKL-OPG-RANK pathway and Wnt pathway) are supported by emerging data, not all epidemiological studies point towards a positive link between these two chronic diseases. To date, a limited number studies demonstrated an associative relationship between NAFLD and osteoporosis; however, conclusive evidence for causative link(s) and their direction are still missing. Further research, both basic/translational and clinical aiming to elucidate the interplay between the liver and bones is essential, including large prospective cohort and interventional studies which could target specific patient populations with NAFLD and osteoporosis.
In this context, recent studies have been further linking sarcopenia with both NAFLD and osteoporosis, thus highlighting sarcopenia as a potential mediating factor between these two diseases. This is also supported by the fact that molecules causatively implicated in sarcopenia, such as sclerostin, RANKL, and 25(OH)-vitamin-D, already constitute therapeutic targets in osteoporosis, whilst are also considered to play a role in the pathophysiology of NAFLD (122–124). Similarly, other therapeutic targets for osteoporosis, such as kathepsin K, also seem to be implicated in NAFLD progression (127, 128). Thus, it can be proposed that there is scope to focus future research in this field among patients with coexisting NAFLD, osteoporosis and sarcopenia since this group may benefit from anti-osteoporotic drugs involved in the overlapping pathophysiological mechanisms underlying these conditions. Since no globally approved pharmacological treatment for NAFLD is available yet, whilst there is an arsenal of approved anti-osteoporotic medication, observational data as well as interventional studies could evaluate the potential impact of these anti-osteoporotic drugs on NAFLD (Figure 1), with focus on certain phenotypic characteristics of the patient population (e.g. sarcopenic or not). Such targeted studies may shed light in the complex and yet not fully clarified links that form the liver-bone axis.
Author contributions
MC: Investigation, Writing – original draft. IK: Writing – review and editing. TA: Methodology, Writing – original draft. C-MF: Writing – original draft. EM: Writing – original draft. KC: Writing – original draft. YA: Writing – original draft. AP: Writing – original draft. EK: Conceptualization, Investigation, Methodology, Supervision, Writing – original draft, Writing – review and editing. HR: Investigation, Methodology, Supervision, Writing – original draft, Writing – review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Teng ML, Ng CH, Huang DQ, Chan KE, Tan DJ, Lim WH, et al. Global incidence and prevalence of nonalcoholic fatty liver disease. Clin Mol Hepatol. (2023) 29:S32–42. doi: 10.3350/cmh.2022.0365
2. Rhee E-J. Nonalcoholic fatty liver disease and diabetes: an epidemiological perspective. Endocrinol Metab. (2019) 34:226. doi: 10.3803/EnM.2019.34.3.226
3. Zachou M, Flevari P, Nasiri-Ansari N, Varytimiadis C, Kalaitzakis E, Kassi E, et al. The role of anti-diabetic drugs in NAFLD. Have we found the Holy Grail? A narrative review. Eur J Clin Pharmacol. (2024) 80:127–50. doi: 10.1007/s00228-023-03586-1
4. El-Kader SMA. Non-alcoholic fatty liver disease: The diagnosis and management. World J Hepatol. (2015) 7:846. doi: 10.4254/wjh.v7.i6.846
5. Chaitanya Thandra K, Barsouk A, Saginala K, Sukumar Aluru J, Rawla P, Barsouk A. Epidemiology of non-alcoholic fatty liver disease and risk of hepatocellular carcinoma progression. Clin Exp Hepatol. (2020) 6:289–94. doi: 10.5114/ceh.2020.102153
6. Younossi ZM. Recent research and insights on the disease burden of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Gastroenterol Hepatol (N Y). (2023) 19:108–10.
7. Hashimoto E, Taniai M, Tokushige K. Characteristics and diagnosis of NAFLD / NASH. J Gastroenterol Hepatol. (2013) 28:64–70. doi: 10.1111/jgh.12271
8. Papatheodoridi M, Cholongitas E. Diagnosis of non-alcoholic fatty liver disease (NAFLD): current concepts. Curr Pharm Des. (2019) 24:4574–86. doi: 10.2174/1381612825666190117102111
9. Younossi ZM. Review article: current management of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis. Aliment Pharmacol Ther. (2008) 28:2–12. doi: 10.1111/j.1365-2036.2008.03710.x
10. Eslam M, Newsome PN, Sarin SK, Anstee QM, Targher G, Romero-Gomez M, et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J Hepatol. (2020) 73:202–9. doi: 10.1016/j.jhep.2020.07.045
11. Sozen T, Ozisik L, Calik Basaran N. An overview and management of osteoporosis. Eur J Rheumatol. (2017) 4:46–56. doi: 10.5152/eurjrheumatol.
12. Tella SH, Gallagher JC. Prevention and treatment of postmenopausal osteoporosis. J Steroid Biochem Mol Biol. (2014) 142:155–70. doi: 10.1016/j.jsbmb.2013.09.008
13. Kim B, Cho Y, Lim W. Osteoporosis therapies and their mechanisms of action (Review). Exp Ther Med. (2021) 22:1379. doi: 10.3892/etm.2021.10815
14. Drapkina OM, Elkina AY, Sheptulina AF, Kiselev AR. Non-alcoholic fatty liver disease and bone tissue metabolism: current findings and future perspectives. Int J Mol Sci. (2023) 24:8445. doi: 10.3390/ijms24098445
15. Vachliotis ID, Anastasilakis AD, Goulas A, Goulis DG, Polyzos SA. Nonalcoholic fatty liver disease and osteoporosis: A potential association with therapeutic implications. Diabetes Obes Metab. (2022) 24:1702–20. doi: 10.1111/dom.14774
16. Flessa C-M, Nasiri-Ansari N, Kyrou I, Leca BM, Lianou M, Chatzigeorgiou A, et al. Genetic and diet-induced animal models for non-alcoholic fatty liver disease (NAFLD) research. Int J Mol Sci. (2022) 23:15791. doi: 10.3390/ijms232415791
17. Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. (2016) 65:1038–48. doi: 10.1016/j.metabol.2015.12.012
18. Flessa C-M, Kyrou I, Nasiri-Ansari N, Kaltsas G, Papavassiliou AG, Kassi E, et al. Endoplasmic reticulum stress and autophagy in the pathogenesis of non-alcoholic fatty liver disease (NAFLD): current evidence and perspectives. Curr Obes Rep. (2021) 10:134–61. doi: 10.1007/s13679-021-00431-3
19. Flessa C, Kyrou I, Nasiri-Ansari N, Kaltsas G, Kassi E, Randeva HS. Endoplasmic reticulum stress in nonalcoholic (metabolic associated) fatty liver disease (NAFLD/MAFLD). J Cell Biochem. (2022) 123:1585–606. doi: 10.1002/jcb.30247
20. Nasiri-Ansari N, Androutsakos T, Flessa C-M, Kyrou I, Siasos G, Randeva HS, et al. Endothelial cell dysfunction and nonalcoholic fatty liver disease (NAFLD): A concise review. Cells. (2022) 11:2511. doi: 10.3390/cells11162511
21. Yang B, Lu L, Zhou D, Fan W, Barbier-Torres L, Steggerda J, et al. Regulatory network and interplay of hepatokines, stellakines, myokines and adipokines in nonalcoholic fatty liver diseases and nonalcoholic steatohepatitis. Front Endocrinol (Lausanne). (2022) 30:1007944/full. doi: 10.3389/fendo.2022.1007944/full
22. Chang M-L, Yang Z, Yang S-S. Roles of adipokines in digestive diseases: markers of inflammation, metabolic alteration and disease progression. Int J Mol Sci. (2020) 21:8308. doi: 10.3390/ijms21218308
23. Burkhardt L-M, Bucher CH, Löffler J, Rinne C, Duda GN, Geissler S, et al. The benefits of adipocyte metabolism in bone health and regeneration. Front Cell Dev Biol. (2023) 11. doi: 10.3389/fcell.2023.1104709
24. Gamberi T, Magherini F, Modesti A, Fiaschi T. Adiponectin signaling pathways in liver diseases. Biomedicines. (2018) 6:52. doi: 10.3390/biomedicines6020052
25. Shabalala SC, Dludla PV, Mabasa L, Kappo AP, Basson AK, Pheiffer C, et al. The effect of adiponectin in the pathogenesis of non-alcoholic fatty liver disease (NAFLD) and the potential role of polyphenols in the modulation of adiponectin signaling. BioMed Pharmacother. (2020) 131:110785. doi: 10.1016/j.biopha.2020.110785
26. Jung TW, Lee YJ, Lee MW, Kim SM, Jung TW. Full-length adiponectin protects hepatocytes from palmitate-induced apoptosis via inhibition of c-Jun NH 2 terminal kinase. FEBS J. (2009) 276:2278–84. doi: 10.1111/j.1742-4658.2009.06955.x
27. Polyzos SA, Kountouras J, Mantzoros CS. Adipokines in nonalcoholic fatty liver disease. Metabolism. (2016) 65:1062–79. doi: 10.1016/j.metabol.2015.11.006
28. Lewis JW, Edwards JR, Naylor AJ, McGettrick HM. Adiponectin signalling in bone homeostasis, with age and in disease. Bone Res. (2021) 9:1. doi: 10.1038/s41413-020-00122-0
29. Deepika F, Bathina S, Armamento-Villareal R. Novel adipokines and their role in bone metabolism: A narrative review. Biomedicines. (2023) 11:644. doi: 10.3390/biomedicines11020644
30. Bacchetta J, Boutroy S, Guebre-Egziabher F, Juillard L, Drai J, Pelletier S, et al. The relationship between adipokines, osteocalcin and bone quality in chronic kidney disease. Nephrol Dial Transplant. (2009) 24:3120–5. doi: 10.1093/ndt/gfp262
31. Jürimäe J, Jürimäe T. Adiponectin is a predictor of bone mineral density in middle-aged premenopausal women. Osteoporos Int. (2007) 18:1253–9. doi: 10.1007/s00198-007-0365-5
32. Roomi AB, Nori W, Al-Badry SH. The value of serum adiponectin in osteoporotic women: does weight have an effect? J Obes. (2021) 2021:1–5. doi: 10.1155/2021/5325813
33. Barbour KE, Zmuda JM, Boudreau R, Strotmeyer ES, Horwitz MJ, Evans RW, et al. Adipokines and the risk of fracture in older adults. J Bone Miner Res. (2011) 26:1568–76. doi: 10.1002/jbmr.361
34. Di Medio L, Brandi ML. Advances in bone turnover markers (2021). Available online at: https://linkinghub.elsevier.com/retrieve/pii/S0065242321000421.
35. Komori T. Functions of osteocalcin in bone, pancreas, testis, and muscle. Int J Mol Sci. (2020) 21:7513. doi: 10.3390/ijms21207513
36. Kassi E, Papavassiliou AG. A possible role of osteocalcin in the regulation of insulin secretion: human in vivo evidence? J Endocrinol. (2008) 199:151–3. doi: 10.1677/JOE-08-0294
37. Kanazawa I. Osteocalcin as a hormone regulating glucose metabolism. World J Diabetes. (2015) 6:1345. doi: 10.4239/wjd.v6.i18.1345
38. Yilmaz Y, Kurt R, Eren F, Imeryuz N. Serum osteocalcin levels in patients with nonalcoholic fatty liver disease: Association with ballooning degeneration. Scand J Clin Lab Invest. (2011) 71:631–6. doi: 10.3109/00365513.2011.604427
39. Yang HJ, Shim SG, Ma BO, Kwak JY. Association of nonalcoholic fatty liver disease with bone mineral density and serum osteocalcin levels in Korean men. Eur J Gastroenterol Hepatol. (2016) 28:338–44. doi: 10.1097/MEG.0000000000000535
40. Luo Y, Ma X, Hao Y, Pan X, Xu Y, Xiong Q, et al. Inverse relationship between serum osteocalcin levels and nonalcoholic fatty liver disease in postmenopausal Chinese women with normal blood glucose levels. Acta Pharmacol Sin. (2015) 36:1497–502. doi: 10.1038/aps.2015.81
41. Fang D, Yin H, Ji X, Sun H, Zhao X, Bi Y, et al. Low levels of osteocalcin, but not CTX or P1NP, are associated with nonalcoholic hepatic steatosis and steatohepatitis. Diabetes Metab. (2023) 49:101397. doi: 10.1016/j.diabet.2022.101397
42. Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys. (2008) 473:139–46. doi: 10.1016/j.abb.2008.03.018
43. Yang YJ, Kim DJ. An overview of the molecular mechanisms contributing to musculoskeletal disorders in chronic liver disease: osteoporosis, sarcopenia, and osteoporotic sarcopenia. Int J Mol Sci. (2021) 22:2604. doi: 10.3390/ijms22052604
44. Vachliotis ID, Polyzos SA. Osteoprotegerin/receptor activator of nuclear factor-kappa B ligand/receptor activator of nuclear factor-kappa B axis in obesity, type 2 diabetes mellitus, and nonalcoholic fatty liver disease. Curr Obes Rep. (2023) 12:147–62. doi: 10.1007/s13679-023-00505-4
45. El Amrousy D, El-Afify D. Osteocalcin and osteoprotegerin levels and their relationship with adipokines and proinflammatory cytokines in children with nonalcoholic fatty liver disease. Cytokine. (2020) 135:155215. doi: 10.1016/j.cyto.2020.155215
46. Mantovani A, Sani E, Fassio A, Colecchia A, Viapiana O, Gatti D, et al. Association between non-alcoholic fatty liver disease and bone turnover biomarkers in post-menopausal women with type 2 diabetes. Diabetes Metab. (2019) 45:347–55. doi: 10.1016/j.diabet.2018.10.001
47. Nikseresht M, Azarmehr N, Arya A, Alipoor B, Fadaei R, Khalvati B, et al. Circulating mRNA and plasma levels of osteoprotegerin and receptor activator of NF-κB ligand in nonalcoholic fatty liver disease. Biotechnol Appl Biochem. (2021) 68:1243–9. doi: 10.1002/bab.2047
48. Hadinia A, Doustimotlagh A, Goodarzi H, Arya A, Jafarinia M. Plasma levels and gene expression of RANK in non-alcoholic fatty liver disease. Clin Lab. (2020) 66(5). doi: 10.7754/Clin.Lab.2019.190913
49. Yilmaz Y, Yonal O, Kurt R, Oral AY, Eren F, Ozdogan O, et al. Serum levels of osteoprotegerin in the spectrum of nonalcoholic fatty liver disease. Scand J Clin Lab Invest. (2010) 70:541–6. doi: 10.3109/00365513.2010.524933
50. Niu Y, Zhang W, Yang Z, Li X, Fang W, Zhang H, et al. Plasma osteoprotegerin levels are inversely associated with nonalcoholic fatty liver disease in patients with type 2 diabetes: A case–control study in China. Metabolism. (2016) 65:475–81. doi: 10.1016/j.metabol.2015.12.005
51. Erol M, Bostan Gayret O, Tekin Nacaroglu H, Yigit O, Zengi O, Salih Akkurt M, et al. Association of osteoprotegerin with obesity, insulin resistance and non-alcoholic fatty liver disease in children. Iran Red Crescent Med J. (2016) 18:e41873. doi: 10.5812/ircmj
52. Ayaz T, Kirbas A, Durakoglugil T, Durakoglugil ME, Sahin SB, Sahin OZ, et al. The relation between carotid intima media thickness and serum osteoprotegerin levels in nonalcoholic fatty liver disease. Metab Syndr Relat Disord. (2014) 12:283–9. doi: 10.1089/met.2013.0151
53. Kostenuik P, Shalhoub V. Osteoprotegerin A physiological and pharmacological inhibitor of bone resorption. Curr Pharm Des. (2001) 7:613–35. doi: 10.2174/1381612013397807
54. Yang M, Liu Y, Zhou G, Guo X, Zou S, Liu S, et al. [Value of serum osteoprotegerin in noninvasive diagnosis of nonalcoholic steatohepatitis]. Zhonghua Gan Zang Bing Za Zhi. (2016) 24:96–101. doi: 10.3760/cma.j.issn.1007-3418.2016.02.005
55. Singh A, Gill G, Kaur H, Amhmed M, Jakhu H. Role of osteopontin in bone remodeling and orthodontic tooth movement: a review. Prog Orthod. (2018) 19:18. doi: 10.1186/s40510-018-0216-2
56. Bertola A, Deveaux V, Bonnafous S, Rousseau D, Anty R, Wakkach A, et al. Elevated expression of osteopontin may be related to adipose tissue macrophage accumulation and liver steatosis in morbid obesity. Diabetes. (2009) 58:125–33. doi: 10.2337/db08-0400
57. Gómez-Santos B, Saenz de Urturi D, Nuñez-García M, Gonzalez-Romero F, Buque X, Aurrekoetxea I, et al. Liver osteopontin is required to prevent the progression of age-related nonalcoholic fatty liver disease. Aging Cell. (2020) 19:e13183. doi: 10.1111/acel.13183
58. Zhao J, Lei H, Wang T, Xiong X. Liver-bone crosstalk in non-alcoholic fatty liver disease: Clinical implications and underlying pathophysiology. Front Endocrinol (Lausanne). (2023) 14:1161402/full. doi: 10.3389/fendo.2023.1161402/full
59. Yao Y, Miao X, Zhu D, Li D, Zhang Y, Song C, et al. Insulin-like growth factor-1 and non-alcoholic fatty liver disease: a systemic review and meta-analysis. Endocrine. (2019) 65:227–37. doi: 10.1007/s12020-019-01982-1
60. Dichtel LE, Corey KE, Misdraji J, Bredella MA, Schorr M, Osganian SA, et al. The association between IGF-1 levels and the histologic severity of nonalcoholic fatty liver disease. Clin Transl Gastroenterol. (2017) 8:e217. doi: 10.1038/ctg.2016.72
61. Wang T-H, Li J-B, Tian Y-G, Zheng J-X, Li X-D, Guo S. Association of TNF-α, IGF-1, and IGFBP-1 levels with the severity of osteopenia in mice with nonalcoholic fatty liver disease. J Orthop Surg Res. (2023) 18:915. doi: 10.1186/s13018-023-04385-1
62. Wang X, Li W, Zhang Y, Yang Y, Qin G. Association between vitamin D and non-alcoholic fatty liver disease/non-alcoholic steatohepatitis: results from a meta-analysis. Int J Clin Exp Med. (2015) 8:17221–34.
63. Asadipooya K, Lankarani KB, Raj R, Kalantarhormozi M. RAGE is a potential cause of onset and progression of nonalcoholic fatty liver disease. Int J Endocrinol. (2019) 2019:1–11. doi: 10.1155/2019/2151302
64. Asadipooya K, Uy EM. Advanced glycation end products (AGEs), receptor for AGEs, diabetes, and bone: review of the literature. J Endocr Soc. (2019) 3:1799–818. doi: 10.1210/js.2019-00160
65. Maeda K, Kobayashi Y, Koide M, Uehara S, Okamoto M, Ishihara A, et al. The regulation of bone metabolism and disorders by wnt signaling. Int J Mol Sci. (2019) 20:5525. doi: 10.3390/ijms20225525
66. Shree Harini K, Ezhilarasan D. Wnt/beta-catenin signaling and its modulators in nonalcoholic fatty liver diseases. Hepatobiliary Pancreat Dis Int. (2023) 22:333–45. doi: 10.1016/j.hbpd.2022.10.003
67. Liu W, Singh R, Choi CS, Lee H-Y, Keramati AR, Samuel VT, et al. Low density lipoprotein (LDL) receptor-related protein 6 (LRP6) regulates body fat and glucose homeostasis by modulating nutrient sensing pathways and mitochondrial energy expenditure. J Biol Chem. (2012) 287:7213–23. doi: 10.1074/jbc.M111.286724
68. Li M, Xu Y, Xu M, Ma L, Wang T, Liu Y, et al. Association between nonalcoholic fatty liver disease (NAFLD) and osteoporotic fracture in middle-aged and elderly Chinese. J Clin Endocrinol Metab. (2012) 97:2033–8. doi: 10.1210/jc.2011-3010
69. Moon S-S, Lee Y-S, Kim SW. Association of nonalcoholic fatty liver disease with low bone mass in postmenopausal women. Endocrine. (2012) 42:423–9. doi: 10.1007/s12020-012-9639-6
70. Purnak T, Beyazit Y, Ozaslan E, Efe C, Hayretci M. The evaluation of bone mineral density in patients with nonalcoholic fatty liver disease. Wien Klin Wochenschr. (2012) 124:526–31. doi: 10.1007/s00508-012-0211-4
71. Cui R, Sheng H, Rui X-F, Cheng X-Y, Sheng C-J, Wang J-Y, et al. Low bone mineral density in Chinese adults with nonalcoholic fatty liver disease. Int J Endocrinol. (2013) 2013:1–6. doi: 10.1155/2013/396545
72. Xia M-F, Lin H-D, Yan H-M, Bian H, Chang X-X, Zhang L-S, et al. The association of liver fat content and serum alanine aminotransferase with bone mineral density in middle-aged and elderly Chinese men and postmenopausal women. J Transl Med. (2016) 14:11. doi: 10.1186/s12967-016-0766-3
73. Lee SH, Yun JM, Kim SH, Seo YG, Min H, Chung E, et al. Association between bone mineral density and nonalcoholic fatty liver disease in Korean adults. J Endocrinol Invest. (2016) 39:1329–36. doi: 10.1007/s40618-016-0528-3
74. Kim G, Kim KJ, Rhee Y, Lim S-K. Significant liver fibrosis assessed using liver transient elastography is independently associated with low bone mineral density in patients with non-alcoholic fatty liver disease. PloS One. (2017) 12:e0182202. doi: 10.1371/journal.pone.0182202
75. Ahn SH, Seo DH, Kim SH, Nam M-S, Hong S. The relationship between fatty liver index and bone mineral density in Koreans: KNHANES 2010–2011. Osteoporos Int. (2018) 29:181–90. doi: 10.1007/s00198-017-4257-z
76. Chen D-Z, Xu Q-M, Wu X-X, Cai C, Zhang L-J, Shi K-Q, et al. The combined effect of nonalcoholic fatty liver disease and metabolic syndrome on osteoporosis in postmenopausal females in eastern China. Int J Endocrinol. (2018) 2018:1–8. doi: 10.1155/2018/2314769
77. Wang Y, Wen G, Zhou R, Zhong W, Lu S, Hu C, et al. Association of nonalcoholic fatty liver disease with osteoporotic fractures: A cross-sectional retrospective study of Chinese individuals. Front Endocrinol (Lausanne). (2018) 9:408/full. doi: 10.3389/fendo.2018.00408/full
78. Chen H-J, Yang H-Y, Hsueh K-C, Shen C-C, Chen R-Y, Yu H-C, et al. Increased risk of osteoporosis in patients with nonalcoholic fatty liver disease. Med (Baltimore). (2018) 97:e12835. doi: 10.1097/MD.0000000000012835
79. Umehara T. Nonalcoholic fatty liver disease with elevated alanine aminotransferase levels is negatively associated with bone mineral density: Cross-sectional study in U.S. adults. PloS One. (2018) 13:e0197900. doi: 10.1371/journal.pone.0197900
80. Sung J, Ryu S, Song Y-M, Cheong H-K. Relationship between non-alcoholic fatty liver disease and decreased bone mineral density: A retrospective cohort study in Korea. J Prev Med Public Heal. (2020) 53:342–52. doi: 10.3961/jpmph.20.089
81. Ciardullo S, Muraca E, Zerbini F, Manzoni G, Perseghin G. NAFLD and liver fibrosis are not associated with reduced femoral bone mineral density in the general US population. J Clin Endocrinol Metab. (2021) 106:e2856–65. doi: 10.1210/clinem/dgab262
82. Lee HJ, Lee DC, Kim CO. Association between 10-year fracture probability and nonalcoholic fatty liver disease with or without sarcopenia in Korean men: A nationwide population-based cross-sectional study. Front Endocrinol (Lausanne). (2021) 12. doi: 10.3389/fendo.2021.599339
83. Xie R, Liu M. Relationship between non-alcoholic fatty liver disease and degree of hepatic steatosis and bone mineral density. Front Endocrinol (Lausanne). (2022) 13:857110/full. doi: 10.3389/fendo.2022.857110/full
84. Yu Z, Wu Y, Zhang R, Li Y, Zang S, Liu J. Increased risk of non-alcoholic fatty liver disease fibrosis is closely associated with osteoporosis in women but not in men with type 2 diabetes. Endocr Connect. (2022) 11:e220174. doi: 10.1530/EC-22-0174
85. Hassan AM, Haridy MA, Shoaeir MZ, Abdel-Aziz TM, Qura MK, Kenawy EM, et al. Non-alcoholic fatty liver disease is associated with decreased bone mineral density in upper Egyptian patients. Sci Rep. (2023) 13:4353. doi: 10.1038/s41598-023-31256-w
86. Liu J, Tang Y, Feng Z, Chen Y, Zhang X, Xia Y, et al. Metabolic associated fatty liver disease and bone mineral density: a cross-sectional study of the National Health and Nutrition Examination Survey 2017–2018. Osteoporos Int. (2023) 34:713–24. doi: 10.1007/s00198-023-06687-w
87. Su Y, Chien K, Yang S, Chia W, Chen J, Chen Y. Nonalcoholic fatty liver disease is associated with decreased bone mineral density in adults: A systematic review and meta-analysis. J Bone Miner Res. (2023) 38:1092–103. doi: 10.1002/jbmr.4862
88. Pan B, Cai J, Zhao P, Liu J, Fu S, Jing G, et al. Relationship between prevalence and risk of osteoporosis or osteoporotic fracture with non-alcoholic fatty liver disease: A systematic review and meta-analysis. Osteoporos Int. (2022) 33:2275–86. doi: 10.1007/s00198-022-06459-y
89. Gregson CL, Armstrong DJ, Bowden J, Cooper C, Edwards J, Gittoes NJL, et al. UK clinical guideline for the prevention and treatment of osteoporosis. Arch Osteoporos. (2022) 17:58. doi: 10.1007/s11657-022-01061-5
90. Song S, Guo Y, Yang Y, Fu D. Advances in pathogenesis and therapeutic strategies for osteoporosis. Pharmacol Ther. (2022) 237:108168. doi: 10.1016/j.pharmthera.2022.108168
91. Grey A, Reid IR. Differences between the bisphosphonates for the prevention and treatment of osteoporosis. Ther Clin Risk Manag. (2006) 2:77–86.
92. Eastell R, Rosen CJ, Black DM, Cheung AM, Murad MH, Shoback D. Pharmacological management of osteoporosis in postmenopausal women: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. (2019) 104:1595–622. doi: 10.1210/jc.2019-00221
93. Hasuzawa N, Tatsushima K, Wang L, Kabashima M, Tokubuchi R, Nagayama A, et al. Clodronate, an inhibitor of the vesicular nucleotide transporter, ameliorates steatohepatitis and acute liver injury. Sci Rep. (2021) 11:5192. doi: 10.1038/s41598-021-83144-w
95. Ettinger B. Reduction of vertebral fracture risk in postmenopausal women with osteoporosis treated with raloxifene. JAMA. (1999) 282:637. doi: 10.1001/jama.282.7.637
97. Yavropoulou MP, Makras P, Anastasilakis AD. Bazedoxifene for the treatment of osteoporosis. Expert Opin Pharmacother. (2019) 20:1201–10. doi: 10.1080/14656566.2019.1615882
98. Takamura T, Shimizu A, Komura T, Ando H, Zen Y, Minato H, et al. Selective estrogen receptor modulator raloxifene-associated aggravation of nonalcoholic steatohepatitis. Intern Med. (2007) 46:579–81. doi: 10.2169/internalmedicine.46.6374
99. Matsumura M, Tashiro K, Miura A, Tajima T, Kinoshita I, Kojima E, et al. [A case of non-alcoholic fatty liver disease (NAFLD) aggravated after treatment with raloxifene]. Nihon Shokakibyo Gakkai Zasshi. (2011) 108:2036–41.
100. Luo F, Ishigami M, Achiwa K, Ishizu Y, Kuzuya T, Honda T, et al. Raloxifene ameliorates liver fibrosis of nonalcoholic steatohepatitis induced by choline-deficient high-fat diet in ovariectomized mice. Dig Dis Sci. (2015) 60:2730–9. doi: 10.1007/s10620-015-3660-6
101. Barrera J, Chambliss KL, Ahmed M, Tanigaki K, Thompson B, McDonald JG, et al. Bazedoxifene and conjugated estrogen prevent diet-induced obesity, hepatic steatosis, and type 2 diabetes in mice without impacting the reproductive tract. Am J Physiol Metab. (2014) 307:E345–54. doi: 10.1152/ajpendo.00653.2013
102. Kim JH, Meyers MS, Khuder SS, Abdallah SL, Muturi HT, Russo L, et al. Tissue-selective estrogen complexes with bazedoxifene prevent metabolic dysfunction in female mice. Mol Metab. (2014) 3:177–90. doi: 10.1016/j.molmet.2013.12.009
103. Kim JH, Kim YJ. Effects of genistein in combination with conjugated estrogens on endometrial hyperplasia and metabolic dysfunction in ovariectomized mice. Endocr J. (2015) 62:531–42. doi: 10.1507/endocrj.EJ15-0056
104. Downs RW, Bell NH, Ettinger MP, Walsh BW, Favus MJ, Mako B, et al. Comparison of alendronate and intranasal calcitonin for treatment of osteoporosis in postmenopausal women*. J Clin Endocrinol Metab. (2000) 85:1783–8. doi: 10.1210/jcem.85.5.6606
105. Gydesen S, Hjuler ST, Freving Z, Andreassen KV, Sonne N, Hellgren LI, et al. A novel dual amylin and calcitonin receptor agonist, KBP-089, induces weight loss through a reduction in fat, but not lean mass, while improving food preference. Br J Pharmacol. (2017) 174:591–602. doi: 10.1111/bph.13723
106. Mathiesen DS, Lund A, Vilsbøll T, Knop FK, Bagger JI. Amylin and calcitonin: potential therapeutic strategies to reduce body weight and liver fat. Front Endocrinol (Lausanne). (2021) 11:617400/full. doi: 10.3389/fendo.2020.617400/full
107. Polymeris A, Papapetrou P, Papandroulaki F, Thanou S. Hyperinsulinemia during oral glucose tolerance test and high normal serum cortisol are associated with increased secretion of calcitonin in normal subjects. Hormones. (2011) 10:304–12. doi: 10.14310/horm.2002.1149
108. Törring O. Effects of denosumab on bone density, mass and strength in women with postmenopausal osteoporosis. Ther Adv Musculoskelet Dis. (2015) 7:88–102. doi: 10.1177/1759720X15579189
109. Bone HG, Wagman RB, Brandi ML, Brown JP, Chapurlat R, Cummings SR, et al. 10 years of denosumab treatment in postmenopausal women with osteoporosis: results from the phase 3 randomised FREEDOM trial and open-label extension. Lancet Diabetes Endocrinol. (2017) 5:513–23. doi: 10.1016/S2213-8587(17)30138-9
110. Kendler DL, Cosman F, Stad RK, Ferrari S. Denosumab in the treatment of osteoporosis: 10 years later: A narrative review. Adv Ther. (2022) 39:58–74. doi: 10.1007/s12325-021-01936-y
111. Xing B, Yu J, Zhang H, Li Y. RANKL inhibition: a new target of treating diabetes mellitus? Ther Adv Endocrinol Metab. (2023) 14:20420188231170754. doi: 10.1177/20420188231170754
112. Zhong L, Yuan J, Huang L, Li S, Deng L. RANKL is involved in runx2-triggered hepatic infiltration of macrophages in mice with NAFLD induced by a high-fat diet. BioMed Res Int. (2020) 2020:1–8. doi: 10.1155/2020/6953421
113. Rinotas V, Niti A, Dacquin R, Bonnet N, Stolina M, Han C-Y, et al. Novel genetic models of osteoporosis by overexpression of human RANKL in transgenic mice. J Bone Miner Res. (2014) 29:1158–69. doi: 10.1002/jbmr.2112
114. Kiechl S, Wittmann J, Giaccari A, Knoflach M, Willeit P, Bozec A, et al. Blockade of receptor activator of nuclear factor-κB (RANKL) signaling improves hepatic insulin resistance and prevents development of diabetes mellitus. Nat Med. (2013) 19:358–63. doi: 10.1038/nm.3084
115. Takeno A, Yamamoto M, Notsu M, Sugimoto T. Administration of anti-receptor activator of nuclear factor-kappa B ligand (RANKL) antibody for the treatment of osteoporosis was associated with amelioration of hepatitis in a female patient with growth hormone deficiency: a case report. BMC Endocr Disord. (2016) 16:66. doi: 10.1186/s12902-016-0148-0
116. Lu N, Shan C, Fu J-R, Zhang Y, Wang Y-Y, Zhu Y-C, et al. RANKL is independently associated with increased risks of non-alcoholic fatty liver disease in Chinese women with PCOS: A cross-sectional study. J Clin Med. (2023) 12:451. doi: 10.3390/jcm12020451
117. Polyzos SA, Goulas A. Treatment of nonalcoholic fatty liver disease with an anti-osteoporotic medication: A hypothesis on drug repurposing. Med Hypotheses. (2021) 146:110379. doi: 10.1016/j.mehy.2020.110379
118. Shakeri A, Adanty C. Romosozumab (sclerostin monoclonal antibody) for the treatment of osteoporosis in postmenopausal women: A review. J Popul Ther Clin Pharmacol. (2020) 27:e25–31. doi: 10.15586/jptcp.v27i1.655
119. Shree Harini K, Ezhilarasan D. Reply to “Wnt/beta-catenin signaling inhibitors and nonalcoholic fatty liver disease: Potential therapeutic implications”. Hepatobiliary Pancreat Dis Int. (2023) 22:439–40. doi: 10.1016/j.hbpd.2023.02.008
120. Kim SP, Frey JL, Li Z, Kushwaha P, Zoch ML, Tomlinson RE, et al. Sclerostin influences body composition by regulating catabolic and anabolic metabolism in adipocytes. Proc Natl Acad Sci. (2017) 114:e11238-47. doi: 10.1073/pnas.1707876115
121. Zhou F, Wang Y, Li Y, Tang M, Wan S, Tian H, et al. Decreased sclerostin secretion in humans and mice with nonalcoholic fatty liver disease. Front Endocrinol (Lausanne). (2021) 12:707505/full. doi: 10.3389/fendo.2021.707505/full
122. Oh H, Park SY, Cho W, Abd El-Aty AM, Hacimuftuoglu A, Kwon CH, et al. Sclerostin aggravates insulin signaling in skeletal muscle and hepatic steatosis via upregulation of ER stress by mTOR-mediated inhibition of autophagy under hyperlipidemic conditions. J Cell Physiol. (2022) 237:4226–37. doi: 10.1002/jcp.30873
123. Polyzos SA, Anastasilakis AD, Kountouras J, Makras P, Papatheodorou A, Kokkoris P, et al. Circulating sclerostin and Dickkopf-1 levels in patients with nonalcoholic fatty liver disease. J Bone Miner Metab. (2016) 34:447–56. doi: 10.1007/s00774-015-0687-x
124. Rhee Y, Kim WJ, Han KJ, Lim SK, Kim SH. Effect of liver dysfunction on circulating sclerostin. J Bone Miner Metab. (2014) 32:545–9. doi: 10.1007/s00774-013-0524-z
125. Feng X, Xiao Y, Guo Q, Peng H, Zhou H-Y, Wang J-P, et al. Parathyroid hormone alleviates non-alcoholic liver steatosis via activating the hepatic cAMP/PKA/CREB pathway. Front Endocrinol (Lausanne). (2022) 13. doi: 10.3389/fendo.2022.899731
126. Jaroenlapnopparat A, Rittiphairoj T, Chaisidhivej N, Walker B, Charoenngam N. High parathyroid hormone level as a marker of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis: A systematic review and meta-analysis. Diabetes Metab Syndr Clin Res Rev. (2023) 17:102827. doi: 10.1016/j.dsx.2023.102827
127. Yu C, Wan Y, Piao L, Wu Cheng X. Can cysteinyl cathepsin activity control diet-induced NAFLD? IJC Hear Vasc. (2020) 28:100516. doi: 10.1016/j.ijcha.2020.100516
Keywords: anti-osteoporotic drugs, non-alcoholic fatty liver disease, denosumab, romosozumab, bisphosphonates, calcitonin, selective estrogen receptor modulators, PTH
Citation: Chondrogianni ME, Kyrou I, Androutsakos T, Flessa C-M, Menenakos E, Chatha KK, Aranan Y, Papavassiliou AG, Kassi E and Randeva HS (2024) Anti-osteoporotic treatments in the era of non-alcoholic fatty liver disease: friend or foe. Front. Endocrinol. 15:1344376. doi: 10.3389/fendo.2024.1344376
Received: 25 November 2023; Accepted: 05 January 2024;
Published: 08 March 2024.
Edited by:
Takefumi Kimura, Shinshu University, JapanReviewed by:
Kamyar Asadipooya, University of Kentucky, United StatesCopyright © 2024 Chondrogianni, Kyrou, Androutsakos, Flessa, Menenakos, Chatha, Aranan, Papavassiliou, Kassi and Randeva. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Harpal S. Randeva, aGFycGFsLnJhbmRldmFAdWhjdy5uaHMudWs=; Eva Kassi, ZWthc3NpQG1lZC51b2EuZ3I=
†These authors have contributed equally to this work and share first authorship
‡These authors have contributed equally to this work and share last authorship