 Jianmei Zhang
Jianmei Zhang Suhong Yang1
Suhong Yang1 Lianshu Han
Lianshu Han
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Endocrinol. , 02 April 2024
Sec. Developmental Endocrinology
Volume 15 - 2024 | https://doi.org/10.3389/fendo.2024.1343977
This article is part of the Research Topic Insights in Developmental Endocrinology: 2023 View all 9 articles
Background: This study aimed to characterize the clinical phenotype and genetic variations in patients with Kallmann syndrome (KS).
Methods: This study involved the collection and analysis of clinical data from an individual with sporadic KS. Following this, peripheral blood samples were obtained from the patient and his parents. Genomic deoxyribonucleic acid was extracted and subjected to whole-exome sequencing and genomic copy number variation (CNV) detection. Finally, Sanger sequencing was performed to validate the suspected pathogenic variants.
Results: Whole-exome sequencing confirmed that the child carried both the IL17RD variant (c.2101G>A, p.Gly701Ser) inherited from the mother and the new CPEB4 variant (c.1414C>T, p.Arg472*). No pathogenic CNVs were identified in CNV testing.
Conclusion: Bioinformatics analysis shows that the IL17RD protein undergoing Gly701Ser mutation and is speculated to be phosphorylated and modified, thereby disrupting fibroblast growth factor signaling. This study also suggested that the CPEB4 might play a crucial role in the key signaling process affecting olfactory bulb morphogenesis. Overall, the findings of this study broaden the gene expression profile of KS-related pathogenic genes. This offers a new avenue for exploring the pathogenic mechanism of KS and provides valuable insights for precise clinical diagnosis and treatment strategies for this condition.
Idiopathic hypogonadotropic hypogonadism (IHH) is a genetically and clinically heterogeneous disorder, primarily due to insufficient activation of the hypothalamic gonadotropin-releasing hormone (GnRH) axis or failure of pituitary gonadotropin synthesis, secretion, or function (1, 2). During embryonic development, several interacting genes are involved in olfaction establishment and the migration of GnRH neurons, implying a shared embryonic origin among these genes (2–4). Typically, IHH is associated with impaired olfactory function (1, 5), which is a characteristic feature of Kallmann syndrome (KS). KS is characterized by delayed pubertal development, inadequate development of secondary sexual characteristics, and hyposmia or loss of olfactory sensation, present in approximately 60% of IHH cases. IHH cases where olfaction remains normal are referred to as normosmic IHH (nIHH) (1, 6, 7). Furthermore, patients with IHH, influenced by various genes, might also manifest a range of non-reproductive symptoms, such as sensorineural deafness, bimanual synkinesia, dental hypoplasia, renal hypoplasia, skeletal abnormalities, and midline hypoplasia (5, 7, 8).
Over the past few decades, more than 50 genes were associated with IHH, and only some of them were strongly associated, but not the majority. To date, approximately 40% of patients with IHH and approximately 10% of patients with KS have definite genetic defects (9–11), among these, classical KS pathogenic genes include KAL1、FGFR1、PROKR2 (1, 12–14). IL17RD has been confirmed to cause KS, however, few reports have been carried out to understand the contribution of IL17RD variants to KS. This case report provides a comprehensive account of the clinical data and diagnostic and therapeutic history of a sporadic KS case attributed to an IL17RD variant. Additionally, a review and summary of the characteristics of patients carrying the IL17RD variant as reported in the existing literature is offered. Finally, through bioinformatics analysis, the molecular mechanism via which the IL17RD variant causes KS is uncovered.
A patient diagnosed with KS in August 2022 was included in this study. The Ethics Committee of our hospital approved this study (approval No.2023-083-01), and informed consent for this clinical research was obtained from the patient’s guardian.
The clinical data of the patients in this study were obtained from the following sources: (1) medical records, encompassing current medical history, past medical history, birth history, and growth and development history; (2) physical examinations involving evaluations of the cranial region, the five sensory organs, and the reproductive system; (3) auxiliary findings, including magnetic resonance imaging (MRI) of the pituitary gland and olfactory bulb, scrotal ultrasonography, digital radiographic (DR) examination of the left wrist joint, and pituitary-related hormone tests.
Peripheral blood samples were collected from the patient and his parents, and these samples were processed to extract genomic deoxyribonucleic acid (DNA) after the anticoagulation process. The whole-genome exon region DNA was captured and enriched using sequence capture technology (Agilent SureSelect series kit) and subjected to high-throughput sequencing (Illumina, PE150). The whole-exome gene assay achieved a coverage rate of 99.7% for the intervals with sequencing depth ≥20×. Following sequencing, the obtained data were aligned with reference databases, including the human reference genome and the 1000 Genomes database. Additionally, candidate variants were verified by Sanger sequencing. Parental origin verification of the variants was performed to determine the presence of new-onset variants using the HGMD、ClinVar、OMIM and existing literature. The pathogenicity of these variant loci was assessed according to the American College of Medical Genetics and Genomics standards and guidelines (15).
Multiple sequence alignment was performed using Clustal Omega (16). Visualization of amino acid conservation within the mutant proteins was accomplished using Jalview and WebLogo. Pathogenicity predictions for the novel variants were made using MutationTaster and Polyphen (17, 18). Furthermore, the three-dimensional spatial structure of the IL17RD protein was established using AlphaFold online software. InBio Map is used to construct a protein-protein interaction network.
A 14.4-year-old male patient presented with a complaint of “having a short penis for over 7 years and reduced olfaction for over 5 years”. He was born at full term with a birth weight of 3.5 kg and had no history of resuscitation, asphyxia, or exposure to specific medications. By approximately 3 years of age, the patient underwent surgery for cryptorchidism and was diagnosed with allergic rhinitis. His parents had a normal clinical phenotype. His mother conceived naturally after marriage, and there was no history of consanguineous marriage or a family history of hereditary disease.
The patient’s physical characteristics included a height of 162 cm, a weight of 58 kg, and a body mass index of 22.1kg/m2. His facial appearance was unremarkable, without café au lait spots, laryngeal nodes, axillary hair or beard growth, and any signs of breast development. His pubertal development is in Tanner stage 1, with a penis length of 2 cm and a penis circumference of 1.2 cm. Prader’s testis measurements indicated a left testis volume of 2 mL and a right testis volume of 1 mL. His external genitalia were infantile. Furthermore, his cardiac, pulmonary, abdominal, and neurological examinations did not reveal any abnormalities. During the olfactory test (water, 75% alcohol, vinegar, and perfume), the patient demonstrated an inability to discern smells. Audiometry confirmed sensorineural hearing loss in the right ear.
The patient’s peripheral blood and urine routine tests, liver and kidney function assessments, lipids levels, electrolytes, myocardial enzyme concentrations, alpha-fetoprotein levels, and carcinoembryonic antigen levels were normal. Pituitary-related hormone tests indicated normal adrenocorticotropic hormone (ACTH) and cortisol rhythms and levels, thyroid function, prolactin, and insulin-like growth factor 1. The basal levels of Luteinising hormone (LH), which was 0.22 IU/L (normal reference range 0.71–6.24, the same below), follicle-stimulating hormone (FSH), which was 1.63 IU/L (0.91–7.25), and testosterone (T) was 0.45 nmol/L(0.71–22.92). Additionally, during the gonadorelin stimulation test (1-day method), LH reached a maximum of only 2.71 IU/L at 60 min. The patient’s chromosomal karyotype analysis confirmed a 46, XY pattern.
DR examination of the left wrist indicated a bone age of 13 years. A pituitary MRI plain scan revealed a small pituitary gland with no significant occupying lesions. Plain MRI imaging of the olfactory bulb indicated bilateral olfactory nerve deficits (Figure 1). Scrotal ultrasonography revealed a left testis measuring 1.2cm×0.6cm×0.6cm (volume: 0.23mL) and a right testis measuring 1.0 cm × 0.9 cm × 0.7 cm (volume: 0.33 mL). Ultrasonography of the penis reported a transverse dimension of approximately 1.5 cm × 0.9 cm in the mid-portion, an extracorporeal length of approximately 1.4 cm, and a length from the penis subcutaneous region to the pubic symphysis of approximately 3.3 cm. Moreover, ultrasonography of the liver, gallbladder, pancreas, spleen, thyroid, adrenal glands, both kidneys, and the heart did not reveal any abnormal findings.
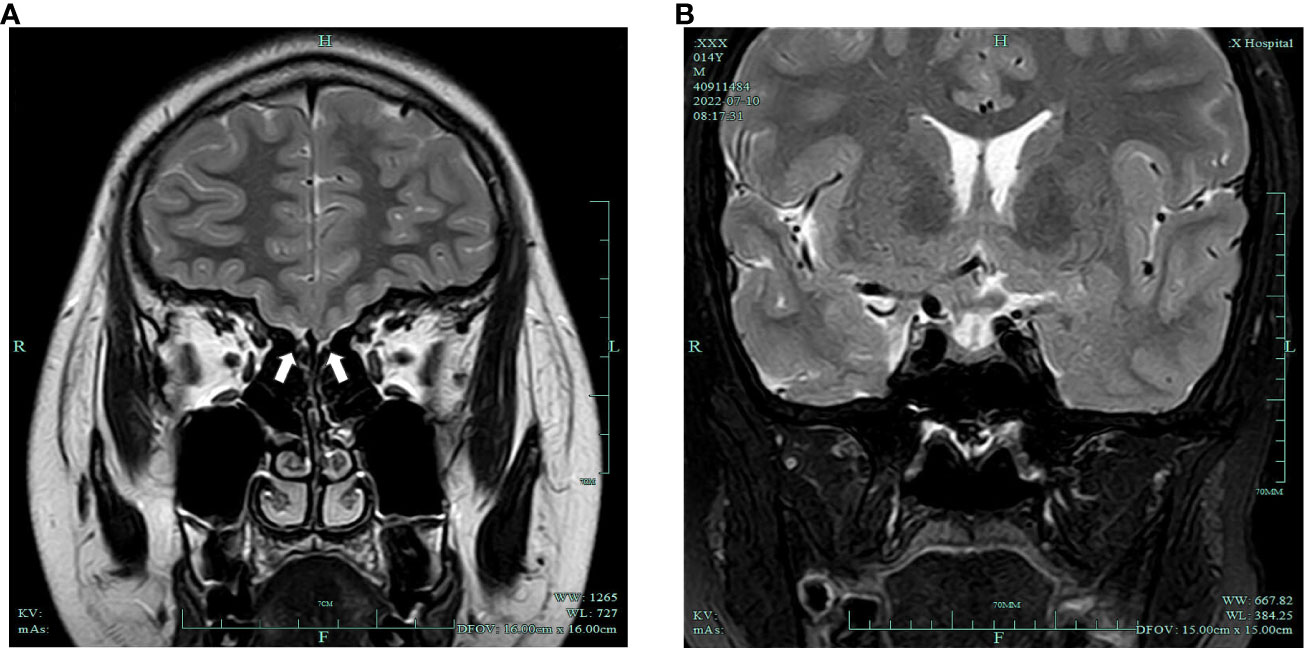
Figure 1 (A) Bilateral olfactory nerve deficits (white arrows); (B) slightly smaller pituitary gland with no obvious occupying lesions.
IL17RD variant (c.2101G>A, p.Gly701Ser) was detected in the patient. His mother carried the same heterozygous variant, while the father carried the wild-type variant. Importantly, this specific variant was absent in several well-established databases, including OMIM, ClinVar, and LOVD, and lacked any documented polymorphic loci. In addition, the patient also detected CPEB4 variant (c.1414C>T, p.Arg472*). A comprehensive analysis of the patient’s family via Sanger DNA sequencing confirmed that neither the father nor the mother carried this variant. The CPEB4 variant was previously unreported in any searches, signifying it as a novel mutation. As for the genomic CNV analysis, no pathogenic or potentially pathogenic CNVs were identified in the patient or his parents.
MutationTaster and Polyphen were employed for pathogenicity prediction of the discovered missense variants, and the results are all harmful. Conservation analysis results indicate that this IL17RD variant (p.Gly701Ser) is not located within the conserved domain of the IL17RD protein (Figure 2).The CPEB4 variant (p.Arg472*) is located in a highly conserved region (Figure 3). Applying Alpha Fold was used to model the IL17RD protein, and no difference was observed in the structures of the wild-type and mutated IL17RD proteins (Figures 4A, B).The impact of the variant on the IL17RD protein structure was examined using AlphaFold protein structure prediction. Notably, this variant did not induce substantial alterations in the IL17RD protein’s structure. But surprisingly, it did induce an amino acid change at position #701, substituting glycine with serine. Serine is a recognized site for phosphorylation, implying that this variant could potentially trigger phosphorylation modifications in the protein. We hypothesized whether this variant might cause phosphorylation modification of the protein and thereby regulate the occurrence of the disease. To this end, we used NetPhos software to predict the phosphorylation of IL17RD protein. The results showed that position 701 on the IL17RD emerged as a potential phosphorylation site, with a predicted probability value of 0.994 (unsp).Thus, the IL17RD protein might undergo phosphorylation modifications after the Gly701Ser mutation. In addition, the CPEB4 variant(c.1414C>T, p.R472*) was detected, and protein structure predictions were conducted for the CPEB4 protein. This revealed a premature termination of the protein sequence at the arginine residue within the β-fold structure for the wild-type and mutant variants. Specifically, the arginine residue did not form an intact β-fold structure thereafter, thus changing the tertiary structure of the protein and presumably affecting the function of the normal protein (Figures 4C, D).
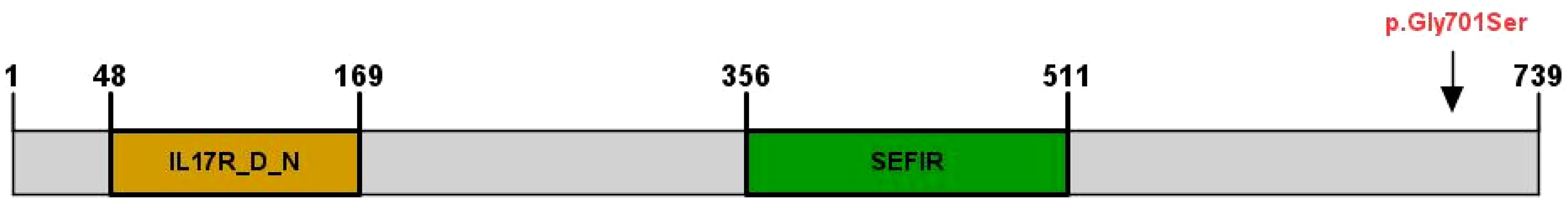
Figure 2 IL17RD protein structural domains and locations of variants observed in this cohort, indicated by arrows (↓).
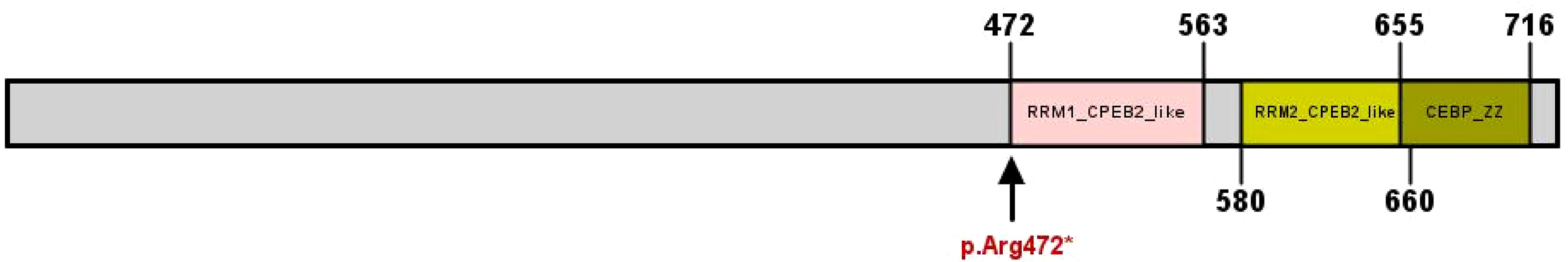
Figure 3 CPEB4 protein structural domains and locations of variants observed in this cohort, indicated by arrows (↓).
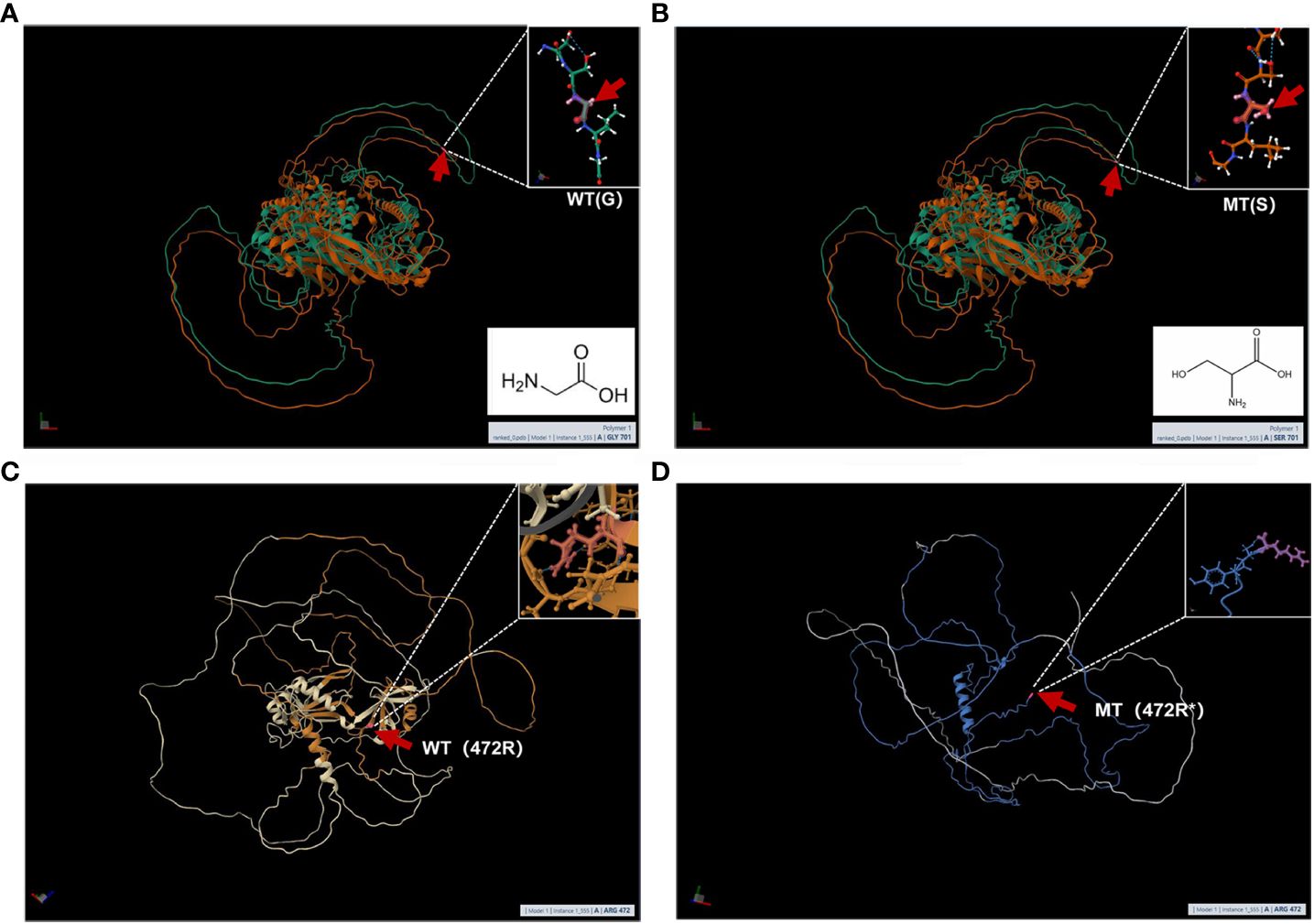
Figure 4 (A, B) are the modeling of wild-type and mutant IL17RD proteins respectively: the red arrow represents the mutation position, (A) is the wild type and (B) is the mutant; (C, D) are the modeling of wild-type and mutant CPEB4 proteins respectively: The red arrow represents the mutation position, (C) is the wild type, and (D) is the mutant.
We used online bioinformatics software to predict that the IL17RD variant (c.2101G>A, p.Gly701Ser) may be the causative mutation of KS. In addition, the CPEB4 variant was also detected in this patient. Except for the prediction of its possible pathogenicity by using bioinformatics software in this study, no definite evidence related to the pathogenesis of KS could be obtained. Notably, prior research conducted by Tseng et al. has suggested a correlation between the CPEB4 and olfactory bulb development (19–21). Therefore, there might be an interaction between the CPEB4 protein and the IL17RD protein, contributing to KS pathogenesis. The IL17RD protein interaction network was predicted using InBio Map to verify this hypothesis. Despite not observing a direct interaction between IL17RD and CPEB4 proteins (Figure 5), between these two proteins and their synergistic effect on regulating KS pathogenesis warrants further investigation.
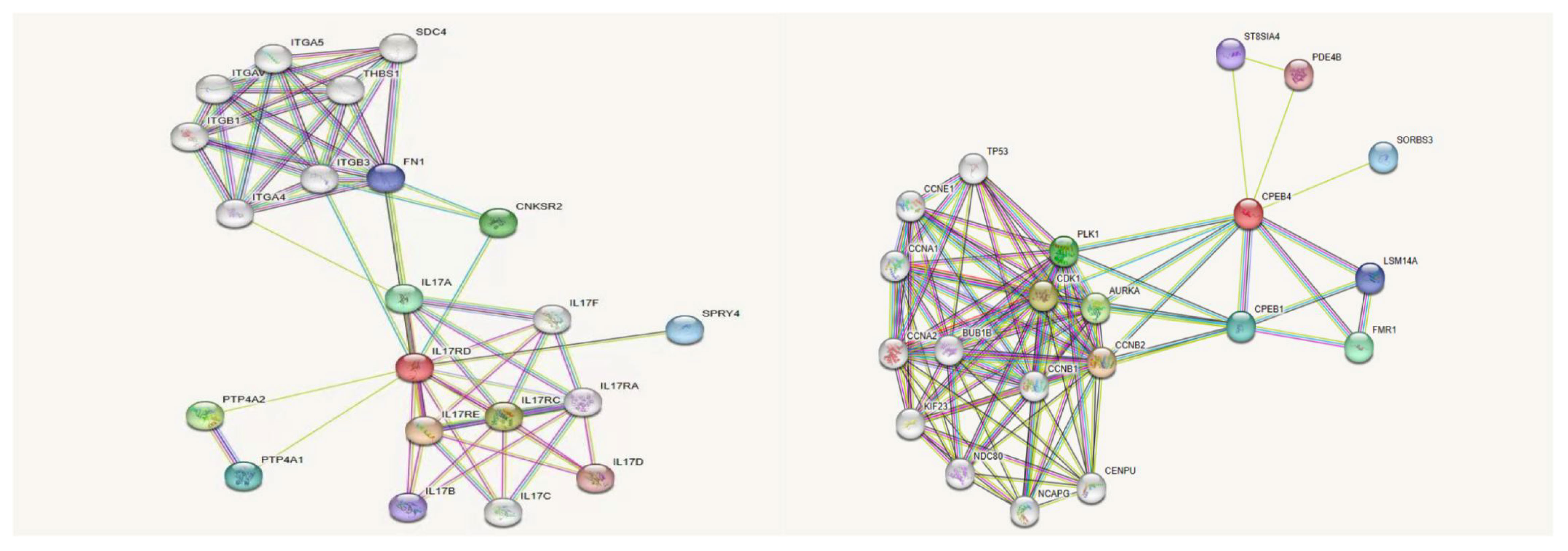
Figure 5 IL17RD and CPEB4 protein interaction network.
After 4 months of gonadorelin pump pulse therapy (10 µg/90 min), LH levels increased to 4.23 IU/L, FSH increased to 10.69 IU/L, and T increased to 4.79 nmol/L. After treatment, the patient exhibited a left testicular volume of 1.58 mL, a right testicular volume of 2.83 mL, and a penile length of 3.4 cm. The patient has been consistently receiving pituitary pump therapy and has been undergoing regular follow-up examinations for the past year. The most recent examination revealed the following results: T levels at 0.69 nmol/L, LH at 1.71 IU/L, penile length at 3.4 cm, left testicular volume at 2.15 mL, and right testicular volume at 4.37 mL. In addition, adnexal torsion near the right testicle occurred during the follow-up period. Currently, the patient is under ongoing regular follow-up.
Situated on chromosome 3p14.3, the IL17RD gene comprises 17 exons. Initially recognized as an inhibitor of fibroblast growth factor (FGF) signaling, IL17RD (also known as Sef) encoded by the IL17RD gene, was believed to function as a negative regulator (22, 23). Within the context of this signaling pathway, FGF8 binds to FGFR1, playing a pivotal role in the development and migration of olfactory and GnRH neurons (23, 24). According to a 2013 study by Miraoui et al., IL17RD serves as a signaling hub in the interaction with components of the ERK1/2 mitogen-activated protein kinase cascade and innate immune signaling pathways. IL17RD exerts an antagonistic effect on FGF8-FGFR1 receptor signaling, with dysfunction in this pathway being a key factor in the pathogenesis of IHH (25–27). In the present study, the IL17RD variant (c.2101G>A, p.Gly701Ser) was examined for its impact on the IL17RD protein structure using AlphaFold protein structure prediction. However, we speculate that the IL17RD protein can be phosphorylated and modified after the G701S mutation. This process could potentially disrupt FGF signaling, thereby affecting the normal development of GnRH neurons and the olfactory system. It is important to note that this hypothesis warrants further confirmation through large-sample experiments.
Currently, the prevalence of IL17RD variants remains unknown, and a definitive correlation between specific IL17RD variant types and clinical phenotypes is yet to be established (26, 28, 29). In a study by Zhou et al. (30), which examined mutation profiles and clinical manifestations of 148 Chinese men with IHH, a mutation rate of 3.38% was identified within the IL17RD. However, this gene exhibited a significant degree of “incomplete explicitness”. Patients with IHH carrying the IL17RD variant from various family lineages displayed varying degrees of hyposmia and hypogonadism. After conducting a literature review, case reports related to KS/IHH induced by this gene variant were summarized after a literature search (Table 1). Among them, KS occurred in 18 out of 22 (81.8%) patients, and 72.2% (13 out of 18) of patients experienced other concomitant manifestations of KS, including cryptorchidism, micropenis, hypospadias, hearing loss, abnormal dentition, and osteoporosis. Hearing loss emerged as the most common concomitant symptom (7 out of 18, 38.9%), as confirmed by our findings. In addition, the Gly701Ser mutation carried by the patient in this study has been reported in 2 cases before. One of the KS patients also carried the PROKR2 (p.Y113H) mutation, which has similar clinical manifestations to the patients in this study (30); the other KS patient who carried both FGFR1 (p.Gly348Glu) and RUVBL2 (p.Arg71Trp) only showed non-reproductive symptoms associated with cleft lip and palate (28, 31).
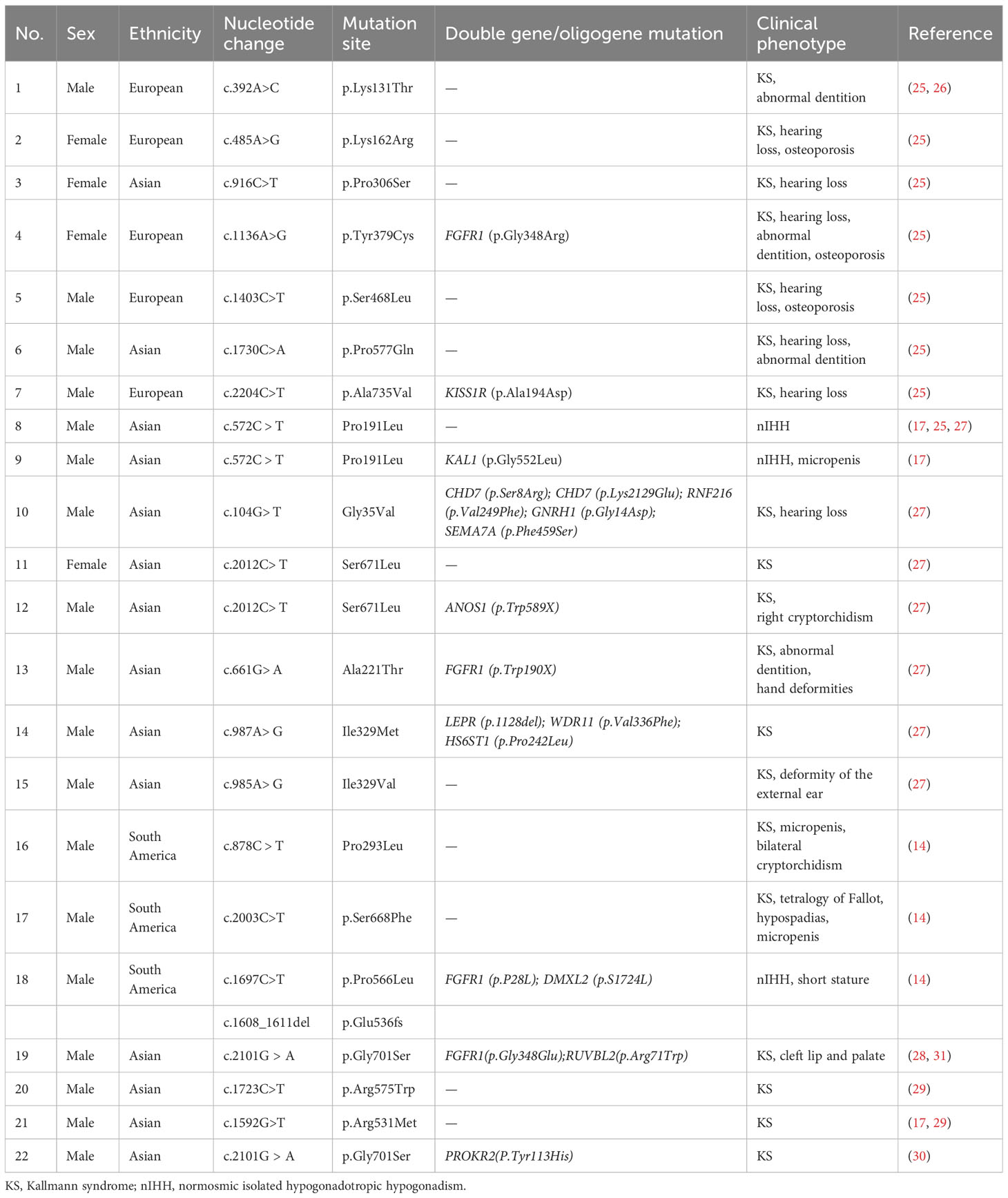
Table 1 Currently reported variant sites in the interleukin 17 receptor D gene and corresponding functions.
Previous studies have demonstrated that the genetic penetrance and phenotypic expressivity of disease symptoms was dependent on factors such as zygosity of the mutation as well as mutations occurring in other genetic loci, suggesting an oligogenic mode of action (26). IL17RD might also function downstream of gonadotropins to regulate the responsiveness of gonadal tissue and other affected neuronal networks (olfactory and auditory), given that IL17RD is expressed in olfactory placodes in mice (26, 32). Alternatively, given its pattern of oligogenic interactions in disease, IL17RD may epistatically regulate the expression of another gene that may influence these functions (26, 32). Reportedly, cases of KS/IHH caused by IL17RD variants had nine out of 21 (42.8%) patients exhibiting double-gene/oligogene inheritance, suggesting that the IL17RD often collaborates with other genes in the manifestation of the disease. for instance, in the case of patient #18, who had nIHH without hearing loss, two variants were identified in the IL17RD (Table 1). These variants included pure missense variants associated with heterozygous frameshifts. Additionally, this patient carried heterozygous variants in the FGFR1 and DMXL2. The IL17RD and FGFR1 variants were categorized as pathogenic, while the DMXL2 variant was categorized as possibly benign. The contribution of each variant to the disease phenotype remains unknown. To date, IL17RD variants have only been reported in patients with KS and nIHH, often exhibiting non-reproductive symptoms such as hearing loss.
Cytoplasmic polyadenylation element binding (CPEB) protein is an RNA binding protein expressed in neuronal tissues including brain and spinal cord that promotes cytoplasmic polyadenylation (33, 34). Vertebrates contain four members of the CPEB family of proteins that are designated CPEB1-4 (19, 20). CPEB1, the founding member of this family, has become an important model for illustrating general principles of translational control by cytoplasmic polyadenylation in gametogenesis, cancer etiology, synaptic plasticity, learning, and memory (35). In studies observing CPEB knockout mice, it was found that CPEB1 plays an important role in mouse gametogenesis (35). It was found that the germ cells from the knockout mice harbored fragmented chromatin, suggesting a possible defect in homologous chromosome adhesion or synapsis (21, 33, 36). CPEB1 has essential functions in mouse gametogenesis. Both male and female CPEB1 knockout mice are sterile (33). Although CPEB4 is more similar to CPEB2 and CPEB3 at the amino acid sequence level, previous studies have shown that CPEB4 is more functionally related to CPEB1 and can in turn promote polyadenylation-induced oocyte RNA translation (36). In this study, the CPEB4 variant (c.1414C>T, p.Arg472*) was detected in this child, resulting in a position change to 472 after translation termination. The alteration disrupts all structural domains and consequently impairs the complete β-folding of the CPEB4 protein, thereby affecting the protein’s structural stability. Bioinformatics revealed that CPEB4 variant might possess pathogenic potential. However, concrete evidence linking CPEB4 variants to KS/IHH or other genetic diseases is currently lacking. In summary, we speculate that changes in CPEB4 protein function caused by CPEB4 variants are likely to affect CPEB1 protein function and thereby disrupt germ cell differentiation, but this needs to be confirmed in future studies.
The mammalian olfactory bulb (OB) is the first relay station for olfactory perception and require continuous replenishment of interneurons (mainly granule cells [GCs]) to support local circuits throughout life (19, 37). Ching-San Tseng et al. found that in the first 2 postnatal weeks in mice, CPEB4 acts as a survival factor exclusively for early postnatal GCs, and olfactory experience initiates CPEB4-activated c-Fos mRNA translation (21). In CPEB4 knockout mice, c-FOS insufficiency reduced neurotrophic signaling to impair GC survival and cause OB hypoplasia (20, 21). Therefore, the CPEB4 might be involved in key signaling processes that affect olfactory bulb morphogenesis. The children in this study had hyposmia, which may be related to changes in protein function caused by mutations in the CPEB4. At present, it remains unclear whether there is a correlation between CPEB4 and KS and may indicate a novel double-gene pathogenesis of KS/IHH. Further investigation is required to explore the interactions between the genes and proteins involved in the KS/IHH pathway to optimize in vitro functional experiments.
In summary, since there is currently unclear molecular mechanism for most KS patients, further research is needed. On this basis, this study provides convincing evidence to elucidate the molecular mechanism of IL17RD variant, and speculates that CPEB4 may be a new causative gene for KS/IHH, providing a scientific basis for precise clinical diagnosis and treatment.
This study was conducted in accordance with the principles of the Declaration of Helsinki. The Ethics Committee of Hangzhou Children's Hospital approved this study. Written informed consent for publication of the participant's clinical details was obtained from the parent.
ZJ: Writing – original draft, Data curation. YS: Data curation, Supervision, Writing – review & editing. ZY: Data curation, Writing – review & editing. LF: Data curation, Investigation, Writing – review & editing. HaoL: Data curation, Supervision, Writing – review & editing. HanL: Conceptualization, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Scientific research Project plan of Shanghai Municipal Health Commission (202140346).
We would like to thank the patients for their participation in the study as well as their family members for their collaboration.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Boehm U, Bouloux PM, Dattani MT, de Roux N, Dodé C, Dunkel L, et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism–pathogenesis, diagnosis and treatment. Nat Rev Endocrinol. (2015) 11:547–64. doi: 10.1038/nrendo.2015.112
2. Bianco SD, Kaiser UB. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat Rev Endocrinol. (2009) 5:569–76. doi: 10.1038/nrendo.2009.177
3. Liu Q, Yin X, Li P. Clinical, hormonal, and genetic characteristics of 25 Chinese patients with idiopathic hypogonadotropic hypogonadism. BMC Endocr Disord. (2022) 22:30. doi: 10.1186/s12902-022-00940-9
4. Teixeira L, Guimiot F, Dodé C, Fallet-Bianco C, Millar RP, Delezoide AL, et al. Defective migration of neuroendocrine GnRH cells in human arrhinencephalic conditions. J Clin Invest. (2010) 120:3668–72. doi: 10.1172/JCI43699
5. Quinton R, Duke VM, Robertson A, Kirk JM, Matfin G, de Zoysa PA, et al. Idiopathic gonadotrophin deficiency: genetic questions addressed through phenotypic characterization. Clin Endocrinol. (2001) 55:163–74. doi: 10.1046/j.1365-2265.2001.01277.x
6. Kim SH. Congenital hypogonadotropic hypogonadism and Kallmann syndrome: past, present, and future. Endocrinol Metab (Seoul Korea). (2015) 30:456–66. doi: 10.3803/EnM.2015.30.4.456
7. Lewkowitz-Shpuntoff HM, Hughes VA, Plummer L, Au MG, Doty RL, Seminara SB, et al. Olfactory phenotypic spectrum in idiopathic hypogonadotropic hypogonadism: pathophysiological and genetic implications. J Clin Endocrinol Metab. (2012) 97:E136–44. doi: 10.1210/jc.2011-2041
8. Poch A, Louden ED, Kim H-G, Ben-Mahmoud A, Kim S-H, Layman LC. Genetics of hypogonadotropic Hypogonadism—Human and mouse genes, inheritance, oligogenicity, and genetic counseling. Mol Cell Endocrinol. (2021) 534, 111334. doi: 10.1016/j.mce.2021.111334
9. Beate K, Joseph N, Nicolas de R, Wolfram K. Genetics of isolated hypogonadotropic hypogonadism: role of GnRH receptor and other genes. Int J Endocrinol. (2012) 2012:147893. doi: 10.1155/2012/147893
10. Quaynor SD, Kim HG, Cappello EM, Williams T, Chorich LP, Bick DP, et al. The prevalence of digenic mutations in patients with normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Fertil Steril. (2011) 96:1424–1430.e6. doi: 10.1016/j.fertnstert.2011.09.046
11. Amato LGL, Montenegro LR, Lerario AM, Jorge AAL, Guerra Junior G, Schnoll C, et al. New genetic findings in a large cohort of congenital hypogonadotropic hypogonadism. Eur J Endocrinol. (2019) 181:103–19. doi: 10.1530/EJE-18-0764
12. Dodé C, Teixeira L, Levilliers J, Fouveaut C, Bouchard P, Kottler ML, et al. Kallmann syndrome: mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PloS Genet. (2006) 2:e175. doi: 10.1371/journal.pgen.0020175
13. Luo H, Zheng R, Zhao Y, Wu J, Li J, Jiang F, et al. A dominant negative FGFR1 mutation identified in a Kallmann syndrome patient. Gene. (2017) 621:1–4. doi: 10.1016/j.gene.2017.04.017
14. Wang Y, Gong C, Qin M, Liu Y, Tian Y. Clinical and genetic features of 64 young male paediatric patients with congenital hypogonadotropic hypogonadism. Clin Endocrinol. (2017) 87:757–66. doi: 10.1111/cen.13451
15. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
16. Thompson JD, Gibson TJ, Higgins DG. Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinf. (2002) Chapter 2:Unit 2.3. doi: 10.1002/0471250953.bi0203s00
17. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. (2010) 7:248–9. doi: 10.1038/nmeth0410-248
18. Schwarz JM, Rödelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. (2010) 7:575–6. doi: 10.1038/nmeth0810-575
19. Tseng CS, Chou SJ, Huang YS. CPEB4-dependent neonate-born granule cells are required for olfactory discrimination. Front Behav Neurosci. (2019) 13:5. doi: 10.3389/fnbeh.2019.00005
20. Tsai LY, Chang YW, Lin PY, Chou HJ, Liu TJ, Lee PT, et al. CPEB4 knockout mice exhibit normal hippocampus-related synaptic plasticity and memory. PloS One. (2013) 8:e84978. doi: 10.1371/journal.pone.0084978
21. Tseng CS, Chao HW, Huang HS, Huang YS. Olfactory-Experience- and Developmental-Stage-Dependent Control of CPEB4 Regulates c-Fos mRNA Translation for Granule Cell Survival. Cell Rep. (2017) 21:2264–76. doi: 10.1016/j.celrep.2017.10.100
22. Girondel C, Meloche S. Interleukin-17 receptor D in physiology, inflammation and cancer. Front Oncol. (2021) 11:656004. doi: 10.3389/fonc.2021.656004
23. Fürthauer M, Lin W, Ang SL, Thisse B, Thisse C. Sef is a feedback-induced antagonist of Ras/MAPK-mediated FGF signalling. Nat Cell Biol. (2002) 4:170–4. doi: 10.1038/ncb750
24. Tsang M, Friesel R, Kudoh T, Dawid IB. Identification of Sef, a novel modulator of FGF signalling. Nat Cell Biol. (2002) 4:165–9. doi: 10.1038/ncb749
25. Fuchs Y, Brunwasser M, Haif S, Haddad J, Shneyer B, Goldshmidt-Tran O, et al. Sef is an inhibitor of proinflammatory cytokine signaling, acting by cytoplasmic sequestration of NF-κB. Dev Cell. (2020) 55:514. doi: 10.1016/j.devcel.2020.10.022
26. Miraoui H, Dwyer AA, Sykiotis GP, Plummer L, Chung W, Feng B, et al. Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. Am J Hum Genet. (2013) 92:725–43. doi: 10.1016/j.ajhg.2013.04.008
27. Ron D, Fuchs Y, Chorev DS. Know thy Sef: a novel class of feedback antagonists of receptor tyrosine kinase signaling. Int J Biochem Cell Biol. (2008) 40:2040–52. doi: 10.1016/j.biocel.2008.03.013
28. Wang D, Niu Y, Tan J, Chen Y, Xu H, Ling Q, et al. Combined in vitro and in silico analyses of FGFR1 variants: genotype-phenotype study in idiopathic hypogonadotropic hypogonadism. Clin Genet. (2020) 98:341–52. doi: 10.1111/cge.13814
29. Men M, Wang X, Wu J, Zeng W, Jiang F, Zheng R, et al. Prevalence and associated phenotypes of DUSP6, IL17RD and SPRY4 variants in a large Chinese cohort with isolated hypogonadotropic hypogonadism. J Med Genet. (2021) 58:66–72. doi: 10.1136/jmedgenet-2019-106786
30. Zhou C, Niu Y, Xu H, Li Z, Wang T, Yang W, et al. Mutation profiles and clinical characteristics of Chinese males with isolated hypogonadotropic hypogonadism. Fertil Steril. (2018) 110:486–495.e5. doi: 10.1016/j.fertnstert.2018.04.010
31. Louden ED, Poch A, Kim HG, Ben-Mahmoud A, Kim SH, Layman LC. Genetics of hypogonadotropic Hypogonadism-Human and mouse genes, inheritance, oligogenicity, and genetic counseling. Mol Cell Endocrinol. (2021) 534:111334. doi:10.1016/j.mce.2021.111334
32. Lin W, Fürthauer M, Thisse B, Thisse C, Jing N, Ang SL. Cloning of the mouse Sef gene and comparative analysis of its expression with Fgf8 and Spry2 during embryogenesis. Mech Dev. (2002) 113:163–8. doi: 10.1016/S0925-4773(02)00018-7
33. Tay J, Richter JD. Germ cell differentiation and synaptonemal complex formation are disrupted in CPEB knockout mice. Dev Cell. (2001) 1:201–13. doi: 10.1016/S1534-5807(01)00025-9
34. Shin J, Salameh JS, Richter JD. Impaired neurodevelopment by the low complexity domain of CPEB4 reveals a convergent pathway with neurodegeneration. Sci Rep. (2016) 6:29395. doi: 10.1038/srep29395
35. Ivshina M, Lasko P, Richter JD. Cytoplasmic polyadenylation element binding proteins in development, health, and disease. Annu Rev Cell Dev Biol. (2014) 30:393–415. doi: 10.1146/annurev-cellbio-101011-155831
36. Novoa I, Gallego J, Ferreira PG, Mendez R. Mitotic cell-cycle progression is regulated by CPEB1 and CPEB4-dependent translational control. Nat Cell Biol. (2010) 12:447–56. doi: 10.1038/ncb2046
Keywords: Kallmann syndrome, delayed pubertal development, gene variant, IL17RD, CPEB4
Citation: Zhang J, Yang S, Zhang Y, Liu F, Hao L and Han L (2024) Clinical phenotype of a Kallmann syndrome patient with IL17RD and CPEB4 variants. Front. Endocrinol. 15:1343977. doi: 10.3389/fendo.2024.1343977
Received: 24 November 2023; Accepted: 21 February 2024;
Published: 02 April 2024.
Edited by:
Christine Rampon, Université Paris Cité, FranceReviewed by:
Leticia Silveira, Federal University of Minas Gerais, BrazilCopyright © 2024 Zhang, Yang, Zhang, Liu, Hao and Han. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lianshu Han, aGFubGlhbnNodUB4aW5odWFtZWQuY29tLmNu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.